
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-boryl mediated organic synthesis
David
Hemming
a,
Russell
Fritzemeier
b,
Stephen A.
Westcott
 c,
Webster L.
Santos
*b and
Patrick G.
Steel
*a
c,
Webster L.
Santos
*b and
Patrick G.
Steel
*a
aDepartment of Chemistry, University of Durham, Science Laboratories South Road, Durham DH1 3LE, UK. E-mail: p.g.steel@durham.ac.uk
bDepartment of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: santosw@vt.edu
cDepartment of Chemistry and Biochemistry, Mount Allison University, Sackville, NB E4L 1G8, Canada
First published on 12th September 2018
Abstract
Organoboron compounds are valuable synthetic intermediates that find application in a diverse variety of processes including both C–X and C–C bond-forming transformations. This has been achieved by using a variety of boron derivatives. Of these, boronate esters are probably the most versatile and, reflecting this, methods for the generation of boronate esters are of considerable current interest. Given the mild reaction conditions, good functional group tolerance, and low cost of the metal catalyst, the use of copper-boryl reagents is particularly attractive. In this review, methodologies in copper-boryl chemistry are discussed and the many different transformations possible are surveyed.
 David Hemming | David Hemming studied chemistry at the University of Edinburgh. During his degree, he undertook a year-long industrial placement at GlaxoSmithKline. He then joined the Steel group, where he investigated synthetic applications of copper-boryl complexes. He obtained his PhD from Durham University in 2017. |
 Russell Fritzemeier | Russell Fritzemeier is currently pursuing his PhD degree at Virginia Tech in Blacksburg, Virginia, where he also earned his BS in Biochemistry in 2014. He currently works under the guidance of Professor Webster Santos in the development of novel transition metal-free borylation protocols. |
 Stephen A. Westcott | Steve Westcott was born in the sixties somewhere around Tecumseh and received his PhD from the University of Waterloo under the joint supervision of Drs Todd B. Marder (now at Universität Würzburg) and R. Tom Baker (now at the University of Ottawa) working on metal-catalyzed hydroborations. He was an NSERC PDF, where he spent one year at Emory University in Atlanta with Dr Lanny Liebeskind, and more than one year working with Dr Maurice Brookhart at the University of North Carolina at Chapel Hill, NC. He has been at Mount Allison University since August 1995 and is currently a Canada Research Chair in Boron Chemistry. His research interests include catalysis and the synthesis and development of biologically active boron compounds. |
 Webster L. Santos | Webster Santos received his PhD in 2002 from the University of Virginia. After training as an NIH postdoctoral fellow with Greg Verdine at Harvard University, he began his independent career in 2006 as an Assistant Professor of Chemistry at Virginia Tech. In 2018, he was promoted to full professor as the A. C. Lilly Professor of Chemistry. His current interests include the development of stereoselective borylation reactions, medicinal chemistry campaigns towards sphingosine kinase and mitochondrial uncouplers, and targeting RNA structures. |
 Patrick G. Steel | Patrick Steel undertook his undergraduate and post-graduate training with Prof Jim Thomas at the University of Oxford. Following a NATO-SERC postdoctoral fellowship with Prof Gilbert Stork at Columbia University NY, he joined the staff at Durham University where he is currently a professor of organic chemistry and chemical biology. His group study problems in organic synthesis (iridium and copper catalysed borylation methodologies) and chemical biology with particular interests in neglected tropical diseases (notably leishmaniasis) and plant chemical biology (herbicide resistance). He is currently an elected council member of the Royal Society of Chemistry, Chemistry Biology Interface Division. |
1 Organoboron compounds
Organoboron compounds are highly important synthetic intermediates that find application in a diverse variety of synthetic processes including both C–X and C–C bond forming transformations exemplified by the Suzuki–Miyaura cross coupling and 1,2-metallate rearrangements including aminations and oxidation reactions (Scheme 1).1–3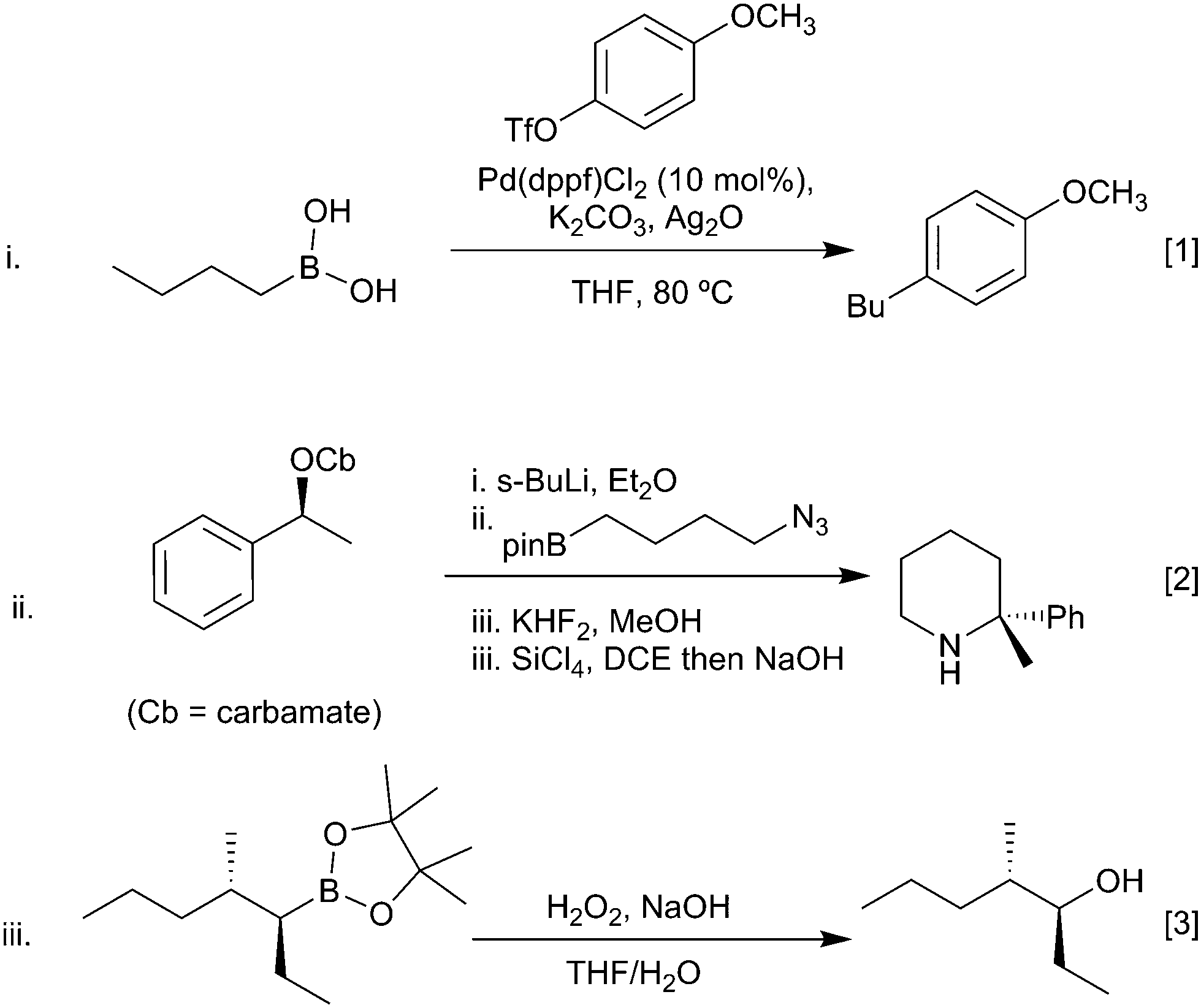 | ||
| Scheme 1 Selected applications of organoboron compounds in synthesis. | ||
These have employed a variety of boron derivatives including alkyl, vinyl and aryl boranes, boronate esters and related compounds. Of these, the boronate esters are probably the most versatile being non-toxic, tolerant to air and water, whilst reacting under relatively mild conditions to produce an inert, benign and easy to separate by-product. Reflecting this, methods for the generation of boronate esters are of considerable current interest. Classically, this is achieved via the reaction of hard organometallic reagents with borate esters or through hydroboration strategies. Such methods, however, have relatively limited substrate scope and, more recently, milder catalysed C–H and C–X borylation strategies have emerged.4–6 Of these, processes mediated by a copper-boryl species are particularly prominent enabled by mild reaction conditions, good functional group tolerance, and low cost of the metal catalyst. In this review, developments in this area are surveyed. Space precludes a comprehensive discussion and we have attempted to focus on aspects that best illustrate the many different possibilities enabled by using these reagents in synthesis.
1.1 Boron reagents
In parallel with the breadth of chemistry possible using copper-boryl reagents, there is a diverse range of boron reagents that have been employed in these transformations. These can be categorised as boranes or boronates and are used as both mono and diboron reagents. The latter can be further subdivided into symmetrical, unsymmetrical and mixed valence diboron species. More recently, preactivated diboron reagents have been developed.7–9 These contain mixed sp2–sp3 hybridised boron atoms in which coordination of a ligating group to one of the boron atoms lengthens the boron–boron bond and, therefore, aids transfer of a boryl group to the catalyst, removing the requirement for an alkoxide base in the reaction mixture, vide infra.7 The structures of some of the most common reagents are shown below (Scheme 2) and shall henceforth be referred to by their abbreviations. The recent commercialisation of many of these diboron reagents has helped to expand the scope of borylation chemistry.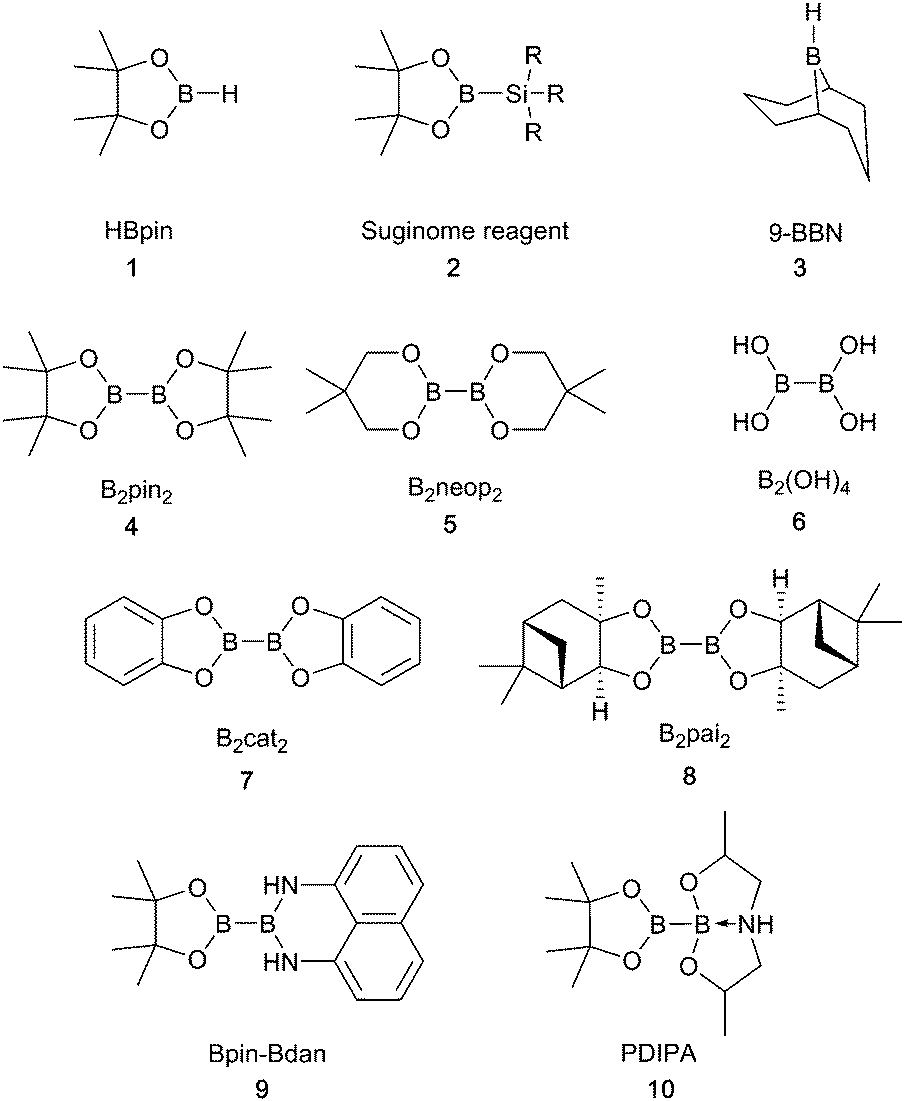 | ||
| Scheme 2 Structures of common boron reagents. | ||
2 Copper-boryl complexes
The first reported examples of copper-boryl complexes in synthesis were the conjugate additions of a Bpin moiety to an enone described by Miyaura and Hosomi, who employed a CuCl/KOAc or a copper(I) phosphine catalyst and a diboron reagent mixture, respectively (Scheme 3).10–12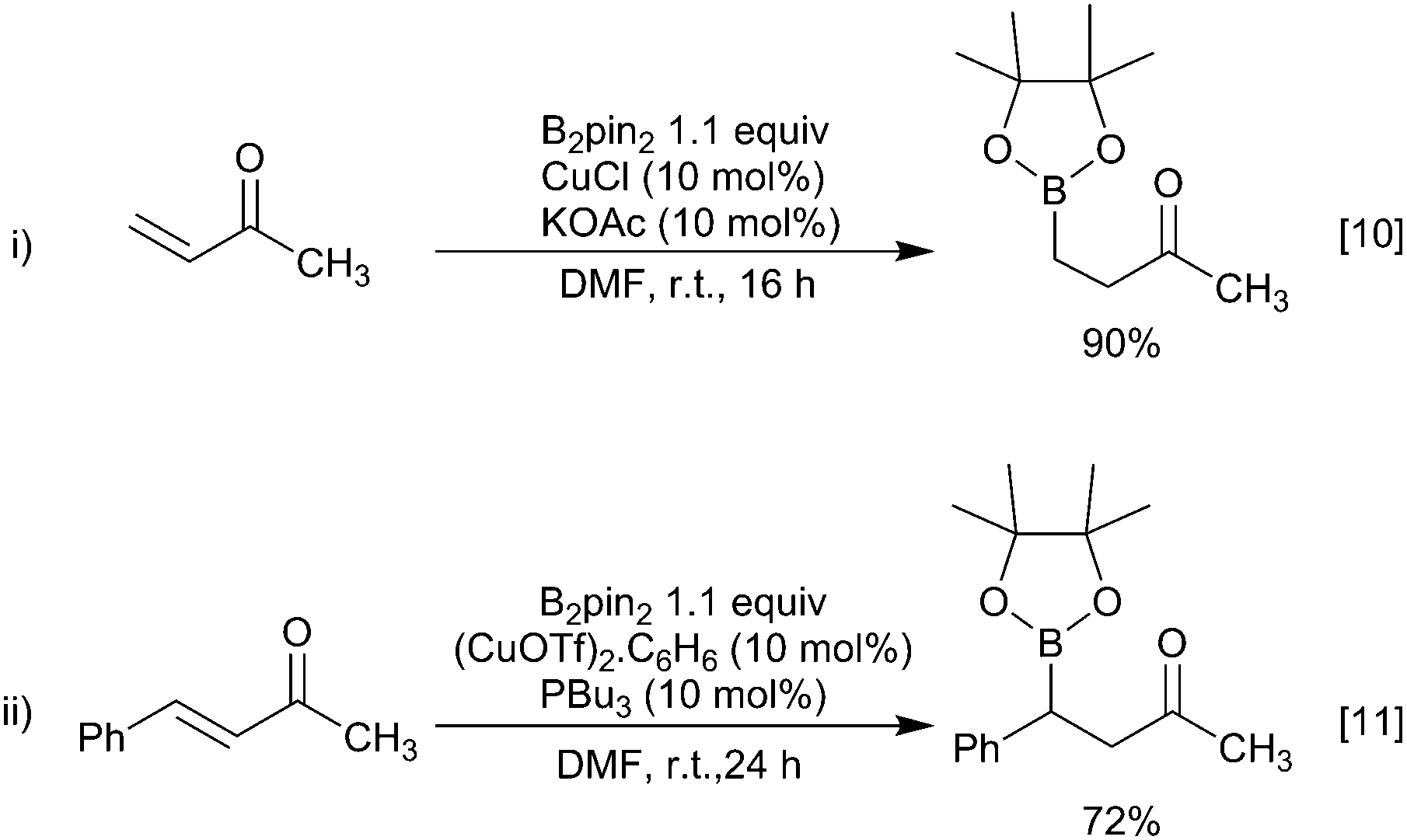 | ||
| Scheme 3 Initial reports of reactions mediated by copper-boryl complexes. | ||
Since these initial reports, the field has expanded rapidly and a range of transformations have been described. Whilst these have been postulated to proceed through the intermediacy of a copper-boryl complex, isolated examples of these species are relatively rare. Predominantly, it is for NHC bound copper-boryls that molecular structures have been reported although a very recent report describes phosphine bound complexes.13–17 For most NHC complexes, a linear arrangement, with the Bpin moiety bound directly opposite to the NHC ligand, was observed (Scheme 4). DFT and in situ11B NMR studies also provide some insight into their formation, with the 11B NMR signal for a copper bound boryl moiety observed at δ = 42 ppm.18–21 It has been calculated that the Cu–B σ* molecular orbital is high in energy, with the filled Cu–B σ-bond being more likely to interact with the substrate consistent with the model of a nucleophilic boron reagent.22 However, the recent studies by Kleeburg suggest that whilst monomeric copper-boryl species cannot be excluded such analysis does require the consideration of higher aggregates which challenge classical mechanistic interpretations.
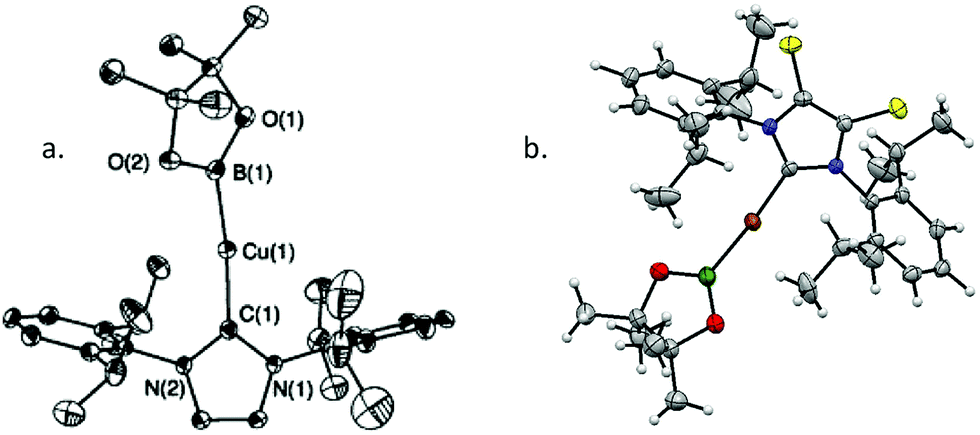 | ||
| Scheme 4 Structures of representative examples of (NHC)CuBpin complexes showing near linear C–Cu–B arrangement. (a) Reprinted with permission from Laitar et al., J. Am. Chem. Soc., 2005, 127, 17196. Copyright 2005 American Chemical Society; (b) Reprinted with permission from Semba et al., Chem. – Eur. J., 2013, 19, 7125. Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Accepting this reservation, in the majority of borylation reactions, the active catalysts are generally formed in situ by the reaction of a copper(I) salt with an alkoxide base.23 A metathesis reaction with the diboron reagent then occurs, generating the active copper-boryl complex. Coordination of the substrate and insertion (Scheme 5, shown with an alkene) then occurs, affording a new borylorganocopper intermediate. Following reaction with an electrophile, ligand exchange with the alkoxide base regenerates the copper alkoxide complex.
