
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective synthesis of the resveratrol analogue 4,4′-dihydroxy-trans-stilbene and stilbenoids modification by fungal peroxygenases†
Carmen
Aranda
a,
René
Ullrich
b,
Jan
Kiebist
c,
Katrin
Scheibner
c,
José C.
del Río
 a,
Martin
Hofrichter
b,
Angel T.
Martínez
d and
Ana
Gutiérrez
*a
a,
Martin
Hofrichter
b,
Angel T.
Martínez
d and
Ana
Gutiérrez
*a
aInstituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, Reina Mercedes 10, E-41012 Seville, Spain. E-mail: anagu@irnase.csic.es
bDepartment of Bio- and Environmental Sciences, TU Dresden, Markt 23, 02763 Zittau, Germany
cJenaBios GmbH, Löbstedter Str. 80, 07749 Jena, Germany
dCentro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, E-28040 Madrid, Spain
First published on 17th April 2018
Abstract
This work gives first evidence that the unspecific peroxygenases (UPOs) from the basidiomycetes Agrocybe aegerita (AaeUPO), Coprinopsis cinerea (rCciUPO) and Marasmius rotula (MroUPO) are able to catalyze the regioselective hydroxylation of trans-stilbene to 4,4′-dihydroxy-trans-stilbene (DHS), a resveratrol (RSV) analogue whose preventive effects on cancer invasion and metastasis have very recently been shown. Nearly complete transformation of substrate (yielding DHS) was achieved with the three enzymes tested, using H2O2 as the only co-substrate, with AaeUPO showing exceptionally higher total turnover number (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) than MroUPO (26000) and rCciUPO (1400). Kinetic studies demonstrated that AaeUPO was the most efficient enzyme catalyzing stilbene dihydroxylation with catalytic efficiencies (kcat/Km) one and two orders of magnitude higher than those of MroUPO and rCciUPO, so that 4-hydroxystilbene appears to be the best UPO substrate reported to date. In contrast, the peroxygenase from the ascomycete Chaetomium globosum (CglUPO) failed to hydroxylate trans-stilbene at the aromatic ring and instead produced the trans-epoxide in the alkenyl moiety. In addition, stilbenoids such as pinosylvin (Pin) and RSV were tested as substrates for the enzymatic synthesis of RSV from Pin and oxyresveratrol (oxyRSV) from both RSV and Pin. Overall, lower conversion rates and regioselectivities compared with trans-stilbene were accomplished by three of the UPOs, and no conversion was observed with CglUPO. The highest amount of RSV (63% of products) and oxyRSV (78%) were again attained with AaeUPO. True peroxygenase activity was demonstrated by incorporation of 18O from H218O2 into the stilbene hydroxylation products. Differences in the number of phenylalanine residues at the heme access channels seems related to differences in aromatic hydroxylation activity, since they would facilitate substrate positioning by aromatic-aromatic interactions. The only ascomycete UPO tested (that of C. globosum) turned out to have the most differing active site (distal side of heme cavity) and reactivity with stilbenes resulting in ethenyl epoxidation instead of aromatic hydroxylation. The above oxyfunctionalizations by fungal UPOs represent a novel and simple alternative to chemical synthesis for the production of DHS, RSV and oxyRSV.
000) than MroUPO (26000) and rCciUPO (1400). Kinetic studies demonstrated that AaeUPO was the most efficient enzyme catalyzing stilbene dihydroxylation with catalytic efficiencies (kcat/Km) one and two orders of magnitude higher than those of MroUPO and rCciUPO, so that 4-hydroxystilbene appears to be the best UPO substrate reported to date. In contrast, the peroxygenase from the ascomycete Chaetomium globosum (CglUPO) failed to hydroxylate trans-stilbene at the aromatic ring and instead produced the trans-epoxide in the alkenyl moiety. In addition, stilbenoids such as pinosylvin (Pin) and RSV were tested as substrates for the enzymatic synthesis of RSV from Pin and oxyresveratrol (oxyRSV) from both RSV and Pin. Overall, lower conversion rates and regioselectivities compared with trans-stilbene were accomplished by three of the UPOs, and no conversion was observed with CglUPO. The highest amount of RSV (63% of products) and oxyRSV (78%) were again attained with AaeUPO. True peroxygenase activity was demonstrated by incorporation of 18O from H218O2 into the stilbene hydroxylation products. Differences in the number of phenylalanine residues at the heme access channels seems related to differences in aromatic hydroxylation activity, since they would facilitate substrate positioning by aromatic-aromatic interactions. The only ascomycete UPO tested (that of C. globosum) turned out to have the most differing active site (distal side of heme cavity) and reactivity with stilbenes resulting in ethenyl epoxidation instead of aromatic hydroxylation. The above oxyfunctionalizations by fungal UPOs represent a novel and simple alternative to chemical synthesis for the production of DHS, RSV and oxyRSV.
Introduction
Resveratrol (RSV, 3,5-4′-trihydroxy-trans-stilbene) has emerged in recent years as a fascinating compound because of its wide spectrum of biological effects including protection against metabolic, cardiovascular and other age-related complications such as neurodegeneration and cancer.1 Therefore, a large number of structure–activity studies have been carried out that have revealed the molecular determinants of RSV necessary for its biological effects, such as the hydroxyl group in position 4′ of the aromatic ring, together with the trans-conformation.2 This information was used for the synthesis of RSV analogues such as 4,4′-dihydroxy-trans-stilbene (DHS) with enhanced cytotoxic, anti-proliferative and anti-tumor properties in in vitro experiments.3 These studies demonstrated as well that both 4- and 4′-OH groups are essential for inducing estrogen receptor down-regulation. DHS was identified for the first time among the metabolites of the trans-stilbene excreted in the urine of guinea pigs, rabbits and mice4 and many years later, the cytochrome P450 isoforms involved in the oxidation of trans-stilbene to DHS were identified. Shortly after, DHS was found as a phytoalexin in the bark of Yucca periculosa.5 More recently, the effects of DHS on cancer invasion and metastasis were demonstrated in vivo.6Usually, DHS can be prepared chemically via several syntheses routes including Perkin condensation.7 However, this chemical synthesis involves a sequence of steps, toxic chemicals, and transition metal catalysts with low yield in DHS. With the purpose of meeting the growing demand for stilbenoids, alternative and sustainable approaches for their production are required. The use of enzymes for organic synthesis of fine chemicals and pharmaceuticals has become a powerful alternative to chemical catalysts. Replacing organic/metal catalysts by biocatalysts has several advantages, the most important being excellent regio- and stereo-selectivity, fewer side products, and reduction in environmental impact.
Biocatalysts that preferably catalyze the transfer of an oxygen atom from peroxide to substrates are known as peroxygenases and are classified in a separate sub-subclass, (EC.1.11.2), which was approved in 2011 and at present comprises five members. Among them, the unspecific peroxygenase (UPO, EC 1.11.2.1) is the most prominent one because of its frequency in fungal organisms and promiscuity for oxygen transfer reactions8 that make it highly attractive as industrial biocatalysts.
