
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin crossover in discrete polynuclear iron(II) complexes†
Ross W.
Hogue‡
 ,
Sandhya
Singh‡
and
Sally
Brooker
*
,
Sandhya
Singh‡
and
Sally
Brooker
*
Department of Chemistry and the MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Otago, PO Box 56, Dunedin 9054, New Zealand. E-mail: sbrooker@chemistry.otago.ac.nz
First published on 20th August 2018
Abstract
Iron(II) spin crossover (SCO) materials have been widely studied as molecular switches with a wide variety of potential applications, including as displays, sensors, actuators or memory components. Most SCO materials have been either monometallic or polymeric, and it is only relatively recently that chemists have really started to focus on linking multiple metal centres together within the one, discrete, molecule in an effort to enhance the SCO properties, such as abrupt, hysteretic, and multistep switching, as well as the potential for quantum cellular automata, whilst still being readily amenable to characterisation. Here we present a review of the ligand designs of the last two decades that have led to self assembly of discrete di- to poly-nuclear iron(II) complexes of helicate, cage, cube, and other supramolecular architectures with rich SCO activity, and to an increased focus on host–guest interactions. Analysis of selected octahedral distortion parameters (Σ, CShM) reveals interesting differences between these structural types, for example that the iron(II) centres in grids are generally significantly more distorted than those in squares or cages, yet are still SCO-active. Of the 127 complexes reviewed (79 published 2012–Feb. 2018), 54% are dinuclear, 10% trinuclear, 31% tetranuclear, and the remaining 5% are penta, hexa and octanuclear. Of the 93 designer ligands utilised in these polynuclear architectures: 60 feature azoles; 55 provide all donors to the Fe(II) centres (no co-ligands coordinated) and form exclusively 5-membered chelate rings via either bidentate azole-imine/pyridine or tridentate heterocycle-imine/amine/thioether/pyridine-heterocycle binding pockets.
 Ross W. Hogue | Dr Ross W. Hogue completed his BSc(Hons) with first class honours in 2012 in Professor Sally Brooker's group at the University of Otago, having scooped many awards during his undergraduate studies, including the Joseph and Emma Mellor Prize for the top final year honours chemistry student. After a gap year travelling the world, he returned to carry out his PhD research, again in the Brooker team (2014–2017). His PhD thesis, “Discrete polynuclear complexes: from spin crossover to hydrogen evolution”, was placed on the list of exceptional theses. Ross is currently a postdoctoral fellow working on inorganic materials for non-aqueous redox flow batteries with Dr Kathryn Toghill at Lancaster University, United Kingdom. |
 Sandhya Singh | Sandhya Singh was awarded a 5 year INSPIRE Fellowship to support her integrated BS-MS studies (2011–2016) at the Indian Institute of Science Education & Research (IISER) Mohali, India. She completed her MS thesis, ‘Synthesis, structural characterization, physico-chemical properties and sensing applications of lanthanide based metal organic frameworks (MOFs)’, in the research group of Professor Sanjay K. Mandal. During this time she qualified CSIR-UGC NET. In 2017 she moved to Dunedin, New Zealand, to take up a University of Otago PhD scholarship in Professor Sally Brooker's research group, working on the self-assembly of discrete polynuclear spin crossover complexes using designer polytopic ligands. |
 Sally Brooker | Professor Sally Brooker (MNZM, FRSNZ, FNZIC, FRSC) studied at the University of Canterbury, New Zealand [BSc(Hons) first class; PhD with Professor Vickie McKee]. After postdoctoral research at Georg-August-Universität Göttingen, Germany with Professor George M. Sheldrick, she took up a Lectureship at the University of Otago where she is now a full Professor. She has been the recipient of numerous awards, most recently including a 2017 Queens Birthday Honour for services to science (MNZM), the 2017 Hector Medal (RSNZ) and 2017 Burrows Award (RACI). Her research interests concern the design, synthesis and full characterisation of, primarily paramagnetic, di- and poly-metallic complexes of transition metal and lanthanide ions with polydentate acyclic and macrocyclic ligands, as these have interesting redox, magnetic, catalytic and photophysical properties (http://otago.ac.nz/brooker). |
Introduction
General introduction to spin crossover
Spin crossover (SCO) complexes are an interesting class of materials exhibiting molecular bistability with potential applications in nanotechnological devices such as memory storage units, sensors, actuators or displays.1–8 The bistability arises from their ability to be switched between two electronic states – high spin (HS) and low spin (LS) – by external stimuli such as a change in temperature or pressure, or light irradiation or guest presence/absence, in a readily detectable and reversible way.For octahedral 3d4–3d7 transition metal ions, there are two ways that electrons can populate the t2g and eg orbitals (Fig. 1), depending on ligand field splitting (ΔO) and the energy cost associated with pairing two electrons in the same orbital, the pairing energy (P). If strong field ligands are present, ΔO is large so the lowest energy configuration involves the pairing of electrons in the t2g set being favoured over population of the eg set (ΔO > P), resulting in the maximum amount of paired electrons and thus a LS state. On the contrary, if weak field ligands are present, ΔO is small and the maximum number of unpaired electrons, i.e. the HS state, is the lowest energy configuration as populating the eg set costs less energy than pairing the electrons in the t2g set (ΔO < P).9
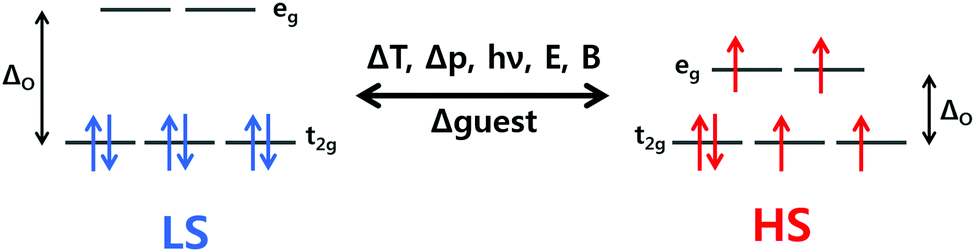 | ||
| Fig. 1 The d orbital splitting diagram for an octahedral Fe(II) ion. When ΔO is large, the LS state is adopted (left); when ΔO is small, the HS state is adopted (right). When ΔO is tuned for SCO, a change in temperature, pressure, magnetic or electric field, light irradiation, or guest molecules, can result in the switching between HS and LS states. | ||
When ΔO is of a comparable magnitude to P, the possibility of inducing switching between the HS and LS states arises, i.e. SCO, by perturbing the system through application of an external stimulus (Fig. 1). SCO can be induced by pressure, magnetic or electric field, light irradiation, and the presence/absence of guest molecules, however, the most common perturbation is a change in temperature, due in large part to facile application and measurement.1
The HS and LS states have different physical properties, which allows for detection and quantification of SCO by many different methods. Fe(II) SCO compounds often exhibit thermochromism, as the LS state absorbs visible light more strongly than the HS state and usually at distinctly different wavelengths, making UV-vis spectroscopy a useful tool for observing SCO.9 The HS state has more electrons in the antibonding eg orbitals than the LS state so has longer metal–ligand bond lengths. Hence X-ray crystallography, often done at more than one temperature, can be used to determine the spin state. This powerful technique can also pinpoint within a structure which ions are HS and which are LS in a mixed spin state system.10 A direct and detailed readout of the spin state of a SCO compound is the molar magnetic susceptibility (χM). In the case of Fe(II) the HS state (S = 2) is paramagnetic, while the LS state (S = 0) is diamagnetic, so for Fe(II) SCO switches the paramagnetism “on” and “off”. The most convenient way to quantify SCO is therefore variable temperature χMT (the product of molar magnetic susceptibility and temperature) measurements, which are routinely performed on modern SQUID, PPMS, or Versalab magnetometers. UV-vis,9 IR,11 Raman,12 NMR,13–16 Mössbauer,17 and fluorescence18 spectroscopies, as well as PXRD19 and DSC,20,21 can also be invaluable tools for monitoring SCO.
Advantages of discrete polynuclear systems for SCO
From a materials perspective, SCO systems were identified as potential molecular switch components in memory storage devices some time ago.22 Hence the development of spin crossover research is heavily influenced by the target of molecular switch and memory applications, with a drive towards SCO which is complete, abrupt, hysteretic, and ideally switchable about room temperature.23,24 Complete transitions are favoured, as they allow for easier and unambiguous detection of distinct fully HS and fully LS states, while abrupt transitions allow for a large change in output (e.g. colour, χMT) for a small input (e.g. small change in temperature). A thermal hysteresis provides a memory effect, where the spin state depends on the thermal history of the complex. Hence at temperatures within the hysteresis loop the system is bistable and there is the potential for binary information storage. The design of ligands which not only provide the right ΔO for SCO, but will also facilitate cooperativity within or between complexes which may in turn facilitate abrupt and/or hysteretic spin transitions, is the main goal of many SCO chemists.25Cooperativity arising from crystallographic disorder, crystal packing interactions, or large molecular size/shape differences between HS and LS states, can have a significant influence on SCO properties of complexes, including mononuclear and polymeric complexes, in the solid state.26 Cooperativity may also be imparted by covalently linking multiple metal centres in a polynuclear or polymeric architecture – but it must be noted that this approach is certainly not guaranteed to lead to cooperativity. That said, a beautiful illustration of the successful application of this ‘linking’ approach is provided by the now famous Fe(II) 4-R-1,2,4-triazole 1-D chain polymers, [Fe(Rtz)3]2+, which feature triply-triazole bridged Fe(II) centres and have remarkably large hysteresis loops around room temperature.22–24,27–29 A single crystal structure of these widely studied [Fe(Rtz)3]2+ chains was only obtained recently, as despite considerable attention these polymers had resisted all attempts at crystallisation until then.30
Discrete polynuclear complexes, in which multiple metal centres are covalently bridged in order to enhance cooperativity, have advantages over polymeric systems in that the design and synthesis can be more controlled, and the characterisation more straightforward.31–35 Polynuclear complexes, discrete or polymeric, have the added bonus of potential polynary, rather than binary, information storage as multiple spin states can be accessed by multi-step SCO.23,36,37 Furthermore the geometric arrangement of HS and LS Fe(II) centres within one complex molecule could be used in quantum cellular automata,38,39 potentially leading to logic gates consisting of only a few molecules.40,41
Scope of this review of SCO-active discrete polynuclear Fe(II)
Iron(II) complexes are by far the most common SCO systems studied1 (of the order of >90%), and almost all subsequent in depth studies by physicists and materials scientists are done on iron(II) systems.7 Most SCO-active Fe(II) complexes are mononuclear or polymeric, while discrete di to poly-nuclear complexes (Fig. 2) are much rarer.7,25,31,35,42–44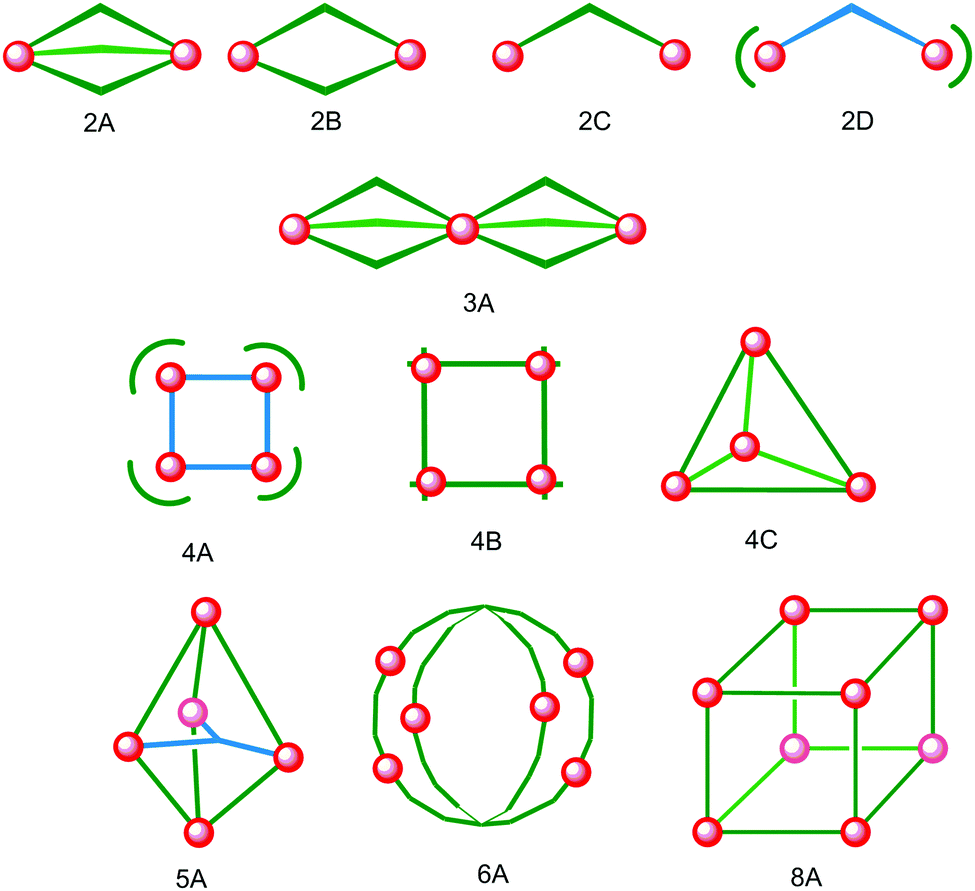 | ||
| Fig. 2 Topologies observed for the discrete polynuclear SCO-active complexes reviewed herein: dinuclear (2A–2D); trinuclear (3A); tetranuclear squares (4A), grids (4B), tetrahedral cages (4C); pentanuclear cage (5A), hexanuclear nanoball (6A) and octanuclear cube (8A). Green = di- to poly-dentate N-donor ligand. Blue = bridging anion. | ||
This is also true for Fe(III), Co(II), and Mn(II), where only a handful of polynuclear SCO systems are known.35 In 201335 we reviewed the development of discrete polynuclear SCO-active complexes, focussing mainly on the 90 new complexes reported over the period 2004–2011: all of the new complexes were either Fe(II) (68) or Fe(III) (22), and the nuclearities ranged from 63 dinuclear to 3 trinuclear to 14 tetranuclear, with another 10 in an assortment of higher nuclearities including mixed metal clusters. Here we review the various classes of structurally characterised, SCO-active, discrete di- to poly-nuclear complexes, specifically of Fe(II), focussing mostly on those published 2012–2017, as there has been considerable growth in this field since our previous review, including an expansion of the range of architectures employed and the setting of a new record for the largest number of metal centres in a discrete SCO-active complex = 8.45 The 127 complexes included in this review were identified by a CSD search (version 5.38, November 2016) for dinuclear and trinuclear iron(II) 1,2,4-triazoles (as structural search fragments) and a Scifinder search (28 February, 2018) for the keywords “Fe(II) or iron(II)” and “dinuclear or trinuclear or tetranuclear or pentanuclear or hexanuclear or septanuclear or nanoball or octanuclear” and “spin crossover or spin-crossover or spin cross-over or spin transition or spin equilibrium”, followed by manually sorting through the resulting ‘hits’. The resulting family of 79 discrete polynuclear SCO-active Fe(II) complexes published from 2012 to February 2018, along with selected examples published before then, brings the total reviewed here to 127. They are reviewed in order of increasing nuclearity: dinuclear (2A–2D), trinuclear (3A), tetranuclear squares (4A)/grids (4B)/cages (4C), pentanuclear (5A), hexanuclear ‘nanoballs’ (6A), and octanuclear cubes (8A) (Fig. 2).
Of these groups, dinuclear remains the most common of the nuclearities seen for discrete SCO-active complexes. A range of structural types can be identified within this dinuclear class: triply bridged triazole [Fe2L5(NCE)4] and helicate [Fe2L3] (Fig. 2, 2A), doubly bridged helicate and PMRT-type and pyridine bridged [Fe2L2] (Fig. 2, 2B), singly ligand linked dinuclear [Fe2L] (Fig. 2, 2C) and assorted anion bridged dinuclear complexes with capping ligands (Fig. 2, 2D).
Dinuclear triply triazole-bridged Fe2L5(NCE)4
Fundamental questions in the field of SCO materials are; what factors influence the spin transition temperature (T1/2) and transition profile.29,46 Therefore it is important to study structurally and magnetically similar series of complexes with minor changes in substituents, counterions, and/or solvent molecules. The triply triazole bridged dinuclear complexes (Fig. 2, 2A) are one type of dinuclear family suitable for such studies, for which crystal structures can be readily determined.As mentioned in the introduction, the use of N4-substituted triazoles to form 1D polymers of triply triazole bridged iron(II) is well known, with wide thermal hysteresis loops and tuneable transition temperatures near room temperature, based on the choice of N4 substituent.47 But it is interesting to note that those 1D-chain complexes resisted single crystal X-ray characterisation until 2011.30 In contrast, dinuclear systems tend to more readily characterised, and facilitate the probing of cooperative effects between the two metal ions.
Here, in the seven examples, plus a solvatomorph, of dinuclear SCO-active [Fe2L5(NCE)4] with L = L1–L7 (Chart 1), three of the 1,2,4-triazoles provide short N1N2, and fairly rigid, bridges between the iron(II) centres, and two more triazoles act as terminal monodentate ligands. The somewhat distorted octahedral geometry of each iron(II) centre is completed by two NCE mono-anions, affording N6 coordination spheres (Fig. 3). All eight of these complexes showed an abrupt one-step incomplete spin transition, with T1/2 in the range of 100–165 K, with no hysteresis loops observed and Fe–Fe distances in same range, 3.6–4.0 Å (Table 1).18,48–50 As expected, the octahedral distortion parameters (Σ, defined as the sum of the absolute values of the difference of each of the 12 cis angles from 90°) for the Fe(II) centres range from 12–19° for LS to the somewhat higher values of 14–35° for HS (Table 1). Some additional details are provided below.
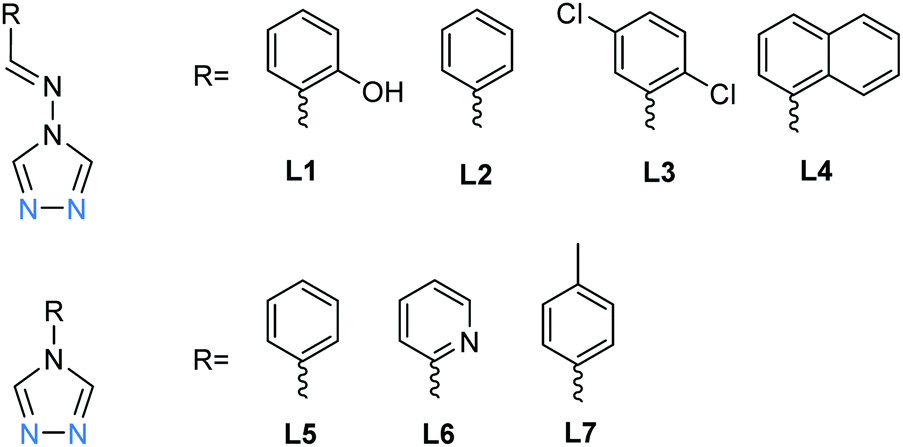 | ||
| Chart 1 The seven ligands, L1–L7, all of which are N4-substituted 1,2,4-triazoles, reported in the literature to form SCO-active triply-triazole bridged dinuclear complexes. | ||
 | ||
| Fig. 3 Schematic representation of [Fe2L5(NCE)4] where L = L1–L7, E = S or Se (L = L4). | ||
| Complex | T (K) | Fe–Fe (Å) | Σ (°) | Spin state | T 1/2 (K) | Lit. |
|---|---|---|---|---|---|---|
| [Fe2L15(NCS)4]·4MeOH | 100 | 3.65 | 15 | 2LS | 150 | 18 |
| 200 | 3.97 | 19 | 2HS | |||
| [Fe2L25(NCS)4]·3.5MeOH | 100 | 3.79 | 17/20 | LS–HS | 115 | 50 |
| 250 | 3.97 | 23/23 | HS–HS | |||
| [Fe2L35(NCS)4]·H2O | 100 | 3.72 | 13/13 | LS–LS | 150 | 48 |
| 180 | 3.96 | 17/35 | LS–HS | |||
| [Fe2L45(NCSe)4]·2DMF·2H2O | 92 | 3.63 | 12/13 | LS–LS | 164 | 49 |
| 280 | 3.95 | 24/20 | HS–HS | |||
| [Fe2L55(NCS)4]·MeOH·EtOH | 100 | 3.92 | 19/24 | LS–HS | 116 | 51 |
| 296 | 3.99 | 16/26 | HS–HS | |||
| [Fe2L55(NCS)4]·2EtOH | 296 | 3.97 | 14/19 | HS–HS | 122 | 51 |
| [Fe2L65(NCS)4]·MeOH·EtOH | 173 | 3.92 | 19/24 | LS–HS | 116 | 52 |
| 2[Fe2L75(NCS)4][FeL72(NCS)2(H2O)2] | 295 | 3.94 | 23/24/8 | HS–HS, HS | 111 | 53 |
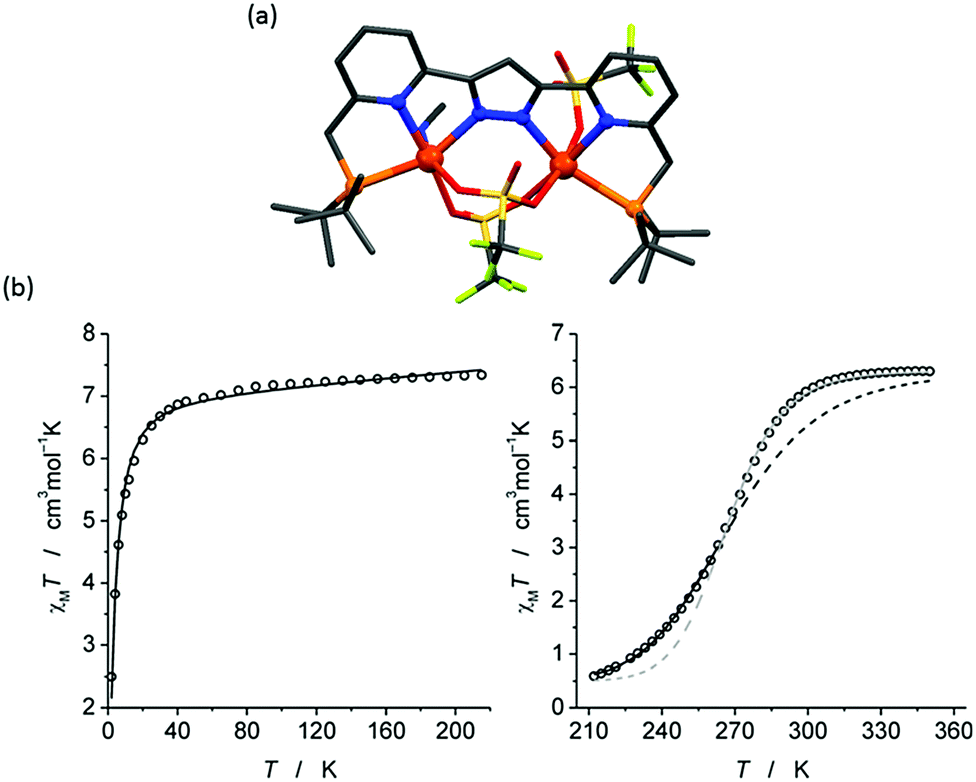
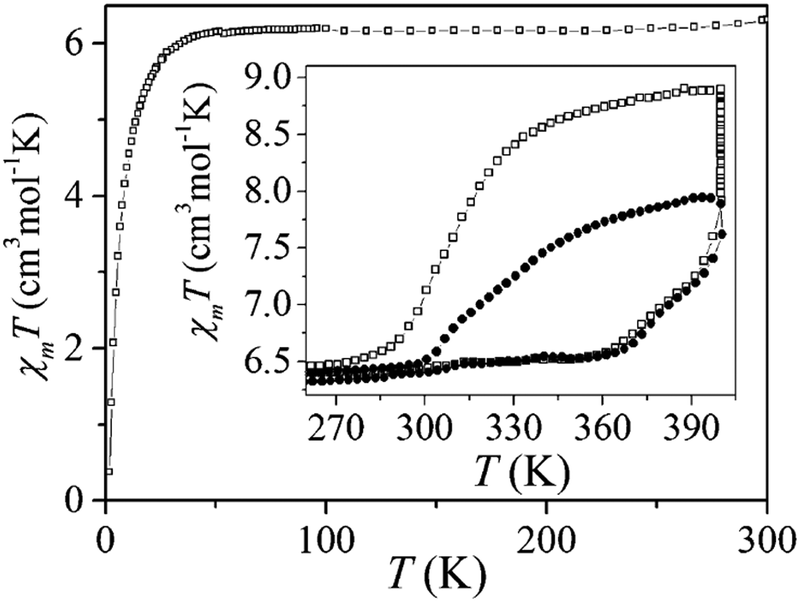
The first of this family of SCO-active dinuclear Fe(II) triply triazole bridged complexes, phenol N4-substituent (L1, Chart 1), [Fe2L15(NCS)4]·4MeOH in the low-spin and high-spin states, was reported in 2011 by Garcia and co-workers.18 SQUID magnetometry revealed a sharp [HS–HS] to [LS–LS] transition with T1/2 = 150 K (Fig. 4a) and the complex was also structurally characterised in both spin states, at 100 and 200 K respectively (Table 1). DSC measurements showed a single phase transition at Tmax = 155 K (Fig. 4a inset). The SCO event was also able to be monitored by variable temperature fluorescence of the enol, as a bathochromic shift of 20 nm is observed in the emission band across the temperature range of 135–179 K (Fig. 4b).
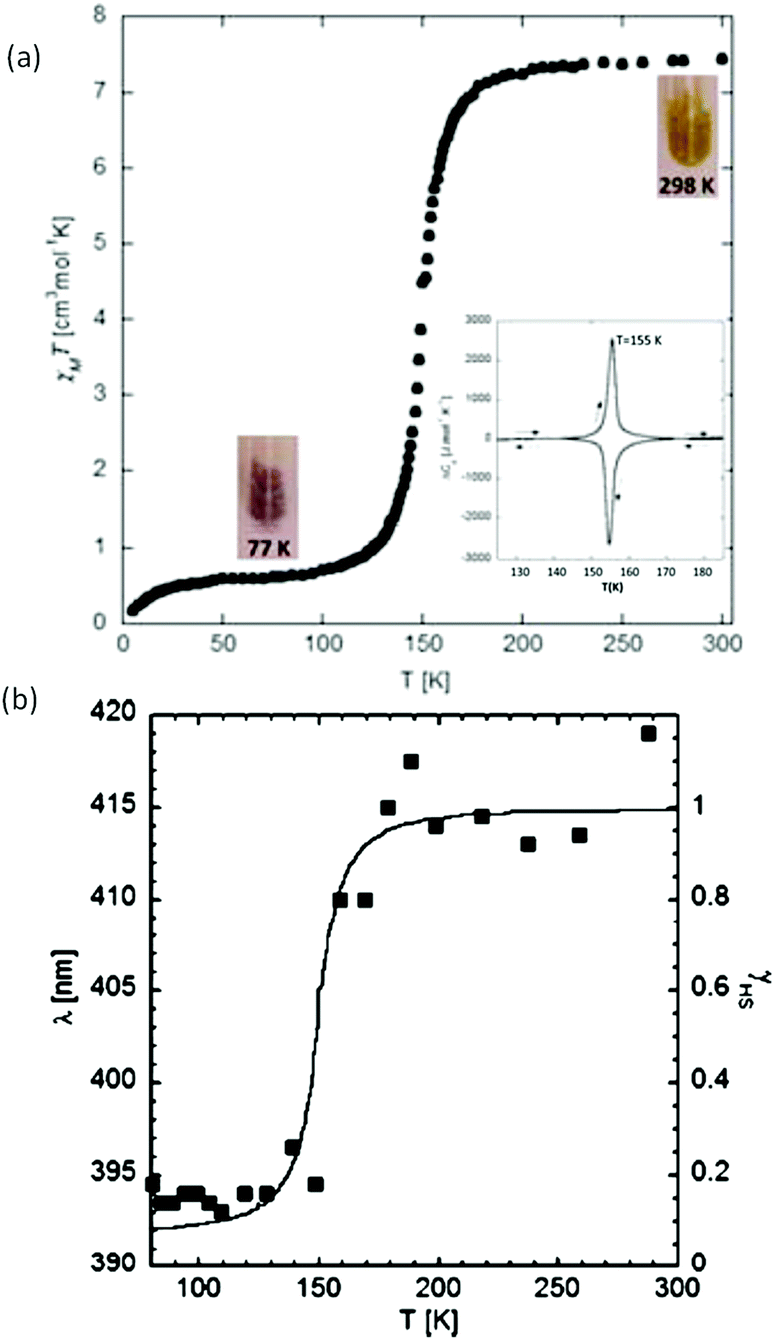 | ||
| Fig. 4 Data obtained on [Fe2L15(NCS)4]·4MeOH: (a) plot of XmT vs. T. The inset shows the DSC peaks, both exothermic and endothermic. (b) λmax of the emission spectrum vs. T (black dots), follows the γHS determined from the SQUID data (solid line). (a) Adapted and (b) reproduced with permission from ref. 18. Copyright 2011 ACS. | ||
Fresh crystals of [Fe2L25(NCS)4]·5MeOH showed a gradual, approximately half, SCO centred at 115 K.50 In contrast, a bulk powder sample of unsolvated [Fe2L25(NCS)4] remained [HS–HS] to low temperature. By introducing electron withdrawing chloride substituents (L3, Chart 1), the ligand field was adjusted, giving a more complete spin transition, with T1/2 = 150 K. The LIESST (light-induced excited spin state trapping) effect was also seen for the [Fe2L35(NCS)4] system: irradiating with 532 nm light at 5 K the χmT value increased abruptly to 4.6 cm3 K mol−1, which was attributed to SCO of the two Fe(II) centres from LS to HS*, with a T(LIESST) of 61 K.48
Liu and co-workers used a naphthalene group as the N4 substituent, generating [Fe2L45(NCSe)4]·2DMF·2H2O (L4, Chart 1) which showed an abrupt [HS–HS] to [LS–LS] SCO with T1/2 = 164 K (Fig. 5a).49 They also studied the photoinduced SCO. Upon irradiation with 532 nm light at 10 K for 2 h, χT increased max. to 2.5 cm3 K mol−1 at 48 K, consistent with the LS Fe(II) centres undergoing partial conversion to metastable HS*. At 10 K, cycles of irradiation at 532 nm followed by irradiation at 808 nm switched the χT value reversibly between 1.23 to 0.47 cm3 K mol−1 implying reversible SCO between metastable HS* and LS (Fig. 5b).49
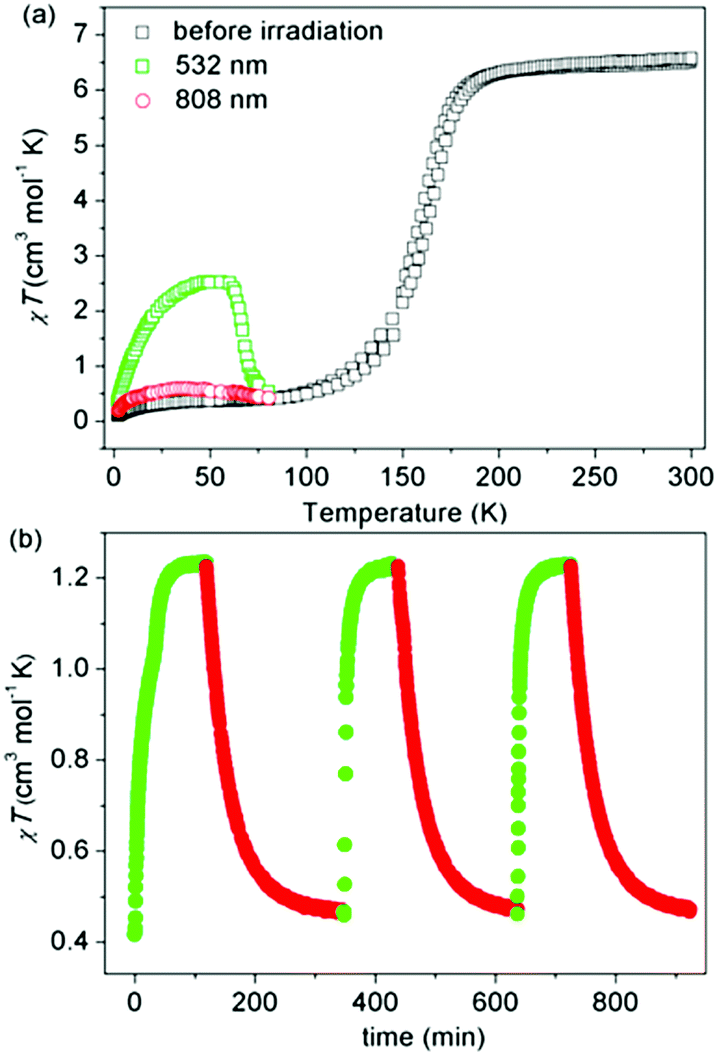 | ||
| Fig. 5 Data obtained on [Fe2L45(NCSe)4]·2DMF·2H2O: (a) χT vs. T (black), including before and after irradiation at 10 K at the stated wavelengths (green and red). (b) χT vs. time during cycles of successive irradiation at 532 nm (green) and 808 nm (red) at 10 K. (a and b) Reproduced with permission from ref. 49. Copyright 2017 ACS. | ||
Wang and co-workers studied the effect of differing solvents of crystallisation, comparing [Fe2L55(NCS)4]·MeOH·EtOH and [Fe2L55(NCS)4]·2EtOH and showing that the T1/2 value for the observed half SCO increased slightly, from 116 K to 122 K.51 Changing the N4 substituent from phenyl (L5) to 2-pyridyl (L6, Chart 1) resulted in a negligible effect magnetically, with the T1/2 remaining at 116 K.52 In contrast, changing from a phenyl (L5) to p-tolyl (L7)53N4 substituent (Chart 1), had a dramatic impact, with two dinuclear Fe2L75(NCS)4 units bridged by hydrogen bonding to the coordinated water molecules of a mononuclear FeL72(NCS)2(H2O)2 unit. But the magnetic behaviour of the resulting material is not much different from other triply triazole bridged dinuclear Fe(II) complexes; it undergoes SCO centred at 111 K, with variable temperature magnetometry and Mössbauer spectroscopy both consistent with four out of five Fe(II) centres transitioning from HS to LS, i.e. the dinuclear Fe2L75(NCS)4 units are SCO-active while the mononuclear FeL72(NCS)2(H2O)2 unit remains HS to low temperature.53
Dinuclear triply stranded helicates Fe2L3
The other type of triply bridged dinuclear Fe(II) SCO-active complex is the helicate architecture, Fe2(L)3 (Fig. 2, 2A). Helicates are made through controlled and predictable self assembly of carefully designed components. They are intrinsically stable, and have wide ranging properties owing to their versatility with respect to choice of metal ion.54–60 Through careful design of the polydentate ligand, the cavity of such helicates can be optimised for favourable host–guest interactions, which can dictate the self-assembly process, and can also be exploited for tuning of the metal ion properties.61–65 In line with the theme of this review, only Fe(II) examples of this architecture which are SCO-active are discussed herein.A reliable route to such helicates is the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 combination of octahedral Fe(II) ions and bis-bidentate linker ligands to give dinuclear Fe2L3 helicates, of which only a handful of examples, 14, have been reported to be SCO-active. Indeed only nine ligands (Chart 2) have been used to produce all of the SCO-active Fe2L3 helicates reported to date,61,66–72 the first of which was reported in 1998.66 In these helicates the three ligands provide all of the donor atoms to the two Fe(II) centres which results in greatly increased Σ values (54–94°, Table 2; 64–159°, Table 3; 59–116°, Table 4) compared to those in the triply bridged triazoles with monodentate co-ligands (12–35°, Table 1).
