
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Au–Pd NPs immobilised on nanostructured ceria and titania: impact of support morphology on the catalytic activity for selective oxidation†
Motaz
Khawaji
 and
David
Chadwick
*
and
David
Chadwick
*
Department of Chemical Engineering, Imperial College London, Exhibition Road, London SW7 2AZ, UK. E-mail: d.chadwick@imperial.ac.uk
First published on 18th April 2018
Abstract
Bimetallic Au–Pd nanoparticles supported on different ceria and titania nanostructures have been prepared by sol-immobilisation, and evaluated in the solvent-less selective oxidation of benzyl alcohol. The catalysts were characterised by TEM, STEM, XRD, XPS, ICP-AES, and nitrogen adsorption–desorption measurements. The activity of the catalysts was found to be strongly related to the morphology, structure and physiochemical properties of the supports. Au–Pd/ceria nanorods exhibited remarkably high catalytic activity (TOF > 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 h−1), and was found to be considerably more active than Au–Pd/titanate nanotubes, and Au–Pd catalysts supported on conventional ceria and titania nanopowders. The outstanding catalytic performance of Au–Pd/ceria nanorods is attributed to the unique surface chemistry of ceria nanorods, and the ability of catalyst preparation method (i.e. sol-immobilisation) to control the metal particle size and the bimetallic alloy formation. The presence of surface defects and high concentration of oxygen vacancies and Ce3+ in ceria nanorods is likely responsible for the stabilisation of Au–Pd NPs during sol-immobilisation, which led to a very small mean particle size (2.1 nm) corresponding to a dispersion of approximately 52%, and a high surface metal concentration.
900 h−1), and was found to be considerably more active than Au–Pd/titanate nanotubes, and Au–Pd catalysts supported on conventional ceria and titania nanopowders. The outstanding catalytic performance of Au–Pd/ceria nanorods is attributed to the unique surface chemistry of ceria nanorods, and the ability of catalyst preparation method (i.e. sol-immobilisation) to control the metal particle size and the bimetallic alloy formation. The presence of surface defects and high concentration of oxygen vacancies and Ce3+ in ceria nanorods is likely responsible for the stabilisation of Au–Pd NPs during sol-immobilisation, which led to a very small mean particle size (2.1 nm) corresponding to a dispersion of approximately 52%, and a high surface metal concentration.
Introduction
Since it was first reported by Haruta1 and Hutchings,2 the unique catalytic properties of supported gold nanoparticles (NPs) have been extensively studied over the past few decades. Gold has been found to be active for numerous reactions including the oxidation of CO,1 the selective oxidation of alcohols and carbohydrates,3 activation of C–H bonds,4 epoxidation of alkenes,5,6 and the direct synthesis of hydrogen peroxide under mild conditions.7 Furthermore, it is well-known that the catalytic activity of gold can be enhanced in various reactions by alloying gold with palladium.8 For instance, in the selective oxidation of alcohols to aldehydes, alloying Au with Pd leads to up to twenty-five-fold enhancement in the activity while retaining the selectivity.9The catalytic activity of supported gold-based catalysts is highly influenced by the nature of the support and its physiochemical properties, the metal particle size, and the catalyst preparation method. It has been demonstrated that highly-dispersed Au NPs ≤ 5 nm in size typically exhibit the highest catalytic activity.9,10 Although Au and Au–Pd NPs supported on Al2O3, SiO2 and carbon are active for selective oxidation, they are notably more active when deposited on reducible metal oxides such as CeO2, TiO2 and Fe2O3,11,12 which has been attributed to the metal–support interaction and the ability of these materials to activate oxygen molecules. In particular, CeO2 is a very reactive support due to its distinctive redox properties, and ability to reversibly exchange lattice oxygen in response to changes in the oxidation state of Ce atoms between Ce4+ and Ce3+.13 As the size of the ceria particle decreases, the formation of oxygen vacancies and concentration of Ce3+ increases.14,15 Ceria nanostructures with different sizes and morphology expose different facets and display differing catalytic activity for a range of reactions.16–19
Ceria either as a support or a catalyst component is able to modify the reactivity of supported metals.5,19,20 This is particularly true for supported gold and palladium catalysts,20 where the morphology and structure of the support material can immensely influence the catalytic activity. For example, Si et al. studied the activity of Au/CeO2 in the water-gas shift reaction using different nanostructured supports, and found that the activity of the catalyst was strongly dependent on the support morphology.21 Hu et al. investigated the effect of the structure and morphology of ceria on the physicochemical and catalytic properties of Pd/ceria for CO and propane oxidation. Palladium supported on ceria nanorods showed excellent catalytic activity for CO oxidation, while Pd/ceria nano-octahedron was more active for propane oxidation. The observed structure sensitivity was attributed to the differing exposed facets and interface interaction between Pd and CeO2.17 Zhang and co-workers prepared Ce-NR under the protection of nitrogen and utilised the strong reductive nature of Ce-NR to reduce Au and Pd ions in situ into metallic form, thus creating a Au–Pd/Ce-NR catalyst that was shown to be active for the selective oxidation of benzyl alcohol under solvent-free conditions.22
We have recently demonstrated that bimetallic Au–Pd NPs prepared by colloidal synthesis can be immobilised on titanate nanotubes to create a very active selective oxidation catalyst.23 The catalyst preparation method is an important factor that can significantly influence the catalytic performance of Au and Au–Pd catalysts. Indeed, different preparation methods can produce different attachments of Au NPs on the support.24 Sol-immobilisation offers a major advantage over traditional preparation methods in that it allows for controlling the metal particle size. Sol-immobilisation has been used extensively to prepare active Au–Pd catalysts on conventional oxide supports25 and carbons.26
In the present paper, we have investigated the influence of the structure and morphology of ceria and titania supports on the catalytic activity for selective oxidation. All the catalysts were prepared by the identical method: sol-immobilisation of Au–Pd NPs on Na-free nanostructured supports (i.e. ceria nanorods and titanate nanotubes) and conventional CeO2 and TiO2 nanopowders. The catalysts were tested in the solvent-less selective oxidation of benzyl alcohol, which is widely used as a model selective oxidation reaction for the study of supported Au–Pd catalysts.9,27 We demonstrate that Au–Pd NPs supported on the nanostructured supports, viz ceria nanorods and titanate nanotubes, are more active for selective oxidation than when supported on their conventional counterpart nanopowders. Moreover, it is shown that the ceria supported catalysts due to their unique surface chemistry have greater activity for this reaction than the titania supported catalysts. Indeed, the use of ceria nanorods as a support is shown to lead to the most active selective oxidation catalyst. The superior catalytic activity of this new catalyst can be attributed to a combination of the physiochemical properties of the ceria nanorod support and the preparation method, which allows a high degree of control over the Au–Pd particle size and facilitates high metal dispersion, and a high concentration of Au–Pd over the exterior surface of the nanorods. In particular, the unique redox properties of Ce-NR surface contribute to the outstanding catalytic activity of Au–Pd/Ce-NR for selective oxidation.
Experimental
Materials
All metal precursors and chemical reagents were purchased from Sigma-Aldrich, and used as received: NaOH (99.99% trace metals basis), H2SO4 (≥97.5% purity), TiO2 (Aeroxide® P25), TiO2 (anatase, 99.7%), Ce(NO3)3·6H2O, cerium(IV) oxide nanopowder (<25 nm particle size), poly(vinyl alcohol) (PVA, Mw 9000–10000, 80% hydrolysed), HAuCl4·3H2O (99.999% purity), PdCl2 (99.999% purity), NaBH4 (Aldrich ≥98.0%), Benzyl alcohol (99.8% purity). O2 (100% pure) for catalytic tests was supplied by BOC.
Catalyst synthesis
Ceria nanorods were prepared by alkaline hydrothermal treatment method reported previously.28 Typically, 0.6 g of Ce(NO3)3·6H2O was added to a 40 mL of NaOH solution of 15 M and stirred for 10 min in a 45 mL PTFE-lined autoclave. The autoclave was subsequently placed in an air-circulating oven at 100 °C for 24 h, allowing for an even temperature distribution throughout the autoclave. Following the hydrothermal synthesis, the autoclave was allowed to cool down to ambient temperature. The powder obtained was filtered, washed several times with both deionised water and a deionised water–ethanol mixture (4:1 ratio in volume), and dried overnight at 120 °C. The dried powder was calcined at 400 °C for 4 h in synthesis air with a flow rate of 100 mL min−1, and a heating rate of 10 °C min−1.