The first enzyme of this type was discovered in the basidiomycete Agrocybe aegerita.9 UPOs are able to catalyze reactions formerly assigned only to P450s,10 using a similar active site and reaction chemistry.11 However, unlike P450s that are intracellular enzymes and whose activation usually requires an auxiliary enzyme and a source of reducing power, UPOs are secreted proteins, therefore far more stable, and only require H2O2 for activation.12 Similar enzymes have also been found in other basidiomycetes, such as Coprinellus radians13 and Marasmius rotula14 and there are strong indications for their widespread occurrence in the fungal kingdom.15 Over one-hundred peroxygenase-type genes (encoding enzymes of the heme-thiolate peroxidase superfamily) have been identified during the analysis of 24 basidiomycete genomes16 including Coprinopsis cinerea.17 The C. cinerea peroxygenase has not been isolated to date from the fungus, but one of the peroxygenase genes from its genome was heterologously expressed by Novozymes A/S (Bagsvaerd, Denmark). Very recently, another peroxygenase has been found in the cellulolytic ascomycete Chaetomium globosum.18 These peroxygenases have been shown to catalyze numerous interesting oxygenation reactions on aromatic compounds19 and later, their ability to oxyfunctionalize diverse aliphatics including linear20–23 and cyclic alkanes as well as complex substrates such as steroids24 and secosteroids25,26 was demonstrated expanding their biotechnological interest.
In this work, the ability of non-recombinant (hereinafter wild) peroxygenases from A. aegerita (AaeUPO), M. rotula (MroUPO) and C. globosum (CglUPO), and recombinant peroxygenase from C. cinerea (rCciUPO), to catalyze the hydroxylation of trans-stilbene is shown. Additionally, the UPO-catalyzed hydroxylation of pinosylvin (Pin, a stilbenoid present in the heartwood of conifers of the family Pinaceae) and RSV was studied.
Experimental
Enzymes
The MroUPO enzyme is a wild peroxygenase isolated from liquid cultures of M. rotula DSM-25031 (a fungus deposited at the German Collection of Microorganisms and Cell Cultures, Braunschweig). MroUPO was purified by fast protein liquid chromatography (FPLC) to apparent homogeneity, and revealed a molecular mass of 32 kDa and an isoelectric point between pH 5.0 and 5.3. The UV-visible spectrum of the enzyme showed a characteristic maximum at 418 nm (Soret band of heme-thiolate proteins).14 All media and columns used for enzyme isolation were purchased from GE Healthcare Life Sciences.The AaeUPO (A. aegerita isoform II, 46 kDa) is another wild peroxygenase, which was isolated from cultures of A. aegerita grown in soybean-peptone medium, and subsequently purified using a combination of Q-Sepharose and SP-Sepharose and Mono-S ion-exchange chromatographic steps.9
rCciUPO corresponds to the protein model 7249 from the sequenced C. cinerea genome available at the DOE JGI (http://genome.jgi.doe.gov/Copci1). It was expressed in Aspergillus oryzae (patent WO/2008/119780), purified using a combination of S-Sepharose and SP-Sepharose ion-exchange chromatography, and provided by Novozymes A/S (Bagsvaerd, Denmark) as a protein preparation. The recombinant peroxygenase is a glycoprotein with a molecular mass around 44 kDa, a typical UV-vis spectrum with the Soret band at 418 nm, and the ability to oxygenate different aromatic compounds including veratryl alcohol with a specific activity of approx. 100 U mg−1 (measured as described below).
CglUPO (36 kDa) is a third wild peroxygenase recently isolated from cultures of C. globosum DSM-62110 (German Collection of Microorganisms and Cell cultures), and purified by ammonium sulfate precipitation and successive FPLC using Q-Sepharose FF (ion exchange), Superdex75 (size exclusion), and Mono Q (ion exchange) columns.18 All chromatographic steps were accomplished with an ÄKTA purifier FPLC system (GE Healthcare).
The purified proteins were electrophoretically homogenous, as shown by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under denaturing conditions, although some minor contaminant proteins were present in CglUPO (ESI† Fig. S1).
One activity unit (U) is defined as the amount of enzyme oxidizing 1 μmol of veratryl alcohol to veratraldehyde (ε310 9300 M−1 cm−1) in 1 min at 24 °C, pH 7.0 (the optimum pH for benzylic hydroxylation by AaeUPO and some other peroxygenases) after addition of 2.5 mM H2O2.
Model compounds
trans-Stilbene ([E]-1,2-diphenylethene) and some stilbenoids, including Pin (3,5-dihydroxy-trans-stilbene) and RSV (3,5,4′-trihydroxy-trans-stilbene) were tested as substrates of the above UPOs. Additionally, 4-hydroxy-trans-stilbene (4HS), 4,4′-dihydroxy-trans-stilbene (DHS), oxyresveratrol (oxyRSV, 3,5,2′,4′-tetrahydroxy-trans-stilbene) and trans-stilbene epoxide were used as standards in GC-MS and HPLC analyses. Chemical structures of these compounds are shown in ESI† (Fig. S2).Enzyme reactions
