
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the mechanism of transition metal-free anti addition to alkynes: the selenoboration case†
Diego
García-López
a,
Marc G.
Civit
a,
Christopher M.
Vogels
b,
Josep M.
Ricart
 a,
Stephen A.
Westcott
b,
Elena
Fernández
*a and
Jorge J.
Carbó
*a
a,
Stephen A.
Westcott
b,
Elena
Fernández
*a and
Jorge J.
Carbó
*a
aDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, C/ Marcel·lí Domingo s/n, Tarragona, Spain. E-mail: mariaelena.ferenandez@urv.cat; j.carbo@urv.cat
bDepartment of Chemistry and Biochemistry, Mount Allison University, Sackville, New Brunswick E4L 1G8, Canada
First published on 22nd June 2018
Abstract
The stereoselective anti-addition of selenoboranes to α,β-acetylenic esters and amides was achieved in a transition metal-free context using catalytic amounts of PCy3. The reaction provides anti-3,4-selenoboration with concomitant delivery of α-vinyl selenides by protodeboronation with MeOH. Interestingly, in the absence of phosphine the selenoboration switches towards the formation of β-vinyl selenides. Theoretical calculations rationalize the regio- and stereoselectivity of the reaction by discovering a new mechanism for the anti-3,4-selenoboration. While the selenoborane is activated via the “push–pull” effect of B, the phosphine interacts with the β position of the alkynoate switching the polarity of the triple bond and favoring 1,3-selenoboration which provides the α-addition of the selenyl group. Then, the autocatalytic action of a second selenoborane reagent, which coordinates to the phosphorus ylide intermediate, determines the stereoselectivity and completes the catalytic process. Finally, the comparison of selenoborane reagents with diboranes and silaboranes, which have exhibited analogous reactivity, shows that the selenium moiety has a larger nucleophilic character favoring the performance of the reaction under mild conditions.
Introduction
The reactivity of diboron reagents and B–interelement systems with organic molecules has become an important source of selective functionalization.1 The addition of these reagents to C–C triple bonds provides an attractive method for the regio- and stereoselective synthesis of functionalized alkenes via the formation of C–B and C–interelement bonds. Much effort has been devoted to transition metal catalysis, which generally generates 1,2-cis-addition products selectively.2 However, some outstanding examples of stereoselective trans-hydroboration of terminal and internal alkynes were achieved by using copper3 and ruthenium4 catalysts, respectively. Also, Miyaura et al. developed the hydroboration of terminal alkynes by Rh and Ir complexes leading selectively to Z-alkenylboranes5via gem-addition of B–H bonds through a vinylidene intermediate.6In recent years several transition metal-free approaches have appeared to yield anti addition products using special substrates or introducing directing groups (Scheme 1).1 Uchiyama et al. utilized propargyl alcohol substrates to provide access to the selective anti-diboration7 and anti-alkynylboration7,8 of triple bonds through the intramolecular reaction of an alkoxide and selective delivery of the nucleophilic boryl unit (Scheme 1a). DFT calculations proposed a mechanism involving deprotonation of propargylic alcohols by nBuLi, coordination of the resulting alkoxide to activate B–X bonds, stereoselective migration of boron or the alkynyl group and Li to form an anti-alkenyllithium intermediate, and final intramolecular capture of the boron.7,8 Santos et al. reported the substrate-assisted anti-diboration of alkynamides using strong bases which deprotonated the amide group (Scheme 1b),9a and then the procedure was extended to silaboration.9b The DFT-derived mechanism indicates that the generated alkoxide activates the diboron reagent intramolecularly resulting in α-addition of one boron, similar to that found for propargyl alcohols, and then a rapid carbon–carbon bond rotation leads to the thermodynamically favored anti product.9a Alternatively, Sawamura et al. achieved the anti-selective carboboration,10 silaboration, and diboration11 of alkynoates using catalytic amounts of PBu3 (Scheme 1c). The proposed mechanism involved the rotation of an enolate double bond as the key step for selectivity-determination, although in this case, it was not supported by computational studies.10 The comparison of Sawamura's reactions with those of Uchiyama and Santos suggests that in the absence of acidic protons alkynoates can be activated by basic phosphines instead of Brønsted bases to produce the alkoxy group, which in turn activates the boron reagent.
 | ||
| Scheme 1 Stereoselective transition metal-free anti addition of diboron and B–interelement reagents to activated alkynes. | ||
Among the potential applications of B–interelement addition to alkynes, processes for the selective synthesis of vinyl selenides, which are important biological scaffolds, are challenging targets.12 β-Vinyl selenides can be obtained from α,β-acetylenic esters and ynones by means of rhodium-catalyzed syn-hydroselenation to generate principally β–E-vinyl selenides (Scheme 2a)13 or via anti-addition of organoselenols, formed in situ from the corresponding diselenides and Zn in acidic media, to provide β–Z-vinyl selenides (Scheme 2b).14,15 Alternatively, phenylselenoborane16 can efficiently be added to alkenes,17 in a transition-metal-free context, taking advantage of the “push–pull” effect of B in the B–interelement reagent. In a previous work, we proved that stereodefined Z-alkenylselenides can be directly formed through the Lewis acid interaction of the carbonyl unit from ynones and the Bpin moiety on PhSe–Bpin, throughout 1,4-selenoborated intermediates.18
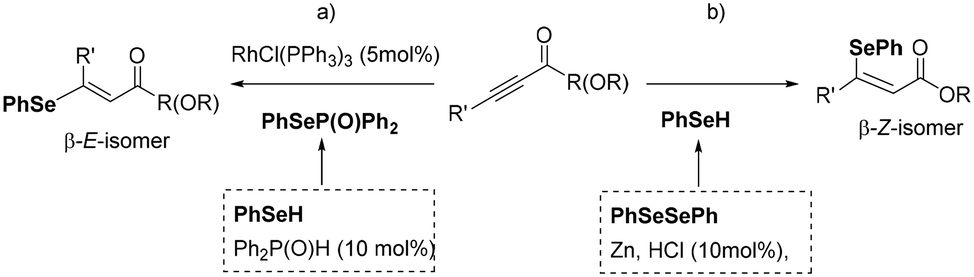 | ||
| Scheme 2 Stereoselective synthesis of β-vinyl selenides: a) with PhSeH and b) with PhSeSePh. | ||
Herein we aim to understand the mechanism of the transition metal-free addition of boron–interelement systems to alkynoates catalyzed by phosphines. To this end we extend our experimental selenoboration work to electron-deficient α,β-acetylenic esters by adding catalytic amounts of phosphine as in Sawamura's report.11 We came across a stereodivergent synthesis of β-vinyl selenides and α-vinyl selenides, depending on the mode of activation of the phenylselenoborane (Scheme 3). Eventually, experimental observations were used to perform a detailed DFT study of the reaction mechanism, including the understanding of the role of the heteroelement nature.