Titanate nanotubes (Ti-NTs) were synthesised by the alkaline hydrothermal treatment method described Kasuga et al.29 In a typical synthesis, 11 g of TiO2 (anatase nanopowder) was added to 185 mL of 10 M NaOH in a 200 mL PTFE-liner and stirred for two hours. The PTFE-lined steel autoclave was then placed in an air-circulating oven at 140 °C for 24 hours. The obtained slurry was then washed with DI water, filtered and dried overnight at 120 °C. The dried powder was washed twice with 0.1 M H2SO4 and several times with distilled water until pH 7 is reached. The obtained powder was subsequently filtered and dried at 100 °C overnight.
The Au–Pd colloid was prepared according to previously reported procedures.23,25 The different supports were acidified to a pH below the point of zero charge (PZC) by the drop-wise addition of 1.0 M solution of H2SO4. Ti-NTs and TiO2 were acidified to pH ∼ 1.8, while CeO2 and Ce-NRs were acidified to pH ∼ 4.0, and 3.0, respectively. The Au–Pd colloid was generated by dissolving calculated amounts of HAuCl4·3H2O and PdCl2 in 100 mL of DI water at 5 °C while stirring vigorously. Subsequently, 2400 mg of 1.0 wt% PVA solution was added to the metal precursors solution and stirred for 15 minutes. PVA was used as a stabiliser and the weight ratio of PVA/(Au + Pd) was 1.2. The metal precursors were reduced by the addition of 7.5 mL of 0.1 M NaBH4 (molar ratio of NaBH4:(Au + Pd) = 5:1). The metal colloid was left stirring at 1500 rpm for one hour before the acidified supports were added to form a slurry. The slurry was stirred for one hour before it was filtered and washed with DI water several times until the final pH of the mother liquor reached ∼7.0. The obtained catalysts were subsequently dried overnight at 100 °C, and then refluxed in hot water (90 °C) for 60 minutes, filtered and dried overnight at 100 °C. The dried catalysts were used as is without any further treatment. All the catalysts prepared in this study had a nominal metal loading of 2 wt% with a Au:Pd weight ratio equal to 1.0 (Au:Pd atomic ratio = 1:1.86).
Reaction procedure
Benzyl alcohol oxidation was carried out in a batch reactor using 25 mL glass-lined minclaves (Buchiglas, Switerland). Typically, 10 g of benzyl alcohol and the requisite amount of catalyst were charged to the reactor before it was purged five times with O2. The reactor was subsequently pressurised with O2 to the required pressure at room temperature. The reactor was heated in an oil bath set at the desired temperature. An oxygen reservoir was connected to the reactor to replenish any oxygen consumed by the reaction. The desired reactor pressure was maintained via a forward-pressure regulator and a series of check-valves. Samples (200 μL) were collected periodically from the reactor via a sampling valve and centrifuged at 5000 rpm for 15 minutes to separate out the catalyst. A sample from the supernatant (100 μL) was taken out, and added to 500 μL acetonitrile and 100 μL of n-butanol (internal standard) prior to product analysis. Analysis of the reaction products was carried out using a GC (Shimadzu GC-2014) fitted with a flame ionization detector (FID), and a wax column (Agilent CP WAX 52 CB UltiMetal, L = 25 m, ID = 0.53 mm, film thinness = 2.0 μm). Carbon balance was 95 ± 5%.Catalyst characterisation
Catalysts were analysed by PANalytical X'Pert Pro multi-purpose diffractometer using Cu-Kα radiation. The analysis was performed over a scan angle of 5–70° 2θ, and a step size of 0.0167° s−1. Nitrogen adsorption–desorption measurements were carried out at −196 °C using a Micromeritics TriStar II. Prior to carrying out any measurements, all samples were degased at 130 °C overnight. Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) were used to measure the specific surface area (SBET), pore volume and pore diameter. The bulk metal loadings were determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES, PE Optima 2000 DV). Typically, 10 mg of catalyst was dissolved in 15 mL of aqua regia, sonicated for 2 hours, and left overnight in order to dissolve completely any remaining solids. The samples were diluted with 20 mL of DI water before analysis. The instrument was calibrated with authenticated standards containing predetermined amounts of each metal. Bright-field TEM and high-angle annular dark-field STEM images were obtained using an FEI Titan ST electron microscope operated at 300 kV, and JEOL JEM-2100F microscope operating at 200 kV. The average particle size and distribution of the metal NPs were determined from the TEM and STEM images by analysing 100 randomly selected metal particles. X-ray photoelectron spectroscopy (XPS) measurements were recorded using a Thermo K-Alpha Spectrometer equipped with Al Kα source gun. Samples were mounted on double-sided adhesive tape, and the spectra were collected using an X-ray spot size of 400 μm and a pass energy of 20 eV with 0.1 eV increments. The binding energies (BE) were referenced to the C 1s peak of adventitious carbon at 284.8 eV. Data analysis and peak fitting was performed using Avantage software from Thermo Scientific. The PZC of the commercial and hydrothermally-synthesised supports was determined by measuring the zeta potential of the materials as a function of pH using Brookhaven ZetaPALS Potential Analyser. The different supports were dispersed in aqueous solutions of HNO3 and KOH at varying pH values ranging from 2 to 11 at 25 °C.Results and discussion
Catalysts preparation and characterization
The as-synthesised Ti-NT and Ce-NR were analysed by XRD, nitrogen adsorption–desorption measurements, and TEM. The characterisation of Ti-NT and Ce-NR supports is given in the ESI.† The formation of Ti-NT is characterised by the emergence of a broad angle peak between 7.2° and 10.3° 2θ in the XRD pattern (Fig. S1†).30 The peaks appearing at 24.5° and 48.6° are also indicative of tri-titanate 1D nanomaterial.31 Ceria nanorods display a crystalline structure with multiple sharp diffraction peaks at 2θ of 28.5°, 33.0°, 47.4°, 56.3°, and 69.6°, which respectively correspond to the (111), (200), (220), (311), (400) crystalline planes of the pure cubic phase (ceria fluorite structure, JCPDS 34-0394).32 The broadening of the XRD diffraction peaks of Ce-NRs relative to CeO2 nanopowder is due to the nanocrystalline nature of Ce-NR.28,33 The nanotubular morphology of Ti-NT and rod-like shape of Ce-NR are clearly visible in the TEM images (Fig. S2†). The selected area electron diffraction (SAED) patterns for the as-synthesised Ce-NR and CeO2 nanopowder are shown in Fig. S3,† and confirms the polycrystalline nature of the materials. The BET surface area, pore volume, and mean pore diameter of the as-synthesised Ti-NT and Ce-NRs are given in Table S1,† and are typical of powders composed of Ti-NTs and Ce-NRs. The commercial CeO2 nanopowder used in the present study is composed predominantly of irregularly shaped polyhedral nanoparticles of variable sizes, and have previously been meticulously characterised by Agarwal et al.33The kinetics of adsorption of Au–Pd sols is largely dependent on the surface charge of the support and the metal NPs – the latter being influenced by the stabiliser.34 Therefore, it is crucial that the surface charge of the support is investigated first, and the preparation conditions are tuned to favour the adsorption of the Au–Pd sol. In the present work, the zeta potential of each support was measured as function of pH, and the PZC was determined (Fig. S4†). Because PVA-stabilised Au–Pd sols carry a negative charge over a wide range of pH values, the supports were positively charged by acidifying them to a pH value lower than PZC.