Enzymatic reactions were performed at 0.1 mM substrate concentration (1 mL reaction volume) and 30 °C, in 50 mM phosphate buffer (pH 7 was selected for comparison). Prior to use, the substrate was dissolved in acetone and added to the buffer to give a final acetone concentration in the reaction of 20% (v/v) (resulting in substrate solubilization and best reaction rate); reactions without acetone were also performed. Ascorbic acid was added to the reaction mixture (except for CglUPO reactions) to prevent further oxidation of hydroxylated aromatic (phenolic) products and subsequent radical coupling,27 the best results being obtained using 8 mM concentration. Enzyme and H2O2 concentration were optimized for each substrate in the following ranges: AaeUPO (6–500 nM) with 1.2–10 mM H2O2,MroUPO (0.2–1 μM) with 0.5–10 mM H2O2, rCciUPO (0.2–1.2 μM) with 0.5–10 mM H2O2, and CglUPO (0.2–10 μM) with 2.5–10 mM H2O2 (and those resulting in maximal conversion were used). Final H2O2 concentrations were selected for maximal transformation and reaction selectivity. In control experiments, substrates were treated under the same conditions (including H2O2) but without enzyme.After 30–60 min reaction, products of stilbene conversion were extracted with methyl tert-butyl ether and dried under N2. Enzymatic reactions with 18O-labeled hydrogen peroxide (H218O2, 90% isotopic content) from Sigma-Aldrich (2% w:v solution) were performed under the same conditions described above. N,O-Bis(trimethylsilyl)trifluoro-acetamide (Supelco) was used to prepare trimethylsilyl (TMS) derivatives of stilbene reaction products that were analyzed by GC-MS. Due to the relatively higher water solubility of RSV and oxyRSV compared with stilbene (which made it difficult to extract them with apolar solvents), products from Pin and RSV conversion were analyzed in a different way. After 30 or 60 min, reactions were stopped with 200 μL of 50 mM sodium azide solution by vigorous shaking and compounds present were analyzed by HPLC. Definitions of some transformation parameters are provided in ESI.†
Enzyme kinetics
The kinetics of stilbene dihydroxylation was studied in two steps. First, stilbene reactions were carried out in 1 mL vials with 3–200 μM substrate; 32 nM MroUPO; 40 nM rCciUPO; 4 nM AaeUPO; 8 mM ascorbic acid; 20% (v/v) acetone. The second hydroxylation step was studied analogously with 3–800 μM of 4-hydroxy-trans-stilbene; 13 nM MroUPO; 120 nM rCciUPO; 4 nM AaeUPO; 8 mM ascorbic acid; 20% (v/v) acetone. The enzyme concentrations were chosen to obtain similar transformation degrees in both reaction steps. The reactions were initiated with 0.5 mM H2O2 and stopped after 10 s by vigorous shaking with 200 μL of 50 mM sodium azide. This short reaction time was selected to: i) obtain maximal turnover numbers; and ii) avoid further oxidation of the first product 4HS enabling the separate analysis of both reactions. All reactions were carried out in triplicate. Product quantification was carried out by GC-MS using external standard curves, and kinetic parameters (kcat, Km) were obtained by fitting the data to the Michaelis–Menten equation using SigmaPlot software (Systat Softwarwe Inc., San Jose, CA, USA). For total turnover number (TTN) determination of trans-stilbene oxidation, reactions were performed with AaeUPO (13 nM), MroUPO (0.2 μM) and rCciUPO (1.2 μM) and substrate concentration was increased up to 5 mM in the presence of 20% (v/v) of acetone.GC-MS analyses
The analyses were performed with a Shimadzu GC-MS QP 2010 Ultra system, using a fused-silica DB-5HT capillary column (30 m × 0.25 mm internal diameter, 0.1 μm film thickness) from J&W Scientific. The oven was heated from 50 °C (1.5 min) to 90 °C (2 min) at 30 °C min−1, and then from 90 °C to 250 °C (15 min) at 8 °C min−1. The injection was performed at 250 °C and the transfer line was kept at 300 °C. Compounds were identified by comparing their mass spectra and retention times with those of the authentic standards (4HS, DHS, trans-stilbene epoxide) and quantified from total-ion peak areas. Substrate conversion rates and relative molar abundances of products were calculated with relative response factors from chromatographic runs of mixtures of authentic standards. Yields of main products were calculated using standard curves.HPLC analyses
HPLC analyses were performed with a Shimadzu LC-2030C 3D system equipped with a photo diode array detector using a reversed phase column Agilent InfinityLab Poroshell 120 Bonus-RP C18 (4.6 mm diameter by 150 mm length, 2.7 μm particle size). The column was isocratically eluted at 40 °C and 1 mL min−1 with 0.1% phosphoric acid (pH 2.2) and acetonitrile, 90:10, for 2 min, followed by an 18 min linear gradient to 70% acetonitrile, finally held for 3 min. Oxidation products were identified by comparing their retention times and spectral data with authentic standards (Pin, RSV and OxyRSV). Quantification was obtained from peak areas recorded at 303 nm, and substrate conversion rates and relative abundances of products were calculated as described above.
UPO molecular structures
Both the A. aegerita and the M. rotula UPOs have been crystallized and their solved molecular structures are available at the Protein Data Bank (www.rcsb.org/pdb) with ID 2YP1 and 5FUJ, respectively, among other entries. In the present study, homology models for the C. cinerea and C. globosum UPOs were obtained at the SWISS-MODEL protein structure homology-modelling server (https://swissmodel.expasy.org).28,29 PyMOL Molecular Graphics System, ver 2.0 Schrödinger, LLC (https://pymol.org) and Swiss-PdbViewer ver 4.1 (https://spdbv.vital-it.ch) were used to examine and illustrate the above UPOs' molecular structures.Results and discussion
Hydroxylation of stilbene by several UPOs