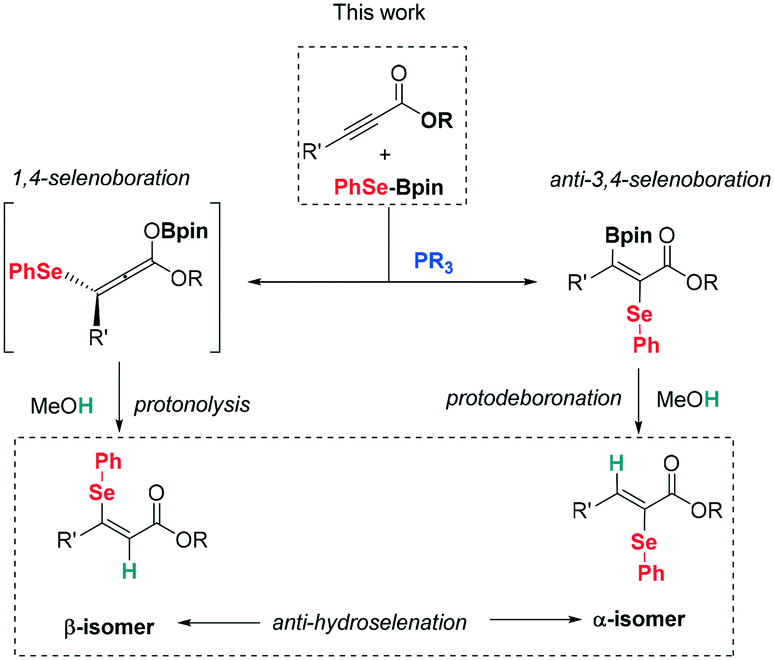 | ||
| Scheme 3 Syn- and anti-hydroselenation towards vinyl selenides. | ||
Results and discussion
The previously determined mechanism for the 1,4-selenoboration of ynones is illustrated in Scheme 4.18 The activation of the boron atom with the oxygen of the carbonyl group in the substrate, leading to intermediate A, enhances the nucleophilic attack of the –SePh moiety on the β carbon of the triple bond yielding the 1,4-selenoborated allene intermediate B which undergoes protonolysis with methanol. Here, we initially analyzed from a computational point of view whether this process can be extended to α,β-acetylenic ester 1-(4-methylphenyl)-3-phenyl-2-propyn-1-one, although the ester functional group is expected to be less efficient than the ketone in activating the B–Se bond through the “push–pull” effect. To evaluate the feasibility of the reaction before attempting it experimentally, we compared the potential free-energy profile for the α,β-acetylenic ester with that previously reported for α,β-acetylenic ketones at the same computational level18 (R = OMe vs. Tol in Scheme 4). In the initial activation of the boron atom with the oxygen of the carbonyl group leading to intermediate A, the free-energy barrier and the relative energy of A are slightly higher (∼2 kcal mol−1) for the ester (R = OMe) than for the ketone (R = Tol). Nevertheless, the lower activation ability of the ester group is mainly manifested in the nucleophilic attack of the –SePh moiety on the β carbon of the triple bond yielding 1,4-selenoborated intermediate B, for which the energy barrier increases by 5.5 kcal mol−1. Finally, the computed free-energy barrier for the protonolysis of intermediate B inverts the trend, which is lowered by 10 kcal mol−1. Thus, although the overall computed free-energy barrier is somewhat higher (27.5 kcal mol−1, from the reactants to the transition state TSA–B) for the α,β-acetylenic ester, the value is still feasible for a reaction occurring at the working temperature, 50 °C, and therefore, there is enough theoretical support to carry out this reaction experimentally. | ||
| Scheme 4 Comparative relative free-energies and barriers (kcal mol−1) for the reaction mechanism of β-selenated α,β-unsaturated ester and ketone (values in parenthesis are taken from ref. 18). | ||
The energy profile for the β-selenation of α,β-acetylenic esters prompted us to determine the expected reactivity with representative substrates that combine aryl and alkyl groups in the β-position, as well as sterically differentiated ester groups. Table 1 shows the moderate to high conversions observed for all the substrates tested, and in particular for methyl-2-nonenoato (Z-9) (Table 1, entry 2), working at 50 °C. Lower temperatures provided only a small amount of desired products. The aryl or alkyl nature of the acetyl substituent did not influence significantly the 1,4-selenoboration followed by protonolysis towards the β-selenated α,β-unsaturated esters. Substrates with electron-withdrawing p-substituents such as CF3 in ethyl (Z)-2-(phenylselanyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-(4-(trifluoromethyl)phenyl)acrylate only converted 16%. The exclusive formation of the Z-isomer was confirmed by spectroscopic data and the full characterization of product Z-8 by X-ray diffraction (Fig. 1), which is in contrast to the Z/E mixture observed in the β-selenation of α,β-acetylenic ketones.18
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | NMR yield (%)b | IY (%)c |
| a Reaction conditions: α,β-acetylenic esters or amides (0.2 mmol), PhSe–Bpin (1.1 equiv.), MeOH (0.15 mL), at 50 °C for 16 h. b Yields were determined by 1H NMR analysis of the crude reaction mixture with naphthalene as an internal standard, which was added after the reaction. c IY = isolated yield. | ||||
| 1 |
 1
1
|
 Z-
8
Z-
8
|
82 | 74 |
| 2 |
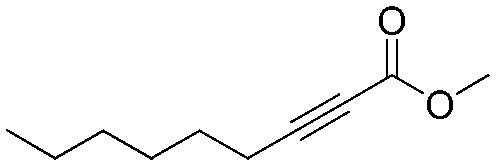 2
2
|
 Z-
9
Z-
9
|
90 | 79 |
| 3 |
 3
3
|
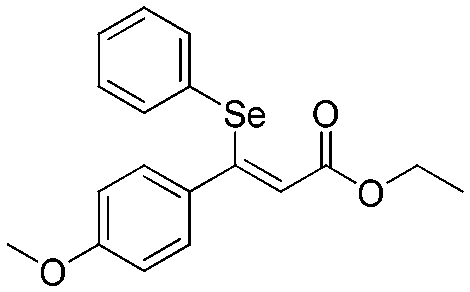 Z-
10
Z-
10
|
89 | 78 |
| 4 |
 4
4
|
 Z-
11
Z-
11
|
75 | 72 |
| 5 |
 5
5
|
 Z-
12
Z-
12
|
88 | 69 |
| 6 |
 6
6
|
 Z-
13
Z-
13
|
89 | 76 |
| 7 |
 7
7
|
 Z-
14
Z-
14
|
75 | 68 |

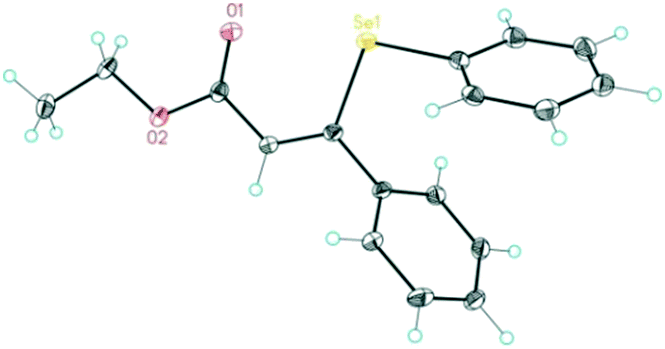 | ||
| Fig. 1 X-ray diffraction structure of product (Z)-3-phenyl-3-(phenylselanyl)acrylate (Z-8) confirming the Z-isomerism. | ||
We extended the study to the β-selenation of α,β-acetylenic amides 3-(4-methoxyphenyl)-N,N-dimethylpropiolamide (6) and 3-(4-methoxyphenyl)-1-(pyrrolidin-1-yl)prop-2-yn-1-one (7), and in both cases the conversion was comparable (Table 1, entries 6 and 7) to the β-selenation of α,β-acetylenic esters, proving the generality of the method despite the functional group.
In the next step, we introduced catalytic amounts of phosphine in order to switch the selenoboration of alkynoates towards the formation of α-vinyl selenides with anti stereoselectivity, as Sawamura et al.11 observed for the diboration or silaboration of α,β-acetylenic esters with PBu3. In our hands, the addition of 10 mol% PBu3 or PPh3 did not convert substrate 1 into any selenated or borylated product, working at 50 °C with 1.1 equiv. of PhSe–Bpin (Table 2, entries 1 and 2). However, the use of PCy3 provided the formation of a new product (61% NMR yield) identified as the corresponding anti-3,4-addition product (Table 2, entry 3). We observed the same yield working at rt, for 8 h, with a variable amount of the PhSe–Bpin reagent, therefore the optimized reaction conditions included only 1.1 equiv. of the PhSe–Bpin reagent and 15 mol% phosphine (Table 2, entries 3–6). Although the reaction works efficiently under neat conditions, the addition of 0.15 mL of THF was required to solubilize the Se–B reagent (Table 2, entries 7–9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | PR3 (mol%) | PhSe–Bpin | T (°C) | t (h)/Sol | NMR yield (%)b |
| a Reaction conditions: 1 (0.2 mmol), PhSe–Bpin (1.1 equiv.), neat or THF (0.15 mL). b Yields were determined by 1H NMR analysis of the crude reaction mixture with naphthalene as an internal standard, which was added after the reaction. | |||||
| 1 | PBu3 (10 mol%) | 1.1 equiv. | 50 °C | 16 h/neat | — |
| 2 | PPh3 (10 mol%) | 1.1 equiv. | 50 °C | 16 h/neat | — |
| 3 | PCy3 (10 mol%) | 1.1 equiv. | 50 °C | 16 h/neat | 61 |
| 4 | PCy3 (10 mol%) | 1.1 equiv. | 50 °C | 8 h/neat | 60 |
| 5 | PCy3 (10 mol%) | 1.1 equiv. | rt | 8 h/neat | 62 |
| 6 | PCy3 (15 mol%) | 1.1 equiv. | rt | 8 h/neat | 62 |
| 7 | PCy3 (15 mol%) | 1.1 equiv. | rt | 8 h/THF | 63 |
| 8 | PCy3 (15 mol%) | 1.5 equiv. | rt | 8 h/THF | 63 |
| 9 | PCy3 (15 mol%) | 2.0 equiv. | rt | 8 h/THF | 65 |

Product 15 was not able to be isolated as a pure product from the reaction media, however, the transformation of substrates 3, 4, and 5 into their corresponding anti-3,4-selenoborated addition products 16, 17 and 18 allowed their isolation (Scheme 5). The addition of the Bpin moiety at the β-position of the substrate is confirmed by NMR data (11B NMR δ: 27–29 ppm). Two interesting points have to be addressed at this moment: a) while the addition of PhSe–Bpin takes place at room temperature, the addition of B2pin2 and PhMe2Si–Bpin requires up to 80 °C; b) while the addition of PhSe–Bpin involves PCy3 as an additive, the addition of B2pin2 and PhMe2Si–Bpin requires the most basic phosphine PBu3. Any attempt to perform the 3,4-addition of PhSe–Bpin, B2pin2 and PhMe2Si–Bpin to α,β-acetylenic ketones proved unsuccessful. The products derived from the anti-3,4-selenoboration were further transformed into their corresponding α-hydroselenated compounds, via the protodeboronation process with K2CO3 (ref. 19) at 70 °C, delivering compounds 19–22 in moderate values (Scheme 5), representing the first attempt to obtain these high value products.20 Similarly, we conducted the same selenoboration to alkynamides 6 and 7, and although anti-3,4-selenoborated products 23 and 24 were quantitatively determined by the NMR yield, their isolation was not successfully accomplished.
