
Highly active, separable and recyclable bipyridine iridium catalysts for C–H borylation reactions†
Hind
Mamlouk
 a,
Jakkrit
Suriboot
b,
Praveen Kumar
Manyam
a,
Ahmed
AlYazidi
a,
David E.
Bergbreiter
*b and
Sherzod T.
Madrahimov
*a
a,
Jakkrit
Suriboot
b,
Praveen Kumar
Manyam
a,
Ahmed
AlYazidi
a,
David E.
Bergbreiter
*b and
Sherzod T.
Madrahimov
*a
aDepartment of Chemistry, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar. E-mail: sherzod.madrahimov@qatar.tamu.edu
bDepartment of Chemistry, Texas A&M University, College Station, TX 77842-3012, USA. E-mail: bergbreiter@tamu.edu
First published on 1st November 2017
Abstract
Iridium complexes generated from Ir(I) precursors and PIB oligomer functionalized bpy ligands efficiently catalyzed the reactions of arenes with bis(pinacolato)diboron under mild conditions to produce a variety of arylboronate compounds. The activity of this PIB bound homogeneous catalyst is similar to that of an original non-recyclable catalyst which allows it to be used under milder conditions than other reported recyclable catalysts. This oligomer-supported Ir catalyst was successfully recovered through biphasic extraction and reused for eight cycles without a loss of activity. Biphasic separation after the initial use of the catalyst led to an insignificant amount of iridium leaching from the catalyst to the product, and no iridium leaching from the catalyst was observed in the subsequent recycling runs. Arylboronate products obtained after extraction are sufficiently pure as observed by 1H and 13C-NMR spectroscopy that they do not require further purification.
The development of highly active, recyclable catalysts for industrially relevant chemical processes is very important for achieving a sustainable future.1 For example, over the past few years, there has been a concerted effort to design recyclable catalysts from known homogeneous systems by tethering them to platforms like metal organic frameworks.2 This latter approach to the development of recyclable catalysts has also been applied to the iridium catalyzed C–H borylation reaction developed by Hartwig, which converts hydrocarbon feedstocks to precursors that are applied for the production of materials and medicinal and commodity chemicals.3 Previous reports that inspired this work employed metal organic framework materials or periodic mesoporous organosilica or organosilica nanotube supports that incorporated iridium bipyridine (bpy) complexes that served as heterogeneous recyclable catalysts.2d,4 These heterogeneous systems were highly recyclable. However, they generally had activities that required higher temperatures than the original homogeneous system. This was presumably due to the electronic constraints associated with tethering the catalyst to the platforms, the heterogeneous nature of the recyclable catalyst, or some combination of these factors.
Herein, we report a highly soluble, recyclable, and homogeneous aromatic C–H borylation catalyst based on a bpy ligand carrying a polyisobutylene (PIB) substituent. The Ir complex formed with this soluble polymer shows a similar activity to that of an original catalyst. We reason that the higher activity of this polyolefin-oligomer bound catalyst vis-à-vis other catalysts immobilized on solid supports stems from its homogeneity and the fact that polyolefin-based ligands form catalysts and metal complexes whose electronic and dynamic properties closely mimic known homogeneous transition metal complexes.5 The results obtained here show that the iridium catalyst formed using a PIB-bpy ligand mimics the original system that employed a 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbpy) ligand (Fig. 1).