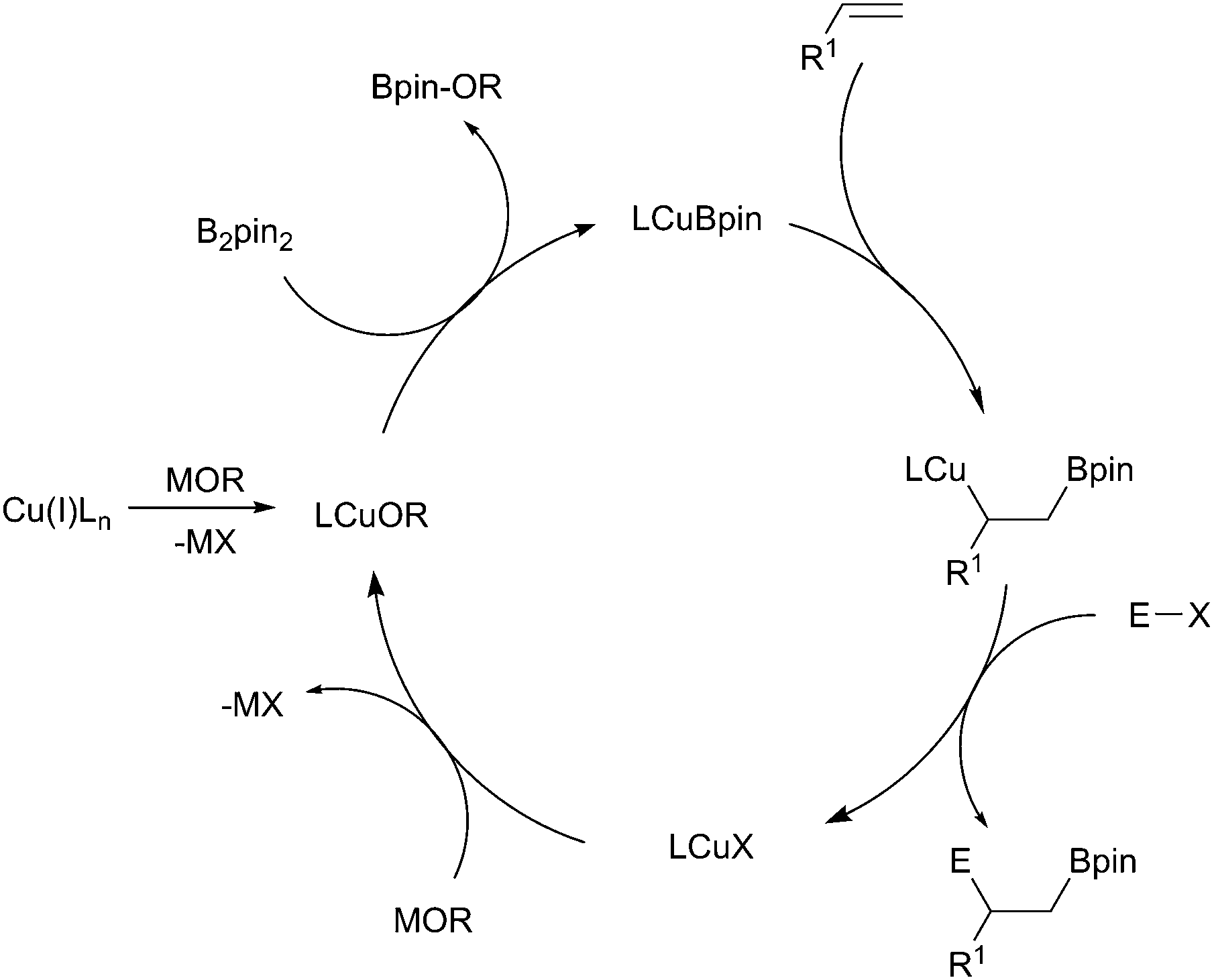 | ||
| Scheme 5 General catalytic cycle for copper-boryl mediated reactions with unsaturated systems. | ||
3 Synthetic transformations mediated by copper-boryl complexes
The mode of reaction of the copper-boryl complex is largely defined by the nature of the substrate structure and this review is organised along these lines, focussing on issues of chemo-, regio- and stereoselectivity. This has been achieved using a diverse range of ligands, with phosphines and NHCs being the most commonly employed. As indicated above, the ultimate boron containing product is readily converted into a range of functional groups and this review will focus on the steps involved in the formation of the organoboron intermediate and not discuss this subsequent step as this has been widely reviewed elsewhere.3.1 Reactions of copper-boryl species with C–C multiple bonds
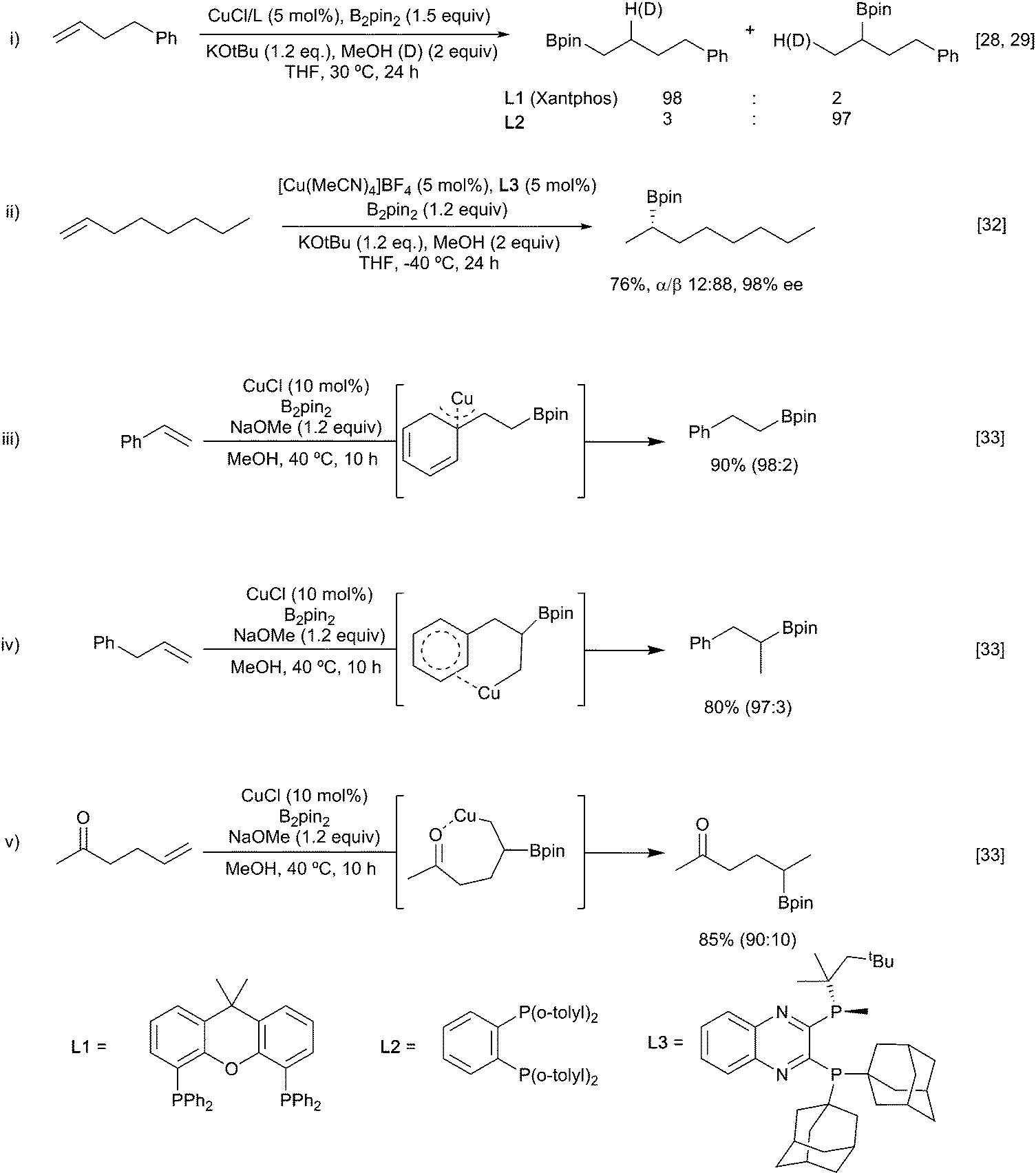
Alkenes, allenes and alkynes have all been used as substrates in reactions with copper-boryl complexes. These reactions involve complexation of the π-system to the copper-boryl species, followed by boryl transfer to afford a borylated organocopper intermediate, which is then trapped by a variety of electrophiles (Scheme 5). Consequently, these processes can be most easily classified by the nature of the overall transformation: hydroboration (E = HX, protonation of the copper complex); diboration (E = B2(OR)4); carboboration (E = R′–X); aminoboration (E = XNR2) etc.With non-polar multiple bond containing substrates, the key issue is the control of regioselectivity. As reactions with simple unactivated alkenes are surprisingly rare, it is difficult to make generic comments as to the intrinsic regioselectivity. For terminal alkenes, reaction with a copper Xantphos complex afforded formal anti-Markovnikov borylation (Scheme 6i)28 whilst use of more sterically demanding ligands, e.g. dppbz derived phosphine L2, lead to the opposite regiochemistry.29,30 In the latter case enantioselection becomes an issue and the use of suitably designed chiral sterically demanding ligands e.g.L3 can permit good levels of absolute control (Scheme 6ii).31,32 Interestingly, the high steric bulk in L3 precludes reactions with internal alkenes enabling crude olefin feedstocks to be used.
 | ||
| Scheme 6 Regiocontrol in the copper catalysed borylation of alkenes. | ||
However, for most substrates, regiochemical control is enabled by inherent structural features. For example, Wen and co-workers reported the copper-catalysed borylation of allyl and vinyl arenes in which the Bpin group added to the terminal position in the case of vinyl arenes (Scheme 6iii).33 In parallel with this observation, dienes preferentially borylate at a terminal position to afford to form the conjugated allyl copper intermediate. As discussed below, Section 3.1.3, for non-symmetrical dienes such as isoprene further regiochemical questions arise. Conversely, allyl arenes were borylated at the more substituted position of the double bond (Scheme 6iv). Such observations could be attributed to the formation of an η3-stabilised benzyl copper complex (Scheme 6iii) or by coordination of the copper to the aromatic π-system, respectively (Scheme 6iv). Further evidence for this coordination directed mode of addition was seen in the regioselective reaction of hex-5-en-2-one (Scheme 6v).
Whilst these substrate-based methods are effective, the ligand/reagent-based control methods, elegantly demonstrated by Ito (Scheme 6i), are inherently more attractive. Enantioselectivity is a further challenge. In 2009, Hoveyda and co-workers described the enantioselective hydroboration of substituted alkenes in the presence of the chiral N-heterocyclic carbene (NHC) ligand L4 to give a range of chiral alkyl boronates in good yields and high ee (Scheme 7).34 This process, which utilises both substrate control (formation of a benzylcopper intermediate) and ligand controlled enantioselectivity, remains a benchmark for this transformation.
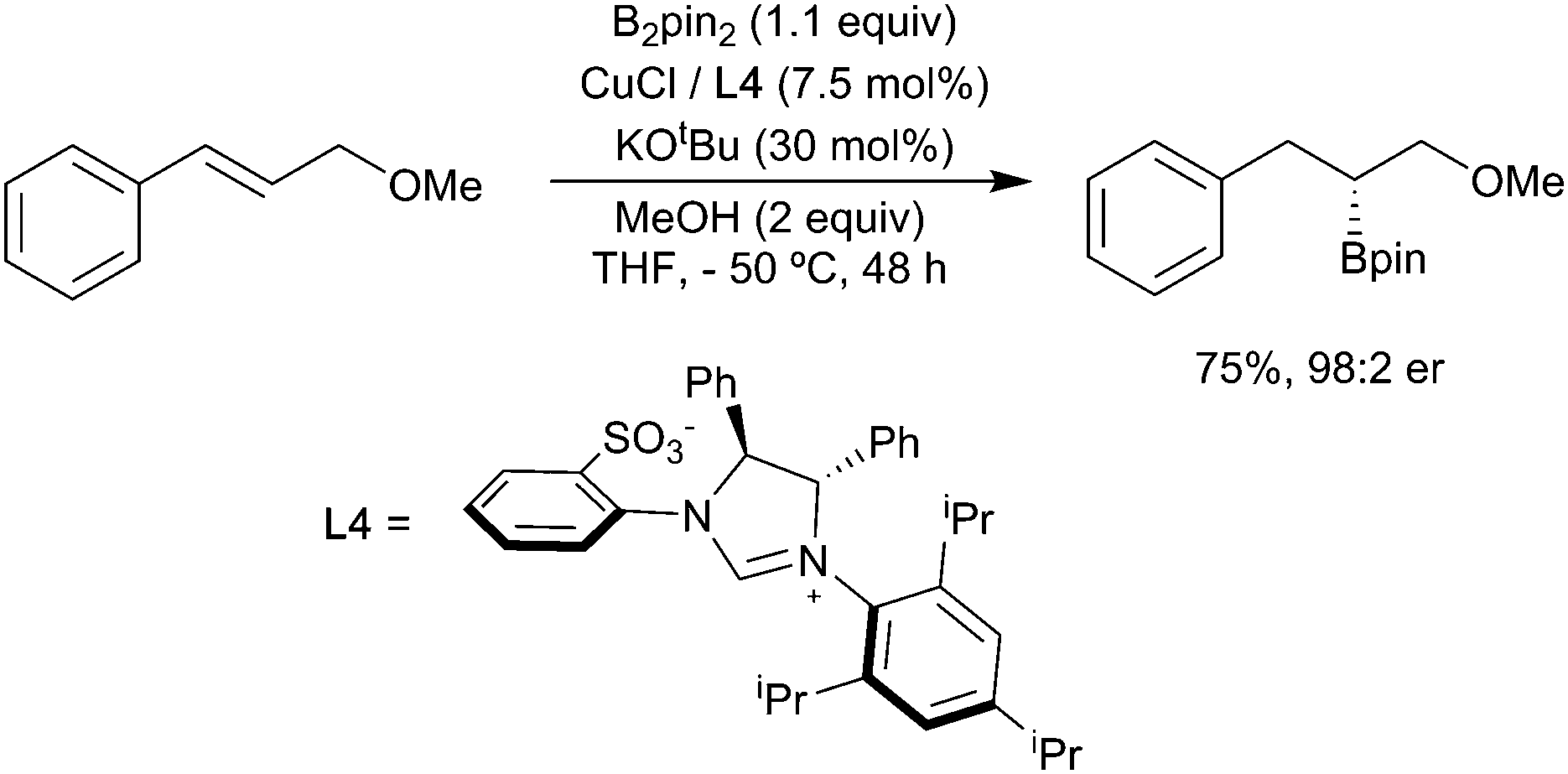 | ||
| Scheme 7 Enantioselective hydroboration reported by Hoveyda and co-workers. | ||
Dienes can also be borylated enantioselectively, with the first copper-catalysed example being reported in 2010.35 In a similar fashion to the styrene systems discussed above, temperature-controlled experiments showed that regiocontrol arose through the kinetically favoured formation of the allylcuprate intermediate A at low temperature via a syn addition across one of the double bonds. SE2′ protonation of this affords the observed allylboronate product 11. At room temperature, this intermediate rapidly isomerised to a more stable 1,4 boryl copper complex B which, on SE2′ protonation, leads to isomeric borylated cyclopentene 12 (Scheme 8).
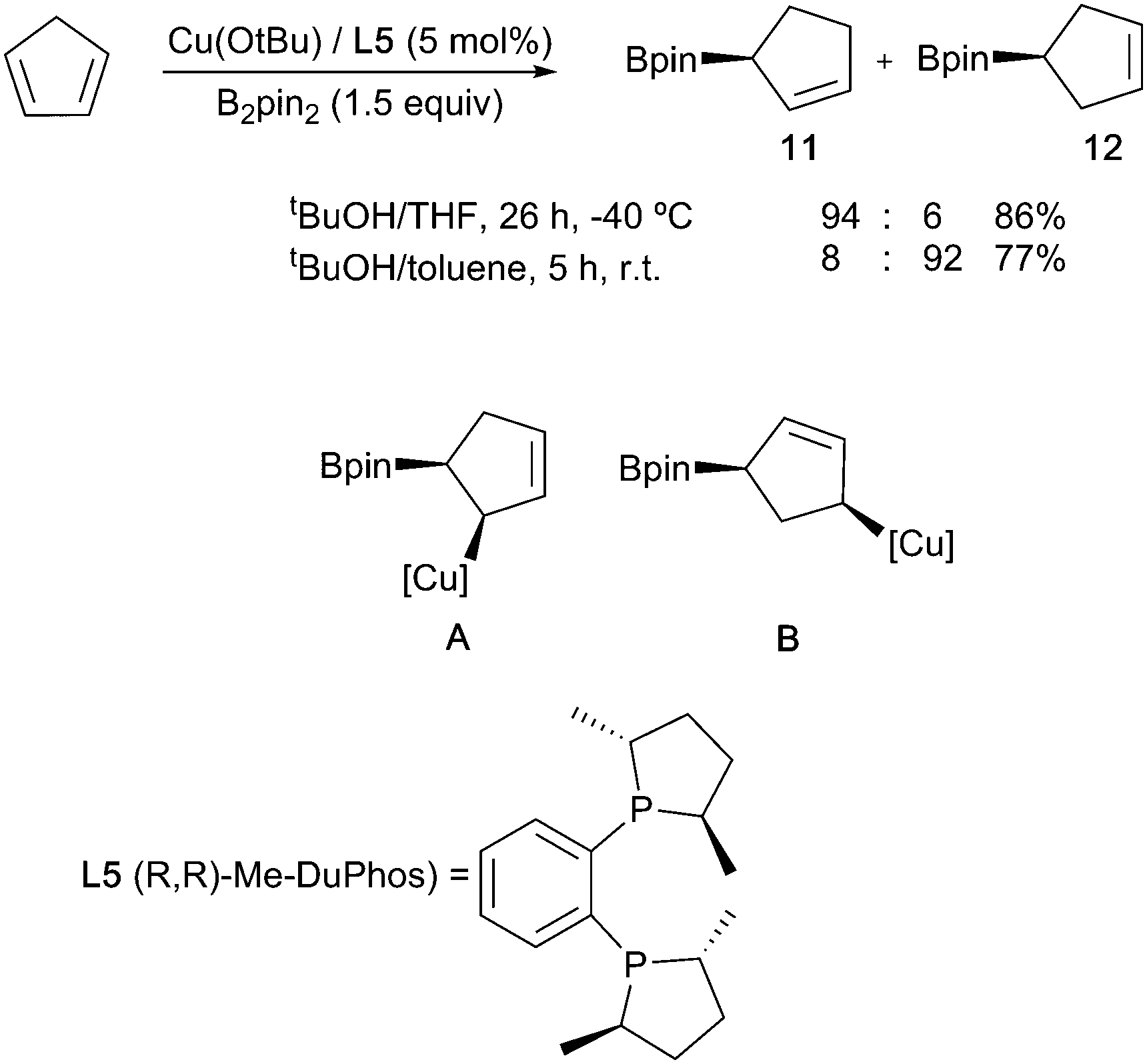 | ||
| Scheme 8 Enantioselective temperature-controlled diene borylation. | ||
A particularly interesting example of enantioselective diene hydroboration is the reaction of cyclic dieneamides, generated through the reduction of pyridinium and quinolinium salts enabling the synthesis of a wide range of chiral cyclic amines (Scheme 9).36,37 As above, the formation of the more stable allyl copper intermediate appears to be a key determinant in the regioselectivity of the process.