In the present work, several fungal peroxygenases namely AaeUPO, rCciUPO, MroUPO and CglUPO, were tested for their ability to hydroxylate trans-stilbene using H2O2 as co-substrate (electron acceptor and oxygen source) (Table 1). Complete conversion (100%) was achieved with all UPOs. However, very different regioselectivities were accomplished by the basidiomycetous and ascomycetous UPOs (Fig. 1A). The former (AaeUPO, rCciUPO and MroUPO) hydroxylated selectively the para-positions of the substrate to form 4,4′-dihydroxy-trans-stilbene (DHS) (about 99% of total products), which was identified by comparing GC-MS data (mass spectrum, retention time) with an authentic standard (ESI,† Fig. S3A and S4A-B, respectively). At shorter incubation times the monohydroxylated 4HS was detected (Fig. 1B). Traces of a trihydroxylated-trans-stilbene (THS), most probably formed by hydroxylation of DHS, were also found (Table 1). In contrast, CglUPO did not hydroxylate the aromatic rings and instead formed exclusively the epoxide (trans-stilbene epoxide) in the aliphatic chain of trans-stilbene. trans-Stilbene epoxide was unambiguously identified by GC-MS, comparing its mass spectrum and retention time with an authentic standard (Fig. S5A and S4C-D,† respectively). Product yields (of both DHS and the epoxide) were over 90% for all the UPOs tested. The yield for DHS is much higher than that of chemical synthesis (only 25%) (Scheme S1†). 18O-labeling studies were performed in trans-stilbene reactions using H218O2 as UPO co-substrate to confirm the source of oxygen incorporated during the formation of both, DHS and the epoxide (Fig. S3B and S5B†). Mass spectral analysis of the resulting dihydroxylated TMS-derivatives (Fig. S3†) showed that the characteristic molecular ion fragments had shifted by approx. 90% from the natural abundance at m/z 356 (found in the reaction with ordinary H216O2; Fig. S3A†) to m/z 360 (Fig. S3B;† 10% of the original fragments remained in the H218O2 reactions owing to the 90% isotopic purity of the labeled peroxide). Additionally, in the mass spectrum of the epoxy derivative from the CglUPO reaction, a 90% shift from the natural abundance at m/z 105, m/z 118, m/z 195 and m/z 196 ([M]+) found in the reaction with unlabeled H2O2 to m/z 107, m/z 120, m/z 197 and m/z 198 was ascertained.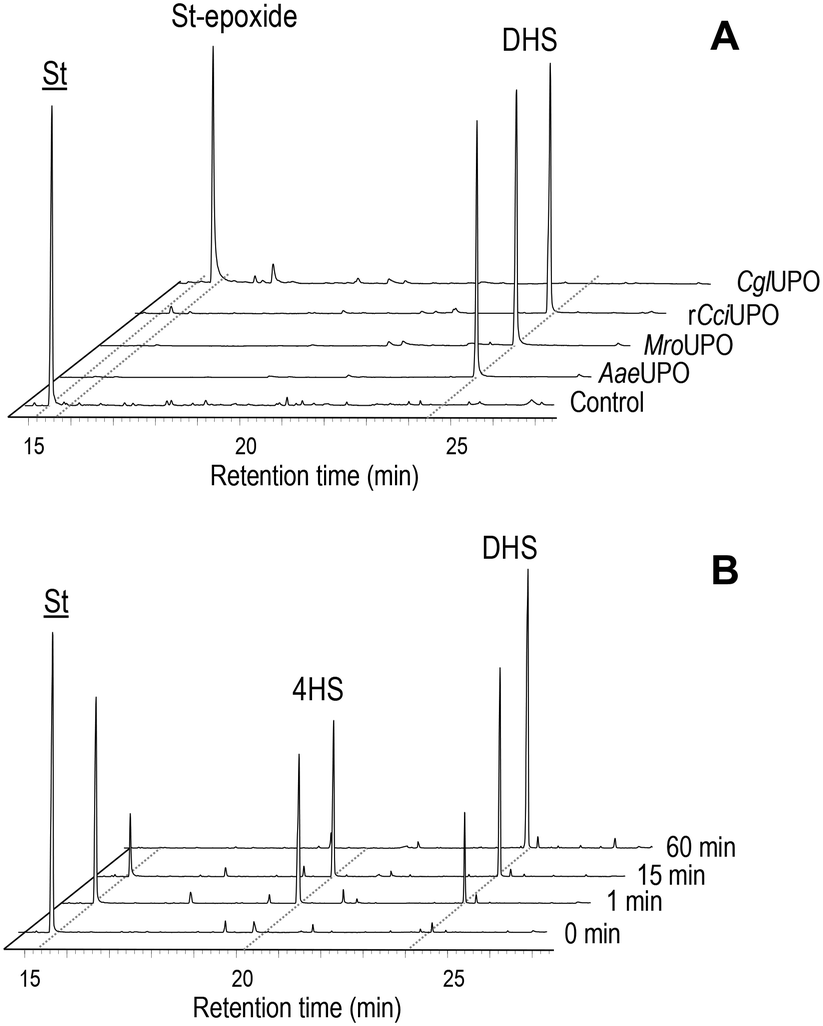 | ||
Fig. 1 GC-MS analysis of trans-stilbene reactions (60 min) with AaeUPO, rCciUPO, MroUPO and CglUPO and control (without enzyme) showing the remaining substrate (![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) ![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif) , trans-stilbene), and the dihydroxylated (DHS, 4,4′-dihydroxy-trans-stilbene) and epoxide (St-epoxide; trans-stilbene epoxide) derivatives (A) and GC-MS analysis of stilbene reactions with AaeUPO at different reaction times (0 min, 1 min, 15 min and 60 min) showing the remaining substrate (), the monohydroxylated (4HS, 4-hydroxy-trans-stilbene) and dihydroxylated (DHS) derivatives (B). , trans-stilbene), and the dihydroxylated (DHS, 4,4′-dihydroxy-trans-stilbene) and epoxide (St-epoxide; trans-stilbene epoxide) derivatives (A) and GC-MS analysis of stilbene reactions with AaeUPO at different reaction times (0 min, 1 min, 15 min and 60 min) showing the remaining substrate (), the monohydroxylated (4HS, 4-hydroxy-trans-stilbene) and dihydroxylated (DHS) derivatives (B). | ||
Kinetics of stilbene oxygenation by UPOs
The estimated kinetic parameters for trans-stilbene hydroxylation by UPOs are summarized in Table 2. AaeUPO was the most efficient enzyme catalyzing the double hydroxylation of trans-stilbene. While the Michaelis–Menten constants (Km) were similar for all enzymes, the turnover numbers (kcat) were much higher for AaeUPO resulting in a 15- and 60-fold increase in the catalytic efficiency (kcat/Km) for the first and second hydroxylation step, respectively. It was also found that 4HS hydroxylation by AaeUPO was more favorable than that of trans-stilbene as it exhibited higher kcat and lower Km values, which increased the catalytic efficiency four times. On the other hand, catalytic efficiencies were roughly in the same range (∼105 M−1 s−1) for both hydroxylation steps in the case of MroUPO and rCciUPO. Compared to most other AaeUPO substrates susceptible to aromatic hydroxylation, the catalytic efficiency for trans-stilbene turned out to be one to two orders of magnitude higher (e.g. for the oxygenation of naphtalene and propranolol, 5 × 105 and 4 × 104 M−1 s−1, respectively, were reported).30,31 This fact can be explained by the higher Km values observed for these substrates reflecting lower affinities to the enzyme compared to trans-stilbene. Similar applies to stilbene hydroxylation by MroUPO, which gave a catalytic efficiency one order of magnitude higher than that reported for naphtalene.14| k cat (s−1) | K m (μM) | k cat/Km (M−1 s−1) | |
|---|---|---|---|
| St → 4HS | |||
| AaeUPO | 85.7 ± 8.5 | 20.8 ± 5.6 | 4.1 ± 1.2 × 106 |
| rCciUPO | 9.1 ± 0.8 | 30.5 ± 7.0 | 3.0 ± 0.7 × 105 |
| MroUPO | 4.2 ± 0.2 | 15.3 ± 2.5 | 2.7 ± 0.5 × 105 |
| 4HS → DHS | |||
| AaeUPO | 225 ± 48.3 | 14.1 ± 7.7 | 1.6 ± 0.9 × 107 |
| rCciUPO | 6.6 ± 1.3 | 18.9 ± 8.5 | 3.5 ± 0.8 × 105 |
| MroUPO | 11.7 ± 0.3 | 46.4 ± 2.3 | 2.5 ± 0.1 × 105 |
The catalytic differences between the UPOs oxyfunctionalizing trans-stilbene were supported by their reaction performance. In the case of AaeUPO, TTN for trans-stilbene hydroxylation (982 μM for DHS and 691 μM for 4HS) raised up to 200000, while using MroUPO and rCciUPO, this number was 8- and 143-fold lower, respectively (Table 3). Thus, AaeUPO displayed a comparably high TTN for the aromatic oxygenation of trans-stilbene as for the benzylic hydroxylation of alkylbenzenes, for which a TTN of 110000 was achieved.14 Very recently, the synthesis of stilbenoid derivatives by a cytochrome P450 monooxygenase (CYP154E1) from Thermobifida fusca and some of its variants has been reported.32 The enzyme showed a similar para-hydroxylating regioselectivity for trans-stilbene and similar conversion rates as reported herein for the UPOs but with considerably lower TTN (4800) as compared with AaeUPO (200000) and MroUPO (25000). Comparing kinetic data, CYP154E1 and its variants exhibited lower affinities (higher Km values) and turnover numbers, and hence a thousand-times lower catalytic efficiency compared to AaeUPO.