After the immobilisation of Au–Pd sol on the supports, the produced catalysts were refluxed in hot water in order to partially remove the stabilising polymer (i.e. PVA), and enhance the surface exposure of Au–Pd NPs. It has been shown previously that the partial removal of the stabiliser by this procedure improves the catalytic performance of supported Au–Pd catalysts.23,35
The XRD diffraction patterns of the catalysts were identical to the diffraction patterns of the blank supports indicating that no significant structural changes were induced in the supports during catalyst preparation (Fig. 1). Due to the relatively low Au and Pd concentration and the small particle size of the metal NPs, no diffraction peaks corresponding to pure Au, Pd or Au–Pd alloy phases were observed in the XRD patterns.
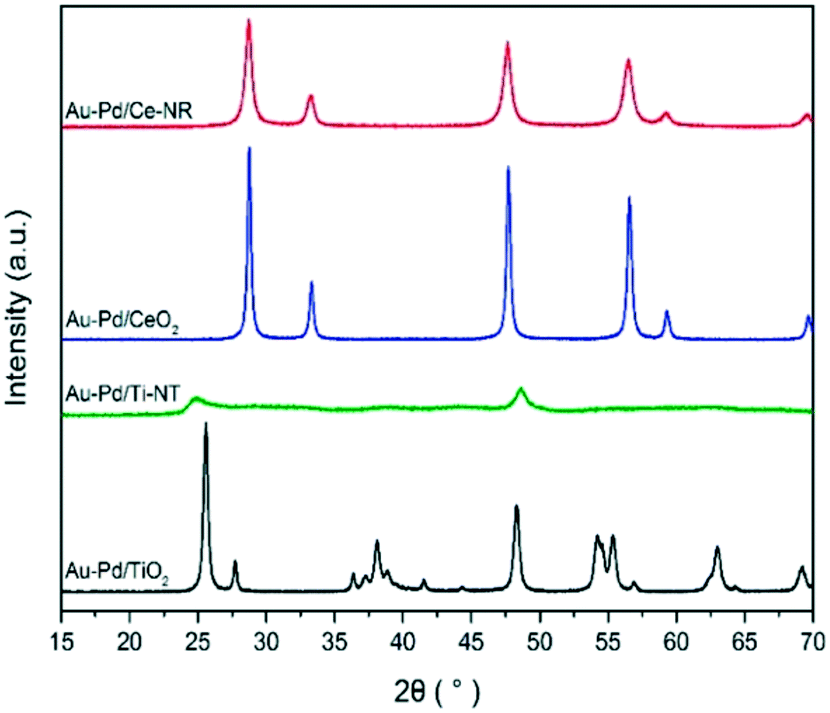 | ||
| Fig. 1 XRD patterns of Au–Pd/Ce-NR, Au–Pd/Ti-NT and Au–Pd/TiO2. | ||
Table 1 reports the textural properties and metal loadings for the different catalysts. As shown in Table 1, the bulk metal loading is slightly and consistently lower than the nominal loading, which is likely due to the leaching of weakly bonded Au–Pd NPs during the washing, refluxing and filtering of the catalysts.
| Catalyst | Au (wt%) | Pd (wt%) | Textural propertiesb | ||||
|---|---|---|---|---|---|---|---|
| Nominal | Actuala | Nominal | Actuala | S BET (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) | |
| a Bulk composition; weight percentage per gram of sample, obtained by ICP-AES analysis. b Determined by nitrogen adsorption–desorption measurement than their PZC. The final metal loadings were found to be close to the nominal loading, which implies that most of metal NPs were efficiently adsorbed by the support. | |||||||
| Au–Pd/TiO2 | 1.0 | 0.75 | 1.0 | 0.70 | 54.4 | 0.33 | 24.5 |
| Au–Pd/Ti-NT | 1.0 | 0.77 | 1.0 | 0.70 | 217.3 | 0.52 | 8.2 |
| Au–Pd/CeO2 | 1.0 | 0.84 | 1.0 | 0.78 | 40.2 | 0.23 | 23.7 |
| Au–Pd/Ce-NR | 1.0 | 0.92 | 1.0 | 0.79 | 57.0 | 0.31 | 21.5 |
The BET surface areas of the prepared Au–Pd catalysts were fairly close to the blank supports, which suggests that the immobilisation of the Au–Pd NPs did not alter the original textural properties of the supports.
Characterisation of the as-synthesised catalysts by TEM and STEM (Fig. 2 and Fig. S5 and S6†) revealed that Au–Pd/Ce-NR exhibited the narrowest particle size distribution and smallest mean particle size (Table 2). Assuming that Au–Pd NPs are face-centred cubic (FCC) cubo-octahedral in shape and using the mean particle size determined from TEM, the metal dispersion was calculated using the approximation method reported by Mori et al.36 As shown in Table 2, catalyst Au–Pd/Ce-NR exhibited a considerably high metal dispersion of approximately 52%. This is in contrast to Au–Pd/CeO2 which displayed a dispersion of ca. 33%. Catalyst sample Au–Pd/Ti-NT and Au–Pd/TiO2 exhibited lower dispersions of ca. 26% and 25%, respectively. Whilst all the prepared catalysts displayed a relatively high metal dispersion, the dispersion of Au–Pd on ceria nanorods was remarkably high. Several factors can define the process by which the metal NPs interact with the support, and ultimately determine the final size of the metal NPs and their dispersion on the support. Among these factors is the surface area of the support, the presence of functionalities and surface defects on the support, and its crystal structure.
 | ||
| Fig. 2 HRTEM and HAADF-STEM images of catalyst sample Au-Pd/Ce-NR (a–d), and Au–Pd/CeO2 (f–i). The inset in figures d and i represents the particle size histogram for Au–Pd/Ce-NR and Au–Pd/CeO2, respectively. | ||
| Catalyst | Pd/Au atomic ratio | (Au + Pd)/Mea | Pd3db | Ce3+b (at%) | Mean particle sized (nm) | Metal dispersiond (%) | ||
|---|---|---|---|---|---|---|---|---|
| ICP | XPSa | Pd0 (%) | Pdδ+c (%) | |||||
| a Surface composition (atomic ratio) determined by XPS; Me = Ti or Ce. b Determined from XPS. c Combined value for Pd2+ and Pd4+ species. d Determined from TEM and STEM images. | ||||||||
| Au–Pd/TiO2 | 2.00 | 2.35 | 0.03 | 69.7 | 30.2 | — | 5.0 ± 1.6 | 25 |
| Au–Pd/Ti-NT | 2.04 | 2.20 | 0.19 | 81.1 | 18.9 | — | 4.7 ± 2.1 | 26 |
| Au–Pd/CeO2 | 2.00 | 2.18 | 0.05 | 60.2 | 39.8 | 32% | 3.7 ± 1.1 | 33 |
| Au–Pd/Ce-NR | 2.17 | 2.95 | 0.18 | 72.8 | 27.2 | 37% | 2.1 ± 0.7 | 52 |
It is generally observed that ceria nanorods grow along the [100] direction with exposed {100} and {110} surfaces28,37 although recent literature suggests that the {110} surfaces of ceria nanorods can reconstruct exposing large fractions of {111} nanofacets on the {110} planes.38 The presence of defects and imperfections in the lattices of ceria nanorods is a consequence of the different plane exposures,37 and play a vital role in the stabilization of metal NPs, which grow preferentially on surface defects sites where the contact area with the support is maximised.39 Hence, the presence of a high number of oxygen vacancies and surface defects in ceria supports, and in Ce-NRs in particular, likely have stabilised the Au–Pd NPs and contributed to high and uniform metal dispersion observed in the TEM and STEM images. This is consistent with the work of Haruta and co-workers who found that the reducibility and density of oxygen vacancies in CeO2 surfaces strongly influenced the size of Au NPs.40 The presence of point surface defects in Au–Pd/Ce-NR is visible in Fig. 2b and d.