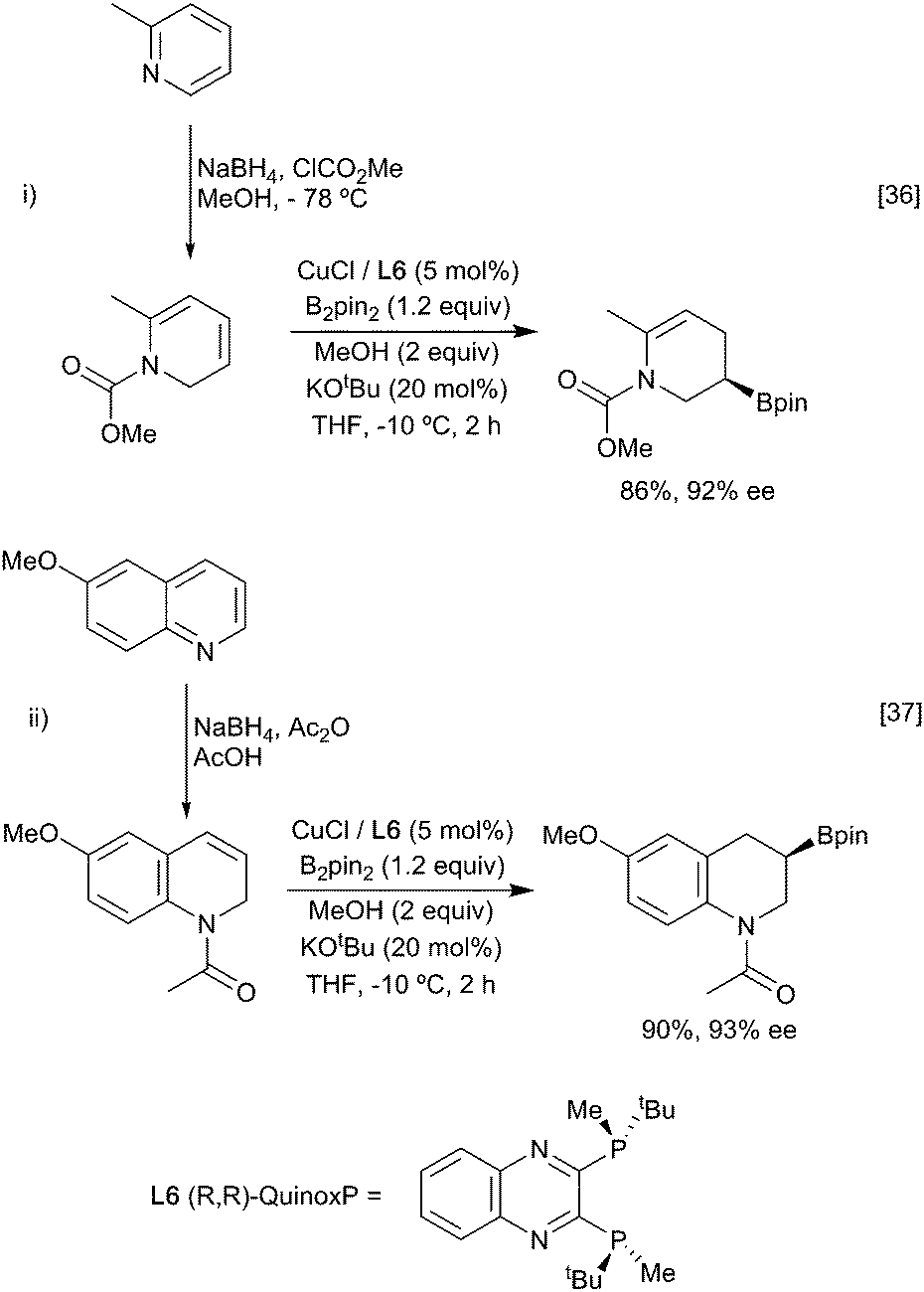 | ||
| Scheme 9 Formation of chiral borylated amines from amino-dienes. | ||
Allyl-copper species are also the favoured intermediates generated in the reaction of a copper-boryl reagent with an allene.38 Regioselective hydroboration of aryl allenes is possible with either a benzylic or allyl copper intermediate being generated depending on the choice of ligand used in conjunction with a copper(I) catalyst (Scheme 10i).39 Alternatively, varying the nature of the boron source allows selection between vinyl and allyl borane products according to the nature of the reactive copper species generated (copper hydride or copper-boryl, respectively) (Scheme 10ii).40
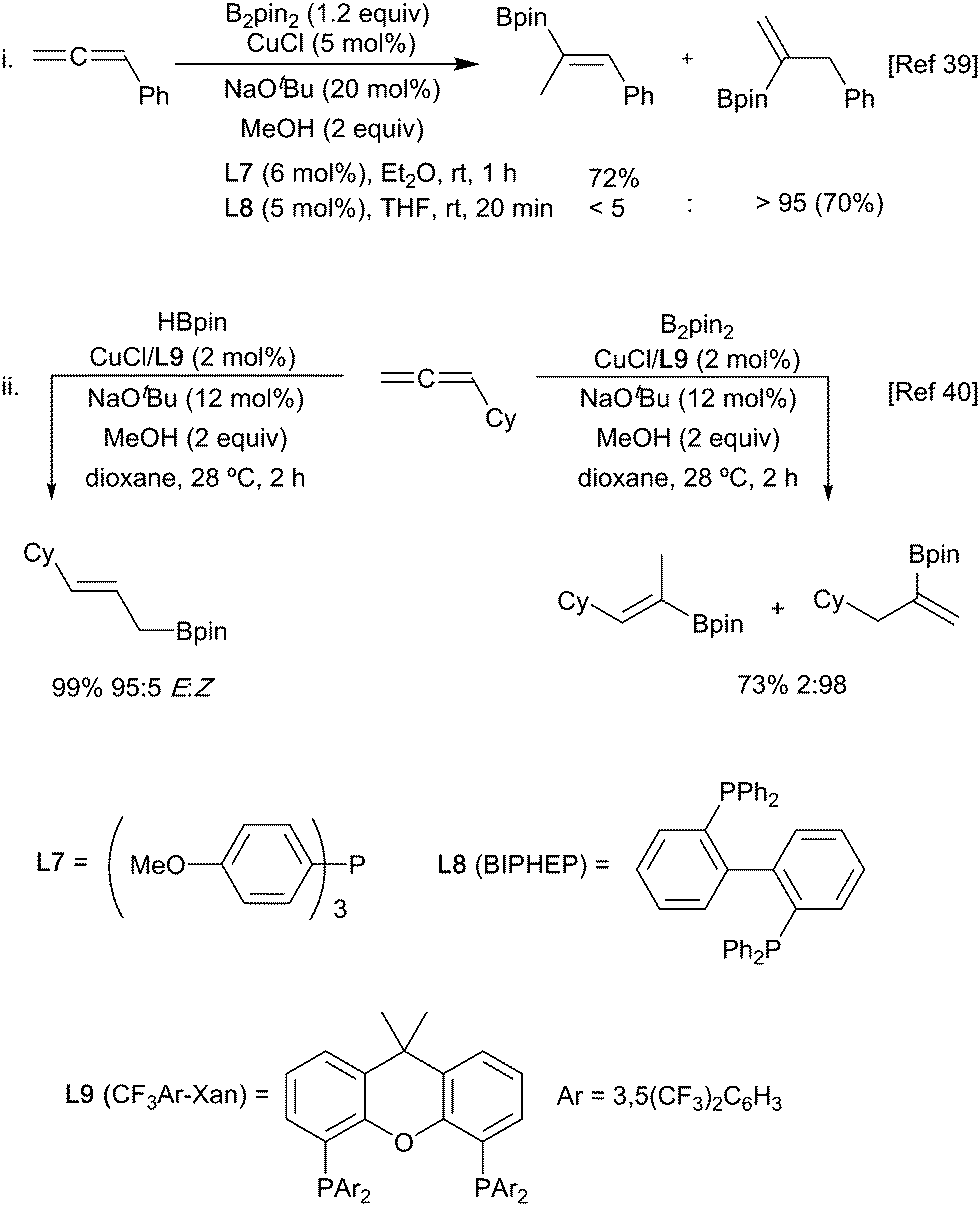 | ||
| Scheme 10 Hydroboration of allenes with different ligands and boryl sources. | ||
Akin to the hydroboration of alkenes, copper-catalysed borylation of alkynes, first reported by Miyaura in 2001,12 is now well established.27,41 Good regioselectivities can be achieved for both simple terminal alkynes and more challenging internal alkynes, with a number of strategies having been described.42 As with the other functional groups discussed above, substrate-based approaches provide the simplest method for achieving regiocontrol.43,44 In general, as with alkenes, regioselectivity in the copper-catalyzed alkyne borylation is more effectively achieved by electronic rather than steric factors. For example, a range of functionalized propargylic silanes undergo a regioselective copper-boryl mediated reaction invoking an α-silyl vinyl cuprate (Scheme 11).45
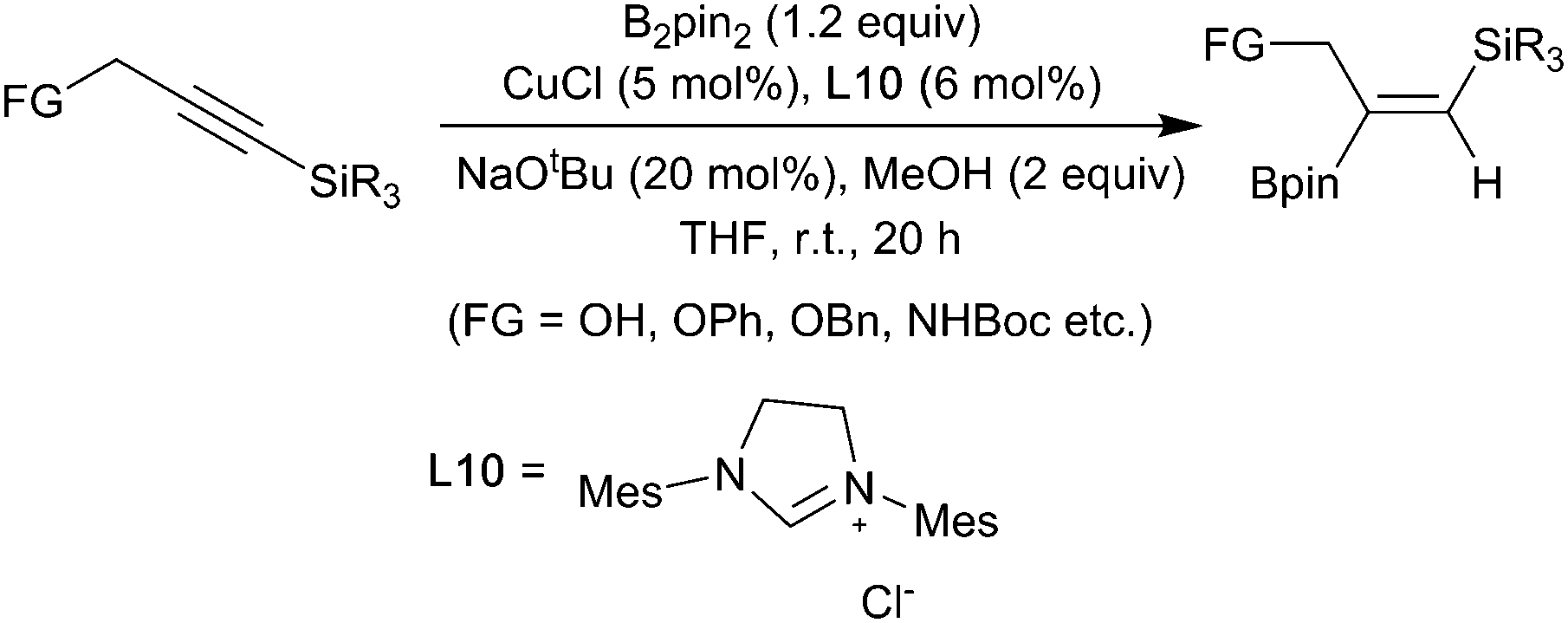 | ||
| Scheme 11 Hydroboration of propargylic silylalkynes. | ||
As with the approaches taken for allenyl substrates, different regioisomers can be obtained from the hydroboration of alkynes depending on whether pinacolborane (HBpin) or bis(pinacolato)diboron (B2pin2) is used as the boryl source (Scheme 12i). As noted above, it was proposed that these reactions were mediated by either a copper hydride or copper-boryl species, respectively.46–48
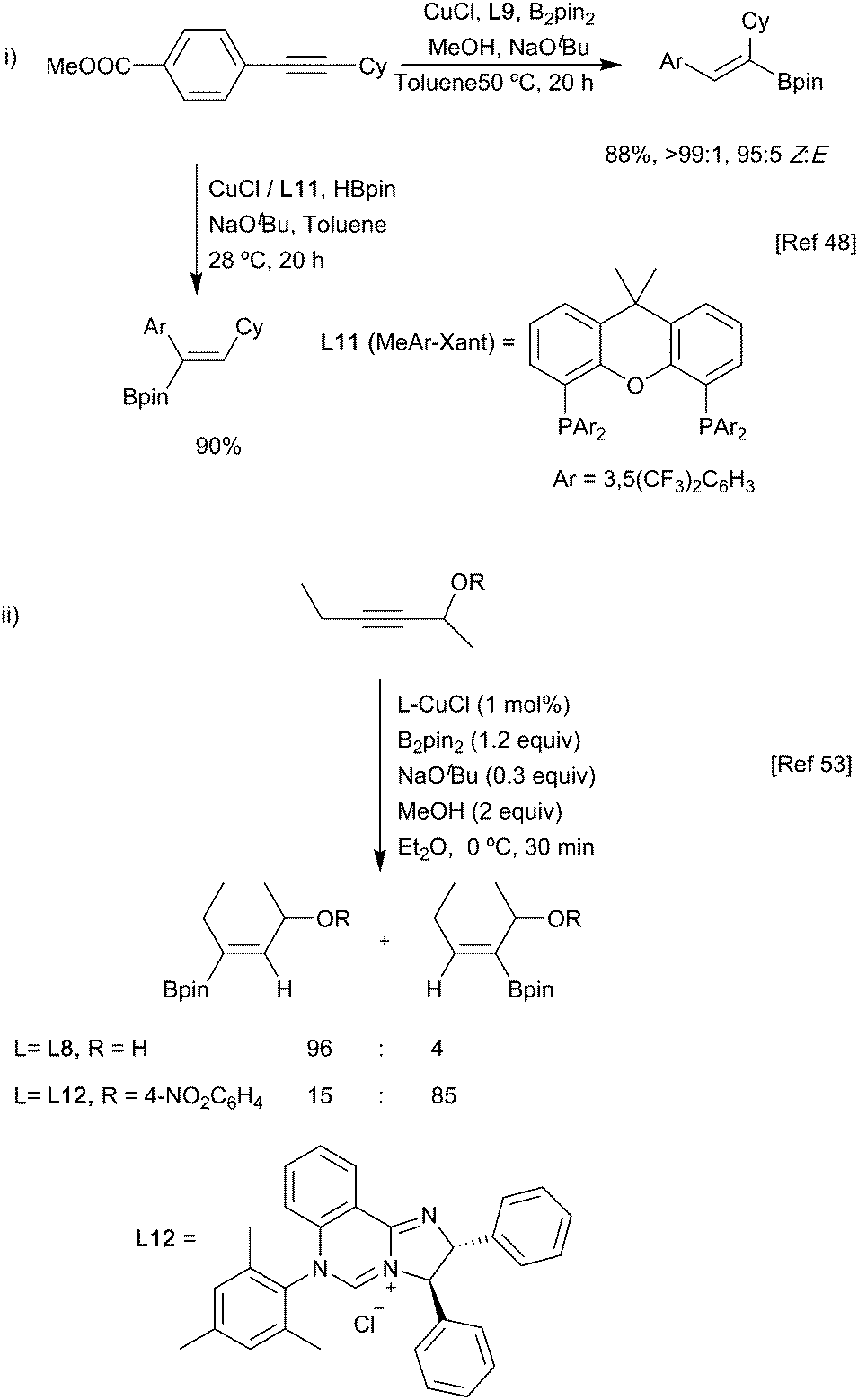 | ||
| Scheme 12 Regiocontrolled alkyne hydroboration. | ||
Ligand-based strategies that provide control are also possible.49–52 One notable example is in the borylation of propargylic alcohols, whereby α- or β-borylation could be controlled depending on the NHC ligand and its interaction with the substrate.53 With a simple NHC, electronic effects in the substrate were the main determinant of regiochemistry and gave β-borylation; with a more sterically hindered NHC ligand L12, the bulk of the catalyst surpassed this electronic bias as the main influence on regiochemistry, yielding α-borylated products (Scheme 12ii).
Although somewhat less developed than Cu(I)-mediated transformations, other copper species can also be used to carry out similar ‘hydroboration’ type reactions but with the benefit of increased stability of the catalyst and ligands.54,55 For example, Cu(II)-catalysed borylation of terminal alkenes and alkynes occurs efficiently to afford β-borylated products with high levels of regioselectivity.56 With the drive towards more sustainable processes, reactions in water have become more attractive and copper-catalysed hydroboration is no exception.57 Building on earlier reports of enone borylation described by Santos and others (vide infra),58,59 there have been reports of alkyne hydroboration under aqueous conditions in which hydroxide rather than alkoxide bases can be used (Scheme 13).60,61
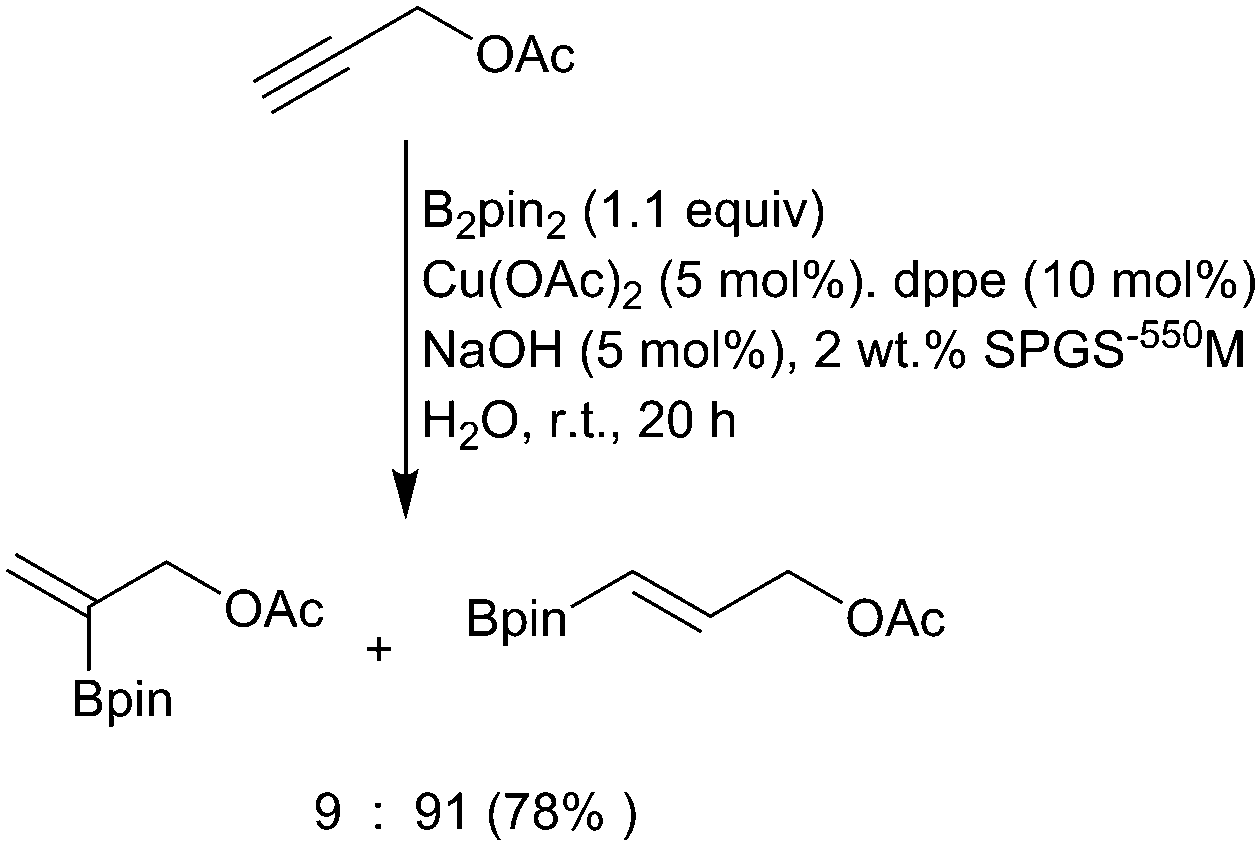 | ||
| Scheme 13 Cu(II)-Catalysed hydroboration of propargylic alkynes in water. | ||
For example, reaction of the borylated cuprate derived from an allene with aldehydes (Scheme 14i)62 proceeds via the more stable primary ‘allyl copper’ intermediate to afford, after metallo-ene reaction of the vinyl borane, the corresponding syn aldol following an oxidative work up step. Imines behave similarly leading to the anti homoallylic amine (Scheme 14ii).63,64 In these reactions with carbonyl based electrophiles, selectivity for activation of the alkene versus the competing direct 1,2-addition of the copper-boryl to the carbonyl group (Section 3.2.2) is key. Control over which pathway is favoured is possible through careful ligand selection.62 For example, with a simple NHC affording extensive 1,2-addition to an aldehyde whilst the less Lewis basic BINAP leads to efficient carboboration of the allene (Scheme 14i). Ketones, being less electrophilic, generally proceed through the carboboration pathway, although this is still substrate dependent. Preferential carboboration to afford the terminal cuprate and metallo-ene reaction is also observed with dienyl derived allyl copper intermediates as observed in the reaction with phosphinoimines (Scheme 14iii).65,66 Whilst boryl-copper addition to enynes occurs regioselectively at the alkene component to afford a propargylic cuprate this rapidly rearranges to generate an allenyl metal intermediate. Subsequent metallo-ene reaction with an aldehyde leads to the homoallylic alcohol (Scheme 14iv).67 Stabilisation of the cuprate can also be via coordination of a proximal heteroatom, as exploited in the intramolecular borylative cyclisation of allenyl ketone. Here, initial boryl cupration with B2neop2 affords the chelated primary allyl copper reagent which following the metallo-ene reaction gives the observed decalinol (Scheme 14v).68 The necessity for an anion stabilising entity is not absolute as simple alkenes, and alkynes have been used to initiate these processes. For