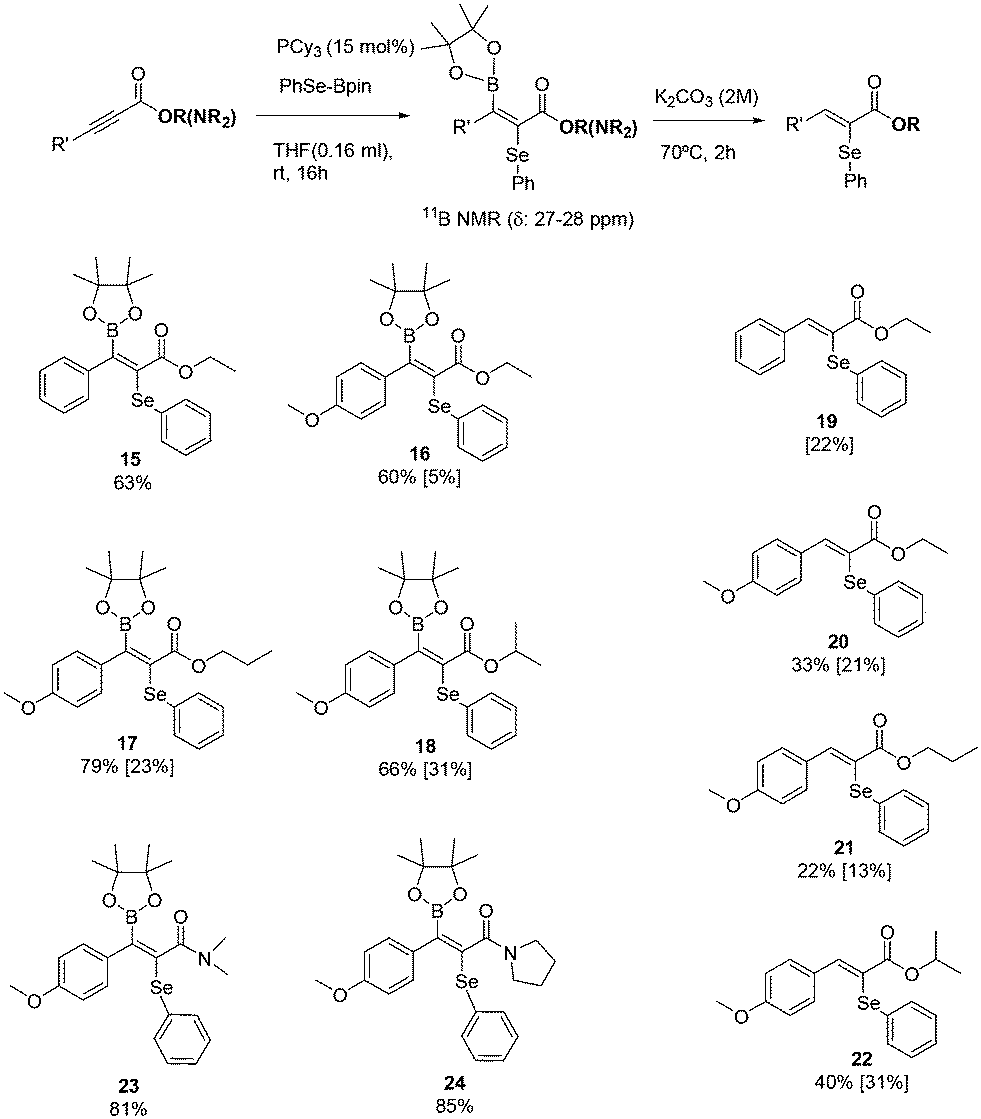 | ||
| Scheme 5 α-Selenation of α,β-acetylenic esters and amides via anti-3,4-selenoboration. Reaction conditions: α,β-acetylenic esters or amides (0.2 mmol), PhSe–Bpin (1.1 equiv.), PCy3 (15 mol%), THF (0.15 mL), rt for 16 h. For compound 15, the reaction time was 8 h. Yields were determined by 1H NMR analysis of the crude reaction mixture with naphthalene as an internal standard, which was added after the reaction. Isolated yield in brackets. | ||
In order to understand the mechanism of the anti-addition to alkynoates using catalytic amounts of phosphines and the switch-like behavior of selenoboration, we carried out a systematic DFT study. We initially explored a catalytic cycle where the phosphine acts as a catalyst, based on previous mechanisms proposed by Sawamura et al.10,11 for anti-selective carboboration, silaboration and diboration (Scheme 6). The phosphine catalyst initiates the reaction by conjugative addition to the alkynoate with the assistance of Lewis acidic boron, which activates the carbonyl group, yielding the zwitterionic allenolate intermediate C. Then, the terminal selenyl undergoes migration to form the ylide intermediate D, in which the enolate double bond has to rotate (D′) to allow the attack of ylidic carbon to the boron atom and to achieve anti-stereochemistry. Finally, from the cyclic borate E, the B–O bond is cleaved and the phosphine is eliminated to yield the product.
 | ||
| Scheme 6 Hypothetical mechanism for anti-3,4-addition of PhSe–Bpin to α,β-acetylenic ester adapted from the proposal for anti-selective carboboration, silaboration and diboration (ref. 10 and 11). Relative free-energies and barriers in kcal mol−1. | ||
We have computed the free-energy profile for the α-addition of PhSe–Bpin to the α,β-acetylenic ester 1-methyl-3-phenyl-2-propyn-1-one catalyzed by PMe3 as model phosphine assuming a Sawamura-type mechanism. The main energy values are depicted in Scheme 6, and the details are provided in the ESI† (Fig. S1). According to our calculations, the overall free-energy barrier for the formation of intermediate C is about 26.5 kcal mol−1, which is mainly associated with the entropic cost of the two consecutive additions to the alkynoate. In intermediate C, the phosphine addition changes the polarity of the triple bond making the α-position the most electrophilic, while the boron coordination to the carbonyl oxygen activates selenium nucleophilicity via a “push–pull” effect.21,22 As a result, the migration of the selenyl to produce D is computed to be a fast process with a low barrier of 4.1 kcal mol−1. The formation of ylide intermediate D is thermodynamically favored, with the structure lying 1.2 kcal mol−1 below the reactants. Then, in order to yield the observed stereoisomer, the bond between the carbonylic- and the α-carbon should rotate 180°. However, the computed free-energy barrier for the rotation is as high as 48.8 kcal mol−1. The D species has the typical structure of α-stabilized phosphonium ylides,23 in which the P–C bond is still covalent but has a significant polar interaction and its ylidic substituent preserves the double bond character. Thus, the Sawamura-type mechanism is less likely for selenoboration due to the kinetic hindrance associated with carbon–carbon bond rotation.
Alternatively, there are two novel mechanistic proposals in recent literature studies for related anti selective addition of boron compounds to alkynes. Nevertheless after close examination, none of them can be used to explain the results of selenoboration. According to the calculations by Santos et al.,9 the stereoselectivity of the transition-metal-free anti-diboration of alkynamides is due to a rapid carbon–carbon bond rotation process (Fig. 2b), which is thermodynamically favored. However, in that case the use of a strong base deprotonates the original amide group generating an intermediate in which the rotating carbon–carbon bond has a single bond character (dC–C = 1.491 Å) instead of the double bond character in the corresponding intermediate D (dC–C = 1.342 Å). Alternatively, Zhang et al.24 characterized computationally the mechanism of anti selective hydroboration of alkyne catalyzed by Ru complexes, showing that the formation of a stable metallacyclopropene intermediate, after hydrogen migration to the alkyne, explains the selectivity (Fig. 2c). Such an intermediate is not possible in our metal-free context. Thus, we propose a novel mechanism, in which the selenoborane auto-catalyzes the reaction.