:3 combination of octahedral Fe(II) ions and bis-bidentate linker ligands to give dinuclear Fe2L3 helicates, of which only a handful of examples, 14, have been reported to be SCO-active. Indeed only nine ligands (Chart 2) have been used to produce all of the SCO-active Fe2L3 helicates reported to date,61,66–72 the first of which was reported in 1998.66 In these helicates the three ligands provide all of the donor atoms to the two Fe(II) centres which results in greatly increased Σ values (54–94°, Table 2; 64–159°, Table 3; 59–116°, Table 4) compared to those in the triply bridged triazoles with monodentate co-ligands (12–35°, Table 1).
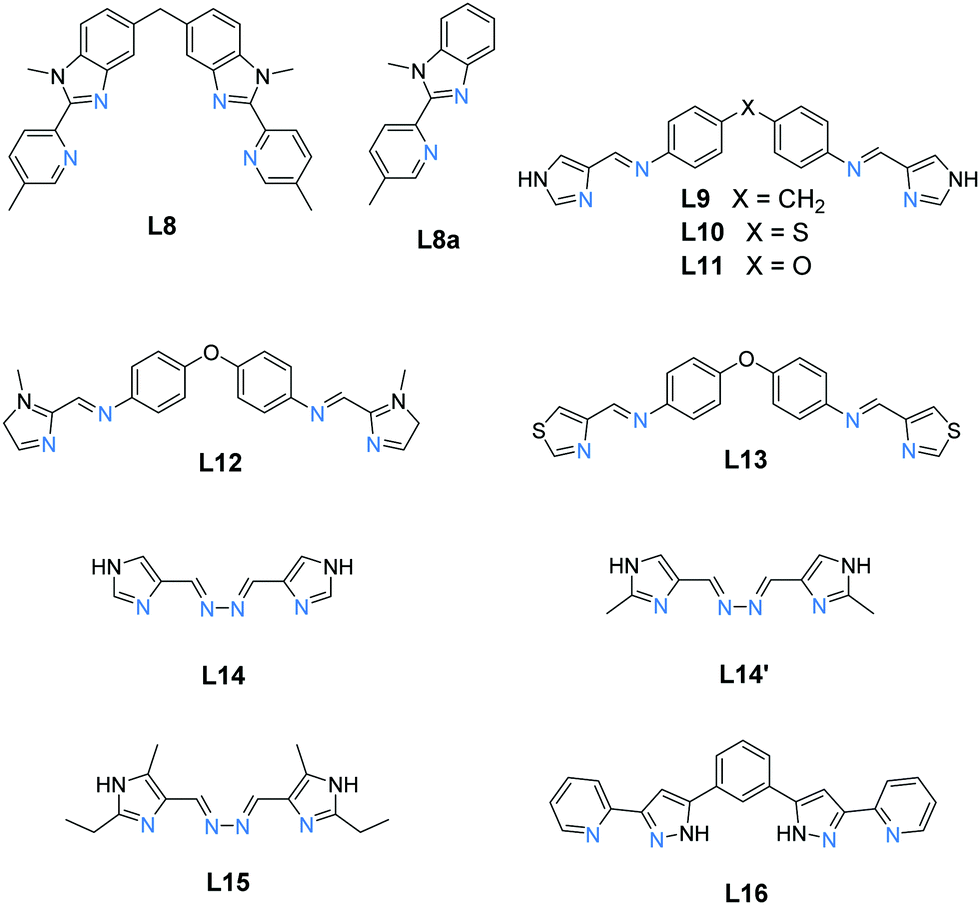 | ||
| Chart 2 The nine ligands, L8–L16, reported in the literature to form SCO-active Fe2L3 helicates. All are bis-bidentate and provide N6 donor sets to the octahedral Fe(II) metal ions. | ||
| T (K) | Σ (°) | Fe–Fe (Å) | Spin state | T 1/2 (K) | Lit. | |
|---|---|---|---|---|---|---|
| [FeII2(L8)3](ClO4)4 | 295 | 62 | 9.16 | 2LS | 341, 402 | 66 and 73 |
| [FeII2(L9)3](PF6)4 | 180 | 64 | 11.40 | 2LS | Not stated | 67 |
| [FeII2(L9)3](BF4)4 | 180 | 63, 83 | 11.56 | LS–HS | Not stated | |
| [FeII2(L9)3](ClO4)4 | 180 | 76, 94 | 11.58 | HS–HS | Not stated | |
| [FeII2(L9)3](BF4)4 | 100 | 76 | 11.71 | 2HS/LS | ↓155 | 71 |
| ↑170 | ||||||
| [FeII2(L10)3](BF4)4 | 100 | 60, 90 | 11.78 | LS–HS | ↓115 | 71 |
| ↑130 | ||||||
| [FeII2(L11)3](BF4)4 | 100 | 84.9, 74.5 | 11.62 | HS–HS/LS | ↓150 | 71 |
| ↑165 | ||||||
| [FeII2(L12)3](ClO4)4·2MeCN | 150 | 65, 62 | 11.45 | LS–LS | 140 | 69 |
| [FeII2(L12)3](ClO4)4·1.5H2O | 150 | 62, 85 | 11.45 | LS–HS | 210–265 | 75 |
| [FeII2(L13)3](BF4)4 | 100 | 54, 65 | 11.33 | LS–LS | 348 | 72 |
| [FeII2(L13)3](BF4)4 | 298 | 58, 70 | 11.36 | LS–LS/HS |
| T (K) | Σ (°) | Fe–Fe (Å) | Spin state | T 1/2 (K) | Lit. | |
|---|---|---|---|---|---|---|
| [FeII2(L14)3](BF4)4·2MeNO2·1H2O | 173 | 66, 159 | 3.87 | LS–HS | ↑190 | 68 |
| ↓183 | ||||||
| [FeII2(L14)3](ClO4)4·5MeNO2 | 103 | 64, 152 | 3.85 | LS–HS | 240 | |
| 293 | 116, 124 | 4.04 | HS–HS | |||
| [FeII2(L15)3](ClO4)4·3MeCN·0.5H2O (plate) | 163 | 138, 65 | 3.84 | HS–LS | 120 | 70 |
| [FeII2(L15)3](ClO4)4·2MeCN (block) | 113 | 132, 135 | 4.178 | HS–HS | HS | 70 |
| T (K) | Σ (°) | Fe–Fe (Å) | Spin state | T 1/2 (K) | Lit. | |
|---|---|---|---|---|---|---|
| Cl@[Fe2(L16)3]Cl(PF6)2·5.7MeOH | 100 | 116, 59 | 9.73 | HS–LS | 302 | 61 |
| 280 | 113, 62 | 9.73 | HS–LS | |||
| Cl@[Fe2(L16)3]Cl(PF6)2·3MeOH·H2O | 90 | 62 | 9.67 | 2LS | 160, 265 | |
| 300 | 95 | 9.80 | 2HS | |||
| Br@[Fe2(L16)3]Br(PF6)2·4MeOH | 100 | 114, 61 | 9.67 | HS–LS | 258 | |
| 280 | 110, 84 | 9.70 | HS–HS | |||
| Br@[Fe2(L16)3]Br(PF6)2·MeOH·H2O | 90 | 64 | 9.69 | 2LS | 200 | |
| 296 | 97 | 9.79 | 2HS |
Williams and co-workers reported the first SCO-active Fe(II) dinuclear triply stranded helicate, based on the bis[2-(pyrid-2′-yl)benzimidazol-5-yl]methane ligand L8 (Chart 2).66,73 The SCO was monitored in CD3CN solution by the Evans method, and revealed a gradual and far from complete spin transition over the measured temperature range (240–330 K). Interestingly, the mononuclear analogue of the 2-(pyrid-2-yl)benzimidazole “half” ligand, [FeL8a3](ClO4)2, was also studied, and it was found that the LS state is better stabilised in the dinuclear [Fe2(L8)3]4+ helicate than in mononuclear [FeL8a3](ClO4)2.
A family of [Fe2(L9)3]X4 helicates prepared using a neutral bis-imidazoleimine ligand which features a central methylene linker (L9, Chart 2; bis-imine made from 4(5)-imidazolecarboxaldehyde and 4,4′-methylenedianiline) was reported by Hannon and co-workers in 2004 (Fig. 6a).67 Variation of the counteranion X (PF6−, BF4−, ClO4−) led to different solid state SCO behaviours being observed (Fig. 6b). While the use of PF6− or BF4− resulted in SCO transitions from fully HS to ∼20% HS, centred at approximately 200 K and 150 K respectively, the ClO4− analogue underwent a HS to ∼50% HS transition, i.e. half SCO, centred around 150 K (Fig. 6b). Subsequent Mössbauer experiments indicated that for [Fe2L93](ClO4)4, the half SCO occurs via half of the helicates undergoing a complete [HS–HS] to [LS–LS] transition and the other half remaining [HS–HS], giving 1:1 [HS–HS]:[LS–LS], as no localised [HS–LS] species was detected.74
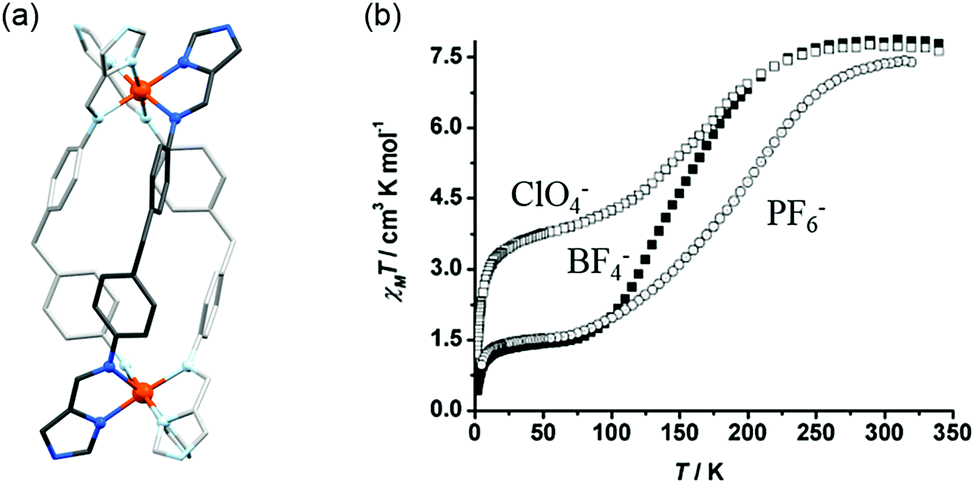 | ||
| Fig. 6 Data obtained on the family of three [Fe2(L9)3]X4 helicates: (a) the structure of the [Fe2(L9)3]4+ cation. (b) χMT vs. T for the [Fe2(L9)3]X4 helicates where X = PF6−, BF4− or ClO4−. Structure redrawn from CCDC data.67 (b) Adapted with permission from ref. 67. Copyright 2004 John Wiley and Sons. | ||
The ditopic ligand scaffold of L9 is well predisposed to forming Fe2L3 helicates, so it is unsurprising that several more recent examples of SCO-active complexes have employed thioether and ether analogues of L9, L10–L13 (Chart 2).
Li and co-workers recently reported the Fe2L3 helicates of the most closely related thioether and ether analogues of L9, L10 and L11, which differ only in the linker (methylene vs. thioether vs. ether), whilst providing the same imidazole-imine binding pocket.71 The magnetic properties of [Fe2(L10)3](BF4)4 and [Fe2(L11)3](BF4)4 were compared to that of a re-synthesised sample of the [Fe2(L9)3](BF4)4 helicate originally reported by Hannon and co-workers. All three samples were found to undergo a [HS–HS] → [HS–LS] transition with a 15 K wide hysteresis loop, but with T1/2 values which varied by 40 K. Changing the linker group of the ligand from methylene to thioether to ether was shown to influence the crystal packing and the intramolecular Fe–Fe separation varied by up to 0.16 Å (Table 2) and the inter-helicate Fe–Fe separation by up to 0.75 Å. But these structural differences were difficult to correlate to the SCO behaviours, which are possibly influenced more heavily by harder to predict inter-helicate interactions e.g. CH–π or hydrogen bonding interactions. This is perhaps illustrated by the differences in the packing and SCO of [Fe2(L9)3](BF4)4 seen in the reports of Hannon versus Li. While Hannon crystallised [Fe2(L9)3](BF4)4 from acetonitrile by diisopropyl ether vapour diffusion and observed a hydrogen bonded 2D network of helicates (through imidazole-NH and BF4− counteranions) which exhibited abrupt SCO,67 Li crystallised the material by diethyl ether vapour diffusion, observed no hydrogen-bonded network, and a less abrupt SCO.71
While most SCO-active Fe2L3 helicates exhibit [HS–HS] to 50% HS transitions (half SCO), a desolvated [Fe2(L12)3](ClO4)4 complex based on the methylimidazole analogue of L11, L12 (Chart 2, bis-imine made from 1-methyl-2-imidazole-carboxaldehyde and 4,4′-oxydianiline), was shown by Kruger and co-workers to undergo a complete SCO (Fig. 7b).69 The χT value at 300 K was 6.6 cm3 K mol−1, consistent with two uncoupled HS Fe(II) ions. At 80 K, all HS transit into LS with T1/2 140 K. At low temperature, a metastable fully [HS–HS] state can be populated by light irradiation, providing a remarkable example of the LIESST effect. Subsequently, when the complex was instead obtained as [Fe2L123](ClO4)4·4H2O, only half SCO was achieved, and the T1/2 value was found to be dependent on the degree of hydration, thus the helicate exhibits moisture-sensitive SCO.75
 | ||
| Fig. 7 Data obtained on [Fe2(L12)3](ClO4)4 (a) structure of the [Fe2(L12)3]4+ cation. (b) Thermal SCO (black data) and LIESST (blue data) behaviour of desolvated [Fe2(L12)3](ClO4)4. Structure redrawn from CCDC data.69 (b) Adapted with permission from ref. 69. Copyright 2009 RSC. | ||
In 2018, Li and co-workers went on to report the [Fe2(L13)3](BF4)4 helicate of a thioazolyl-imine analogue, L13, of the ligand L11 (Chart 2).72 The complex, [Fe2(L13)3](BF4)4, was structurally characterised in the [LS–LS] state at 100 K, and again at 298 K at which temperature it showed slightly increased Fe–N bond lengths and octahedral distortion parameter at one of the Fe(II) centres, consistent with slight population of a [HS–LS] state. Indeed, the magnetic data show that at 298 K there is an onset of a SCO, which becomes complete above room temperature, with T1/2 = 348 K. This complete [LS–LS] to [HS–HS] SCO was also observed by X-ray photoelectron spectroscopy (XPS). This report provides a nice example of deliberately tuning the T1/2 of a SCO-active complex, in this case by ∼200 K, by changing the ligand field strength of these ether linked ditopic ligands upon changing the terminal imidazole group of the L11 and L12 ligands to the stronger ligand field thiazole group of L13 (Chart 2).
Abrupt SCO was achieved for two complexes of a ‘tight’ Fe2L3 helicate architecture derived from the relatively short L14 ligand (Chart 2) reported by Sunatsuki, Kojima, and co-workers.68 This short, conjugated, ditopic L14 ligand is prepared by the 2:1 condensation of imidazole-4-carbaldehyde with hydrazine hydrate. In order to bind in a bis-bidentate manner, and provide a two-atom NN bridge between the two iron(II) centres, the ligand twists significantly away from being flat, forming the triple helicate complex, [Fe2(L14)3](ClO4)4 (Fig. 8a) with very short Fe–Fe distances ∼4 Å (Table 3) for a helicate (Tables 2 and 4). This undergoes an abrupt [HS–HS] to 50% HS transition at 240 K (Fig. 8b, blue data), while the BF4− analogue has a lower T1/2, at 190 K, with a hysteresis loop 7 K wide (2 K min−1 scan rate) (Fig. 8c). The methylimidazole analogue of L14, L14′ results in a [LS–LS] complex (no SCO) in the analogous perchlorate complex, [Fe2(L14′)3](ClO4)4 (Fig. 8b, red data), which the authors attribute to being a direct result of tuning of the ligand field by introduction of an electron donating group to the ligand scaffold.
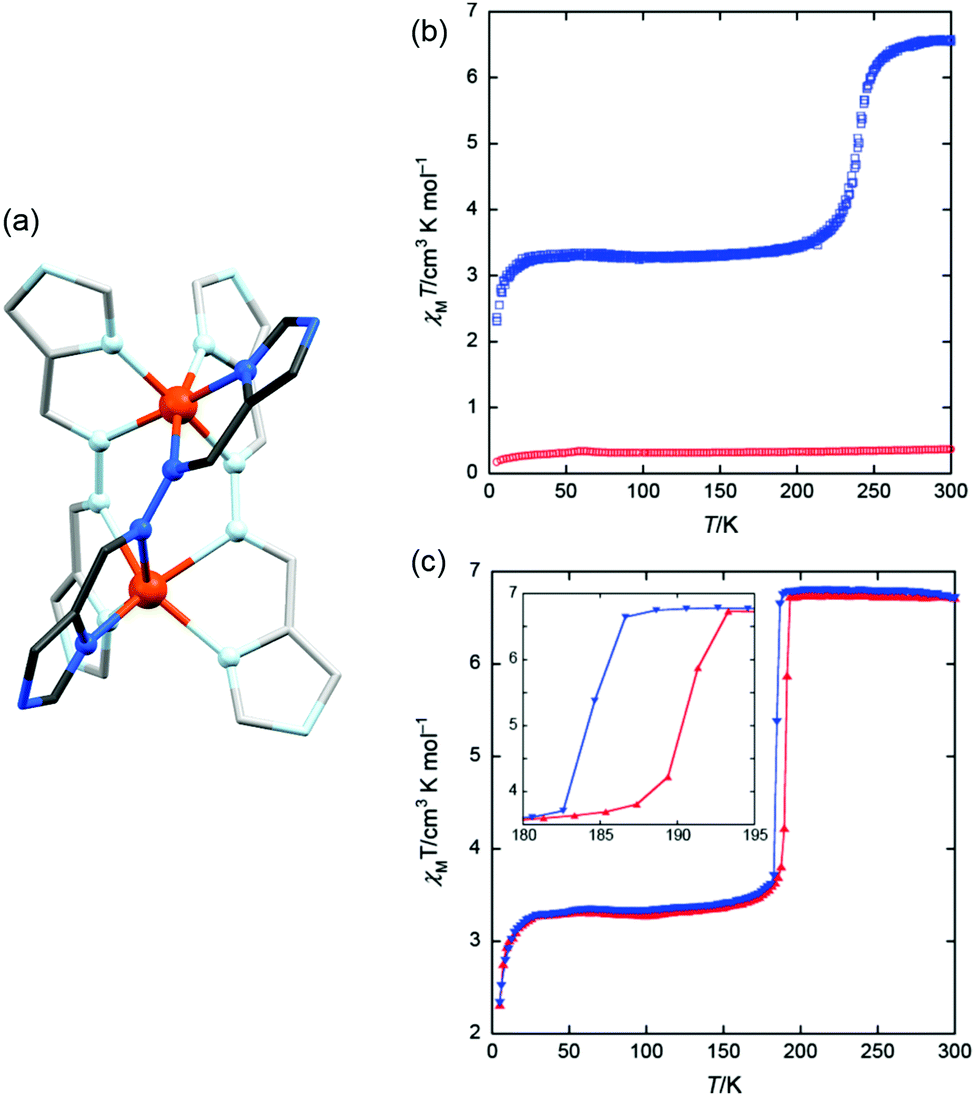 | ||
| Fig. 8 Data obtained on [Fe2(L14)3](ClO4)4, [Fe2(L14)3](BF4)4 and [Fe2(L14′)3](ClO4)4: (a) structure of the cation of the [Fe2(L14)3]4+ helicate. (b) χMT vs. T for [Fe2(L14)3](ClO4)4 in blue, and the methylimidazole analogue, [Fe2(L14′)3](ClO4)4 helicate, in red. (c) χMT vs. T for [Fe2(L14)3](BF4)4. Structure redrawn from CCDC data.68 (b and c) Reproduced with permission from ref. 68. Copyright 2009 ACS. | ||
A follow-up publication from the same authors reported the SCO-active dinuclear helicate [Fe2(L15)3](ClO4)4·solvents based on the (electron donating) ethyl/methyl substituted analogue, L15, of the bis-imidazole ligand L14 (Chart 2).70 [Fe2(L15)3](ClO4)4·solvents crystallised as two solvatomorphs, block shaped and plate crystals, both of which were structurally characterised (Table 3). The plate crystals, [Fe2(L15)3](ClO4)4·H2O, undergo a half SCO from [HS–HS] to [HS–LS] at 120 K, with a localised [HS–LS] state confirmed by X-ray crystallography, and no [LS–LS] state is accessible. On the other hand, the block shape crystals, [Fe2(L15)3](ClO4)4·½CH3CN, remain [HS–HS] to low temperature, as observed by SQUID magnetometry and X-ray crystallography. These results highlight the large influence that crystal packing and/or lattice solvent content can have on the SCO properties of a system, in this case overruling the expected electronic influence of methyl/ethyl substituents.
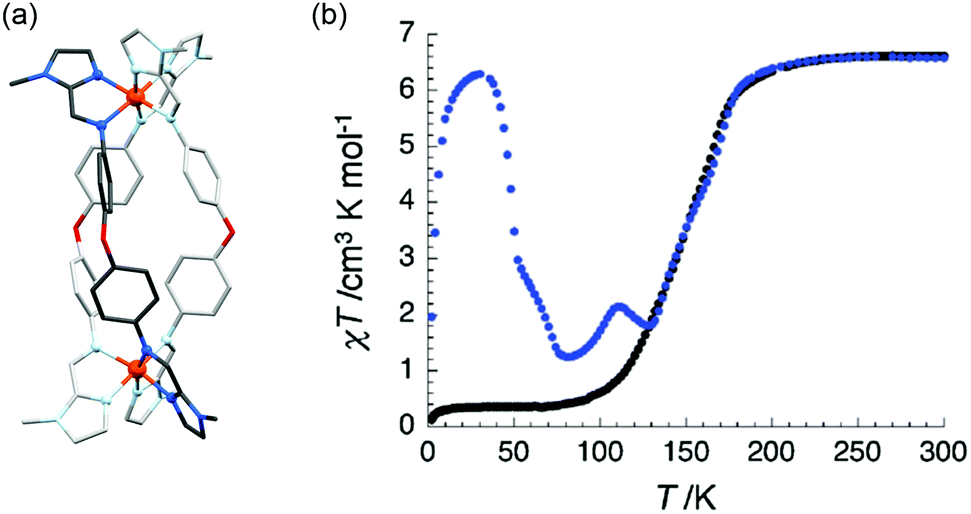
An example of a SCO-active Fe2L3 helicate which is triply switchable – by temperature, light, and guests – was reported by Aromi and co-workers in 2016.61 Here the ligand strand, L16 (Chart 2), consists of two bidentate pyrazole-pyridine units, linked by a central 1,3-benzene group, and features two internally facing ‘spare’ NH moieties. Due to these NH groups, the resulting triply stranded dinuclear helicates, {X@[Fe2(L16)3]}X(PF6)2 (X = Cl− or Br−), can encapsulate a halide counteranion. One halide ion is encapsulated inside the cavity defined by the three ligand strands and the other is external (Fig. 9a). For the as-synthesised helicate structures, the encapsulated halide ion interacts with the six pyrazole rings through hydrogen bonding to the internally facing NH moieties, and is closer to one of the two Fe(II) centres. The external halide counteranion is closer to the same Fe(II) centre, and has one X⋯H–N interaction with a pyrazole ring. These out-of-sphere interactions stabilise this Fe(II) centre in the HS state down to low temperatures, and, as such, only a half SCO, to a localised [HS–LS] state, as determined by an X-ray structure determination at 100 K (Fig. 9a), is observed (Table 4).
 | ||
| Fig. 9 (a) [HS–LS] {Cl@[Fe2(L16)3]}Cl(PF6)2·5.7MeOH structure (PF6− anions and solvents omitted), showing the encapsulated Cl− (spacefill) nearer the HS Fe(II) (red) than the LS Fe(II) (purple), and also the external Cl− which interacts with one pyrazole NH closer to HS Fe(II) (red dashed line). (b) Thermal SCO and LIESST effect of the helicates {Cl@[Fe2(L16)3]}Cl(PF6)2·5.7MeOH (1 in this figure) and {Br@[Fe2(L16)3]}Br(PF6)2·4MeOH (2 in this figure), and the corresponding SCSC generated helicates {Cl@[Fe2(L16)3]}Cl(PF6)2·3MeOH·H2O (1a in this figure) and {Br@[Fe2(L16)3]}Br(PF6)2·MeOH·H2O (2a in this figure). Structure redrawn from CCDC data.61 (b) Reproduced with permission from ref. 61. Copyright 2016 John Wiley and Sons. | ||
The X = Cl− and X = Br− helicates are isostructural, with the only difference being the nature of the encapsulated/external halide ions, yet the T1/2 of the SCO is shifted down by 40 K on changing from X = Cl− to Br−, highlighting the significant impact of the out-of-sphere interactions on the ligand field strength and hence SCO properties. In both compounds, a single crystal to single crystal (SCSC) transformation of the helicate, by partial methanol solvent loss and uptake of water, results in the encapsulated halide ions being equidistant to the two Fe(II) centres, thus breaking the structural non-equivalence, and full [HS–HS] to [LS–LS] SCO is observed (Fig. 9b and Table 4). Attempts to encapsulate the larger I− anion were unsuccessful and instead yielded complexes with external I3− counteranions. These persisted in the [HS–HS] state to low temperatures.
Doubly stranded dinuclear helicates, Fe2L2
While there are numerous examples of SCO-active dinuclear Fe(II) triply stranded helicates made from bis-bidentate ligands, doubly stranded systems are rare (Fig. 2, 2B). For just two ligand strands to complete the octahedral coordination sphere of two octahedral Fe(II) centres in a helicate complex, the ditopic ligand must be bis-terdentate. To date, only two such ligands, the neutral 1,2,3-triazole-based ligands L17 and L18 (Chart 3), have been reported to give SCO-active Fe2L2 helicates.76,77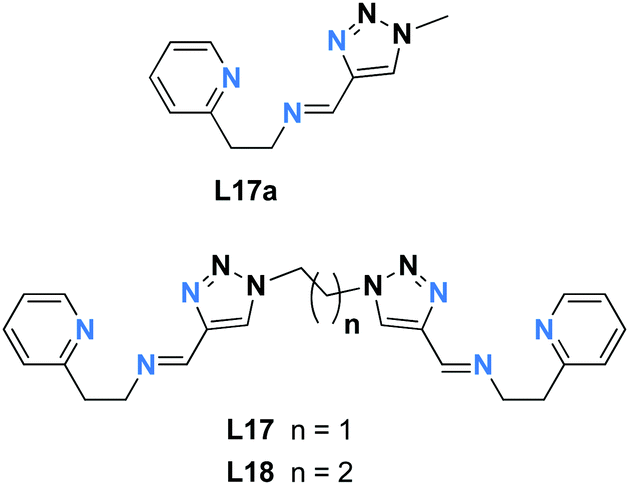 | ||
| Chart 3 The two ditopic 1,2,3-triazole-based ligands, L17 and L18, known to give dinuclear Fe(II) doubly stranded double helicates. Both are bis-terdentate and provide N6 donor sets to the octahedral Fe(II) metal ions. | ||
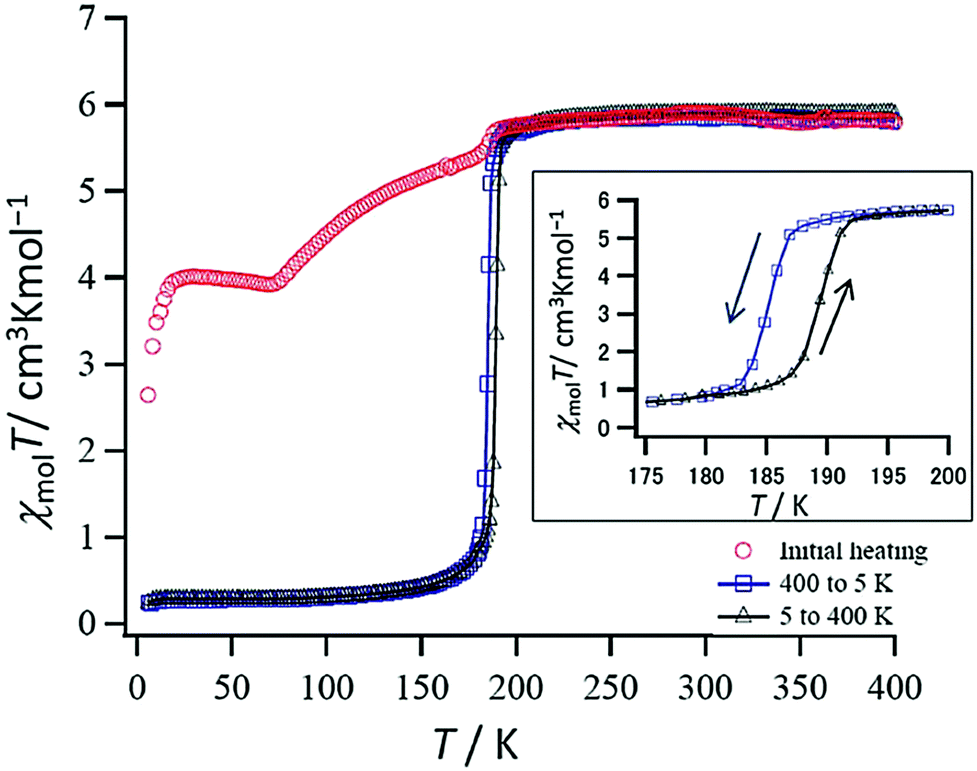
In 2016 Hagiwara, Tanaka and Hora reported the first SCO-active dinuclear double helicate, [Fe2(L17)2](PF6)4·5H2O·MeCN (Fig. 10a).76 X-ray structural analysis at 120 K revealed two identical LS Fe(II) centres in the dication, and no higher temperature structures could be obtained due to loss of crystallinity upon desorption of lattice solvent molecules. SQUID magnetometry on [Fe2(L17)2](PF6)4·5H2O·MeCN showed that the helicate remains [LS–LS] to 300 K, but measurements above room temperature revealed an unusual two-step SCO. Upon warming from 300 K to 435 K, the χMT value increases steadily from 0.1 to 2.6 cm3 K mol−1, consistent with a gradual SCO to ∼40% HS (Fig. 10b, red data). An abrupt second step is then observed between 435 and 443 K, with χMT increasing to 5.6 cm3 K mol−1, before again increasing gradually to 6.5 cm3 K mol−1 at 470 K (Fig. 10b, red data), consistent with a [HS–HS] state. In cooling mode (Fig. 10b, blue data), the abrupt transition occurs at 426 K giving the double helicate an 11 K wide hysteresis loop: importantly this behaviour is also shown to be reversible over three cycles (Fig. 10b). Another key component of this report was the comparison of these SCO properties with those of the analogous mononuclear Fe(II) complex, [Fe(L17a)2](PF6)2, where L17a is a monotopic terdentate ligand with a CH3 group in place of the ethylene linker of L17. [Fe(L17a)2](PF6)2 undergoes a gradual SCO from LS to HS that is very similar to the first SCO step of [Fe2(L17)2](PF6)4 between 300 and 435 K, however in the mononuclear compound there is no abrupt second step (Fig. 10b green/orange data). This example clearly demonstrates the enhanced SCO properties that can be achieved by linking two Fe(II) centres together in a dinuclear complex, in this case by simply joining two SCO-active mononuclear units through an ethylene linker. The resulting dinuclear double helicate is also remarkable for the high temperature SCO which occurs (T1/2 ↑437 and T1/2 ↓426 K) with an 11 K thermal hysteresis loop.
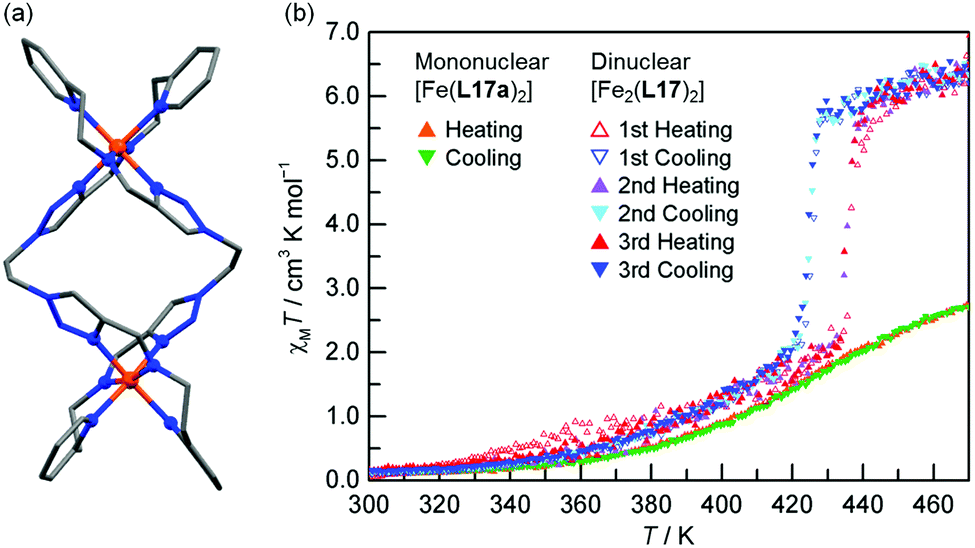 | ||
| Fig. 10 (a) Structure of the cation [Fe2(L17)2]4+ of [Fe2(L17)2](PF6)4·5H2O·MeCN (b) χMT vs. T for [Fe2(L17)2](PF6)4·5H2O·MeCN and the mononuclear analogue [Fe(L17a)2](PF6)2. Structure redrawn from CCDC data.76 (b) Adapted, with permission, from ref. 76. Copyright 2016 RSC. | ||
In a follow-up study, Hagiwara and Hora changed the linker length in the ligand from ethylene in L17 to propylene in L18, and assembled the second example of an SCO-active double helicate, [Fe2(L18)2](AsF6)4·5H2O·MeCN.77 The change in linker from ethylene (L17) to propylene (L18) has changed the Fe–Fe distance from 7.85 to 10.87 Å (Table 5). This new double helicate is also [LS–LS] at 300 K, but above room temperature it undergoes an abrupt SCO to [HS–HS] with a remarkably wide hysteresis loop of 84 K (T1/2↑ = 485 K, T1/2↓ = 401 K). But in this case, upon further cycling in the SQUID, the SCO becomes more gradual and the hysteresis loop becomes narrower, due to lattice solvent loss and/or loss of crystallinity at high temperatures – once again highlighting the sensitivity of polynuclear SCO compounds to crystal packing and solvent content.
The use of these bis-terdentate ligands (Chart 3) results in higher T1/2 values and less distorted Fe(II) centres in the resulting doubly-stranded helicates (43–48°, Table 5), than in the triply-stranded helicates (54–159°, Tables 2–4) formed using bis-bidentate ligands (Chart 2).