| TTN | TOF (s−1) | |
|---|---|---|
| AaeUPO | 200000 |
55.6 |
| rCciUPO | 1400 | 0.4 |
| MroUPO | 26000 |
7.2 |
Hydroxylation of stilbene analogs by several UPOs
In addition to DHS synthesis from trans-stilbene, other stilbenoids (trans-stilbene hydroxylated derivatives) such as Pin and RSV were also tested as UPO substrates (Table 4).| Enzyme | Substrate | Conversion (%) | Products (relative molar %) | ||
|---|---|---|---|---|---|
| RSV | oxyRSV | oxyPin | |||
| a 30 min reactions with 2.5 mM H2O2. b 60 min reactions with 5 mM H2O2. c 30 min reactions with 5 mM H2O2. | |||||
| AaeUPO | |||||
| 0.025 μM | Pina | 96.8 | 62.8 | 35.2 | 2.0 |
| 0.050 μM | Pina | 94.0 | 37.3 | 62.7 | — |
| 0.100 μM | Pinb | 95.9 | 21.8 | 78.2 | — |
| 0.144 μM | RSVa | 71.9 | — | 100 | — |
| rCciUPO | |||||
| 0.1 μM | Pinc | 80.4 | 39.6 | 27.0 | 33.5 |
| 0.1 μM | RSVb | 22.2 | — | 100 | — |
| MroUPO | |||||
| 1 μM | Pinc | 10.7 | 100 | — | — |
| 1 μM | RSVb | 0 | — | — | — |
| CglUPO | |||||
| 10 μM | Pina | 0 | — | — | — |
| 10 μM | RSVb | 0 | — | — | — |
Substrate conversion and product identification was followed/achieved by HPLC (Fig. 2) comparing respective data with authentic standards (Fig. S6†). In Pin reactions, different conversion rates and product patterns were observed in dependence of the particular UPO. While AaeUPO almost completely converted Pin (up to 97%) and rCciUPO did it to large extent (80%), just a low conversion rate (11%) was obtained with MroUPO and no conversion was observed with CglUPO. The conversion of Pin by AaeUPO and rCciUPO yielded RSV that was in turn oxidized to oxyRSV (up to 78% of oxidized product when the highest AaeUPO dose was used) but, in the case of rCciUPO, a significant amount of another monohydroxylated compound tentatively assigned as 2′-hydroxypinosylvin (oxypinosylvin, oxyPin) was detected (being very minor in the AaeUPO reactions). This suggest that AaeUPO is more selective towards the position 4′ of Pin yielding RSV, while similar amounts of RSV and oxyPin are formed by rCciUPO hydroxylation of Pin at the 4′ and 2′ positions, respectively. With MroUPO, RSV was the only product obtained. When using RSV as substrate, up to 72% and 22% conversion was attained with AaeUPO and rCciUPO, respectively, but no conversion was observed with CglUPO and MroUPO. The reaction of RSV with the two former enzymes generated oxyRSV.
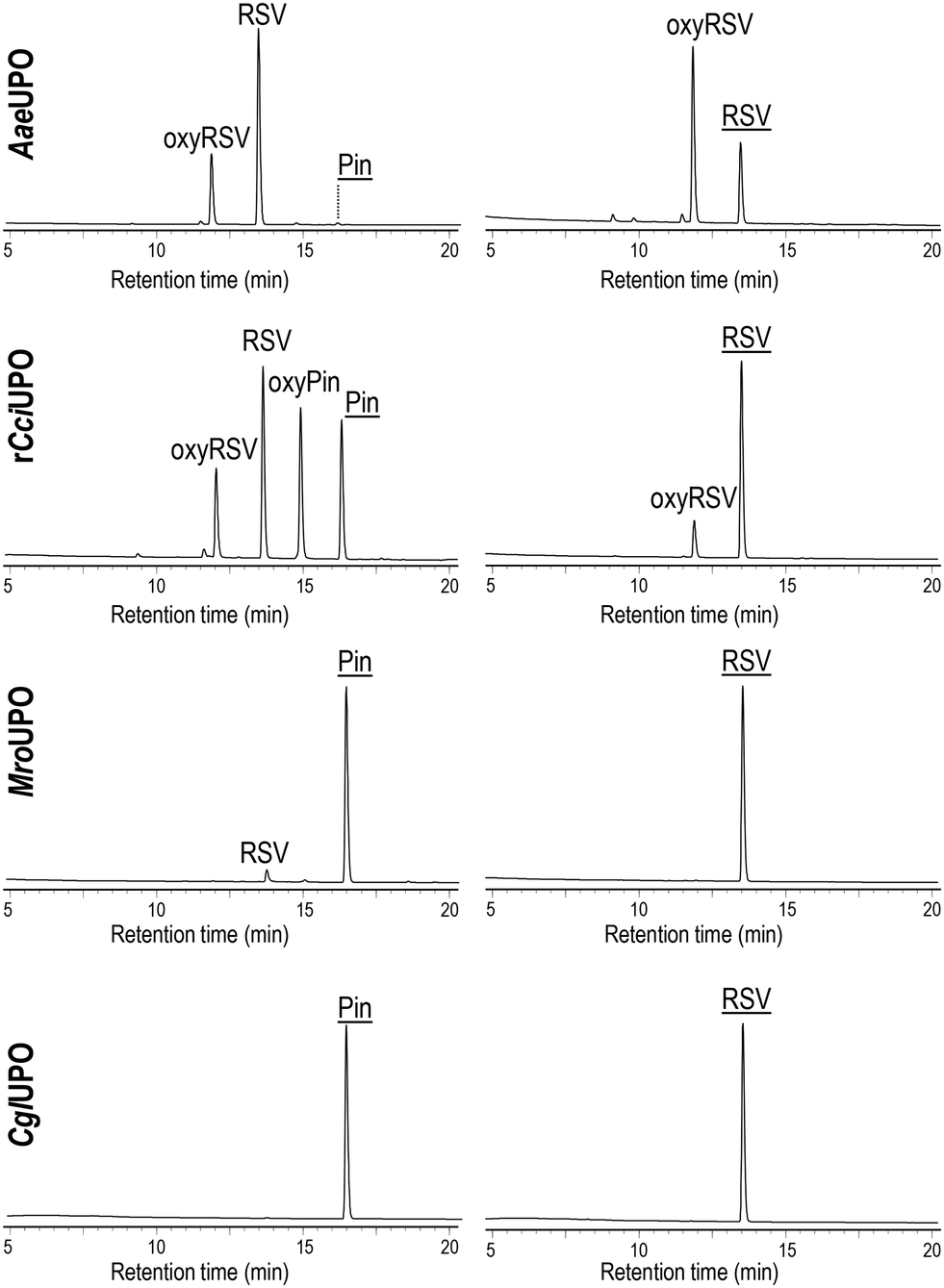 | ||
Fig. 2 HPLC analysis of pinosylvin (Pin, left) and resveratrol (RSV, right) reactions with AaeUPO, rCciUPO, MroUPO and CglUPO showing the remaining substrates (![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) ![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif) , , ![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) ![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif) ) and the products, RSV, oxyresveratrol (oxyRSV), and the trihydroxylated derivative of Pin (oxyPin). ) and the products, RSV, oxyresveratrol (oxyRSV), and the trihydroxylated derivative of Pin (oxyPin). | ||
Molecular architecture vs. UPO activity
The above differences in the regioselectivity and kinetic constants for stilbene oxidation (Tables 1 and 2) should be relatable to differences in the UPOs' molecular structure affecting the specific residues putatively involved in catalysis. These would be located at: i) the access channel leading to the buried heme cofactor and its surface opening; and ii) the enzyme active site itself, i.e. the heme pocket that follows the access channel.Besides the obvious difference in molecular size – ‘long’ AaeUPO and rCciUPO (Fig. 3 left) consist of 325 and 337 amino acids (mature protein), respectively, while ‘short’ MroUPO and CglUPO (Fig. 3 right) only comprise 236 and 242 residues, respectively8 – the most pronounced difference is the distribution of solvent-exposed phenylalanines. AaeUPO possesses four phenylalanine residues (Phe69, Phe191, Phe199 and Phe274) at the entrance of the heme access channel, which make it rather hydrophobic and affine to aromatic rings, while MroUPO and rCciUPO only have one (Fig. 3). It is conceivable that aromatic interactions between the substrate rings and these four phenylalanine residues may contribute to optimal positioning of stilbene (and later of 4HS) with the para-position at reaction distance of the ferryl oxygen (Fe4+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of activated heme (UPO compound I). The same would apply to other stilbenoids investigated in the present study. Less ‘perfect’ positioning of the substrate in the more aliphatic environment of rCciUPO and MroUPO channels would result in lower hydroxylating efficiency both for stilbene (14–15 times lower) and 4HS (45–65 times lower) (Table 2). For MroUPO, the catalytic efficiencies were similar to those reported for other UPO substrates like veratryl alcohol or benzyl alcohol,14 but the catalytic efficiencies of AaeUPO for the oxyfunctionalization of stilbene and especially of 4HS turned out to be much better than the values reported for any other ‘two-electron oxidation’ substrate.9 The oxygenation results also show that UPOs hydroxylate the phenyl moiety of stilbenoids with preference respect to the 4-OH–phenyl moiety. This is revealed by the production of DHS in the stilbene reaction. The complete reaction most probably involves 4HS release from the enzyme and re-entering with the second aromatic ring pointing towards the heme cofactor.