example, γ- and ∂-alkenyl ketones react under CuCl/Xantphos (L1) conditions to selectively give cyclobutanols and cyclopentanols, respectively (Scheme 14vi) when an aryl ketone is used as a substrate, although alkyl and alkenyl analogues gave complex mixtures. Presumably the carbonyl group can assist in stabilising the initially formed boryl copper complex in a 6-membered ring chelate prior to ring closure.69 Enones are viable partners for reaction with the organocopper intermediate and can react by both 1,2 or 1,4-conjugate addition pathways depending on the relative reactivity of the electrophilic carbon atoms and any steric constraints (Scheme 15). Consistent with this, enoates and dienoates, having a less electrophilic carbonyl group, react by 1,4- or 1,6-conjugate addition pathways, respectively to enable complex structures to be rapidly assembled in a multicomponent fashion (Scheme 15iii).66,70,71
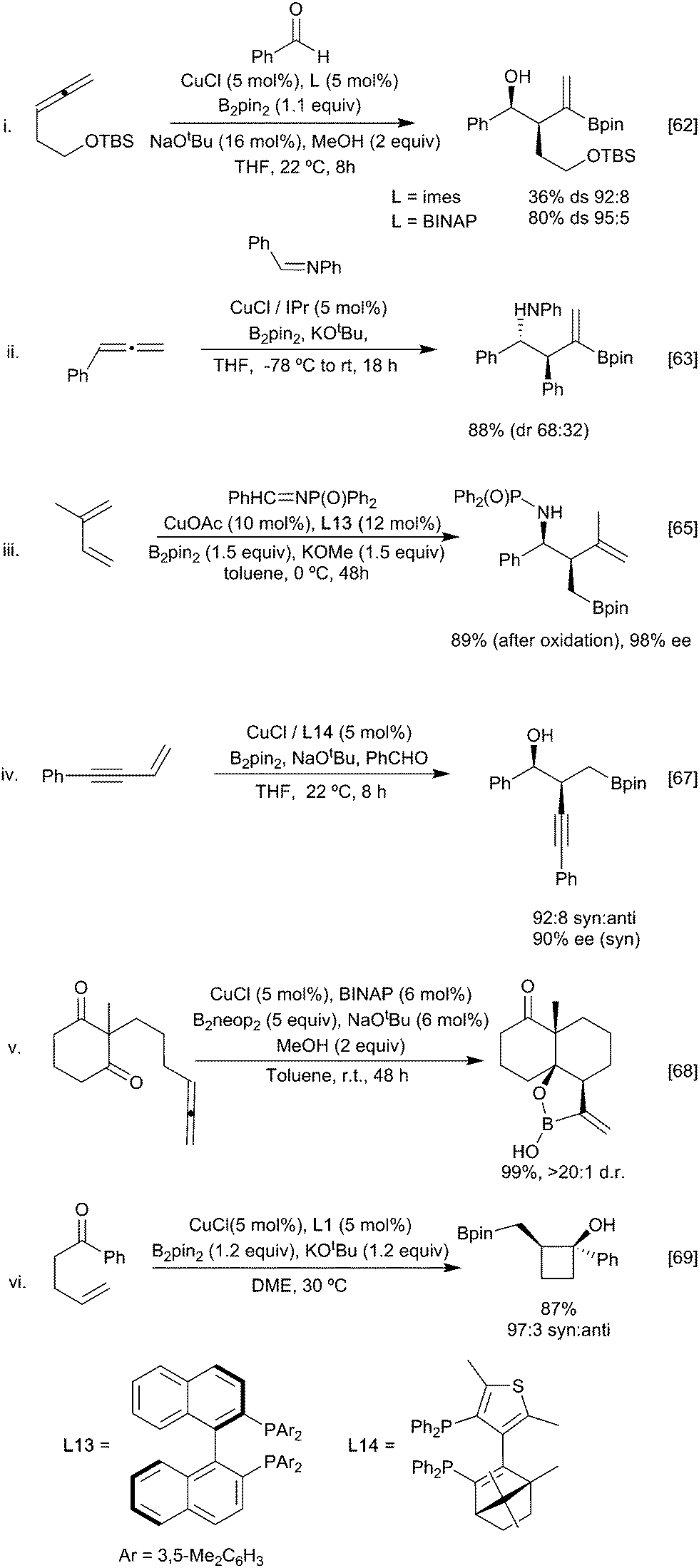 | ||
| Scheme 14 Carboboration via trapping with a carbonyl compound. | ||
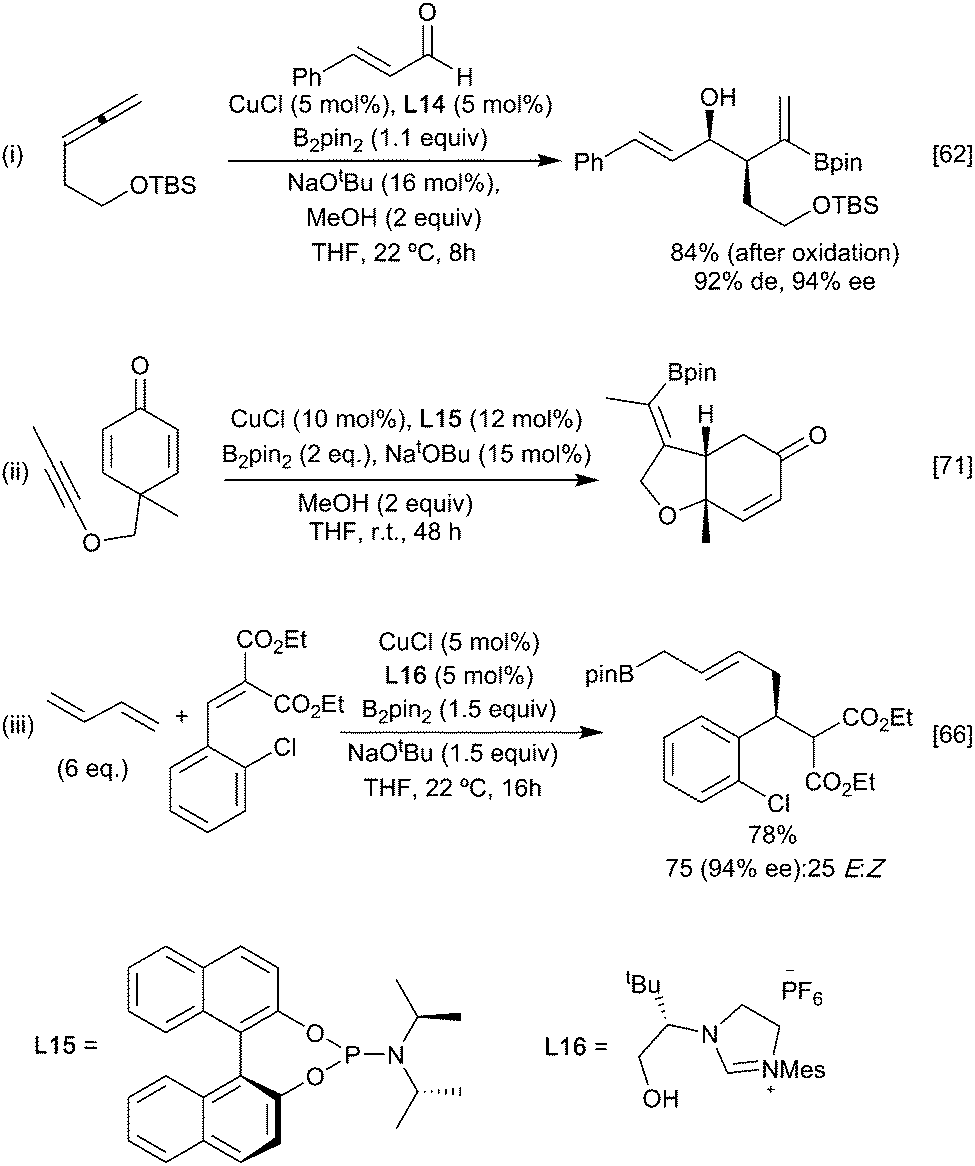 | ||
| Scheme 15 Carboboration with enones. | ||
Carboboration involving capture of the initial boryl copper species with sp3 (alkyl) electrophiles is also feasible although, again, there is a possibility for competitive direct borylation of the alkyl ‘halide’ (see Section 3.3). Higher yields have traditionally been associated with three structural features: a more stable organocopper intermediate, more reactive methyl, allylic and benzylic electrophiles,72–74 and an intramolecular ‘alkylation’ step.75–77 For example, allene-derived boracuprates can be coupled with allylic phosphates in an SN2′ process (Scheme 16i), and a range of cycloalkyl boronate esters (Scheme 16ii–iv) have been synthesised by the intramolecular bora-alkylation of vinylsilanes and styrenes, in which the anion stabilising group facilitates and directs the boracupration step.77–79 A larger range of primary alkyl halides can be used in intermolecular alkylation of butenyl ethers wherein the heteroatom can coordinate and stabilise the organocopper intermediate (Scheme 16v).80 Interestingly, through choice of ligand (Xantphos L1 or Cy-Xantphos L18), both the α- and β-borylated regioisomers could be obtained from the same starting material. DFT calculations suggest that the regioselectivity is determined during the addition of the alkene to the copper-boryl species.81 Cy-Xantphos, which has a larger steric demand than Xantphos, requires that the less hindered, terminal, end of the alkene orientates closer to the ligand in the active species. Conversely, with Xantphos the alkene orients to minimise unfavourable interactions with the Bpin substituents leading to the alternative product. Ligand induced enantioselectivity is also possible with a recent report describing the enantioselective methyl borylation of non-stabilised alkenes suggesting that the scope of this transformation will continue to grow (Scheme 16vi).82
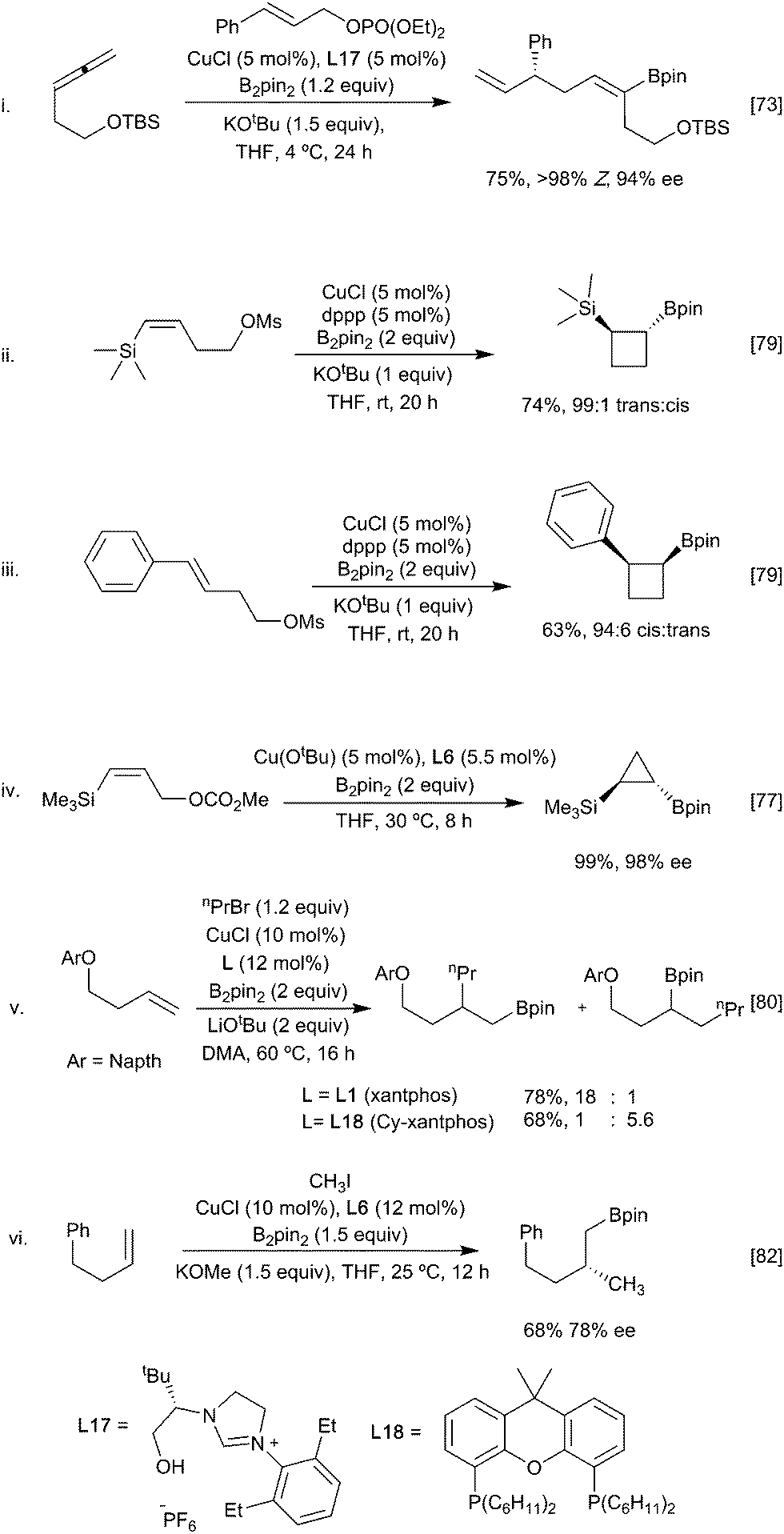 | ||
| Scheme 16 Alkylborylation of alkenes. | ||
Alkynes being more reactive do not require the stabilisation to promote the addition of the copper-boryl reagent. A broad range of alkyl (and aryl) halide electrophiles can be coupled with the initially formed vinyl copper complex, although the use of secondary alkyl electrophiles remains challenging.83,84 In a similar fashion as described for alkenes above, sterically derived, ligand-controlled regioselective bora-alkylations are possible starting from terminal and non-symmetrical internal alkynes (Scheme 17).52,85 With terminal alkynes the use of the bulkier borane B2(pai)28 in conjunction with DMAP as a ligand leads to enhanced selectivity for the terminal vinyl boronate product. When coupled with a subsequent hydrogenation step, alkyne carboboration provides an alternative route to regioselective intermolecular alkene carboboration.
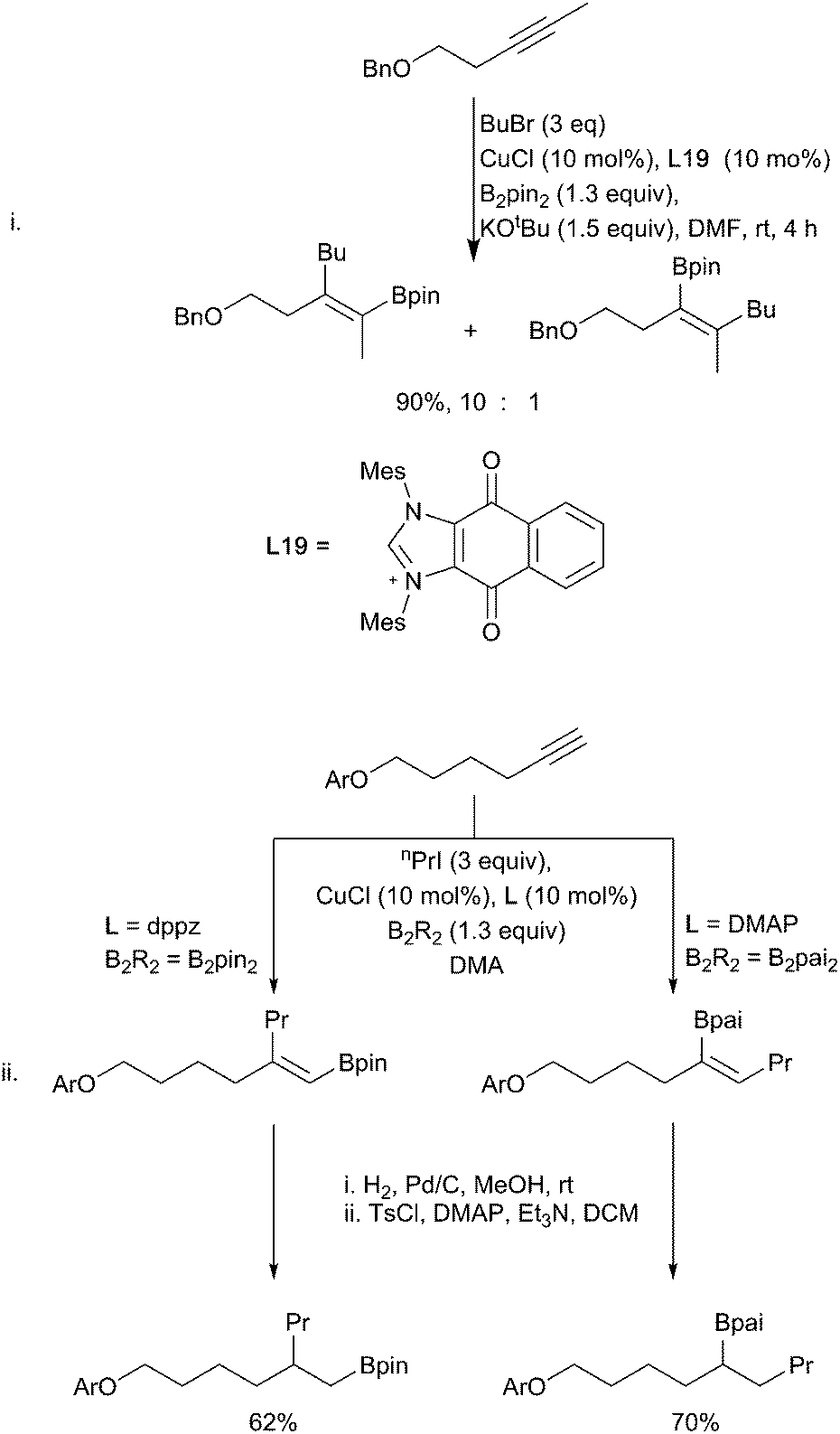 | ||
| Scheme 17 Regioselective alkylboration of alkynes. | ||
One approach to enhance the scope of the electrophile is to use mixed catalysis, as exemplified in various reports describing cooperative copper and palladium or nickel catalysed arylboration of alkenes, dienes, allenes, and alkynes.86–88 In this process, the initially generated β-borylalkylcopper intermediate undergoes fast transmetallation with an in situ generated ArPd(II)(halide) complex, followed by reductive elimination to give the arylboration product. By varying the nature of the phosphine ligand and base, it is possible to change the nature of the transmetallation step from stereoretentive to an invertive pathway, thus enabling either syn or anti-selective arylboration (Scheme 18i). With chiral ligands, for the borylcupration step, good control of enantioselectivity can be achieved.89,90 As noted earlier, dienes preferentially borylate at the terminal position leading to a 1,4 butenyl boryl copper intermediate. In the presence of a Pd catalyst these normally react with an aryl halide with retention of regiochemistry. For non-symmetrical dienes, such as isoprene, preferential formation of the 1,4-arylborylation product occurs; a situation reinforced by more sterically demanding ligands e.g. It-Bu. However, in the presence of DMAP the alternative 2,1 aryl-borylation product results (Scheme 18ii).91 The reasons underlying this change in selectivity remain to be elucidated. Recent studies suggest that both regioisomers of the 1,4-butenyl boryl copper intermediate are formed but can interchange, by an as yet unidentified mechanism,92 favouring the formation of the less hindered boryl product.