Dinuclear Fe2L2: PMRT and related ligands
Grouped together here are a range of bis-terdentate ligands, generated since the first SCO-active complex of such a PMRT-type ligand was reported in 2005,10 that provide an N1N2-bridge (of a five-membered heterocycle) between the binding pockets (Chart 4) forming doubly-bridged Fe2L2 complexes (Fig. 2, 2B).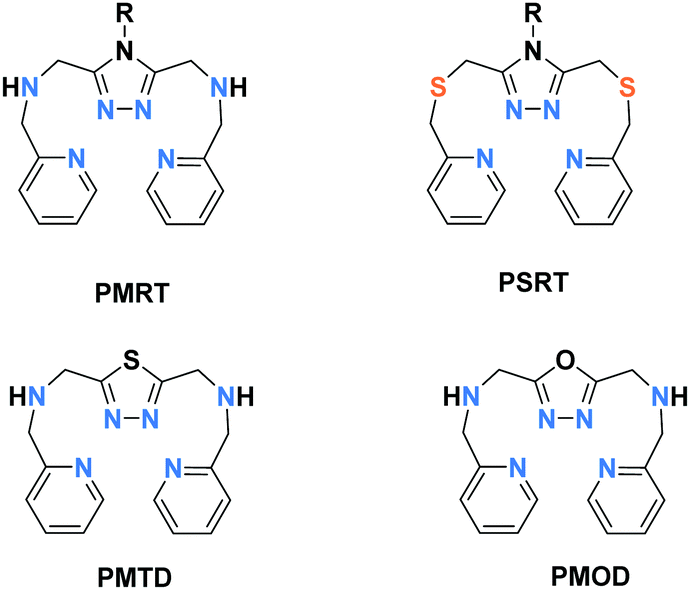 | ||
| Chart 4 Bis-terdentate PMRT and PSRT (which vary in the choice of R, including PMAT where R = NH2) and PMTD and PMOD ligands. | ||
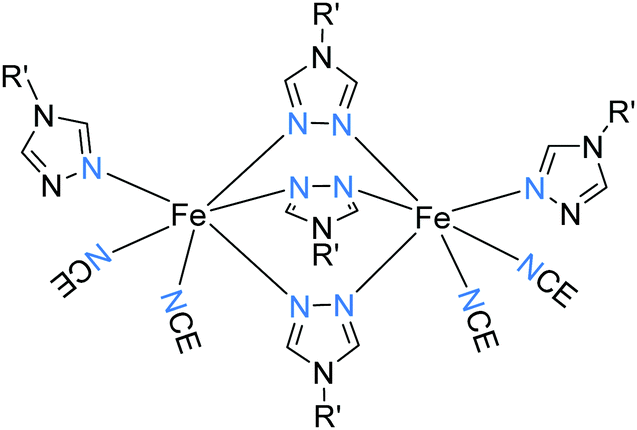
Of these ligands (Chart 4), the bis-terdentate 4-substituted-3,5-bis{[(2-pyridylmethyl)-amino]methyl}-4H-1,2,4-triazole ligands (PMRT, Chart 4)10,78–82 have been widely studied within the present authors group since we first reported them in 2005.10,78 The bis-terdentate ligand scaffold is strongly predisposed to forming dinuclear complexes. Key features include: two PMRT ligands provide all 12 N donors to the two octahedral metal ions, and double triazole bridges between them, and the R group can be widely varied (Fig. 2, 2B). The [FeII2(PMAT)2](BF4)4·DMF (PMRT with R = NH2) complex undergoes an abrupt half-SCO at 224 K. The half SCO was determined to be to a localised [HS–LS] state,10 rather than a 50:50 mixture of complexes in the [HS–HS] and [LS–LS] states,83 by X-ray crystallography. The LS Fe(II) centre has a more regular octahedral coordination sphere, and hence a smaller octahedral distortion parameter, Σ, as well as shorter metal–ligand bond lengths (Fig. 11 and Table 6). This was the first report of a crystal structure of a localised mixed spin state dinuclear complex.10 The localised mixed spin state was later further confirmed by standard Mössbauer spectroscopy, as the HS ion in the [HS–LS] state gave a distinct signal to that of the HS ions in the [HS–HS] state – the first time such a distinction had been made for a dinuclear complex without the use of an applied field.79
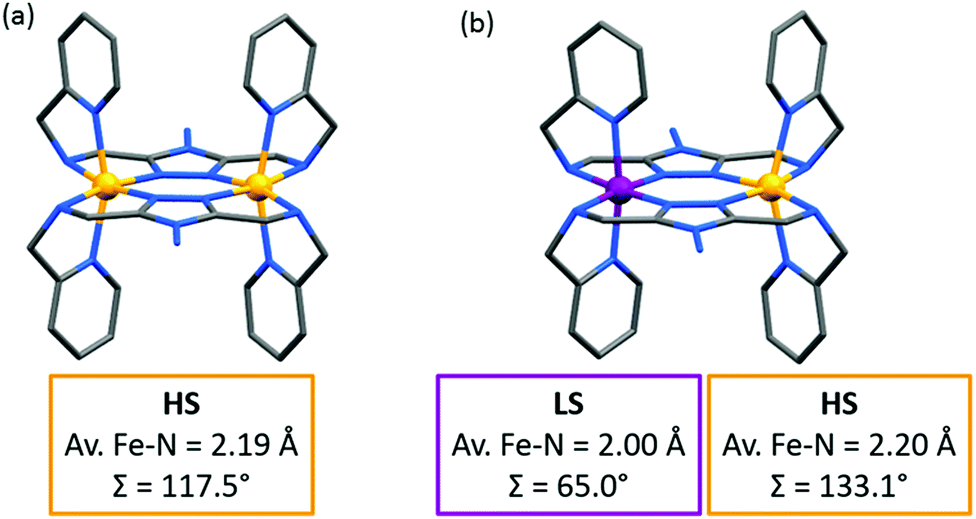 | ||
| Fig. 11 (a) The [HS–HS] 298 K structure and (b) [HS–LS] 123 K structure of [FeII2(PMAT)2](BF4)4·DMF. Structures redrawn from CCDC data.10 | ||
| T (K) | Σ (°) | Fe–Fe (Å) | Spin state | T 1/2 (K) | Lit. | |
|---|---|---|---|---|---|---|
| [FeII2(PMAT)3](BF4)4·DMF | 123 | 65, 133 | 4.212 | LS–HS | 224 | 10 |
| 298 | 117 | 4.296 | 2HS | |||
| [FeII2(PMBzT)2](BF4)4·2CH3CN | 91 | 99 | 4.178 | 2HS | 147 | 84 |
| [FeII2(PMPhtBuT)2](BF4)4·3CH3CN·0.5Et2O | 91 | 118 | 4.262 | 2HS | ↑217 | 85 |
| ↓194 | ||||||
| [FeII2(PSPhT)2](BF4)4·2MeCN·H2O | 100 | 110 | 4.216 | 2HS | 265, 210 and 87 | 86 |
| [FeII2(PSPhT)2](BF4)4·2.5MeCN·0.5H2O·THF | 100 | 105 | 4.231 | 2HS | Not stated | |
| [FeII2(PSMePhT)2](BF4)4·2MeCN | 100 | 88 | 4.189 | 2LS/HS | 109 | |
| [FeII2(PMTD)2](BF4)4·0.75MeOH·0.5H2O | 173 | 58, 65 | 3.946 | LS–LS | Not stated | 88 |
| [FeII2(PMTD)2](ClO4)4·0.75MeOH·0.5H2O | 173 | 59, 66 | 3.952 | LS–LS | Not stated | |
| [FeII2(PMTD)2](OTf)4·1.5MeOH·0.5CH2Cl2 | 173 | 61, 58 | 3.948 | LS–LS | Not stated | |
| [FeII2(PMTD)2](BF4)4·4DMF | 173 | 59, 60 | 3.950 | LS–LS | 380 | 89 |
| [FeII2(PMTD)2](ClO4)4·4DMF | 193 | 59, 60 | 3.953 | LS–LS | 380 | |
| [FeII2(PMOD)2](ClO4)4·4MeCN | 193 | 121 | 4.402 | 2HS | 150 | 90 |
| 100 | 70, 137 | 4.326 | LS–HS | |||
| [FeII2(PMOD)2](BF4)4·2MeCN | 173 | 126 | 4.378 | 2HS | Not stated | |
| [FeII2(PMOD)2](OTf)4·2MeCN | 193 | 125 | 4.361 | 2HS | 150 |
Variation of the N4 substituent, R, to give other PMRT ligands was shown to greatly affect the SCO properties of the resulting dinuclear iron(II) complexes, [FeII2(PMRT)2]X4, despite R being physically and electronically (when R is a ring it is usually almost at right angles to the triazole, switching off any possible resonance effects) remote from the donor atoms (Fig. 12).84
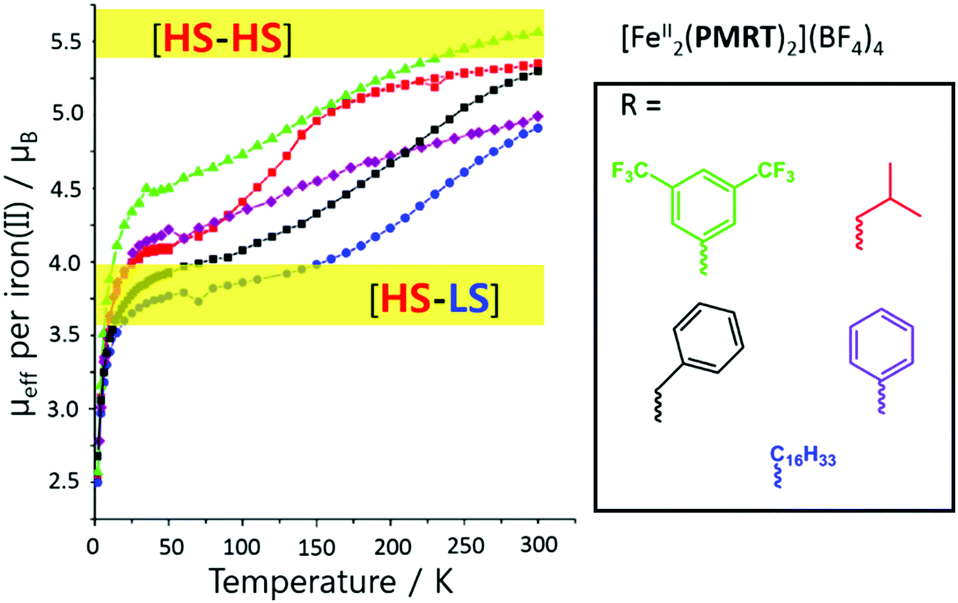 | ||
| Fig. 12 Effective magnetic moment per iron(II) centre vs. T for the SCO-active complexes [FeII2(PMRT)2](BF4)4, where R is one of the five moieties shown in the box. Figure adapted with permission from ref. 84. Copyright 2013 ACS. | ||
When the R group of PMRT was the 4-(tert-butyl)phenyl group, and specifically only for the solvatomorph [FeII2(PMt-BuPhT)2](BF4)4·3½H2O, an abrupt half SCO was observed with a wide thermal hysteresis loop of 22 K – at the time a record equalling hysteresis loop width for a dinuclear complex.85 Hysteresis loops are a kinetic phenomenon so typically become narrower with a decreasing rate of change of temperature,6 ultimately closing entirely (but potentially only after a very long time). However, remarkably, upon decreasing the temperature scan rate of the SQUID measurements on [FeII2(PMt-BuPhT)2](BF4)4·3½H2O only the cooling mode narrows, while the T1/2 of the warming mode is unchanged (Fig. 13).
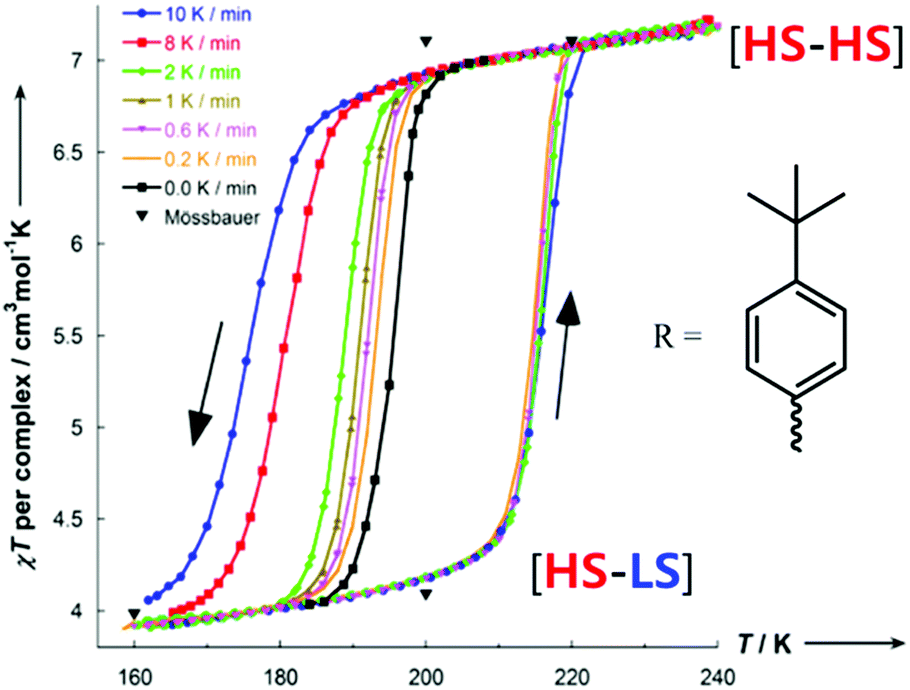 | ||
| Fig. 13 Scan rate dependence of the thermal hysteresis loop of [FeII2(PMt-BuPhT)2](BF4)4·3½H2O. Figure adapted with permission from ref. 85. Copyright 2014 ACS. | ||
The family of dinuclear iron(II) complexes of PMRT has revealed many interesting SCO features, with highlights including the first example of a localised [HS–LS] state to be identified by X-ray crystallography (Fig. 11) and Mössbauer spectroscopy (no applied field), and an example of unique thermal hysteresis behaviour (Fig. 12). However, in no case could the second potential SCO event, to the [LS–LS] state, be accessed, most probably because of the steric restraint of having all twelve donors to the two iron(II) centres, and two N1N2-triazole bridges between them, provided by just two ligands. These features lead to a significantly increased distortion at the HS centre in the [HS–LS] state, over that in the [HS–HS] state, which stabilises the mixed spin state and prevents the second HS centre from undergoing SCO (Table 6). Indeed, even under the immense pressure of 1.03 GPa and at the extremely low temperature of 4 K, the [LS–LS] state was inaccessible for [FeII2(PMAT)2](BF4)4·DMF.82 It is clear that the very constraining PMRT ligand family is extremely good at communicating the structural effects that SCO from HS to LS at the first metal ion induces, to the second metal ion such that it becomes even more distorted away from octahedral and therefore remains HS (Table 6).
The [LS–LS] state was recently accessed by changing the amino linkages to the pyridine side-arms of PMRT to thioether linkages in the PSRT family (Chart 4).86 This change resulted in a more flexible ligand framework, due to longer C–S bonds compared to C–N bonds. In the corresponding [Fe2(PSRT)2](BF4)4 complexes (Fig. 14a), not only was SCO retained upon changing from the N6 donor sphere provided by the PMRT family to N4S2, but, for the first time in complexes of this kind, all three of the possible spin states – [HS–HS], [HS–LS] and [LS–LS] – were accessible.
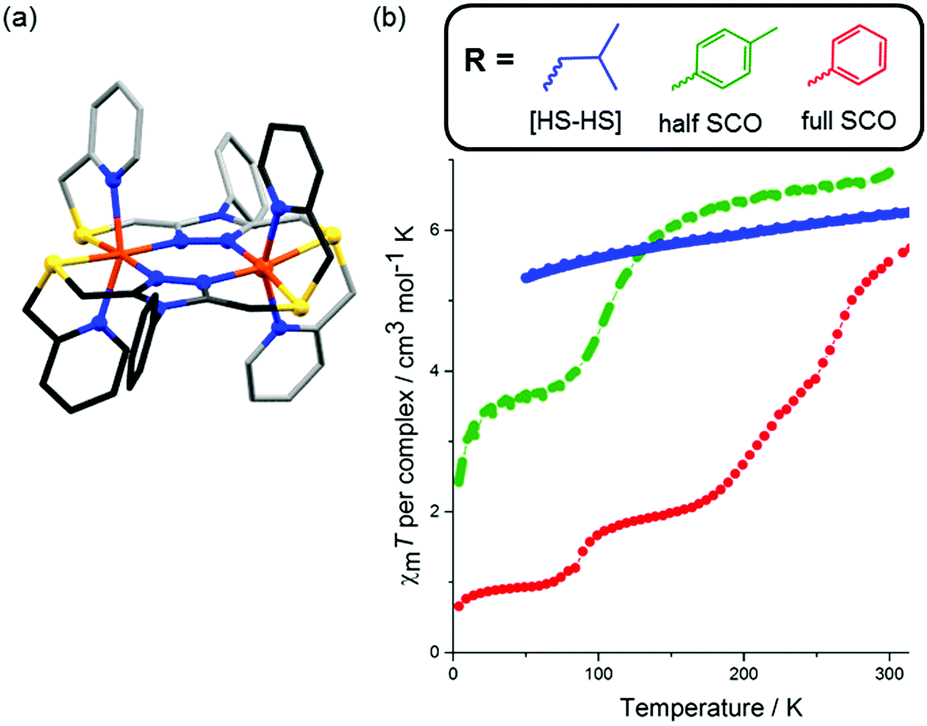 | ||
| Fig. 14 (a) Structure of the complex cation of [Fe2(PSPhT)2](BF4)4, with carbons shaded dark/light grey on opposite ligands to highlight the trans-axial binding mode. (b) Solid state SCO of [Fe2(PSRT)2](BF4)4 complexes with varying R. Structure redrawn from CCDC data.86 (b) Adapted with permission from ref. 86. Copyright 2016 ACS. | ||
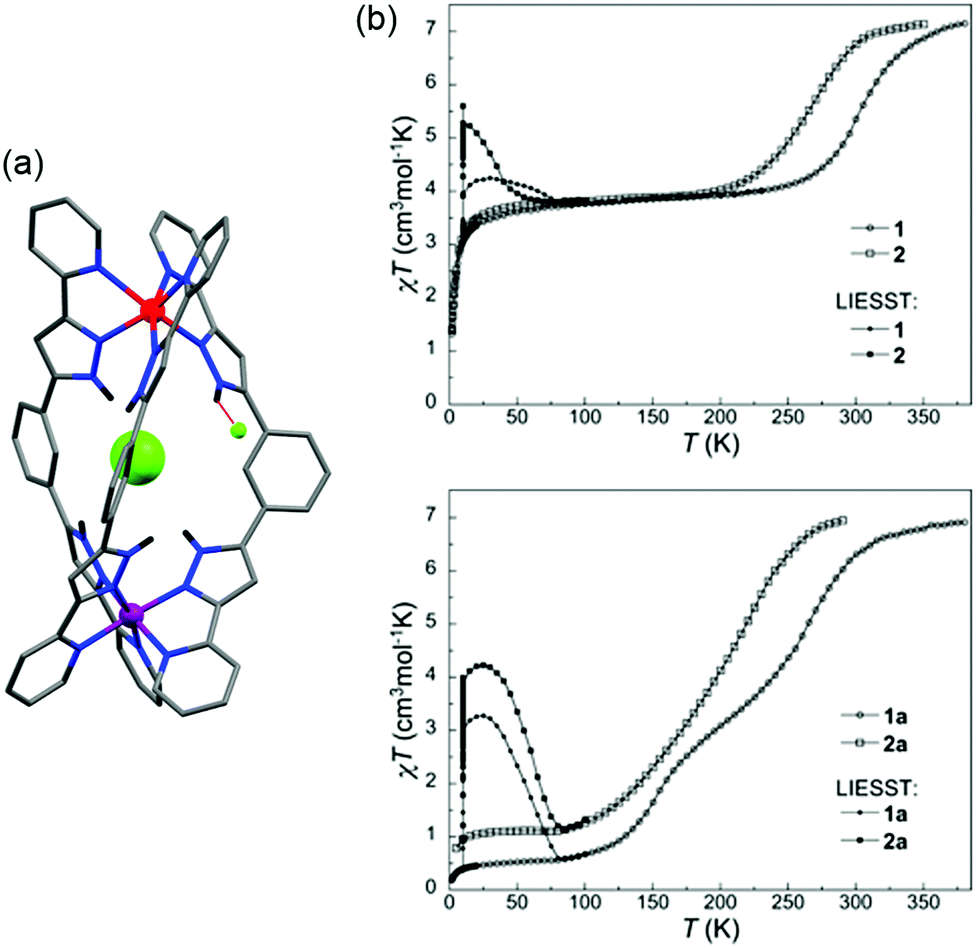
The complex [Fe2(PSPhT)2](BF4)4 in particular had interesting SCO properties, undergoing a multistep [HS–HS] → [HS–LS] → [LS–LS] transition (Fig. 14b). Unfortunately, the [Fe2(PSRT)2](BF4)4 complexes are only SCO active after MeCN solvent loss, which is associated with a loss of crystallinity, and no structural observations of the multistep SCO could be made. From the [HS–HS] structures (MeCN solvated), however, one can observe the enhanced flexibility of these PSRT dinuclear systems, over the PMRT analogues, and this is believed to facilitate the observed complete [HS–HS] → [LS–LS] SCO. The Fe–N bond lengths in the PSRT structures are completely within the range expected for HS Fe(II) (Table 6). Furthermore, while PMRT ligands have always been observed to bind two Fe(II) metals in a bis-terdentate fashion and in a cis-axial mode (both pyridine arms up, but note that an alternative binding mode has been seen for an Fe(III) oxo product87), the more flexible PSRT ligands were observed to bind in bis-terdentate fashion and either a cis-axial mode or trans-axial mode (pyridine arms up/down, Fig. 14a). In addition to solid state SCO-activity, the [Fe2(PSRT)2](BF4)4 complexes exhibit SCO in MeCN solution, induced by both temperature and pressure, with more electron donating R groups predictably favouring LS more.16,86
The Rentschler group has reported an interesting family of PMRT-like complexes of a thiadiazole analogue of PMRT, 2,5-bis[(2-pyridylmethyl)amino]methyl-1,3,4-thiadiazole (PMTD, Chart 4). The bis-terdentate PMTD ligand differs from PMRT ligands in that there is a non-coordinating sulphur atom in place of the non-coordinating N4-R moiety of PMRT. It was shown to produce [FeII2(PMTD)2]X4 complexes with the trans-axial binding mode (Fig. 15a, like the thioether PSRT systems), revealing more flexibility than in the analogous PMRT complexes (always cis-axial mode), and thus giving access to the fully [LS–LS] state.
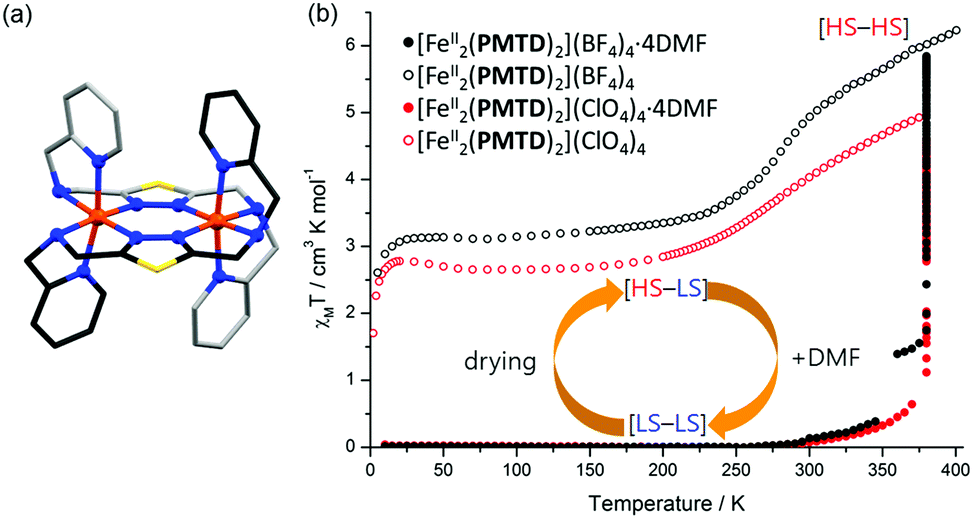 | ||
| Fig. 15 (a) Structure of the cation of [FeII2(PMTD)2]X4, with carbons shaded dark/light grey on opposite ligands to highlight the trans-axial binding mode. (b) [FeII2(PMTD)2]X4·solvent χMT vs. T data sets showing the DMF sensing ability of the complexes. Structure redrawn from CCDC data.88 (b) Adapted with permission from ref. 89. Copyright 2016 ACS. | ||
The impact of the larger S heteroatom in the central five membered ring of PMTD (over the N atom in PMRT) on the overall flexibility can also be seen in the angle at which the amino side arms protrude from the thiadiazole/triazole, with Nazole–Nazole–Namino angles averaging 176.04° for LS PMTD complexes and 172.30° for [HS–LS] [Fe2(PMAT)2](BF4)4. Indeed, all three [FeII2(PMTD)2]X4 complexes highly favour the [LS–LS] state and undergo a gradual SCO starting above 250 K, not reaching the [HS–LS] state (or the [HS–HS] state) at the limit of the measurement, 380 K.88 In 2016, the DMF solvates of [FeII2(PMTD)2](BF4)4 and [FeII2(PMTD)2](ClO4)4, which are both [LS–LS], were shown to undergo a transition to the [HS–HS] state upon thermal desolvation of the DMF molecules, and to subsequently undergo a half-SCO to the [HS–LS] state upon cooling (Fig. 15b and Table 6).89
The Rentschler group further investigated the effect of changing the non-coordinated-heteroatom in the five-membered ring, this time to an oxygen atom, generating the oxadiazole analogue, 2,5-bis{[(2-pyridylmethyl)amino]-methyl}-1,3,4-oxadiazole (PMOD, Chart 4). The more electronegative oxygen (cf. the sulphur atom of PMTD) was anticipated to decrease the ligand field strength at the N donor atoms of the heterocycle, and indeed the HS state is stabilised in comparison to the thiadiazole PMTD complexes: all three [Fe2(PMOD)2]X4 (X = ClO4−, BF4−, or CF3SO3−) complexes are [HS–HS] at room temperature. While the X = BF4− complex remains HS to low temperatures, the X = ClO4− and X = CF3SO3− complexes undergo abrupt [HS–HS] to [HS–LS] SCO centred about 150 K, with a 26 K wide hysteresis loop for [Fe2(PMOD)2](CF3SO3)4. The [LS–LS] state was inaccessible due to steric constraints imposed by the rigid ligand: the smaller oxygen heteroatom tightens the angle of the amino side arms relative to that in PMTD, as seen by a reduction of the Nazole–Nazole–Namino angle from 177 to 168°, and by the cis-axial binding mode (Fig. 16). As was seen with the PMRT ligands, the transition to the [HS–LS] state for [Fe2(PMOD)2](ClO4)4 is associated with an increase in octahedral distortion parameter at the residual HS Fe(II) centre ([HS–HS] @ 193 K, ΣHS = 121°; [HS–LS] @ 100 K, ΣHS = 137° Fig. 16 yellow, ΣLS = 70° Fig. 16 purple; Table 6). Crystal packing appears to have a large influence on the SCO behaviour. A hydrogen bonding network for one of the compounds, linking amino NH groups on neighbouring [Fe2(PMOD)2]4+ cations via CF3SO3− counteranions, infers high cooperativity and results in the hysteresis loop. Also, all three compounds contain two or four solvent MeCN molecules, and although no significant interactions to the cations are obvious, SCO is lost upon desolvation.
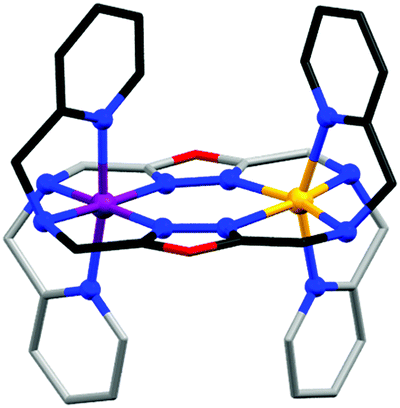 | ||
| Fig. 16 Structure of the cation of [FeII2(PMOD)2]X4, (yellow HS and purple LS FeII centre) with carbons shaded dark/light grey on opposite ligands to highlight the cis-axial binding mode.90 Structure redrawn from CCDC data. | ||
Dinuclear Fe2L2 formed by assorted other ligands
Further examples of dinuclear Fe2L2 type complexes (Fig. 2, 2B) are formed by the diverse family of ligands L19–L21 (Chart 5).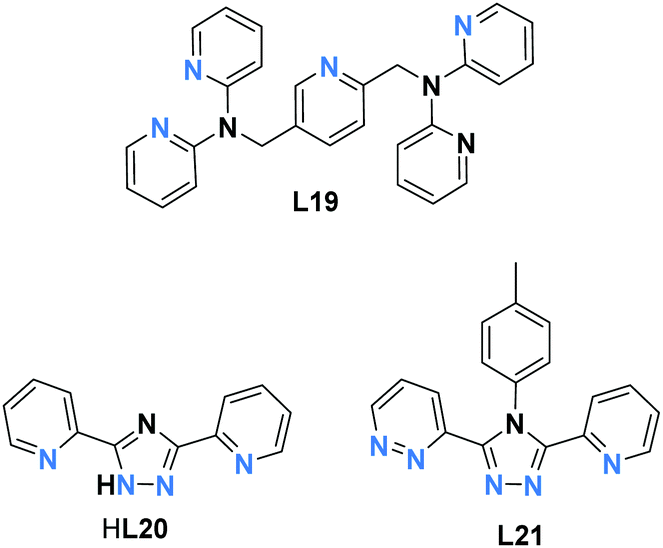 | ||
| Chart 5 Assorted other ligands, L19–L21, forming SCO-active Fe2L2 type dinuclear Fe(II) complexes (Fig. 2, 2B). | ||
Murray, Kepert and co-workers have reported a pyridyl rich ligand, L19 (Chart 5), which binds in a bis-bidentate manner, forming a SCO active dinuclear Fe2L2 complex, [Fe2(L19)2(NCS)4]·4CH2Cl2 (Fig. 17a).91 It undergoes a two-step SCO with all three spin states accessed: [HS–HS], [HS–LS], and [LS–LS]. The plateaus occurred at 110 and 140 K with T1/2 = 80 and 180 K (Fig. 17b, a circle). The authors traced the magnetic behaviour with Σ angle at different temperatures (Table S1, ESI†). At 25 K both Fe(II) centres were in the LS state with Σ = 28.4° and 35.2°. The intermediate [HS–LS] state observed at 123 K has FeLSΣ = 27.7° (Fig. 17a, purple) and FeHSΣ = 45.9° (Fig. 17a, yellow). Furthermore, heating from 123 K to 250 K the geometry around Fe2 is slightly changed from 45.9° to 46.7°, Fe1 = 41.8°. The partially de-solvated compound underwent a one-step SCO with increased transition temperature, T1/2 of 200 K (Fig. 17b, b square). Furthermore, immersion of partially de-solvated compound into dichloromethane reverted the two-step SCO (Fig. 17b, c triangle). The fully de-solvated compound lost its SCO nature and remained HS all over the temperature range, χmT = 3.15 cm3 K mol−1 at 300 K (Fig. 17b, d rhombus).
 | ||
| Fig. 17 (a) [FeII2(L19)2NCS4] structure, with carbons shaded dark/light grey. (b) Plots of χmT per FeIIvs. T, for complex [Fe2(L19)2(NCS)4]·xCH2Cl2 (a) x = 4 (circle), (b) x = 1 (square) (c) re-solvated, x = 4 (triangle) (d) x = 1 (rhombus). Structure redrawn from CCDC data and (b) adapted from ref. 91 with permission. Copyright 2006 John Wiley and Sons. | ||
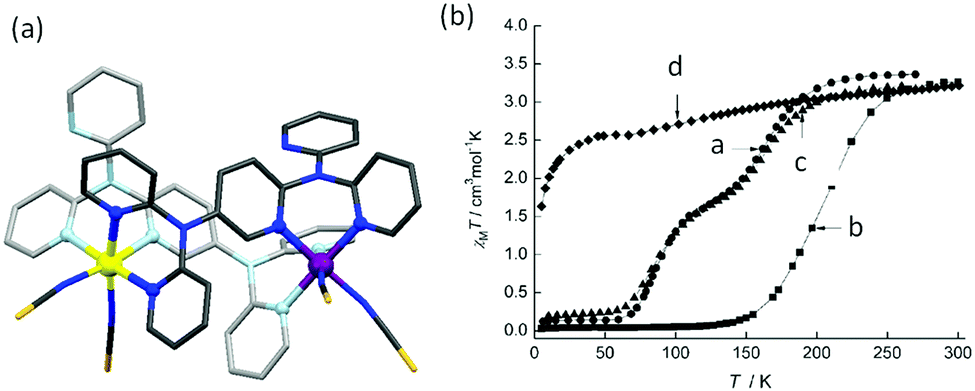
Murray and co-workers also reported, in 2013, a triazolate bridged dinuclear complex of anionic L20, Chart 5 (Fig. 18a). Single crystal and powder forms of [Fe2(L20)2(NCBH3)2(py)2] showed distinctly different degrees of thermally induced SCO behaviour with different T1/2 values. The powder sample showed a two-step complete SCO at T1/2 = 194 and 151 K (Fig. 18b) whereas the crystal form showed an abrupt half step SCO at T1/2 102 K (Fig. 18c).92
 | ||
| Fig. 18 (a) The crystal structure of [Fe2(L20)2(NCBH3)2(py)2]. (b) χmT vs. T plot for powder sample and (c) crystal form. Structure redrawn from CCDC data. (b and c) Adapted from reference, with permission.92 Copyright 2016 John Wiley and Sons. | ||
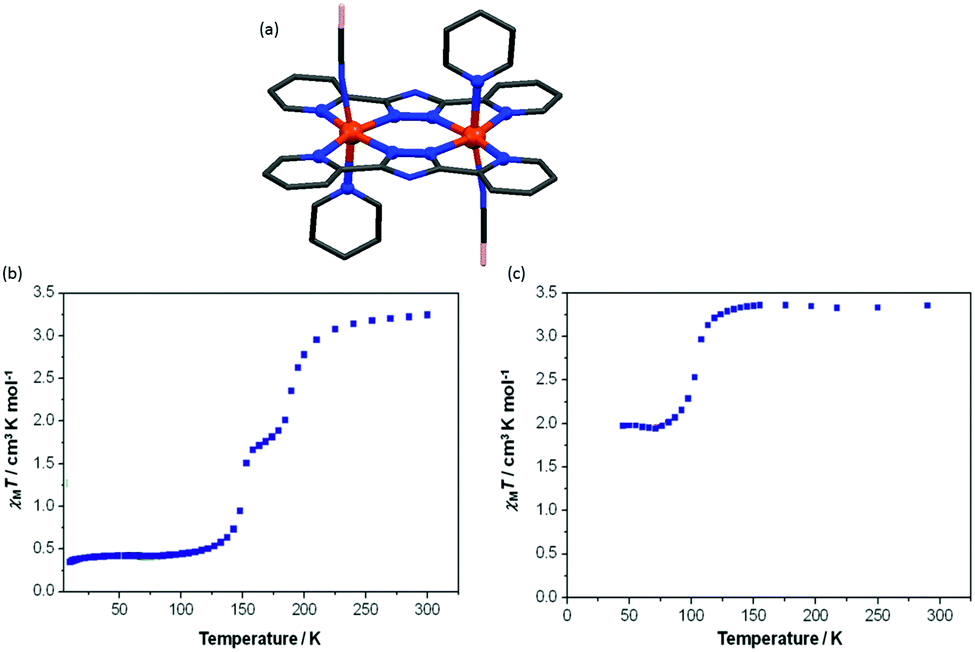
Brooker and co-workers reported another kind of Fe2L2 type SCO–Fe(II) complex in 2016, by employing L21 (Chart 5).93 The resulting dark red dinuclear complex [Fe2(L21)2(MeCN)4](BF4)4·2MeCN was shown by a crystal structure determination to be [LS–LS] at 100 K (Fig. 19a). It underwent a gradual SCO with a 13 K wide thermal hysteresis loop, at T1/2↓ = 356 K and T1/2↑ = 369 K (Fig. 19b, maroon curve). The χmT was 2.45 emu mol−1 K at 400 K, indicating about 80% conversion from [LS–LS] to [HS–HS]. Guest exchange, by reversible SCSC transformations, gave two other complexes, orange [Fe2(L21)2(EtOH)4](BF4)4 and yellow polymer {[Fe2(L21)2(H2O)4](BF4)4}∞, both of which were [HS–HS] at all temperatures 50–400 K (Fig. 19b, orange and yellow curves, respectively).
 | ||
| Fig. 19 Data for [Fe2(L21)2(MeCN)4](BF4)4·2MeCN: (a) structure of the tetracation. (b) χmT (per Fe) vs. T plot (maroon). Structure redrawn from CCDC data and (b) adapted from reference, with permission.93 Copyright 2013 John Wiley and Sons. | ||
Dinuclear singly N-ligand linked Fe2L
Grouped together in this section are the Fe2L dinuclear complexes in which the two Fe(II) centres are linked by a single ligand L that provides some, or all, of the N-donors (Fig. 2, 2C). The ligands employed vary considerably, ranging from bis-monodentate, to bis-bi-, bis-tri-, bis-tetra- and bis-hexa-dentate (Chart 6), but all give rise to dinuclear M2L complexes of type 2C (Fig. 2). | ||
| Chart 6 Ligands used to link two Fe(II) ions forming SCO-active singly-ligand linked dinuclear M2L complexes (Fig. 2, 2C). | ||
In 2015, Meyer and co-workers reported a dinuclear Fe2L complex (Fig. 2, 2C) of a bis-terdentate pyrazolate ligand, HL22 (Chart 6).94 HL22 is deprotonated and provides a single pyrazolate bridge between the two Fe(II) centres as well as meridionally coordinated N2P donors to each of them, in [Fe2L22(OTf)3(CH3CN)] (Fig. 20a). In addition to the bridging pyrazolate, the Fe(II) centres are also bridged by two triflate anions, and the octahedral geometry of HS Fe1 is completed by a terminal triflate (O3N2P), and that of Fe2 by a MeCN ligand (O2N3P). The magnetic behaviour of this complex was studied in both solid and solution (Fig. 20b). In the solid state it remains fully high spin, due to the O-donors. In contrast, in MeCN solution, the complex exchanges the triflate anions for MeCN solvent molecules, becoming N5P-coordinated and enabling it to undergo a reversible and complete thermally induced SCO (Fig. 20b).94
 | ||
| Fig. 20 (a) The solid state structure of [Fe2L22(OTf)3(CH3CN)]. (b) χmT vs. T plot in the solid state (left) and MeCN solution (right). Structure redrawn from CCDC data. (b) Reproduced from reference, with permission.94 Copyright 2015 John Wiley and Sons. | ||
Sato and co-workers95 studied the pyrazine-bridged Fe2L dinuclear complexes (Fig. 2, 2C) [Fe2L(NCS)4]·DMF·2H2O, of the bis-tetradentate pyrazine ligands L = L23, L24 (Chart 6). The dinuclear complex of the 2,5-substituted pyrazine ligand L23, [Fe2L23(NCS)4]·DMF·2H2O, underwent a one-step SCO at around 180 K (Fig. 21, red circles). After being heated to 400 K, desolvated [Fe2L23(NCS)4] showed a different SCO curve, with an abrupt transition (T1/2↑ = 189, T1/2↓ = 184 K) and 5 K wide hysteresis loop (Fig. 21, inset). In the case of the 2,3-substituted pyrazine ligand L24, the resulting complex displayed a gradual two step SCO, [HS–HS] ↔ [LS–HS] ↔ [LS–LS].95 At 300 K, χmT value is 5.58 cm3 K mol−1, consistent with the [HS–HS] state, and decreases gradually to 0.71 cm3 K mol−1 at 70 K, consistent with the [LS–LS] state, however with a change in slope occurring at approximately χmT = 3 cm3 K mol−1, 185 K, indicating that the Fe(II) centres undergo SCO at differing temperatures. The LIESST studies on this pair of complexes gave pairs of thermal transition temperature (T1/2) and photo-induced transition temperature (TLIESST) values for desolvated [Fe2L23(NCS)4] (192, 70 K) and for [Fe2L24(NCS)4]·DMF·2H2O (210 and 56 K; 130 and 71 K). These fall close to the T0 = 120 K line of the correlation, TLIESST = T0 − 0.3T1/2, earlier identified by Létard and co-workers.96
 | ||
| Fig. 21 X mol T vs. T for [Fe2L23(NCS)4]·DMF·2H2O (initial heating: red circles; cooling cycle: blue squares, heating cycles: black triangles (on heated sample)). Figure reproduced from ref. 95. Copyright 2013 John Wiley and Sons. | ||
In 2013, Petzold and co-workers reported a dinuclear Fe2L complex of a bis-hexadentate, biphenyl-linked, L25 (Chart 6), in which the two Fe(II) centres are linked by this single ligand.97 Remarkably L25 provides all twelve N-donors to the two octahedral Fe(II) centres in [Fe2(L25)](BPh4)4. The structure determination shows the complex to be fully LS at 153 K (Fig. 22a) whereas mononuclear [Fe(L25)](BPh4)2 was paramagnetic at room temperature. In MeCN solution there is a very gradual, partial SCO towards HS as the temperature is raised from 298 (25 °C) to 348 K (75 °C), with the fraction HS rising from 0.05 to 0.15 (Fig. 22b).