It is important to emphasise that even with sol-immobilisation, the morphology and physiochemical properties of the support can significantly influence the metal particle size,34 and that the metal–support interaction plays a key role in determining the final metal dispersion on the support. Characterisation of the catalysts with X-ray photoelectron spectroscopy provided information about the surface composition of the catalyst and oxidation state of Au, Pd and Ce in the case of Ce-NR and CeO2. Fig. 3a and b show the XPS spectra for Au 4f and Pd 3d. The binding energy (B.E.) of the Au (4f7/2) component for a pure metallic gold film (Au0) is typically observed at 84.0 eV. However, initial and final state effects can produce a shift in the B.E. of Au 4f on various supports. The initial and final state effects can be influenced by charge transfer, which depends on the ability of the support material to neutralise the metal NPs during the lifetime of the core-hole created.41,42 Hence, we measured the Au 4f XPS signal of monometallic Au NPs supported on Ce-NR, Ti-NT, TiO2 and CeO2 in order to determine the exact B.E. of Au supported on these materials. The B.E. of the Au (4f7/2) component for Au/Ce-NR, Au/CeO2, Au/Ti-NT and Au/TiO2 were measured to be 83.8, 83.3, 83.6 and 83.3 eV, respectively (Fig. S8†). It can be seen in Fig. 3a that all the Au–Pd catalysts display Au (4f7/2) peaks lower than that of the corresponding monometallic gold catalysts. This negative peak shift in the B.E. can be attributed to the electronic modification of Au species by Pd, and is indicative of the close interaction between the Au and Pd atoms, and the formation of Au–Pd alloys.43 It can also be observed that the magnitude of the shift in the B.E. varies between the different catalyst samples. In principle, this could be attributed to the differing metal–support interaction, the chemical environment, and the atomic ratio of Pd to Au in each sample. Indeed, the size and direction of the binding energy shift have been shown to be largely dependent on the catalyst support and the ratio of Au and Pd present in the alloy phase.43 The difference in the B.E. shift observed in this study mainly reflects the effect of the support, and the difference in metal dispersion, and perhaps a different degree of surface segregation in the nanoparticles.
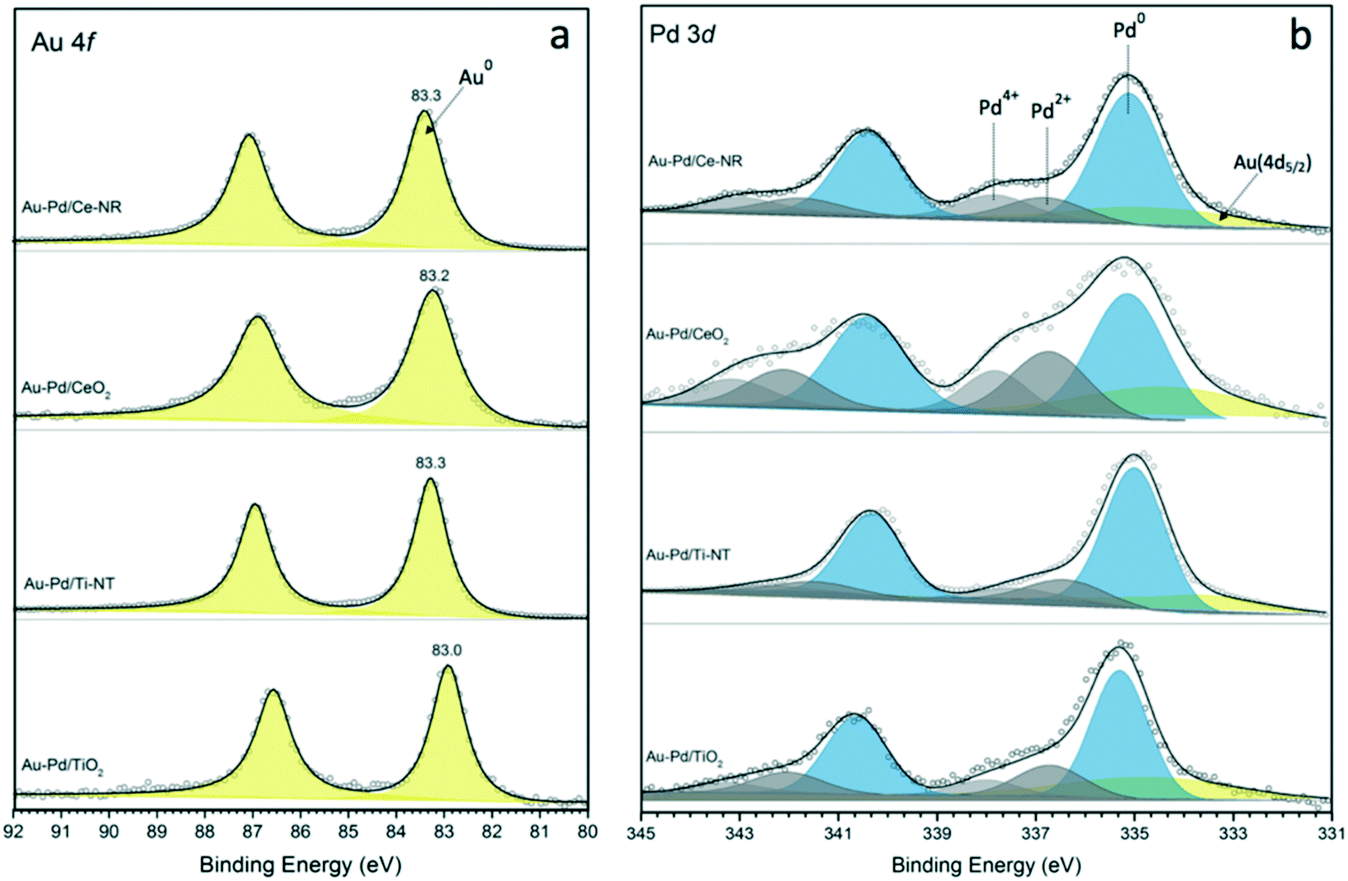 | ||
| Fig. 3 XPS spectra of (a) Au 4f, and (b) Pd 3d for catalysts Au–Pd/Ce-NR, Au–Pd/CeO2, Au–Pd/Ti-NT and Au–Pd/TiO2. | ||
The Au 4f XPS data also suggests that while gold is mostly present as Au0 in these catalyst samples, the slight broadening to higher B.E. may indicate the presence of either oxidised gold species (Auδ+) or very small gold nanoparticles not seen in the HDAAF-STEM images.43,44 The Pd (3d5/2) spectra clearly indicates that both metallic palladium (Pd0) and Pdδ+ species are present in the analysed catalyst samples. As shown in Fig. 3b, the binding energies for Pd0 fall between ca. 334.9 and 336.3 eV, whereas Pdδ+ species appear at higher binding energies between 336.4 and 338.7 eV. These results are in agreement with previous studies showing the binding energy of Pd0 in the range of 334.8–336.2 eV, and between 336.2–338.7 for Pdδ+ species.43,45–47 The proportion of Pd0 and Pdδ+ species present in each catalyst was determined from the fitted Pd3d spectra, and is given in Table 2. In principle, the formation of Pdδ+ and Auδ+ species could be attributed to surface oxidation arising from drying, transfer and storage of the catalysts. However, the presence of noticeably higher amounts of Pdδ+ in catalyst samples Au–Pd/Ce-NR and Au–Pd/CeO2 suggests that it may also be related to the ability of the support to supply oxygen to the metal NPs (i.e. oxygen spillover), thus facilitating the formation of Pdδ+ species, and perhaps some Pd surface segregation. Interestingly, the Pd spectra for the conventional supports show clear formation of Pdδ+, especially in Au–Pd/CeO2. This suggests the possibility of the existence of unalloyed Pd in these catalysts which is very susceptible to oxidation upon exposure to air. The existence of Pdδ+ and Auδ+ species may also in part be due to the electron transfer from the metal NPs to the support, a phenomenon which has recently been shown to take place in many catalytic systems including Au–Pd supported on ceria.48,49
While the analysis of the XPS spectra of Ce 3d is very complex due to the presence of several overlapping peaks, a reasonably accurate deconvolution of the peaks can be made following previously reported methods.50,51 The Ce 3d XPS spectra for Au–Pd/Ce-NR and Au–Pd/CeO2 are presented in Fig. 4, (see Fig. S7† for the deconvolution of Ce 3d in Ce-NR and CeO2 supports). The two primary peaks associated with Ce4+ appear at ∼882.5 and 901.1 eV, and correspond to Ce 3d5/2 and Ce (3d3/2), respectively. Four additional satellite peaks are attributed to the ionization of Ce4+, and observed at ∼889.1, 989.8, 907.6, and ∼916.9 eV. The peaks appearing at ∼880.5, 885.6, 898.5, and 903.1 eV are assigned to Ce3+.15 The concentration of Ce3+ in the ceria supports and catalysts was determined from the deconvoluted XPS spectra, and is given in Table S2† and Table 2, respectively. The as-synthesised ceria nanorods displayed a higher concentration of Ce3+ (31%) in comparison with the ceria nanopowder (27%). After the immobilisation of Au–Pd NPs on the ceria supports, a minor increase in the concentration of Ce3+ was observed in the supported catalysts, Table 2. This marginal increase in Ce3+ could be attributed to surface reduction by NaBH4 (by hydrogen spillover) during the preparation of the catalyst, and/or electron transfer from the Au–Pd NPs to the ceria supports. We note that surface reduction of ceria nanoparticles and nanorods by gaseous hydrogen in the presence of supported metals occurs at 150–300 °C,52 whereas the reduction by NaBH4 used here, was done at 5 °C.