O) of activated heme (UPO compound I). The same would apply to other stilbenoids investigated in the present study. Less ‘perfect’ positioning of the substrate in the more aliphatic environment of rCciUPO and MroUPO channels would result in lower hydroxylating efficiency both for stilbene (14–15 times lower) and 4HS (45–65 times lower) (Table 2). For MroUPO, the catalytic efficiencies were similar to those reported for other UPO substrates like veratryl alcohol or benzyl alcohol,14 but the catalytic efficiencies of AaeUPO for the oxyfunctionalization of stilbene and especially of 4HS turned out to be much better than the values reported for any other ‘two-electron oxidation’ substrate.9 The oxygenation results also show that UPOs hydroxylate the phenyl moiety of stilbenoids with preference respect to the 4-OH–phenyl moiety. This is revealed by the production of DHS in the stilbene reaction. The complete reaction most probably involves 4HS release from the enzyme and re-entering with the second aromatic ring pointing towards the heme cofactor.
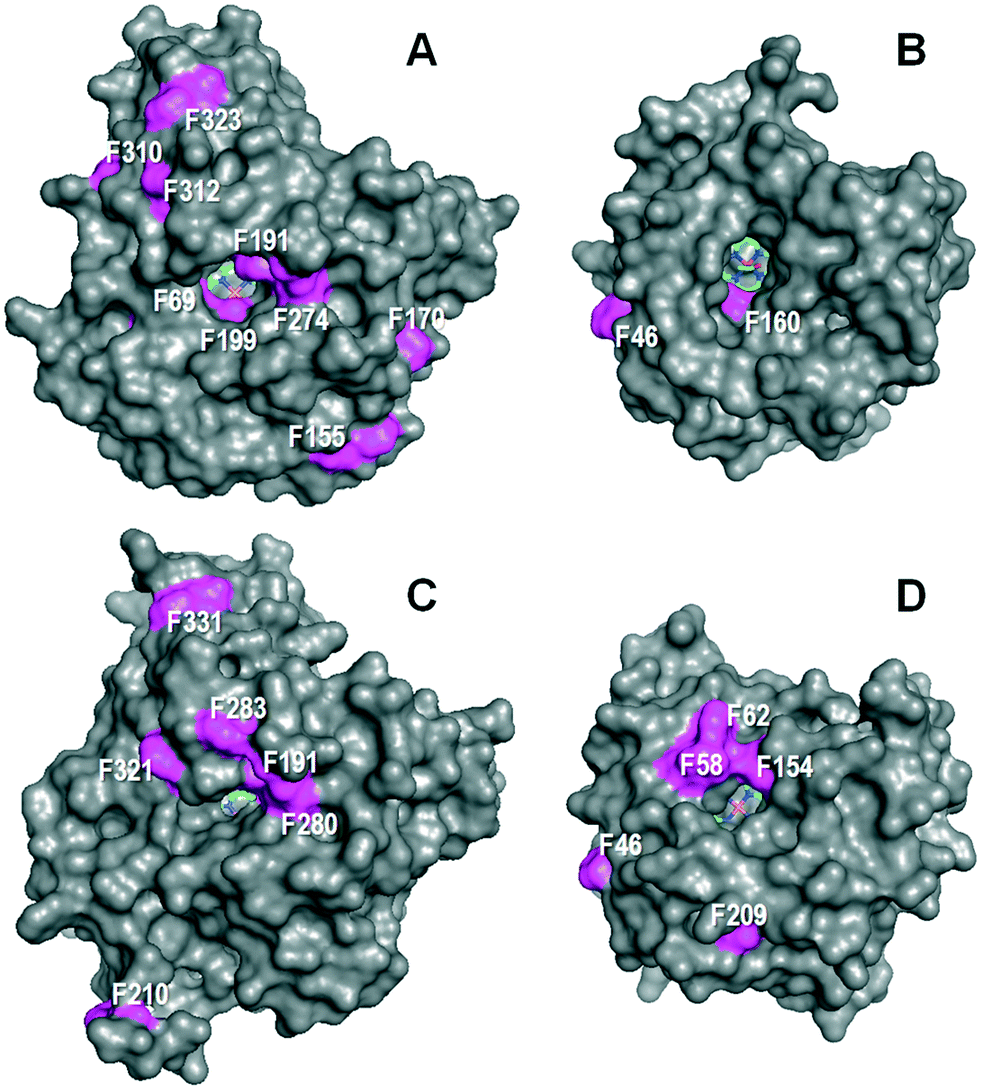 | ||
| Fig. 3 Solvent access surface in AaeUPO (A), MroUPO (B), rCciUPO (C) and CglUPO (D) showing different: i) sizes of the channel giving access to the heme cofactor (sticks); and ii) phenylalanine residues (magenta surface) at the channel edge and other surface regions. Cenital views showing the channels giving access to the distal side of the cofactor pocket on the heme iron atom (one acetate molecule is shown inside the MroUPO channel). From 2YP1 (A), 5FUJ (C) and two homology models (B and D). Residue numbering does not includes the signal peptide. | ||
The regioselectivity of CglUPO in stilbene oxygenation radically differed from that of aromatic oxygenation discussed above, since ethenyl epoxidation was produced. Interestingly, the UPO of the ascomycete C. globosum (A. aegerita, M. rotula and C. cinerea are basidiomycetes), has a noticeably different active site. Of the five residues above the heme plane (Fig. 4), it only shares with AaeUPO the glutamic residue (CglUPO Glu158) putativelly involved in the binding and fission of H2O2 (ref. 8) (also conserved in the other two UPOs). An adjacent residue is also thought to contribute to this reaction (as charge stabilizer), being either an arginine (in AaeUPO and CciUPO; Arg189) or a histidine (in MroUPO and CglUPO; His86 and His88, respectively). CglUPO also shares with MroUPO the absence of a phenylalanine near the heme edge (homologous to AaeUPO Phe121). However, the main peculiarities of CglUPO's active site are: i) the absence of a conserved phenylalanine (homologous to AaeUPO Phe199, although Phe154 occupies a neighbor position) and in particular, ii) the presence of a unique and potentially reactive tyrosine (Tyr162) in the middle of the active site. Whether this tyrosine is responsible for the preferred epoxidation of isolated double bonds within complex molecules – in contrast to other UPOs, CglUPO also specifically epoxidized testosterone18 – will have to be clarified in future mutagenesis studies. Although a yeast expression system for AaeUPO has been developed after enzyme directed evolution,33 new procedures and hosts for expressing wild-type genes of this and other UPOs are required for investigating active-site residues and engineering UPO biocatalysts for selective oxygenation reactions of biotechnological interest.34