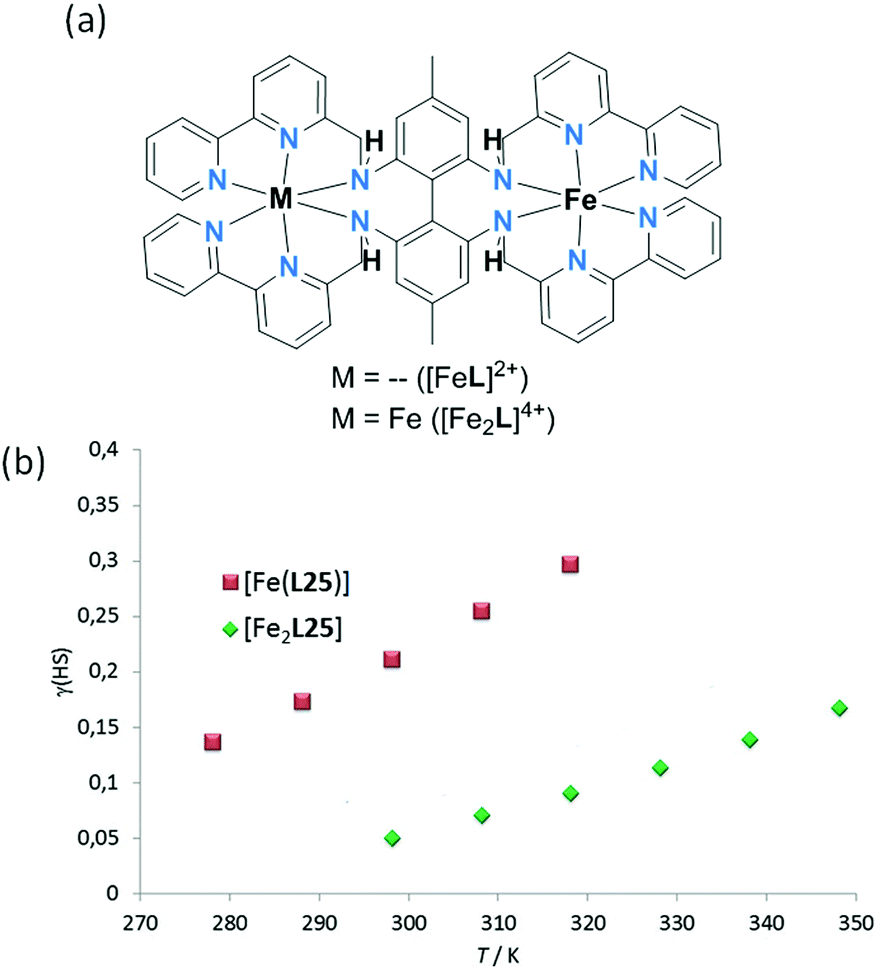 | ||
| Fig. 22 (a) Binding sites in mononuclear and dinuclear Fe(II) complexes of hexadentate L25; dinuclear singly-ligand linked (Fig. 2, 2C). (b) Evans measurements on mononuclear [Fe(L25)](BPh4)2 and dinuclear [Fe2(L25)](BPh4)4. Figure adapted from reference, with permission.97 Copyright 2013 John Wiley and Sons. | ||
Some of the 4,4′-bi-pyridine-like bridged SCO-active Fe2L complexes were covered in our previous review,35 but for completeness are also included here. Matouzenko and co-workers studied a series of dinuclear Fe2L complexes, [{Fe(L26)(NCX)2}2(L)]·nMeOH (X = S and BH3; L = L27–L30, Chart 6) varying the length of the bipyridyl-like bridges L, between the Fe(II) centres (Fig. 2, 2C), and employing L26 capping ligands (Chart 6). They first reported this kind of complex in 2009, with X = S and L27.98
Then in 2011 they replaced the L27 bridge by L29 (Chart 6), retaining X = S, generating three forms of [{Fe(L26)(NCS)2}2(L29)]·nMeOH, a pair of solvent-free polymorphs (n = 0) and a solvatomorph (n = 2), which displayed different SCO behaviours. The two solvent-free polymorphs were dark vinous (red-wine coloured) prismatic versus brown crystals. The crystal structures of the vinous polymorph at 300 K, 183 K and 90 K, revealed [HS–HS], localised [HS–LS], and [LS–LS] spin states, respectively. This is consistent with the occurrence of a two-step SCO, separated by a subtle inflection point with T = 182 K as revealed by the magnetic study. The brown crystals remained high spin. In contrast, the red-brown crystals of the methanol solvate underwent a complete one step transition, without hysteresis, at 159 K.99
The following year they instead employed a L30 bridge (Chart 6), again retaining X = S, and generating two solvatomorphs, n = 0 and n = 2 (Fig. 23a), which differed in SCO behaviour. The solvent free complex underwent a two-step SCO, with 50% spin conversion and short plateau at 145 K, with a 17 K wide hysteresis loop for the lower temperature SCO event (T1/2↓ = 126 and T1/2↑ = 143 K) (Fig. 23b). The methanol solvate, [{Fe(L26)(NCS)2}2(L30)]·2MeOH, undergoes a one-step SCO with a narrow 4 K hysteresis loop (T1/2↓ = 150 and T1/2↑ = 154 K; Fig. 23c).100
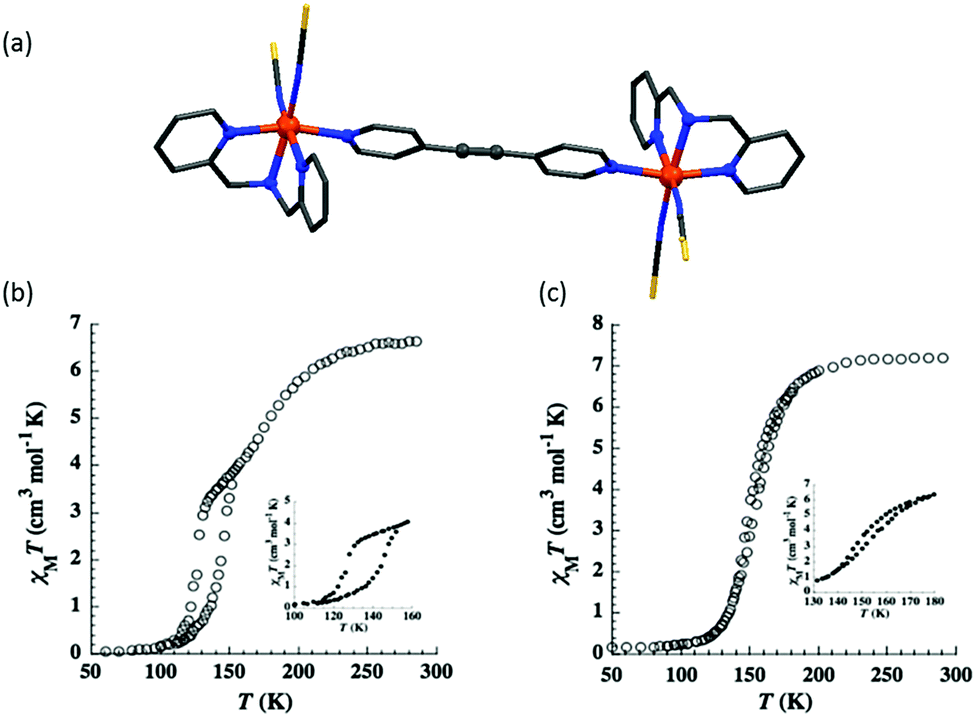 | ||
| Fig. 23 (a) Crystal structure of [{Fe(L26)(NCS)2}2(L30)]. (b) χMT vs. T plot for n = 0 and (c) n = 2. Structure redrawn from CCDC data. (b and c) Reproduced with permission.100 Copyright 2012 John Wiley and Sons. | ||
Then in 2013 they changed to using cyanoborohydride anions, generating a set of three complexes which varied in the choice of L (L = L27, L28, L30), [{Fe(L26)(NCBH3)2}2(L)]. The complex with an L = L27 bridge showed abrupt two step SCO with an inflection point at 210 K whereas with the complexes with an L = L28 or L30 bridge showed gradual one step transitions with T1/2 of 241 and 290 K, respectively.101
Pressure induced SCO is less commonly studied. A key early study carried out on bipymidine-bridged SCO-active Fe2L complexes {[Fe(L)(NCE)2]2L31}, where E = S or Se and the capping ligands were L = L31 or L32 (Chart 6), was reported in 2001 by Gutlich, Real and co-workers (Fig. 24).102
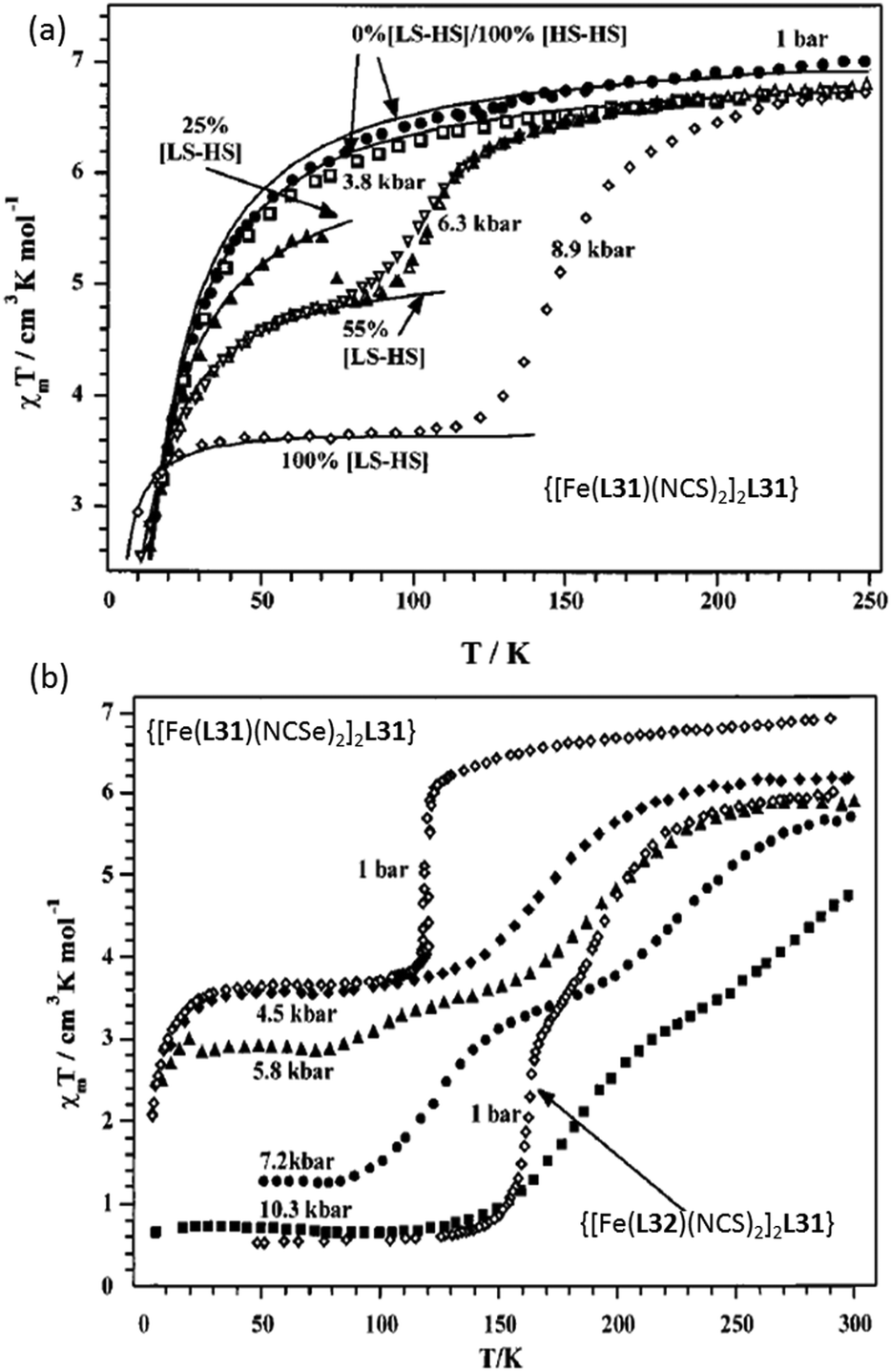 | ||
| Fig. 24 (a) χmT vs. T plot of [Fe(L31)(NCS)2]2L31 and (b) [Fe(L31)(NCSe)2]2L31 and [Fe(L32)(NCS)2]2L31 at various pressures. (a and b) Adapted with permission from ref. 102. Copyright 2001 ACS. | ||
At ambient pressure {[Fe(L31)(NCS)2]2L31} was paramagnetic 4–300 K (Fig. 24a). In contrast, at 8.9 kbar it showed incomplete one-step SCO behaviour, T1/2 ≈ 150 K. At ambient pressure (1 bar) the X = Se analogue, {[Fe(L31)(NCSe)2]2L31}, undergoes a 50% spin transition at T1/2 = 120 K (Fig. 24b).102 The complex featuring bis-thiazole capping ligands (L32, Chart 6), {[Fe(L32)(NCS)2]2L31}, underwent an almost complete two step SCO at ambient pressure (Fig. 24b).
In 2012, Molnár, Guionneau and co-workers reported the effect of pressure on the SCO curve of the 4,4′-bipyridine (L27, Chart 6) bridged Fe2L complex, [{Fe(L33)(NCS)2}2(L27)]·2MeOH, which features L33 capping ligands (Chart 6).103 This complex undergoes thermally and photo-induced incomplete SCO, which was ascribed to a purely structural phase transition and only partial conversion to LS at low temperature.104 But under the influence of high pressure, this complex showed no crystallographic phase transition. High pressure X-ray diffraction and Raman spectroscopic studies showed that a fully LS state is realised at low temperature, and that SCO is very gradual at pressures between 7 and 25 kbar.103
Assorted anion bridged dinuclear complexes
Non-heterocyclic anion bridged (Chart 7) SCO-active dinuclear Fe(II) complexes (Fig. 2, 2D) are rarely reported, with only four cases in the literature, all of which employ polydentate N-donor capping ligands (Chart 8). | ||
| Chart 7 Anions used as bridges in SCO-active dinuclear Fe(II) complexes (Fig. 2, 2D). | ||
 | ||
| Chart 8 Capping ligands used in anion bridged SCO-active Fe(II) dinuclear complexes (Fig. 2, 2D). | ||
In the first case, dicyanamide (dca, Chart 7) acts as the anionic bridge between the two Fe(II) centres, with the N6 coordination spheres completed by L34 capping ligands (Chart 8).105 In the second case, cyanocarbanions (A1, Chart 7) doubly bridge the two Fe(II) centres, with the N6 coordination spheres completed by L35 and L36 capping ligands (Chart 8), giving a neutral complex.106 In the third case, NCS anions doubly bridge the Fe(II) centres with N5S coordination environments and L37 capping ligands (Chart 8).107 The fourth example features a diiminoquinonoid-bridges (A2–A5, Chart 7) with L35 capping ligands (Chart 8).108
Real and co-workers reported the first example, a dicyanamide bridged Fe(II) SCO-active dinuclear complex, in 2005,105 [Fe2L342(dca)](PF6)3·nH2O (n = 1 and 0). When n = 1 it remains HS with a constant χmT value of 6.7 cm3 K mol−1 from 350 K to 50 K. In contrast, n = 0 [Fe2L342(dca)](PF6)3 showed a two-step spin transition: at 400 K, χmT = 7.0 cm3 K mol−1 indicating all iron centres are HS, then χmT decreased gradually to 3.65 cm3 K mol−1 at 250 K with a broad plateau from 285–208 K, then in a second step it decreased gradually to 0.21 cm3 K mol−1 at 50 K. This SCO response was attributed to 50% spin conversion in each step. Magnetic studies in D6-acetone solution showed the one-step incomplete SCO-behaviour of both solvatomorphs.105
In 2016, Triki and co-workers used two types of tetradentate capping ligands, L35 and L36 (Chart 8), to obtain a pair of A1-bridged complexes (A1, Chart 7), [Fe2L2(A1)2]·nMeOH (n = 0.8 and 2, respectively). The small difference between these tetradentate capping ligands, L35 and L36 (Chart 8), leads to a large change in T1/2 (ΔT1/2 > 150 K), from 365 to 180 K, respectively (Fig. 25), which they attributed to changes in crystal packing. After desolvation of this pair of complexes, the T1/2 values were slightly different, 352 and 196 K, respectively.106
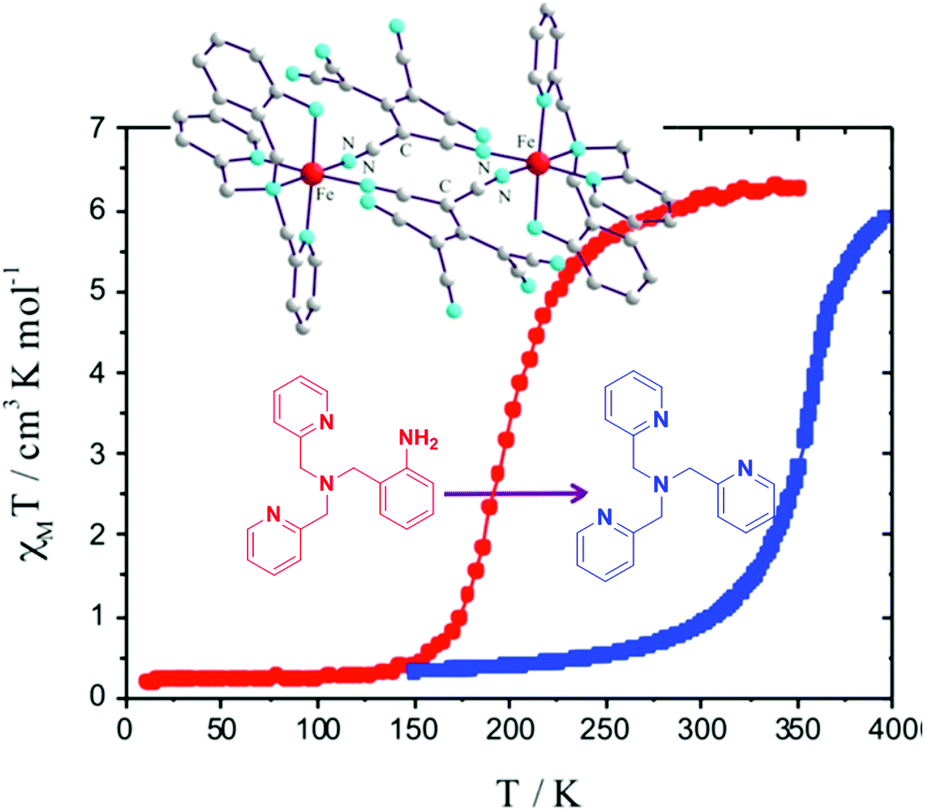 | ||
| Fig. 25 Plot of χmT vs. T of desolvated [Fe2L2(A1)2] [L35 (blue points) and L36 (red points)]. Figure adapted from reference with permission.106 Copyright 2016 ACS. | ||
Harris and co-workers also employed the tetradentate capping ligand L35, but utilised a series of diiminoquinonoid anion bridges, A2–A5 (Chart 7) to generate dinuclear [Fe2(L35)2(A)](B(PhCF3)4)2, with A2–A5, complexes in order to systematically study the effect of varying the electronic effects of substituents on the bridge A on the SCO behaviour (Fig. 26a).108 They were able to correlate T1/2 and ΔH (the latter obtained by fitting the magnetic data to an ideal solution model) (Fig. 26b). They showed that both the T1/2 (from 160–110 K) and ΔH (from 11.4–7.5 kJ mol−1) values decreased with increasingly electron-withdrawing groups X on the bridging anion A2–A5 (–H to –Br to –Cl to –F), as measured by the Pauling electronegativity (χP).108 The incorporation of electron withdrawing groups in A increases the π-back donation from the L35 pyridyl capping ligand and stabilises the LS state, hence shifting T1/2 towards higher temperatures.
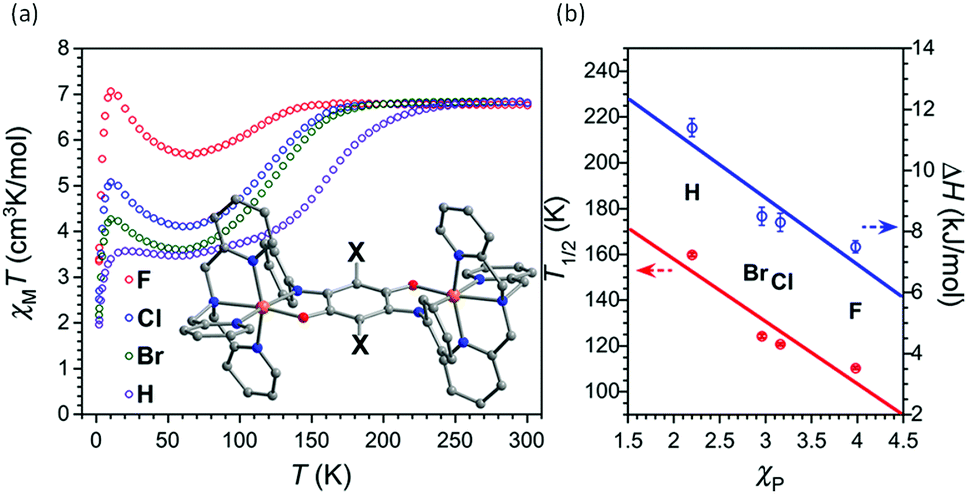 | ||
| Fig. 26 (a) χmT vs. T plot of [Fe2(A)(L35)2](B(PhCF3)4)2, A2–A5. (b) Correlation between T1/2 (red points) and ΔH (blue points) as a function of Pauling electronegativity (χP) of the X substituent (F, Cl, Br, H). (a and b) Adapted with permission from ref. 108. Copyright 2015 ACS. | ||
Another example features bridging NCS− anions, and employs L37 as capping ligands (Chart 8). The complex [{Fe(L37)(NCS)(μ-NCS)}2] showed gradual one step SCO, [LS–LS] ↔ [HS–HS], at 207 K, with no hysteresis loop.107
Trinuclear: triply triazole bridged, Fe3L6
All of the structurally characterized SCO-active trinuclear Fe(II) complexes after 2011 are of one structural type: triply triazole-bridged (Fig. 2, 3A). Like the dinuclear analogues, they are readily structurally characterised, in contrast to the related 1D-chain polymers. To date, 10 different N4-substituted 1,2,4-triazoles (Chart 9) have been employed in generating these complexes. In all cases, the six R-triazole ligands provide triple N1N2 bridges between the central Fe(II) and each of the two terminal Fe(II) ions, resulting in an N6 octahedral central Fe(II) ion, and providing 3N donors to each of the terminal Fe(II) ions with their octahedral coordination completed by three mono-anions and/or solvent molecules (Fig. 27). Depending on coordinated or non-coordinated anions, the three Fe(II) centres are afforded either N3O3–N6–N3O3109–116 or N6–N6–N6117 or N5O–N6–N5O118 coordination environments. In all of these trinuclear complexes, the central Fe(II) is always N6, being triply triazole bridged to each of the other two Fe(II) centres. Hence the central Fe(II) centre in these complexes is the only one that undergoes SCO – except in the case of [Fe3L466A64]117 (N6–N6–N6) where all three of the FeII centres underwent SCO. These complexes have shown a range of gradual to abrupt SCO, with T1/2 varying with the R group, anions and solvent molecules present.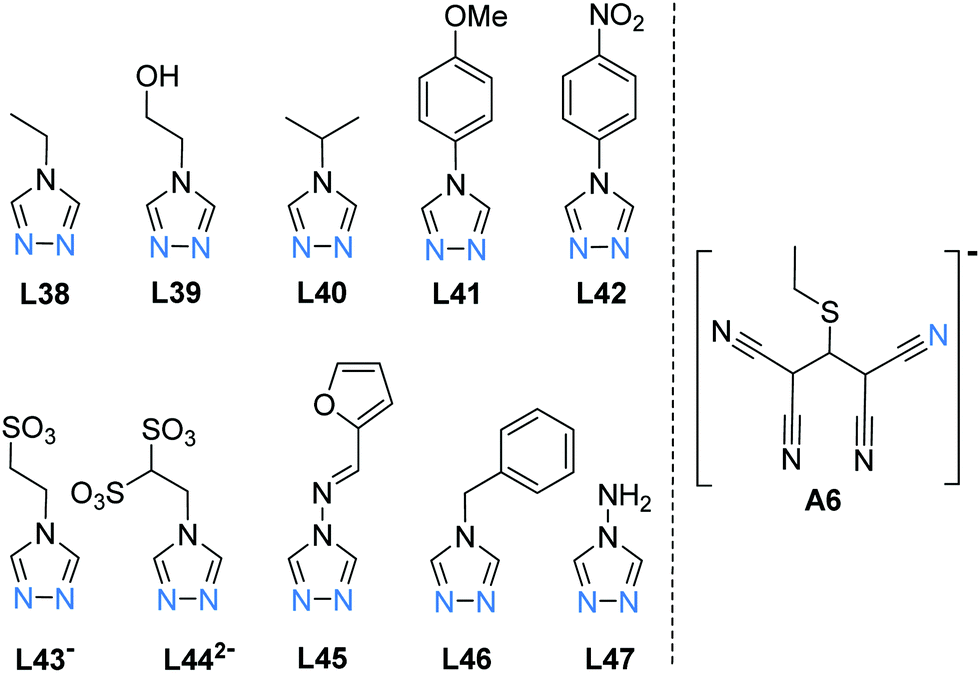 | ||
| Chart 9 Ligands L38–L47 reported in the literature to form SCO-active FeII trinuclear complexes (Fig. 2, 3A): in all cases only the central Fe(II) site is SCO active – except for L46 and A6 which provide all three Fe(II) sites with SCO active N6 coordination environments. | ||
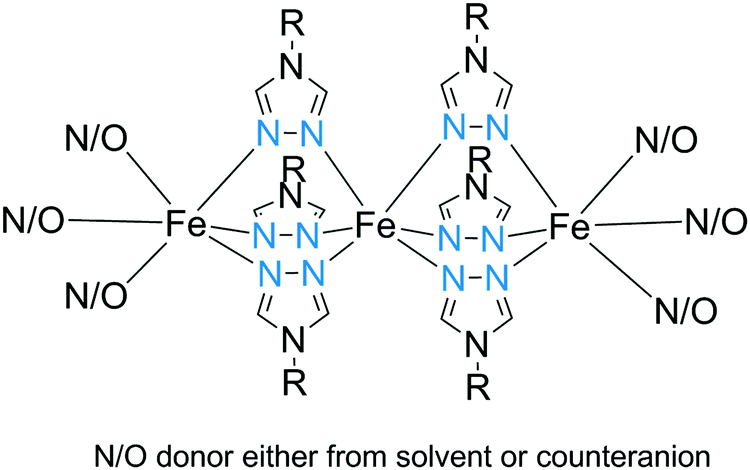 | ||
| Fig. 27 Schematic representation of trinuclear triply-triazole bridged SCO-active Fe(II) complexes (Fig. 2, 3A). | ||
In 1983, the first structurally characterized trinuclear SCO-active complex, [(Fe3(L38)6(H2O)6)](CF3SO3)6, was reported by Reedijk and co-workers.109 This complex showed an abrupt SCO of one third of the Fe(II) ions, as seen by Faraday balance at 203 K and confirmed by Mössbauer studies. By incorporating a hydroxyl group at C2 of the ethyl group (i.e. use of L39, Chart 9), the T1/2 was tuned to room temperature (290 K) which was attributed to the incorporation of hydrogen bonding into the system which increased the ligand field strength at the central metal atom.112
The effect of anion variation on the complex of L40 (Chart 9, 4-(4-isopropyl)triazole) was studied by Haasnoot and co-workers.110 [Fe3L406(H2O)6]6+ showed a gradual spin transition at 242 K and 187 K, for the tosylate and triflate anions, respectively. By varying the R group from L41111 to L42113 with tosylate anions, the T1/2 values changed slightly to 245 K, which was similar to the case of L40, and 148 K, respectively.
Galán-Mascarós and co-workers reported [Fe3(L43)6(H2O)6]·8H2O with hysteresis in its partially dehydrated form, where L43− is anionic in nature with a highly polar functional group at the 4-position. It showed three crystallographic phases with three different magnetic behaviours (Fig. 28a). The original compound had a single step spin transition below 250 K with T1/2 at 150 K. Upon partial dehydration at 320 K the second phase occurred and showed spin transition with hysteresis. The third phase, observed after heating at 393 K, was paramagnetic down to low temperature. For the first phase, the χmT value at room temperature was 9.25 cm3 K mol−1, as expected for three independent HS iron(II) centres. On heating, the χmT value remained constant from 300 K to 320 K, then decreased to 6.2 cm3 K mol−1 at 330 K due to partial solvent loss to form the second phase. This then showed an abrupt increase to 9.44 cm3 K mol−1 at 360 K, and a 14 K wide hysteresis (T1/2↑↓: 357, 343 K) on cooling down again (Fig. 28b).114
 | ||
| Fig. 28 (a) Plots of χmT versus T for [Fe3L436(H2O)6]·8H2O; magnetic behaviour of all three crystallographic phases, (b) for [Fe3L436(H2O)6]·8H2O (5 in this figure) upon heating above room temperature (320 K) showing the transformation into phase partially de-solvated [Fe3L436(H2O)6] (5b in this figure). Figure adapted from ref. 114 with permission. Copyright 2014 John Wiley and Sons. | ||
The same group subsequently reported the trinuclear complex of anionic L442− (Chart 9), (Me2NH2)6[Fe3(L44)6(H2O)6]·6H2O, in which the N4-substituent is ethanedisulphonate. This compound showed a one step SCO above room temperature, with a 65 K wide thermal hysteresis loop (Fig. 29, inset filled squares). The χmT value was 6.37 cm3 K mol−1 at 300 K, corresponding to [HS–LS–HS], and remains constant down to 50 K. On heating above 360 K it increased, and reached 7.92 cm3 K mol−1 at 400 K, consistent with [HS–HS–HS]. Maintaining the sample at 400 K, χmT kept increasing over time and saturated at 8.89 cm3 K mol−1 and the cooling cycle showed a 90 K wide and reproducible hysteresis, with T1/2↑↓ = 400 K and 310 K (Fig. 29, inset empty squares). They have also studied the temperature induced excited spin transition (TIESST) effect. The excited spin state (metastable-HS*) was thermally trapped and remained HS* at high temperature, TTIESST = 250 K, which was the highest value ever reported.115
 | ||
| Fig. 29 Plot of χmT vs. T for (Me2NH2)6[Fe3(L44)6(H2O)6]·6H2O in the range of 2–300 K. Inset: 270–400 K range, empty squares show the saturated magnetic behaviour of (Me2NH2)6[Fe3L446(H2O)6]·6H2O when maintained at 400 K. Figure is reproduced from ref. 115. Copyright 2015 ACS. | ||
Neville and co-workers have recently reported [Fe3(L45)6(p-tol)2(MeOH)4](p-tol)4 (p-tol = p-tolylsulfonate), which showed a gradual one step spin transition with no hysteresis. The χT value at 300 K was 10.2 cm3 K mol−1 indicating fully HS Fe(II), then decreased gradually to 7.8 cm3 K mol−1 at 50 K, which corresponds to one third of Fe(II) undergoing SCO to LS,116 as is usual in these trinuclear complexes (see above).