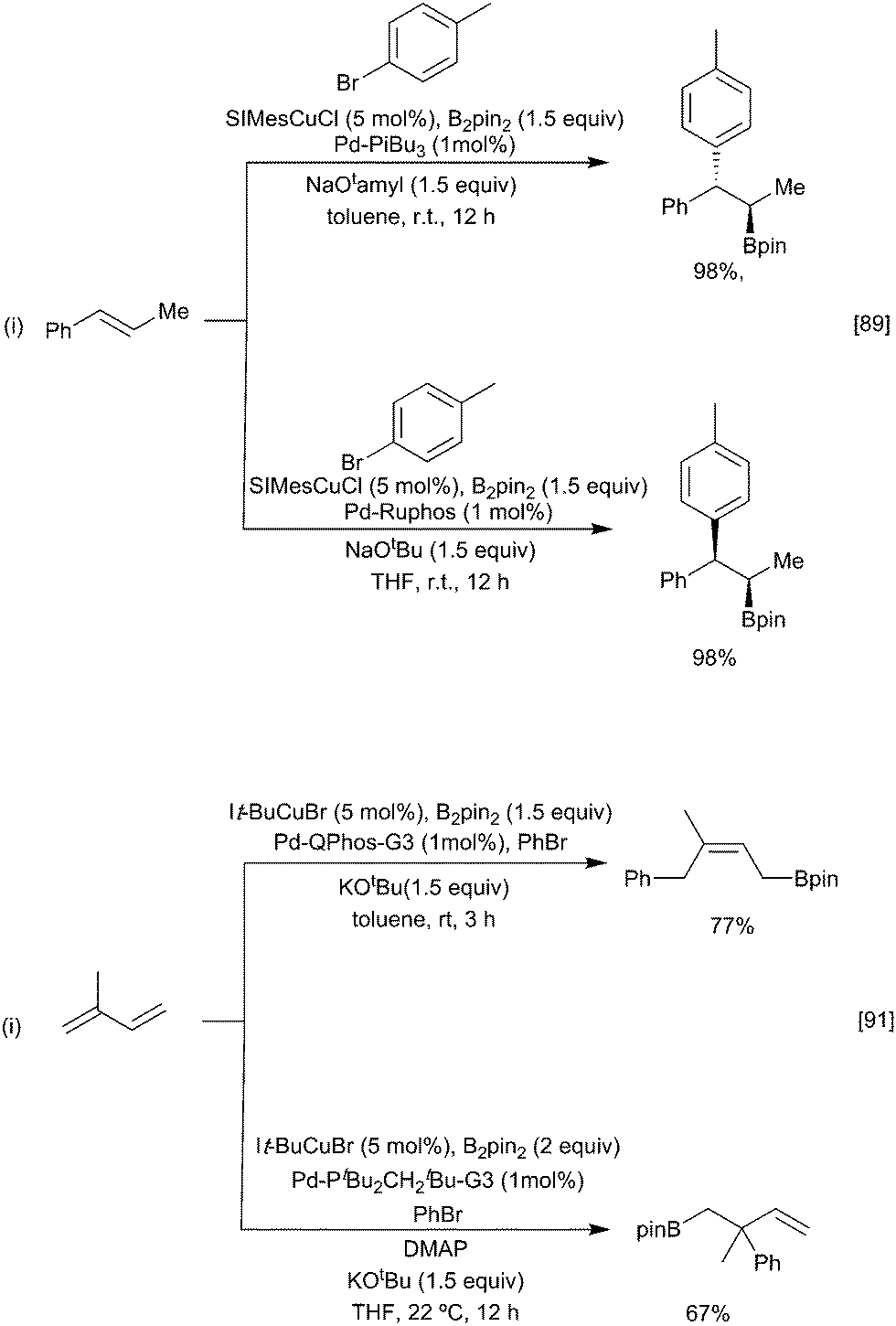 | ||
| Scheme 18 Selective aryl borylation of alkenes via cooperative catalysis. | ||
Finally, whilst the use of B2pin2 is dominant in these processes, other boranes are also viable and can offer advantages. For example, as noted earlier, use of the mixed diboron Bpin–Bdan leads to exclusive transfer of the Bdan reagent in the bora-alkylation of vinylsilanes (Scheme 19).93
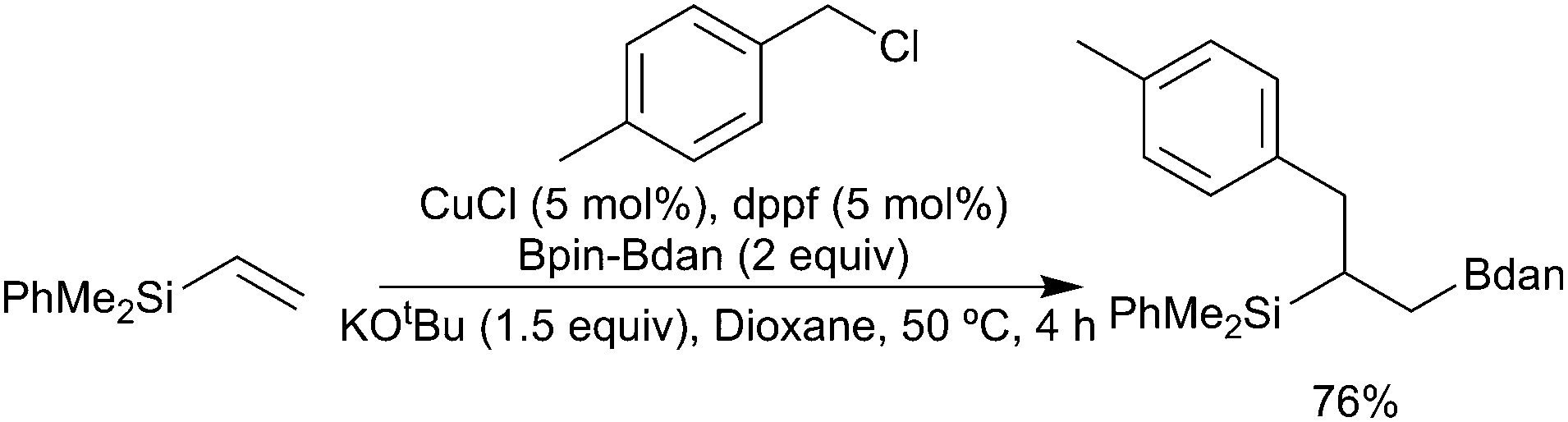 | ||
| Scheme 19 Carboboration of silylalkenes with Bpin–Bdan. | ||
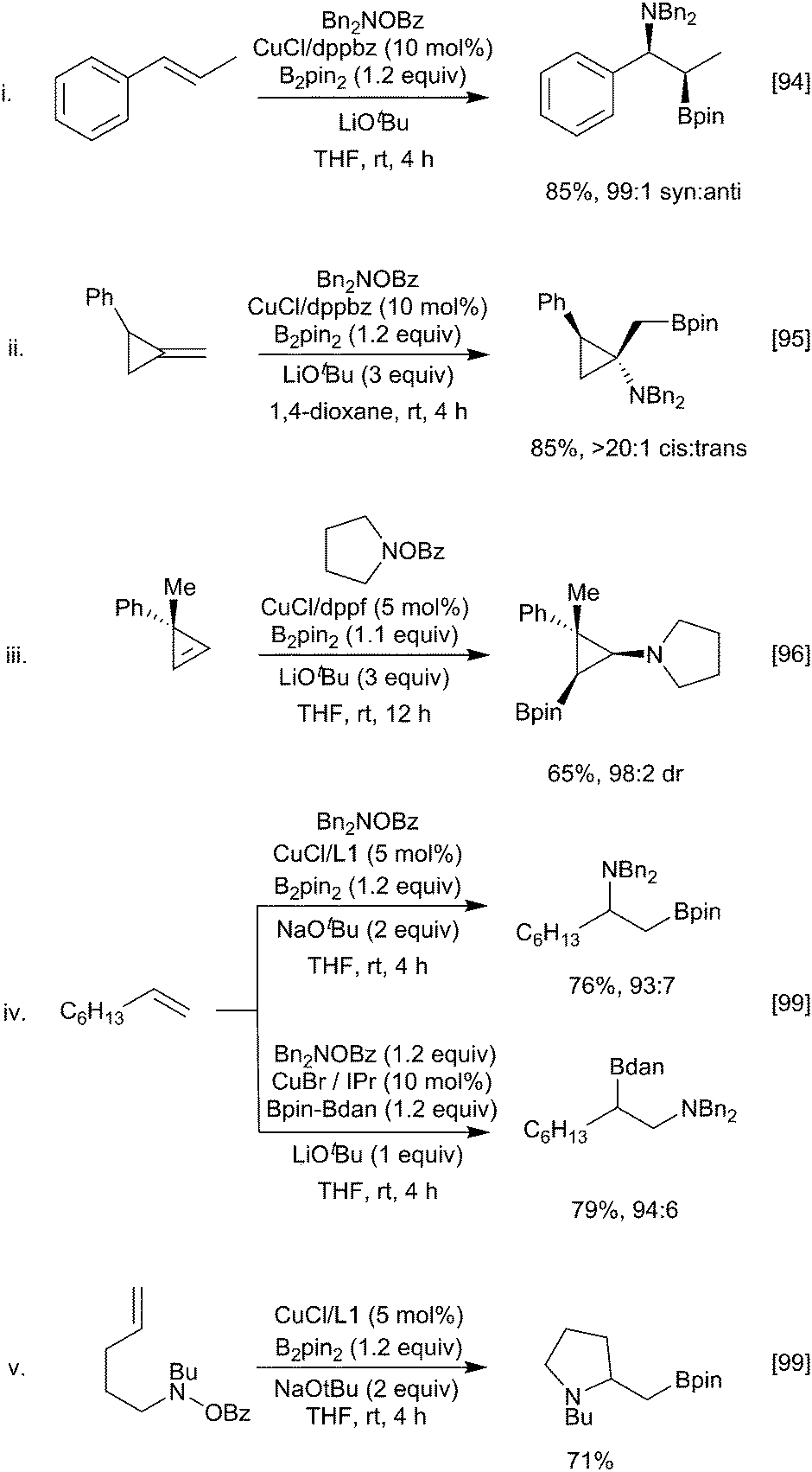 | ||
| Scheme 20 Aminoboration. | ||
Unactivated terminal alkenes are also viable substrates and, in parallel with alkene hydroboration (Scheme 6), different regiochemistry is possible depending on the catalyst and diboron reagent combination used (Scheme 20iv).98,99 The rationale for this reversal of regioselectivity is not fully understood, but one possibility is that the bulky NHC ligated Cu complex is only tolerated at the terminal position. Intramolecular variants to give borylated cyclic amines are also possible (Scheme 20v).
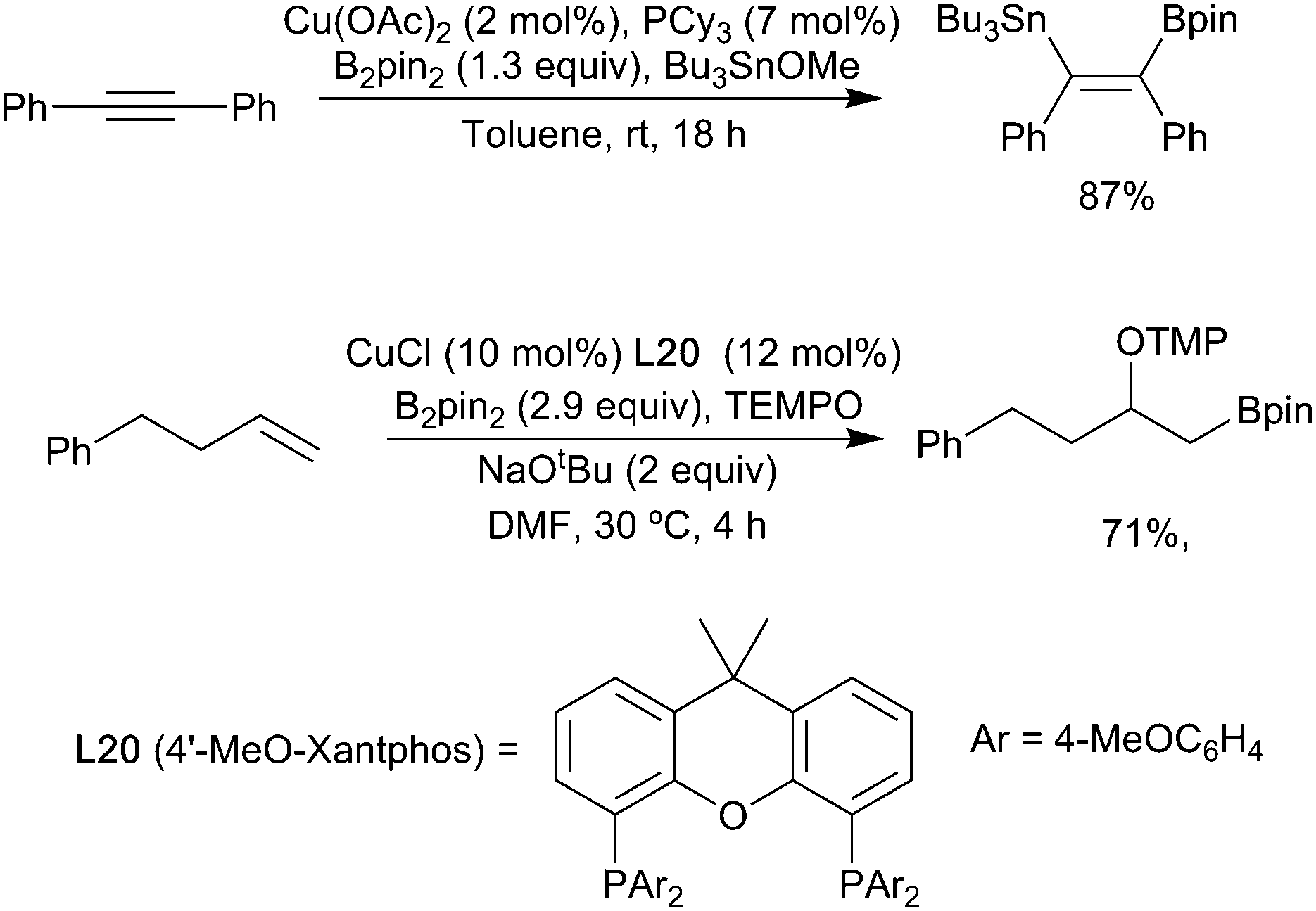 | ||
| Scheme 21 Selected examples of heteroboration. | ||
3.2 Reactions of copper-boryl complexes with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) X multiple bonds
X multiple bonds
While high degrees of regioselectivity of additions to alkenes, alkynes, and allenes relies on a balance of steric factors between ligand and substrate and the stability of the resultant organocopper intermediate, reactions with more polar electron deficient unsaturated bonds, such as carbonyl derivatives, proceed with excellent regioselectivity.
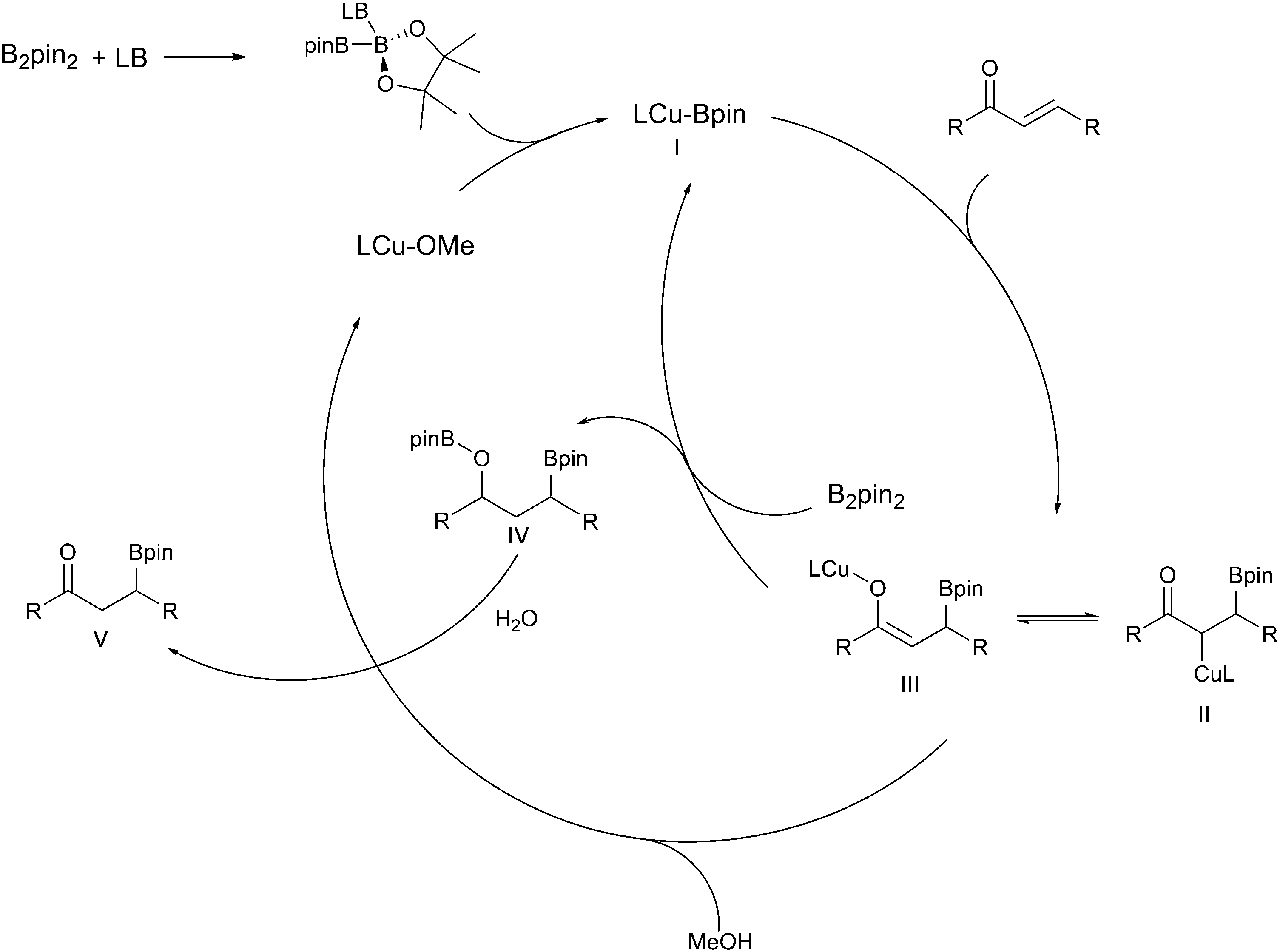 | ||
| Scheme 22 Catalytic cycle for the copper-catalysed β-borylation of α,β-unsaturated carbonyl compounds.18,107 | ||
Under these conditions the β-borylation of α,β-unsaturated ketones, esters, nitriles and amides occurs efficiently, albeit with amides requiring the use of bulky and electron rich ligands such as dtpf and dppbz (Scheme 23ii).108 Introduction of asymmetry can be realised through the use of chiral ligands. Early examples were described by Yun using Josiphos and MandyPhos ligands.107–109 Interestingly for ketone substrates, but not for esters, a stoichiometric amount of methanol was crucial with either none or excess proving to be detrimental. These challenges appear to be relatively substrate specific and it is possible to choose from a diverse set of chiral ligands that show good selectivity for distinct subsets of Michael acceptors. A full list of all ligands is beyond the scope of this review and a selected set of examples is shown in (Scheme 23).110–113
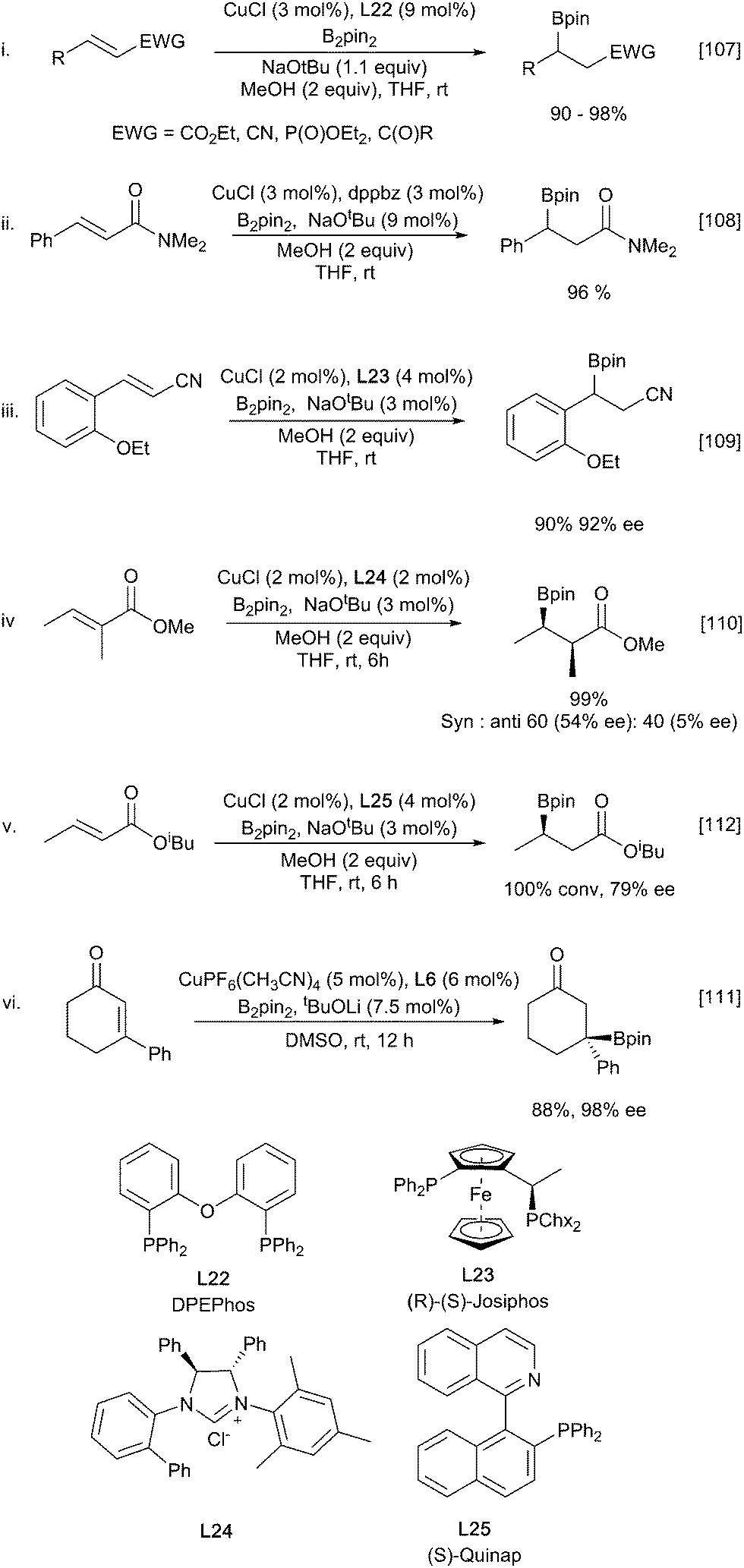 | ||
| Scheme 23 Selected examples of asymmetric β-borylation of enones and enoates. | ||
Propiolate derivatives also readily undergo copper-catalysed borylation to afford the corresponding vinylboronates. A Xantphos/CuCl catalyst system promoted the formation of (Z)-β-boryl acrylates with complete regio- and stereoselectivity and in excellent yields (Scheme 24i).114 In a complementary approach to enantioselective β-borylation described above, subsequent asymmetric reduction of the corresponding vinylboronate to yield enantiopure β-boryl esters could be performed in a one-pot synthesis from the propiolate starting material (Scheme 24ii).101 As with alkene hydroboration (Scheme 6), using HBpin can lead to the formation of a copper hydride species and the reversal of regioselectivity affording the (Z)-α-borylated enoate with complete regioselectivity and high stereoselectivity (Scheme 24iii).115
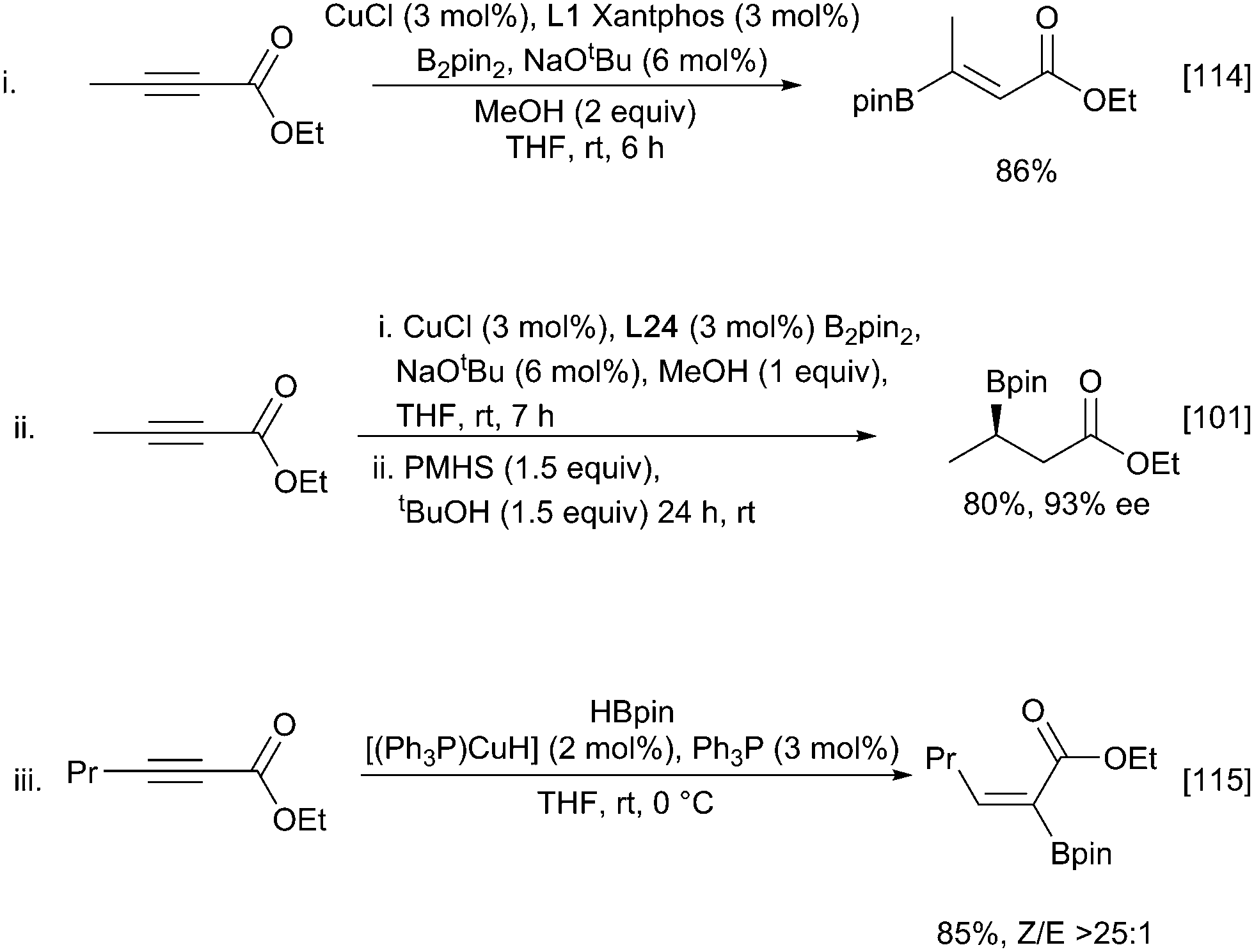 | ||
| Scheme 24 Borylation of propiolate esters. | ||
The borylation of allenoates provides a route to the homologous vinylboronates with alkyl and phenyl substitutions being tolerated at the α and β positions affording the (Z)-isomer exclusively (Scheme 25).116 The use of the sp2–sp3 mixed diboron PDIPA (10) allowed for the base-free reaction to occur under mild conditions with trifluoroethanol (TFE) as an additive.