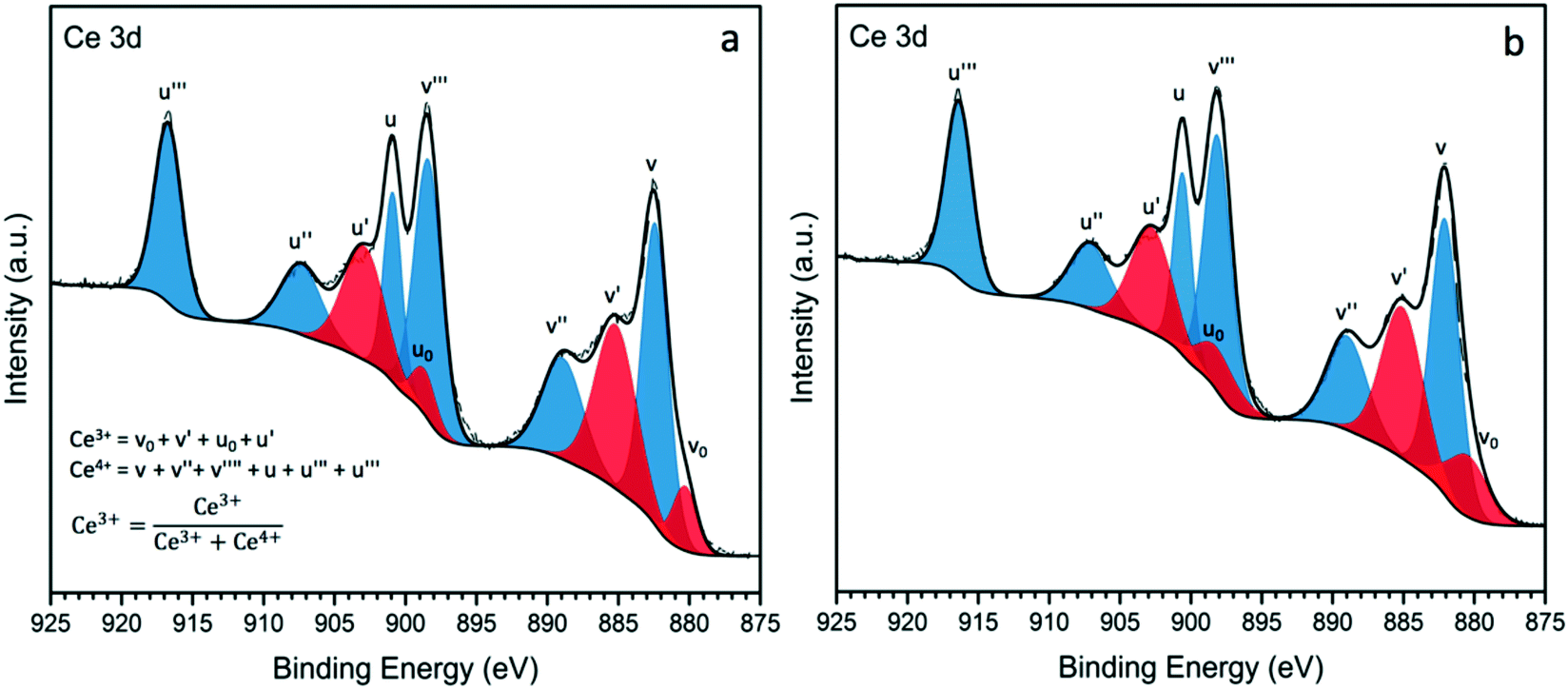 | ||
| Fig. 4 XPS spectra of Ce 3d for catalyst samples a) Au–Pd/Ce-NR and b) Au–Pd/CeO2. | ||
The presence of high amounts of Ce3+ is associated with surface oxygen vacancies and surface defects, and as noted above plays a crucial role in the catalytic activity of ceria-based materials.53 Au–Pd/Ce-NR was found to display a higher amount of the non-stoichiometric cerium Ce3+ compared to Au–Pd/CeO2. Although the difference is not too large, it is consistent with the higher activity observed for this catalyst as discussed in the next section.
The O 1s XPS spectra for ceria nanorods and nanopowder supports are shown in Fig. 5. Three states of oxygen appears in the spectra: Oα (529.2) is attributed to lattice oxygen, Oβ (531.0) is typically assigned to oxygen vacancies, and Oγ (533.0) is attributed to weakly bound oxygen species.17,51 The ratio of Oβ/Oα can be used to estimate the concentration of surface oxygen vacancies (Table S2†), which are important for stabilising metal NPs, and promoting catalytic activity. The Oβ/Oα for ceria nanorods and nanopowder supports are 0.57, and 0.26, respectively. This result is consistent with higher amount of Ce3+ observed in ceria nanorods.
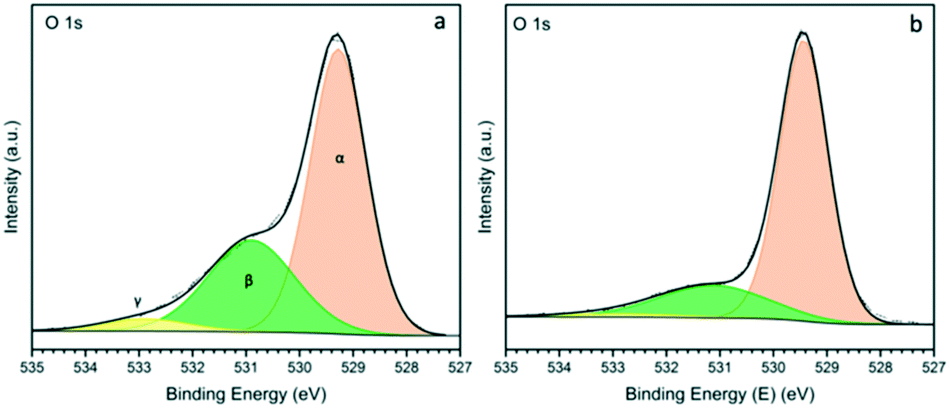 | ||
| Fig. 5 XPS spectra of O 1s for a) Ce-NR and b) CeO2 nanopowder. | ||
The atomic surface composition of the catalysts was calculated from the fitted XPS data. Table 2 gives the atomic ratio of Pd/Au, (Au + Pd)/Ti and (Au + Pd)/Ce for the different catalysts. The Pd/Au ratio derived from XPS was found to be larger than the bulk ratio determined by ICP, the difference being greater for the Ce-NR catalyst. This can be attributed to the variation in XPS analysis depths between Au and Pd. For the Au 4f signal, the analysis depth is reported to be between 5.4 and 5.8 nm; while it is between 4.6–5.0 nm for the Pd 3d signal.54,55 For catalyst sample Au–Pd/Ce-NR, the ratio of Pd/Au on the surface appears to be slightly higher than expected, which might imply surface enrichment in Pd. Concepción et al. observed an increase in the surface concentration of Pd and the re-dispersion of Au and Pd on CeO2 surfaces during reduction with H2.54 A few other authors have also observed the occurrence of surface Pd-enrichment in supported bimetallic Au–Pd catalysts.55,56
As shown in Table 2, catalyst samples Au–Pd/Ti-NT and Au–Pd/Ce-NR exhibited the highest Au–Pd surface concentration. The XPS surface concentration can be influenced by several factors such as the analysis depth of the elements, dispersion of the metals on the support, and the surface area and pore volume of the support. TiO2 P25 and CeO2 nanopowders are mesoporous materials with large pore volumes. It is likely that a large fraction of the Au–Pd NPs occupies pores deep within these two supports, and therefore become undetectable by XPS surface analysis. For Ti-NTs, we have previously conducted detailed analysis using TEM, STEM and XPS and demonstrated that the Au–Pd NPs are largely deposited on the external surfaces of Ti-NTs.23 Ceria nanorods lack any porous structures and possess exposed surfaces upon which Au–Pd NPs are anchored, thereby giving rise to the observed high surface concentration.
Lastly, no residual sodium, boron or chloride were detected by XPS in any of the catalyst samples indicating the full reduction of the metals and removal of chlorides and sodium.
Catalytic activity measurement
The selective oxidation of benzyl alcohol to benzaldehyde has been widely used as a model oxidation reaction for studying supported Au–Pd catalysts.9,55 The synergetic effect between Au and Pd in the selective oxidation of benzyl alcohol is well-documented in literature.12,57 The two primary products in the selective oxidation of benzyl alcohol are benzaldehyde and toluene. Other minor products such benzene, benzoic acid and benzyl benzoate can also be produced during the oxidation of benzyl alcohol. Consequently, this reaction is very useful for evaluating the catalytic performance of Au–Pd supported catalysts. In this study, we investigated the solvent-less selective oxidation performance of the different catalysts prepared, and examined the influence of the support material on the activity and selectivity. Fig. 6 shows the conversion as a function of reaction time. Catalysts were tested under the same reaction conditions (T = 120 °C, pO2 = 2 bar, stirring rate = 1000 rpm, molar ratio of benzyl alcohol/metal = 50000). Preliminary studies showed conversion to be independent of the stirring speed in the range of 600–1000 rpm. A study of the conversion as a function of time showed the reaction to be first order up to 2 hours, and free of any mass transfer limitations. Catalyst sample Au–Pd/Ce-NR showed very high activity achieving a remarkable 38% conversion after only 0.5 hour, and nearly 100% conversion after 3 hours. This is in contrast to a conversion of 82% achieved on Au–Pd/CeO2, 74% on Au–Pd/Ti-NT and 54% on Au–Pd/TiO2. Indeed, Au–Pd/TiO2 was found to be the least active among the catalysts investigated in this study, and appeared to deactivate very rapidly after 2 hours. The deactivation of Au–Pd/TiO2 catalyst prepared by sol-immobilisation using PVA has been reported previously, and was attributed to the strong adsorption of benzoic acid and benzoate species by the catalyst.58
 | ||
| Fig. 6 Benzyl alcohol conversion with time obtained for the different catalysts. Reaction conditions: T = 120 °C, pO2 = 2 bar, stirring rate = 1000 rpm, molar ratio of benzyl alcohol/metal = 50000. | ||
Ceria-supported catalysts (Au–Pd/Ce-NR and Au–Pd/CeO2) displayed higher selectivities to benzaldehyde and lower selectivity to toluene than the titania-supported catalysts (Fig. 7). Furthermore, Au–Pd/Ce-NR was slightly more selective to benzaldehyde than Au–Pd/CeO2, especially at short reaction times. Although Au–Pd/Ti-NT was more active than Au–Pd/TiO2, the latter exhibited a higher selectivity to benzaldehyde, which might be ascribed to the surface acidity of the support since titanate nanotubes are known to possess a large number of Brønsted and Lewis acid sites.59 High surface acidity is reported to promote the disproportion of benzyl alcohol to toluene and benzaldehyde.60
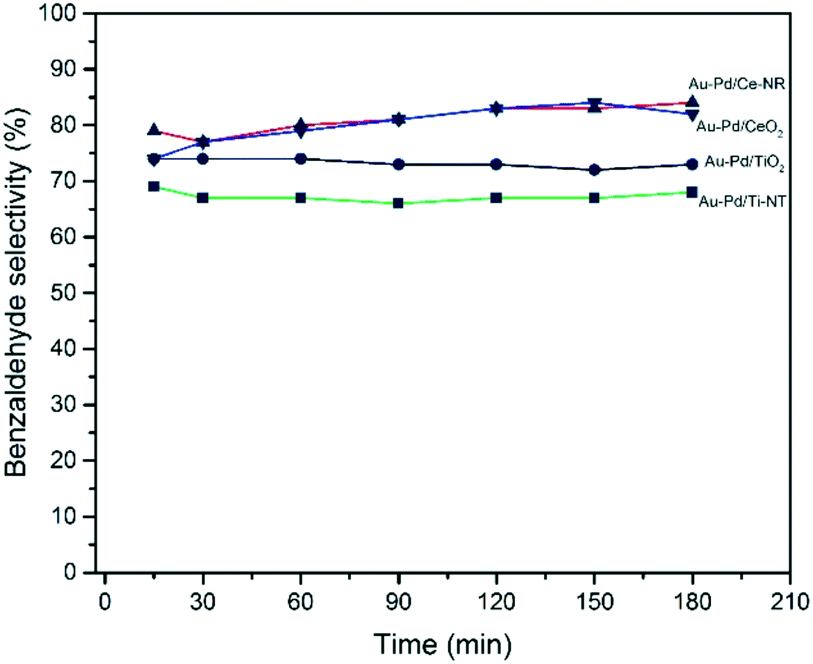 | ||
| Fig. 7 Benzyl alcohol selectivity with time obtained for the different catalysts. Reaction conditions: T = 120 °C, pO2 = 2 bar, stirring rate = 1000 rpm, molar ratio of benzyl alcohol/metal = 50000. | ||
The initial TOF calculated at 0.5 hour is essentially analogous to the initial rate of reaction. Therefore, the catalytic performance of the different catalysts was compared at 0.5 hour. Fig. 8 compares the catalytic activity in terms of turnover frequency (moles of benzyl alcohol converted per moles of metal per hour), and the total benzaldehyde yield in wt% after three hours. The catalyst prepared using ceria nanorods (Au–Pd/Ce-NR) exhibits the highest catalytic activity with TOF > 35900 h−1, and the highest benzaldehyde yield (ca. 78%). On the other hand, Au–Pd supported on commercial CeO2 nanopowders displayed a significantly lower activity (TOF ∼ 26300 h−1) and a benzaldehyde yield of only 66%. As expected Au–Pd supported on titania was less active than ceria in this reaction. The superior catalytic activity of Au–Pd/Ce-NR is due to its unique physicochemical and morphological properties. It is likely that the high surface concentration of Ce3+, and the presence of oxygen vacancies and surface defects led to the stabilisation of the Au–Pd NPs, and facilitated high and uniform metal dispersion on Ce-NR. This finding is in agreement with previous work in which the reducibility of ceria (i.e. Ce3+ concentration) was found to be directly related to the size of Au NPs.61 Furthermore, oxygen vacancies have been found to play a highly important role in oxidation reactions over ceria and metal NPs supported on ceria.37 For example, in the oxidation of toluene oxygen vacancies contribute to the activation of the substrate via surface oxygen vacancies, and the migration of bulk oxygen to the surface.53 The concentration of Ce3+ has also been correlated with the adsorption of molecular oxygen by ceria and formation of superoxide species on the surfaces of ceria, and has been linked to the observed high catalytic oxidation activity of ceria in some oxidation reactions.62 Interestingly we note, in regard to the difference in TOF between Au–Pd/Ce-NR and Au–Pd/CeO2, that in addition to the proportion of Ce3+ being slightly higher in the former, the difference in surface areas, Table 1, implies that there are a much greater number of Ce3+ and surface oxygen vacancies in Au–Pd/Ce-NR giving a greater potential for oxygen activation. The combination of this and the smaller Au–Pd particle size in Au–Pd/Ce-NR, Table 1, may in part explain the observed difference in TOF.