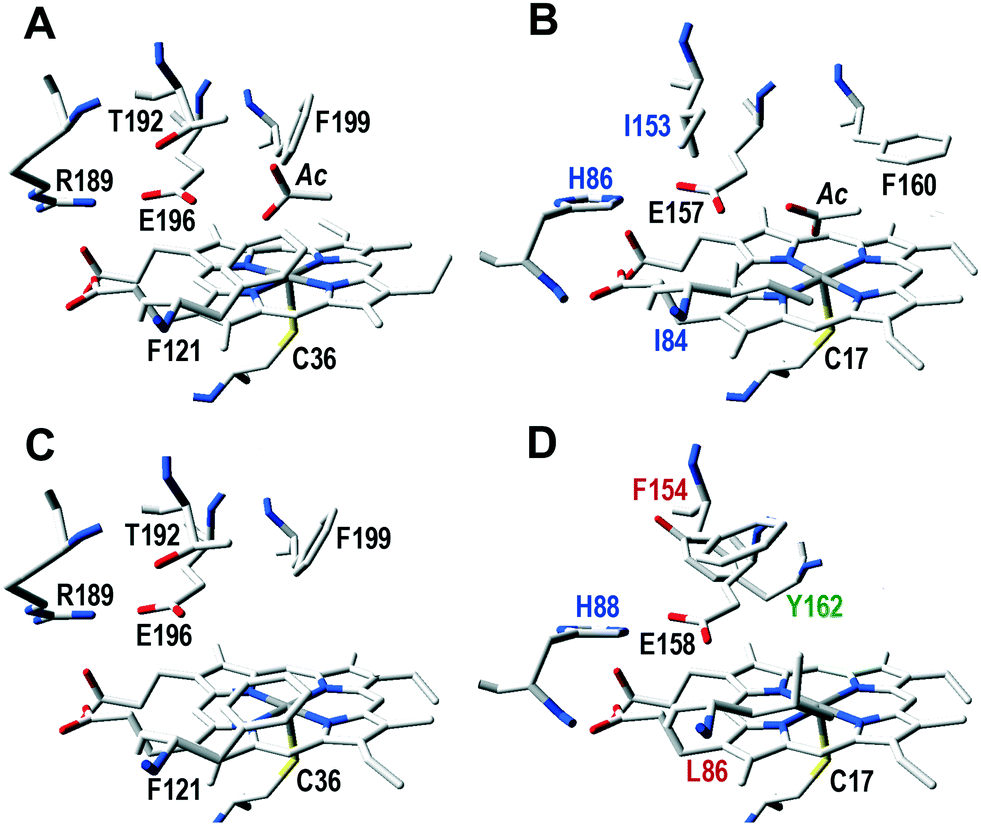 | ||
| Fig. 4 Active site residues, including those putatively involved in catalysis, in AaeUPO (A), and homologous residues in MroUPO (B), rCciUPO (C) and CglUPO (D): five residues above the cofactor (heme pocket “distal” side) plus the cysteine acting as ligand of the heme iron below the cofactor (pocket “proximal” side) are shown, together with an acetate molecule (Ac) at the substrate position in AaeUPO and MroUPO. Black labels correspond to residues as found in AaeUPO, while varying residues have color labels (blue, red or green). See Fig. 3 for model references. | ||
Conclusions
We report here on a new enzymatic route for the production of DHS from trans-stilbene and RSV from Pin (Scheme 1) in one-step by fungal peroxygenases (UPOs). The potent effect of DHS in inhibition of cancer invasion and metastasis has recently been reported “in vivo”.6 Usually, DHS is obtained by chemical synthesis, which involves several-steps, the use of metal catalysts, etc. (Scheme S1†). The enzymatic reaction with UPOs represents a smart and environmentally sound alternative to chemical synthesis. In addition, the UPO-catalyzed synthesis of the potent antioxidants and free radical scavengers RSV (and oxyRSV) from Pin (a natural constituent of pine wood) is also reported here for the first time. A preliminary analysis of the active site cavities and access channels has revealed differences that may explain and affect the reactivity and regioselectivity of the different UPOs in the stilbene oxygenation, but more structural and functional information is required for reaction understanding and enzyme engineering. | ||
| Scheme 1 Enzymatic synthesis of DHS from trans-stilbene (St) and RSV from Pin. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the EnzOx2 (H2020-BBI-PPP-2015-2-1-720297) EU-project, and the BIORENZYMERY (AGL2014-53730-R) project of the Spanish MINECO (co-financed by FEDER). H. Lund (Novozymes) is acknowledged for rCciUPO.References
- S. S. Kulkarni and C. Canto, Biochim. Biophys. Acta, Mol. Basis Dis., 2015, 1852, 1114–1123 CrossRef CAS PubMed.
- L. A. Stivala, M. Savio, F. Carafoli, P. Perucca, L. Bianchi, G. Maga, L. Forti, U. M. Pagnoni, A. Albini, E. Prosperi and V. Vannini, J. Biol. Chem., 2001, 276, 22586–22594 CrossRef CAS PubMed.
- M. Savio, T. Coppa, L. Bianchi, V. Vannini, G. Maga, L. Forti, O. Cazzalini, M. C. Lazze, P. Perucca, E. Prosperi and L. A. Stivala, Int. J. Biochem. Cell Biol., 2009, 41, 2493–2502 CrossRef CAS PubMed.
- J. E. Sinsheimer and R. V. Smith, Biochem. J., 1969, 111, 35–41 CrossRef CAS PubMed.
- P. Torres, J. G. Avila, A. R. de Vivar, A. M. Garcia, J. C. Marin, E. Aranda and C. L. Cespedes, Phytochemistry, 2003, 64, 463–473 CrossRef CAS PubMed.
- M. Savio, D. Ferraro, C. Maccario, R. Vaccarone, L. D. Jensen, F. Corana, B. Mannucci, L. Bianchi, Y. H. Cao and L. A. Stivala, Sci. Rep., 2016, 6, 19973 CrossRef CAS PubMed.
- G. A. Locatelli, M. Savio, L. Forti, I. Shevelev, K. Ramadan, L. A. Stivala, V. Vannini, U. Hubscher, S. Spadari and G. Maga, Biochem. J., 2005, 389, 259–268 CrossRef CAS PubMed.
- M. Hofrichter, H. Kellner, M. J. Pecyna and R. Ullrich, Adv. Exp. Med. Biol., 2015, 851, 341–368 CrossRef CAS PubMed.