In 2017, Marchivie and co-workers reported that [Fe3L466(A6)6] (Chart 9, A6 = 1,1,3,3-tetracyano-2-thioethylpropenide), with N3N3–N6–N3N3 coordination spheres around each Fe(II), undergoes an abrupt one-step complete SCO at 318 K, 3LS → 3HS.117 At 380 K, the χmT value was 9.48 cm3 K mol−1, attributed to three uncoupled HS Fe(II) centres. The SCO curve decreased sharply at 320 K to 0.0 cm3 K mol−1, consistent with complete SCO to fully LS. The structural transition was also monitored by single crystal X-ray crystallography and DSC studies (Fig. 30).
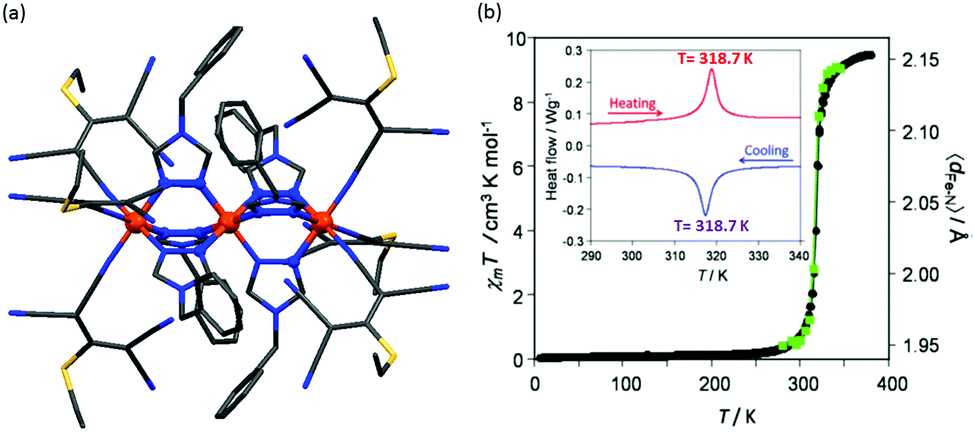 | ||
| Fig. 30 Data for [Fe3L466(A6)6]: (a) crystal structure (b) plot of χmT vs. T (black circle) and the average Fe–N distances (green squares) around the transition region. Inset: DSC study in both exo- and endo thermic modes. Structure redrawn from CCDC data. Figure (b) is adapted from ref. 117 with permission. Copyright 2017 RSC. | ||
Most recently, Tong, Dong, and co-workers used 4-amino-1,2,4-triazole, L47 (Chart 9), to generate the trinuclear complex [Fe3(L47)6(SCN)4(H2O)2](SCN)2·H2O.118 Heating at 400 K caused the complex to undergo a thermally induced single crystal-to-single crystal transformation, losing two water molecules and becoming [Fe3(L47)6(SCN)5(H2O)](SCN). This changed the SCO T1/2 from 202 to 160 K, with the SCO being gradual and, as usual (see above), only being observed for the central Fe(II) ion in both cases, i.e. [HS–HS–HS] ↔ [HS–LS–HS] (Fig. 31).118
 | ||
| Fig. 31 χ m T vs. T plots for as synthesised [Fe3(L47)6(SCN)4(H2O)2](SCN)2·H2O (blue, labelled 1 in this figure) and de-solvated [Fe3(L47)6(SCN)5(H2O)](SCN) (red, labelled 1a in this figure). Figure adapted with permission from ref. 118. Copyright 2018 RSC. | ||
Tetrametallic anion-bridged Fe4 squares
The metal ions in cyanometallates like [Fe(CN)6]4− are LS due to the coordination of six strong field π-acceptor CN− anions.119 Such cyanometallate polyanions can be used to bridge other metal ions, as both the C and N of the cyanide anion can coordinate, and this has been a profitable line of enquiry for the generation of Single Chain Magnets (SCMs).120 But to generate SCO-active complexes requires a significant reduction in the number of CN− ligands, with weaker field co-ligands being used in place of some of them.121In this section we consider CN− or (dca−, Chart 7) bridged tetranuclear Fe(II) squares (Fig. 2, 4A), featuring an Fe4(μ-mono-/di-cyano)4 core, with capping ligands (Chart 10) completing the, mixed N4C2–N6 with cyano bridges or all –N6 with dicyano bridges, octahedral coordination environments around the Fe(II) centres,122–125i.e. two cis-sites are occupied by N/C of two mono-/di-cyanide ions and the remaining four sites are occupied by either one tetradentate or two bidentate ‘capping’ ligands (Chart 10). In the case of μ-CN bridged squares, the Fe⋯Fe distance is ∼5 Å, whereas in dca− bridged squares this distance is ∼8 Å (Table 7). The octahedral distortion Σ of the Fe(II) centres in these squares is modest: LS in the range 31–70° and HS in the range 74–109° (Table 7).
 | ||
| Chart 10 Capping ligands used in anion bridged SCO-active Fe(II) tetrametallic square complexes (Fig. 2, 4A). | ||
| T (K) | Fe–Fe (Å) | Σ (°) | Spin state | T 1/2 (K) | Lit. | |
|---|---|---|---|---|---|---|
| [Fe2L35(L48)2(μ-NC)2]2(PF6)4 | 100 | 5.00, 4.95. 4.99, 4.99 | 52, 61, 47, 45 | LS–LS–LS–LS | 160, 380 | 122 |
| 200 | 5.04, 5.02, 4.99, 4.99 | 50, 108, 48, 45 | LS–HS–LS–LS | |||
| 293 | 5.05, 5.02, 5.0, 4.99 | 49, 109, 46, 47 | LS–HS–LS–LS | |||
| [Fe2L312(L48)2(NC)2]2(PF6)4·6MeOH·4H2O | 200 | 4.96, 4.98, | 49, 56 | 2LS–2LS | Not stated | 123 |
| [Fe(L35)(CN)]4(BF4)4·MeCN·H2O | 100 | 4.94, 4.97, 4.94, 4.95 | 68, 49, 55, 70 | LS–LS–LS–LS | Not stated | 124 |
| [Fe(L35)(CN)]4(BF4)4·0.75MeCN·H2O | 210 | 4.59, 4.99, 4.96, 4.98 | 69, 74, 47, 79 | LS–HS–LS–HS | ||
| [Fe(L35){dca}]4(BF4)4·2(H2O) | 150 | 8.23, 8.40 | 54, 60 | 2LS–2LS | 302, 194 | 126 |
| 250 | 8.44, 8.36 | 57, 91 | 2LS–2HS | |||
| [Fe(L35){dca}]4(BF4)4 | 350 | 8.51, 8.49 | 88, 98 | 2HS–2HS | ||
| [Fe4(L49)4(L35)2(CN)4](PF6)4·1MeCN·H2O | 100 | 4.98, 4.99 | 44, 43 | 2LS–2LS | Not stated | 125 |
| [Fe4(L49)4(L35)2(CN)4](PF6)4 | 370 | 5.07, 5.04 | 31, 86 | 2LS–2HS |
Oshio and co-workers have reported several such squares. The first, reported in 2005, featured a mixture of tetradentate L35 and bidentate L48 capping ligands (Chart 10). Initially, [Fe2L35(L48)2(μ-NC)2]2(PF6)4 was studied, in which [FeL35]2+ and [Fe(L48)2]2+ centres are alternately arranged in the square, and bridged by four cyanide groups – with the carbon atoms of CN− always binding to [Fe(L48)2]2+. This leads to alternating N4C2 and N6 coordination of the four Fe(II) centres in the square. The magnetic studies showed thermally induced two step SCO, with T1/2 = 160 K and 380 K. Up to 100 K all FeII sites are LS (χmT = 0.3 cm3 mol−1 K), then upon warming to 200 K, χT increases to 3.2 cm3 mol−1 K, corresponding to one HS iron site. X-ray crystallography at 200 K revealed that this SCO event occurs at one of the two [Fe(L35)]2+ units (where both CN bridges bond through the N atom). Upon further increasing the temperature, the χmT remained constant until 300 K, then increased to 4.9 cm3 mol−1 K at 400 K, which corresponds to 57% of the second [Fe(L35)]2+ unit becoming HS (Fig. 32a).122
 | ||
| Fig. 32 (a) Plot of χmT vs. T for [Fe2L35(L48)2(u-CN)2]2(PF6)4; (b) reflectance spectra; R = reflectivity, E = photon energy. Figure adapted with permission from ref. 122. Copyright 2005 John Wiley and Sons. | ||
The authors also monitored SCO by reflectance studies on crystals at 100 K and 300 K on the (001) plane with unpolarised light. At 100 K, the spectra consisted of one sharp peak at 2.13 eV (582 nm) and two broad peaks at 2.3 eV (539 nm) and 2.6 eV (477 nm) (Fig. 32b). These peaks were assigned to d–d transitions of the LS Fe(II) centres in the [Fe(L48)2]2+ and [Fe(L35)]2+ units respectively. At 300 K, two broad peaks merged at 2.40 eV (517 nm) and one new sharp peak appeared at 1.47 eV (843 nm) which was assigned to a d–d transition of a HS Fe(II) of a [Fe(L35)]2+ moiety (Fig. 32b).
The same group has reported a related heteroleptic tetranuclear cyanide bridged square, again using bidentate L48 but replacing tetradentate L35 by two bidentate L31 ligands (Chart 10), [Fe4(L48)2(L31)2(CN)4](PF6)4·6MeOH·4H2O (Fig. 33a). Again the corners alternate between [Fe(L48)2]2+ and [Fe(L31)2]2+ around the square. The complex remained LS up to 250 K, then the χmT product increased gradually to 3.42 cm3 mol−1 K at 400 K. The [Fe(L48)2]2+ corners remain LS throughout, whereas the [Fe(L31)2]2+ corners undergo SCO to HS in one step (Fig. 33b).123
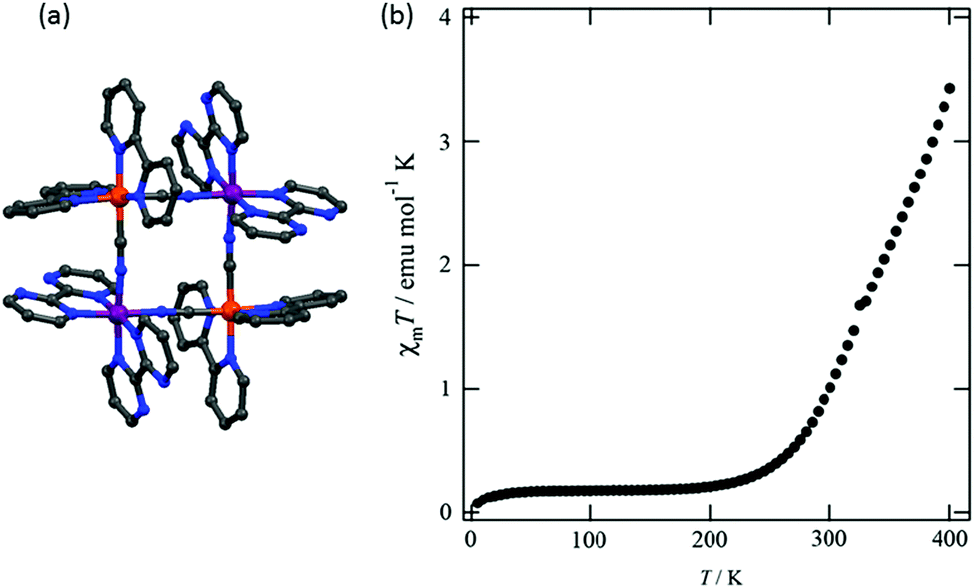 | ||
| Fig. 33 (a) Crystal structure of the cation of [Fe4(L48)2(L31)2(CN)4](PF6)4, SCO active Fe(II) centres violet (b) plot of χmT vs. T for [Fe4(L48)2(L31)2(CN)4](PF6)4·6MeOH·4H2O. Structure redrawn from CCDC data. (b) Reproduced with permission from ref. 123. Copyright 2009 Elsevier. | ||
Shatruk and co-workers prepared homoleptic capped FeII4 squares using the tetradentate capping ligand L35 (Chart 10). [Fe(L35)(CN)]4X4·nMeOH (X = BF4−, ClO4−), revealed SCO curves which were strongly influenced by the presence of solvent MeOH molecules, while counteranion variation had little effect.124 The χmT value of both complexes covered in a small amount of MeOH mother liquor at 300 K was ∼7.5 cm3 mol−1 K which was consistent with two HS and two LS Fe(II) centres in each cluster. The wet samples showed abrupt SCO with complete one step spin conversion of two N6 Fe(II) HS ions to the LS state at low temperature. Dry powder samples showed more gradual incomplete one-step spin transition (Fig. 34).124
 | ||
| Fig. 34 Plot of χT and the high spin fraction (γHS) vs. T for the two HS Fe(II) sites attributed to being in N6 coordination environments: red curves, as synthesized (wet) samples; blue curves, powder samples (a) [Fe(L35)(μ-CN)]4(BF4)4 and (b) [Fe(L35)(μ-CN)]4(ClO4)4. Figures adapted with permission from ref. 124. Copyright 2014 ACS. | ||
Zheng and co-workers have also used the tetradentate L35 capping ligand (Chart 10) to prepare a homoleptic capped square, but this time featuring dicyanamide bridges, [Fe(L35)(dca)2]4(BF4)4·2(H2O). In this square all of the Fe(II) centres have N6 coordination (Fig. 35a). It exhibits both thermal and light irradiation induced two step, complete SCO in both the hydrated and dehydrated forms. The as-synthesized sample was heated at 350 K to SCSC transform the hydrated crystals to the dehydrated form. The asymmetric unit of the as-synthesised hydrated form contained two crystallographically different Fe(II) centres. At 150 K, Σ = 60° and 54°, which changed to 91° and 57° at 250 K (Table 7), which suggested that two identical iron(II) centres have undergone SCO to HS. At 350 K, solvent molecules are removed but crystal packing remained intact and Σ changed to 98.0 and 88.2° corresponding to all Fe(II) in the HS state. For the two-step SCO of the hydrated square T1/2 = 302 and 194 K, and for the dehydrated form T1/2 = 294 and 211 K. The χmT vs. T curve follows the same route for the cooling and heating processes, except for a 6 K wide hysteresis (Fig. 35b) in the 4LS to 2HS–2LS step. The LIESST effect was studied at 5 K with 457 nm irradiation: the maximum χmT observed was 7.08 cm3 K mol−1, which was assigned to 47.5% photo-excited HS population. When the light source is switched-off, the metastable HS state relaxed steeply between 5–13 K, then smoothly from 13–33 K, with a plateau at approximately 3 cm3 mol−1 K which corresponds to relaxation of half of the metastable HS population (Fig. 35b, inset).126
 | ||
| Fig. 35 (a) Crystal structure of the cation of [Fe(L35)(dca)2]4(BF4)4·2H2O (b) plots of χmT vs. T for [Fe(L35)(dca)2]4(BF4)4; in two successive temperature cycles 300–100–380 K (square), and 380–100–380 K (circle). Inset: LIESST studies. Structure redrawn from CCDC data. (b) Reproduced with permission from ref. 126. Copyright 2011 John Wiley and Sons. | ||
Real and co-workers reported a heteroleptic capped square of L35 and L49 (Chart 10), [Fe4(μ-CN)4(L49)4(L35)2](PF6)4. Again the cyanide bridges are ordered, binding via N to the [Fe(L35)]2+ corners and via C to the [Fe(L49)2]2+ corners. This complex undergoes incomplete and gradual one-step SCO. The χmT value at 400 K was 5.3 cm3 K mol−1, which corresponds to a value just below that expected for a 2HS–2LS state, and decreased to 0.5 cm3 K mol−1 at 4 K, i.e. fully LS. The crystal structure investigation showed that the two N6-coordinated [Fe(L35)]2+ units are SCO active, while the two N4C2-coordinated [Fe(L49)2]2+ units remain LS up to 370 K.125 The authors also studied the magnetic behaviour on changing the ligand field strength by varying the extent of methyl substitution at the 6-position of one or two of the pyridine groups of L35. They observed that these methyl substituents increased the steric constraint which led to longer Fe–N bond distances, weakening the ligand strength and stabilising the HS state of [FeN6] and LS [FeN4C2] centres all over the temperature range.125
Tetranuclear [2 × 2] grids Fe4L4
Like the tetrametallic squares, tetrametallic [2 × 2] grids (Fig. 2, 4B) are an appealing architecture for SCO design, with the possibility of multistability being exhibited in an array of four addressable metal ion sites.42,43 The combination of octahedral Fe(II), the most commonly studied SCO metal ion, with bis-terdentate rigid linear bridging ligands to, in a controlled way, self-assemble Fe4L4 grids is therefore a very attractive pursuit in SCO research. Again this is a relatively young area of investigation, with few reported Fe4L4 grids in the literature, and even fewer examples that exhibit SCO. In fact only 21 ligands (Chart 11) have been reported to produce SCO-active Fe4 grids,39,127–137 the first of which was reported in 2000 – this was also the first example of an SCO-active tetranuclear complex,127 and held the record for the largest nuclearity discrete SCO-active complex until 2009 (see the ‘nanoball’ later). Interestingly, 16 of the ligands employed to date feature pyrimidine-bridging moieties L50–L65− (only 6 of the resulting grids have been structurally characterised, one in two different spin states, Table 8), whilst the other 5 ligands, L66− to L70 (all of the resulting grids have been structurally characterised, Table 9), present a wide range of different bridging moieties (Chart 11). | ||
| Chart 11 The 21 bis-terdentate ligands known to form SCO-active Fe4L4 grids (Fig. 2, 4B). All ligands provide N6 donor sets to the octahedral Fe(II) centres, except (L66)− which provides a N4O2 donor set, to the octahedral Fe(II) centres, so no co-ligands are bound. | ||
| T (K) | Fe–Fe (Å) | Σ (°) | Spin state | T 1/2 (K) | Lit. | |
|---|---|---|---|---|---|---|
| [FeII4(L50)4](ClO4)8·9MeCN·1.25H2O | 100 | 6.42, 6.35, 6.32, 6.42 | 88, 87, 91, 146 | HS–3LS | Not stated | 127 |
| 293 | 6.43, 6.47, 6.50, 6.47 | 137, 109, 143, 146 | 3HS–LS | |||
| [FeII4(L53)4](ClO4)8·3MeCN·5H2O | 120 | 6.27, 6.33, 6.42, 6.39 | 160, 146, 125, 98 | 2HS–2LS | Not stated | 128 |
| [FeII4(L54)4](ClO4)7(Cl)·7MeNO2·6H2O | 120 | 6.30, 6.35 | 85, 84 | 2LS–2LS | Not stated | 128 and 129 |
| [FeII4(L65)4]Cl4·9H2O | 153 | 5.93, 5.98 | 88, 91 | 2LS–2LS | Not stated | 135 |
| [FeII4(L65)4](BF4)4·7H2O | 153 | 6.07, 6.06 | 86 | 4LS | Not stated | 135 |
| T (K) | Fe–Fe (Å) | Σ (°) | Spin state | T 1/2 (K) | Lit. | |
|---|---|---|---|---|---|---|
| [FeII4(L66)4](BF4)4·MeOH·2H2O | 123 | 3.77 | 105 | 2LS–2HS | 175 | 134 |
| 3.84 | 129 | |||||
| 3.87 | 149 | |||||
| 3.75 | 113 | |||||
| [FeII4(L66)4](BF4)4·MeOH·2H2O | 283 | 3.84 | 149 | HS–HS–HS–HS | ||
| 3.73 | 147 | |||||
| 3.86 | 146 | |||||
| 3.88 | 150 | |||||
| [FeII4(L67)4](BF4)4·4DMF | 133 | 4.52 | 145 | 3HS–LS | Not stated | 39 |
| 4.51 | 97 | |||||
| 4.42 | 144 | |||||
| 4.57 | 156 | |||||
| [FeII4(L67)4](BF4)4·4DMF | 233 | 4.88 | 151 | HS–HS–HS–HS | ||
| 4.45 | 137 | |||||
| 4.45 | 149 | |||||
| 4.51 | 152 | |||||
| [FeII4(L67)4](BF4)6·3CH3CN | 133 | 4.53 | 92 | 2LS–2HS | LS–HS | 39 |
| 4.59 | 148 | |||||
| [FeII3(HL67)2(L67)2](BF4)4·2CH3CN | 133 | 4.59 | 95 | 2LS–HS | ↑↓355, 329 | 138 |
| 4.54 | 94 | |||||
| 142 | ||||||
| [FeII4(L68)4](PF6)4·DMF·THF | 133 | 4.50 | 99 | 2LS–2HS | 2LS–2HS | 133 |
| 4.48 | 170 | |||||
| [FeII4(L68)4](PF6)4·4DMF | 133 | 4.42 | 150 | HS–HS–HS–HS–HS–HS–HS–HS | HS | 133 |
| 4.35 | 153 | |||||
| 4.36 | 153 | |||||
| 4.40 | 159 | |||||
| 4.36 | 159 | |||||
| 4.39 | 139 | |||||
| 4.40 | 148 | |||||
| 4.37 | 155 | |||||
| [FeII4(L68)4](ClO4)4·0.25DMF·DME | 133 | 4.41 | 152 | 6HS–2LS | 3HS–1LS | 133 |
| 4.48 | 142 | |||||
| 4.48 | 150 | |||||
| 4.50 | 90 | |||||
| 4.50 | 91 | |||||
| 4.51 | 173 | |||||
| 4.46 | 136 | |||||
| 4.47 | 159 | |||||
| [FeII4(L68)4](BF4)4·THF·4H2O | 293 | 4.44 | 150 | 4HS | 250 | 133 |
| [FeII4(L68)4]Br4·4DMF·2H2O | 133 | 4.53 | 105 | LS–HS–HS–HS | Not stated | 133 |
| 4.41 | 176 | |||||
| 176 | ||||||
| 160 | ||||||
| [FeII4(L69)4](BF4)4·2MeCN | 293 | 6.48 | 131 | HS–HS–HS–HS | Not stated | 136 |
| 6.44 | 145 | |||||
| 6.51 | 144 | |||||
| 6.42 | 147 | |||||
| [FeII4(L70)4](CF3SO3)8·12MeNO2·C6H14O | 180 | 9.16 | 87 | 2LS–2LS | 310 | 137 |
| 9.05 | 85 | |||||
| [FeII4(L70)4](CF3SO3)5(F)3·2MeNO2·H2O | 180 | 7.33 | 167 | 2HS–2HS | HS | 137 |
| 7.22 | 152 |
In each case the ligand is bis-terdentate and as such the four ligands in the corresponding Fe4L4 grid complexes provide all of the donor atoms to the Fe(II) centres with no co-ligands bound. This, coupled with the fact that all ligands contain either a heterocyclic or single atom bridging moiety, results in a very rigid coordination environment and significant octahedral distortions at the Fe(II) centres. As a result, these grids impose much larger Σ values on the Fe(II) centres (LS 84–125°, HS 129–176°; Tables 8 and 9) than those observed in the other tetranuclear architectures, squares (LS 31–70°, HS 79–109°; Table 7) and cages (LS 43–75°, HS 69–121°; see later, Tables 10 and 11).
| T (K) | Fe–Fe (Å) | Σ (°) | Spin state | Lit. | |
|---|---|---|---|---|---|
| [FeII4(L71)4](BF4)8·14.75MeCN·4.5C6H6·3H2O | 153 | 14.28, 14.28, 14.28, 13.99, 14.14, 13.98 | 61, 60 | 2LS–2LS | 140 |
| [FeII4(L71)4](BF4)8·14.75CH3CN·4.5C6H6·3H2O | 293 | 14.46, 14.29, 14.11, 14.52, 14.46, 14.11 | 66, 69 | 2LS/HS–2HS | 140 |
| FeII4(L73)(CF3SO3)8 | 100 | 6 × 11.85 | 65 | 4LS | 141 |
| [FeII4(L74)4](BF4)4·16CH3CN | 100 | 3 × 14.61, 3 × 14.78, 3 × 14.54, 3 × 14.94, 3 × 14.56, 3 × 15.14 | 2 × 82, 2 × 67, 2 × 70, 2 × 121, 2 × 86, 2 × 75 | 6LS–6HS | 142 |
| T (K) | Fe–Fe (Å) | Σ (°) | Spin state | Lit. | |
|---|---|---|---|---|---|
| [FeII4(L75)6](ClO4)8 (R) | 150 | 3 × 9.56, 3 × 9.67 | 61, 49 | LS–LS | 143 |
| [FeII4(L75)6](ClO4)8 (S) | 123 | 3 × 9.66, 3 × 9.48 | 60, 42 | LS–LS | 143 |
| [FeII4(L76)6](ClO4)8·11.59MeCN·2C4H10O·H2O (R) | 123 | 9.66, 9.45, 9.70, 9.59, 9.64, 9.79 | 56, 58, 56, 64 | 4LS | 143 |
| [FeII4(L76)6](ClO4)8·6MeCN (S) | 123 | 9.74, 9.70, 9.68, 9.41, 9.77, 9.76 | 56, 55, 60, 63 | 4LS | 143 |
| [FeII4(L77)6](ClO4)8·2MeCN (S) | 123 | 3 × 9.53, 3 × 9.81 | 55, 57 | 2LS | 143 |
| [FeII4(L78)6](ClO4)8·2MeCN | 173 | 11.35, 11.04, 11.90, 11.73, 11.95, 11.18, 3 × 11.97, 3 × 11.78 | 43, 45, 49, 43, 53, 50 | 6LS | 144 |
In 2000, Lehn, Gütlich and co-workers reported the first SCO-active Fe(II) [2 × 2] grid.127 The grid was assembled from the linear bis-terdentate ligand 4,6-bis(2,2′-bipyridin-6-yl)-2-phenyl-pyrimidine (L50, Chart 11), and the structure of [Fe4(L50)4]8+ was determined by X-ray crystallography (Fig. 36a). Analysis of the Fe–N bond lengths at 293 K clearly revealed three of the Fe(II) centres were HS while the fourth had bond lengths intermediate between those typical for HS and LS states. In contrast, at 100 K three of the Fe(II) centres have intermediate Fe–N bond lengths (but closer to LS) and one is HS, indicating that a thermal SCO has occurred. Variable temperature magnetic susceptibility showed that there was indeed a SCO event, which was very gradual and incomplete over 300–30 K. The authors performed Mössbauer spectroscopy studies to yield further evidence of SCO (Fig. 36b) and were able to induce SCO by varying the pressure, and by the LIESST effect, making this a triply switchable SCO [2 × 2] grid.
 | ||
| Fig. 36 (a) Reaction of 4,6-bis(2,2′bipyrid-6-yl)-2-phenyl-pyrimidine (L50) with iron(II) perchlorate in acetonitrile at room temperature to produce the corresponding [Fe4(L50)4](ClO4)8 [2 × 2] grid and (b) evidence of the SCO, by Mössbauer spectroscopy, from 80% HS at 300 K (top) to 46% HS at 4.2 K (bottom). Structure redrawn from CCDC data.127 (b) Figure adapted, reproduced with permission, from ref. 127. Copyright 2000 John Wiley and Sons. | ||
Modifications to the bis-terdentate ligand, through substitution of peripheral groups at the pyrimidine or bipyridine groups, allowed for a family of closely related grids to be investigated. Substitution of the phenyl group at the pyrimidine-2-position for a methyl group (L51, Chart 11) accessed the second example of an Fe(II) SCO grid, whereas H or OH groups yielded diamagnetic analogues.128 Further SCO-active grids were obtained by either the attachment of peripheral groups at the pyrimidine or bipyridine groups (X or Y, L52–L55, Chart 11)128,129 or the use of bipyridine-like groups (L56, Chart 11).128 Lehn and co-workers have since produced several more SCO-active Fe(II) grids of related pyrimidine ligands. In fact they have produced more than half of the known SCO-active Fe(II) grids in the literature. The other ligands used by them are also based on the pyrimidine bridging unit, but these ligands contain functionalised bis-hydrazone side arms and either pyridine130,131 [(L57)− and L58–L62, Chart 11] or imidazole/thiazole132 (L63 and L64, Chart 11) rings, and various substitutions thereof, to complete the terdentate binding pockets. In each case where SCO was observed the transition was very gradual and incomplete across a wide thermal range.
In 2013 Kou and co-workers reported two SCO-active [Fe4(L65)4]X4 grid complexes based on a bis-terdentate Schiff base ligand [(L65)−, Chart 11].135 Similar to the ligands employed by Lehn and co-workers, the ligand scaffold also features a central pyrimidine bridging moiety. The SCO for the X = Cl− grid is incomplete, with the transition taking place above room temperature between (approximately) the [2HS–2LS] and [HS–3LS] states (Fig. 37, black data points). The transition is noticeably more abrupt than the pyrimidine based grids of Lehn and co-workers, and that of the X = BF4− grid is even more abrupt again and appears to be centred at about 400 K, the high temperature limit of the measurement (Fig. 37, red data points). Interestingly, the analogous grid with less bulky methyl groups in place of the bromine atoms on the ligand scaffold is fully LS to high temperature.
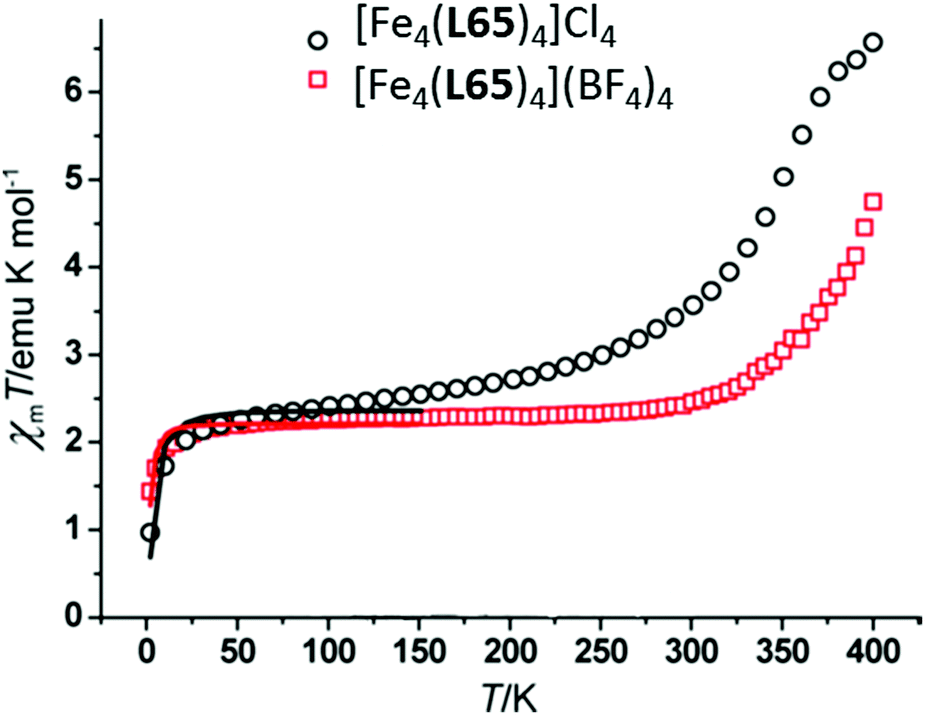 | ||
| Fig. 37 χ m T vs. T for the [2 × 2] grids of [Fe4(L65)4]Cl4·9H2O (black circles) and [Fe4(L65)4](BF4)4·6H2O (red squares).135 Figure adapted, reproduced with permission, from ref. 135. Copyright 2013 ACS. | ||
Whilst nearly all of the SCO-active Fe(II) [2 × 2] grids reported in the literature to date have iron(II) ions with an N6 coordination sphere, there is one exception, the N4O2 coordinated grid from Sato and co-workers.134 Here (L66)− (Chart 11) features two terdentate pockets which share one bridging oxygen atom, and the corresponding O-bridged [Fe4(L66)4](BF4)4 grid complex undergoes an abrupt SCO from [4HS] to [2HS–2LS] centred around 175 K.
Another bis-terdentate bridging ligand family which has provided a wealth of Fe4L4 [2 × 2] grids are the bis-bipyridine pyrazolate family (Chart 11) from the group of Meyer. In 2010 they reported the synthesis of the [Fe4(L67)4](BF4)4 [2 × 2] grid (Fig. 38a) of the simple 3,5-bis{6(2,2′-dipyridyl)}pyrazolate ligand [(L67)−, Chart 11].39
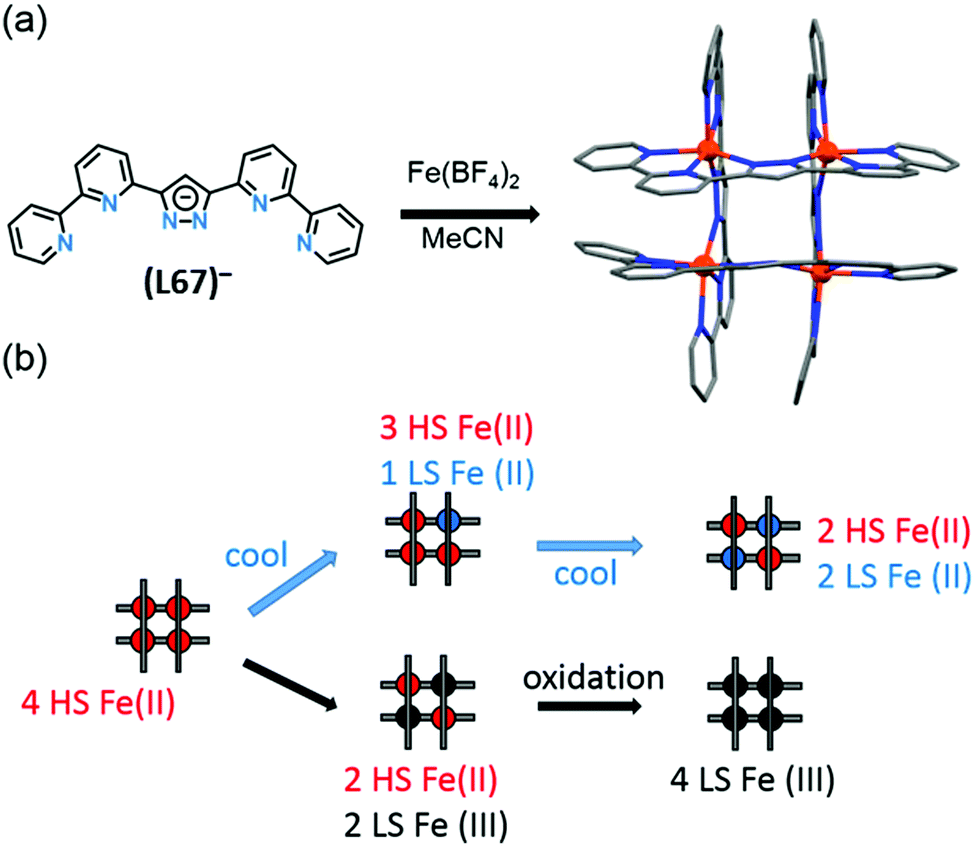 | ||
| Fig. 38 (a) Synthesis of the [Fe4(L67)4](BF4)4 [2 × 2] grid from 3,5-bis{6(2,2′-dipyridyl)}pyrazolate (L67)− and iron(II) tetrafluoroborate in MeCN. (b) Overview of the physical (SCO) and chemical (redox) transformations of the grid complex [Fe4(L67)4](BF4)4. Structure redrawn from CCDC data.39 Figure adapted, reproduced with permission, from ref. 39. Copyright 2010 John Wiley and Sons. | ||
At room temperature both magnetic measurements and structural analysis indicate a fully HS state. Upon cooling to 133 K the structure is consistent with one LS Fe(II) and three HS Fe(II) ions in the grid complex and this is confirmed by a [3HS–LS] plateau in the χMT vs. T data. Cooling even further allows for the beginning of a second SCO event to be observed, the SCO of a second Fe(II) to the LS state, thus the complex almost reaches [2HS–2LS] at 50 K. Cyclic voltammetry revealed two separate Fe(II)/Fe(III) redox events, meaning the complex is doubly switchable, by SCO or redox, with multistep events observed for both phenomena (Fig. 38b). By stoichiometric control, Meyer and co-workers have been able to access the “corner” unit of their [Fe4(L67)4]4+ grids, i.e. [Fe(HL67)2]2+ which crystallises as hydrogen-bonded dimers.139
Here the Fe(II) ion of each FeL2 unit of the dimer is in the LS state, as evidenced by the short Fe–N bond lengths and regular octahedral geometry (Σ = 85.24°) as well as by Mössbauer spectroscopy. The LS [Fe(HL67)2]2+ complex suggests that the bipyridine-pyrazole binding pocket of HL67 is strong field and not suited to SCO. However, when the ligand is pyrazolate and bridging two Fe(II) ions, in addition to the steric constraints of four rigid ligands holding four Fe(II) ions in a [2 × 2] grid, the binding environment facilitates SCO in the tetranuclear [Fe4(L67)4]4+ complex.