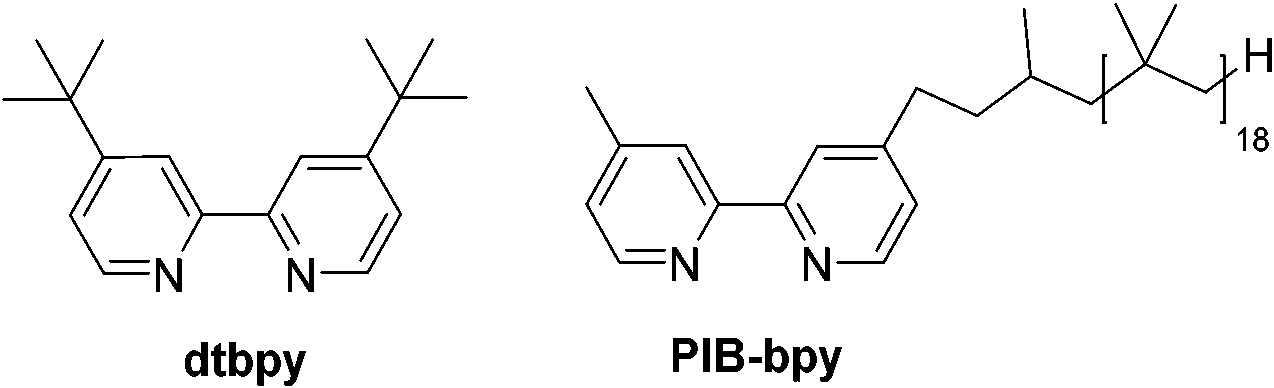 | ||
| Fig. 1 4,4′-Di-tert-butyl-2,2′-dipyridyl (dtbpy) and polyisobutylene functionalized bpy (PIB-bpy). | ||
Based on a prior work that used PIB-bound bpy ligands,6 we synthesized a PIB-bpy ligand from PIB oligomers as part of a broader project developing PIB-ligands that could increase the solubility of nanoparticles in organic solvents.7 This synthesis afforded PIB-bpy ligands with a molecular weight of 1690 g mol−1, as estimated by 1H-NMR spectroscopy (ESI,† Fig. S1). These ligands contain predominantly one PIB group. As noted in the prior work, this PIB-bpy ligand contains a small amount of bpy with PIB groups at the 4 and 4′ positions. While the bpy ligands formed did not prove to be the most optimal ligands for magnetic nanoparticle solubilization, we reasoned that these highly soluble ligands might be useful in iridium-catalyzed C–H activation chemistry. In addition, unlike other common soluble organic polymer supports like polystyrene or poly(ethylene glycol), polyisobutylene only has secondary and primary C–H bonds, so this support is less likely to be a substrate for these iridium C–H activation catalysts.
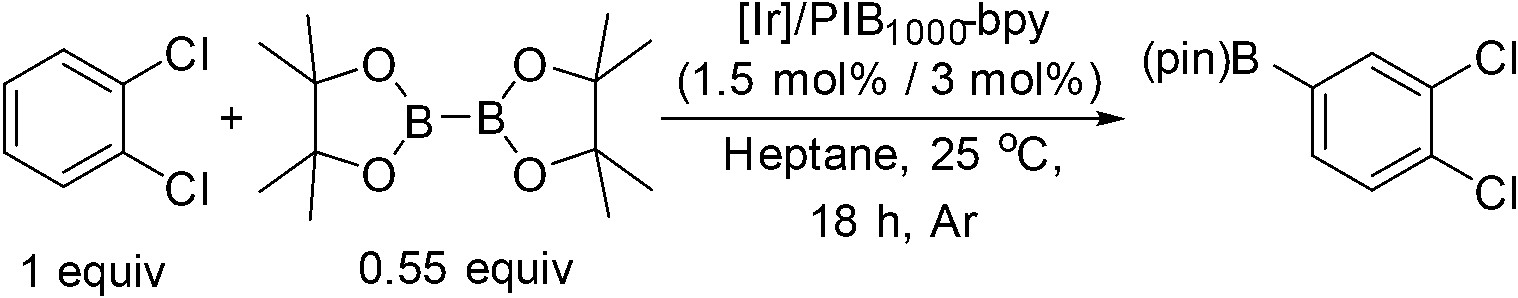
To investigate this possibility, we started our studies by optimizing the iridium precursor for the C–H borylation of 1,2-dichlorobenzene. The optimization reactions were con)ducted in a heptane solution at ambient temperature with a 3 mol% catalyst loading and stopped after 18 h. The [Ir(OMe)(COD)]2 complex was found to be the best iridium source for aromatic C–H borylation among the three iridium precursors that were tested, as shown in Table 1, affording a 90% yield of the isolated product. This 90% yield was later further improved to 95% with an optimized procedure for biphasic extraction (Table 2, entry 1). This result paralleled what was seen to be optimal for the original system that employed a dtbpy ligand.3a Since the synthesis of PIB-bpy produces a minor amount of 2,2′-bipyridine bearing two PIB-derived substituents at the 4- and 4′-positions, we isolated this bpy with two PIB groups by chromatography and examined it as the ligand in our reaction.6 The reactions with this ligand did not display any improvement over the reactions with PIB-bpy and showed that an extra PIB functionalization does not impact the steric and electronic properties of the ligand and the presence of one PIB functionalization is sufficient for efficient recycling of the catalyst. This result is consistent with our prior observations that noted the equivalence of a Ru catalyst with either one or two PIB groups on a bpy ligand.6
|
|
||
|---|---|---|
| Entry | [Ir] | Yield (%) |
| 1 | [IrCl(COE)2]2 | 83 |
| 2 | [Ir(OMe)(COD)]2 | 90 |
| 3 | [IrCl(COD)]2 | 68 |
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Substrate | T (°C) | Yield (%) | Substrate | T (°C) | Yield (%) | ||
a Conditions: A mixture of PIB-bpy (0.03 mmol), Pin2B2 (1.1 mmol), [Ir(OMe)(COD)2] (0.015 mmol) and the substrate (2 mmol) was stirred in dry heptane (3 mL) at 25 °C or at 80 °C under Ar, for 18 h.