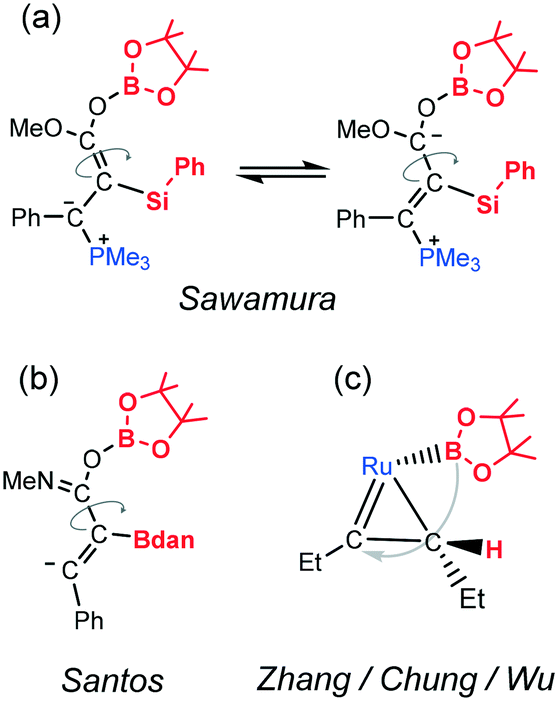 | ||
| Fig. 2 Comparison of key stereo-determining intermediates of different mechanistic proposals for anti selective addition of borane compounds to alkynes from ref. 10 (a), ref. 9 (b) and ref. 24 (c). | ||
The new mechanism is schematically presented in Scheme 7, while Fig. 3 and 4 show the free-energy profile and the key intermediates and transition states, respectively. The first part of the mechanism is analogous to Sawamura's proposal. Then, once intermediate D is formed, the catalytic process could be completed in three new steps: (1) the ylidic carbon of D acts as a Lewis base and coordinates to a second selenoborane molecule to yield intermediate F, (2) the terminal selenyl undergoes 1,4-migration to stereoselectively form intermediate G, and (3) the 1,2-elimination of PhSe–Bpin occurs at the carboxylic group to yield the final product and regenerate PhSe–Bpin species.
 | ||
| Scheme 7 Novel mechanistic proposal for anti-3,4-addition of PhSe–Bpin to α,β-acetylenic ester implying autocatalysis of one of the selenoborane substrates. | ||
 | ||
| Fig. 3 Free-energy profile (kcal mol−1) for the anti-3,4-addition of PhSe–Bpin to α,β-acetylenic ester via autocatalysis of selenoborane. | ||
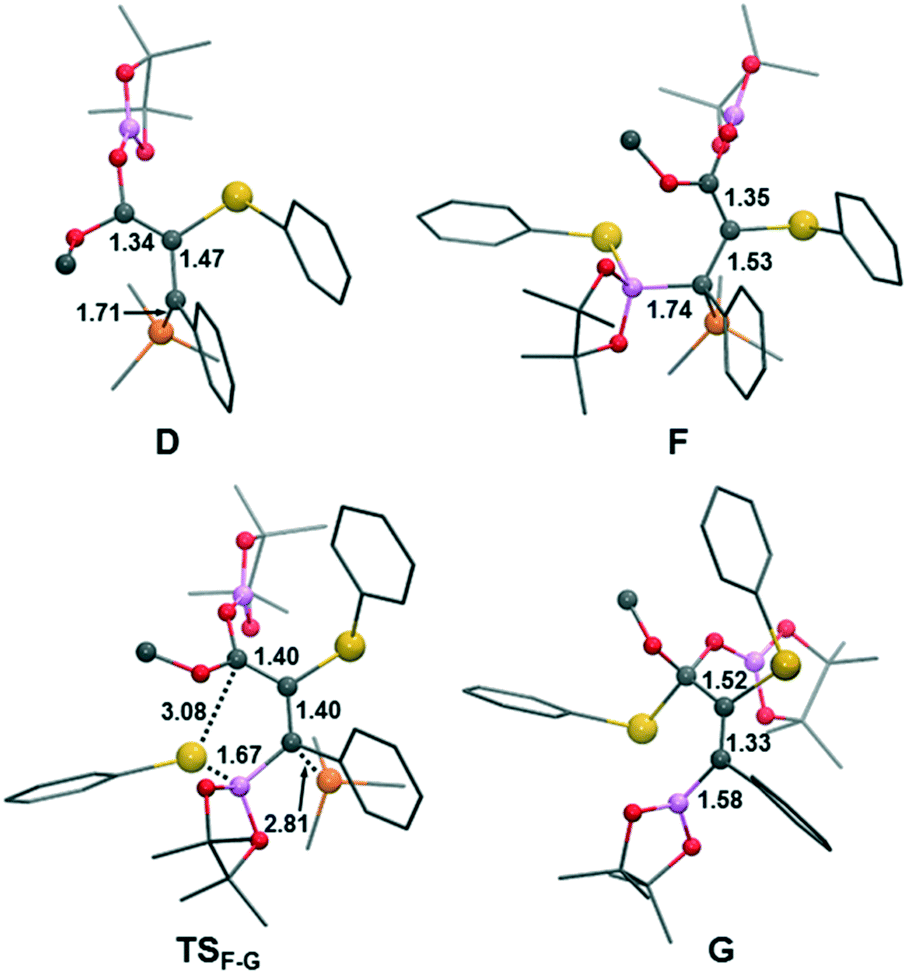 | ||
| Fig. 4 Molecular structures and main geometric parameters (Å) of the intermediates and transition states for the novel mechanism of anti-3,4-addition of PhSe–Bpin to α,β-acetylenic ester. | ||
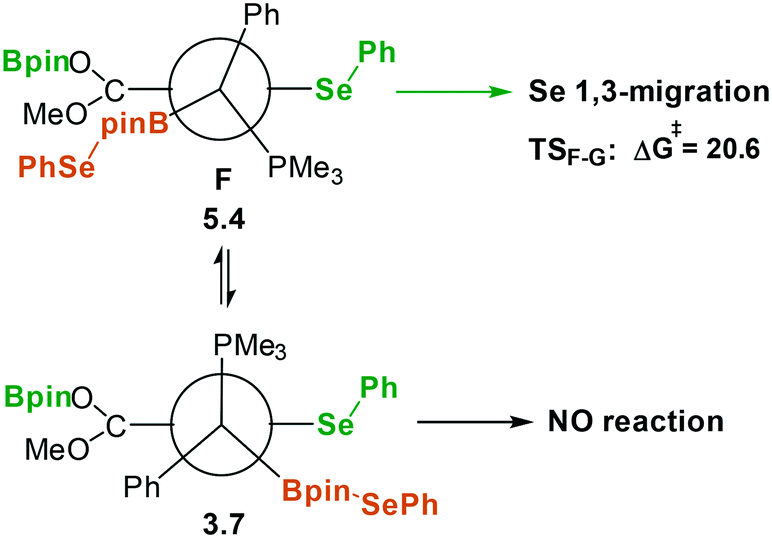
As shown in Fig. 3, the coordination of the second PhSe–Bpin molecule to the ylidic carbon of D yielding intermediate F is somewhat endergonic (+6.6 kcal mol−1) and it has a moderate free-energy barrier, 20.2 kcal mol−1. Analogous to the first selenyl migration, the boron coordination enhances the nucleophilic character of the –SePh moiety via the “push–pull” effect and it promotes the selenyl 1,4-migration to the electrophilic carboxylic carbon resulting in intermediate G and the concomitant release of the phosphine. Thus, the migration of the activated selenyl group has an accessible free energy barrier (20.6 kcal mol−1 from F to TSF–G). This step is exergonic by 17.1 kcal mol−1, with intermediate G lying 11.7 kcal mol−1 below the reactants.