Meyer and co-workers have since introduced a bromo substituent to the C4 position of the pyrazolate ring, i.e. (L68)− (Chart 11), and subsequently synthesised the corresponding [Fe4(L68)4]X4 grids.133 Variation of the counteranion (X = PF6−, ClO4−, BF4−, Br−) gave access to a variety of stable spin states, from [4HS] to [3HS–LS] to [2HS–2LS], with gradual thermal SCO between the states seen for some of the grids. The analogous grid of the C4-methyl-substituted pyrazolate bis-bipyridine ligand is SCO inactive and persists in the [2HS–2LS] state up to 350 K; however, the authors note that the observed stability of the mixed spin state is favourable for potential use as a component for quantum cellular automata.38 Finally, whilst technically not a [2 × 2] grid, the controlled formation of a trinuclear defect grid [Fe3(HL67)2(L67)2](BF4)4·MeCN is nevertheless highly relevant work.138 It exists in the [HS–2LS] state at room temperature, but after loss of the lattice solvent MeCN it can undergo thermal SCO to the [2HS–LS] state above room temperature.
Another prominent example of a SCO-active Fe(II) grid was reported by Oshio and co-workers, using a bis-(pyrazole-pyridyl) ligand with a central bridging imidazolate, (L69)− (Chart 11).136 Here, a two-step hysteretic SCO occurs for the Fe(II) grid, [Fe4(L69)4](BF4)4, from the [4HS] state via the [3HS–LS] state to the [2HS–2LS] state (Fig. 39). At low temperatures, the [3HS–LS] state was also accessible by the LIESST effect, meaning the tri-stable [2 × 2] grid is doubly switchable by thermal variation and light irradiation. The corresponding mixed valent [FeII2FeIII2(L69)4](BF4)6 grid is fully LS at low temperatures and only undergoes an incomplete gradual SCO upon warming at one of the Fe(III) centres (Fig. 39).
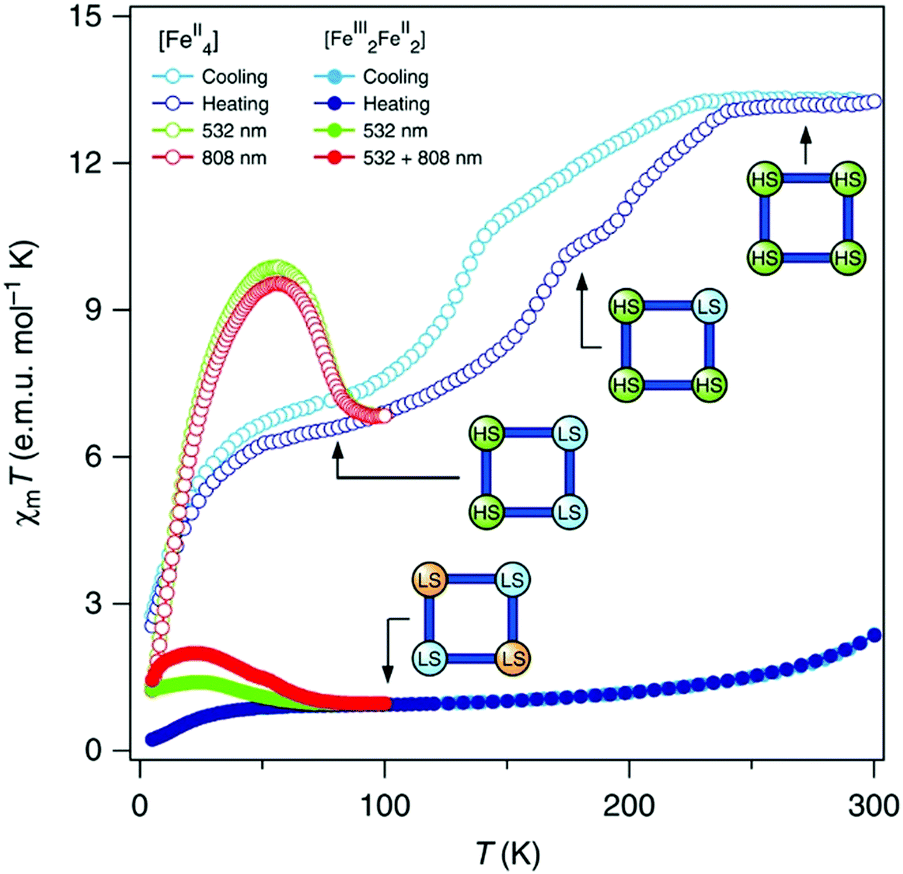 | ||
| Fig. 39 χ m T vs. T for the two [2 × 2] grids of the imidazolate-bridging ligand L69−. [Fe4(L69)4](BF4)4·2MeCN undergoes multistep hysteretic SCO, [4HS] to [2HS–2LS], and also LIESST at low temperature. [FeII2FeIII2(L69)4](BF4)6 shows incomplete gradual Fe(III) SCO and modest LIESST effects. Figure reproduced with permission from ref. 136. Copyright 2014 Springer Nature. | ||
An interesting case of two tauto-conformer Fe(II) [2 × 2] grids, where only one is SCO-active, was reported by Ruben and co-workers.137 Here the bis-terdentate ligand L70 (Chart 11) consists of a benzo-diimidazole bridge, with two imidazole-pyridyl side-arms. The central bridging unit can undergo tautomerisation which, along with rotation of the single bonds between aromatic side-arm groups, results in the two terdentate pockets being in either a cis or trans arrangement (Fig. 40). This gives rise to the parallel formation of two isomeric grid complexes [Fe4(L70)4](CF3SO3)8, which can be separated by fractional crystallisation – only one of which undergoes SCO. This is the direct result of the impact of the ligand conformations on the octahedral geometries at the Fe(II) centres: the cis conformation gives a highly distorted octahedral geometry (Σ = 160°) which locks the Fe(II) ions in the HS state, while the trans conformation reduces the octahedral distortion (Σ = 86°) and facilitates SCO to the LS state. This highlights that the geometric strain imposed by highly chelating ligands can have a larger influence on the spin state of the bound metal ions than the donor atoms alone.
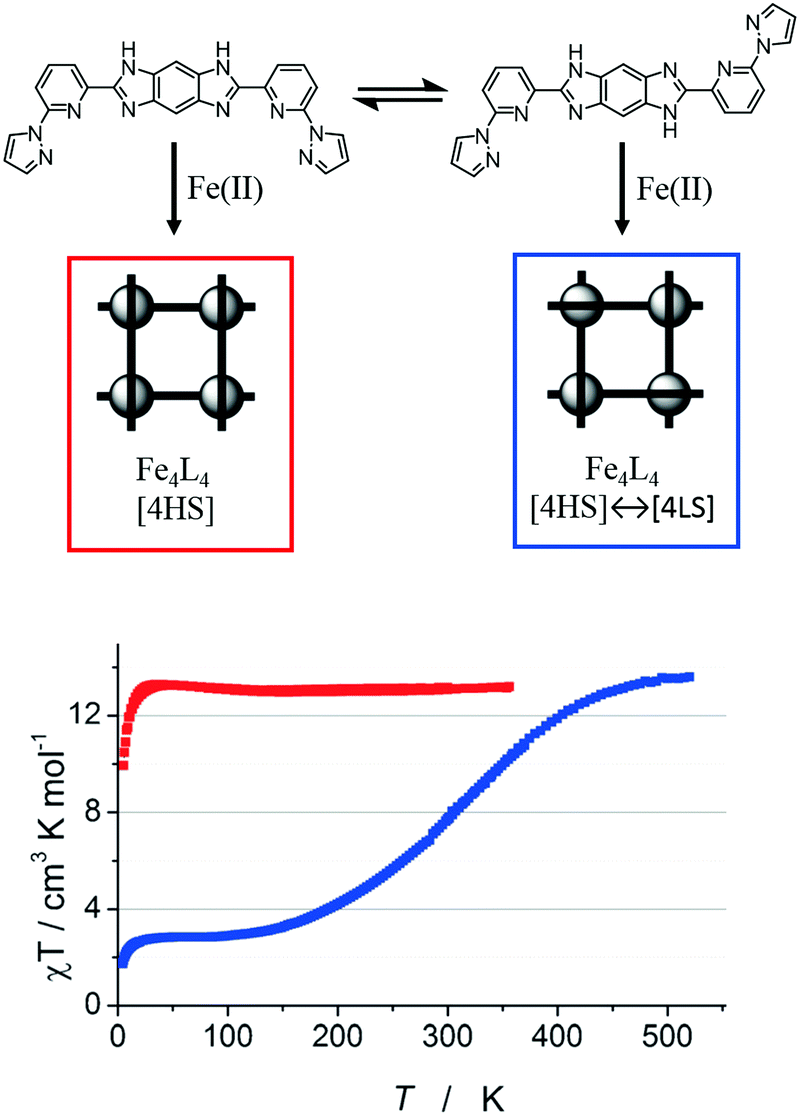 | ||
| Fig. 40 (top) Tautomerisation of ligand L70 gives rise to parallel formation of a HS grid (red box) where the ligands bind two metals in a cis mode, and a SCO-active grid (blue box) where ligands bind in a trans mode, and (bottom) χMT vs. T for the HS cis-grid (red) and SCO trans-grid (blue). Figure adapted, reproduced with permission, from ref. 137. Copyright 2016 John Wiley and Sons. | ||
Tetranuclear tetrahedral cage Fe4L4 and Fe4L6 complexes
In 2013, tetrametallic SCO-active architectures were expanded from [2 × 2] grids and squares to include Fe4 tetrahedral cages (Fig. 2, 4C).140–144 The SCO-active Fe4 cage architectures have been mostly prepared via self-assembly of Fe(II) with tritopic bidentate ligands which define the four faces of the Fe4L4 tetrahedral (Td) cages, i.e. four face-capping ligands (L71–L74, Chart 12), but examples of ditopic bidentate ligands which define the six edges of Fe4L6 Td cages are also known (L75–L78, Chart 12). In all cases, all of the donors to the bound Fe(II) centres are provided by the ligands. But, as these are bidentate, and quite long, they are reasonably flexible, so the Fe(II) octahedral distortion parameters Σ are smaller for these cages (42–121°, Tables 10 and 11) than for the tetranuclear grids (84–176°, Tables 8 and 9). The advent of these SCO-active 3D cages has also led to increased interest in studying SCO in solution,14,16,145 in particular due to the possibility of guest recognition by the cage interior modifying the SCO-response.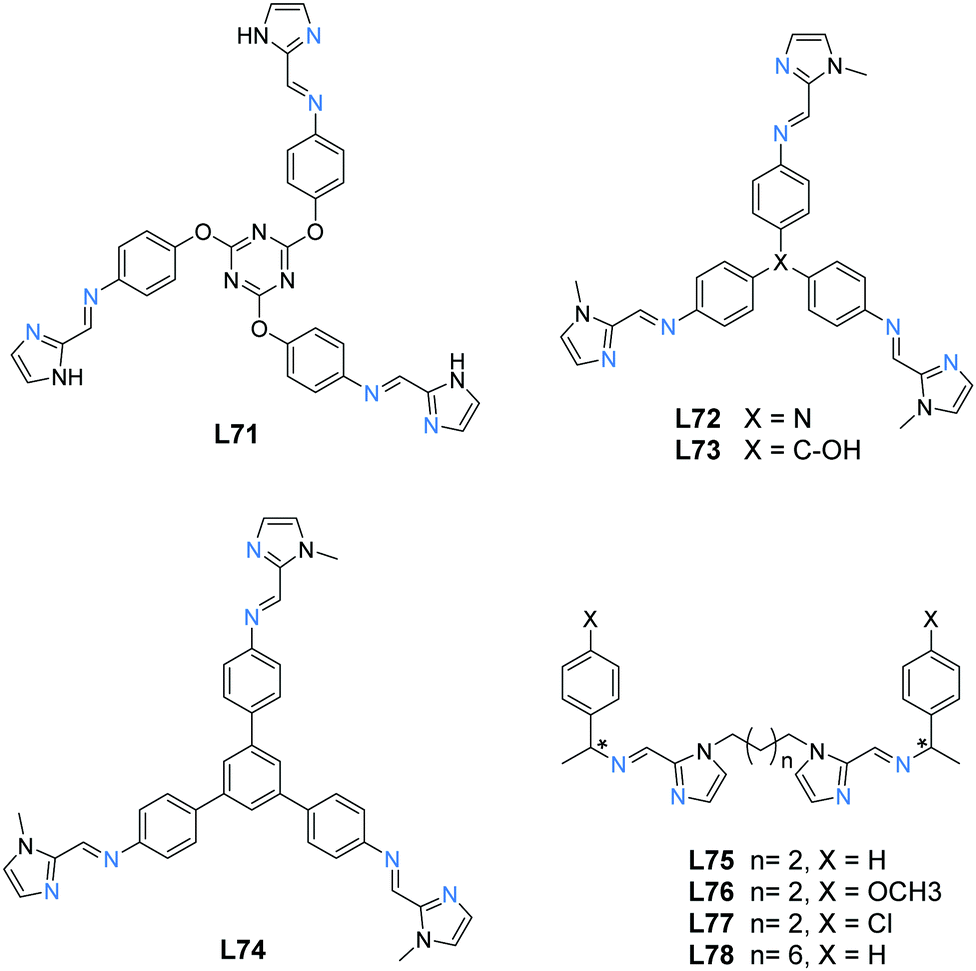 | ||
| Chart 12 The eight ligands reported in the literature to form SCO-active Fe4 tetrahedral cages. Four are tri-topic (face bridging, to give [Fe4(L)4]8+) and other four are di-topic (edge bridging to give [Fe4(L)6]8+). All provide N6 donor sets to the octahedral Fe(II) metal ions through the binding of three sets of bidentate binding pockets. For L75–L77 both the (R,R) and (S,S) configurations at the chiral (*) centre have been used to form cages. | ||
The first example of an SCO-active Td cage was reported by Kruger, Clérac and co-workers in 2013, with a face-capping ligand that was assembled from a triamine with a substituted triazine central moiety and 2-imidazolecarboxaldehyde (L71, Chart 12).140 Coordination to Fe(II) resulted in a [Fe4(L71)4](BF4)8 tetrahedral cage (Fig. 41), which exhibits SCO in both the solid state and acetone solution.
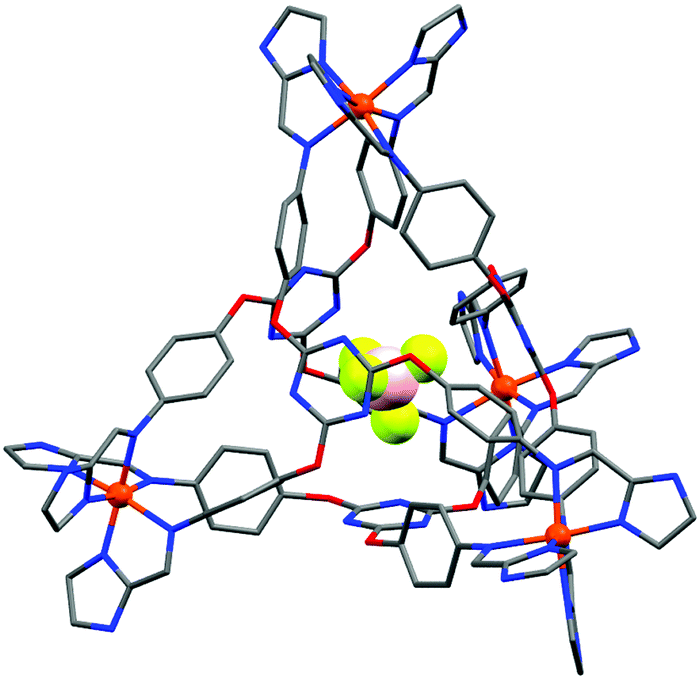 | ||
| Fig. 41 Structure of the complex cation of [Fe4(L71)4](BF4)8 showing the encapsulated BF4− counteranion in spacefill.140 Structure redrawn from CCDC data. | ||
Also in 2013, Nitschke, Brooker and co-workers reported a pair of self-assembled cages, [Fe4(L72)4](OTf)8 and [Fe4(L73)4](OTf)8 (Fig. 42), featuring smaller face-capping ligands (Chart 12).141 While only [Fe4(L72)4](OTf)8 was SCO-active in the solid state (approximately [4HS] to [HS–3LS] transition), both were SCO-active in nitromethane solution. Furthermore, the solution SCO properties were able to be tuned by encapsulation of Br− or CS2, with the T1/2 lowering from 336 K for the empty cage [Fe4(L73)4](OTf)8 to 328 K for Br− or 324 K for CS2, thus highlighting the exciting possibilities of synergistic host–guest chemistry and magnetic properties that arise when SCO is extended to such large architectures.
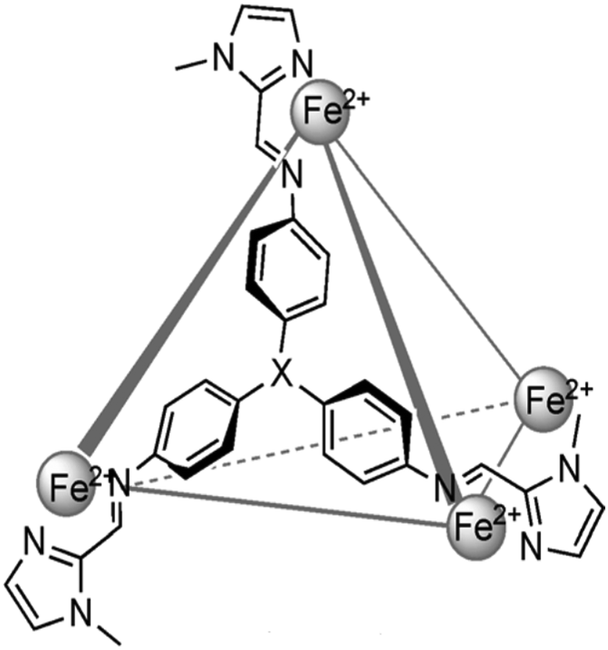 | ||
| Fig. 42 Representation of the molecular structure of [Fe4(L72)4](OTf)8 and [Fe4(L73)4](OTf)8. Figure adapted, reproduced with permission, from ref. 141. Copyright 2013 John Wiley and Sons. | ||
A third face-capped SCO-active [Fe4(L74)4](BF4)8 tetrahedral cage (Fig. 43), also self-assembled from an imidazole carboxaldehyde and a triamine (Chart 12), was reported by Li and co-workers in 2015.142 It exhibits a very gradual SCO from fully HS at 300 K to approximately 75% HS at 50 K in the solid state.
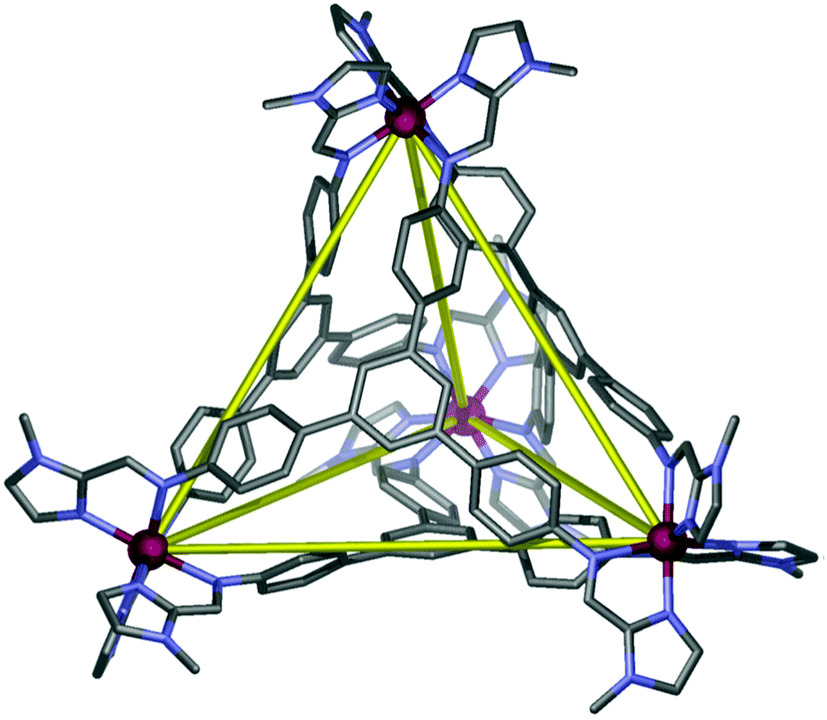 | ||
| Fig. 43 Complex cation structure of [Fe4(L74)4](BF4)8. Figure reproduced, with permission, from ref. 142. Copyright 2015 RSC. | ||
An Fe(II) tetrahedral cage system which instead involves di-topic Schiff base bidentate ligands (L75–L77, Chart 12) that define the six edges of the tetrahedron, giving [Fe4(L)6]8+ cages, was reported by Gu and co-workers in 2015 (Fig. 44).143 Here the intrinsic at-metal chirality of the tetrahedral cage architecture is exploited to produce enantiopure homochiral cages by self-assembly involving either the (R) or (S) optical isomer of the amine. The enantiomers, of course, exhibit identical magnetic properties. R group variation (H, OCH3, Cl) on the ligand, as well as solvent desorption, subtly affecting the (gradual and incomplete) SCO behaviour observed in the solid state. Specifically, below 200 K the cages are 3–18% HS when solvated or 17–37% HS when desolvated, and transition to 74–87% HS at 400 K (desolvated), with the LS state best stabilised when R = Cl.
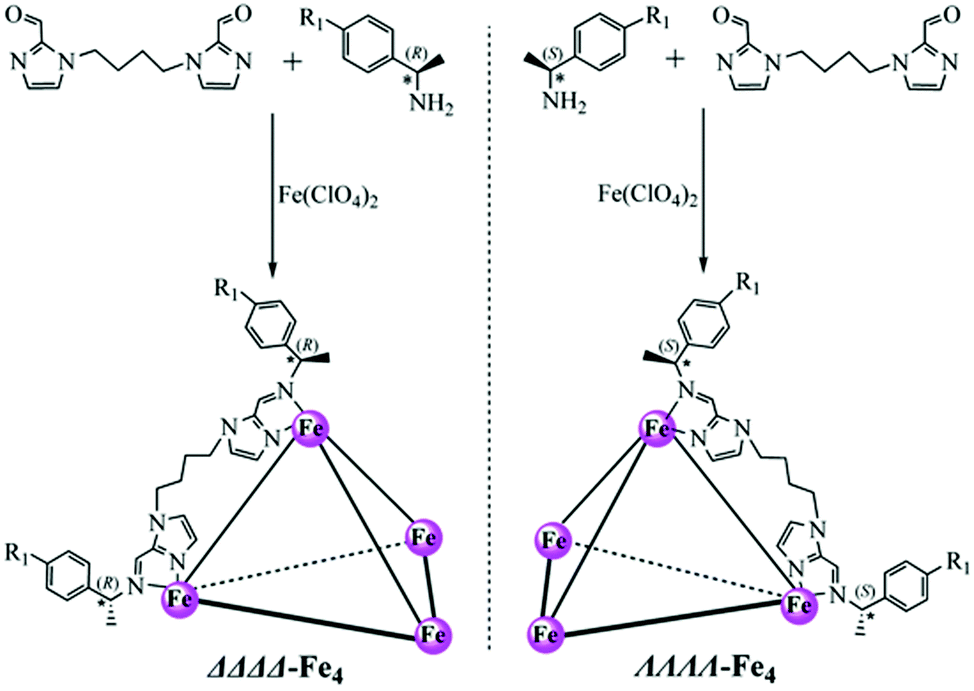 | ||
| Fig. 44 Synthesis of enantiopure chiral tetrahedral [Fe4L6]8+ cages, where L is L75–L77, by use of either (R)- or (S)-phenylethylamine. Figure reproduced with permission from ref. 143. Copyright 2015 RSC. | ||
The same group reported a FeII4L786 (Chart 12) cage exhibiting gradual and incomplete SCO. The χmT value at 400 K was 10.64 cm3 K mol−1 which indicated about 88.7% of Fe(II) were HS. The SCO curve falls abruptly from 400 K to 300 K, then gradually from 300 to 10 K (Fig. 45a). They have also studied the effect of varying the Fe(II) content of a NiII4L6 cage on the SCO behaviour. Through SCSC transformations, they successfully manipulated the Fe(II) proportion in a colourless Ni(II)4 cage from 20% (yellow) to 33% (red) and 50% (dark purple), which was confirmed via atomic absorption spectroscopy. The former two did not show any SCO, but at 50% Fe(II), the Ni2Fe2 cage showed a gradual transition at 300 K (Fig. 45e).144
 | ||
| Fig. 45 χ m T vs. T plots for cage [Fe4L786](BF4)8 (a) FeII cage (b) NiII cage (c) 20% FeII into NiII cage (d) 33% FeII content into NiII cage (e) 50% FeII content in NiII cage. Figure adapted, reproduced with permission from ref. 144. Copyright 2016 RSC. | ||
Penta-, hexa- and octa-nuclear complexes
SCO has rarely been seen in Fe(II) supramolecular architectures with more than four metal centres, but this is an active area of research, and since 2009 this has been achieved in an Fe6 “nanoball”,146 then in an Fe5 “helical cluster” with a planar [FeII3(μ3-O)]4+ core,147 and in 2017 in a pair of Fe8 cubes45 (Fig. 2, 6A, 5A and 8A, respectively), using the ligands shown in Chart 13. | ||
| Chart 13 Ligands used in penta-, hexa- and octa-nuclear SCO-Fe(II) complexes (Fig. 2, 5A, 6A and 8A). | ||
The first SCO-active cluster helicate, [{FeII(μ-L20)3}2FeII3(μ3-O)][FeII2(μ-Cl)(μ-L20)(NCS)4(H2O)] (HL20, Chart 13), was reported by Tong and Ni and co-workers in 2016 (Fig. 46a).147 Closely related cationic cluster helicates had already been reported by Kawata and Kaizaki and co-workers in 2006, where two LS triple stranded [FeL3]− units wrapped a planar HS [Fe3(μ3-O)]4+ core, but with different counter anion, NCS−,148 and in 2010 Oshio and co-workers varied the anions: the two apical [FeL3]− units were LS in the case of NCS−, ClO4−, I− and HS in the case of FeIII2(μ-O)Cl62−. But no SCO behaviour was observed with any of these complexes.149
 | ||
| Fig. 46 (a) Structure of [{FeII(μ-L20)3}2FeII3(μ3-O)]. (b) χMT plot for the single SCO-active site (Fe2, at the bottom in these images) in the pentanuclear cluster complex, [{FeII(μ-L20)3}2FeII3(μ3-O)][FeII2(μ-Cl)(μ-L20)(NCS)4(H2O)]·2H2O·C2H5OH, after subtraction from the contribution of [FeII3(μ3-O)]4+ core, LS [FeL3]− and dinuclear counteranion. (b) Reproduced from reference with permission.147 Copyright 2016 ACS. | ||
Tong and Ni and co-workers instead crystallised this cationic pentanuclear cluster helicate by use of a complex dinuclear dianion, [FeII2(μ-Cl)(μ-L20)(NCS)4(H2O)]2−, to induce SCO behaviour. In this modified cluster helicate, which again features a planar HS [FeII3(μ3-O)]4+ core, the average Fe–N bond lengths (and Σ values), at 150 and 293 K, clearly showed that one of the two apical [FeL3]− moieties was SCO-active (Fe2) whilst the other (Fe1) remained LS up to 300 K (Fig. 46b). All other Fe(II) centres, in the cluster and in the anion, remained HS. The Σ for two apical [FeL3]− moieties, Fe1 changed from 60.9 to 63.7° and Fe2 changed from 65.8 to 93.7°, at 150 and 293 K respectively.
In 2009, Batten and co-workers reported the first SCO-active Fe6 supramolecular architecture. The six Fe(II) centres are bridged by eight neutral tritopic tris-monodentate Cu(I) metalloligands, {CuIL79(MeCN)} (Chart 13), forming a Fe6L8 cluster which they termed a “nanoball”, [FeII6(NCS)28/3(MeCN)8/3{CuIL79(MeCN)}8](ClO4)8/3.146 The equatorial plane of each of the octahedral N6-coordinated Fe(II) centres comprises four 4-pyridyl groups from four Cu(I) metalloligands, and the axial sites are occupied by either NCS or NCMe. The six Fe(II) ions are arranged octahedrally, so, along with the cubic arrangement of the eight Cu(I) ions, this forms a distorted rhombic dodecahedron with a diameter of ca. 3 nm (Fig. 47). The as-synthesised complex undergoes approximately a 50% HS to LS SCO in the solid state, which is switched off, reversibly, by MeCN solvent loss. Owing to the poor lattice packing of the nanoball, large voids between complex cations were exploited for guest solvent molecule sorption/desorption, and this was found to tune the SCO properties. The same group went on to report the analogues with different anions, NCE− (E = Se, BH3) in 2012, i.e. [FeII6(NCSe)28/3(MeCN)8/3{CuIL79(MeCN)}8](ClO4)8/3 and [FeII6(NCBH3)28/3(MeCN)8/3{CuIL79(MeCN)}8](ClO4)8/3.150 These new family members, with NCSe− and NCBH3− co-ligands, which provide stronger field strengths than NCS−, did not change the geometry significantly but, as expected, increased the T1/2 from 124 (NCS) to 162 (NCSe) to 173 K (NCBH3). The authors also studied the LIESST effect on all three nanoballs and correlated the T1/2 with TLIESST. These nanoballs held the record for the largest nuclearity discrete Fe(II) complex to undergo SCO until 2017.
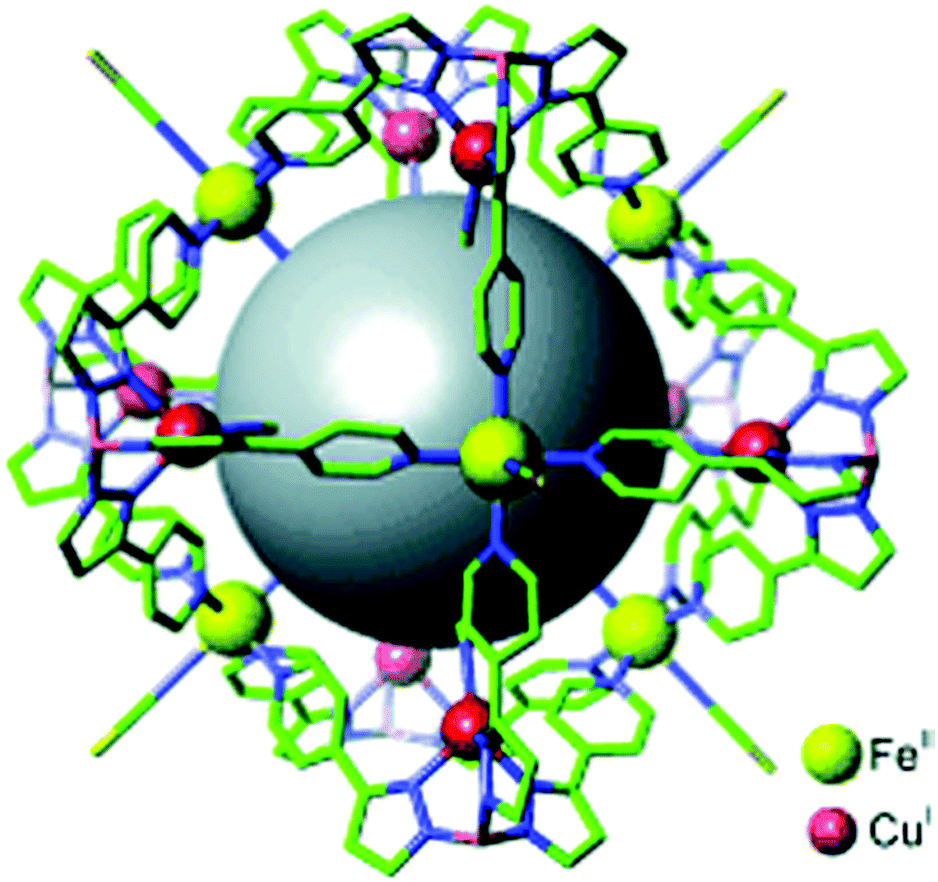 | ||
| Fig. 47 Fe6[CuIL79(MeCN)]8 shell of nanoball structure featuring eight Cu(I) metalloligands. Figure reproduced with permission from ref. 146. Stick colours: blue = nitrogen, green = carbon, yellow = sulphur, pink = boron. Copyright 2009 John Wiley and Sons. | ||
In 2017, Lützen and co-workers reported two [Fe8L6]16+ cubes, [Fe8(H2L80)6](CF3SO3)16 and [Fe8(ZnL81)6](CF3SO3)16 which exhibit SCO in methanol solution.45 The eight Fe(II) centres define the corners of the supramolecular cube, and six tetra-topic porphyrin ligands (Chart 13), where the porphyrins are all either uncoordinated (H2L80) or bound to Zn(II) (ZnL81), act as face-capping units (Fig. 48). Zn(II) coordination to the porphyrin faces of the Fe8 cube appears to very slightly stabilise the HS state. Evans method studies from 298–203 K in deuterated-methanol solution revealed that the Zn-coordinated cube transitions from ∼85% HS to ∼30% HS (Fig. 49), while the cube without Zn(II) transitions from ∼80% HS to ∼15% HS. In a nice example of guest influence on SCO, encapsulation of C70, facilitated by the appropriately sized internal cavity, was found to stabilise the HS state and lowered the T1/2 of the solution based SCO by 15 K and 20 K for the Zn and Zn-free Fe8 cubes respectively.