- R. Ullrich, J. Nuske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
- R. Bernhardt, J. Biotechnol., 2006, 124, 128–145 CrossRef CAS PubMed.
- K. Piontek, E. Strittmatter, R. Ullrich, G. Gröbe, M. J. Pecyna, M. Kluge, K. Scheibner, M. Hofrichter and D. A. Plattner, J. Biol. Chem., 2013, 288, 34767–34776 CrossRef CAS PubMed.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- D. H. Anh, R. Ullrich, D. Benndorf, A. Svatos, A. Muck and M. Hofrichter, Appl. Environ. Microbiol., 2007, 73, 5477–5485 CrossRef CAS PubMed.
- G. Gröbe, M. Ullrich, M. Pecyna, D. Kapturska, S. Friedrich, M. Hofrichter and K. Scheibner, AMB Express, 2011, 1, 31–42 CrossRef PubMed.
- M. J. Pecyna, R. Ullrich, B. Bittner, A. Clemens, K. Scheibner, R. Schubert and M. Hofrichter, Appl. Microbiol. Biotechnol., 2009, 84, 885–897 CrossRef CAS PubMed.
- D. Floudas, M. Binder, R. Riley, K. Barry, R. A. Blanchette, B. Henrissat, A. T. Martínez, R. Otillar, J. W. Spatafora, J. S. Yadav, A. Aerts, I. Benoit, A. Boyd, A. Carlson, A. Copeland, P. M. Coutinho, R. P. de Vries, P. Ferreira, K. Findley, B. Foster, J. Gaskell, D. Glotzer, P. Górecki, J. Heitman, C. Hesse, C. Hori, K. Igarashi, J. A. Jurgens, N. Kallen, P. Kersten, A. Kohler, U. Kües, T. K. A. Kumar, A. Kuo, K. LaButti, L. F. Larrondo, E. Lindquist, A. Ling, V. Lombard, S. Lucas, T. Lundell, R. Martin, D. J. McLaughlin, I. Morgenstern, E. Morin, C. Murat, M. Nolan, R. A. Ohm, A. Patyshakuliyeva, A. Rokas, F. J. Ruiz-Dueñas, G. Sabat, A. Salamov, M. Samejima, J. Schmutz, J. C. Slot, F. St. John, J. Stenlid, H. Sun, S. Sun, K. Syed, A. Tsang, A. Wiebenga, D. Young, A. Pisabarro, D. C. Eastwood, F. Martin, D. Cullen, I. V. Grigoriev and D. S. Hibbett, Science, 2012, 336, 1715–1719 CrossRef CAS PubMed.
- J. E. Stajich, S. K. Wilke, D. Ahren, C. H. Au, B. W. Birren, M. Borodovsky, C. Burns, B. Canbäck, L. A. Casselton, C. K. Cheng, J. X. Deng, F. S. Dietrich, D. C. Fargo, M. L. Farman, A. C. Gathman, J. Goldberg, R. Guigo, P. J. Hoegger, J. B. Hooker, A. Huggins, T. Y. James, T. Kamada, S. Kilaru, C. Kodira, U. Kües, D. Kupfert, H. S. Kwan, A. Lomsadze, W. X. Li, W. W. Lilly, L. J. Ma, A. J. Mackey, G. Manning, F. Martin, H. Muraguchi, D. O. Natvig, H. Palmerini, M. A. Ramesh, C. J. Rehmeyer, B. A. Roe, N. Shenoy, M. Stanke, V. Ter Hovhannisyan, A. Tunlid, R. Velagapudi, T. J. Vision, Q. D. Zeng, M. E. Zolan and P. J. Pukkila, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11889–11894 CrossRef CAS PubMed.
- J. Kiebist, K. U. Schmidtke, J. Zimmermann, H. Kellner, N. Jehmlich, R. Ullrich, D. Zänder, M. Hofrichter and K. Scheibner, ChemBioChem, 2017, 18, 563–569 CrossRef CAS PubMed.
- M. Hofrichter, R. Ullrich, M. J. Pecyna, C. Liers and T. Lundell, Appl. Microbiol. Biotechnol., 2010, 87, 871–897 CrossRef CAS PubMed.
- E. D. Babot, J. C. del Río, L. Kalum, A. T. Martínez and A. Gutiérrez, Biotechnol. Bioeng., 2013, 110, 2332 CrossRef PubMed.
- A. Olmedo, C. Aranda, J. C. del Río, J. Kiebist, K. Scheibner, A. T. Martínez and A. Gutiérrez, Angew. Chem., Int. Ed., 2016, 55, 12248–12251 CrossRef CAS PubMed.
- A. Gutiérrez, E. D. Babot, R. Ullrich, M. Hofrichter, A. T. Martínez and J. C. del Río, Arch. Biochem. Biophys., 2011, 514, 33–43 CrossRef PubMed.
- S. Peter, M. Kinne, X. Wang, R. Ulrich, G. Kayser, J. T. Groves and M. Hofrichter, Rev. Geophys., 2011, 278, 3667–3675 CAS.
- E. D. Babot, J. C. del Río, M. Cañellas, F. Sancho, F. Lucas, V. Guallar, L. Kalum, H. Lund, G. Gröbe, K. Scheibner, R. Ullrich, M. Hofrichter, A. T. Martínez and A. Gutiérrez, Appl. Environ. Microbiol., 2015, 81, 4130–4142 CrossRef CAS PubMed.
- E. D. Babot, J. C. del Río, L. Kalum, A. T. Martínez and A. Gutiérrez, ChemCatChem, 2015, 7, 283–290 CrossRef CAS.
- F. Lucas, E. D. Babot, J. C. del Río, L. Kalum, R. Ullrich, M. Hofrichter, V. Guallar, A. T. Martínez and A. Gutiérrez, Catal. Sci. Technol., 2016, 6, 288–295 Search PubMed.
- M. Kinne, M. Poraj-Kobielska, E. Aranda, R. Ullrich, K. E. Hammel, K. Scheibner and M. Hofrichter, Bioorg. Med. Chem. Lett., 2009, 19, 3085–3087 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. G. Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
- N. Guex, M. C. Peitsch and T. Schwede, Electrophoresis, 2009, 30, S162–S173 CrossRef PubMed.
- M. Poraj-Kobielska, J. Atzrodt, W. Holla, M. Sandvoss, G. Gröbe, K. Scheibner and M. Hofrichter, J. Labelled Compd. Radiopharm., 2013, 56, 513–519 CrossRef CAS PubMed.
- M. G. Kluge, R. Ullrich, K. Scheibner and M. Hofrichter, Appl. Microbiol. Biotechnol., 2007, 75, 1473–1478 CrossRef CAS PubMed.
- A. Rühlmann, D. Antovic, T. J. J. Müller and V. B. Urlacher, Adv. Synth. Catal., 2017, 359, 984–994 CrossRef.
- P. Molina-Espeja, S. Ma, D. M. Maté, R. Ludwig and M. Alcalde, Enzyme Microb. Technol., 2015, 73–74, 29–33 CrossRef CAS PubMed.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy00272j |
| This journal is © The Royal Society of Chemistry 2018 |