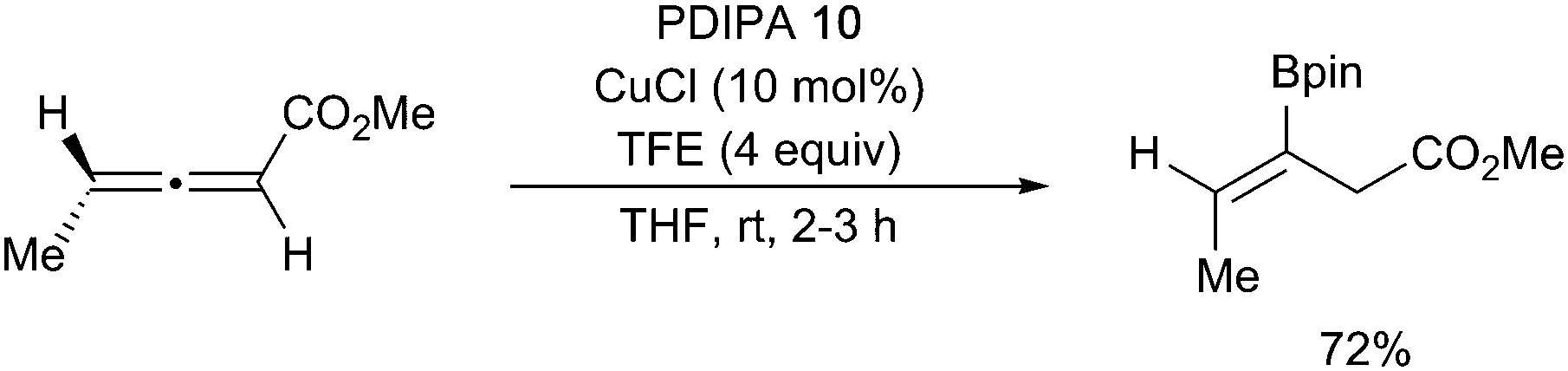 | ||
| Scheme 25 β-Borylation of allenoates with an sp2–sp3 mixed diboron. | ||
In analogous fashion to carboborylation, the initially formed copper enolate can be trapped by a range of electrophiles leading to multiple bond constructions in a single process. This is nicely exemplified by the copper-boryl enabled enantioselective aldol cyclisation of enone diones in the presence of Josiphos described by Lam (Scheme 26).117
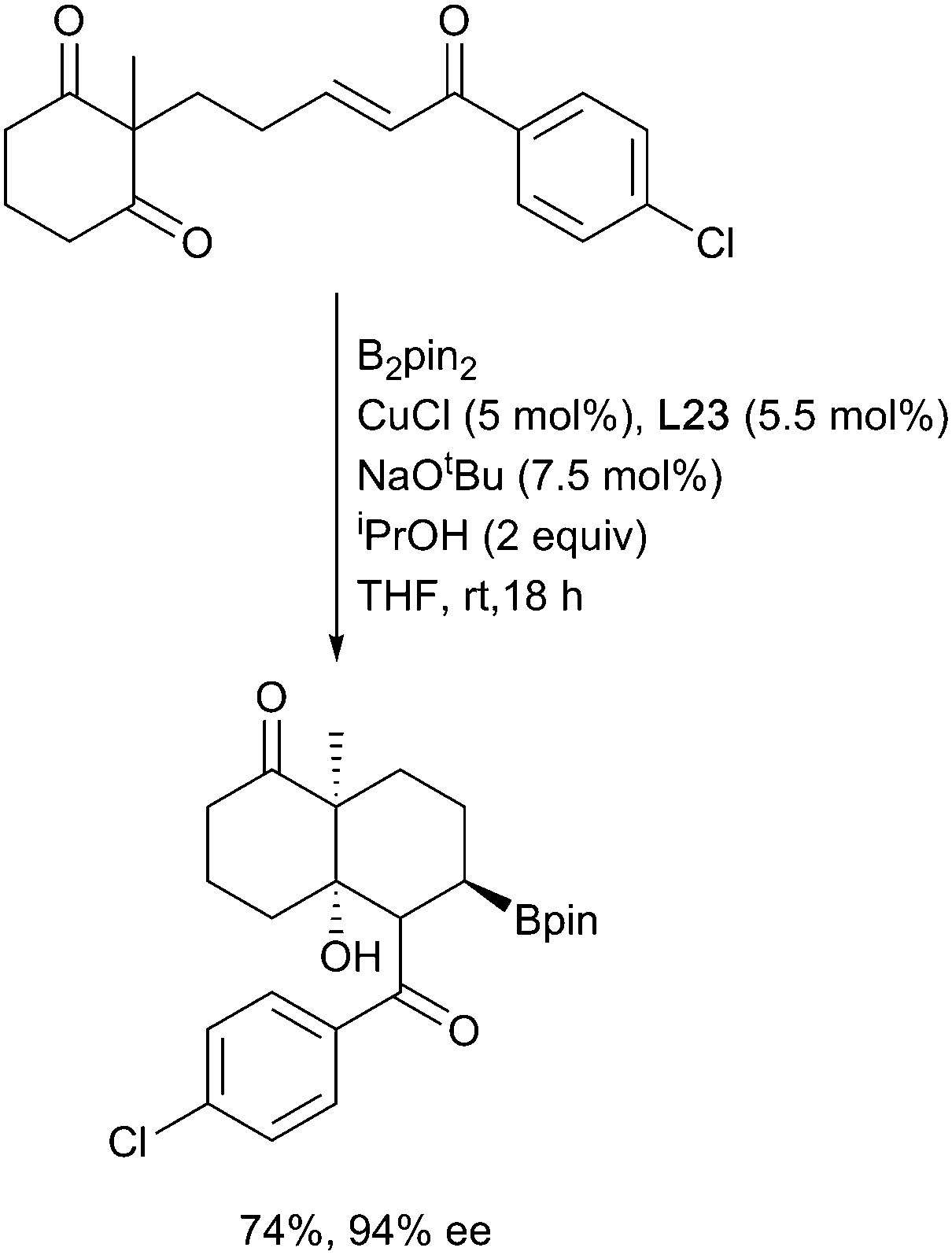 | ||
| Scheme 26 Borylative aldol cyclizations of enone diones. | ||
While all the above methods are efficient at β-borylation of a variety of α,β-unsaturated carbonyl substrates, copper(I) catalysis has disadvantages due to air and moisture stability. These can be avoided with the use of copper(II) sulfate which enables the borylation of α,β-unsaturated carbonyl compounds in mild, aqueous conditions in the presence of a weak base (Scheme 27i).59 Observation of a primary kinetic isotope effect in D2O and the nonreactive nature of pinacolboronic acid supports a pathway involving amine-assisted water activation and subsequent transmetallation to form the nucleophilic borylcopper complex. Santos and co-workers extended aqueous the β-borylation process to propiolate esters with great success to afford the β-borylated product with complete regio- and stereoselectivity (Scheme 27ii).118 Substrates containing additional unsaturation proceed with complete chemoselectivity for the propiolate alkyne. Replacing the B2pin2 with the mixed diboron danBBpin (9), allows selective transfer of the Bdan moiety and hence complementary chemistry to the normal Bpin products (Scheme 27iii).119 However, the use of 10% ethanol in water or 2% TPSG detergent is required to solubilize the diboron reagent.
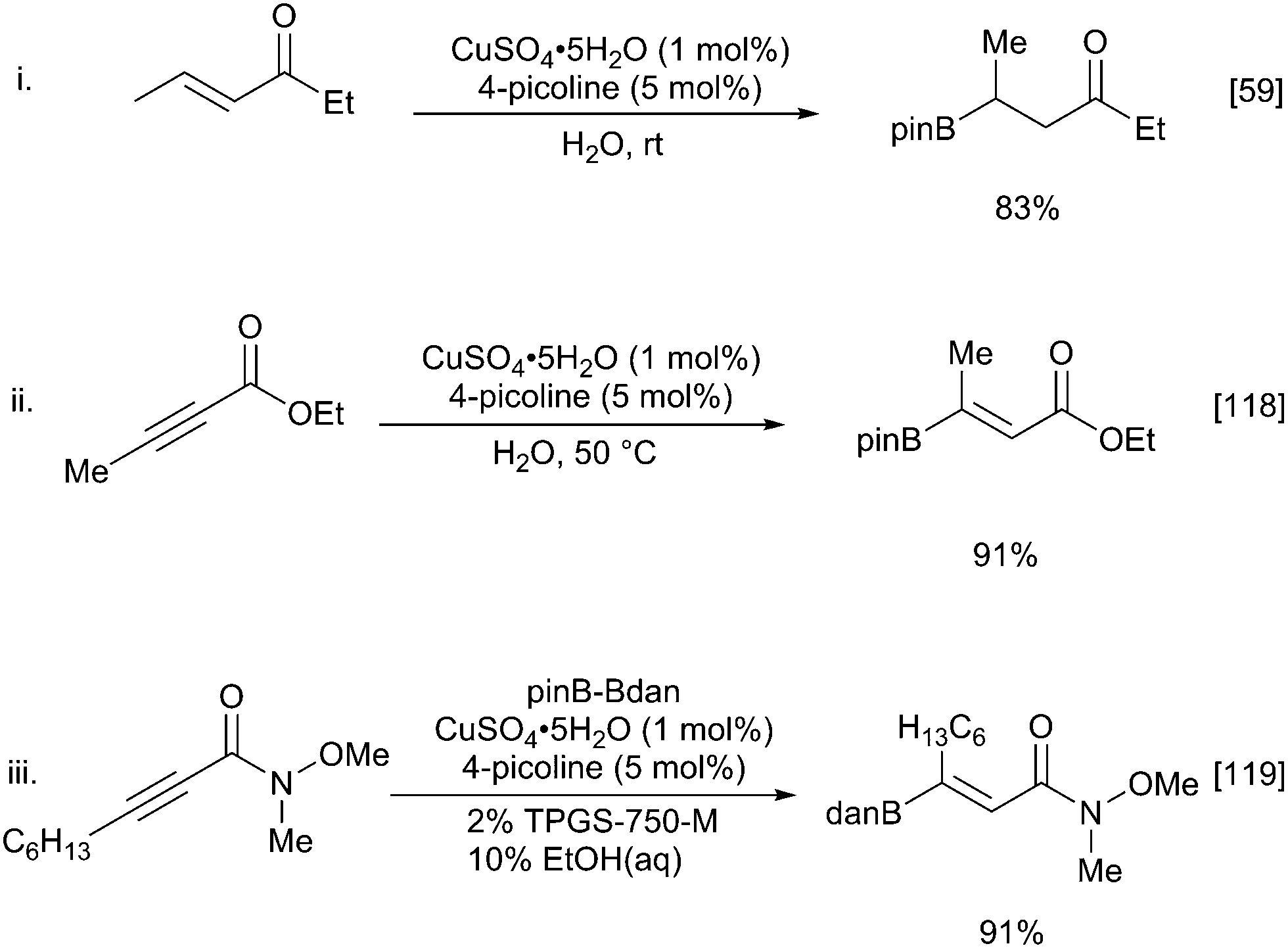 | ||
| Scheme 27 Aqueous β-borylation of α,β-unsaturated carbonyl compounds. | ||
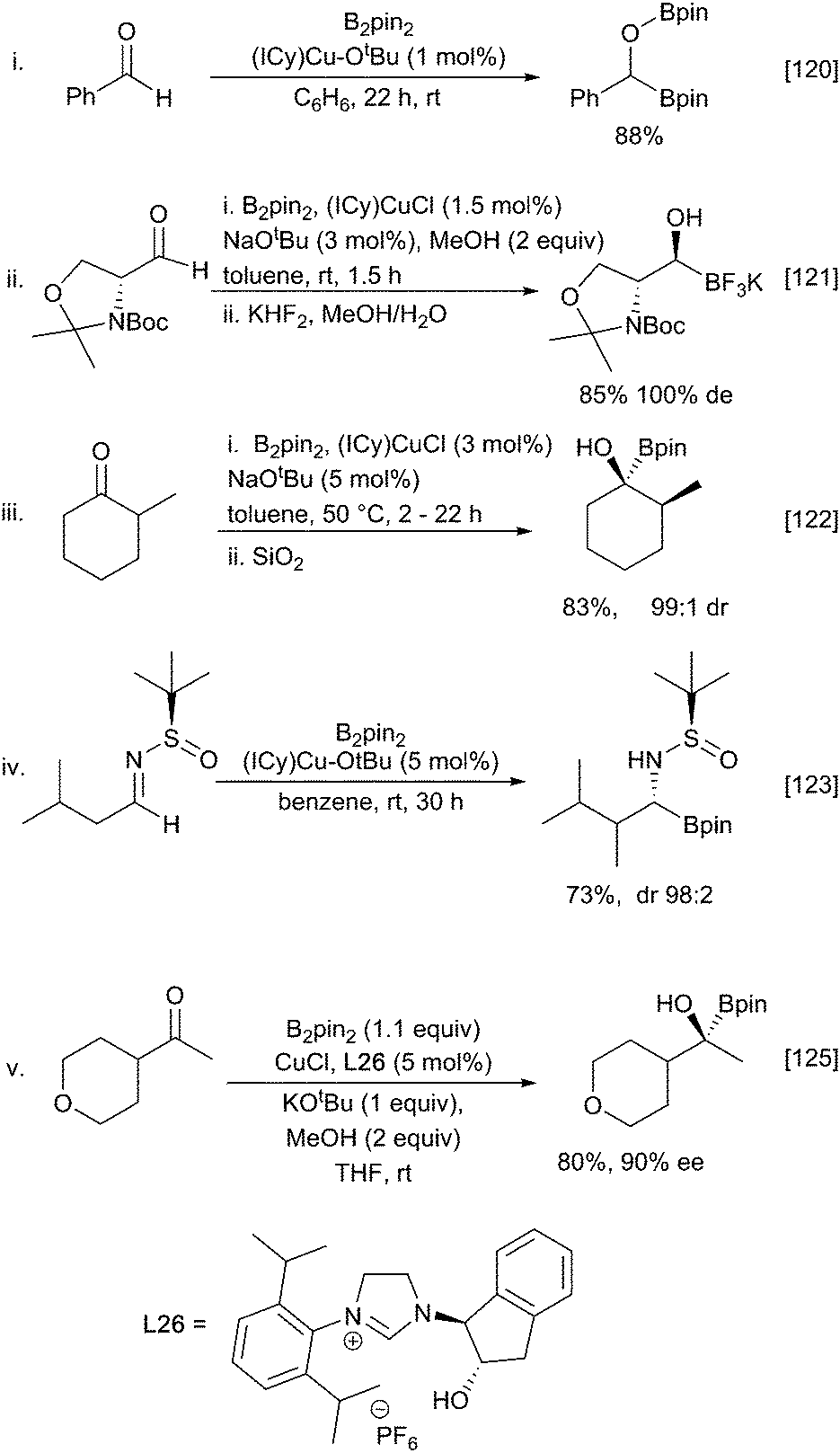 | ||
Scheme 28 Copper-catalysed 1,2-addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds. X bonds. | ||
 | ||
| Scheme 29 Catalytic cycle for the copper-catalysed 1,2-diboration. | ||
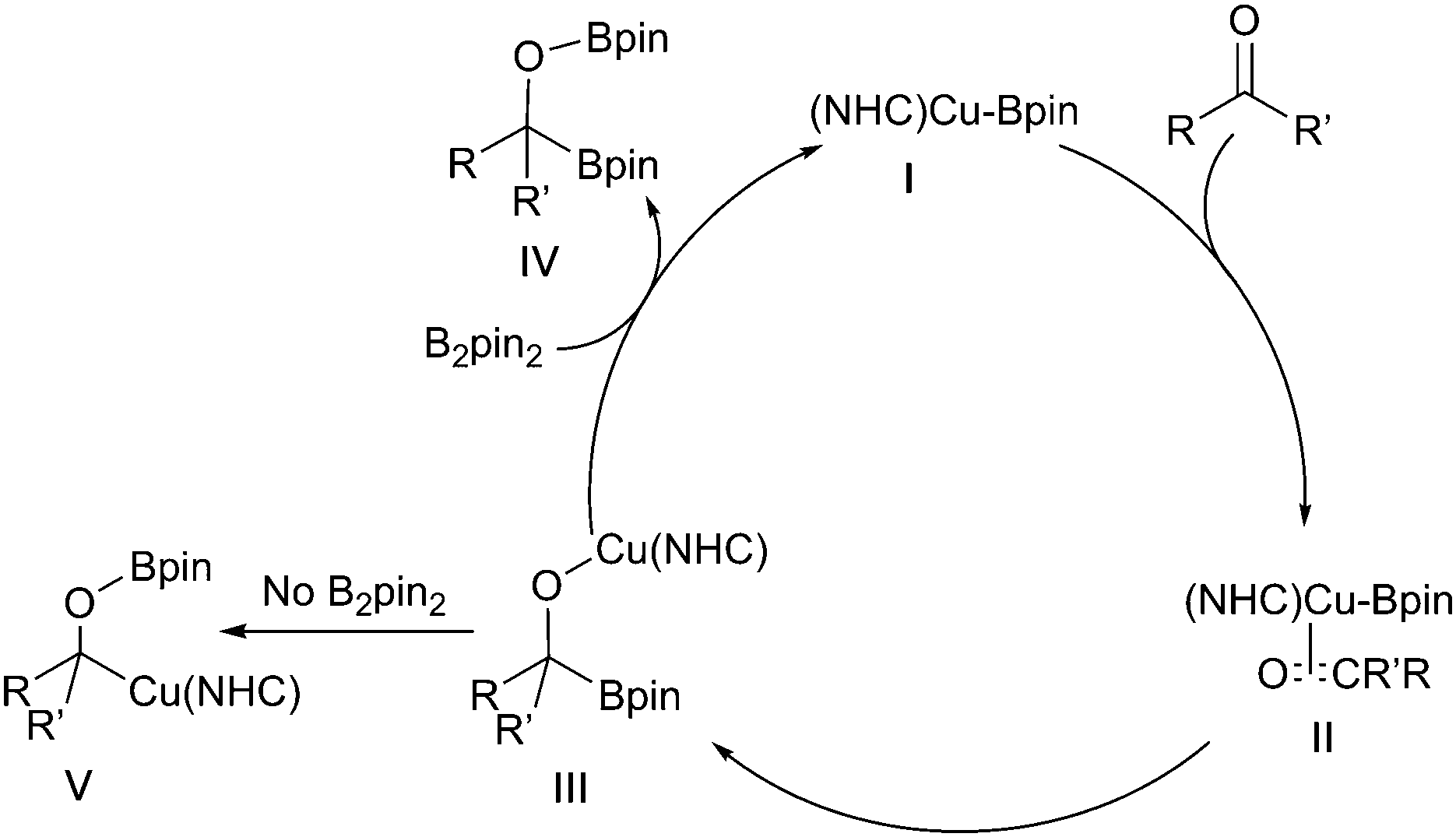
Similar 1,2-diborations occur with ketones, although an in situ catalyst generation from (NHC)CuCl and excess NaOtBu rather the pre-formed Sadihgi catalyst [(NHC)Cu-OtBu] was required for efficient reaction. It was proposed that the excess NaOtBu was required to promote rapid regeneration of the active borylcopper reagent.122 Consistent with rate limiting nucleophilic addition of a ‘boryl anion’ reactions with chiral ketones proceed with high diastereoselectivity, likely driven by the steric bulk of the inserting pinacolboronate in accordance with the Felkin–Ahn model (Scheme 28iii).122 Similar stereoselective additions have also been described for reactions with chiral aldimines providing an entry to α-aminoboronates (Scheme 28iv).123
3.3 Copper catalysed C–X borylation
Building on the analogous palladium catalysed Miyaura borylation, Zhu and Ma demonstrated that aryl iodides could be borylated by HBpin in the presence of copper iodide and sodium hydride (Scheme 30i).127 Subsequently, Marder & Lin described the synthesis of aryl boronates via the copper-catalysed borylation of aryl bromides and iodides and a diboron reagent (Scheme 30ii).19 Marder & Lin proposed that the reaction occurred via oxidative addition of the aryl halide to a Cu–Bpin complex. Reductive elimination of the aryl-Bpin moiety then generates the product, with the active copper-boryl species being regenerated via halide ligand exchange to give a copper-alkoxide species, followed by metathesis with B2pin2.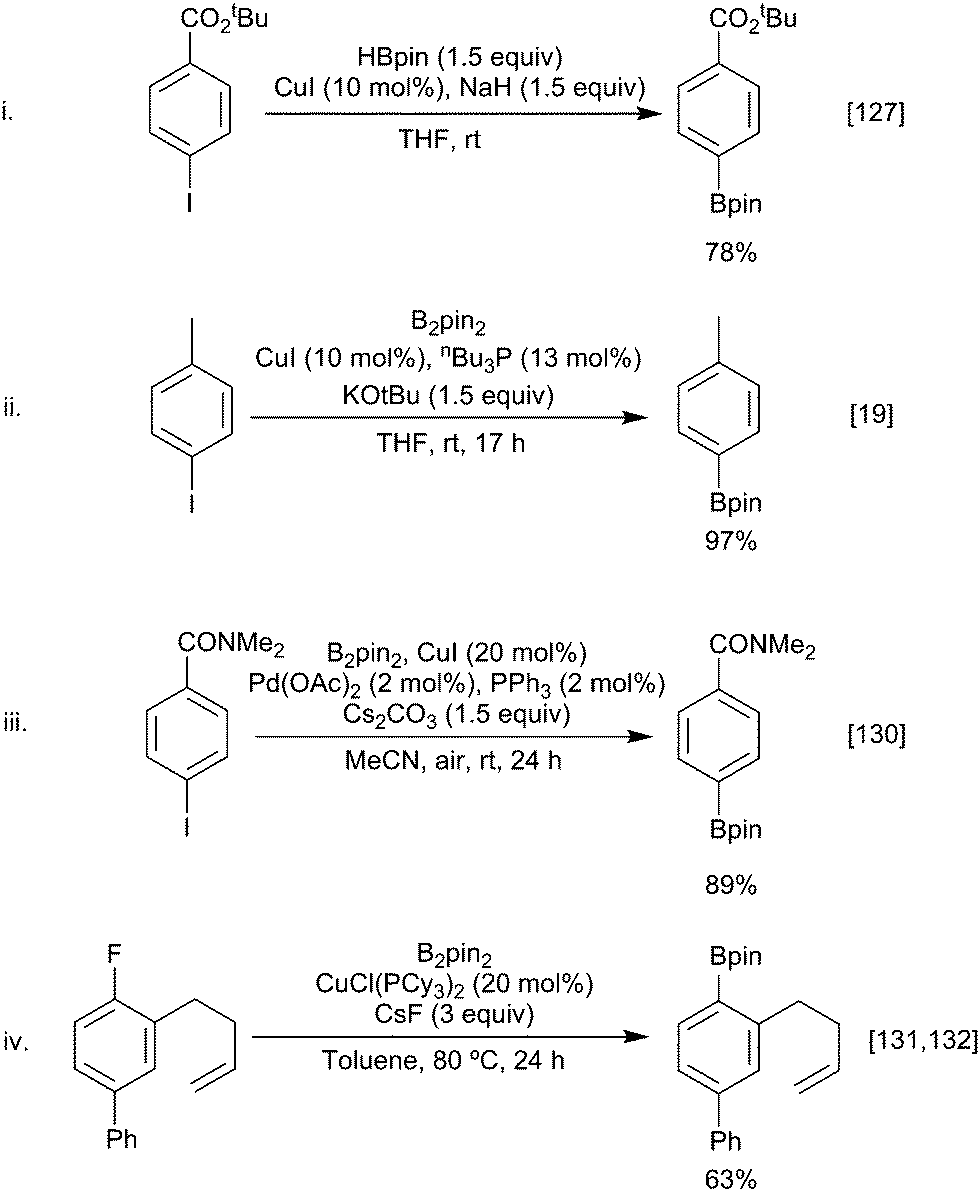 | ||
| Scheme 30 Aryl halide borylation. | ||
Since then further developments of this copper variant of the classical palladium catalysed Miyaura boryation have included borylations in shorter reaction times using designer bicyclic NHC ligands,128 the use of alternative diboron reagents (e.g. Bin–Bdan) leading to aryl-Bdan esters,129 co-catalyst combinations that give even greater functional group tolerance enabling phenols, amides, esters and ketones to be used in reactions undertaken open to the air (Scheme 30iii),130 and applications to other nucleofuges, including arylfluorides, although this last transformation probably proceeds by an SRN1 pathway (Scheme 30iv).131,132