More interestingly, the overall reaction leads to the anti-addition of selenyl and boryl moieties to the triple bond of the substrate (see Fig. 4). The stereoselectivity can be explained by analyzing the conformations of the species involved in the F → G transformation (see Fig. 5). Note that several conformational and enantiomeric paths can be considered. However, for the simplicity of our analysis, we have only discussed the conformational isomers connecting directly with the lowest-energy path, which progresses through transition state TSF–G. A detailed description of all the possible conformational and enantiomeric isomers is provided in Fig. S2 of the ESI.† As illustrated in Fig. 5, to reach the transition state for the selenyl migration, TSF–G, the PhSe–Bpin moiety coordinated to the ylidic carbon and the carboxylic group need to be syn to each other, forcing the initially added selenyl moiety (green color) and the subsequently added boryl moiety (orange color) to be anti to each other. Thus, although the coordination of PhSe–Bpin through transition state TSD–F can yield different conformers of F species, only those with the syn carboxyl–PhSeBpin arrangement would be reactive, and consequently, the formation of products with a cis arrangement of selenyl and boryl moieties is not possible (Fig. 5, bottom). Finally, intermediate G undergoes 1,2-elimination of PhSe–Bpin, which is built from the fragments of two different PhSe–Bpin molecules yielding the anti 3,4-selenoborated product.
 | ||
| Fig. 5 Schematic representation of the stereoselectivity-determining step yielding anti isomers. Relative free-energies and barrier in kcal mol−1. | ||
The computed free-energy profile of the overall mechanism in Fig. 4 indicates that the rate-determining process corresponds to the coordination of the second PhSe–Bpin molecule to the ylidic carbon and the subsequent 1,3-migration of the selenyl moiety to the carboxylic carbon (D + PhSe–Bpin → TSF–G) with an overall computed free energy barrier about 27 kcal mol−1. In the mechanism, the phosphine might play a dual role. First, it coordinates to the β-acetylenic carbon of the alkynoate inverting the polarity of the triple bond and directing the selenyl addition to the α-carbon of the alkyne. Second, it generates a nucleophilic ylide which is able to coordinate to a second PhSe–Bpin molecule through the Lewis acidic boron atom. Interestingly, the PhSe–Bpin acts as an autocatalyst allowing the overall anti addition of selenyl and boryl units and regenerating from the fragments of two different PhSe–Bpin species.
Experimentally, while the addition of PhSe–Bpin takes place at room temperature, the addition of B2pin2 and PhMe2Si–Bpin requires up to 80 °C. Thus, it would be interesting to rationalize the higher reactivity of the selenoborane reagent and to relate this to its stereoelectronic properties. Recently, we identified electronic and steric descriptors of trivalent boron compounds, which can be qualitatively and quantitatively related to their nucleophilic reactivity.25 Similarly, here we compare these descriptors in activated B–interelement compounds using the methoxy group as a model of the Lewis base: MeO− → Bpin–X (X = –SePh, –Bpin and –SiMe3). Replacing the interelement fragment from Bpin and SiMe to SePh, its negative charge (q[X]), measured as the sum of all the natural bond order (NBO) atomic charges, increases significantly from 0.00 and −0.12 to −0.56 a.u. Likewise, the interelement p/s population ratio of the atomic orbital in the B–X σ bond increases (from 2.1 and 1.1 for X = SePh and Bpin to 5.3 for X = SePh), which could indicate that the more the p character the more reactive the fragment as a nucleophile. On the other hand, we did not appreciate significant differences in the measurement of the steric bulkiness of the molecular environment using the distance-weighted volume (VW) parameter.26 Thus, both electronic descriptors, q[B] and p/s, indicate that selenoborane activated via a “push–pull” effect has an enhanced nucleophilic character. This might favor key reaction steps such as the migration of the selenyl moiety explaining why the reaction can take place at room temperature.
Finally, we carried out kinetic simulations in order to compare the mechanistic proposal (Fig. 3) with the experimental results, taking into account the differences in concentrations of the catalyst, the reagents and the solvent. The ESI† provides details on the simulation, in which rate constants emerged from DFT free-energy results via transition state theory. The initially estimated yield for the α-selenoborated product at room temperature after 8 h is very low (<1%), indicating that the computed barriers are too high. Besides the strong dependence of rate constants on computed energy values, one possible reason could be the limitations of phosphine modelling. In fact if we replace the model PMe3 phosphine by the experimental PCy3, the free-energy barrier for the first step (TS1P) is reduced by 3 kcal mol−1. Applying this correction factor to all the steps involving phosphine, the estimated yield of 15 becomes 50% in 8 h, which is in good agreement with the experimental one for the formation of 15 (63%, Scheme 5).
Additionally, we performed an experimental kinetic analysis of the selenoboration of 1 by monitoring the reaction by NMR spectroscopy (Fig. S1 and S2†). NMR conditions with a larger amount of THF solvent containing some quantity of water (∼0.021%) resulted in the formation of β-selenoboration product Z-8 using water as a protonation source. The overall free-energy barrier for α-vinyl selenide 15 is somewhat lower (27.2 or 24.2 kcal mol−1 for corrected and non-corrected values) than that for β-vinyl selenide Z-8 (27.5 kcal mol−1). However, at the early stages of the reaction, in which the concentration of the reactants is high, the formation of the β-isomer is faster because the α-isomer formation is slowed down by the low concentration of the phosphine catalyst. The kinetic simulation under NMR conditions derived from the energy values of the proposed mechanisms reproduces the main experimental features including the sigmoidal profile of α-selenated product 15 (see Fig. S3 and S4†). Thus, we can conclude that the kinetic simulations further support the proposed mechanism and that the nature of the solvent is important to determine the selectivity of the reaction.
Conclusions
We have shown that the regio- and the stereoselectivity of the addition of selenoboranes to α,β-acetylenic esters and amides can be controlled through the activation mode of the substrates in the absence of transition metal complexes. The non-catalyzed reaction provides access to β-vinyl selenides though 1,4-selenoboration promoted by the “push–pull” effect of B by the ester group. The addition of catalytic amounts of PCy3 phosphine switches the reaction to the anti-3,4-selenoboration with delivery of α-vinyl selenides by protodeboronation with MeOH. Computational studies discovered a novel mechanism (Scheme 7) which differed from previous mechanistic proposals for analogous anti-selective carboboration, silaboration and diboration (Scheme 6). The phosphine addition to the β position of the alkynoate modifies the regioselectivity favoring the 1,3-selenoboration and the production of the α-selenated phosphorus-ylide intermediate. Then, the autocatalytic action of a second PhSe–Bpin substrate determines the stereoselectivity and completes the anti-3,4-selenoboration reaction. Thus, the ylidic carbon coordinates to a second selenoborane molecule, which is activated via the “push–pull” effect to provide the 1,4-migration of the selenyl moiety to electrophilic carboxylic carbon. The conformational arrangement of the reacting selenyl and carboxyl groups imposes the resulting anti-stereoselectivity of the boryl and selenyl groups. Finally, the 1,2-elimination of PhSe–Bpin occurs at the carboxylic group to yield the final product and to regenerate PhSe–Bpin. The proposed mechanism is further supported by kinetic modelling using DFT free-energy values to obtain rate constants. The larger nucleophilic character of the SePh moiety compared to those of related B2pin2 and PhMe2Si–Bpin species allows the reaction to be performed under mild conditions.Experimental