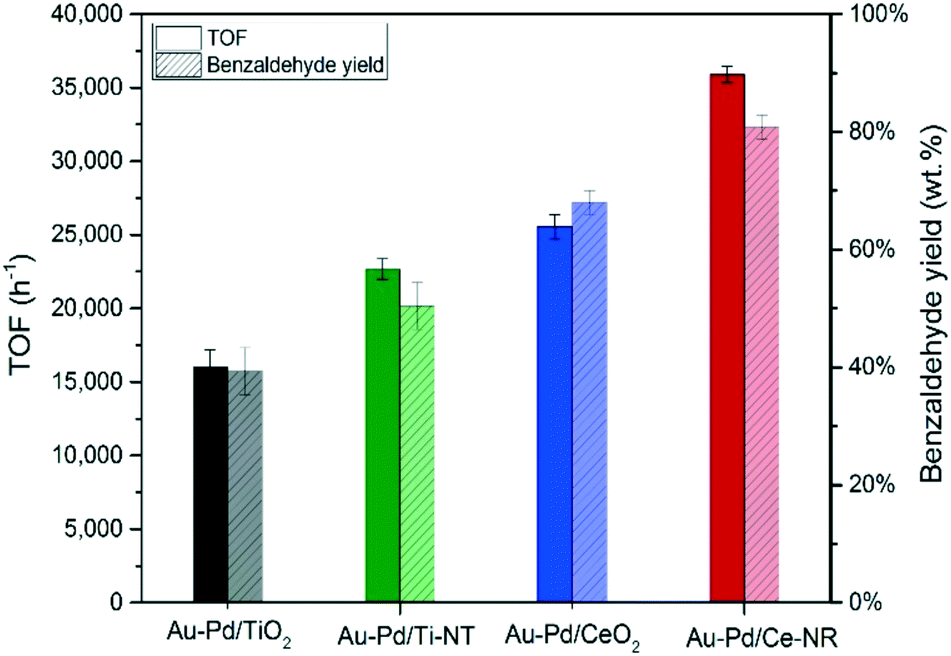 | ||
| Fig. 8 TOF (h−1) and benzaldehyde yield obtained for the different catalysts. Reaction conditions: T = 120 °C, pO2 = 2 bar, stirring rate = 1000 rpm, molar ratio of benzyl alcohol/metal = 50000. TOF calculated after 0.5 hour, and yield after 3 hours. | ||
In addition, the high catalytic activity of Au–Pd/Ce-NR and Au–Pd/Ti-NT could be associated with the high Au–Pd surface concentration, and the ability of the reactants to easily access the highly dispersed active metal sites on the external surfaces of the supports, and the ability of the products to escape. On the other hand, the relatively high porosity of TiO2 and CeO2 nanopowders could slow the diffusion of the reactants to the active metal NPs inside the pores.
It is important to highlight that the TOF observed for Au–Pd/Ce-NR (TOF > 35900 h−1) is markedly higher than the TOF of similar Au–Pd catalysts reported in literature. For example, Miedziak et al. prepared Au–Pd catalysts supported on CeO2 nanoparticles synthesised by supercritical anti-solvent precipitation, and tested these catalysts in the solvent-less selective oxidation of benzyl alcohol and reported a TOF of up to 17700 h−1 at 160 °C.63 Perhaps, the closest study to our present work was that conducted by Zhang et al. in which Au–Pd/Ce-NR catalyst with a similar Pd:Au molar ratio to ours (i.e. Pd:Au = 2:1) was tested in the solvent-less selective oxidation of benzyl alcohol at 120 °C and pO2 = 10 bar. A TOF of 9360 h−1 was reported for this catalyst.22
The higher catalytic activity of the present catalyst compared to the latter study, can be ascribed to the smaller metal particle size and therefore higher dispersion, the degree of alloying, and the quality of the ceria nanorods used in the present catalysts (TEM images of the catalyst reported in the previous study show Ce-NR with varying sizes and morphologies). Moreover, the catalyst in the previously mentioned study displayed a Ce3+ content of only 30%, as opposed to 37% for the Au–Pd/Ce-NR reported in the present study. Lastly, the selectivity reported by Zhang et al. was higher than in the present work. However, these were obtained under pO2 = 10 bar. We have shown previously that higher oxygen partial pressures lead to a significant enhancement in benzaldehyde selectivity from suppression of toluene formation.23
The synergic effect of Au–Pd alloys in the selective oxidation of benzyl alcohol was also confirmed in the present study whereby Au/Ce-NR and Pd/Ce-NR catalysts were tested under the same reaction conditions as in Fig. 8, and exhibited a TOF of 1664 and 12300 h−1, respectively (see Fig. S9†).
Lastly, for comparison purposes, a benchmark Au–Pd/Ce-NR catalyst was prepared by dry impregnation as previously described,23 and tested under the same reaction conditions used throughout this study. The catalyst displayed a relatively low activity (TOF < 6000 h−1), and benzaldehyde yield of only 27% after three hours of reaction time. These results are in agreement with our previous work23 in which Au–Pd/Ti-NT prepared by sol-immobilisation were found to be significantly more active than Au–Pd/Ti-NT prepared by impregnation. More importantly, these findings highlight the role of alloying and metal particle size in the catalytic activity since it is well-known that sol-immobilisation leads to a high degree of alloying, and small and regular particle size distribution.
Preliminary tests of catalyst recycle showed only mild deactivation of Au–Pd/Ce-NR upon reuse. Although one might expect that Ce3+ would be oxidised under the present reaction conditions, analysis of the spent catalyst by XPS showed virtually no change in the concentration of Ce3+. The mild deactivation observed in the recycling tests might have been caused by a combination of the adsorption of the products by the catalyst,23 minor leaching of the metals and possibly sintering. Further investigations are needed in order to fully understand the deactivation mechanism of Au–Pd/Ce-NR and its regeneration.
The catalytic tests demonstrate that the physical properties and structural features of the support material significantly influence the catalytic activity and selectivity in the selective oxidation of benzyl alcohol. The catalytic performance can be optimised by controlling the morphology and the structure of the support, as well as the catalyst preparation method.
Conclusion
This work has demonstrated that immobilising Au–Pd sols on different nanostructured ceria and titania supports creates highly active selective oxidation catalysts compared to the corresponding nanopowder supports. The new Au–Pd/Ce-NR catalyst introduced in the present paper exhibited a TOF > 35900 h−1, and was found to be notably more active in comparison to similar Au–Pd catalysts reported in literature. The morphology and physiochemical properties of the support were found to play a crucial role in determining the final size of the Au–Pd NPs and the dispersion of metals on the support. The high Au–Pd surface concentration is related to the ability of the Ce-NR to stabilise finely dispersed Au–Pd NPs on its exposed facets and defect sites. The superior catalytic activity Au–Pd/Ce-NR is also associated with a high concentration of Ce3+ and oxygen vacancies in Ce-NR, which can promote the activity of the Au–Pd nanoparticles by oxygen activation. In principle, Au–Pd/Ce-NR should be active for several other important selective oxidation reactions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Motaz Khawaji gratefully acknowledges the financial support of Saudi Aramco. This work was funded in part by Saudi Aramco and by EPSRC under grant EP/K014749/1. We are grateful to Dr. Ecaterina Ware for her assistance with TEM.References
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- R. Zhao, D. Ji, G. Lv, G. Qian, L. Yan, X. Wang and J. Suo, Chem. Commun., 2004, 904–905 RSC.
- S. Carrettin, P. Concepción, A. Corma, J. M. López Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS PubMed.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058–2059 RSC.
- A. Villa, D. Wang, D. S. Su and L. Prati, Catal. Sci. Technol., 2015, 5, 55–68 CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- Y. Y. Wu, N. A. Mashayekhi and H. H. Kung, Catal. Sci. Technol., 2013, 3, 2881 CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- D. I. Enache, D. Barker, J. K. Edwards, S. H. Taylor, D. W. Knight, A. F. Carley and G. J. Hutchings, Catal. Today, 2007, 122, 407–411 CrossRef CAS.
- T. X. T. Sayle, S. C. Parker and D. C. Sayle, Phys. Chem. Chem. Phys., 2005, 7, 2936–2941 RSC.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed.