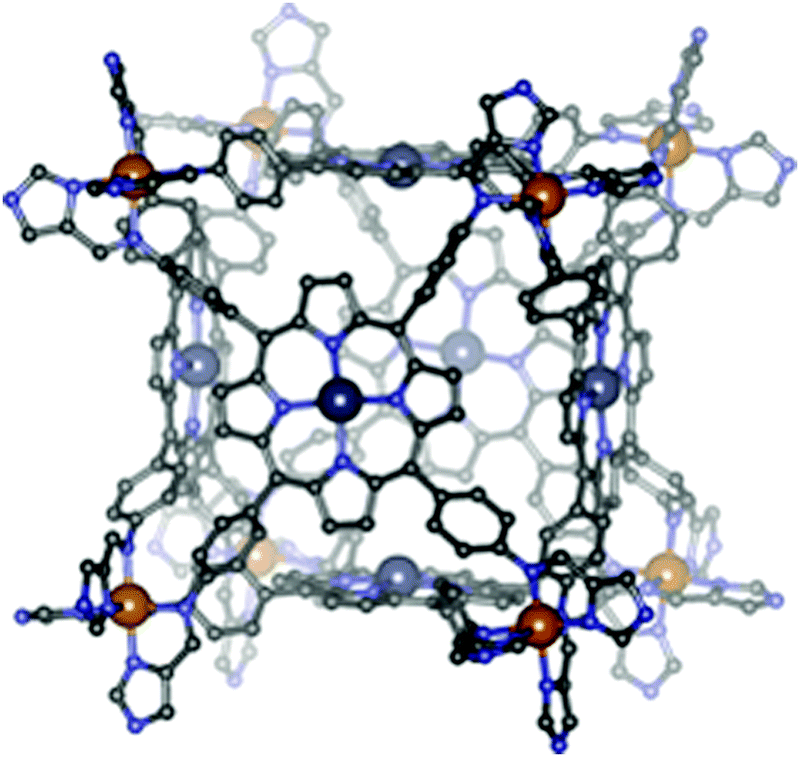 | ||
| Fig. 48 [Fe8(ZnL81)6]16+ cube structure featuring six face capping Zn(II) porphyrin metalloligands. Figure reproduced with permission from ref. 45. Copyright 2017 John Wiley and Sons. | ||
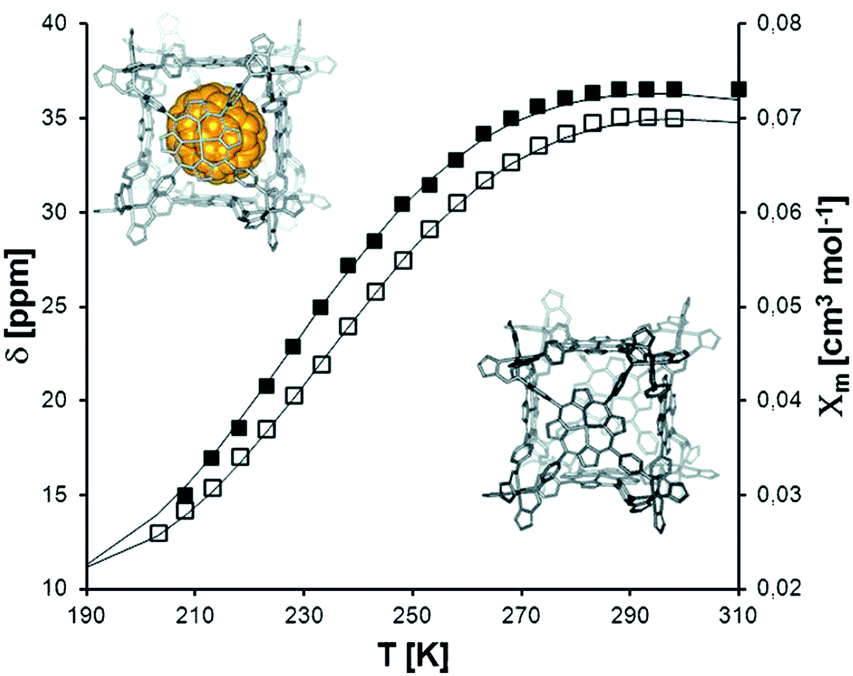 | ||
| Fig. 49 VT-1H NMR in [D4] MeOH, empty squares: [Fe8(ZnL81)6]16+ cube; filled square C70@[Fe8(ZnL81)6]16+; black line calculated molar susceptibility χm based on the ideal solution model. Figure reproduced with permission from ref. 45. Copyright 2017 John Wiley and Sons. | ||
Summary
We have reviewed all 79 of the new discrete polynuclear SCO-active Fe(II) complexes reported between 2012 to February 2018, as well as selected examples from before then, which brings the total to 127 complexes. Of these 127 complexes, 54% are dinuclear, 10% trinuclear, 31% tetranuclear, and the remaining 5% are penta, hexa and octanuclear (Fig. 50 and 51).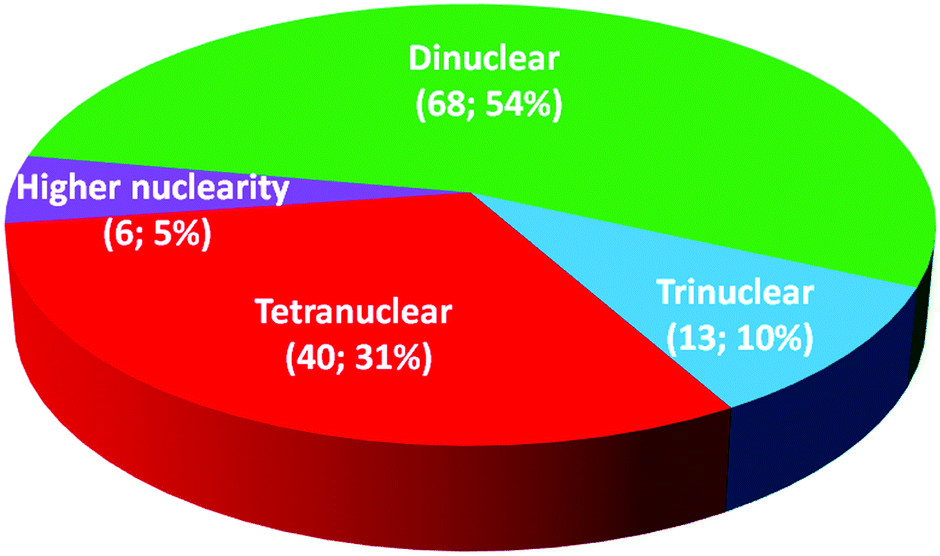 | ||
| Fig. 50 Distribution of the 127 discrete polynuclear Fe(II) SCO-active complexes reviewed herein, as a function of nuclearity. | ||
For the design of Fe(II) SCO-active complexes, the first and foremost consideration is providing the right ligand field strength through careful choice of donor groups, and this also applies when constructing polynuclear architectures. As for mononuclear Fe(II) SCO-active materials, most commonly a N6 coordination sphere is targeted, by the use of exclusively N-donor ligands. Of the 93 ligand and 8 bridging anion designs featured here, only 11 of them provide anything other than purely N-donors: three PSRT ligands (N4S2 coordination sphere), ligand L66− (N4O2), ligand HL22 (N5P) and anions A2–A5 (N5O, Chart 7), μ-NCS (N5S), and μ-CN− (N4C2) feature some S, O, P, or C donor atoms. Amongst the types of N-donor moieties, azoles feature strongly across a range of architectures, with them being incorporated into 60 of the 93 designer ligands, including those that generated the numerous examples of simple triply-triazole-bridged dinuclear and trinuclear SCO-active Fe(II) complexes (Charts 1 and 9).
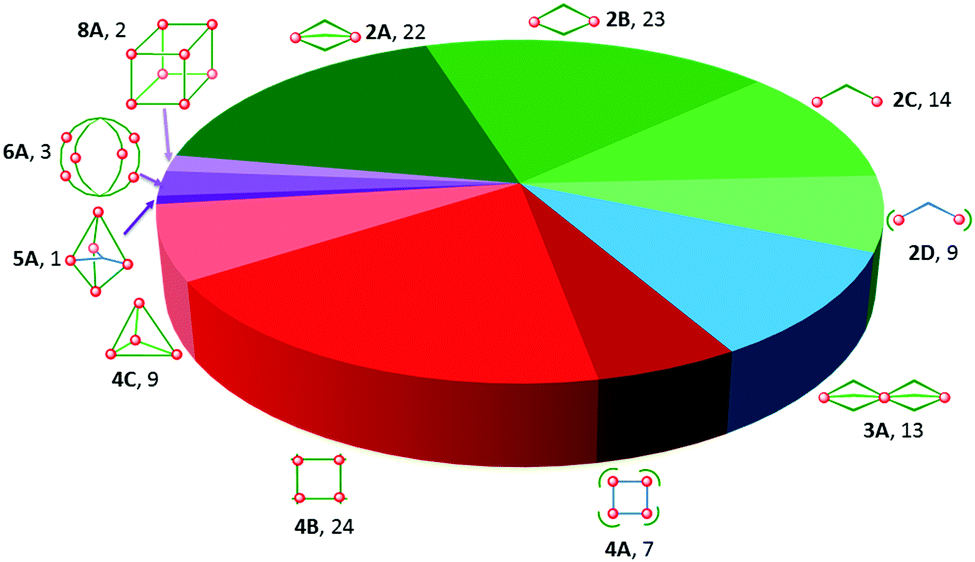 | ||
| Fig. 51 The distribution of the 127 SCO complexes studied in this review across the different categories from dinuclear (2A triply bridged, 2B doubly bridged, 2C single bridged, 2D anion bridged), to trinuclear (3A triply triazole bridged), to tetranuclear (4A squares, 4B grids, 4C tetrahedral cages), to pentanuclear (5A), to hexanuclear (6A ‘nanoball’) to octanuclear cube (8A). | ||
Of the 93 designer ligands presented here, 55 provide all of the donor atoms to the Fe(II) centres (no co-ligands are coordinated) in the polynuclear architectures. These ligands are constructed to have a particular number of bi- or tri-dentate binding pockets that are arranged carefully relative to each other to predictably result in the self-assembly of the desired architecture (Fig. 52). Specific examples include ditopic ligands with bidentate pockets (bis-bidentate), with a central linker which directs the angle of the two pockets (L8–L16), to give dinuclear Fe2L3 helicates (Fig. 52a). When the linker is flexible (L75–L78), such ligands can instead generate edge-bridged tetranuclear Fe4L6 tetrahedral cages (Fig. 52b). Carefully designed ditopic-tridentate ligands (Fig. 52c) result in either (a) dinuclear Fe2L2 complexes, in the case of flexible facially coordinating pockets (PMRT and PSRT families, PMTD, PMOD, L17, L18), or (b) tetranuclear Fe4L4 grids, all but one (L66−, O− bridge) featuring heterocycle-bridged, meridionally coordinated, binding pockets (L50–L70). Moving up another level of complexity, tritopic-bidentate (tris-bidentate) ligands (L71–L74) can be used to generate face-capped tetranuclear Fe4L4 tetrahedral cages (Fig. 52d). Finally, taking this to the maximum level of complexity reported to date, the first examples of octanuclear SCO-active complexes were generated using designer tetratopic-bidentate ligands (H2L80, ZnL81) to form face-bridged Fe8L6 cubes (Fig. 52e).
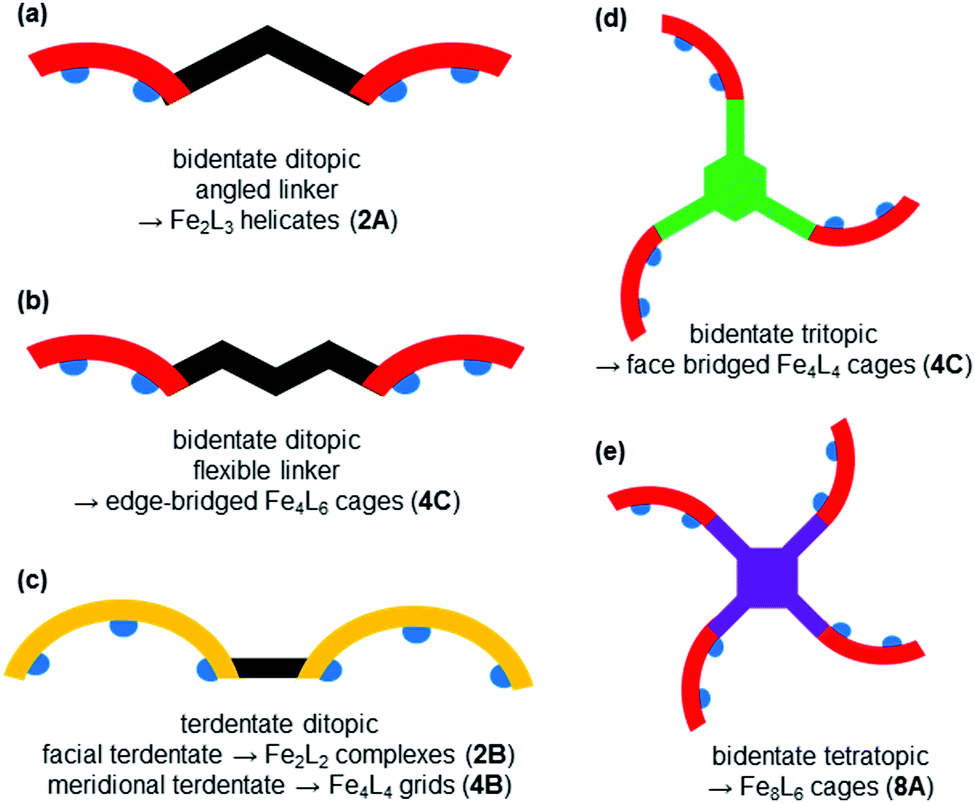 | ||
| Fig. 52 Examples of ligand designs which provide the right number and topology of binding pockets, each with appropriate denticity, for the controlled self-assembly of discrete polynuclear Fe(II) complexes. | ||
It is worth noting that amongst these 55 designer ligands, that the 19 which provide bidentate pockets all generate 5-membered chelate rings, and comprise either azole-imine (17 ligands; 22 complexes) or azole-pyridine (2 ligands; 5 complexes) moieties (Fig. 53) – so clearly the combination of three such azole-imine/pyridine pockets around Fe(II) is a winner for providing about the right ligand field to support SCO.
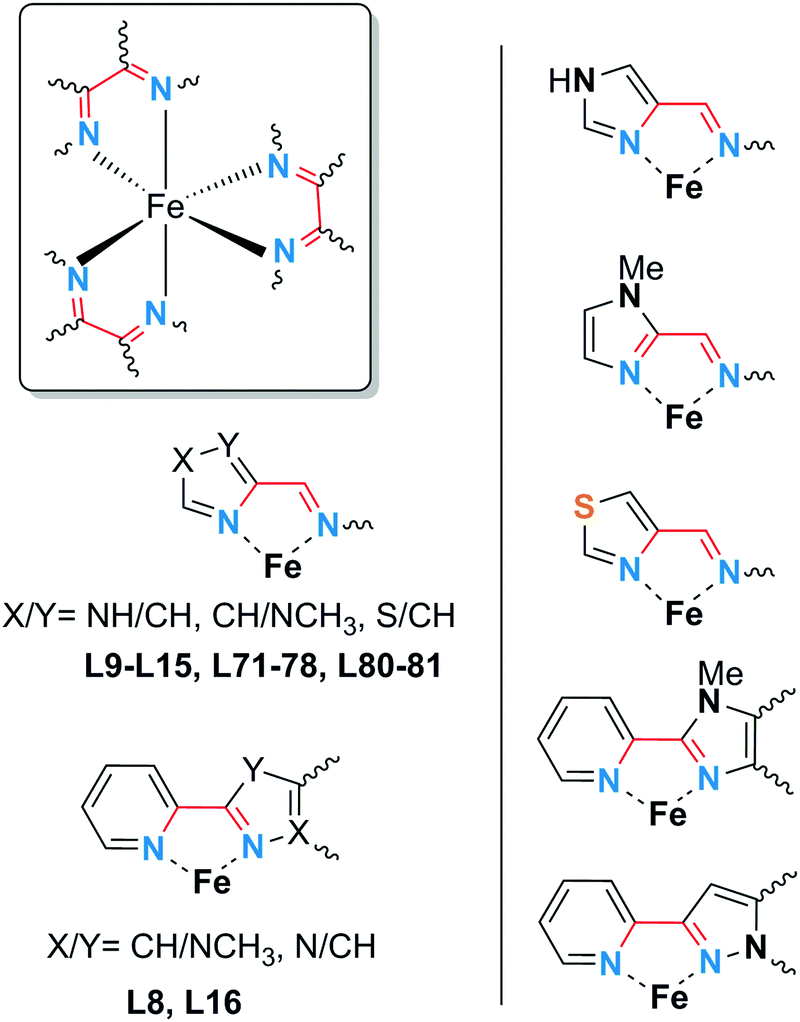 | ||
| Fig. 53 Binding motifs common to the 19 bidentate ligands out of the 55 designer ligands that provide all of the donors to the SCO-active octahedral Fe(II) centres (no co-ligands bound) – by binding of three identical bidentate pockets (box). Right: The 5 specific binding moieties employed in all 19 of these polytopic ligands. | ||
Similarly, out of these 55 designer ligands, 35 provide tridentate pockets, all of which generate a pair of fused 5-membered chelate rings (Fig. 54). These 35 poly-tridentate ligands all comprise either (a) azole-amine–pyridine (9 ligands) or azole-thioether–pyridine (3 ligands) binding facially (Fig. 54a) to give dinuclear PMRT-like complexes, or (b), with the exception of oxygen-bridging L66−, a combination of three conjugated azines and/or azoles (11 ligands) or two azines or azine/azole joined by a conjugated central imine (12 ligands), all of which bind meridionally to give Fe4L4 grids (Fig. 54b). So clearly the coordination of two such pockets, heterocycle-imine/amine/thioether/pyridine-heterocycle, provides about the right ligand field to support SCO at Fe(II).
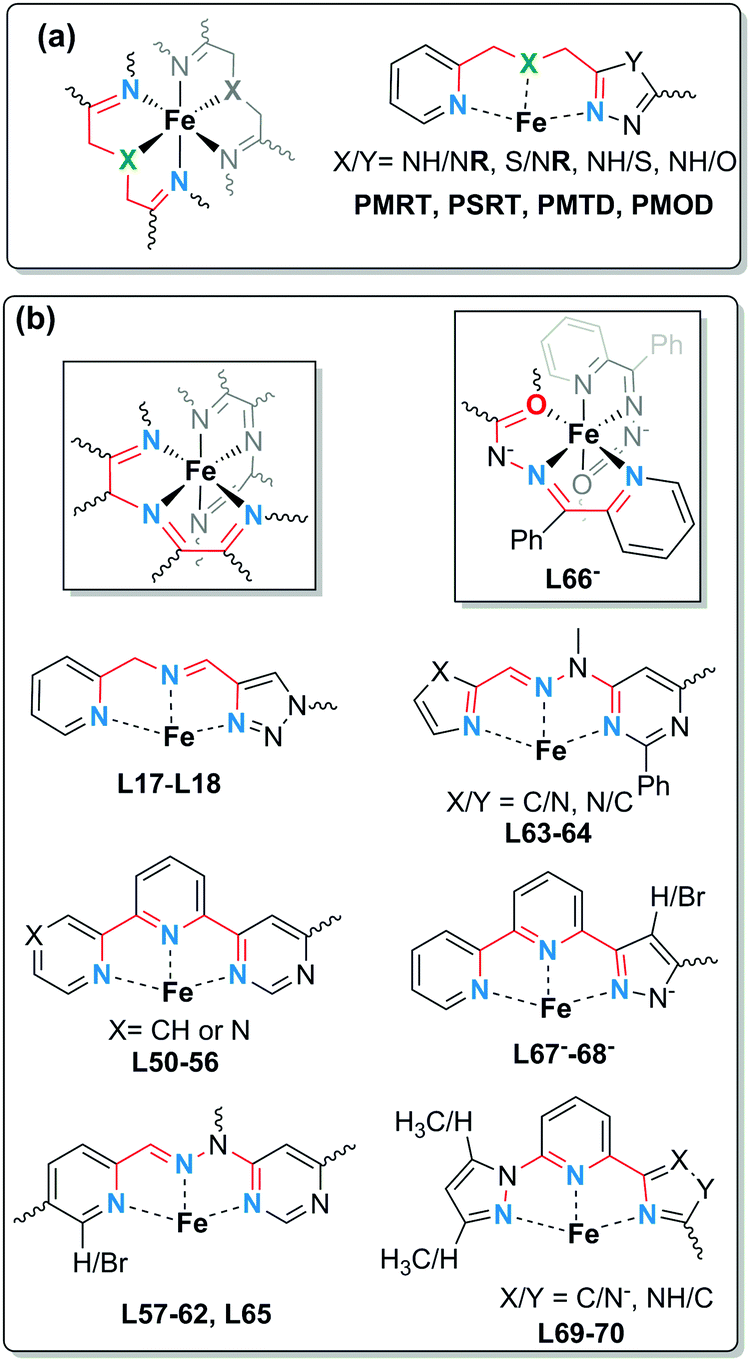 | ||
| Fig. 54 Binding motifs common to the 35 tridentate ligands, out of the 55 designer ligands that provide all of the donors to the SCO-active octahedral Fe(II) centres (no co-ligands bound) by binding of two identical tri-dentate pockets in either (a) facial or (b) meridional binding mode. | ||
The remaining ligand of this group of 55 designer ligands is the unique L25 (Chart 6), which is bis-hexadentate and provides all 12 N-donor atoms to two Fe(II) centres in an Fe2L complex.
On the other hand, of the 38 ligands designed to work in conjunction with co-ligands, 28 of the resulting complexes involve NCE anions (NCS, NCSe, NCBH3, dca−) completing the coordination sphere and helping to provide the right overall field strength for SCO at Fe(II). Overall, 22% of the 127 complexes reviewed herein contain NCE− anions as co-ligands, so it is clear that the use of NCE co-ligands remains another key way to obtain ligand fields that are in the right ballpark to promote the formation of SCO-active Fe(II) complexes.
Another factor that must be taken into consideration is the degree of distortion of the Fe(II) centres away from perfect octahedral geometry, as this also modifies the ligand field. There are a number of ways to probe and quantify this distortion. Here we focus on the octahedral distortion parameter Σ, the sum of the deviations from 90° of the 12 cis angles, and the SChM,151–154 the continuous shape measure of the degree of distortion from an ideal geometry, here octahedral (Tables 1–11 and Fig. 55–59; see also the ESI†). As expected, in general the Fe(II) centres are less distorted when in the LS state than when in the HS state. Another key finding from this analysis of the structurally characterised discrete polynuclear complexes reviewed herein is that for the 55 designer ligands which provide all of the donors to the Fe(II) centres, the octahedral distortion is generally higher than for the 38 ligands which are designed to work in conjunction with co-ligands. This is perhaps not surprising, as the latter systems will have greater flexibility due to involving a mixture of ligands, often including some monodentate ligands. Clearly the same arguments, regarding the degree of distortion away from octahedral being to some extent a function of whether the metal ion is coordinated by (a) only polydentate ligands or (b) a mixture of ligands including monodentate, also apply to non SCO-active complexes – of iron(II) or indeed other metal ions.
 | ||
| Fig. 55 Distribution of octahedral distortion parameter, Σ (°), for the Fe(II) centres in dinuclear complexes of type 2A (Fig. 2): triply triazole bridged [Fe2L5(NCE)4] (green) and triple helicates [Fe2L3] (violet). Dark colour = LS; light colour = HS. | ||
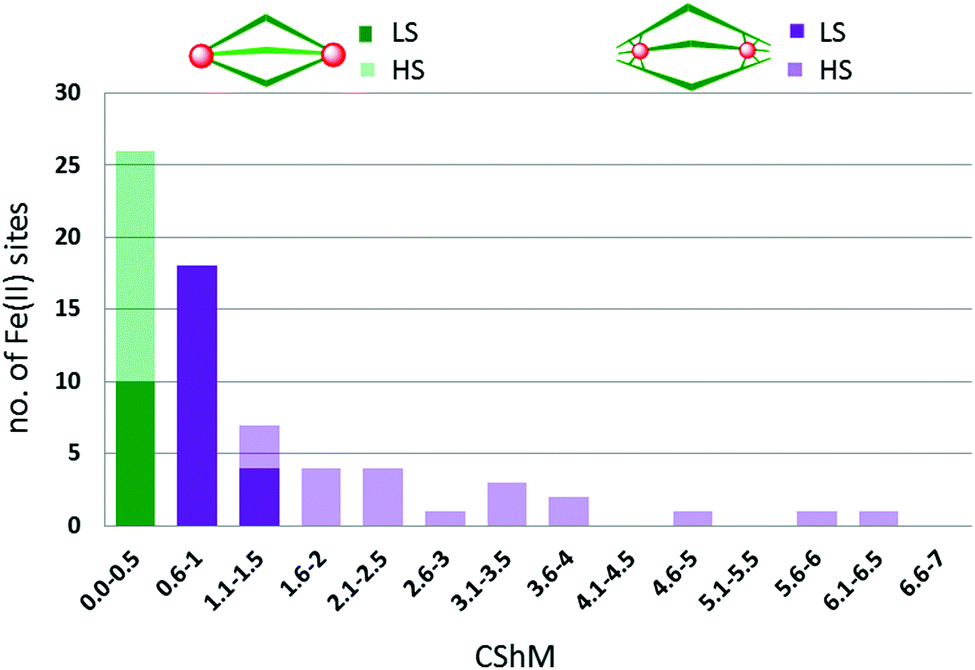 | ||
| Fig. 56 Distribution of continuous shape measured value, CShM, of octahedral distortion, for the Fe(II) centres in dinuclear complexes of type 2A (Fig. 2): triply triazole bridged [Fe2L5(NCE)4] (green) and triple helicates [Fe2L3] (violet). Dark colour = LS; light colour = HS. | ||
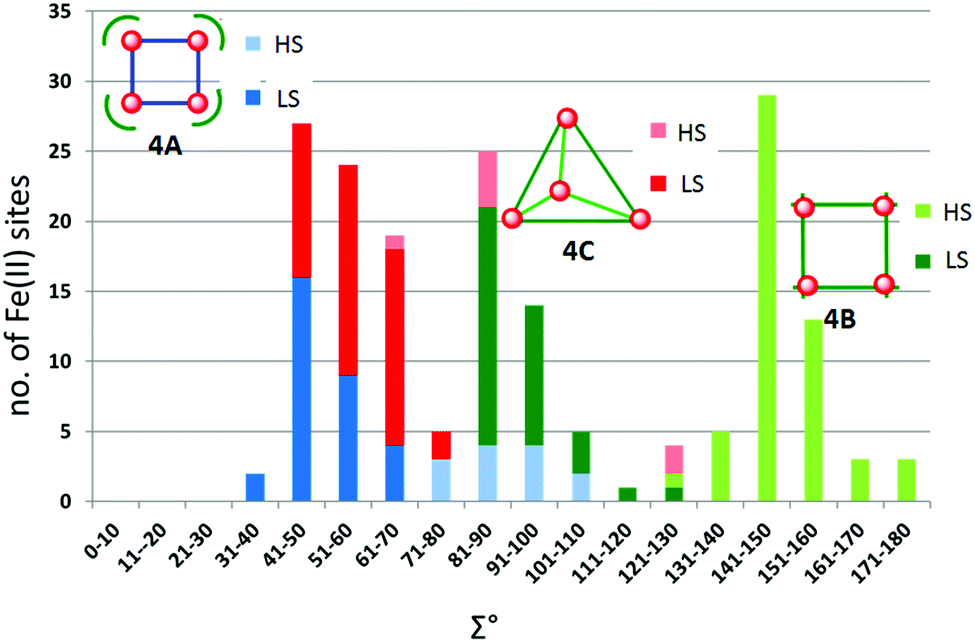 | ||
| Fig. 57 Distribution of octahedral distortion parameter, Σ (°), for the Fe(II) centres in tetranuclear complexes: types 4A squares (blue), 4B grids (green) and 4C tetrahedral cages (red) (Fig. 2). | ||
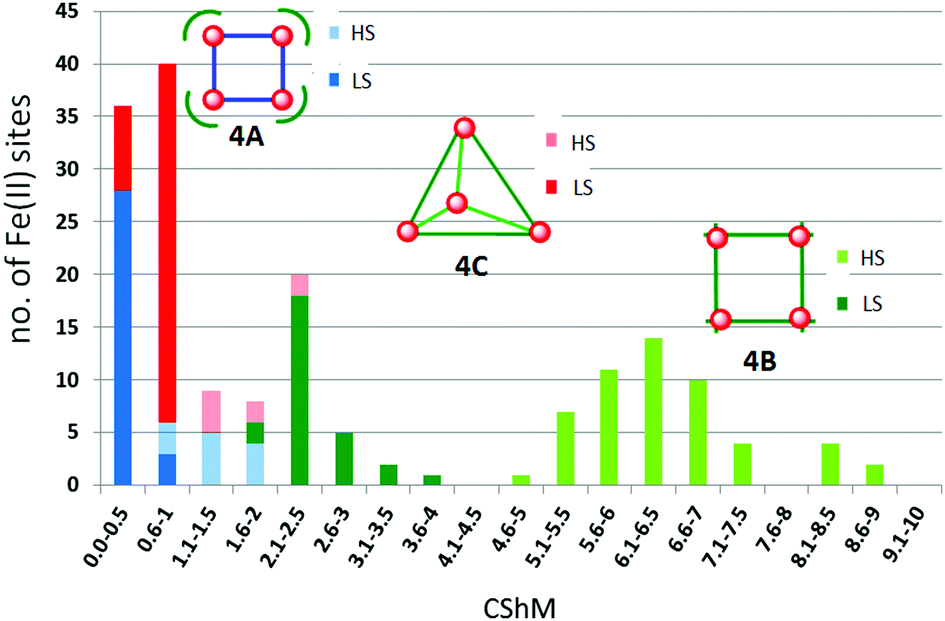 | ||
| Fig. 58 Distribution of continuous shape measured value, CShM, of octahedral distortion, for the Fe(II) centres in tetranuclear complexes: types 4A squares (blue), 4B grids (green) and 4C tetrahedral cages (red) (Fig. 2). | ||
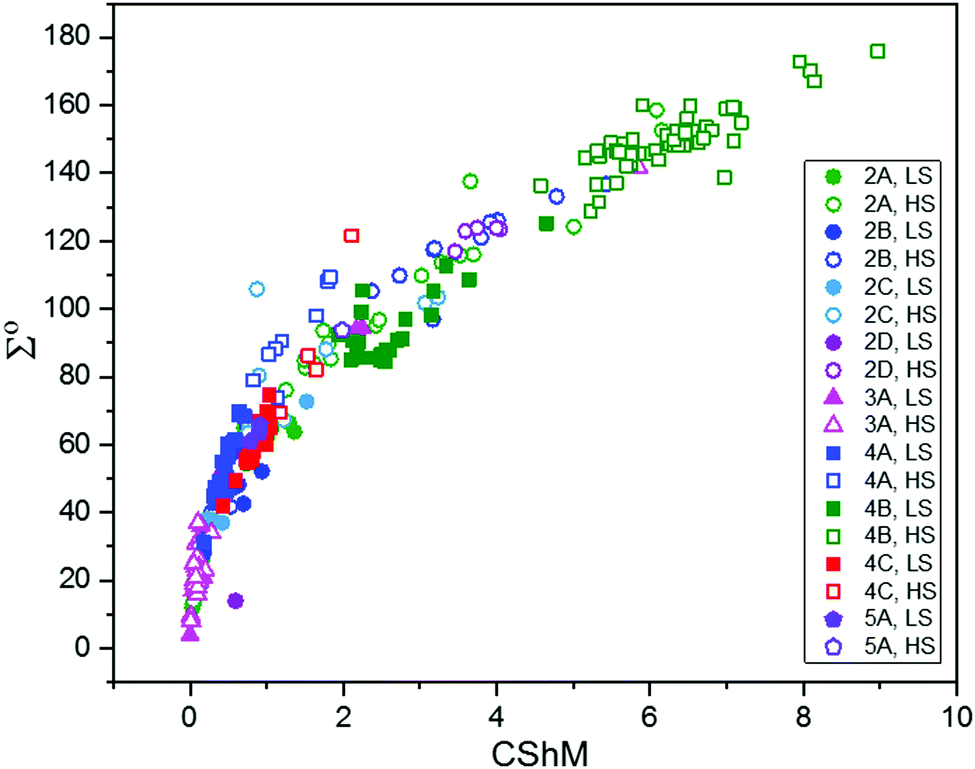 | ||
| Fig. 59 Comparison of two measures of octahedral distortion, Σ (°) and CShM, for structurally characterised discrete polynuclear SCO-active Fe(II) complexes reviewed herein (see also ESI,† Table S1). | ||
Examples of this variation, within SCO active iron(II) complexes, of the octahedral distortion as a function of complex type, are clearly seen in comparing the triply bridged dinuclear complexes (2A), which fall into two distinct categories: triply-triazole bridged with co-ligands [Fe2L5(NCE)4] (Fig. 3 and Chart 1) for which Σ is consistently lower (Table 1) than it is for the triple helicates [Fe2L3] (Fig. 6–9, Chart 2 and Tables 2–4), as is illustrated in Fig. 55. Indeed the Σ values for the Fe(II) centres in the triply-triazole bridged complexes (Fig. 55, green) are in the range 11–20° for LS and 11–40° for HS, with the octahedral geometry close to ideal in both cases (CShM < 0.5, Fig. 56, green). In contrast, for Fe2L3 helicates, the Σ values are far higher, at 51–70° for LS versus 71–160° for HS (Fig. 55, violet), with correspondingly high CShM values (LS = 0.6–1.5, HS = 1.1–6.5; Fig. 56, purple).
Similarly, the Fe(II) centres in tetranuclear squares with co-ligands (Fig. 2, 4A) have smaller Σ values (Fig. 57, blue) than those in grids (Fig. 2, 4B, Fig. 57 green), as the latter systems comprise just four ligands that provide all of the 24 donors to the four Fe(II) centres. Interestingly, the tetranuclear tetrahedral cages (Fig. 2, 4C, Fig. 57 red) also comprise a single type of ligand which provides all of the donors, and yet the distortions are smaller than in the grids, falling in a similar range to those seen for the squares (Fig. 57 blue). Specifically, in the LS state, the Fe(II) centres in the squares and cages have consistently much lower Σ (31–70° and 43–75° respectively, Fig. 57 dark blue and red) and CShM (0.183–0.826 and 0.433–1.035 respectively, Fig. 58 dark blue and red) values, than in the grids (Σ = 84–125°, Fig. 57 dark green; CShM = 1.935–5.686; Fig. 58, dark green). Indeed the Σ values for the LS grids (Fig. 57, dark green) are in the same range as for the HS squares (Σ = 79–109°, Fig. 57 light blue).
The highly constrained coordination environment imposed by the four bis-terdentate ligands in the Fe4L4 grids, as evidenced by large Σ and CShM values, may be providing a barrier to complete SCO to the [4LS] state, with a regular octahedral LS coordination environment for all four of the Fe(II) centres perhaps geometrically impossible in some cases. In most of the grids reviewed here, the fully LS state is inaccessible, with only the grids of L54, L65, and L70 (only the trans isomer, which is unique among the grids reviewed here) being structurally characterised in the [4LS] state. On the other hand, all of the square complexes, which contain co-ligands and hence a more readily accessible regular octahedral geometry, have been structurally characterised as [4LS]. For the tetrahedral cages, all but two complexes, [FeII4(L74)4](BF4)4·16CH3CN and [FeII4(L71)4](BF4)8·14.75CH3CN·4.5C6H6·3H2O, have been observed in the [4LS] state by X-ray crystallography, despite having all donors to the Fe(II) centres provided by just the one type of ligand. The key difference, however, for the tetrahedral cages is that the octahedral environment at the Fe(II) centres is completed by three bidentate donors, unlike in the grid complexes where just two terdentate donors coordinate to each Fe(II) centre.