Synthesis of alkynoates
An oven-dried Schlenk flask equipped with a magnetic stir bar was charged with 2 mmol of the appropriate alkyne which was dissolved in 3 mL of THF. At −78 °C, 1.05 eq. of nBuLi (1.6 M) was added dropwise. After stirring for 30 min, 1 eq. of chloroformate or carbamoyl chloride was added dropwise. After 8 h stirring the reaction was allowed to increase to room temperature and left to react overnight. The reaction was quenched with 2 mL of saturated Na2S2O4 and extracted 3 times with 5 mL of DCM. All the organic layers were collected, dried over MgSO4 and evaporated to dryness. The crude residue was analysed by 1H NMR. Then the products were purified by silica gel flash chromatography.Synthesis of α,β-acetylenic amides
An oven-dried Schlenk flask equipped with a magnetic stir bar was charged with 2 mmol of the appropriate alkyne which was dissolved in 3 mL of THF. At −78 °C, 1.05 eq. of nBuLi (1.6 M) was added dropwise. After stirring for 30 min, 1 eq. of carbamoyl chloride was added dropwise. After 8 h stirring the reaction was allowed to increase to room temperature and left to react overnight. The reaction was quenched with 2 mL of saturated Na2S2O4 and extracted 3 times with 5 mL of DCM. All the organic layers were collected, dried over MgSO4 and evaporated to dryness. The crude residue was analysed by 1H NMR. Then the products were purified by silica gel flash chromatography.Spectral data of ethyl 3-(4-methoxyphenyl)propiolate (3)
Flash column chromatography yielded 3 (367.6 mg, 90%) as a colourless oil. 1H NMR (CDCl3, 400 MHz) δ 7.53–7.49 (m, 2H), 6.87–6.84 (m, 2H), 4.26 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 1.32 (t, J = 7.1 Hz, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 161.5, 154.4, 135.0, 114.3, 111.4, 86.9, 80.2, 62.0, 55.4, 14.2. HRMS (ESI) for C12H13O3 [M + H]+: calculated: 205.0865, found: 205.0862. Characterization data matches the literature.27Spectral data of propyl 3-(4-methoxyphenyl)propiolate (4)
Flash column chromatography yielded 4 (388.5 mg, 89%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 7.55–7.51 (m, 2H), 6.89–6.85 (m, 2H), 4.17 (t, J = 6.8 Hz, 2H), 3.82 (s, 3H), 1.77–1.68 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 161.5, 154.6, 135.0, 114.3, 111.4, 87.0, 80.2, 67.6, 55.5, 22.0, 10.5. HRMS (ESI) for C13H15O3 [M + H]+: calculated: 219.1021, found: 219.1018.Spectral data of isopropyl 3-(4-methoxyphenyl)propiolate (5)
Flash column chromatography yielded 5 (379.8 mg, 87%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 7.56–7.52 (m, 2H), 6.89–6.86 (m, 2H), 5.15 (hept, J = 6.3 Hz, 1H), 3.83 (s, 3H), 1.33 (d, J = 6.3 Hz, 6H). 13C {1H} NMR (CDCl3, 100 MHz) δ 161.5, 154.1, 135.0, 114.4, 111.6, 86.6, 80.6, 69.9, 55.5, 21.9. HRMS (ESI) for C13H15O3 [M + H]+: calculated: 219.1021, found: 219.1017.Spectral data of 3-(4-methoxyphenyl)-N,N-dimethylpropiolamide (6)
Flash column chromatography yielded 6 (281.1 mg, 92%) as a colourless oil. 1H NMR (CDCl3, 400 MHz) δ 7.54–7.43 (m, 2H), 6.93–6.80 (m, 2H), 3.83 (s, 3H), 3.28 (s, 3H), 3.02 (s, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 161.0, 155.1, 134.2, 114.3, 112.6, 90.8, 81.0, 77.2, 55.5, 38.6, 34.3. HRMS (ESI) for C12H14NO2 [M + H]+: calculated: 204.1025, found: 204.1023.Spectral data of 3-(4-methoxyphenyl)-1-(pyrrolidin-1-yl)prop-2-yn-1-one (7)
Flash column chromatography yielded 7 (330.3 mg, 96%) as a colourless oil. 1H NMR (CDCl3, 400 MHz) δ 7.52–7.45 (m, 2H), 6.90–6.84 (m, 2H), 3.83 (s, 3H), 3.76–3.68 (m, 2H), 3.56–3.49 (m, 2H), 2.02–1.90 (m, 4H).13C {1H} NMR (CDCl3, 100 MHz) δ 161.0, 152.7, 134.3, 114.3, 112.7, 89.3, 82.1, 77.2, 55.5, 48.3, 45.5, 25.6, 24.9. HRMS (ESI) for C14H15NNaO2 [M + Na]+: calculated: 252.1000, found: 252.1001.General procedure for β-selenation of α,β-acetylenic esters and amides
In a glove-box, an oven-dried resealable vial equipped with a magnetic stir bar was charged with 0.2 mmol of the alkynoate or ynamide compound. Then, the vial was charged with 1.1 eq. of pinB–SePh dissolved in 0.15 mL of dry MeOH. After 16 h at 50 °C the reaction was evaporated to dryness. The crude residue was analysed by GC-MS and 1H NMR using naphthalene as an internal standard. Then the products were purified by silica gel flash chromatography.Spectral data of ethyl (Z)-3-phenyl-3-(phenylselanyl)acrylate (Z-8)
Flash column chromatography yielded Z-8 (49.0 mg, 74%) as a yellowish solid.1H NMR (CDCl3, 400 MHz) δ 7.25–7.20 (m, 2H), 7.11–6.96 (m, 8H), 6.32 (s, 1H), 4.30 (q, J = 7.1 Hz, 2H), 1.35 (t, J = 7.1 Hz, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 166.9, 161.4, 139.2, 136.4, 129.3, 128.7, 128.5, 128.1, 128.0, 127.6, 117.1, 60.7, 14.5. HRMS (ESI) for C17H17O2Se [M + H]+: calculated: 333.0394, found: 333.0391.Spectral data of methyl (Z)-3-(phenylselanyl)non-2-enoate (Z-9)
Flash column chromatography yielded Z-9 (51.4 mg, 79%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 7.69–7.65 (m, 2H), 7.43–7.38 (m, 1H), 7.36–7.32 (m, 2H), 6.16 (s, 1H), 3.76 (s, 3H), 2.17–2.13 (m, 2H), 1.34–1.26 (m, 2H), 1.17–1.10 (m, 2H), 1.05–0.97 (m, 4H), 0.79 (t, J = 7.3 Hz, 3H).13C {1H} NMR (CDCl3, 100 MHz) δ 167.7, 164.4, 137.7, 129.3, 129.2, 127.6, 113.2, 51.5, 37.9, 31.4, 29.8, 28.5, 22.5, 14.1.Spectral data of ethyl (Z)-3-(4-methoxyphenyl)-3-(phenylselanyl)acrylate (Z-10)
Flash column chromatography yielded Z-10 (52.0 mg, 72%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 7.24–7.21 (m, 2H), 7.11–7.07 (m, 1H), 7.04–6.95 (m, 4H), 6.58–6.54 (m, 2H), 6.30 (s, 1H), 4.28 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 1.34 (t, J = 7.1 Hz, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 166.9, 161.0, 159.6, 136.1, 131.8, 130.2, 129.7, 128.5, 127.9, 116.7, 113.0, 60.6, 55.3, 14.5. HRMS (ESI) for C18H19O3Se [M + H]+: calculated: 363.0499, found: 363.0495.Spectral data of propyl (Z)-3-(4-methoxyphenyl)-3-(phenylselanyl)acrylate (Z-11)
Flash column chromatography yielded Z-11 (54.0 mg, 72%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 7.24–7.22 (m, 2H), 7.11–7.07 (m, 1H), 7.03–6.95 (m, 4H), 6.57–6.54 (m, 2H), 6.31 (s, 1H), 4.19 (t, J = 6.7 Hz, 2H), 3.68 (s, 3H), 1.78–1.69 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H).13C {1H} NMR (CDCl3, 100 MHz) δ 167.0, 160.9, 159.5, 136.1, 131.7, 130.2, 129.7, 128.5, 127.8, 116.7, 113.0, 66.2, 55.3, 22.2, 10.6. HRMS (ESI) for C19H21O3Se [M + H]+: calculated: 377.0656, found: 377.0651.Spectral data of isopropyl (Z)-3-(4-methoxyphenyl)-3-(phenylselanyl)acrylate (Z-12)
Flash column chromatography yielded Z-12 (51.8 mg, 69%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 7.23–7.21 (m, 2H), 7.11–7.07 (m, 1H), 7.03–6.95 (m, 4H), 6.57–6.53 (m, 2H), 6.27 (s, 1H), 5.17 (hept, J = 6.2 Hz, 1H), 3.67 (s, 3H), 1.32 (d, J = 6.3 Hz, 6H). 13C {1H} NMR (CDCl3, 100 MHz) δ 166.5, 160.6, 159.5, 136.1, 131.8, 130.2, 129.7, 128.5, 127.8, 117.2, 113.0, 67.9, 55.3, 22.2. HRMS (ESI) for C19H21O3Se [M + H]+: calculated: 377.0656, found: 377.0648.Spectral data of (Z)-3-(4-methoxyphenyl)-N,N-dimethyl-3-(phenylselanyl)acrylamide (Z-13)
Flash column chromatography yielded Z-13 (54.8 mg, 76%) as a colourless oil. 1H NMR (CDCl3, 400 MHz) δ 7.26–7.20 (m, 2H), 7.09–6.95 (m, 5H), 6.60 (s, 1H), 6.58–6.54 (m, 2H), 3.68 (s, 3H), 3.08 (brs, 6H). 13C {1H} NMR (CDCl3, 100 MHz) δ 167.1, 159.3, 156.1, 135.9, 132.6, 130.8, 130.2, 128.3, 127.4, 117.1, 113.0, 55.3, 24.9. HRMS (ESI) for C18H20NO2Se [M + H]+: calculated: 362.0659, found: 362.0651.Spectral data of (Z)-3-(4-methoxyphenyl)-3-(phenylselanyl)-1-(pyrrolidin-1-yl)prop-2-en-1-one (Z-14)
Flash column chromatography yielded Z-14 (49.2 mg, 69%) as a colourless oil. 1H NMR (CDCl3, 400 MHz) δ 7.25–7.20 (m, 2H), 7.08–7.03 (m, 1H), 7.01–6.93 (m, 4H), 6.58–6.52 (m, 2H), 6.45 (s, 1H), 3.67 (s, 3H), 3.55 (brs, 4H), 1.93 (brs, 4H). 13C {1H} NMR (CDCl3, 100 MHz) δ 165.3, 159.2, 156.9, 136.2, 132.6, 130.9, 130.2, 128.3, 127.5, 117.6, 112.9, 77.2, 55.3, 46.4 (brs), 25.7 (brs). HRMS (ESI) for C20H22NO2Se [M + H]+: calculated: 388.0816, found: 388.0811.General procedure for the insertion of pinB-SeR into α,β-acetylenic esters and amides
In the glove-box, an oven-dried resealable vial equipped with a magnetic stir bar was charged with 0.2 mmol of the alkynoate or ynamide compound. Then, 15 mol% tricyclohexylphosphine in 0.15 ml of dry THF was added. After stirring in the glove-box for 5 min the vial was charged with 1.1 eq. of pinB–SePh. After 16 h at room temperature the reaction was evaporated to dryness. The crude residue was analysed by GC-MS and 1H NMR using naphthalene as an internal standard. Then the products were purified by silica gel flash chromatography.Spectral data of ethyl (Z)-3-(4-methoxyphenyl)-2-(phenylselanyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)acrylate (16)
Flash column chromatography yielded 16 (4.9 mg, 5%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 7.29–7.35 (m, 3H), 7.19–7.16 (m, 3H), 6.92–6.88 (m, 2H), 4.01 (q, J = 7.2 Hz, 2H), 3.82 (s, 3H), 1.29 (s, 12H), 0.94 (t, J = 7.1 Hz, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 169.4, 159.8, 134.5, 132.9, 131.6, 131.0, 129.9, 129.0, 127.2, 113.5, 83.9, 62.8, 55.4, 25.0, 13.7. 11B NMR (128.3 MHz, CDCl3) δ 28.3. HRMS (ESI) for C24H30BO5Se [M + H]+: calculated: 489.1352, found: 489.1358.Spectral data of propyl (Z)-3-(4-methoxyphenyl)-2-(phenylselanyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)acrylate (17)
Flash column chromatography yielded 17 (23.1 mg, 23%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 7.40–7.33 (m, 3H), 7.17–7.14 (m, 3H), 6.91–6.88 (m, 2H), 3.93 (t, J = 6.7 Hz, 2H), 3.82 (s, 3H), 1.41–1.32 (m, 2H), 1.29 (s, 12H), 0.70 (t, J = 7.4 Hz, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 169.9, 159.8, 134.3, 132.3, 131.5, 131.2, 129.9, 129.0, 128.3 (bs), 127.0, 113.4, 83.8, 68.6, 55.3, 25.0, 21.6, 10.3. 11B NMR (128.3 MHz, CDCl3) δ 27.9. HRMS (ESI) for C50H62B2NaO10Se2 [2M + Na]+: calculated: 1027.2757, found: 1027.2789.Spectral data of isopropyl (Z)-3-(4-methoxyphenyl)-2-(phenylselanyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)acrylate (18)
Flash column chromatography yielded 18 (31.1 mg, 31%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 7.41–7.36 (m, 3H), 7.18–7.15 (m, 3H), 6.92–6.89 (m, 2H), 4.85 (hept, J = 6.3 Hz, 1H), 3.82 (s, 3H), 1.29 (s, 12H), 0.95 (d, J = 6.3 Hz, 6H). 13C {1H} NMR (CDCl3, 100 MHz) δ 169.6, 159.9, 132.6, 131.5, 131.4, 130.0, 129.0, 128.3, 127.1, 113.4, 83.6, 71.3, 55.3, 25.0, 21.3. 11B NMR (128.3 MHz, CDCl3) δ 27.8. HRMS (ESI) for C25H31BNaO5Se [M + Na]+: calculated: 525.1327, found: 525.1332.Spectral data of (Z)-3-(4-methoxyphenyl)-N,N-dimethyl-2-(phenylselanyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)acrylamide (23)
No isolated product 23 was obtained. 1H NMR (CDCl3, 400 MHz) δ 7.25–7.21 (m, 2H), 7.19–7.13 (m, 2H), 7.09–6.98 (m, 3H), 6.63–6.57 (m, 2H), 3.69 (s, 3H), 3.09 (brs, 6H), 1.06 (s, 13H). 11B NMR (128.3 MHz, CDCl3) δ 27.3.Spectral data of (Z)-3-phenyl-2-(phenylselanyl)-1-(pyrrolidin-1-yl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)prop-2-en-1-one (24)
No isolated product 24 was obtained. 1H NMR (CDCl3, 400 MHz) δ 7.24–7.19 (m, 2H), 7.14–7.09 (m, 2H), 7.08–6.95 (m, 3H), 6.60–6.54 (m, 2H), 3.68 (s, 3H), 3.56 (brs, 4H), 1.92 (brs, 4H), 1.04 (s, 12H).General procedure for one-pot α-selenation of alkynoates
In the glove-box, an oven-dried resealable vial equipped with a magnetic stir bar was charged with 0.2 mmol of the alkynoate or ynamide compound. Then, 15 mol% tricyclohexylphosphine in 0.15 ml of dry THF was added. After stirring in the glove-box for 5 min the vial was charged with 1.1 eq. of pinB–SePh. After 16 h at room temperature the reaction was evaporated to dryness. The crude residue was dissolved in 2 mL of THF and 0.2 mL of K2CO3 (2 M) water solution was added dropwise. The reaction was heated to reflux for 2 h. Then the reaction was extracted with DCM and filtered through a pad of Celite and MgSO4. The solvent was removed and the residue was analysed by GC-MS and 1H NMR using naphthalene as an internal standard. Then the products were purified by silica gel flash chromatography.Spectral data of ethyl (Z)-3-phenyl-2-(phenylselanyl)acrylate (19)
Flash column chromatography yielded 19 (14.6 mg, 22%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 7.66–7.64 (m, 2H), 7.42–7.37 (m, 5H), 7.22–7.19 (m, 3H), 4.07 (q, J = 7.1 Hz, 2H), 1.04 (t, J = 7.1 Hz, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 166.8, 145.0, 138.4, 135.3, 134.4, 131.9, 130.4, 129.2, 128.3, 127.2, 124.5, 62.0, 13.9. HRMS (ESI) for C17H17O2Se [M + H]+: calculated: 333.0394, found: 333.0392.Spectral data of ethyl (Z)-3-(4-methoxyphenyl)-2-(phenylselanyl)acrylate (20)
Flash column chromatography yielded 20 (15.2 mg, 21%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 8.17 (s, 1H), 7.75–7.72 (m, 2H), 7.40–7.37 (m, 2H), 7.22–7.18 (m, 3H), 6.93–6.90 (m, 2H), 4.08 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 1.06 (t, J = 7.1 Hz, 3H). 13C {1H} NMR (CDCl3, 100 MHz) δ 167.0, 161.0, 145.8, 132.7, 131.3, 129.8, 129.2, 127.7, 127.0, 113.8, 70.7, 61.9, 55.5, 14.0. HRMS (ESI) for C18H19O3Se [M + H]+: calculated: 363.0499, found: 363.0507.Spectral data of propyl (Z)-3-(4-methoxyphenyl)-2-(phenylselanyl)acrylate (21)
Flash column chromatography yielded 21 (9.7 mg, 13%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 8.20 (s, 1H), 7.77–7.73 (m, 2H), 7.39–7.36 (m, 2H), 7.22–7.18 (m, 3H), 6.93–6.89 (m, 2H), 4.00 (t, J = 6.7 Hz, 2H), 3.83 (s, 3H), 1.53–1.44 (m, 2H), 0.81 (t, J = 7.4 Hz, 3H).13C {1H} NMR (CDCl3, 100 MHz) δ 167.1, 161.0, 146.2, 136.0, 132.7, 131.0, 129.2, 127.7, 126.8, 120.4, 113.8, 67.6, 55.5, 21.9, 10.5. HRMS (ESI) for C19H21O3Se [M + H]+: calculated: 377.0656, found: 377.0651.Spectral data of isopropyl (Z)-3-(4-methoxyphenyl)-2-(phenylselanyl)acrylate (22)
Flash column chromatography yielded 22 (23.3 mg, 31%) as a yellowish oil. 1H NMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 7.73–7.70 (m, 2H), 7.40–7.37 (m, 2H), 7.23–7.17 (m, 3H), 6.94–6.90 (m, 2H), 4.91 (hept, J = 6.2 Hz, 1H), 3.84 (s, 3H), 1.04 (d, J = 6.3 Hz, 6H). 13C {1H} NMR (CDCl3, 100 MHz) δ 166.5, 160.9, 145.3, 132.6, 131.4, 130.2, 129.2, 127.8, 126.9, 121.4, 113.8, 69.5, 55.5, 21.5. HRMS (ESI) for C19H21O3Se [M + H]+: calculated: 377.0656, found: 377.0651.Computational details
Geometry optimizations and transition state searches were performed with the Gaussian09 package.28 The quantum mechanics calculations were performed within the framework of density functional theory (DFT)29 by using the hybrid M06-2X functional30 and a standard 6-311G(d,p) basis set.31 Full geometry optimizations were performed without constraints. The nature of the stationary points encountered was characterized either as minima or transition states by means of harmonic vibrational frequency analysis. The zero-point, thermal, and entropy corrections were evaluated to compute the Gibbs free energies (T = 298 K, p = 1 bar). The selected method is analogous to that reported in ref. 18 and it allows a straightforward comparison of the results. The kinetic simulation was carried out with Acuchem software32 and all rate constants were calculated using the Eyring approximation and transition state theory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present research was supported by the Spanish Ministerio de Economia y Competitividad (MINECO) through projects CTQ2016-80328-P (EF) and CTQ2014-52774-P (JJC), the NSERC (SAW), and the Generalitat de Catalunya through project 2014SGR199 (JJC).Notes and references
- For recent reviews in boron chemistry and its synthetic applications, see: (a) L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848 RSC; (b) E. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef PubMed; (c) A. B. Cuenca, R. I. Shishido, H. Ito and E. Fernandez, Chem. Soc. Rev., 2017, 46, 415 RSC.