- X. Liu, K. Zhou, L. Wang, B. Wang and Y. Li, J. Am. Chem. Soc., 2009, 131, 3140–3141 CrossRef CAS PubMed.
- K. Zhou, X. Wang, X. Sun, Q. Peng and Y. Li, J. Catal., 2005, 229, 206–212 CrossRef CAS.
- Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo and G. Lu, ACS Catal., 2016, 6, 2265–2279 CrossRef CAS.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS PubMed.
- G. Vilé, S. Colussi, F. Krumeich, A. Trovarelli and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2014, 53, 12069–12072 CrossRef PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935 CrossRef CAS PubMed.
- R. Si and M. Flytzani-Stephanopoulos, Angew. Chem., Int. Ed., 2008, 47, 2884–2887 CrossRef CAS PubMed.
- H. Zhang, Y. Xie, Z. Sun, R. Tao, C. Huang, Y. Zhao and Z. Liu, Langmuir, 2011, 27, 1152–1157 CrossRef CAS PubMed.
- M. Khawaji and D. Chadwick, ChemCatChem, 2017, 9, 4353–4363 CrossRef CAS.
- N. Dimitratos, A. Villa, C. L. Bianchi, L. Prati and M. Makkee, Appl. Catal., A, 2006, 311, 185–192 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Bethell and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 5142–5153 RSC.
- N. Dimitratos, F. Porta and L. Prati, Appl. Catal., A, 2005, 291, 210–214 CrossRef CAS.
- J. Wang, S. A. Kondrat, Y. Wang, G. L. Brett, C. Giles, J. K. Bartley, L. Lu, Q. Liu, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 3575–3587 CrossRef CAS.
- H.-X. Mai, L.-D. Sun, Y.-W. Zhang, R. Si, W. Feng, H.-P. Zhang, H.-C. Liu and C.-H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS PubMed.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef CAS.
- D. L. Morgan, H.-Y. Zhu, R. L. Frost and E. R. Waclawik, Chem. Mater., 2008, 20, 3800–3802 CrossRef CAS.
- E. Morgado, M. A. S. de Abreu, G. T. Moure, B. A. Marinkovic, P. M. Jardim and A. S. Araujo, Chem. Mater., 2007, 19, 665–676 CrossRef CAS.
- L. Torrente-Murciano, A. Gilbank, B. Puertolas, T. Garcia, B. Solsona and D. Chadwick, Appl. Catal., B, 2013, 132, 116–122 CrossRef.
- S. Agarwal, L. Lefferts, B. L. Mojet, D. A. J. M. Ligthart, E. J. M. Hensen, D. R. G. Mitchell, W. J. Erasmus, B. G. Anderson, E. J. Olivier, J. H. Neethling and A. K. Datye, ChemSusChem, 2013, 6, 1898–1906 CrossRef CAS PubMed.
- L. Prati and A. Villa, Acc. Chem. Res., 2014, 47, 855–863 CrossRef CAS PubMed.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS PubMed.
- A. Trovarelli and J. Llorca, ACS Catal., 2017, 7, 4716–4735 CrossRef CAS.
- C. Yang, X. Yu, S. Heißler, A. Nefedov, S. Colussi, J. Llorca, A. Trovarelli, Y. Wang and C. Wöll, Angew. Chem., Int. Ed., 2017, 56, 375–379 CrossRef CAS PubMed.
- J. C. González, J. C. Hernández, M. López-Haro, E. del Río, J. J. Delgado, A. B. Hungría, S. Trasobares, S. Bernal, P. A. Midgley and J. J. Calvino, Angew. Chem., Int. Ed., 2009, 48, 5313–5315 CrossRef PubMed.
- T. Akita, M. Okumura, K. Tanaka, M. Kohyama and M. Haruta, Catal. Today, 2006, 117, 62–68 CrossRef CAS.
- B. R. Cuenya, S.-H. Baeck, T. F. Jaramillo and E. W. McFarland, J. Am. Chem. Soc., 2003, 125, 12928–12934 CrossRef PubMed.
- A. Zwijnenburg, A. Goossens, W. G. Sloof, M. W. J. Crajé, A. M. van der Kraan, L. Jos de Jongh, M. Makkee and J. A. Moulijn, J. Phys. Chem. B, 2002, 106, 9853–9862 CrossRef CAS.
- J. H. Carter, S. Althahban, E. Nowicka, S. J. Freakley, D. J. Morgan, P. M. Shah, S. Golunski, C. J. Kiely and G. J. Hutchings, ACS Catal., 2016, 6, 6623–6633 CrossRef CAS PubMed.
- C. M. Olmos, L. E. Chinchilla, J. J. Delgado, A. B. Hungría, G. Blanco, J. J. Calvino and X. Chen, Catal. Lett., 2016, 146, 144–156 CrossRef CAS.
- Z. Bastl, Collect. Czech. Chem. Commun., 1995, 60, 383–392 CrossRef CAS.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568–16577 CrossRef CAS PubMed.
- S. Marx and A. Baiker, J. Phys. Chem. C, 2009, 113, 6191–6201 CAS.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. r. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310 CrossRef CAS PubMed.
- A. S. Thill, A. S. Kilian and F. Bernardi, J. Phys. Chem. C, 2017, 121, 25323–25332 CAS.
- A. Galtayries, R. Sporken, J. Riga, G. Blanchard and R. Caudano, J. Electron Spectrosc. Relat. Phenom., 1998, 88–91, 951–956 CrossRef.
- J. M. López, A. L. Gilbank, T. García, B. Solsona, S. Agouram and L. Torrente-Murciano, Appl. Catal., B, 2015, 174–175, 403–412 CrossRef.
- B. Ouyang, W. Tan and B. Liu, Catal. Commun., 2017, 95, 36–39 CrossRef CAS.
- A. I. Y. Tok, S. W. Du, F. Y. C. Boey and W. K. Chong, Mater. Sci. Eng., A, 2007, 466, 223–229 CrossRef.
- P. Concepción, S. García, J. C. Hernández-Garrido, J. J. Calvino and A. Corma, Catal. Today, 2016, 259, 213–221 CrossRef.
- C. M. Olmos, L. E. Chinchilla, E. G. Rodrigues, J. J. Delgado, A. B. Hungría, G. Blanco, M. F. R. Pereira, J. J. M. Órfão, J. J. Calvino and X. Chen, Appl. Catal., B, 2016, 197, 222–235 CrossRef CAS.
- A. A. Herzing, A. F. Carley, J. K. Edwards, G. J. Hutchings and C. J. Kiely, Chem. Mater., 2008, 20, 1492–1501 CrossRef CAS.
- A. Villa, N. Janjic, P. Spontoni, D. Wang, D. S. Su and L. Prati, Appl. Catal., A, 2009, 364, 221–228 CrossRef CAS.
- E. Nowicka, J. P. Hofmann, S. F. Parker, M. Sankar, G. M. Lari, S. A. Kondrat, D. W. Knight, D. Bethell, B. M. Weckhuysen and G. J. Hutchings, Phys. Chem. Chem. Phys., 2013, 15, 12147–12155 RSC.
- R. Li, X. Zhu, X. Yan, D. Shou, X. Zhou and W. Chen, RSC Adv., 2016, 6, 100103–100107 RSC.
- E. H. Cao, M. Sankar, E. Nowicka, Q. He, M. Morad, P. J. Miedziak, S. H. Taylor, D. W. Knight, D. Bethell, C. J. Kiely, A. Gavriilidis and G. J. Hutchings, Catal. Today, 2013, 203, 146–152 CrossRef CAS.
- B. Acosta, E. Smolentseva, S. Beloshapkin, R. Rangel, M. Estrada, S. Fuentes and A. Simakov, Appl. Catal., A, 2012, 449, 96–104 CrossRef CAS.
- Z. Wu, M. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- P. J. Miedziak, Z. Tang, T. E. Davies, D. I. Enache, J. K. Bartley, A. F. Carley, A. A. Herzing, C. J. Kiely, S. H. Taylor and G. J. Hutchings, J. Mater. Chem., 2009, 19, 8619–8627 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy02329d |
| This journal is © The Royal Society of Chemistry 2018 |