Unsurprisingly a positive, but not linear, correlation is observed between these two measures of distortion, Σ and CShM (Fig. 59 and Table S1, ESI†). It is interesting to note that when the distortion away from octahedral is relatively small, Σ is perhaps a more informative measure than the CShM. Indeed it is pertinent to note that a CShM value of just 2 corresponds to a Σ value of approximately 90°. Finally, as expected, regardless of the measure used, Σ or CShM, lower distortion parameters are observed for the LS Fe(II) centres (Fig. 59, solid points) than for the HS Fe(II) centres (Fig. 59, hollow points).
Generally, the Fe⋯Fe distances in the helicates vary from 9–12 Å, except in case of L14 and L15 (Chart 2), where distance is far lower, ∼4 Å, because of the short NN bridge between them and the resultant twist of the ligand to bind, which also leads to a very high distortion Σ and CShM for the Fe(II) centres (158.5° and 6.084, respectively, Table S1, ESI†). The Fe4 grids have a very rigid geometry around the metal centres and their Fe⋯Fe distances (4–7 Å) are much lower than those seen in the Fe4 tetrahedral cages (with face capping ligands, 11–15 Å; edge capping ligands 9–11 Å). This is mostly because the bis-terdentate ligands of the grids also provide a short bridge (e.g. pyrimidine-bridge) between the two binding pockets and hence Fe(II) centres, whereas for the cages, longer, non-coordinating linkers (e.g. 1,3,5-triphenylbenzene) are utilised.
An important feature of many of the SCO-active polynuclear architectures reviewed here is the presence of a guest-accessible internal cavity which may be exploited for SCO tuning and/or guest sensing. The effects on SCO of guest inclusion into the cavity in Fe2L3 helicates, Fe4L4 cages, and Fe8L6 cubes have been probed in a handful of studies. The largest of the internal cavities to date occur in the Fe8L6 cubes, where the uptake of the large C70 into the 1300 Å3 cavity tuned the SCO T1/2 by 15–20 K in the solution phase. For the face-capped Fe4L4 cages, smaller internal cavities of 61–183 Å3 are reported, and in the case of [Fe4(L73)4](OTf)8 inclusion of the small guests Br− and CS2 tuned the SCO T1/2 by 8–12 K in the solution phase. Interestingly, the smallest of the cavities probed for guest tuning of SCO, the 28–33 Å3 cavities in the {X@[Fe2(L16)3]}X(PF6)2 (X = Cl− or Br−) helicates, had the largest observed shift in T1/2. Upon changing the encapsulated Cl− to the larger Br− anion, the T1/2 in the solid state was decreased by 40 K. Furthermore, the position of the halide inside the small cavity was seen to greatly affect the nature of the SCO, with half vs. full SCO observed when the halide is positioned closer to one Fe(II) centre vs. equidistant to both Fe(II) centres. Clearly, large, easily observable effects on the SCO properties of these complexes can be induced by the encapsulation of various guests, and as more examples of large cage-like SCO-active complexes are developed one can anticipate a wealth of host–guest chemistry, including sensing applications, being reported in the near future.
Finally, the number of discrete polynuclear SCO-active Fe(II) complexes has increased significantly over the period from 2012 to 2017 (Fig. 60): the 76 new examples reported more than doubles the number reported in the literature, 68, over the period 2004–2011 covered by our previous review.35
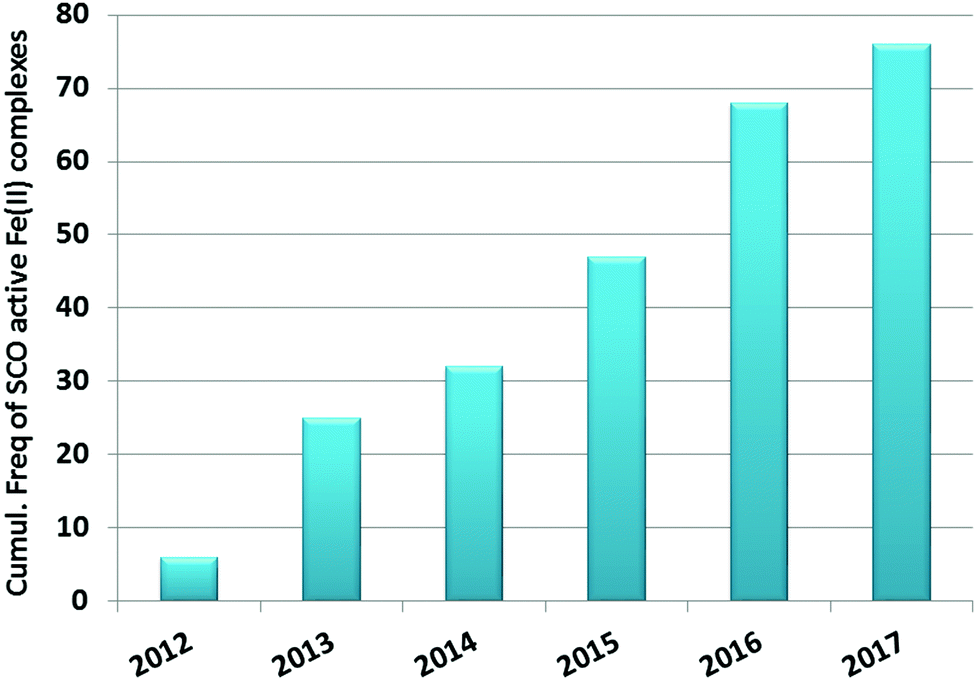 | ||
| Fig. 60 Increasing number of discrete di- to poly-nuclear SCO-active Fe(II) complexes reported in the period from 2012 to 2017 (76 in total). Our previous review35 covered the 68 examples reported 2004–2011. | ||
Concluding remarks
By applying supramolecular self-assembly strategies,43,58,155–158 and linking multiple donor pockets of the right ligand field strength and denticity into carefully structured ligands (Fig. 52), chemists have created a wealth of SCO-active discrete polynuclear Fe(II) complexes, ranging to date from dinuclear to octanuclear. The development of such complexes is a flourishing field, as illustrated by the fact that the 8 years from 2004–2011, 68 complexes were reported, and in the 7 subsequent years another 76 were reported. The architectural diversity of these complexes has also expanded considerably since 2011, with the first examples of SCO-active tetranuclear cages reported in 2013, and then a new record nuclearity for a discrete SCO complex was set in 2017 when the first examples of SCO-active octanuclear cube cages were reported. These trends are likely to continue, with many more reports of SCO-active discrete supramolecular architectures anticipated.Covalently linking Fe(II) centres into polynuclear architectures has led to access to multistep SCO with stable intermediate spin states, e.g. [HS–LS], which opens up the possibility of multi-state molecular switches. In some cases linking SCO-appropriate binding pockets has led to more abrupt and even hysteretic SCO being observed in comparison to mononuclear analogues of the same immediate coordination environment. In many cases, the polymetallic architectures are highly strained, with steric and intermolecular crystal packing or solvent interactions often dominating the SCO properties. In other cases, less strained ligands in same family induced different SCO behaviour through very small modification in ligand or counter-anion/solvent molecules.
Whilst a number of winning ligand design features for inducing SCO at Fe(II) are identified in the previous section (e.g. Fig. 53 and 54), it remains very difficult to predictably fine-tune or control SCO behaviour (including T1/2 or hysteresis) in advance of synthesis and characterisation.159–163 The effects of intermolecular crystal packing and/or counter anion/solvent interactions can dominate, over ligand field effects, the SCO properties in the solid state.
Solution state studies13–16,145 are of increasing prevalence and promise to facilitate the development of more detailed understanding of the ligand field effects, as packing effects no longer apply. Solution studies are also of key importance in the case of SCO-active systems that can recognise guests, such as cages, as modifications in the SCO behaviour could facilitate guest sensing. But solution studies warrant two notes of caution: (a) the potential for speciation to be an issue must be addressed, otherwise it could confound such studies, and (b) the influence of solvent choice on the SCO should also be explored.164 Nevertheless, the solution stability of many of these supramolecular assemblies is such that exploiting the internal cavities of SCO-active helicates, cages and new types of discrete supramolecules for guest sensing applications is another rapidly evolving area of interest in this field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Otago (PhD scholarships to RWH and SS) for supporting this research.References
- P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, 233, 1–47 CrossRef.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC.
- M. A. Halcrow, Spin-Crossover Materials: Properties and Applications, John Wiley & Sons, Ltd, Chichester, 1st edn, 2013 Search PubMed.
- P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef PubMed.
- H. J. Shepherd, I. A. Gural'skiy, C. M. Quintero, S. Tricard, L. Salmon, G. Molnár and A. Bousseksou, Nat. Commun., 2013, 4, 2607 CrossRef PubMed.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC , and front cover feature.
- K. Senthil Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef.
- O. Kahn, Molecular Magnetism, VCH Publishers Inc., New York, 1993 Search PubMed.
- A. Hauser, Top. Curr. Chem., 2004, 233, 49–58 CrossRef.
- M. H. Klingele, B. Moubaraki, J. D. Cashion, K. S. Murray and S. Brooker, Chem. Commun., 2005, 987–989 RSC and front cover feature.
- J. A. Kitchen, N. G. White, C. Gandolfi, M. Albrecht, G. N. L. Jameson, J. L. Tallon and S. Brooker, Chem. Commun., 2010, 46, 6464–6466 RSC.
- H. L. C. Feltham, C. Johnson, A. B. S. Elliot, K. C. Gordon, M. Albrecht and S. Brooker, Inorg. Chem., 2015, 54, 2902–2909 CrossRef PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- B. Weber and F. A. Walker, Inorg. Chem., 2007, 46, 6794–6803 CrossRef PubMed.
- B. Weber, E. S. Kaps, J. Obel, K. Achterhold and F. G. Parak, Inorg. Chem., 2008, 47, 10779–10787 CrossRef PubMed.
- R. W. Hogue, C. P. Lepper, G. B. Jameson and S. Brooker, Chem. Commun., 2018, 54, 172–175 RSC.
- P. Gütlich, A. Bhattacharjee, M. Seredyuk and A. B. Gaspar, Hyperfine Interact., 2009, 189, 3–19 CrossRef.
- Y. Garcia, F. Robert, A. D. Naik, G. Zhou, B. Tinant, K. Robeyns, S. Michotte and L. Piraux, J. Am. Chem. Soc., 2011, 133, 15850–15853 CrossRef PubMed.
- M. J. Murphy, K. A. Zenere, F. Ragon, P. D. Southon, C. J. Kepert and S. M. Neville, J. Am. Chem. Soc., 2017, 139, 1330–1335 CrossRef PubMed.
- O. Roubeau, M. Castro, R. Burriel, J. G. Haasnoot and J. Reedijk, J. Phys. Chem. B, 2011, 115, 3003–3012 CrossRef PubMed.
- M. Sorai and S. Seki, J. Phys. Chem. Solids, 1974, 35, 555–570 CrossRef.
- O. Kahn, J. Kröber and C. Jay, Adv. Mater., 1992, 4, 718–728 CrossRef.
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef.
- J. Krober, E. Codjovi, O. Kahn, F. Groliere and C. Jay, J. Am. Chem. Soc., 1993, 115, 9810–9811 CrossRef.
- J. A. Real, A. B. Gaspar, V. Niel and M. C. Muñoz, Coord. Chem. Rev., 2003, 236, 121–141 CrossRef.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- O. Kahn, L. Sommier and E. Codjovi, Chem. Mater., 1997, 9, 3199–3205 CrossRef.
- E. Codjovi, L. Sommier, O. Kahn and C. Jay, New J. Chem., 1996, 20, 503–505 Search PubMed.
- J. Kroeber, J.-P. Audiere, R. Claude, E. Codjovi, O. Kahn, J. G. Haasnoot, F. Groliere, C. Jay, A. Bousseksou, J. Linares, F. Varret and A. Gonthier-Vassal, Chem. Mater., 1994, 6, 1404–1412 CrossRef.
- A. Grosjean, N. Daro, B. Kauffmann, A. Kaiba, J.-F. Létard and P. Guionneau, Chem. Commun., 2011, 47, 12382–12384 RSC.
- K. S. Murray, Eur. J. Inorg. Chem., 2008, 3101–3121 CrossRef.
- J. A. Real, A. B. Gaspar, M. C. Muñoz, P. Gütlich, V. Ksenofontov and H. Spiering, Top. Curr. Chem., 2004, 233, 167–193 CrossRef.
- Y. Garcia, V. Niel, M. C. Muñoz and J. A. Real, Top. Curr. Chem., 2004, 233, 229–258 CrossRef.
- A. B. Gaspar, M. C. Muñoz and J. A. Real, J. Mater. Chem., 2006, 16, 2522–2533 RSC.
- J. Olguín and S. Brooker, in Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Ltd, 1st edn, 2013, pp. 77–120 Search PubMed.
- J.-F. Létard, P. Guionneau and L. Goux-Capes, Top. Curr. Chem., 2004, 235, 221–249 CrossRef.
- J. A. Real, H. Bolvin, A. Bousseksou, A. Dworkin, O. Kahn, F. Varret and J. Zarembowitch, J. Am. Chem. Soc., 1992, 114, 4650–4658 CrossRef.
- B. Schneider, S. Demeshko, S. Neudeck, S. Dechert and F. Meyer, Inorg. Chem., 2013, 52, 13230–13237 CrossRef PubMed.
- B. Schneider, S. Demeshko, S. Dechert and F. Meyer, Angew. Chem., Int. Ed., 2010, 49, 9274–9277 CrossRef PubMed.
- I. Amlani, A. O. Orlov, G. Toth, G. H. Bernstein, C. S. Lent and G. L. Snider, Science, 1999, 284, 289–291 CrossRef PubMed.
- C. S. Lent, Science, 2000, 288, 1597–1599 CrossRef.
- M. Ruben, J. Rojo, F. J. Romero-Salguero, L. H. Uppadine and J.-M. Lehn, Angew. Chem., Int. Ed., 2004, 43, 3644–3662 CrossRef PubMed.
- J. G. Hardy, Chem. Soc. Rev., 2013, 42, 7881–7899 RSC.
- A. B. Gaspar, M. C. Munoz and J. A. Real, J. Mater. Chem., 2006, 16, 2522–2533 RSC.
- N. Struch, C. Bannwarth, T. K. Ronson, Y. Lorenz, B. Mienert, N. Wagner, M. Engeser, E. Bill, R. Puttreddy, K. Rissanen, J. Beck, S. Grimme, J. R. Nitschke and A. Lützen, Angew. Chem., Int. Ed., 2017, 56, 4930–4935 CrossRef PubMed.
- S. B. Erenburg, N. V. Bausk, V. A. Varnek and L. G. Lavrenova, J. Magn. Magn. Mater., 1996, 157-158, 595–596 CrossRef.
- O. Roubeau, Chem. – Eur. J., 2012, 18, 15230–15244 CrossRef PubMed; L. G. Lavrenova and O. G. Shakirova, Eur. J. Inorg. Chem., 2013, 670–682 CrossRef.
- X. Cheng, Q. Yang, C. Gao, B.-W. Wang, T. Shiga, H. Oshio, Z.-M. Wang and S. Gao, Dalton Trans., 2015, 44, 11282–11285 RSC.
- J.-L. Wang, Q. Liu, Y.-S. Meng, H. Zheng, H.-L. Zhu, Q. Shi and T. Liu, Inorg. Chem., 2017, 56, 10674–10680 CrossRef PubMed.
- O. Roubeau, P. Gamez and S. J. Teat, Eur. J. Inorg. Chem., 2013, 934–942 CrossRef.
- X. X. Wu, Y. Y. Wang, P. Yang, Y. Y. Xu, J. Z. Huo, B. Ding, Y. Wang and X. Wang, Cryst. Growth Des., 2014, 14, 477–490 CrossRef.
- B. Ding, Y. Y. Liu, Y. Wang, J.-G. Ma, Z. Niu, W. Shi and P. Cheng, Inorg. Chem. Commun., 2013, 31, 44–48 CrossRef.
- J. J. A. Kolnaar, M. I. deHeer, H. Kooman, A. L. Spek, G. Schmitt, V. Ksenofontov, P. Gütlich, J. G. Haasnoot and J. Reedijk, Eur. J. Inorg. Chem., 1999, 881–886 CrossRef.
- A. Williams, Chem. – Eur. J., 1997, 3, 15–19 CrossRef.
- C. Piguet, G. Bernadinelli and G. Hopfgarter, Chem. Rev., 1997, 97, 2005–2062 CrossRef PubMed.
- C. Piguet, M. Borkovec, J. Hamacek and K. Zeckert, Coord. Chem. Rev., 2005, 249, 705–726 CrossRef.
- C. R. K. Glasson, L. F. Lindoy and G. V. Meehan, Coord. Chem. Rev., 2008, 252, 940–963 CrossRef.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef PubMed.
- M. J. Hannon and L. J. Childs, Supramol. Chem., 2004, 16, 7–22 CrossRef.
- M. Albrecht, Chem. Rev., 2001, 101, 3457–3498 CrossRef PubMed.
- M. Darawsheh, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromí, Chem. – Eur. J., 2016, 22, 8635–8645 CrossRef PubMed.
- S. Goetz and P. E. Kruger, Dalton Trans., 2006, 1277–1284 RSC.
- F. Cui, S. Li, C. Jia, J. S. Mathieson, L. Cronin, X.-J. Yang and B. Wu, Inorg. Chem., 2012, 51, 179–187 CrossRef PubMed.
- J. Xu, T. N. Parac and K. N. Raymond, Angew. Chem., Int. Ed., 1999, 38, 2878–2882 CrossRef PubMed.
- C. R. K. Glasson, G. V. Meehan, C. A. Motti, J. K. Clegg, P. Turner, P. Jensen and L. F. Lindoy, Dalton Trans., 2011, 40, 12153–12159 RSC.
- L. J. Charbonnière, A. F. Williams, C. Piguet, G. Bernardinelli and E. Rivara-Minten, Chem. – Eur. J., 1998, 4, 485–493 CrossRef.
- F. Tuna, M. R. Lees, G. J. Clarkson and M. J. Hannon, Chem. – Eur. J., 2004, 10, 5737–5750 CrossRef PubMed.
- Y. Sunatsuki, R. Kawamoto, K. Fujita, H. Maruyama, T. Suzuki, H. Ishida, M. Kojima, S. Iijima and N. Matsumoto, Inorg. Chem., 2009, 48, 8784–8795 CrossRef PubMed.
- D. Pelleteret, R. Clérac, C. Mathonière, E. Harte, W. Schmitt and P. E. Kruger, Chem. Commun., 2009, 221–223 RSC.
- S. Yukinari, M. Hisashi, F. Kunihiro, S. Takayoshi, K. Masaaki and M. Naohide, Bull. Chem. Soc. Jpn., 2009, 82, 1497–1505 CrossRef.
- A. Craze, N. Sciortino, M. Badbhade, C. Kepert, C. Marjo and F. Li, Inorganics, 2017, 5, 62 CrossRef.
- L. Li, A. R. Craze, R. Akiyoshi, A. Tsukiashi, S. Hayami, O. Mustonen, M. M. Bhadbhade, S. Bhattacharyya, C. E. Marjo, Y. Wang, L. F. Lindoy, J. R. Aldrich-Wright and F. Li, Dalton Trans., 2018, 47, 2543–2548 RSC.
- S. G. Telfer, B. Bocquet and A. F. Williams, Inorg. Chem., 2001, 40, 4818–4820 CrossRef PubMed.
- Y. Garcia, M. Grunert, S. Reiman, O. van Campenhoudt and P. Gütlich, Eur. J. Inorg. Chem., 2006, 3333–3339 CrossRef.
- R. J. Archer, C. S. Hawes, G. N. L. Jameson, V. McKee, B. Moubaraki, N. F. Chilton, K. S. Murray, W. Schmitt and P. E. Kruger, Dalton Trans., 2011, 40, 12368–12373 RSC.
- H. Hagiwara, T. Tanaka and S. Hora, Dalton Trans., 2016, 45, 17132–17140 RSC.
- S. Hora and H. Hagiwara, Inorganics, 2017, 5, 49 CrossRef.
- M. H. Klingele, B. Moubaraki, K. S. Murray and S. Brooker, Chem. – Eur. J., 2005, 11, 6962–6973 CrossRef PubMed.
- C. M. Grunert, S. Reiman, H. Spiering, J. A. Kitchen, S. Brooker and P. Gütlich, Angew. Chem., Int. Ed., 2008, 47, 2997–2999 CrossRef PubMed , and front cover feature.
- A. Bhattacharjee, V. Ksenofontov, J. A. Kitchen, N. G. White, S. Brooker and P. Gütlich, Appl. Phys. Lett., 2008, 92, 174104 CrossRef.
- J. A. Kitchen, N. G. White, G. N. L. Jameson, J. L. Tallon and S. Brooker, Inorg. Chem., 2011, 50, 4586–4597 CrossRef PubMed.
- A. Bhattacharjee, M. Roy, V. Ksenofontov, J. A. Kitchen, S. Brooker and P. Gütlich, Eur. J. Inorg. Chem., 2013, 843–849 CrossRef.
- K. Nakano, S. Kawata, K. Yoneda, A. Fuyuhiro, T. Yagi, S. Nasu, S. Morimoto and S. Kaizaki, Chem. Commun., 2004, 2892–2893 RSC.
- J. A. Kitchen, J. Olguín, R. Kulmaczewski, N. G. White, V. A. Milway, G. N. L. Jameson, J. L. Tallon and S. Brooker, Inorg. Chem., 2013, 52, 11185–11199 CrossRef PubMed.
- R. Kulmaczewski, J. Olguín, J. A. Kitchen, H. L. C. Feltham, G. N. L. Jameson, J. L. Tallon and S. Brooker, J. Am. Chem. Soc., 2014, 136, 878–881 CrossRef PubMed.
- R. W. Hogue, H. L. C. Feltham, R. G. Miller and S. Brooker, Inorg. Chem., 2016, 55, 4152–4165 CrossRef PubMed.
- N. G. White and S. Brooker, Polyhedron, 2016, 103, 283–287 CrossRef.
- C. F. Herold, L. M. Carrella and E. Rentschler, Eur. J. Inorg. Chem., 2015, 3632–3636 CrossRef.
- C. F. Herold, S. I. Shylin and E. Rentschler, Inorg. Chem., 2016, 55, 6414–6419 CrossRef PubMed.
- C. Köhler and E. Rentschler, Eur. J. Inorg. Chem., 2016, 1955–1960 CrossRef.
- J. J. M. Amoore, C. J. Kepert, J. D. Cashion, B. Moubaraki, S. M. Neville and K. S. Murray, Chem. – Eur. J., 2006, 12, 8220–8227 CrossRef PubMed.
- C. J. Schneider, J. D. Cashion, N. F. Chilton, C. Etrillard, M. Fuentealba, J. A. K. Howard, J. F. Létard, C. Milsmann, B. Moubaraki, H. A. Sparkes, S. R. Batten and K. S. Murray, Eur. J. Inorg. Chem., 2013, 850–864 CrossRef.
- S. Rodríguez-Jiménez, H. L. C. Feltham and S. Brooker, Angew. Chem., Int. Ed., 2016, 55, 15067–15071 CrossRef PubMed , and back cover.
- S. Samanta, S. Demesko, S. Dechert and F. Meyer, Angew. Chem., Int. Ed., 2015, 54, 583–587 Search PubMed.
- S. Kanegawa, S. Kang and O. Sato, Eur. J. Inorg. Chem., 2013, 725–729 CrossRef.
- J.-F. Létard, P. Guionneau, O. Nguyen, J. S. Costa, S. Marcén, G. Chastanet, M. Marchivie and L. Goux-Capes, Chem. – Eur. J., 2005, 11, 4582–4589 CrossRef PubMed.
- S. Heider, H. Petzold and G. Teucher, Eur. J. Inorg. Chem., 2013, 2382–2388 CrossRef.
- A. Y. Verat, N. Ould-Moussa, E. Jeanneau, L. G. Boris, A. Boussekou, S. A. Borshch and G. S. Matouzenko, Chem. – Eur. J., 2009, 15, 10070–10082 CrossRef PubMed.
- G. S. Matouzenko, E. Jeanneau, A. Y. Verat and A. Bousseksou, Dalton Trans., 2011, 40, 9608–9618 RSC.
- G. S. Matouzenko, E. Jeanneau, A. Y. Verat and Y. d. Gaetano, Eur. J. Inorg. Chem., 2012, 969–977 CrossRef.
- Y. d. Gaetano, E. Jeanneau, A. Y. Verat, L. Rechignat, A. Bousseksou and G. S. Matouzenko, Eur. J. Inorg. Chem., 2013, 1015–1023 CrossRef.
- V. Ksenofontov, A. B. Gaspar, J. A. Real and P. Gütlich, J. Phys. Chem. B, 2001, 105, 12266–12271 CrossRef.
- H. J. Shepherd, P. Rosa, L. Vendier, N. Casati, J.-F. Létard, A. Bousseksou, P. Guionneau and G. Molnár, Phys. Chem. Chem. Phys., 2012, 14, 5265–5271 RSC.
- D. Fedaoui, Y. Bouhadja, A. Kaiba, P. Guionneau, J.-F. Létard and P. Rosa, Eur. J. Inorg. Chem., 2008, 1022–1026 CrossRef.
- N. Ortega-Villar, A. Thompson, M. C. Muñoz, V. M. Ugalde-Saldívar, A. E. Goeta, R. Moreno-Esparza and J. A. Real, Chem. – Eur. J., 2005, 11, 5721–5734 CrossRef PubMed.
- E. Milin, S. Belaïd, V. Patinec, S. Triki, G. Chastanet and M. Marchivie, Inorg. Chem., 2016, 55, 9038–9046 CrossRef PubMed.
- M. Yamasaki and T. Ishida, Chem. Lett., 2015, 44, 920–921 CrossRef.
- J. G. Park, I.-R. Jeon and T. D. Harris, Inorg. Chem., 2015, 54, 359–369 CrossRef PubMed.
- G. Vos, R. A. le Fèbre, R. A. G. de Graaff, J. G. Haasnoot and J. Reedijk, J. Am. Chem. Soc., 1983, 105, 1682–1683 CrossRef.
- J. J. A. Kolnaar, G. v anDijk, H. Kooijman, A. L. Spek, V. G. Ksenofontov, P. Gütlich, J. G. Haasnoot and J. Reedijk, Inorg. Chem., 1997, 36, 2433–2440 CrossRef PubMed.
- M. Thomann, O. Kahn, J. Guilhem and F. Varret, Inorg. Chem., 1994, 33, 6029–6037 CrossRef.
- Y. Garcia, P. Guionneau, G. Bravic, D. Chasseau, J. A. K. Howard, O. Kahn, V. Ksenofontov, S. Reiman and P. Gütlich, Eur. J. Inorg. Chem., 2000, 1531–1538 CrossRef.
- D. Savard, C. Cook, G. D. Enright, I. Korobkov, T. J. Burchell and M. Murugesu, CrystEngComm, 2011, 13, 5190–5197 RSC.
- V. Gómez, J. Benet-Buchholz, E. Martin and J. R. Galán-Mascarós, Chem. – Eur. J., 2014, 20, 5369–5379 CrossRef PubMed.
- V. Gómez, C. Sáenz de Pipaón, P. Maldonado-Illescas, J. C. Waerenborgh, E. Martin, J. Benet-Buchholz and J. R. Galán-Mascarós, J. Am. Chem. Soc., 2015, 137, 11924–11927 CrossRef PubMed.
- Y. Klein, N. Sciortino, C. Housecroft, C. Kepert and S. Neville, Magnetochemistry, 2016, 2, 7 CrossRef.
- N. Pittala, F. Thetiot, C. Charles, S. Triki, K. Boukheddaden, G. Chastanet and M. Marchivie, Chem. Commun., 2017, 53, 8356–8359 RSC.
- W.-B. Chen, Y.-C. Chen, M. Yang, M.-L. Tong and W. Dong, Dalton Trans., 2018, 47, 4307–4314 RSC.
- H. Vahrenkamp, A. Gei and G. N. Richardson, J. Chem. Soc., Dalton Trans., 1997, 3643–3652 RSC.
- S. Dhers, H. L. C. Feltham and S. Brooker, Coord. Chem. Rev., 2015, 296, 24–44 CrossRef.
- C. P. Berlinguette, A. Dragulescu-Andrasi, A. Sieber, J. R. Galán-Mascarós, H.-U. Güdel, C. Achim and K. R. Dunbar, J. Am. Chem. Soc., 2004, 126, 6222–6223 CrossRef PubMed.
- M. Nihei, M. Ui, M. Yokota, L. Han, A. Maeda, H. Kishida, H. Okamoto and H. Oshio, Angew. Chem., Int. Ed., 2005, 44, 6484–6487 CrossRef PubMed.
- M. Nihei, M. Ui and H. Oshio, Polyhedron, 2009, 1718–1721 CrossRef.
- O. Hietsoi, P. W. Dunk, H. D. Stout, A. Arroyave, K. Kovnir, R. E. Irons, N. Kassenova, R. Erkasov, C. Achim and M. Shatruk, Inorg. Chem., 2014, 53, 13070–13077 CrossRef PubMed.
- I. Boldog, F. J. Muñoz-Lara, A. B. Gaspar, M. C. Muñoz, M. Seredyuk and J. A. Real, Inorg. Chem., 2009, 48, 3710–3719 CrossRef PubMed.
- R.-J. Wei, Q. Huo, J. Tao, R.-B. Huang and L.-S. Zheng, Angew. Chem., Int. Ed., 2011, 50, 8940–8943 CrossRef PubMed.
- E. Breuning, M. Ruben, J.-M. Lehn, F. Renz, Y. Garcia, V. Ksenofontov, P. Gütlich, E. Wegelius and K. Rissanen, Angew. Chem., Int. Ed., 2000, 39, 2504–2507 CrossRef PubMed.
- M. Ruben, E. Breuning, J.-M. Lehn, V. Ksenofontov, F. Renz, P. Gütlich and G. B. M. Vaughan, Chem. – Eur. J., 2003, 9, 4422–4429 CrossRef PubMed.
- M. Ruben, U. Ziener, J.-M. Lehn, V. Ksenofontov, P. Gütlich and G. B. M. Vaughan, Chem. – Eur. J., 2005, 11, 94–100 CrossRef PubMed.
- L. H. Uppadine, J.-P. Gisselbrecht, N. Kyritsakas, K. Nättinen, K. Rissanen and J.-M. Lehn, Chem. – Eur. J., 2005, 11, 2549–2565 CrossRef PubMed.
- A. R. Stefankiewicz, G. Rogez, J. Harrowfield, M. Drillon and J.-M. Lehn, Dalton Trans., 2009, 5787–5802 RSC.
- A. R. Stefankiewicz, G. Rogez, J. Harrowfield, A. N. Sobolev, A. Madalan, J. Huuskonen, K. Rissanen and J.-M. Lehn, Dalton Trans., 2012, 41, 13848–13855 RSC.
- M. Steinert, B. Schneider, S. Dechert, S. Demeshko and F. Meyer, Inorg. Chem., 2016, 55, 2363–2373 CrossRef PubMed.
- D.-Y. Wu, O. Sato, Y. Einaga and C.-Y. Duan, Angew. Chem., Int. Ed., 2009, 48, 1475–1478 CrossRef PubMed.
- Y.-T. Wang, S.-T. Li, S.-Q. Wu, A.-L. Cui, D.-Z. Shen and H.-Z. Kou, J. Am. Chem. Soc., 2013, 135, 5942–5945 CrossRef PubMed.
- T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 3865 CrossRef PubMed.
- B. Schäfer, J.-F. Greisch, I. Faus, T. Bodenstein, I. Šalitroš, O. Fuhr, K. Fink, V. Schünemann, M. M. Kappes and M. Ruben, Angew. Chem., Int. Ed., 2016, 55, 10881–10885 CrossRef PubMed.
- M. Steinert, B. Schneider, S. Dechert, S. Demeshko and F. Meyer, Angew. Chem., Int. Ed., 2014, 53, 6135–6139 CrossRef PubMed.
- B. Schneider, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chem., 2012, 51, 4912–4914 CrossRef PubMed.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2013, 49, 1597–1599 RSC.
- R. A. Bilbeisi, S. Zarra, H. L. C. Feltham, G. N. L. Jameson, J. K. Clegg, S. Brooker and J. R. Nitschke, Chem. – Eur. J., 2013, 19, 8058–8062 CrossRef PubMed.
- L. Li, N. Saigo, Y. Zhang, D. J. Fanna, N. D. Shepherd, J. K. Clegg, R. Zheng, S. Hayami, L. F. Lindoy, J. R. Aldrich-Wright, C.-G. Li, J. K. Reynolds, D. G. Harman and F. Li, J. Mater. Chem. C, 2015, 3, 7878–7882 RSC.
- D.-H. Ren, D. Qiu, C.-Y. Pang, Z. Li and Z.-G. Gu, Chem. Commun., 2015, 51, 788–791 RSC.
- F.-L. Zhang, J.-Q. Chen, L.-F. Qin, L. Tian, Z. Li, X. Ren and Z.-G. Gu, Chem. Commun., 2016, 52, 4796–4799 RSC.
- M. P. Shores, C. M. Klug and S. R. Fiedler, Spin-Crossover Materials: Properties and Applications, John Wiley & Sons Ltd, Chichester, 2013, pp. 281–301 Search PubMed.
- M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J.-F. Létard, K. S. Murray, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 2549–2552 CrossRef PubMed.
- Z. Yan, W. Liu, Y.-Y. Peng, Y.-C. Chen, Q.-W. Li, Z.-P. Ni and M.-L. Tong, Inorg. Chem., 2016, 55, 4891–4896 CrossRef PubMed.
- K. Yoneda, K. Adachi, K. Nishio, M. Yamasaki, A. Fuyuhiro, M. Katada, S. Kaizaki and S. Kawata, Angew. Chem., Int. Ed., 2006, 45, 5459–5461 CrossRef PubMed.
- X. Bao, J.-D. Leng, Z.-S. Meng, Z. Lin, M.-L. Tong, M. Nihei and H. Oshio, Chem. – Eur. J., 2010, 16, 6169–6174 CrossRef PubMed.
- M. B. Duriska, S. M. Neville, B. Moubaraki, K. S. Murray, C. Balde, J.-F. Létard, C. J. Kepert and S. R. Batten, ChemPlusChem, 2012, 77, 616–623 CrossRef.
- S. Alvarez, Chem. Rev., 2015, 115, 13447–13483 CrossRef PubMed.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE (2.1), University of Barcelona, Spain, 2013.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef.
- S. Alvarez, D. Avnir, M. Llunell and M. Pinsky, New J. Chem., 2002, 26, 996–1009 RSC.
- J.-M. Lehn, Supramolecular chemistry: concepts and perspectives, VCH, Weinheim, 1995 Search PubMed.
- S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef PubMed.
- H. S. Scott, R. W. Staniland and P. E. Kruger, Coord. Chem. Rev., 2018, 362, 24–43 CrossRef.
- A. J. McConnell, Supramol. Chem., 2018, 30, 858–868 CrossRef.
- K. Nakano, N. Suemura, K. Yoneda, S. Kawata and S. Kaizaki, Dalton Trans., 2005, 740–743 RSC.
- L. J. Kershaw Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem., Int. Ed., 2016, 55, 4327–4331 CrossRef PubMed.
- H. Phan, J. J. Hrudka, D. Igimbayeva, L. M. Lawson Daku and M. Shatruk, J. Am. Chem. Soc., 2017, 139, 6437–6447 CrossRef PubMed.
- S. Rodríguez-Jiménez, M. Yang, I. Stewart, A. L. Garden and S. Brooker, J. Am. Chem. Soc., 2017, 139, 18392–18396 CrossRef PubMed.
- A. Kimura and T. Ishida, ACS Omega, 2018, 3, 6737–6747 CrossRef.
- S. Rodríguez-Jiménez, A. S. Barltrop, N. G. White, H. L. C. Feltham and S. Brooker, Inorg. Chem., 2018, 57, 6266–6282 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cs00835j |
| ‡ Equal contributions from these authors. |
| This journal is © The Royal Society of Chemistry 2018 |