- H. Yoshida, ACS Catal., 2016, 6, 1799 CrossRef.
- W. J. Jang, W. L. Lee, J. H. Moon, J. Y. Lee and J. J. Yun, Org. Lett., 2016, 18, 1390 CrossRef PubMed.
- B. Sundararaju and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 15050 CrossRef PubMed.
- T. Ohmura, Y. Yamamoto and N. Miyaura, J. Am. Chem. Soc., 2000, 122, 4990 CrossRef.
- (a) C. Gunanathan, M. Holscher, F. Pan and W. Leitner, J. Am. Chem. Soc., 2012, 134, 14349 CrossRef PubMed; (b) J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2012, 18, 1512 CrossRef PubMed.
- Y. Nagashima, K. Hirano, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2014, 136, 8532 CrossRef PubMed.
- M. Nogami, K. Hirano, M. Kanai, C. Wang, T. Saito, K. Miyamoto, A. Muranaka and M. Uchiyama, J. Am. Chem. Soc., 2017, 139, 12358 CrossRef PubMed.
- (a) A. Verma, R. F. Snead, Y. Dai, C. Slebodnick, Y. Yang, H. Yu, F. Yao and W. L. Santos, Angew. Chem., Int. Ed., 2017, 56, 5111 CrossRef PubMed; (b) R. Fritzemeier and W. L. Santos, Chem. – Eur. J., 2017, 23, 15534 CrossRef PubMed.
- K. Nagao, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2014, 136, 10605 CrossRef PubMed.
- K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 1304 CrossRef PubMed.
- (a) G. Perin, E. J. Lenardão, R. G. Jacob and R. B. Panatieri, Chem. Rev., 2009, 109, 1277 CrossRef PubMed; (b) P. H. Menezes and G. Zeni, Vinyl Selenides in Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd., Hoboken, USA, 2011 Search PubMed; (c) M. Palomba, L. Bagnoli, F. Marini, C. Santi and L. Sancineto, Phosphorous, sulfur and silicon, 2016, vol. 191, p. 235 Search PubMed.
- S. Kawaguchi, M. Kotani, S. Atobe, A. Nomoto, M. Sonoda and A. Ogawa, Organometallics, 2011, 30, 6766 CrossRef.
- B. Battistelli, L. Testaferri, M. Tiecco and C. Santi, Eur. J. Org. Chem., 2011, 10, 1848 CrossRef.
- (a) M. Renard and L. Hevesi, Tetrahedron, 1985, 41, 5939 CrossRef; (b) J. V. Comasseto and C. A. Brandt, Synthesis, 1987, 146 CrossRef; (c) O. S. D. Barros, E. S. Lang, C. A. F. de Oliveira, C. Peppe and G. Zeni, Tetrahedron Lett., 2002, 43, 7921 CrossRef; (d) G. Perin, R. G. Jacob, F. de Azambuja, G. V. Botteselle, G. M. Siqueira, R. A. Freitag and E. J. Lenardao, Tetrahedron Lett., 2005, 46, 1679 CrossRef; (e) E. J. Lenardao, M. S. Silva, S. R. Mendes, F. de Azambuja, R. G. Jacob, P. C. S. dos Santos and G. Perin, J. Braz. Chem. Soc., 2007, 18, 943 CrossRef.
- (a) S. A. Westcott, J. D. Webb, D. I. McIsaac and C. M. Vogels, WO Pat., 2006/089402A1 Search PubMed; (b) J. A. Fernández-Salas, S. Manzini and S. P. Nolan, Chem. Commun., 2013, 49, 5829 RSC.
- X. Sanz, Ch. M. Vogels, A. Decken, C. Bo, S. A. Westcott and E. Fernández, Chem. Commun., 2014, 50, 8420 RSC.
- M. G. Civit, X. Sanz, Ch. M. Vogels, C. Bo, S. A. Westcott and E. Fernández, Adv. Synth. Catal., 2015, 357, 3098 CrossRef.
- W. Torres-Delgado, F. Shahin, M. J. Ferguson, R. Mc Donald, G. He and E. Rivard, Organometallics, 2016, 35, 2140 CrossRef.
- To the best of our knowledge, there is only one example of the preparation of methyl-3-phenyl-2-(phenylselano)acrylate as a mixture of the E/Z isomers in ref. 14.
- (a) J. Cid, H. Gulyás, J. J. Carbó and E. Fernández, Chem. Soc. Rev., 2012, 41, 3558 RSC; (b) R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594 RSC.
- (a) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158 CrossRef PubMed; (b) C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyás and E. Fernández, Chem. – Eur. J., 2012, 18, 112 CrossRef PubMed; (c) X. Sanz, G. M. Lee, C. Pubill-Ulldemolins, A. Bonet, H. Gulás, S. A. Westcott, C. Bo and E. Fernández, Org. Biomol. Chem., 2013, 11, 7004 RSC; (d) J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2014, 20, 3616 CrossRef PubMed; (e) N. Miralles, J. Cid, A. B. Cuenca, J. J. Carbó and E. Fernández, Chem. Commun., 2015, 51, 1693 RSC.
- (a) A. Lledós, J. J. Carbó and E. P. Urriolabeitia, Inorg. Chem., 2001, 40, 4913 CrossRef; (b) E. Serrano, C. Vallés, J. J. Carbó, A. Lledós, T. Soler, R. Navarro and E. P. Urriolabeitia, Organometallics, 2006, 25, 4653 CrossRef; (c) L. R. Falvello, J. C. Ginés, J. J. Carbó, A. Lledós, R. Navarro, T. Soler and E. P. Urriolabeitia, Inorg. Chem., 2006, 45, 6803 CrossRef PubMed.
- L. J. Song, T. W. Wang, X. Zhang, L. W. Chung and Y.-D. Wu, ACS Catal., 2017, 7, 1361 CrossRef.
- (a) J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2012, 18, 12794 CrossRef PubMed; (b) D. García-López, J. Cid, R. Marqués, E. Fernández and J. J. Carbó, Chem. – Eur. J., 2017, 23, 5066 CrossRef PubMed.
- (a) S. Aguado-Ullate, S. Saureu, L. Guasch and J. J. Carbó, Chem. – Eur. J., 2012, 18, 995 CrossRef PubMed; (b) S. Aguado-Ullate, M. Urbano, I. Villaba, E. Pires, J. I. García, C. Bo and J. J. Carbó, Chem. – Eur. J., 2012, 18, 14026 CrossRef PubMed , MolQuO application: http://rodi.urv.es/~carbo/quadrants/index.html accessed November 2017.
- P. R. Andrews, R. I. Brinkworth, A. C. Partridge and J. A. Reiss, Aust. J. Chem., 1988, 41, 1717 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02; Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. G. Parr and W. Yang, in Density Functional Theory of Atoms and Molecule, Oxford University Press, Oxford, UK, 1989 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- (a) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef; (b) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef; (c) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef.
- W. Braun, J. T. Herron and D. K. Kahaner, Int. J. Chem. Kinet., 1988, 20, 51 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, X-ray data and computational details. CCDC 1517230. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cy02295f |
| This journal is © The Royal Society of Chemistry 2018 |