In a similar fashion, alkyl (sp3) halides can undergo copper-mediated borylation. First reported in 2012 by Liu, Marder and Steel mediated by a CuI/PPh3 catalytic system (Scheme 31i),133 and, independently, by Ito using a CuCl/Xantphos catalytic system (Scheme 31ii),134 a broad range of functional groups are tolerated including esters, amides, silyl ethers and alcohols. Other diboron reagents can be used for this process135 and a range of leaving groups are viable. The most common are iodides, bromides and other pseudohalides but even the direct displacement of alcohols has been reported.136,137 When the unsaturated moiety (alkene or alkyne) is suitably positioned to enable facile cyclisation (n ≤ 7) a variety of cyclic products (Scheme 31iii–vi) can be obtained.28,133,138,139 Whilst these transformations are formally an intramolecular carbo-borylation of the alkene (see Section 3.1.3) the mechanism of the process remains an interesting question as simple hex-1-ene was not hydroborated under the reaction conditions, suggesting that activation of the C–X bond is a prerequisite. Further studies by Ito have suggested that different mechanisms are in operation depending on the catalyst/ligand combination.28 With the CuI/PPh3 system, a radical generated from the copper catalyst results in cyclisation although attempts to suppress reaction with radical scavengers had no adverse effect. Alternatively, with the CuCl/Xantphos conditions an ionic metallocycle was invoked as the key intermediate with cyclisation occurring upon reductive elimination. Cyclisation of larger chain lengths (>7) would be disfavoured in both cases, accounting for the preferential formation of the linear product with these substrates.
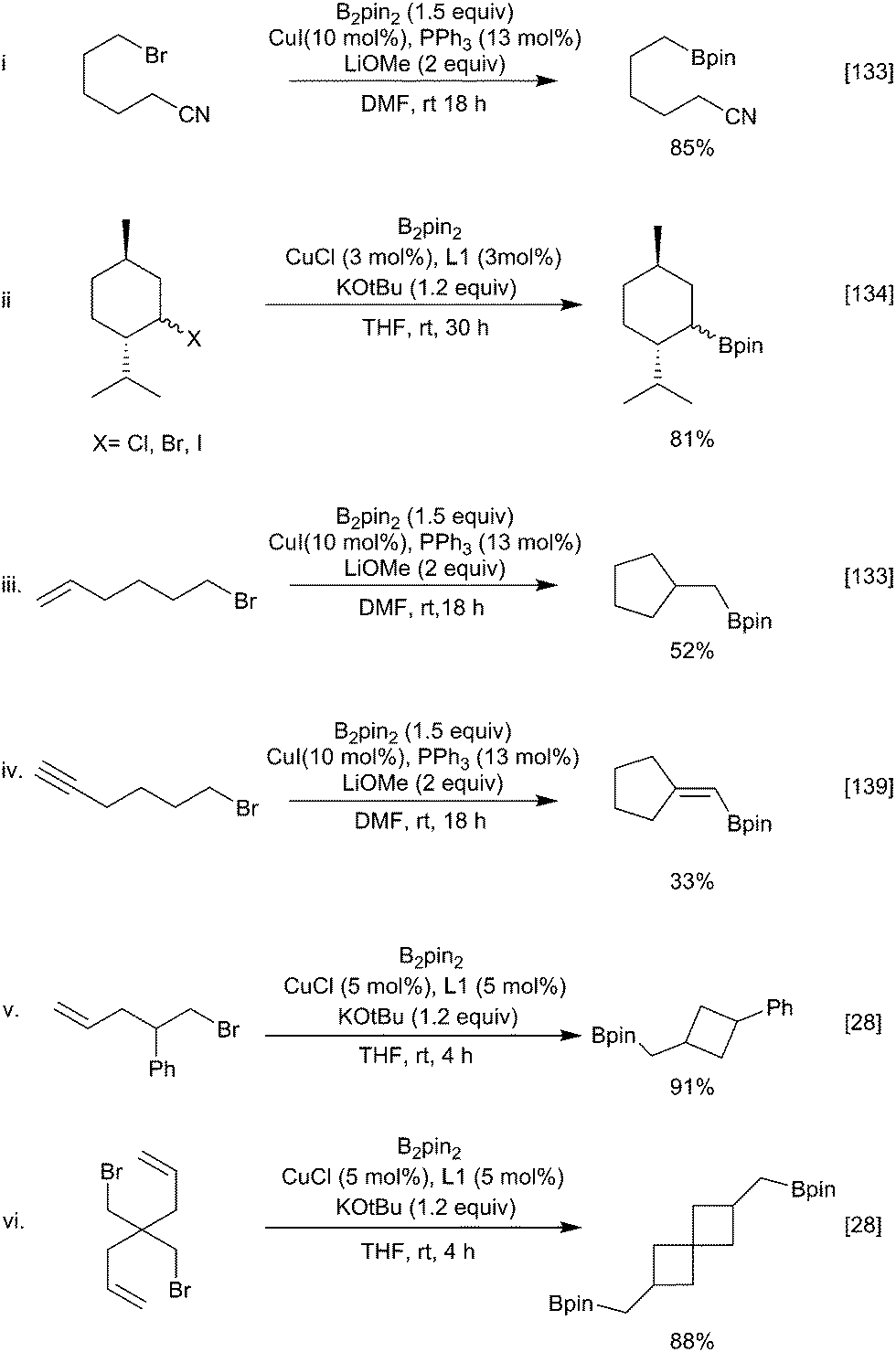 | ||
| Scheme 31 Copper catalysed borylation of alkyl halides. | ||
Based on observations from the use of chiral substrates (Scheme 31ii) in neither case is the displacement reaction an SN2 process. This contrasts with the reaction of copper boryls with allylic substrates where the ability of the soft copper centre to interact with the alkene π system can facilitate an SN2′ pathway. Following the first example of a stoichiometric copper-boryl mediated SN2′ reaction reported by Ramachandran (Scheme 32i),140 this strategy has since been expanded to include a variety of nucleofuges (phosphates,141 carbonates,142 acetals,143 ethers,53,144 esters140 alcohols136,137) and unsaturated moieties (alkenes,145 alkynes146 and allenes)40,78 (Scheme 32). Depending on the ligand and substrate both linear or cyclic products can be generated with high enantioselectivities.147
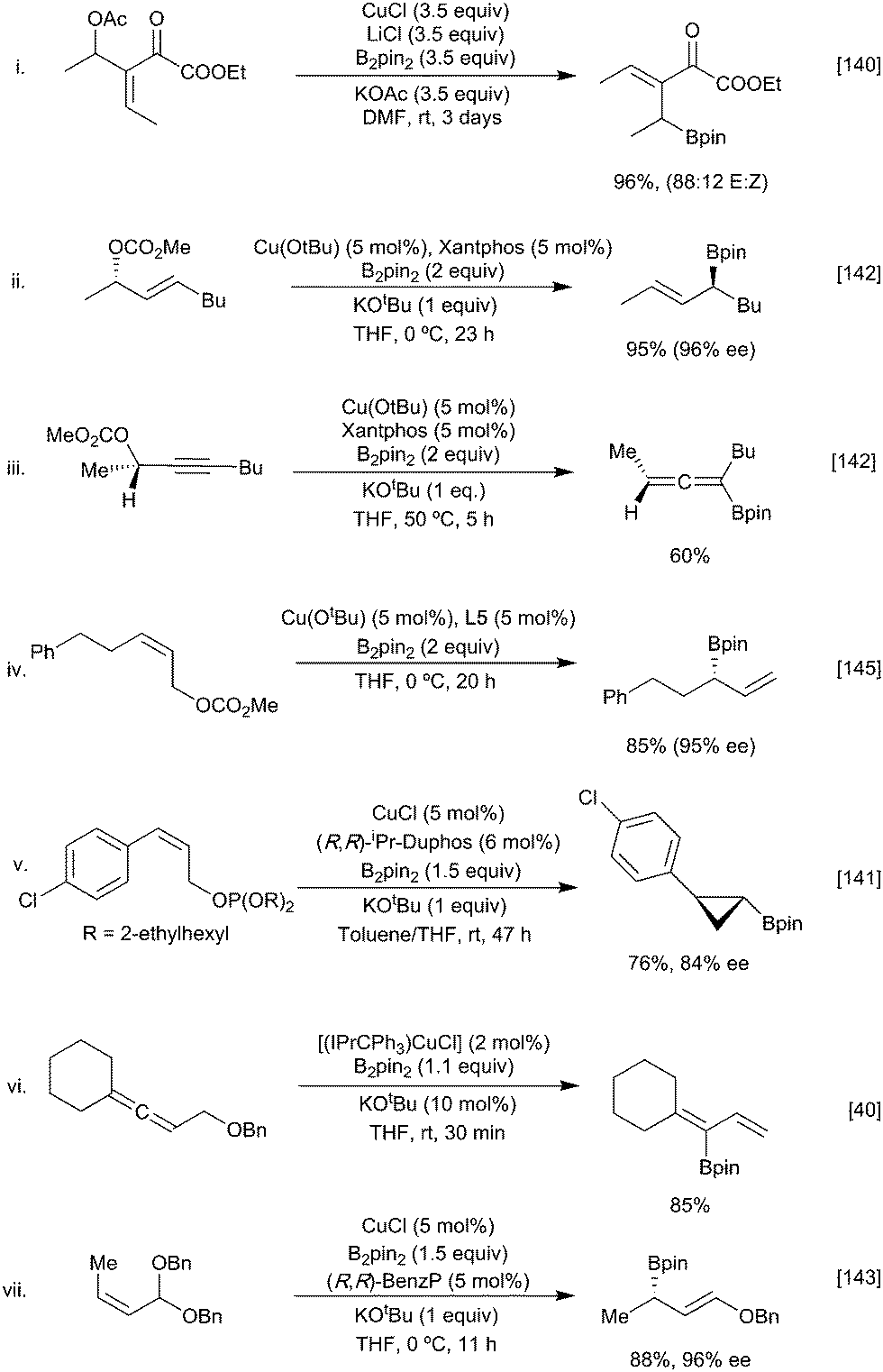 | ||
| Scheme 32 Examples of SN2′ borylative substitution with different leaving groups. | ||
Finally, as with other processes involving copper-boryl species, there are advantages to the use of Cu(II) salts as these enable the reaction to be carried out open to the atmosphere. Interestingly under these conditions, alkyl chlorides are viable substrates (Scheme 33i). The observation of significant cyclisation of a hexenyl halide substrate is again indicative of a radical mediated process (Scheme 33ii).148
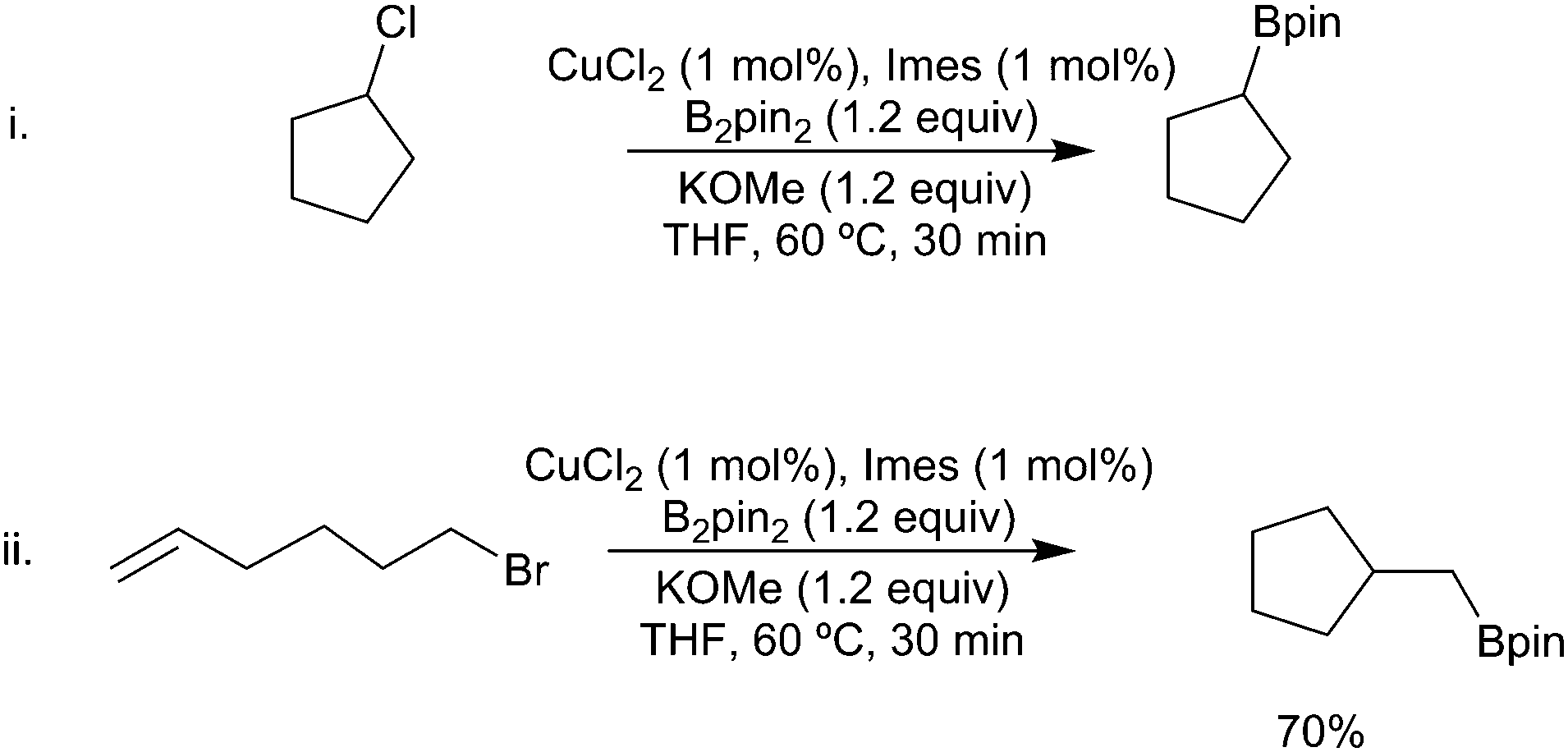 | ||
| Scheme 33 Copper(II)-catalysed borylation of alkyl halides. | ||
4 The future of copper-boryl mediated transformations
From the initial reports of copper-catalysed enone borylation, a vast number of substrate classes have now been shown to react with copper-boryl complexes including alkenes, alkynes, allenes, carbonyl derivatives, and aryl, alkenyl and alkyl halides. The breadth of chemistry possible with this simple reagent combination is quite stunning and, with ligand mediated control of reaction regiochemistry and stereochemistry, this has become a significant tool in the synthetic chemist's lexicon of methodology. Numerous elegant exemplars already exist, and the authors apologise to their colleagues in this field whose many excellent variants and applications that space did not allow room to present. Given the rapid expansion of the field over the past 17 years, it is likely that further exciting discoveries (e.g. copper-catalysed C–H borylation?) are imminent. Watch this space!Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the EPSRC (EP/L504762/1) and GlaxoSmithKline, PhD studentship (DH); Natural Science and Engineering Research Council (SAW) and The Royal Society of Chemistry, International 5+5+5 Collaboration Award (SAW, WLS and PGS) for support of our programmes.References
- G. Zou, Y. K. Reddy and J. R. Falck, Tetrahedron Lett., 2001, 42, 7213–7215 CrossRef.