b
p-isomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :m-isomer = 40:60.
c
m-isomer:3,5-bis-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)toluene:p-isomer = 60:5:35.
d
m-isomer:3,5-bis-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)anisole:p-isomer = 65:20:15.
e A mixture of PIB-bpy (0.03 mmol), Pin2B2 (1.1 mmol), [Ir(OMe)(COD)2] (0.015 mmol) and benzene (30 mmol), neat conditions, at 80 °C, Ar, for 18 h. :m-isomer = 40:60.
c
m-isomer:3,5-bis-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)toluene:p-isomer = 60:5:35.
d
m-isomer:3,5-bis-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)anisole:p-isomer = 65:20:15.
e A mixture of PIB-bpy (0.03 mmol), Pin2B2 (1.1 mmol), [Ir(OMe)(COD)2] (0.015 mmol) and benzene (30 mmol), neat conditions, at 80 °C, Ar, for 18 h.
|
|||||||
| 1 |
|
25 | 95 | 6 |
|
80 | 82c |
| 2 |
|
80 | 92b | 7 |
|
80 | 81 |
| 3 |
|
25 | 70 | 8 |

|
80 | 92d |
| 4 |
|
80 | 86 | 9 |
|
25 | 84 |
| 5 |
|
80 | 87 | 10 |
|
80 | 92e |
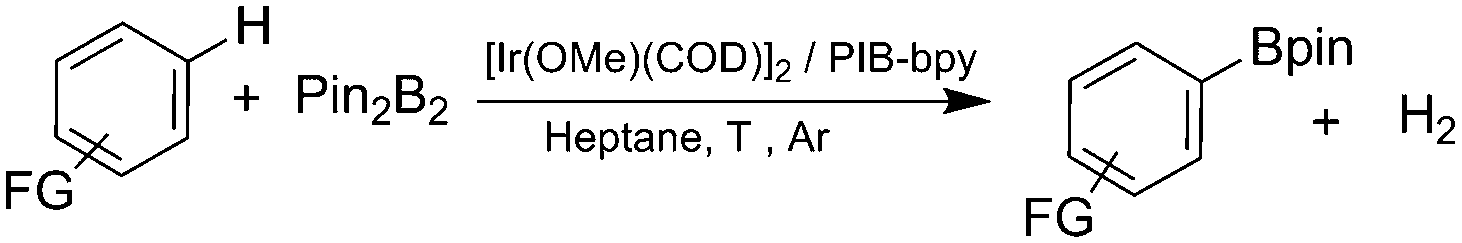
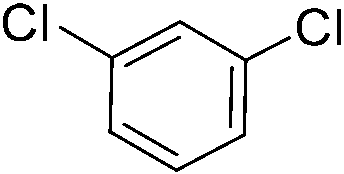
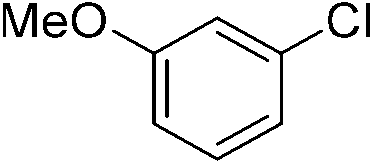
Once we identified the most suitable catalyst precursor to conduct the C–H borylation reactions, we started testing the PIB-bpy based catalyst for recyclability. We carried out the catalyst recycling tests for the C–H borylation of 1,2-dichlorobenzene with PIB-bpy and [Ir(OMe)(COD)]2 under the same conditions used for the catalyst precursor optimization as described above. Once the reactions were complete, the solvent was evaporated from the red solution of the catalyst, leaving a small amount of heptane in the reaction mixture. The product was extracted with acetonitrile, taking advantage of the high phase-selective solubility of the PIB bound species in heptane versus moderately polar solvents to insure catalyst/product separation.7b The extraction was performed by shaking the reaction mixture with 2 mL of acetonitrile in a 15 mL centrifuge tube. After centrifugation, the nonpolar phase containing the red catalyst was physically separated from the acetonitrile phase containing the C–H activation borylation product by simple decantation (Fig. 3). We repeated the extraction four times to insure full recovery of the product. The product, isolated after removing acetonitrile under reduced pressure, was >97% pure. The only contaminant was a trace (<3%) of the starting material remaining based on 1H-NMR spectroscopy. The catalyst was also fully recovered with this extraction procedure. This recovered catalyst was dried and reused in subsequent recycling runs without the addition of any more iridium precursors or ligands. This recycled catalyst maintains the same dark red color. We found that the catalyst activity was conserved for at least 8 catalytic runs, giving a ca. 90% isolated yield of the product in each of the runs (Fig. 2). We also tested the catalyst recyclability for the reaction of 1,3-dichlorobenzene which maintained a ca. 84% yield over 5 cycles (ESI,† section S3), which showed the generality of the recycling procedure.
 | ||
| Fig. 2 Recyclability studies of the [PIB-bpy-Ir] catalyst. Recycling reactions were conducted under the same conditions as described in Table 1. | ||
 | ||
| Fig. 3 Recycling process of the PIB-bpy Ir catalyst. | ||
A separate experiment left a catalyst sample recovered after 4 cycles on a benchtop exposed to the atmosphere for three weeks to test the catalyst stability over time. A fifth recycling run conducted with this catalyst afforded the product in 85% yield. This shows that in addition to recyclability, the catalyst functionalized with the PIB oligomer has excellent stability.
Iridium leaching from the PIB-bpy supported Ir catalyst during the recycling process was tested by inductively coupled plasma atomic emission spectroscopy (ICP-OES) to measure the loss of the Ir catalyst or Ir from the catalyst during the recycling process. This study was performed on aqueous solutions of samples of both the recovered catalyst and borylation products that were digested in acid. The tests showed that the amount of iridium in the recovered and acid digested catalysts goes from 49 ppm after the first reaction to a constant value of ca. 43 ppm after the 2nd and 3rd recycling runs, which matches the theoretical value of 43.5 ppm for the iridium content of the PIB-bpy supported activated catalyst calculated based on molecular weights (ESI,† part S3). We reason that this initial loss of iridium is due to the leaching of the portion of the added iridium precursor that did not form an activated catalyst with the PIB-bpy ligand.
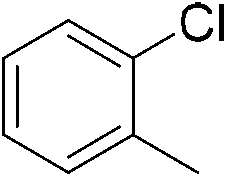
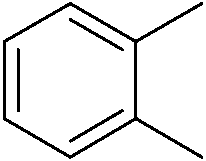
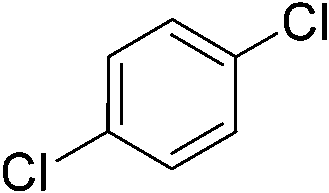
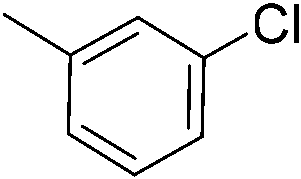
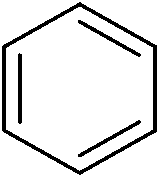
We then proceeded to evaluate the utility of the PIB-based catalyst with a select variety of substrates. In general, this soluble recyclable C–H activation catalyst afforded yields and selectivities, at the most sterically accessible C–H position, that were similar to the parent dtbpy ligand based catalyst. The catalyst was equally active for substrates with electron donating or electron withdrawing groups. Aromatic compounds with a single substitution gave a mixture of meta-substituted, para-substituted and 3,5-disubstituted products and no reaction at the ortho position was observed, due to steric hindrance, as seen for toluene (meta:3,5-disubstituted:para = 60:5:35, Table 2, entry 6) and anisole (meta:3,5-disubstituted:para = 65:20:15, Table 2, entry 8). Disubstituted compounds with a 1,2 substitution underwent a reaction at the 4 or 5 position which led to the formation of single products for 1,2-dichlorobenzene and 1,2-dimethylbenzene in 95% and 80% yield, respectively (Table 2, entries 1 and 7). A mixture of products substituted at the 4 (para-isomer) and 5 (meta-isomer) positions in the ratio of 40/60 was obtained for the reactions of 2-chlorotoluene in 92% yield (Table 2, entry 2). 1,3-substituted aromatic compounds only underwent substitution at the most sterically accessible 5 position and furnished borylated products in high yields as observed for 3-chlorotoulene, 3-chloroanisole and 1,3-dichlorobenzene (Table 2, entries 4, 5 and 9). We obtained a moderate yield of ortho substituted products for the borylation reaction of 1,4-dichlorobenzene (Table 2, entry 3), despite the steric hindrance of the substituent, which has a chlorine atom ortho to each of its C–H groups. Finally, the borylation reaction of benzene occurred under neat conditions and the final product was obtained with a high yield (Table 2, entry 10).
In summary, the direct borylation reactions of arenes gave very good yields of arylboronates using Pin2B2 as the borylating reagent and a recyclable catalyst generated from [IrOMe(COD)]2 and PIB-bpy is described. The catalyst/product separation is simple, requiring only the extraction of the borylated product with acetonitrile to afford a >97% pure product in ca. 95% isolated yield. This separation affords highly pure arylboronates with only trace amounts of iridium. The recyclable catalyst was reused up to 8 times without a significant loss of activity or catalyst.
Notes
The support of this research by the NPRP award (NPRP 7-1263-1-230) from the Qatar National Research Fund is gratefully acknowledged as is the gift of samples of polyisobutylene from the TPC Group. JS from the Texas A&M University College Station was supported by the National Science Foundation (CHE-1362735). STM also acknowledges the Texas A&M University at Qatar and the Qatar Foundation for generous start-up funding.Conflicts of interest
The authors declare no competing financial interest.References
- (a) M. Benaglia, Recoverable and Recyclable Catalysts, Wiley, 2009 Search PubMed; (b) J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS; (c) M. Rimoldi, A. J. Howarth, M. R. DeStefano, L. Lin, S. Goswami, P. Li, J. T. Hupp and O. K. Farha, ACS Catal., 2017, 7, 997–1014 CrossRef CAS; (d) K. Schroder, K. Matyjaszewski, K. J. T. Noonan and R. T. Mathers, Green Chem., 2014, 16, 1673–1686 RSC.
- (a) T. Sawano, Z. Lin, D. Boures, B. An, C. Wang and W. Lin, J. Am. Chem. Soc., 2016, 138, 9783–9786 CrossRef CAS PubMed; (b) S. T. Madrahimov, J. R. Gallagher, G. Zhang, Z. Meinhart, S. J. Garibay, M. Delferro, J. T. Miller, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2015, 5, 6713–6718 CrossRef CAS; (c) T. Zhang, K. Manna and W. Lin, J. Am. Chem. Soc., 2016, 138, 3241–3249 CrossRef CAS PubMed; (d) K. Manna, T. Zhang, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2015, 137, 2665–2673 CrossRef CAS PubMed.
- (a) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (b) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed; (c) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (d) M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287–4299 CrossRef CAS PubMed; (e) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (f) T. Ishiyama and N. Miyaura, Pure Appl. Chem., 2006, 78, 1369–1375 CrossRef CAS; (g) S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS PubMed; (h) N. G. Leonard, M. J. Bezdek and P. J. Chirik, Organometallics, 2017, 36, 142–150 CrossRef CAS.
- (a) M. I. Gonzalez, E. D. Bloch, J. A. Mason, S. J. Teat and J. R. Long, Inorg. Chem., 2015, 54, 2995–3005 CrossRef CAS PubMed; (b) Y. Maegawa and S. Inagaki, Dalton Trans., 2015, 44, 13007–13016 RSC; (c) K. Manna, T. Zhang and W. Lin, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed; (d) S. Zhang, H. Wang, M. Li, J. Han, X. Liu and J. Gong, Chem. Sci., 2017, 8, 4489–4496 RSC.
- (a) D. E. Bergbreiter and R. Chandran, J. Am. Chem. Soc., 1987, 109, 174–179 CrossRef CAS; (b) J. Suriboot, Y. Hu, T. J. Malinski, H. S. Bazzi and D. E. Bergbreiter, ACS Omega, 2016, 1, 714–721 CrossRef CAS; (c) D. E. Bergbreiter and Y.-C. Yang, J. Org. Chem., 2010, 75, 873–878 CrossRef CAS PubMed.
- (a) N. Priyadarshani, Y. Liang, J. Suriboot, H. S. Bazzi and D. E. Bergbreiter, ACS Macro Lett., 2013, 2, 571–574 CrossRef CAS; (b) D. Rackl, P. Kreitmeier and O. Reiser, Green Chem., 2016, 18, 214–219 RSC.
- (a) C.-G. Chao and D. E. Bergbreiter, Catal. Commun., 2016, 77, 89–93 CrossRef CAS; (b) C.-G. Chao, M. P. Kumar, N. Riaz, R. T. Khanoyan, S. T. Madrahimov and D. E. Bergbreiter, Macromolecules, 2017, 50, 1494–1502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data and detailed experimental procedures. See DOI: 10.1039/c7cy01641g |
| This journal is © The Royal Society of Chemistry 2018 |