- D. S. Matteson, Tetrahedron, 1998, 54, 10555–10607 CrossRef.
- S. Essafi, S. Tomasi, V. K. Aggarwal and J. N. Harvey, J. Org. Chem., 2014, 79, 12148–12158 CrossRef PubMed.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef PubMed.
- W. K. Chow, O. Y. Yuen, P. Y. Choy, C. M. So, C. P. Lau, W. T. Wong and F. Y. Kwong, RSC Adv., 2013, 3, 12518–12539 RSC.
- M. Gao, S. B. Thorpe and W. L. Santos, Org. Lett., 2009, 11, 3478–3481 CrossRef PubMed.
- M. Gao, S. B. Thorpe, C. Kleeberg, C. Slebodnick, T. B. Marder and W. L. Santos, J. Org. Chem., 2011, 76, 3997–4007 CrossRef PubMed.
- R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC.
- K. Takahashi, T. Ishiyama and N. Miyaura, Chem. Lett., 2000, 982–983 CrossRef.
- H. Ito, H. Yamanaka, J.-I. Tateiwa and A. Hosomi, Tetrahedron Lett., 2000, 41, 6821–6825 CrossRef.
- K. Takahashi, T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2001, 625, 47–53 CrossRef.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef PubMed.
- T. Kajiwara, T. Terabayashi, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2008, 47, 6606–6610 CrossRef PubMed.
- K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2013, 19, 7125–7132 CrossRef PubMed.
- C. Borner, L. Anders, K. Brandhorst and C. Kleeberg, Organometallics, 2017, 36, 4687–4690 CrossRef.
- L. Dang, Z. Lin and T. B. Marder, Organometallics, 2008, 27, 4443–4454 CrossRef.
- C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef PubMed.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, Organometallics, 2006, 25, 2405–2408 CrossRef.
- J. H. Moon, H. Y. Jung, Y. J. Lee, S. W. Lee, J. Yun and J. Y. Lee, Organometallics, 2015, 34, 2151–2159 CrossRef.
- L. Dang, H. Zhao, Z. Lin and T. B. Marder, Organometallics, 2007, 26, 2824–2832 CrossRef.
- H. Ito, T. Miya and M. Sawamura, Tetrahedron, 2012, 68, 3423–3427 CrossRef.
- A.-M. Carroll, T. P. O'Sullivan and P. J. Guiry, Adv. Synth. Catal., 2005, 347, 609–631 CrossRef.
- D. A. Evans, G. C. Fu and A. H. Hoveyda, J. Am. Chem. Soc., 1988, 110, 6917–6918 CrossRef.
- D. Männig and H. Nöth, Angew. Chem., Int. Ed. Engl., 1985, 24, 878–879 CrossRef.
- K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Tetrahedron, 2015, 71, 2183–2197 CrossRef.
- K. Kubota, E. Yamamoto and H. Ito, J. Am. Chem. Soc., 2013, 135, 2635–2640 CrossRef PubMed.
- H. Iwamoto, K. Kubota and H. Ito, Chem. Commun., 2016, 52, 5916–5919 RSC.
- H. A. Kerchner and J. Montgomery, Org. Lett., 2016, 18, 5760–5763 CrossRef PubMed.
- Y. Cai, X. T. Yang, S. Q. Zhang, F. Li, Y. Q. Li, L. X. Ruan, X. Hong and S. L. Shi, Angew. Chem., Int. Ed., 2018, 57, 1376–1380 CrossRef PubMed.
- H. Iwamoto, T. Imamoto and H. Ito, Nat. Commun., 2018, 9, 2290 CrossRef PubMed.
- Y. Wen, J. Xie, C. Deng and C. Li, J. Org. Chem., 2015, 80, 4142–4147 CrossRef PubMed.
- Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3160–3161 CrossRef PubMed.
- Y. Sasaki, C. Zhong, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 1226–1227 CrossRef PubMed.
- K. Kubota, Y. Watanabe, K. Hayama and H. Ito, J. Am. Chem. Soc., 2016, 138, 4338–4341 CrossRef PubMed.
- K. Kubota, Y. Watanabe and H. Ito, Adv. Synth. Catal., 2016, 358, 2379–2384 CrossRef.
- W. Yuan, X. Zhang, Y. Yu and S. Ma, Chem. – Eur. J., 2013, 19, 7193–7202 CrossRef PubMed.
- W. Yuan and S. Ma, Adv. Synth. Catal., 2012, 354, 1867–1872 CrossRef.
- K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2013, 52, 12400–12403 CrossRef PubMed.
- H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef.
- Y. Sasaki, Y. Horita, C. Zhong, M. Sawamura and H. Ito, Angew. Chem., Int. Ed., 2011, 50, 2778–2782 CrossRef PubMed.
- H. Jang, A. R. Zhugralin, Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7859–7871 CrossRef PubMed.
- A. L. Moure, R. Gómez Arrayás, D. J. Cárdenas, I. Alonso and J. C. Carretero, J. Am. Chem. Soc., 2012, 134, 7219–7222 CrossRef PubMed.
- Y. E. Kim, D. Li and J. Yun, Dalton Trans., 2015, 44, 12091–12093 RSC.
- Y. D. Bidal, F. Lazreg and C. S. J. Cazin, ACS Catal., 2014, 4, 1564–1569 CrossRef.
- T. Fujihara, K. Semba, J. Terao and Y. Tsuji, Catal. Sci. Technol., 2014, 4, 1699–1709 RSC.
- K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2012, 18, 4179–4184 CrossRef PubMed.
- H. R. Kim, I. G. Jung, K. Yoo, K. Jang, E. S. Lee, J. Yun and S. U. Son, Chem. Commun., 2010, 46, 758–760 RSC.
- H. R. Kim and J. Yun, Chem. Commun., 2011, 47, 2943–2945 RSC.
- D. Li, Y. E. Kim and J. Yun, Org. Lett., 2015, 17, 860–863 CrossRef PubMed.
- W. Su, T.-J. Gong, Q. Zhang, Q. Zhang, B. Xiao and Y. Fu, ACS Catal., 2016, 6, 6417–6421 CrossRef.
- J. K. Park, B. A. Ondrusek and D. T. McQuade, Org. Lett., 2012, 14, 4790–4793 CrossRef PubMed.
- A. Grirrane, A. Corma and H. Garcia, Chem. – Eur. J., 2011, 17, 2467–2478 CrossRef PubMed.
- Y. Zhou, W. You, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2014, 53, 3475–3479 CrossRef PubMed.
- S. Liu, X. Zeng and B. Xu, Tetrahedron Lett., 2016, 57, 3706–3710 CrossRef.
- G. Stavber and Z. Casar, ChemCatChem, 2014, 6, 2162–2174 CrossRef.
- T. Kitanosono and S. Kobayashi, Asian J. Org. Chem., 2013, 2, 961–966 CrossRef.
- S. B. Thorpe, J. A. Calderone and W. L. Santos, Org. Lett., 2012, 14, 1918–1921 CrossRef PubMed.
- J. S. da Costa, R. K. Braun, P. A. Horn, D. S. Ludtke and A. V. Moro, RSC Adv., 2016, 6, 59935–59938 RSC.
- Z.-J. Yao, S. Hong, W. Zhang, M. Liu and W. Deng, Tetrahedron Lett., 2016, 57, 910–913 CrossRef.
- F. Meng, H. Jang, B. Jung and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 5046–5051 CrossRef PubMed.
- J. Rae, K. Yeung, J. J. W. McDouall and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 1102–1107 CrossRef PubMed.
- K. Yeung, R. E. Ruscoe, J. Rae, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 11912–11916 CrossRef PubMed.
- L. Y. Jiang, P. Cao, M. Wang, B. Chen, B. Wang and J. Liao, Angew. Chem., Int. Ed., 2016, 55, 13854–13858 CrossRef PubMed.
- X. B. Li, F. K. Meng, S. Torker, Y. Shi and A. H. Hoveyda, Angew. Chem., Int. Ed., 2016, 55, 9997–10002 CrossRef PubMed.
- F. K. Meng, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 11304–11307 CrossRef PubMed.
- W. X. Zhao and J. Montgomery, J. Am. Chem. Soc., 2016, 138, 9763–9766 CrossRef PubMed.
- E. Yamamoto, R. Kojima, K. Kubota and H. Ito, Synlett, 2016, 272–276 Search PubMed.
- F. K. Meng, X. B. Li, S. Torker, Y. Shi, X. Shen and A. H. Hoveyda, Nature, 2016, 537, 387–393 CrossRef PubMed.
- P. Liu, Y. Fukui, P. Tian, Z. T. He, C. Y. Sun, N. Y. Wu and G. Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700–11703 CrossRef PubMed.
- I. Kageyuki, H. Yoshida and K. Takaki, Synthesis, 2014, 1924–1932 Search PubMed.
- F. K. Meng, K. P. McGrath and A. H. Hoveyda, Nature, 2014, 513, 367–374 CrossRef PubMed.
- H. Yoshida, I. Kageyuki and K. Takaki, Org. Lett., 2013, 15, 952–955 CrossRef PubMed.
- E. Bunuel and D. J. Cardenas, Eur. J. Org. Chem., 2016, 5446–5464 CrossRef.
- K. Kubota, H. Iwamoto and H. Ito, Org. Biomol. Chem., 2017, 15, 285–300 RSC.
- H. Ito, Y. Kosaka, K. Nonoyama, Y. Sasaki and M. Sawamura, Angew. Chem., Int. Ed., 2008, 47, 7424–7427 CrossRef PubMed.
- H. Ito, Y. Sasaki and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 15774–15775 CrossRef PubMed.
- H. Ito, T. Toyoda and M. Sawamura, J. Am. Chem. Soc., 2010, 132, 5990–5992 CrossRef PubMed.
- W. Su, T.-J. Gong, X. Lu, M.-Y. Xu, C.-G. Yu, Z.-Y. Xu, H.-Z. Yu, B. Xiao and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 12957–12961 CrossRef PubMed.
- Z.-Y. Xu, Y.-Y. Jiang, W. Su, H.-Z. Yu and Y. Fu, Chem. – Eur. J., 2016, 22, 14611–14617 CrossRef PubMed.
- B. Chen, P. Cao, Y. Liao, M. Wang and J. Liao, Org. Lett., 2018, 20, 1346–1349 CrossRef PubMed.
- R. Alfaro, A. Parra, J. Alemán, J. L. García Ruano and M. Tortosa, J. Am. Chem. Soc., 2012, 134, 15165–15168 CrossRef PubMed.
- J. Zhao, Z. Niu, H. Fu and Y. Li, Chem. Commun., 2014, 50, 2058–2060 RSC.
- T. Itoh, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2016, 138, 7528–7531 CrossRef PubMed.
- K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 7567–7570 CrossRef PubMed.
- K. Semba, Y. Ohtagaki and Y. Nakao, Org. Lett., 2016, 18, 3956–3959 CrossRef PubMed.
- K. B. Smith, K. M. Logan, W. You and M. K. Brown, Chem. – Eur. J., 2014, 20, 12032–12036 CrossRef PubMed.
- K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 851–855 CrossRef PubMed.
- K. M. Logan, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 5228–5231 CrossRef PubMed.
- K. B. Smith and M. K. Brown, J. Am. Chem. Soc., 2017, 139, 7721–7724 CrossRef PubMed.
- K. B. Smith, Y. Huang and M. K. Brown, Angew. Chem., Int. Ed., 2018, 57, 6146–6149 CrossRef PubMed.
- I. Kageyuki, I. Osaka, K. Takaki and H. Yoshida, Org. Lett., 2017, 19, 830–833 CrossRef PubMed.
- N. Matsuda, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2013, 135, 4934–4937 CrossRef PubMed.
- R. Sakae, N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1228–1231 CrossRef PubMed.
- A. Parra, L. Amenós, M. Guisán-Ceinos, A. López, J. L. García Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833–15836 CrossRef PubMed.
- H.-C. Jiang, X.-Y. Tang and M. Shi, Chem. Commun., 2016, 52, 5273–5276 RSC.
- K. Kato, K. Hirano and M. Miura, J. Org. Chem., 2017, 82, 10418–10424 CrossRef PubMed.
- R. Sakae, K. Hirano and M. Miura, J. Am. Chem. Soc., 2015, 137, 6460–6463 CrossRef PubMed.
- V. Lillo, M. R. Fructos, J. Ramírez, A. A. C. Braga, F. Maseras, M. M. Díaz-Requejo, P. J. Pérez and E. Fernández, Chem. – Eur. J., 2007, 13, 2614–2621 CrossRef PubMed.
- H.-Y. Jung, X. Feng, H. Kim and J. Yun, Tetrahedron, 2012, 68, 3444–3449 CrossRef.
- H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita and K. Takaki, Angew. Chem., Int. Ed., 2012, 51, 235–238 CrossRef PubMed.
- T. Itoh, T. Matsueda, Y. Shimizu and M. Kanai, Chem. – Eur. J., 2015, 21, 15955–15959 CrossRef PubMed.
- Y. Takemoto, H. Yoshida and K. Takaki, Chem. – Eur. J., 2012, 18, 14841–14844 CrossRef PubMed.
- Y. Takemoto, H. Yoshida and K. Takaki, Synthesis, 2014, 3024–3032 Search PubMed.
- H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2015, 51, 6297–6300 RSC.
- S. Mun, J.-E. Lee and J. Yun, Org. Lett., 2006, 8, 4887–4889 CrossRef PubMed.
- H. Chea, H.-S. Sim and J. Yun, Adv. Synth. Catal., 2009, 351, 855–858 CrossRef.
- J.-E. Lee and J. Yun, Angew. Chem., Int. Ed., 2008, 47, 145–147 CrossRef PubMed.
- V. Lillo, A. Prieto, A. Bonet, M. M. Diaz-Requejo, J. Ramirez, P. J. Perez and E. Fernandez, Organometallics, 2009, 28, 659–662 CrossRef.
- I. H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664–11665 CrossRef PubMed.
- W. J. Fleming, H. Muller-Bunz, V. Lillo, E. Fernandez and P. J. Guiry, Org. Biomol. Chem., 2009, 7, 2520–2524 RSC.
- V. Lillo, A. Bonet and E. Fernandez, Dalton Trans., 2009, 2899–2908 RSC.
- J.-E. Lee, J. Kwon and J. Yun, Chem. Commun., 2008, 733–734 RSC.
- B. H. Lipshutz, Ž. V. Bošković and D. H. Aue, Angew. Chem., Int. Ed., 2008, 47, 10183–10186 CrossRef PubMed.
- S. B. Thorpe, X. Guo and W. L. Santos, Chem. Commun., 2011, 47, 424–426 RSC.
- A. R. Burns, J. S. Gonzalez and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 10827–10831 CrossRef PubMed.
- C. L. Peck, J. A. Calderone and W. L. Santos, Synthesis, 2015, 2242–2248 Search PubMed.
- A. K. Nelson, C. L. Peck, S. M. Rafferty and W. L. Santos, J. Org. Chem., 2016, 81, 4269–4279 CrossRef PubMed.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036–11037 CrossRef PubMed.
- G. A. Molander and S. R. Wisniewski, J. Am. Chem. Soc., 2012, 134, 16856–16868 CrossRef PubMed.
- M. L. McIntosh, C. M. Moore and T. B. Clark, Org. Lett., 2010, 12, 1996–1999 CrossRef PubMed.
- M. A. Beenen, C. An and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6910–6911 CrossRef PubMed.
- K. Kubota, E. Yamamoto and H. Ito, J. Am. Chem. Soc., 2015, 137, 420–424 CrossRef PubMed.
- K. Kubota, S. Osaki, M. Jin and H. Ito, Angew. Chem., Int. Ed., 2017, 56, 6646–6650 CrossRef PubMed.
- H. Zhao, L. Dang, T. B. Marder and Z. Lin, J. Am. Chem. Soc., 2008, 130, 5586–5594 CrossRef PubMed.
- W. Zhu and D. Ma, Org. Lett., 2005, 8, 261–263 CrossRef PubMed.
- S. Ando, H. Matsunaga and T. Ishizuka, J. Org. Chem., 2015, 80, 9671–9681 CrossRef PubMed.
- H. Yoshida, Y. Takemoto, S. Kamio, I. Osaka and K. Takaki, Org. Chem. Front., 2017, 4, 1215–1219 RSC.
- J. Ratniyom, N. Dechnarong, S. Yotphan and S. Kiatisevi, Eur. J. Org. Chem., 2014, 1381–1385 CrossRef.
- T. Niwa, H. Ochiai and T. Hosoya, ACS Catal., 2017, 7, 4535–4541 CrossRef.
- H. Sakaguchi, Y. Uetake, M. Ohashi, T. Niwa, S. Ogoshi and T. Hosoya, J. Am. Chem. Soc., 2017, 139, 12855–12862 CrossRef PubMed.
- C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528–532 CrossRef PubMed.
- H. Ito and K. Kubota, Org. Lett., 2012, 14, 890–893 CrossRef PubMed.
- X. Lou, Z. Q. Zhang, J. H. Liu and X. Y. Lu, Chem. Lett., 2016, 45, 200–202 CrossRef.
- L. J. Mao, R. Bertermann, K. Emmert, K. J. Szabo and T. B. Marder, Org. Lett., 2017, 19, 6586–6589 CrossRef PubMed.
- L. J. Mao, K. J. Szabo and T. B. Marder, Org. Lett., 2017, 19, 1204–1207 CrossRef PubMed.
- K. Kubota, H. Iwamoto, E. Yamamoto and H. Ito, Org. Lett., 2015, 17, 620–623 CrossRef PubMed.
- D. Hemming and P. G. Steel, unpublished results, 2018.
- P. V. Ramachandran, D. Pratihar, D. Biswas, A. Srivastava and M. V. Ram Reddy, Org. Lett., 2004, 6, 481–484 CrossRef PubMed.
- C. Zhong, S. Kunii, Y. Kosaka, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 11440–11442 CrossRef PubMed.
- H. Ito, C. Kawakami and M. Sawamura, J. Am. Chem. Soc., 2005, 127, 16034–16035 CrossRef PubMed.
- E. Yamamoto, Y. Takenouchi, T. Ozaki, T. Miya and H. Ito, J. Am. Chem. Soc., 2014, 136, 16515–16521 CrossRef PubMed.
- D. S. Hemming, E. P. Talbot and P. G. Steel, Tetrahedron Lett., 2017, 58, 17–20 CrossRef.
- H. Ito, S. Ito, Y. Sasaki, K. Matsuura and M. Sawamura, J. Am. Chem. Soc., 2007, 129, 14856–14857 CrossRef PubMed.
- H.-Y. Bin, X. Wei, J. Zi, Y.-J. Zuo, T.-C. Wang and C.-M. Zhong, ACS Catal., 2015, 5, 6670–6679 CrossRef.
- H. Iwamoto, K. Kubota, E. Yamamoto and H. Ito, Chem. Commun., 2015, 51, 9655–9658 RSC.
- S. K. Bose, S. Brand, H. O. Omoregie, M. Haehnel, J. Maier, G. Bringmann and T. B. Marder, ACS Catal., 2016, 6, 8332–8335 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |