
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral catalysts immobilized on achiral polymers: effect of the polymer support on the performance of the catalyst
Belén
Altava
 ,
M. Isabel
Burguete
,
Eduardo
García-Verdugo
* and
Santiago V.
Luis
*
,
M. Isabel
Burguete
,
Eduardo
García-Verdugo
* and
Santiago V.
Luis
*
Department of Inorganic and Organic Chemistry, University Jaume I, Avda Sos Baynat s/n, E-12071, Castellón, Spain. E-mail: cepeda@uji.es; luiss@uji.es
First published on 26th March 2018
Abstract
Positive effects of the polymeric support on the performance of supported chiral catalysts, in terms of activity, stability and selectivity–enantioselectivity, have been reported when the support is properly selected and optimized opening the way to the design of more efficient catalytic systems.
 Belén Altava | Belén Altava graduated in Chemistry at the University of Zaragoza (1992), and completed her PhD at the Universitat Jaume I (1996) under the supervision of Prof. S. V. Luis. After postdoctoral stays at the University of Düsseldorf (Prof. G. Wulff) and Novartis (Dr H. U. Blaser), she returned to the Universitat Jaume I (2001), where she is currently a member of the research group of Supramolecular and Sustainable Chemistry with her research focusing on supported asymmetric catalysis, ionic liquids, green chemistry and supramolecular chemistry. For the last 12 years she has been involved in the Spanish Interuniversity Master and PhD Programs in Sustainable Chemistry. |
 M. Isabel Burguete | Maria Isabel Burguete graduated in Chemistry at the University of Zaragoza in Spain and after a stay at the University of Pittsburgh (USA) under the supervision of Dr J. Rebek, she completed her PhD at the University of Valencia in 1989 under the direction of Dr F. Gaviña working on regulated crown ethers and convergent diacids. In 1989 she took an academic appointment at the University Jaume I in Castellón (Spain), where she is currently Professor of Organic Chemistry. Her main field of research is developing new tools, in particular homogeneous and supported catalysis approaches, in Green and Sustainable Chemistry. |
 Eduardo García-Verdugo | Eduardo García-Verdugo obtained his chemistry degree from the University of Valencia (1995) and his PhD from the University Jaume I (2001). After working as a researcher at Nottingham University (M. Poliakoff, until 2004), he returned to Spain as a Ramon y Cajal fellow at the University Jaume I until 2009, and then obtained a permanent position at the CSIC-ICP (Madrid, 2009–2010). In 2010, he moved to a permanent academic position at the University Jaume I, working on the integration of different enabling techniques (catalysis, polymers, continuous flow processes, microreactors, bio-catalysis, neoteric solvents) to develop efficient and greener organic transformations. |
 Santiago V. Luis | Santiago V. Luis studied Chemistry at the University of Zaragoza and completed his PhD at the University of Valencia in 1983 working on mechanistic studies. After a postdoctoral work at the University of Pittsburgh under the supervision of Dr J. Rebek, in Supramolecular chemistry, he obtained an academic appointment at the University Jaume I in Castellón (Spain) where he is currently Professor of Organic Chemistry and Head of the research group in Surpramolecular and Sustainable Chemistry. His research areas involve Supramolecular and Biomimetic chemistry and new tools in Sustainable and Green Chemistry, with emphasis on catalysis and flow chemistry. |
Introduction and scope
Chirality plays a major role in chemistry. The physicochemical and biological properties of enantiomeric compounds can significantly differ, for instance due to stereospecific interactions with enzymes and receptors in biological systems. Enantiomers of chiral drugs can present differences in pharmacological activity and accordingly in toxicity. In this regard, new therapeutic formulations, driven by both regulatory and therapeutic motivations, are moving towards the use of enantiopure ingredients.1 Indeed, the chiral drug industry constitutes approximately one-third of all drug sales worldwide. It is expected that nearly 95% of the pharmaceutical drugs would be chiral by 2020.2 Enantiopure chiral compounds are also gaining importance in agrochemicals, such as fungicides, herbicides, and insecticides.3 Improvements in performance and the elimination or reduction of harmful effects can be achieved by switching from racemic pesticides to single enantiomers.4–6 Among the different methodologies for the preparation of enantiopure chiral compounds, asymmetric catalysis can be envisioned as an ideal methodology where a small amount of a well-designed chiral catalyst is able to transform achiral substrates into enriched chiral compounds stereoselectively in a large quantity.7,8 Since the early days of this field thousands of chiral ligands and their transition metal complexes have been developed and many of them have been proved to be highly effective in the asymmetric formation of C–H, C–C, C–O, and C–N bonds.9 The award, in 2001, of the Nobel Prize in Chemistry to W. S. Knowles,10 R. Noyori,11 and K. B. Sharpless12 recognised the impact of this field on modern chemistry. Furthermore, in the last few years, organocatalytic asymmetric methods have blossomed as an alternative way to produce enriched chiral molecules.13–15Since the early developments in homogeneous asymmetric catalysts, their immobilisation onto inorganic or organic supports has been targeted.16,17 The supported versions of reagents and catalysts present, ideally, several distinct advantages in terms of their practical use, relative to their homogeneous analogues.18,19 In particular, we should mention the facilitation of the work up, with the possibility of easy separation and recovery of the supported systems from the reaction mixture and the concomitant facilitation of recycling. These advantages are even more pronounced when enantioselective reagents and catalysts are considered. For those cases, the possibility of recovering and reusing the chiral component, which is generally the most expensive component of the system, can be a critical factor for assessing their practical utility.
A large number of efforts have been devoted in recent years to the heterogenisation of systems that have been proved useful in solution in particular as enantioselective reagents and catalysts.20–23 However, in spite of the well-stabilised advantages of a supported catalyst combining the best of both homogeneous and heterogeneous worlds, after 40 years of research and development enough examples of large scale processes using chiral supported catalysts are still lacking.
Usually, the immobilisation is carried out on commercially available polymeric supports, which are not optimised for immobilisation, thus leading sometimes to deceiving catalytic performances. It is often reported that results obtained with the supported systems, even successfully enabling the separation and recovery of the catalysts, are quite different from those expected from solution studies, and can suffer from lower activities and selectivities. Moreover, it is generally concluded, not necessarily a correct idea, that “the best homogeneous catalyst will remain the best upon immobilization”, assuming that the polymeric matrix can have either no effect, in the best case, or a negative one, leading to less efficient catalysts upon immobilisation.
Hence, when analysing data from the literature, many authors conform to the idea that enantioselective supported reagents and catalysts are usually less efficient than the homogeneous ones in terms of the three critical catalytic parameters: activity, stability and selectivity, in particular enantioselectivity.24
Fortunately, this is not always true. Many results clearly reveal how the presence of the matrix can be used advantageously to increase the efficiency of the supported species over that of the related homogeneous ones.
The present critical review intends to highlight significant recent examples of how the matrix can really contribute to improve the global efficiency of chiral immobilized catalysts. The period from 2000 to 2017 has been mainly covered, although some selected older references have also been included when their relevance to a given aspect made it appropriate. This work does not intend to cover exhaustively the literature in this field for the above mentioned period but to present examples able to illustrate the main concepts discussed here. The challenge is not anymore the immobilisation of catalysts to facilitate their recovery and reuse, but the development of methodologies and new advanced materials to enhance their catalytic properties as to surpass those of the soluble counterparts. These effects will be analysed taking into account the above mentioned three main factors: activity, stability and selectivity. For the sake of clarity and to provide well identified elements of comparison, this review will be focused on the use of organic polymeric matrices as supports. The use of silica and other inorganic materials as supports for the asymmetric catalysts is out of the scope of this work and will only be mentioned as illustration in a few cases. Some reviews dealing with the effect of the inorganic support on the efficiency of immobilised asymmetric catalysts can be also found in the literature.25–28 Some examples of non-chiral systems or from supported reagents (stoichiometric instead of catalytic systems) will also be presented, when appropriate, to highlight the effects of the support.
A very particular case regarding the influence of the polymer matrix on the efficiency of the catalyst is given by the immobilisation of the catalyst onto chiral polymer supports.29,30 Although examples of such systems are still limited, attempts at using the chirality of the polymer matrix to exert control over catalytic efficiency, in particular over enantioselectivity, are gaining great success. Immobilised catalysts containing either chiral catalytic pendant units31 or achiral ones have been developed.32,33 In the latter class, the asymmetric induction solely relies on the chirality of the polymer matrix, in particular through the adoption of helical structures. Although these new types of catalysts supported on chiral polymer matrices are undergoing very rapid development in recent years, they are not discussed in depth in this review article as they lie out of its main scope.
The polymer matrix as a design vector
In the initial steps of the work with polymers as supports, it is sometimes assumed that the polymer, if appropriately selected, would play no other role than that of an inert matrix supporting the active sites and facilitating their simple isolation and reuse. This is a naive and wishful thinking and in most of the cases distinct polymer effects can be detected.34–37 This is even true when the nature of the support is carefully chosen as to avoid the presence of any other interfering functional group or when the active site is separated from the matrix through a long spacer. Clearly, the support determines, to a good extent, not only the diffusion of the reagents and products to and from the active sites but also the actual conditions and microenvironment of the reaction taking place. Therefore, the selection of the nature of the polymeric support and how the catalyst is immobilised are key factors for developing efficient polymeric supported asymmetric catalysts.At least four elements should be considered when designing an immobilized catalyst: (i) the nature of the attachment of the catalytic sites to the support, (ii) the nature and length of the spacer between the catalytic sites and the polymeric network, (iii) the density of catalytic sites and their location along the polymeric network and (iv) the physicochemical nature of the polymeric backbone. Thus, the methodology employed for the preparation of an immobilised catalyst defines, to a great extent, its resulting properties and potential applications. In this regard, the challenge is to develop methodologies where both ligand and polymeric matrix are specifically designed to obtain a given supported catalyst. Accordingly, through the optimisation of the ligand and support, the same matrix effects leading to negative effects after immobilization of a good homogeneous catalyst might be able to improve those obtained with a less optimal one. Some results based on systematic studies clearly reflect how the presence of a well-designed support can be advantageous to increase the efficiency, especially in terms of activity and selectivity, of the supported species relative to that of the homogeneous ones.
There are a wide range of classical and new methodologies to develop chiral polymer-supported catalytic systems and this section briefly summarises these approaches. Different strategies have been evaluated for immobilisation: (i) covalent bonding, (ii) ion-pair formation, (iii) encapsulation or (iv) entrapment.38–40 Without any doubt, covalent binding is by far the most frequently used strategy, significantly limiting the potential leaching of catalytically active components. Two main possibilities exist for carrying out this approach. The first one relies on the chemical modification of preformed polymeric supports. Alternatively, the polymerization of functional monomers with the desired functionality, by themselves or in the presence of additional monomers, has also been exploited.
Among the different types of polymeric supports, a clear differentiation can be established regarding their solubility in the reaction medium. Both soluble and insoluble polymeric materials have been investigated as supports. Linear soluble polymers have been early envisioned as suitable media for catalyst immobilisation.41,42 They can significantly limit the diffusional problems found in crosslinked systems. However, their recovery and recycling is not so straightforward, requiring tuneable solubility properties.43–45 With the advances made in macromolecular chemistry in recent years, it is now possible to design and synthesize complex soluble polymers with new architectures and well-defined catalyst location and loading along the polymeric chain. Besides, the control of the polymer structure allows not only the immobilisation of the catalyst but also mimicking of the form and function of Nature's nanostructures using, for instance, amphiphilic block copolymers that spontaneously self-assemble in selective solvents into specific assemblies.46
Dendrimers have emerged as alternative soluble supports for the immobilisation of different types of catalytic units filling the gap between homogeneous and heterogeneous systems.47 They are highly branched macromolecules with the larger ones having precisely defined globular structures. It is possible to control the incorporation of the catalytic sites within the dendrimer either at the core, at the periphery, or at intermediate positions.48 The different locations can be used to define and tune their catalytic performance. Dendrimer recovery can be achieved by precipitation from solution or, if large enough, using membrane filters.49
Polystyrene and styrene/divinylbenzene (PS–DVB) copolymers (Merrifield type resins) are by far the most common insoluble polymeric supports exploited for the preparation of immobilised catalysts.50,51 This type of supports, especially the ones with a low crosslinking degree (1–2% DVD gel-type) initially developed for solid-phase peptide synthesis,52 have been widely used as functionalized polymers because of their stability and compatibility with a wide range of reaction conditions, their good swelling properties or their reasonably high loading capacity (>1 mmol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g−1), providing access to a large number of postfunctionalization processes.53 The swelling or solvation, the effective pore size, the surface area and the chemical and mechanical stability of the polymer are important factors that control the diffusion of reagents and substrates into the active sites. The degree of crosslinking of the resin, the functional fragments introduced and the conditions employed during the preparation of the polymers allow fine-tuning of these properties. Therefore, along with the originally used microporous or gel-type resins, a variety of macroporous/macroreticular resins have also been developed to improve the performance of these polymers as catalyst supports. They can be produced, for example, by phase separation, emulsion polymerization, or by templating methods.54,55 In the same way, in addition to the classical PS–DVB polymeric networks of Merrifield resins, other organic polymers like polyvinyls, polyacrylates, cellulose or PS–DVB copolymerised with other polar monomers or modified with hydrophilic flexible units such as polyglycol chains acting as crosslinking units or linkers have been developed and tested as alternative polymeric supports.56–60
g−1), providing access to a large number of postfunctionalization processes.53 The swelling or solvation, the effective pore size, the surface area and the chemical and mechanical stability of the polymer are important factors that control the diffusion of reagents and substrates into the active sites. The degree of crosslinking of the resin, the functional fragments introduced and the conditions employed during the preparation of the polymers allow fine-tuning of these properties. Therefore, along with the originally used microporous or gel-type resins, a variety of macroporous/macroreticular resins have also been developed to improve the performance of these polymers as catalyst supports. They can be produced, for example, by phase separation, emulsion polymerization, or by templating methods.54,55 In the same way, in addition to the classical PS–DVB polymeric networks of Merrifield resins, other organic polymers like polyvinyls, polyacrylates, cellulose or PS–DVB copolymerised with other polar monomers or modified with hydrophilic flexible units such as polyglycol chains acting as crosslinking units or linkers have been developed and tested as alternative polymeric supports.56–60
In the last decade, a whole new range of different porous networks, including crystalline and amorphous structures, have blossomed as suitable alternatives to traditional organic polymers.61 In some instances, they can combine high surface areas, good physicochemical stability and an increasing degree of functional modularity.
Among these materials based on an organic skeleton, the design and synthesis of chiral Metal–Organic Frameworks (MOF) or self-supported systems offer a new strategy for the generation of robust immobilized asymmetric catalysts combining supramolecular chemistry, coordination chemistry and catalysis concepts. These systems are obtained in a straightforward way through the coordination of a polytopic ligand and metal species leading to the self-assembly of the metal–ligand components to generate an extended organometallic/coordination network.
Porous Organic Polymers (POPs) with high permanent porosity, high accessible specific surface areas and very robust chemical properties have been developed by using extensive crosslinked covalent bonds between organic building blocks. Conceptually, the component organic fragments in POPs are connected in a similar way as in MOFs but using strong covalent bonds with the linkers acting as connectors, instead of the relatively weak coordinative bonds to metal ions and clusters present in MOFs.62–64 They can be obtained by a variety of synthetic routes offering new types of functionalised polymeric networks. Their topology and porosity can be tailored by varying the connectivity, size, and geometry of the building blocks by a bottom-up approach leading ideally to a perfect control of the size and shape of the catalytic environment formed inside the cavities of the material or on the surface of the solid, enhancing the control of the reaction.61,65 Within this category of supports (POPs) it is possible to consider Hyper-Cross-linked Polymers (HCPs), which are obtained by post-crosslinking of polystyrene-type precursors in their swollen state (Davankov's hypercrosslinked polystyrene polymers) or from the condensation of small building blocks (Webster's poly(arylcarbinol) networks).66 They present a very rigid non-collapsible polymeric matrix with small pore sizes and high surface areas, and micropore volumes.67 Alternatively, different synthetic strategies have been developed to bring together numerous organic building blocks directly or with the aid of suitable linkers onto predesigned skeletons of porous networks. Depending on the nature of the synthetic motifs and methodologies, these systems can be defined as Covalent Organic Frameworks (COFs),68,69 or Polymer Organic Frameworks (POFs),70,71 which can be Conjugated Microporous Polymers (CMPs),72,73 Polymers with Intrinsic Microporosity (PIMs),74 or Porous Polymeric Aromatic Frameworks.75
Evaluating the matrix effect
Different factors should be considered to rationalize the effect of the polymeric matrix on the efficiency of immobilised catalysts leading to enhanced turnover numbers and frequencies, stability, and selectivity. The first one is related to the modifications needed, on the catalytic structure, to provide a way for the efficient immobilization onto the polymeric matrix. It is well known that for stoichiometric or catalytic enantioselective transformations, even minor structural modifications in the chiral system can have important effects on the results obtained.76,77The second factor is related to the effect of the polymeric matrix itself. In order to detect and understand experimental results directly connected with the nature of the matrix, a very systematic approach is required. This involves the preparation of the appropriate homogeneous counterpart having exactly the same structural features as the supported one, including the presence of the additional groups required for anchoring, and carrying out the corresponding reaction strictly under the same experimental conditions with both the homogeneous and the heterogeneous system. For instance, Larionov et al. have recently reported the immobilization of a chiral-at-metal iridium Lewis acid catalyst (1) on polystyrene macro beads using different linkers (2, 3) for the attachment (Fig. 1).78 When the resulting immobilised Ir–chiral complexes were tested as catalysts for the Friedel–Crafts alkylation of indole (6) with the α,β-unsaturated 2-acyl imidazole 7, the supported catalysts (2–3) provided, in comparison to the homogeneous original catalyst (1), higher enantioselectivities (97% ee vs. 93% ee) while featuring lower catalytic activities. However, when the results were evaluated in comparison with their structural homogeneous homologues (4, 5), the activity and enantioselectivity were comparable and the results could be correlated with the structural modification required for the immobilisation. Many times, this is not taken into account and claims about the preparation of supported reagents or catalysts performing differently than the homogeneous ones need to be considered with caution and do not provide any hint towards the understanding of the effects of the matrix and for the design of new supported systems in which the polymeric backbone contributes to improve their efficiency.
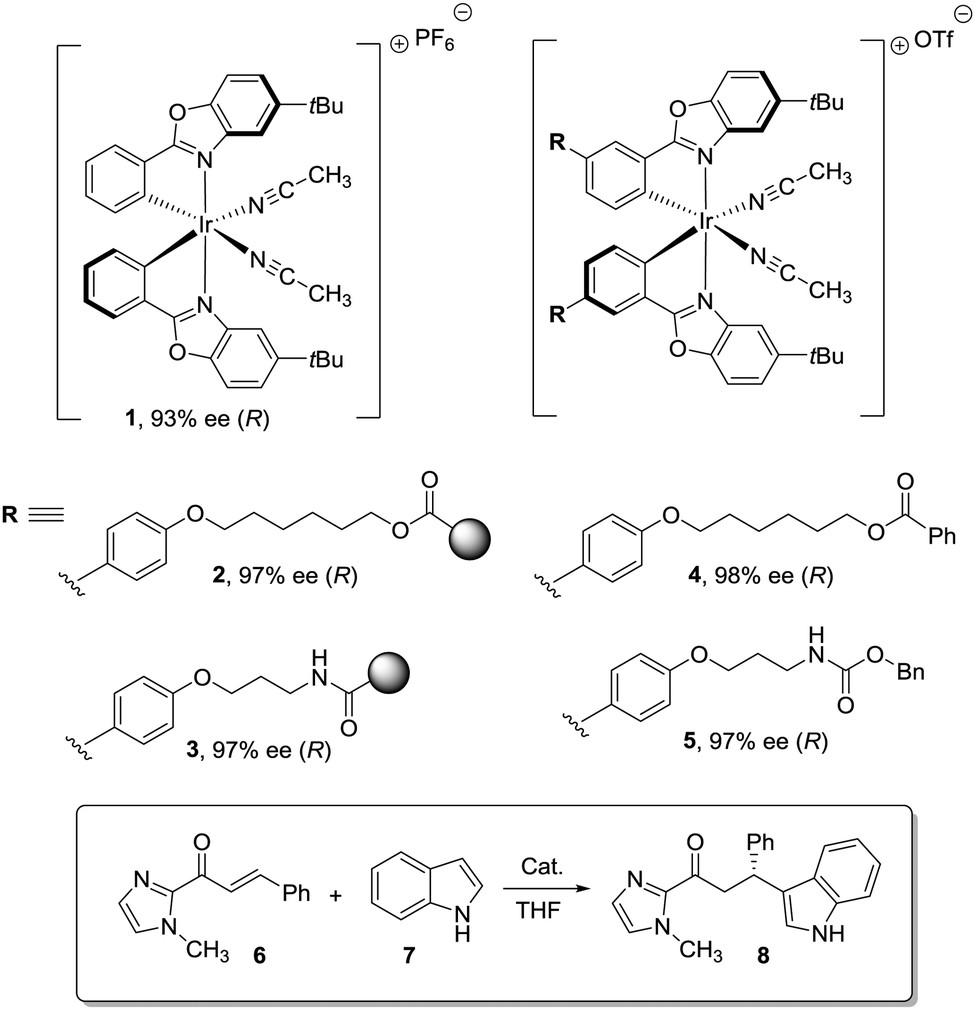 | ||
| Fig. 1 Effect of structural modification and immobilization on the performance of chiral-at-metal iridium catalysts. | ||
A good understanding of the mechanistic role of the catalyst, i.e. the presence of monometallic vs. multi-metallic species in the transition state and how this affects the reaction efficiency, should be also taken into account to design the adequate immobilisation protocol either favouring the site isolation or the possible interaction between catalytic sites. Besides, to properly assess recyclability, stability and the mechanisms of deactivation, kinetic data should be provided in each cycle (i.e. initial rates or TOF at low conversion degrees).†
For the correct evaluation of the efficiency of a supported enantioselective catalyst, three different aspects need to be addressed: (i) activity, i.e. the effect of immobilization on the rate of the reaction under study; (ii) stability, i.e. the analysis of how much actually the heterogenisation process contributes to stabilize the reagent/catalyst and to increase the possibilities of reusing and recycling the reagents and/or catalysts, by performing systematic studies of the stability and deactivation of catalysts; and (iii) selectivity, and in particular, in the present context, enantioselectivity. Energy differences between the transition states leading to the formation of either one or the other enantiomer, with the help of a chiral reagent or catalyst, are usually small and, accordingly, they can be very much affected by the microenvironment.
Activity
The catalyst immobilisation is usually carried out on commercially available polymeric supports, which are not optimised for immobilisation (Fig. 2). As a result, often the reactions taking place with the participation of polymer-supported species are reported to be slower than those using the homogeneous analogues and require much longer reaction times. In many cases, the morphology of the polymer network is one of the main factors determining the accessibility of the active catalytic sites and, accordingly, the activity of the corresponding supported systems.79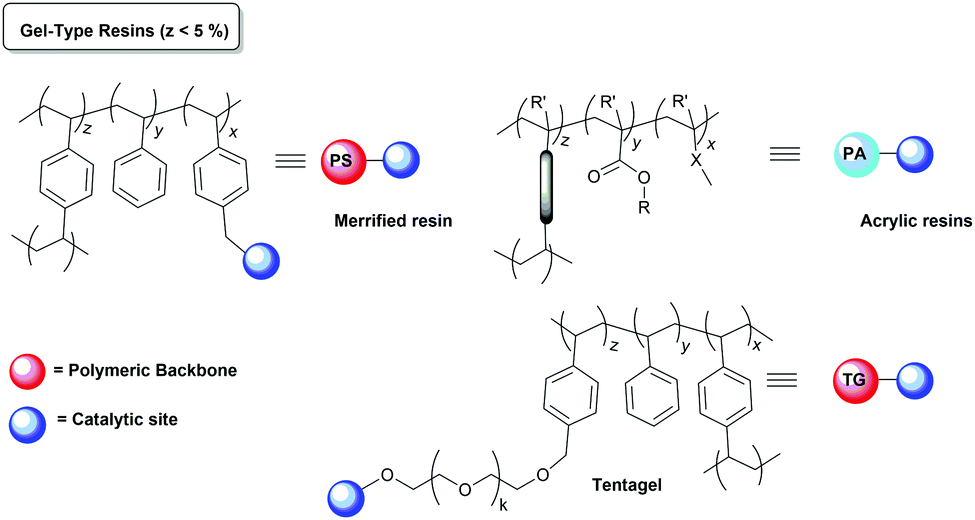 | ||
| Fig. 2 Examples of polymeric supports commonly used for immobilization of chiral catalysts. | ||
Gel-type polymeric supports
Most examples of insoluble polymeric reagents and catalysts are related to the use of gel-type PS–DVB resins commercially available in the form of beads. Those gel-type resins do not have any permanent porosity and need to be swollen in an appropriate solvent in order to make accessible the functional sites inside the bead.80 On the other hand, some crosslinking degree is necessary to obtain insoluble materials with the appropriate morphologies. In the case of polystyrene resins (PS), which are the most standard materials in this field, 1–4% of divinylbenzene (DVB, a commercial mixture of different isomers) is generally used as the crosslinking agent. Swelling is always less effective at the crosslinked regions of the polymer and accordingly functional sites located at those positions become less accessible.81Accordingly, an increase in crosslinking for this type of polymers is usually accompanied by a lower swelling and by a decrease in the accessibility of the functional sites.82 This is always reflected in slower reaction rates. Thus, when working with this kind of materials, it is important to keep in mind that the activity of a given supported species will be very much dependent on the solvent used and the crosslinking degree of the polymer.83–85 This has been studied in detail in the case of different families of functional, catalytic resins.86–88
In this regard, the introduction of long aliphatic or ether spacers favouring compatibility with a wide range of solvents58–60,92,93 or the substitution of DVB by more flexible crosslinkers has been widely explored.84 Thus, Montanari and coworkers found that the use of long aliphatic spacers (up to 10 methylene units) and low crosslinking degrees (1%) increased the corresponding activities 2–4 times in polymer-supported phase transfer catalysts. This classical effect was based on the higher flexibility of the functional chain and the increase in the lipophilicity of the polymer facilitating its swelling.93–96
Dyer et al. also illustrated nicely those concepts in the case of Ti–diol complexes supported using spacer-modified gel-type resins (Fig. 3).97 In this work, functional resins 9 were used as catalysts for the Diels–Alder reaction between cyclopentadiene (10) and methyl acrylate (11). The catalyst 9c with the larger spacer (–(CH2)9–) was the most active one, suggesting that the use of a long spacer improves the accessibility to the Lewis acid sites. Rather surprisingly, introducing a modified spacer unit (13, X = –(CH2)5–C6H4–CH2) led to markedly slower catalysts. This was associated with the poor swelling behaviour of those polymers. No improvement in activity relative to the homogeneous system was reported in this case, although the homogeneous counterpart selected presented some structural differences with the polymer-bound system. Similar trends were found in the case of 2% crosslinked polymer-bound lithium dialkylamides 14.98
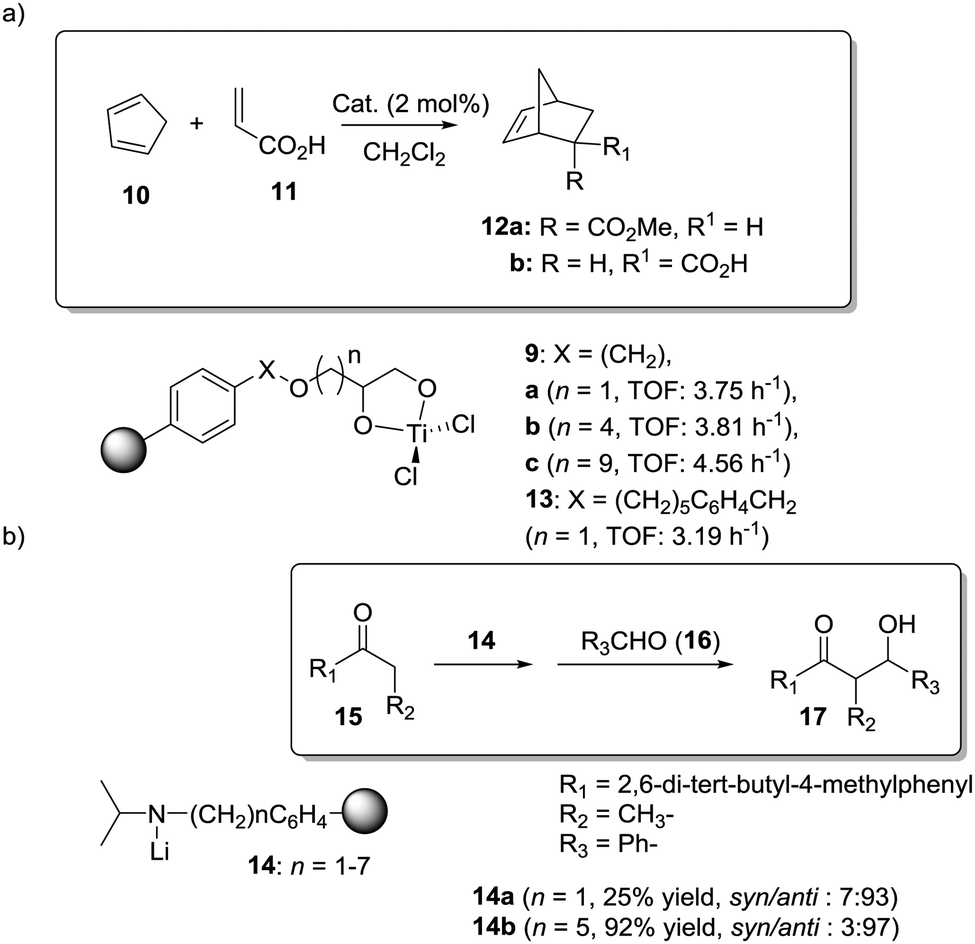 | ||
| Fig. 3 Examples of supported catalysts where the length and nature of the linker enable modification of the activity. | ||
The introduction of spacer chains between the polymeric backbone and the lithium dialkylamide fragment enhanced the yields observed for the crossed aldol reaction of various carbonyl compounds with aldehydes. An increase in the size of the spacer seems to have a favourable effect up to n = 5. For n > 5, no further improvement was observed. Given the complex nature of many lithium derivatives, it is reasonable to assume that the small increases in activity found in some instances, relative to LDA, for polymer-bound dialkylamides 14, can be associated with the same phenomenon.98 Nevertheless, data given need to be used with some caution as no kinetic data were provided, with just yields of isolated compounds given as proof of activity.
Toy and coworkers have also demonstrated a direct relationship between catalytic efficiency, crosslinker nature, catalyst loading and swelling in the aza-Baylis–Hillman reactions catalysed by JandaJel-PPh3.99,100 Similar effects were observed in the work of Pedrosa, Andres and coworkers with a series of chiral ureas and thioureas immobilized on PS–DVB resins by either grafting and copolymerisation methodologies and studied for the aza-Henry reaction between N-Boc-benzaldimine (18) and nitromethane (19) under solvent free conditions (Fig. 4).101,102 Their results suggested that the linker connecting the catalytic moieties with the polymer framework led to marginal effects on enantioselectivity while the effects on activity were more relevant.
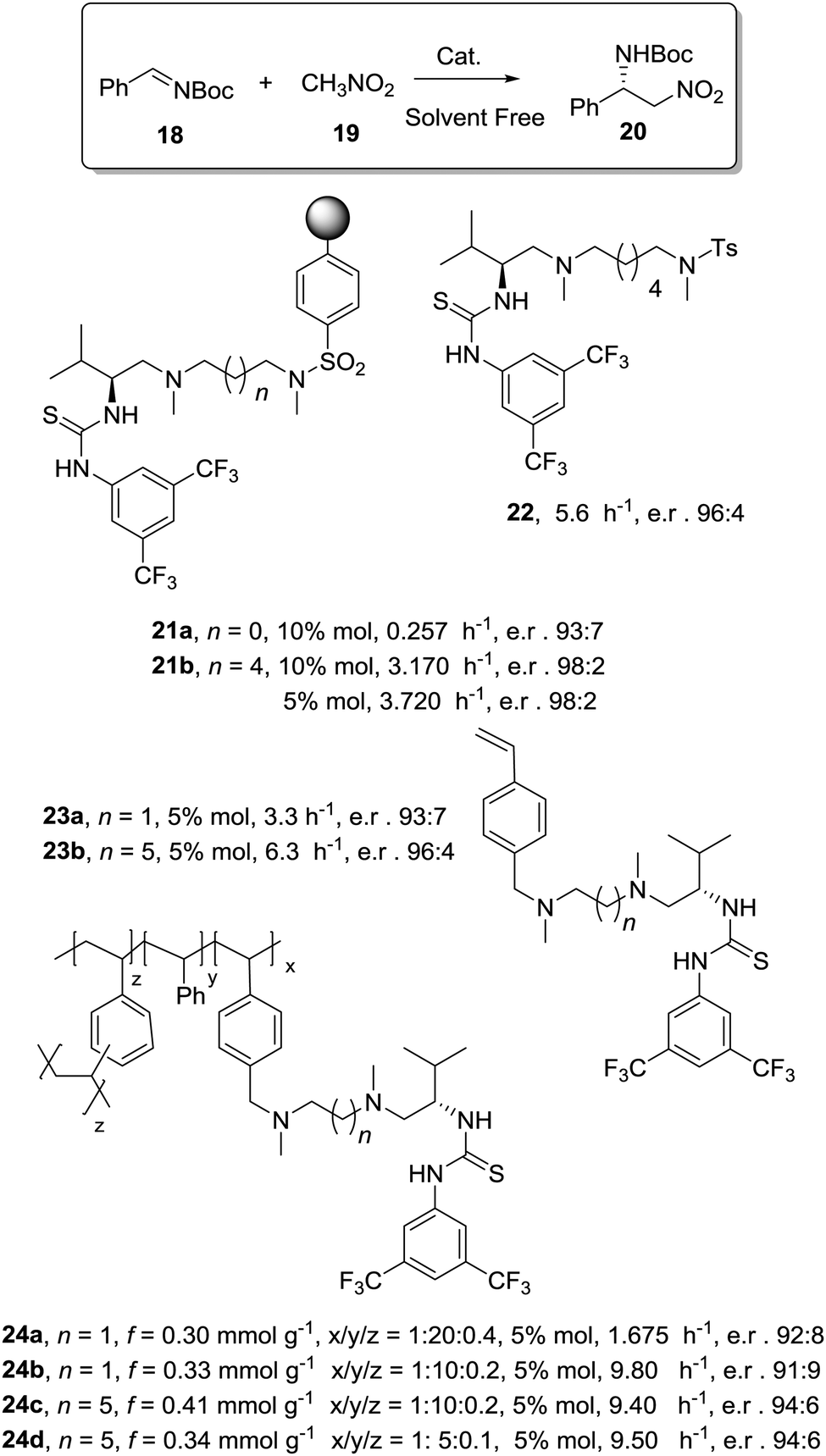 | ||
| Fig. 4 Activity of some supported thioureas for the aza-Henry reaction between N-Boc-benzaldimine (18) and nitromethane. | ||
Thus, for the systems prepared by grafting using sulfonamide groups for the attachment, up to a 12 times enhancement in activity was observed with the length of the spacer (3.17 h−1 for 21b with six methylene units vs. 0.257 h−1 for 21a with two methylene units). In good agreement with previous reports in this field presented above, the longer spacers locating the active sites away from the polymeric backbone provide more “solution-like” and active systems by improving the accessibility to the functional sites inside the bead. However, the extent of this effect is not enough to surpass the activity found for the homogeneous counterpart (5.6 h−1 for 22). A different situation was found for the catalysts obtained by a bottom-up synthesis of chiral bifunctional thioureas by copolymerization of styrene, 4-vinyl benzylamine derivatives and divinylbenzene as the crosslinker. Some of these systems (24a–b) showed an enhancement of the catalytic activity of 2.9–1.5 times in comparison with the homogeneous counterpart (23a–b). Noteworthily, in this case the role of the spacer had a minimal effect. These results highlight the importance of the methodology used in the preparation of the catalysts (grafting vs. polymerisation). When immobilisation takes place by polymerisation in the presence of the corresponding chiral monomers a more uniform and accessible distribution of the chiral catalytic sites within the polymeric network can be achieved. Hence, a bottom-up methodology usually provides better accessibility of the catalytic sites, minimizing the role played by the spacer. It must be noted that using a different attachment group in the linker (–CH2– instead of –SO2–) led to some changes in the activity, highlighting once again the importance of selecting the appropriate homogeneous counterpart in order to evaluate the performance of the immobilised catalysts.
The exact origin of the positive effects of the matrix on the rates is not so well defined in other cases. In general, they can be ascribed to the local microenvironment of the active site and this includes effects such as the hydrophobic/hydrophilic balance, the conformational flexibility of the functional sites or the interaction with functional groups or fragments present in the polymeric network. In many senses, the heterogenisation process can affect the rates (and likely the whole reactivity) very much in the same way a change in the solvent does.103 In this context, Pericàs and coworkers have recently reported the immobilization of a modified version of the triflate ammonium salt of the chiral vicinal diamine 25 and its use as catalyst for the Robinson annulation reaction.104 The substitution of one of the ethyl groups on the tertiary amine by a hydroxyethyl group allowed both the preparation of the polymer supported system (27) via the nucleophilic substitution of the chlorine atoms of a commercially available microporous Merrifield resin and the synthesis of the homogeneous counterpart 26 by reaction with benzyl bromide (Fig. 5). Important effects on activity were observed for the reaction between the commercially available diketone (28) and methyl vinyl ketone (29) in 2-MeTHF. Under identical experimental conditions, the immobilised catalyst (27, TOF 8.8 h−1, 91% ee) was 2.8 times more active than its homogeneous counterpart (26, TOF 3.1 h−1, 90% ee). Larger enhancements, up to 18.7 times, were found in comparison with unmodified homogeneous catalyst 25 (TOF 0.47 h−1, 89% ee). The polymer efficiently provided a suitable microenvironment to favour the local concentration of the substrates within the polymeric network, and in this case, the reaction inside the polymeric network takes place much more efficiently than in solution.80 While this effect could be somewhat masked by mass transfer limitations at room temperature, it became fully operational at 55–60 °C with a tenfold enhancement of the activity on going from room temperature to 55 °C.
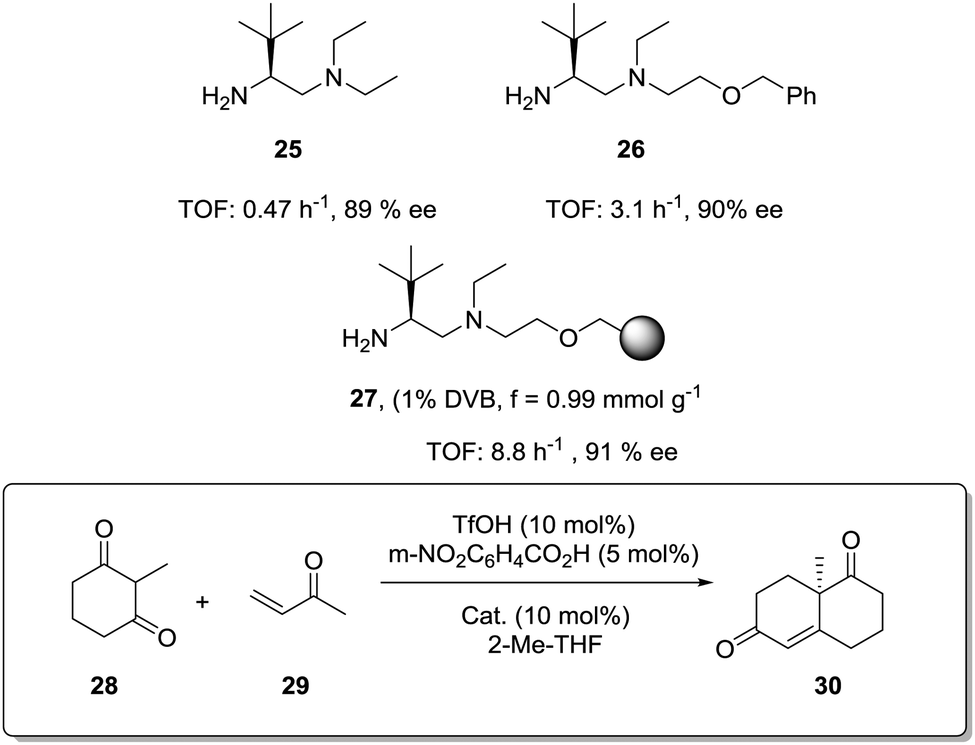 | ||
| Fig. 5 Enhancement in activity for a PS-supported chiral vicinal diamine. | ||
An important point to take into account for understanding the activity of insoluble polymer-supported systems is that, as rates can be much affected by diffusion, the temperature can have a very important effect on activity. A marked decrease in activity is often found on lowering the temperature, and this, sometimes, puts some limits on the use of low temperatures that are usually employed for enantioselective transformations under homogeneous conditions.105–107
As mentioned before, spacers have been used to attach the ligand to the polymer by a point remote from the active site (tail-tie), reducing any possible “polymeric (negative) effect” associated with the bulky polymeric matrix. However, in some cases, the spacer and/or linker themselves can play a non-innocent role in catalytic activity. These effects can be related to the modification of the hydrophobic/hydrophilic balance and/or to the presence of an additional functional group. Alexandratos and co-workers have systematically studied how the microenvironment surrounding the active sites in polymer-supported systems can be used to enhance the reactivity of the intermediates and the rate of product formation, using the Mitsunobu reaction as the benchmark reaction.108
The potential of the 1,2,3-triazole moiety as the attachment point in the linker has been extensively exploited. Its use has facilitated the immobilization of catalytic units with a wide range of structural diversity using a clean 1,3-dipolar cycloaddition reaction (Fig. 6). This triazole linker allows not only the grafting of catalytic moieties but also the incorporation of spacers drawing them away from the polystyrene chain, in an attempt to improve the mass transfer and to accelerate the reaction. By using several variations of this click-chemistry strategy, different derivatives of 4-hydroxyproline have been grafted onto several commercially available resins (Merrified, ArgoPore and PS-PEG NovaBioSyn) (Fig. 6).109–111 The results observed with the resulting supported proline organocatalysts for the aldol reaction between cyclohexanone and benzaldehyde in water at room temperature suggest the non-innocent nature of this linker/spacer.
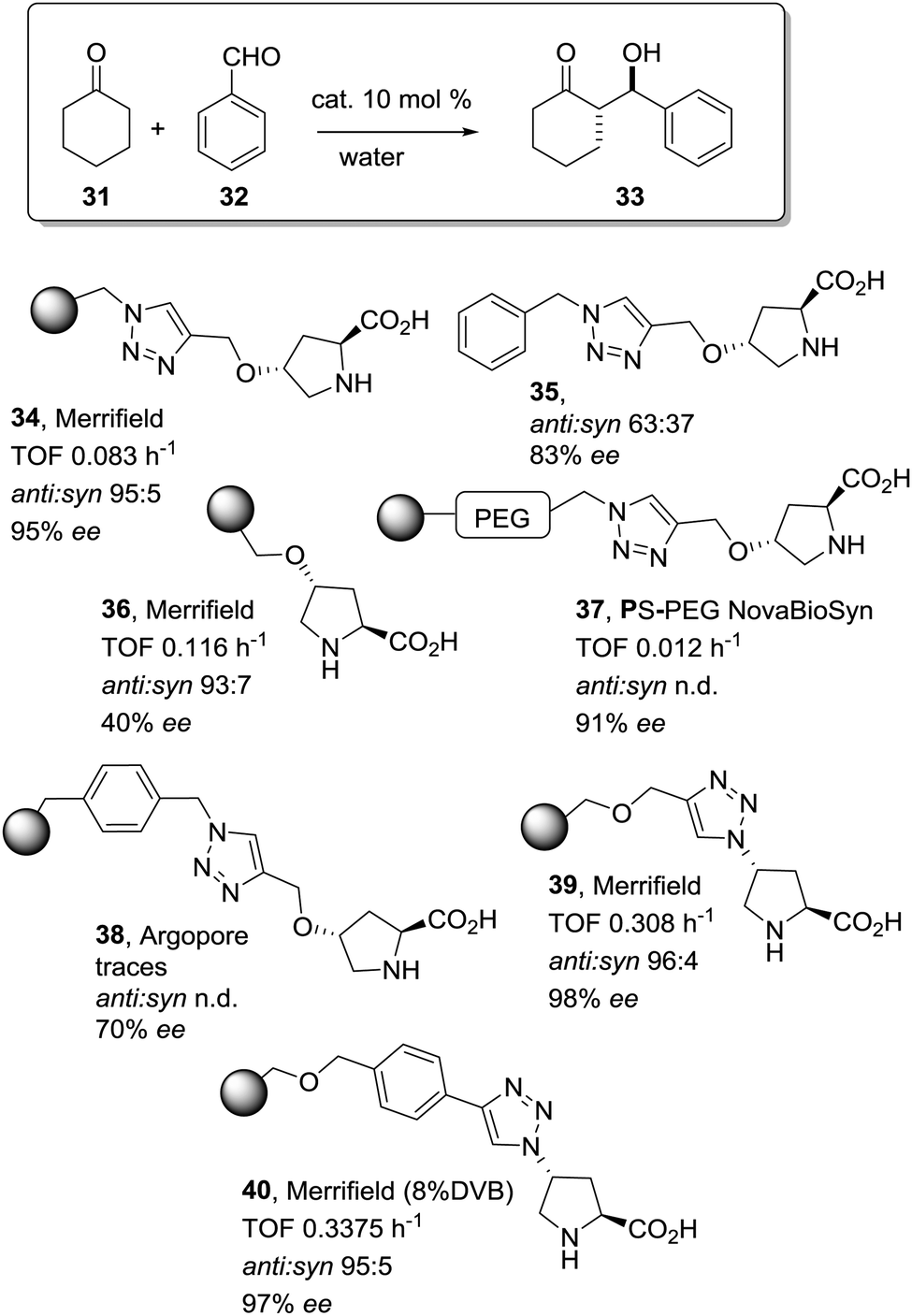 | ||
| Fig. 6 Immobilization of 4-hydroxyproline fragments on different polymeric supports using a click strategy with the formation of linkers containing the 1,2,3-triazole moiety. | ||
Noteworthily, the polymer 34 exhibited a much better catalytic performance than its monomeric counterpart in terms of selectivity. However, this resin still presents low reaction rates in water (TOF 0.083 h−1). The use of an analogous macroporous resin in substitution of a gel-type support did not improve the situation and only traces of the product were found for this catalyst (38). However, a controlled manipulation of the linker and spacer between the catalytic unit and the polymeric backbone allowing tuning the improvement of the catalytic performance. The lack of the 1,2,3-triazole moiety in the linker (36) led to similar activity but much poorer enantioselectivity. The use of a resin bearing more flexible and hydrophilic ethylene glycol units (37) rendered a significant activity drop. Polymer 39, where the 1,2,3-triazole moiety is directly grafted to the proline ring, exhibited an optimal catalytic performance, with a notable rate acceleration over those of the other studied resins (up to 3.7 by comparing 34 and 39).
Noticeably, and in sharp contrast with other resins, polymer 39 showed a perfect swelling behaviour in water. The observed activity enhancement can be related to the interaction of water molecules connecting the amino acid and the 1,2,3-triazole moieties building an aqueous microenvironment around the catalytic sites resembling the effect of essential water on some natural enzymes used in organic media. The introduction of an additional spacer unit (4-ethynylbenzyl ether, 40) increased the separation between the hydrophilic, catalytically active moiety and the hydrophobic polymer backbone. This catalyst, based on a PS polymer with 8% of DVB, showed a higher activity and diastereoselectivity than the homogeneous counterpart for all the solvents tested (homogeneous vs. heterogeneous: H2O: 0.3125 h−1anti:syn 93:7, 97% ee vs. 0.3375 h−1, anti:syn 95:5, 97% ee; DMF:H2O (1:1) 0.321 h−1, anti:syn 79:21, 97% ee vs. 0.383 h−1, anti:syn 95:5, 97% ee; CH2Cl2 0.187 h−1, anti:syn 80:2, 77% ee vs. 0.354 h−1, anti:syn 95:5, 85% ee).
The same group has also observed similar results in Michael reactions regarding the role of the p-phenylene spacer for a chiral pyrrolidine PS-support tethered by the 1,2,3-triazole moiety.112
In this regard, Itsuno has exploited this phenomenon to design highly active supported catalysts for the transfer hydrogenation in neat water and in organic solvents.115 The supported catalysts were based on the chiral ruthenium(II)–N-toluenesulfonyl-1,2-diphenylethylene-diamine (TsDPEN) complex developed by Ikariya and Noyori,116 and prepared as micro-reticular resins by copolymerisation (Fig. 7). These authors have systematically evaluated the influence of the polymer composition in terms of catalyst loading, degree of crosslinking and the content and nature of pendant groups on the efficiency of the Rh, Ru and Ir catalysts derived from this supported chiral ligand for the asymmetric transfer hydrogenation of different substrates.117–120 The balance between hydrophilicity and hydrophobicity induced by the presence of additional polar groups on the polymer backbone was the most important factor controlling the efficiency of the catalyst in the transfer hydrogenation of ketones and imines. The presence of additional polar groups not only allows the reaction in water to be carried out, but also allows adjustment of the activity of the catalyst through the selection of the appropriate hydrophilic, hydrophobic, or amphiphilic moieties. The most dramatic activity effect was found for the hydrogenation of cyclic sulfonimine 41 in neat water (Fig. 7a). The soluble catalyst TsDPEN showed no catalytic activity due to its hydrophobic character. The polystyrene-immobilized chiral catalyst (43a), which consists of a gel-type hydrophobic polymer chain, was also not suitable for the reaction. However, the introduction of polar functional groups in the polymeric network turned the inactive supported catalyst into an active one in water (see 44a and 45a, Fig. 7a).117
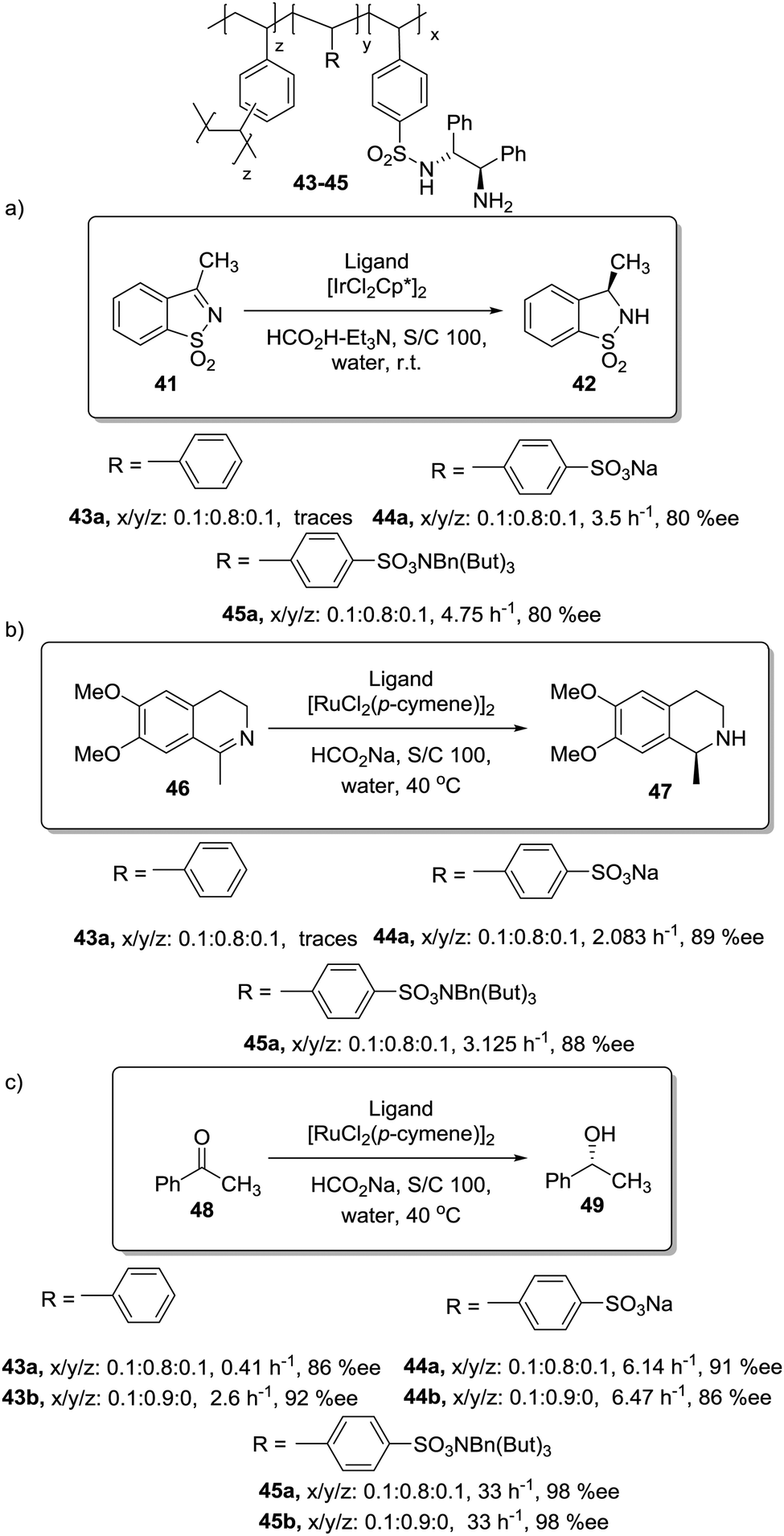 | ||
| Fig. 7 Polymer supported TsDPEN ligand for the Ir or Ru catalysed transfer hydrogenation of sulfonimines (a), imines (b) and ketones (c). | ||
The pendent polar functional groups in the multifunctional polymeric framework can induce proper polymer swelling and solvation. Indeed, styrene based polymer 43a swelled in traditional good solvents for crosslinked polystyrene such as DMF, THF and toluene, but completely shrunk in water due to its hydrophobic character. In contrast, polymers bearing sulfonate groups (i.e.44a and 45a) were found to swell in polar solvents such as dimethylsulfoxide and water. This effect was also confirmed when the asymmetric transfer hydrogenation of cyclic imine 46 was studied (Fig. 7b).118 Catalysts 44a and 45a bearing either sodium or benzyltributylammonium sulfonate groups were active in water (2.08 h−1, 89% ee and 3.125 h−1, 89% ee, respectively), but became inactive in methylene chloride, leading to only traces of the product, while the hydrophobic catalyst 43a (not active in water) displayed a good activity and enantioselectivity (3.83 h−1, 92% ee) in this organic solvent.
The lack of catalytic activity of the hydrophobic resin 43a for the asymmetric transfer hydrogenation of acetophenone (48) was clearly related to the limited accessibility of the catalytic sites in water (Fig. 7c). In fact, the soluble polymeric counterpart of this polymer (43b), where diffusion issues encountered in the crosslinked polymer 43a due to the lack of swelling are minimised, was active for this asymmetric transfer hydrogenation in water. Interestingly, the same catalytic activity was found for the two soluble polymers 44b and 45b having polar pendent groups in the polymeric network and their crosslinked counterparts (44a and 45a). These results highlight that diffusional limitations in gel-type resins can be minimised by the presence in the polymeric network of functional sites able to tune the swelling of the polymer in the presence of a given solvent.
This is the case, for instance, of supported pseudopeptides 55 and 56 prepared by grafting onto gel-type resins,122–125 whose copper complexes were studied for the catalytic cyclopropanation of styrene (Fig. 8).126 When the copper complexes of the homogeneous analogues 53 and 54 were evaluated for this reaction, the presence of an induction period was observed. No conversion of 50 to 52 was detected for a period of 1 to 3 hours. Such an induction time was, however, absent for the supported polymeric catalysts derived from 53 and 54 that, accordingly, showed a higher activity at shorter reaction times. The pseudodilution effect seems to be responsible for this behaviour.81 All data suggest that in CH2Cl2 soluble copper complexes of 53 and 54 tend to form aggregates that present a very low activity. Non-catalysed decomposition of ethyl diazoacetate results in the formation of polar species that finally favour disruption of the aggregates. Aggregation is inhibited or, at least, very much diminished for the supported species and, accordingly, the inhibition period is not observed.
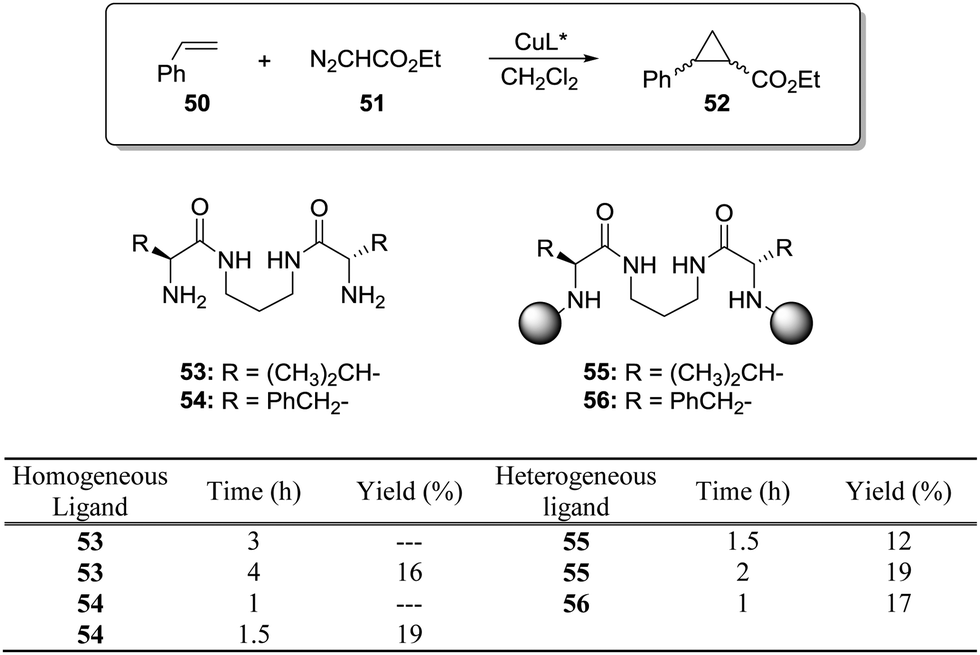 | ||
| Fig. 8 Homogeneous and supported pseudopeptidic ligands assayed for the enantioselective cyclopropanation of styrene with ethyl diazoacetate. | ||
Similar effects were also found for the supported copper–2-(pyridine-2-yl)imidazolidine-4-thione complex in the asymmetric Henry reactions (Fig. 9).127 The homogeneous catalysts based on the copper complexes with 59 and 60 were ca. 3 times less active than the copper complex obtained with the immobilized ligand obtained through grafting onto a commercially available JandaJel™ polymeric resin (61). The site-isolation on the polymer matrix reduces the possible dimer/oligomer adducts found in the homogeneous case,128 enhancing the catalytic activity.
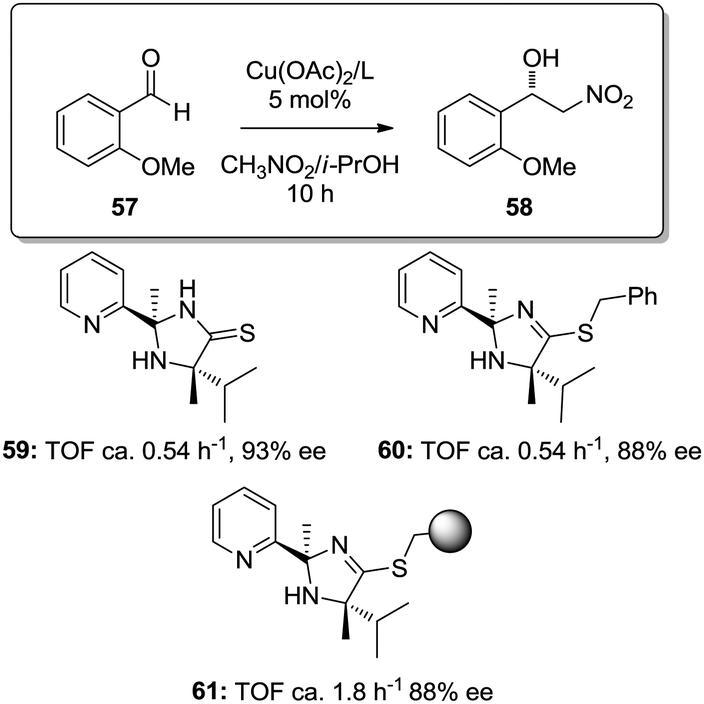 | ||
| Fig. 9 Homogeneous and supported 2-(pyridine-2-yl)imidazolidine-4-thione ligands assayed for the enantioselective Henry reaction. | ||
Some increases in activity, relative to the homogeneous phase, have been also reported by Irurre et al. using polymer-supported Ti-TADDOLates.129,130 The high potential of TADDOL ligands (α,α,α′,α′-tetraaryl-1,3-dioxolane-4,5-dimethanol) as chiral auxiliaries in catalysis and separation processes131 has led different groups to study their immobilization on a variety of supports.132–139 In the case of supported Ti-TADDOLates 62 (Fig. 10) prepared by grafting and studied for the Diels–Alder cycloaddition of cyclopentadiene and 3-crotonoyl-1,3-oxazolidin-2-one using a 10:1 catalyst dienophile ratio,129,130 no clear rationale was given to explain the observed increase in activity, but the presence of changes in the mechanism can be envisaged, in particular taking into account the potential participation in this reaction of several complexes with different molecularities.140
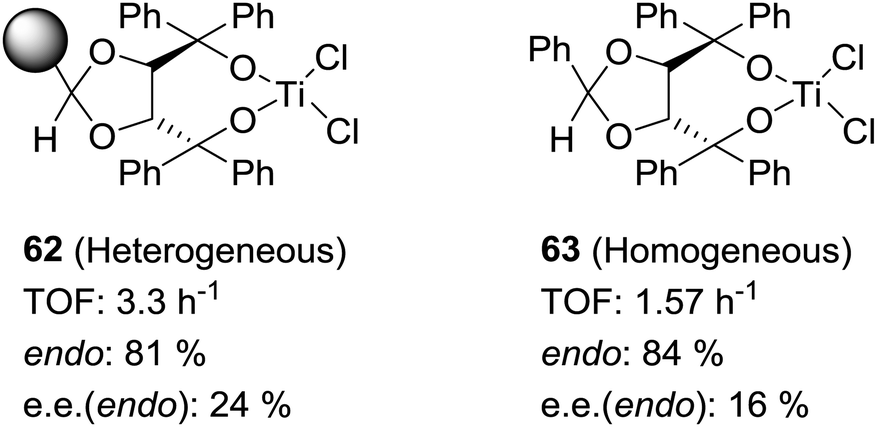 | ||
| Fig. 10 General structures of supported Ti-TADDOLates and of supported β-aminoalcohols and their N-benzylated homogeneous analogues. | ||
An even more dramatic effect was observed in the case of some β-amino alcohol derivatives (Fig. 11).141 Heterogeneous derivatives of (1R,2S)-ephedrine (64) and (R)-3-pyrrolidinol (65) were easily prepared by direct reaction of the corresponding β-amino alcohol with a Merrifield resin, while prolinols 66 and 67 were better prepared by initial reaction of the proline methyl ester with a chloromethylated resin followed by treatment with an excess of lithium aluminium hydride (66) or PhMgBr (66).142–144 The corresponding N-benzylated derivatives (68–71) were prepared as the homogeneous analogues. The related chiral Lewis acids were obtained by the reaction of 64–71 with EtAlCl2 (see general structure, Fig. 11) and evaluated as catalysts for the Diels–Alder reaction of methacrolein with cyclopentadiene (Fig. 11).
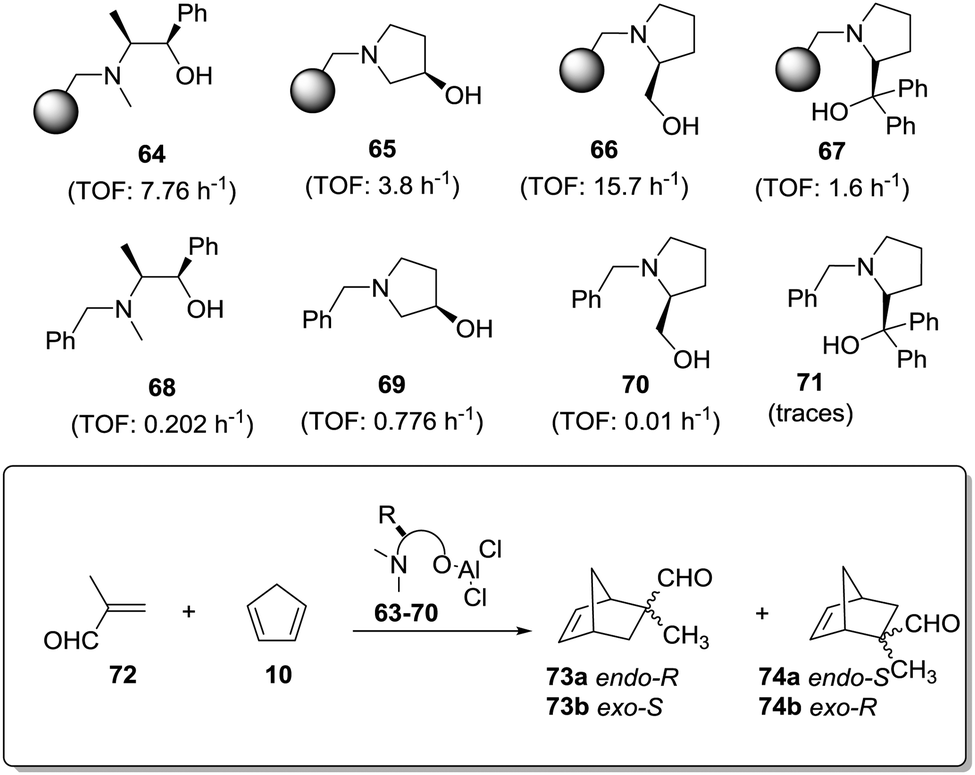 | ||
| Fig. 11 General structure of supported chiral Lewis acids and the results for their application as catalysts for the Diels–Alder reaction of methacrolein with cyclopentadiene. | ||
The results presented in Fig. 11 clearly reveal that reaction times required to achieve the same chemical yields are much shorter always for the polymer-supported systems. Thus, for instance, in the case of (R)-3-pyrrolidinol (65), the supported system allows obtaining a 95% conversion after 60 min (TOF: 3.8 h−1), whilst the catalyst derived from the N-benzylated analogue 69 requires a fivefold increase in time (300 min) to achieve a similar conversion (TOF: 0.776 h−1). In the case of the ephedrine derivatives 64 and 68, the homogeneous system catalyses the reaction 38 times slower than the heterogeneous one. For the prolinol derivatives, it can be seen that, essentially, only the supported species are active (66 and 67). As could be expected, the more hindered derivatives (catalysts derived from 67 and 71) are less active both in solution and in the supported species. Again, the formation of aggregates in solution, which should be greatly inhibited on the heterogeneous systems, seems to be responsible for this behaviour.
A similar effect has been observed in the case of α-amino amides derived from natural amino acids. They can be considered as structural equivalents of amino alcohols providing an acidic N-fragment instead of the hydroxyl group. Accordingly, their Ni(II) complexes were shown to be efficient chiral catalysts for the addition of Et2Zn to aldehydes.145 However, in the absence of Ni(II) these ligands were unable to catalyse this reaction in opposition to the general behaviour observed for amino alcohols. Interestingly, when these amino amides were prepared attached to polystyrene matrices, the supported systems were found to be very active also in the absence of Ni(II), leading to the formation of 1-phenyl ethanol from benzaldehyde and Et2Zn in almost quantitative yields and >90% ee.146
Dendrimers as supports
The use of dendrimers as supports is a suitable alternative to mitigate some of the diffusional problems ascribed to the gel-type resins, while maintaining some of the advantages found for the gel-type polymeric supports related to site isolation and suitable hydrophilic–hydrophobic balance.147–149Their soluble nature and the possibility to locate the catalytic species in well-defined positions within the dendrimer can be used to tune the catalytic activity of a given catalyst (Fig. 12).150–152 Different examples in non-asymmetric transformations have demonstrated significant enhancements in yields and activity in comparison with their homogeneous counterparts.153,154 Such effects can be observed for the original monomeric component and/or for larger dendrimer generations and show a strong dependence on both the generation used and the localisation of the active sites. Indeed, the localization of the catalytic sites in the core of the dendrimer may lead to “positive dendrimer effects” originating either from the influence of the branches on the core, modifying the polarity of the environment, or from site isolation effects.
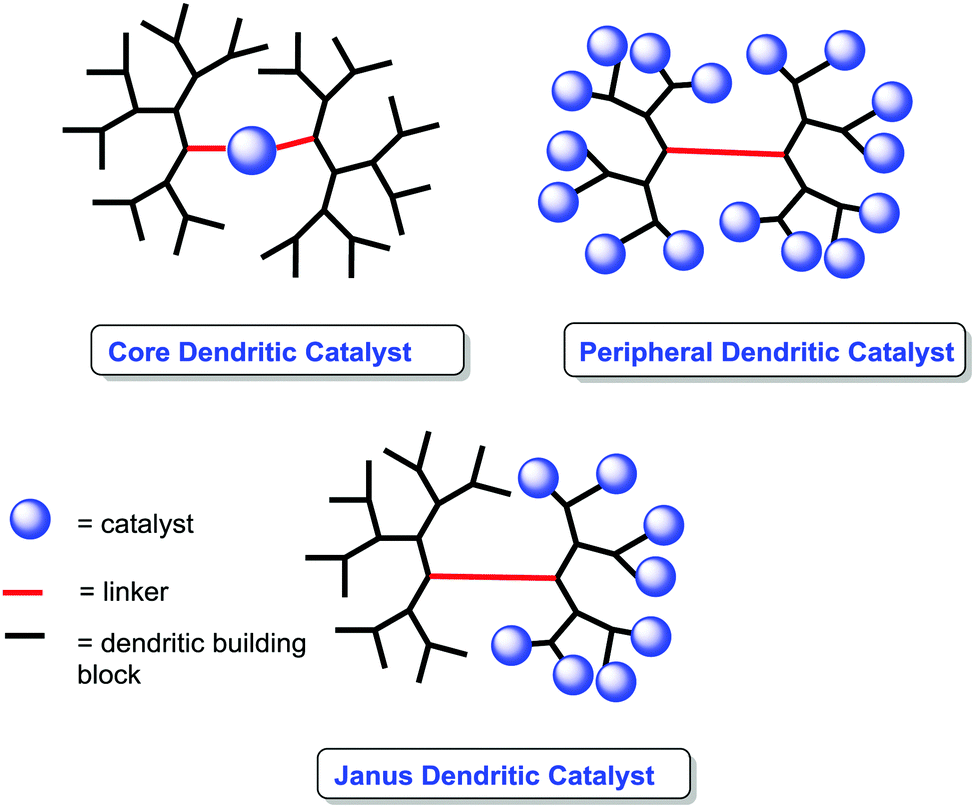 | ||
| Fig. 12 General representation for dendritic catalysts. | ||
This can be illustrated in the case of 5,5′-modified BINAP ligands immobilized on the core of Frechet type dendrimers (Fig. 13). This supported catalyst provided impressive results for the Ir-catalyzed asymmetric hydrogenation of quinolones (77),155 with an important enhancement in the catalytic activity from a TOF of 1 × 10−3 h−1 for the first generation to 19 × 10−3 h−1 for the fourth one without compromising the enantioselectivity. Noteworthily, the supported catalyst (75) was more active than the related homogeneous BINAP Ir complex (TOF 430 h−1). Indeed, the best catalytic dendrimer, under optimized conditions, behaved as a highly active catalyst with a maximum initial TOF of 3450 h−1 and with a TON of 43000, highlighting the non-innocent effect played by the support. This effect is attributed to the efficient site isolation reducing the formation of dimers observed for the unsupported complex.
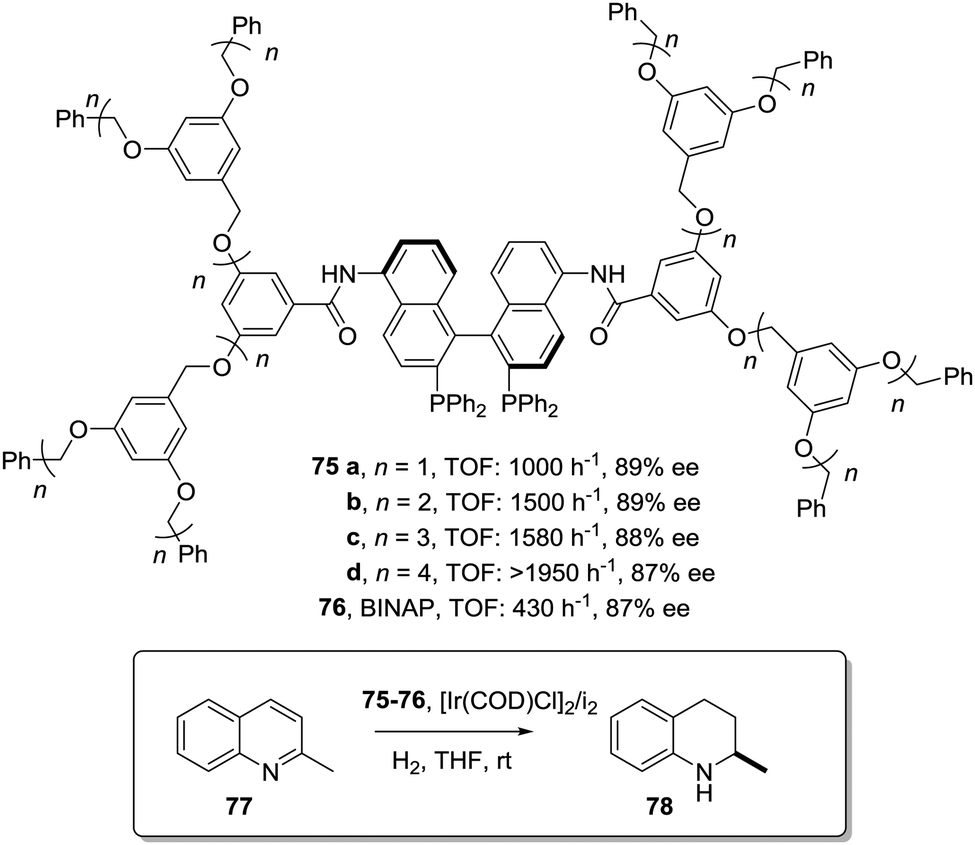 | ||
| Fig. 13 Modified BINAP ligands at the core of dendrimeric structures and their application to the Ir-catalysed asymmetric hydrogenation of quinolines. | ||
Similar positive effects were also found for the asymmetric hydrogenation of 2-(4-isobutylphenyl)acrylic acid (79) catalysed by the same dendritic BINAP systems (75) complexed this time with Ru (Fig. 14).150,156 Again the higher generation dendrimers showed an enhanced activity (up to a 3 times increase) than the unsupported homogeneous counterpart. Such a rate enhancement was not observed for the mono-substituted dendrimer and the activity was by far higher in toluene than in toluene:methanol (1:1, v/v).157,158 These facts suggest that the dendrimers contribute to generate a suitable polar micro-environment to induce a pseudoconcentration effect of the substrates on the proximity of the catalytic sites. The same effect on activity was found when the Ru–BINAP homogeneous complex was encapsulated inside an analogous third generation Fréchet dendrimer lacking the catalytic center.
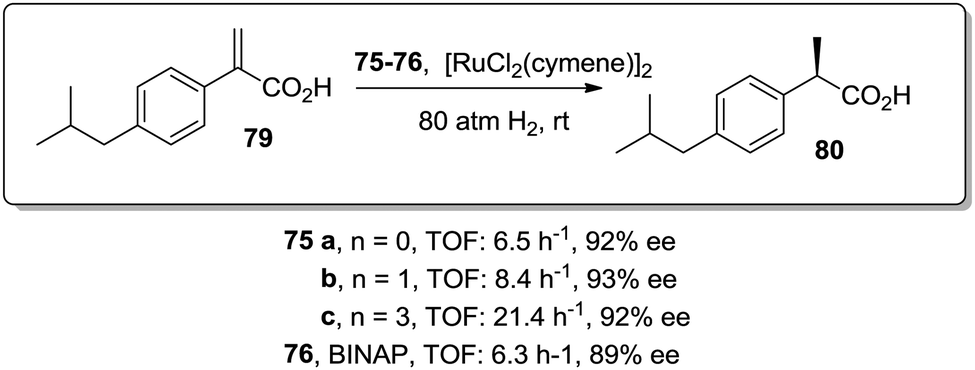 | ||
| Fig. 14 Ru-Catalysed asymmetric hydrogenation in the presence of the modified BINAP ligand at the core of dendrimeric structures. | ||
The activity observed for chiral Ir–phosphinooxazoline (PHOX) complexes immobilized onto Frechet type dendrimers also surpassed the activity found for the homogeneous systems in the hydrogenation of diaryl-1,5-benzodiazepines (83). (Fig. 15).159 Usually, the activity of the Ir–PHOX complexes is highly affected by the formation of polynuclear complexes,160 but in the dendrimer supported systems (81) the sterically demanding dendrimer branches facilitate suitable site isolation reducing such adverse effect. In this way, the second-generation dendritic catalyst (81c) was ca. 4 times more active than the non-dendrimeric counterpart (82). However, the third-generation dendritic catalyst (81d) was only 2 times more active. This suggests that a compromise between site isolation and mass transfer is required to design a suitable catalytic dendrimer.
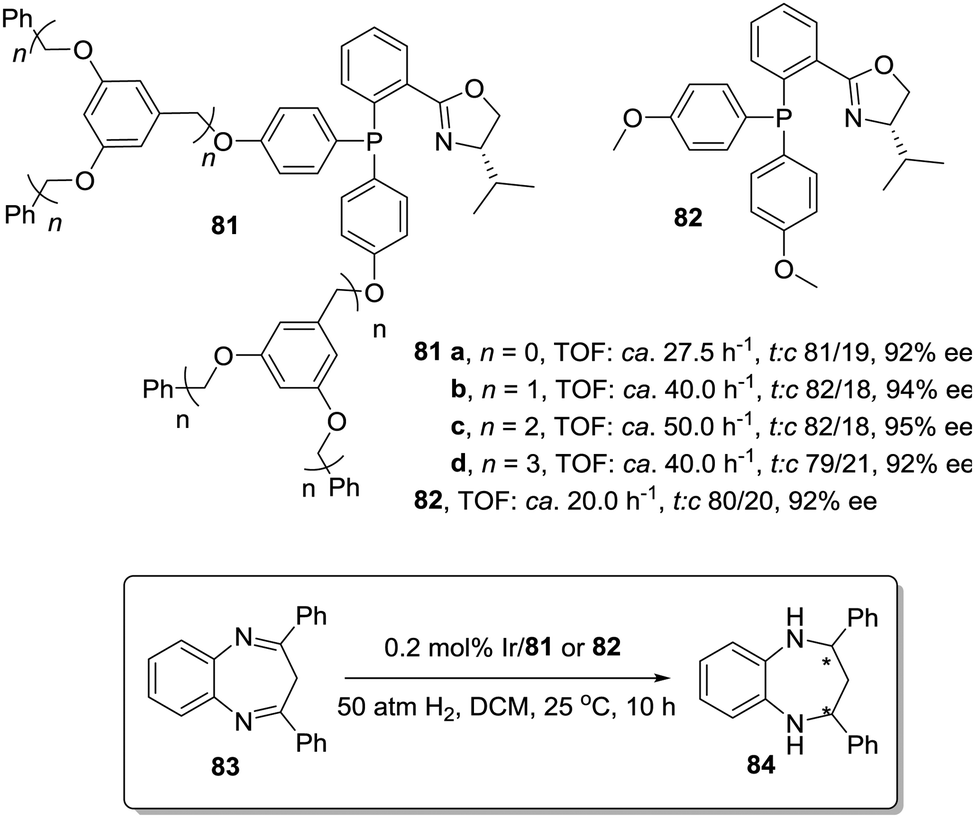 | ||
| Fig. 15 Modified phosphinooxazoline ligands immobilized onto Frechet type dendrimers for the asymmetric Ir-catalysed hydrogenation of diaryl-1,5-benzodiazepines. | ||
A similar reasoning has been used to explain the changes in activity found for dendritic systems containing 4-(dialkylamino)pyridines supported onto gel-type polymers as catalysts for acylation reactions.161 From the experimental results, it was concluded that the nano-environment created by the dendrimer within the polymeric network played a dominant role in determining the activity of those supported catalysts.
A second type of “dendrimer effect” can be found when the catalytic units are located in the dendrimer periphery. In those cases, cooperativity between catalytic units is expected due to the high local concentration of these sites within well-defined nanoscopic peripheral space, which, in some cases, may result in a positive effect enhancing the catalytic properties. This can be illustrated, for instance, by the application of BINAP Janus-type dendrimers 85 formed by a Fréchet-type polyether dendrimeric unit fused to a second one having up to 16 peripheral BINAP moieties.162 These BINAP subunits were able to form the corresponding ruthenium complexes for the asymmetric hydrogenation of 2-aryl-acrylic acid 79. The generation of the dendrimer influenced the reactivity of these catalysts with increasing reaction rates (more than 3.5 times increase) for the higher generations (Fig. 16).
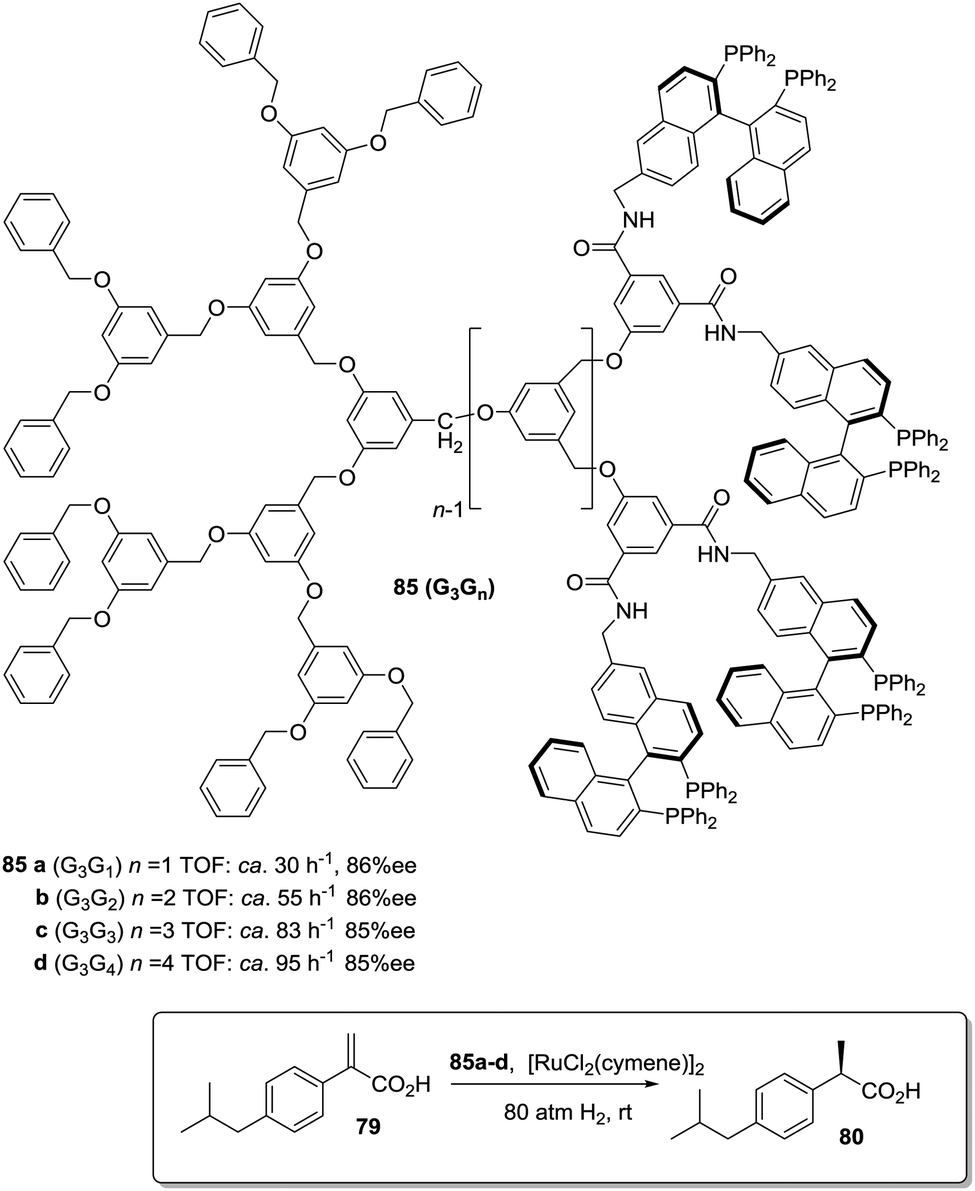 | ||
| Fig. 16 Janus-type dendrimer structure containing BINAP peripheral subunits on one of the faces. | ||
An even more dramatic increase was found by Jacobsen for Co–salen complexes immobilized onto NH2-terminated PAMAM dendrimers (86) in the hydrolytic kinetic resolution of terminal epoxides (88; Fig. 17).163 Mechanistic studies had suggested an increase in the catalytic activity by a cooperative interaction between catalyst units.164 The presence of Co–salen units in the periphery of the dendrimer led to important cooperative interactions between [Co–(salen)] units due to their higher local concentration, affording an up to 24 times increase of the relative rate per [Co(salen)] unit in comparison with the homogeneous system 87. Hence, it is clear that dendritic supports offer very interesting opportunities to tune the activity of different types of catalytic systems, although an important drawback is the need for complex synthetic and purification processes in their preparation.
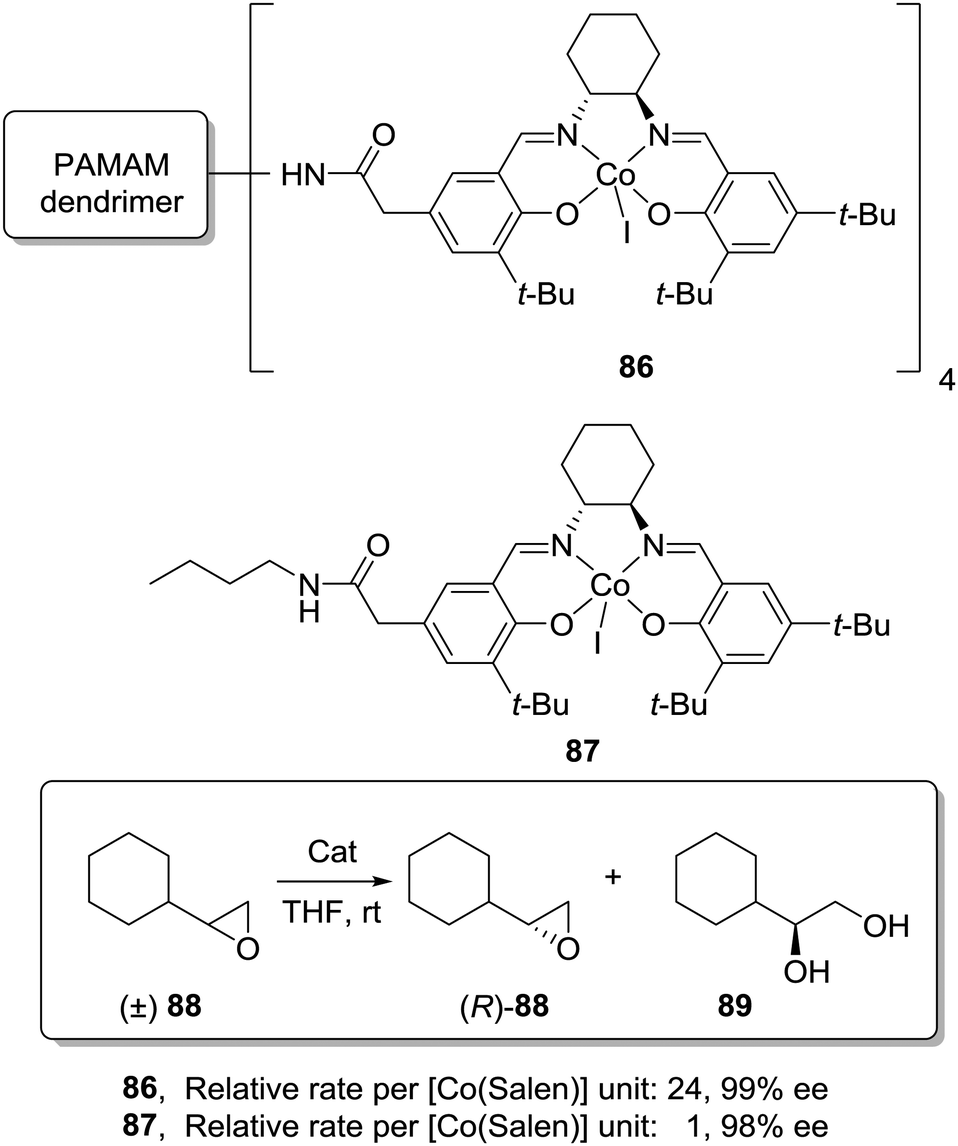 | ||
| Fig. 17 General structure of a Co–salen complex immobilized onto NH2-terminated PAMAM dendrimers used for the hydrolytic kinetic resolution of terminal epoxides. | ||
Macroporous polymers as supports
A different possibility to minimise diffusional limitations is the use of macroporous polymers with permanent porosity. In this case, the functional sites are distributed both at the surface and at the interior of the dense polymer particles. Accessibility of sites located on the surfaces is greatly enhanced and does not depend very much on the solvent. In contrast, the functional groups located on the interior of the particles may not be easily accessible. The balance between surface and non-surface sites determines the activity of the resulting species and very often, this can be regulated through an appropriate selection of the polymerisation conditions.This can be illustrated by the work of Abu-Elfotoh et al. with a macroporous polymer-supported chiral ruthenium(II)/phenyloxazoline (Ru–PHEOX) complex (90; Fig. 18), prepared by copolymerization of the corresponding monomeric complex with styrene and 1,4-divinyl benzene (DVB) in water:CH2Cl2 (7:2).165 The resulting catalyst displayed, in the cyclopropanation of styrene (50) with EDA (51), a higher activity than the homogeneous catalyst 93 (TOF of 3.034 h−1, cis:trans: 88:12, 94% ee vs. TOF of 2.694 h−1cis:trans: 90:10, 98% ee). Interestingly, the polymer 92, obtained under the same conditions as 90, but by initial polymerisation of the phenyloxazoline ligand followed by complexation with the metal, suffered from a significant drop in catalytic efficiency in terms of both activity and enantioselectivity (TOF of 1.102 h−1, cis:trans: 83:17, 74% ee). Furthermore, the polymeric catalyst 90 was also more active than the one based on a macroreticular polymer (91, ca. 50% DVB) (TOF of 1.265 h−1, cis:trans: 82:18, 84% ee). The difference in activity can be explained based on the macroporous structure of the different materials. The direct polymerisation of the Ru-complex, in the presence of water:CH2Cl2, afforded a uniform distribution of the catalytic sites in the polymeric matrix, which also presented a larger internal surface area than the macroreticular-type catalyst 91.
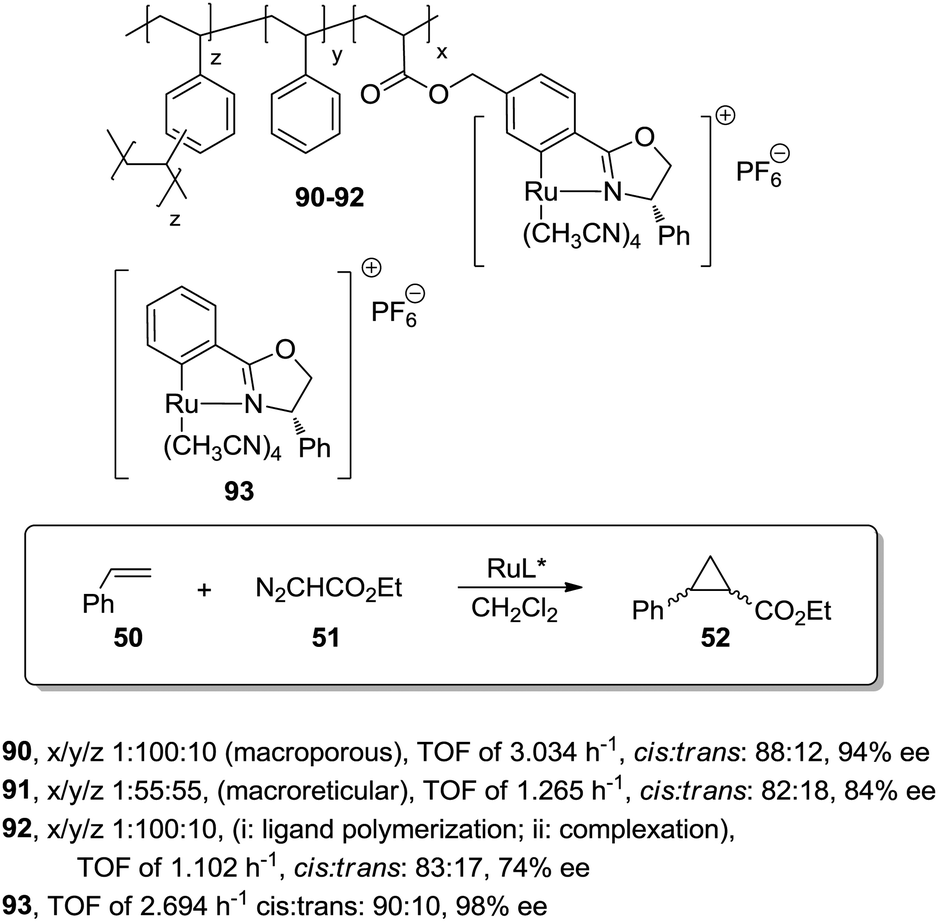 | ||
| Fig. 18 Structure of Ru–PHEOX polymers studied as catalysts for the cyclopropanation of styrene. | ||
Petri et al. have studied how the asymmetric dihydroxylation of alkenes, catalysed by cinchona alkaloid derivatives (96–98; Fig. 19), is strongly affected by the appropriate design of the polymeric matrix.166 The rate of dihydroxylation of styrene depends on the hydrophobic/hydrophilic properties of the polymeric backbone. The activity increases for supports with a more hydrophilic backbone (98) (introduction of HEMA and EGDMA as comonomers), showing similar results to those obtained under homogeneous conditions (96). Only one hour is required to afford an 80% conversion with 96 and 98. With a PS–DVB matrix (97), however, four hours are required to get a similar conversion.
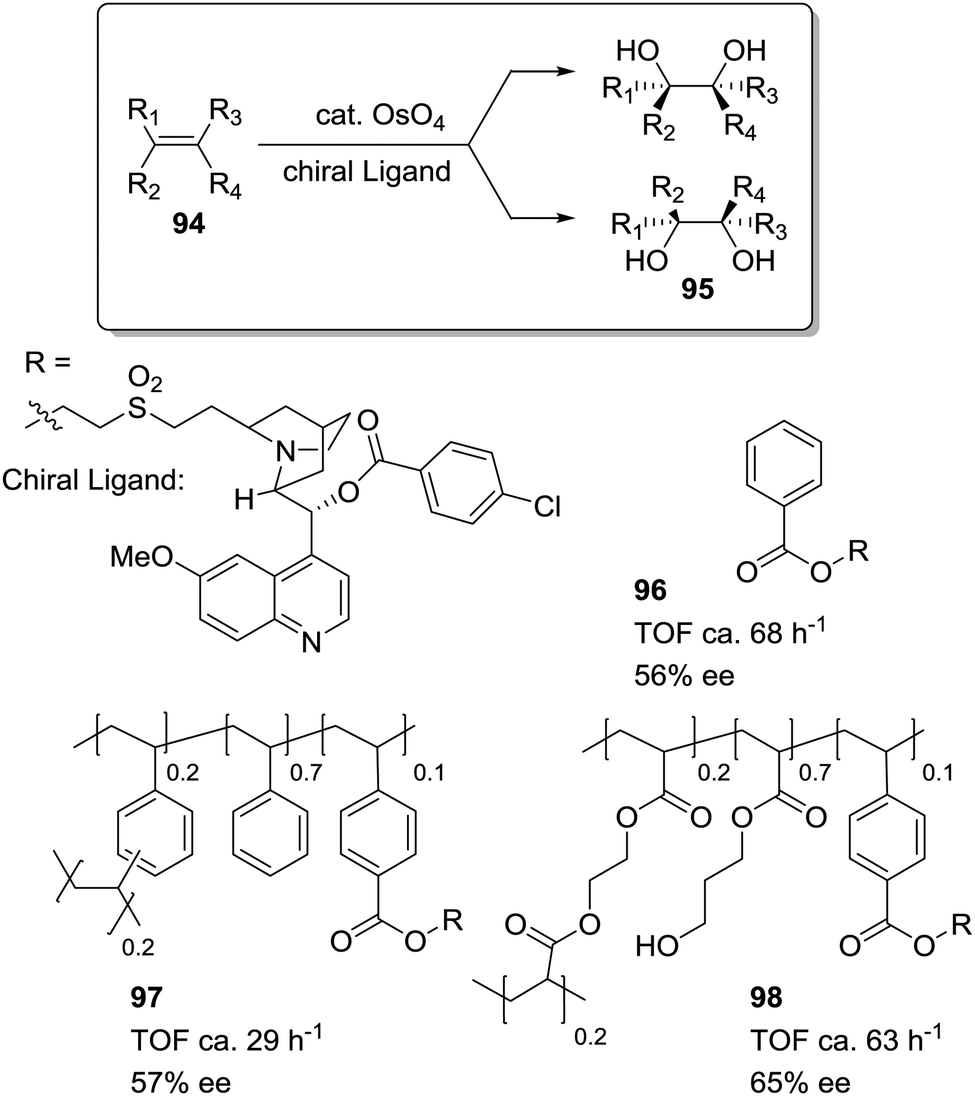 | ||
| Fig. 19 Cinchona alkaloids supported on polymeric matrices of different polarity for the dihydroxylation of alkenes. | ||
Ti-TADDOLates also provide a useful example. In fact, when the kinetics for the Et2Zn addition to PhCHO using supported Ti-TADDOLate catalysts such as 102 (Fig. 20) was initially considered, the measured rates were significantly slower than for the homogeneous analogue, and this was ascribed to the reduced accessibility to the catalytic sites.167 One of the strategies considered to overcome this limitation, improving the accessibility of the active sites, was the incorporation of the chiral ligand at the core of a polymerizable dendrimer (99).168,169 Only for the polymer prepared from 99 and styrene, the rate of the heterogeneous reaction increased relative to that in the homogeneous phase using directly 99.
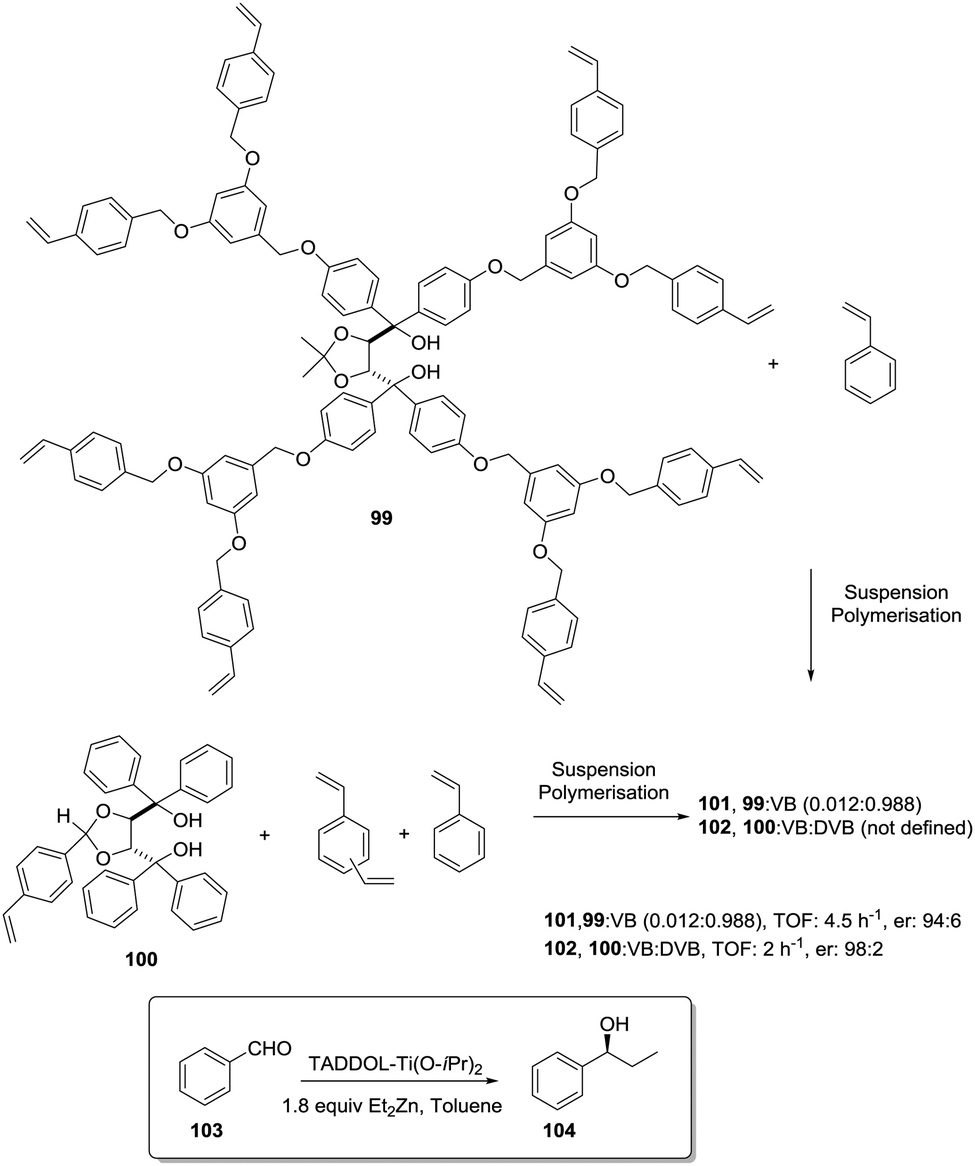 | ||
| Fig. 20 Preparation of polymer-supported TADDOLs via copolymerization of the corresponding vinylic TADDOL derivatives. | ||
The rate depended on the loading, with slower rates observed for higher loadings. This is reasonable, as this functional monomer acts as the crosslinker and, accordingly, higher loadings increase crosslinking and would make it difficult to access the reactive sites. Several reasons are given to explain this behaviour. They include considering that the attachment of the dendritic arms of 99 to the polymeric backbone could hinder them from wrapping around the TADDOL core, allowing, in this way, a better diffusion of reactants to and from the catalytic center.
On the other hand, the “oxygen-rich” dendritic branches could provide a more polar environment around the active sites within the apolar polystyrene matrix. This could, for instance, favour the accumulation of polar substrates (PhCHO, Et2Zn) in the proximity of the catalytic centres, increasing the local concentrations and, hence, leading to higher rates.
As mentioned, a possible alternative to obtain supports with good diffusional properties is the use of polymeric macroporous monoliths, which can be moulded in different shapes according to the required uses.170 In a polymeric monolith,171 the resin can be described as a continuous material integrated with highly crosslinked particles and presenting a complex network of permanent pores. Usually, those materials are prepared by polymerisation of a mixture containing a functional monomer along with styrene (VB) and divinylbenzene (DVB) (or the corresponding crosslinking agent, i.e.106 or 107 in Fig. 21) in the presence of an appropriate porogen.
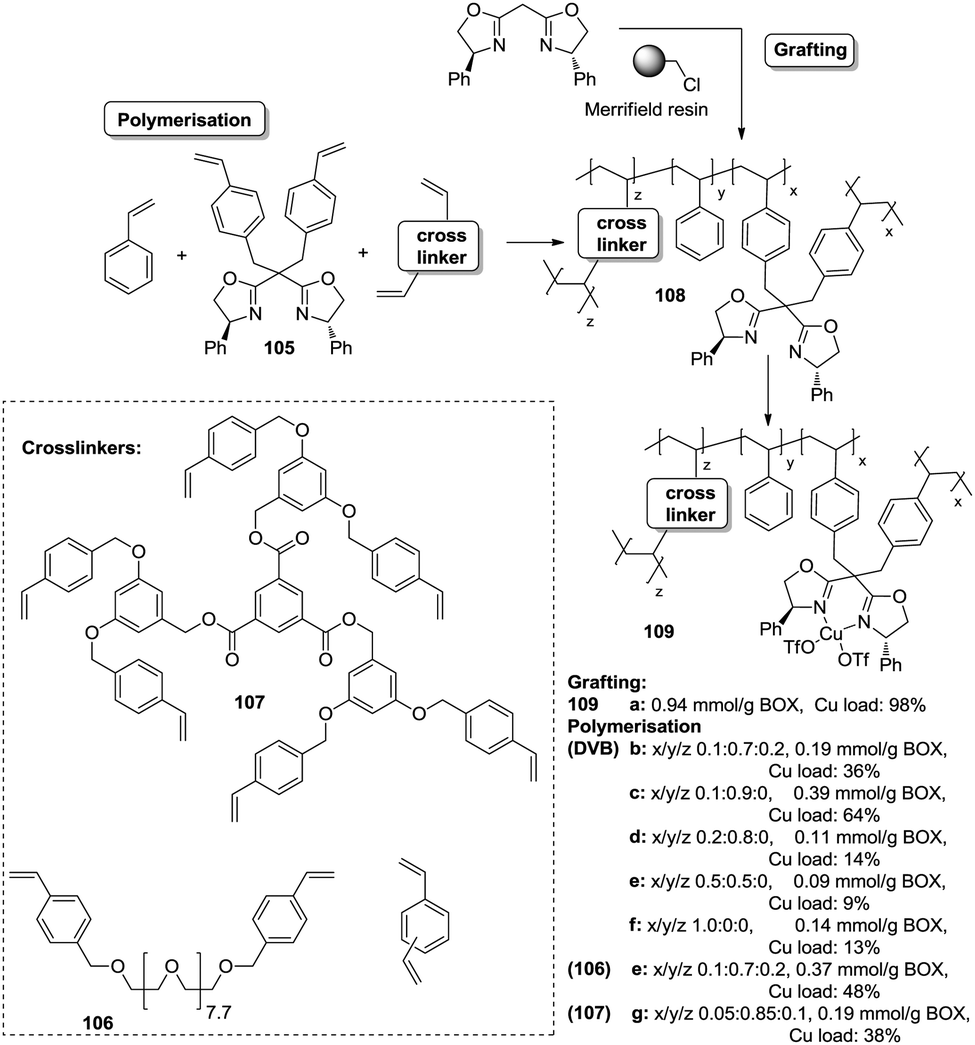 | ||
| Fig. 21 Preparation of monolithic polymers by polymerization of vinylic BOX derivatives. | ||
Polymer-supported bis(oxazolines) (BOX) can be prepared following this strategy as monolithic polymers. The accessibility of the functional sites can be assessed through the analysis of the copper uptake after treatment with Cu(OTf)2 (Fig. 21).172–175 Several resins were prepared using monomeric mixtures containing variable amounts of the functional monomer 105, styrene and DVB. As can be seen from the data depicted in Fig. 21, the accessibility of the bis(oxazoline) groups on the polymer seems to be related to the loading and the crosslinking degree. The number of non-accessible sites increases, in general, with both parameters. The increase with loading is reasonable, as the functional monomer acts as a crosslinker and, accordingly, higher loadings increase crosslinking and would make it difficult to access the reactive sites. Nevertheless, this is not a simple relationship, and thus the copper uptake of the homopolymer 109f (100% of functional monomer, 100% of crosslinking) is comparable with that obtained for a polymer containing only 20 molar% of the functional monomer and 80% of styrene (109d). As also shown by the use of JandaJel and related resins,58,176,177 the use of more flexible crosslinking agents, such as the vinylic derivative 106 (Fig. 20) containing a PEG chain, favours the accessibility of the bis(oxazoline) moieties. Thus, for one of the monomeric compositions used for polymer 109e (10:70:20), but using 106 instead of DVB, 48% of the reactive sites were loaded with Cu.172,178 Some increase in the Cu uptake was also observed for the polymer 109g where first generation dendrimers containing at least six vinylic groups (107) were used as the crosslinkers (up to 38% of functional groups loaded with copper).179
Finally, the exact balance between accessible and non-accessible sites is clearly dependent on the polymerisation conditions and here the porogen can play a central role. Thus, for the homopolymers prepared from 105, the use of toluene/dodecanol as the porogenic mixture produces a polymer in which about 13% of the functional groups are accessible, whilst only 1% of the groups are accessible when toluene, which has been described to form small pores,85,180,181 is the only component of the porogenic mixture.
As the BOX monomer conserving the C2-symmetry acts itself as a crosslinking agent and can reduce the accessibility of some of the active sites, a “C2-disruptive” design for the BOX monomeric derivative may provide a suitable alternative to reduce to a minimum the structural perturbation induced by polymerization, enhancing accessibility. Mandoli et al. have exploited this concept to immobilize the BOX ligand by either copolymerisation or grafting (Fig. 22).182,183 Furthermore, and to minimise the backbone effect, an alkyl flexible linker was also used to tether the ligand to the support.
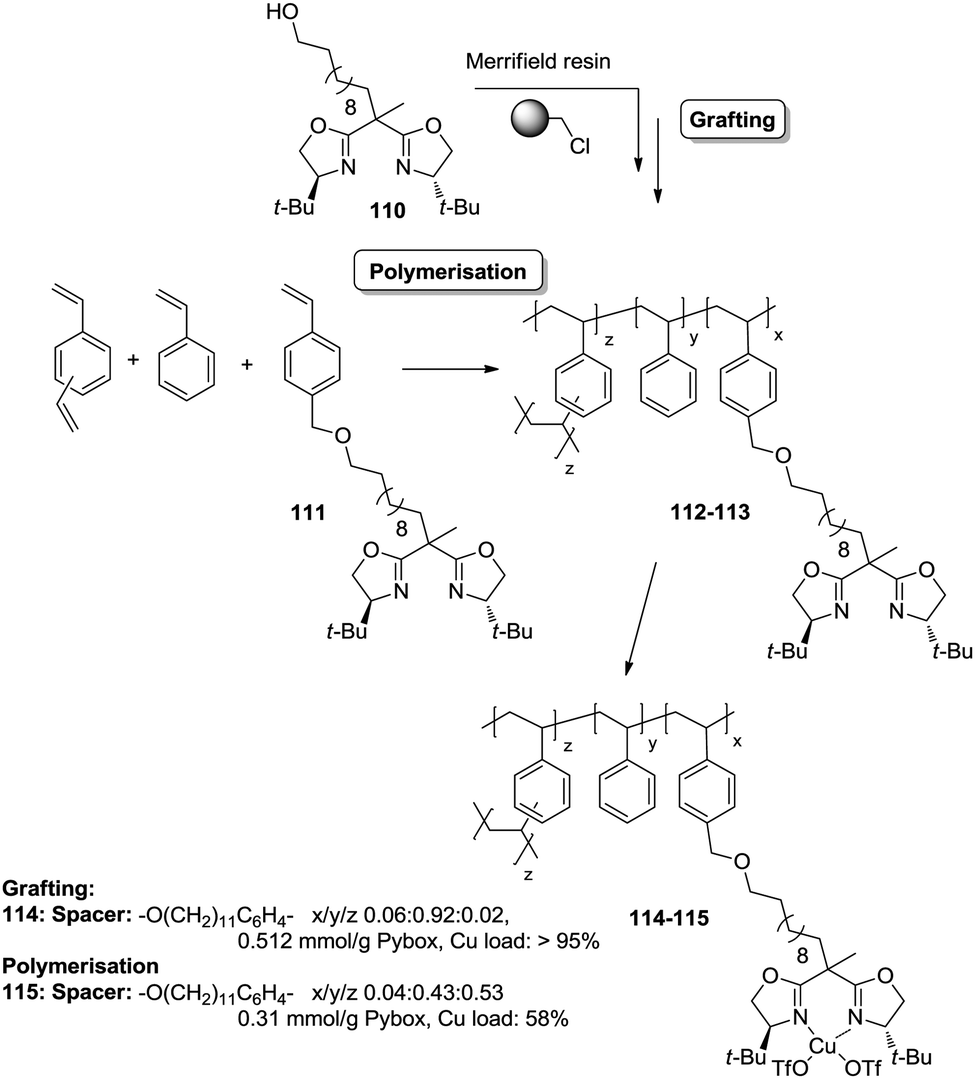 | ||
| Fig. 22 Preparation of polymer supported BOX derivatives through a C2-disruptive strategy for improving the accessibility of the active sites. | ||
The copolymerisation of the corresponding functionalised monomer (111) with toluene as the porogenic agent rendered a support with ca. half of the functional sites accessible (115). This represents a significant accessibility improvement over the above mentioned C2-BOX systems. In the case of the functionalised polymer prepared by grafting, all the ligands appeared to be accessible.183
However, the use of long linkers does not always represent an optimal approach. This can be illustrated by the differences in functional site accessibility observed for the case of polymer supported pyridinebis(oxazoline) ligands 116–117 (PYBOX) prepared by copolymerisation as crosslinked monoliths or by grafting onto commercially available Merrifield resins (Fig. 23).184,185 It should be noted that Ru–PYBOX complexes, depending on the concentration, can lead to dimer formation. Thus, especially for resins with low crosslinking degrees, an increase in the spacer length could enhance accessibility at the expense of site–site isolation, favouring the formation of dimeric species, which will be also favoured by a pseudo-concentration effect on the polymeric network. The accessibility was estimated taking into account the Ru uptake relative to the ligand loading. For the resins obtained by polymerisation (119d–g), the Ru uptake steadily increased from 62% to 82% with the length of the spacer. The larger spacers reduce the steric hindrance provided by the polymeric backbone improving PYBOX accessibility. For these highly crosslinked resins (51% DVB, 119d–g), the crosslinking helps to reduce site–site interactions. In the case of supported PYBOX ligands obtained by grafting, a positive effect of length of the spacer, facilitating good accessibility to the ligand, was observed for resins 119a and 119b, with Ru-uptakes of 85% and 93%, respectively. However, when the length of the spacer was further increased (119c), the Ru-uptake was only 60%, most likely due to the formation of dimeric species.
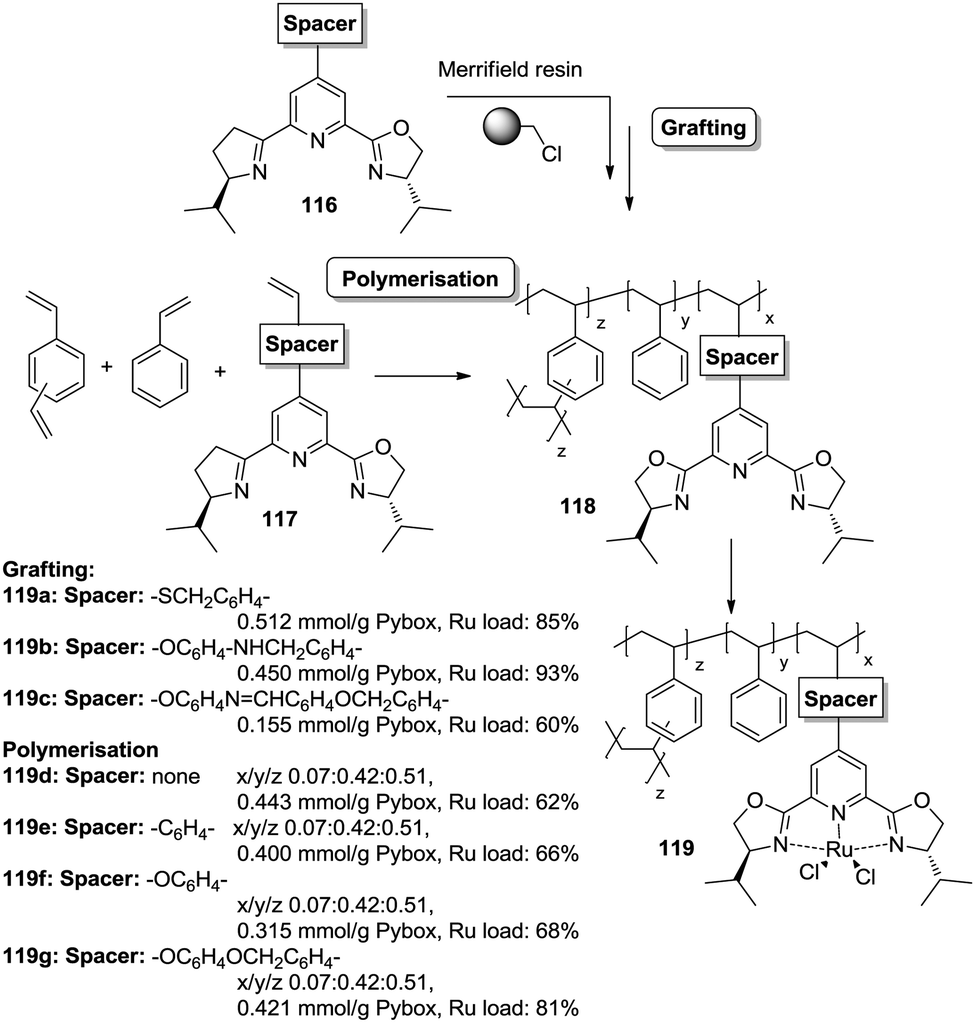 | ||
| Fig. 23 Preparation of polymer supported PYBOX derivatives through different strategies and with different linkers. | ||
These examples illustrate the effects of the methodology used in the immobilisation of a given catalyst on the accessibility of the catalytic sites, especially for systems based on the polymerisation of the corresponding chiral ligand and/or complex. Different aspects like type and amount of porogen, type and degree of crosslinking, use of spacers, etc., should always be carefully considered for optimization of the accessibility to the catalytic sites.
An illustrative example of how supported species on highly crosslinked polymeric frameworks can be more active, with better reaction rates, than the corresponding homogeneous analogues is provided by some of the polymer-supported bis(oxazolines) mentioned above. For the immobilized BOX ligand prepared using a dendrimeric crosslinking agent, the total TON was 70.8 moles cyclopropanes per mol BOX, while for the homogeneous analogue the TON observed under the same conditions was 3.2. The effect is more significant taking into account that less than 40% of the box moieties are able to form the corresponding Cu–BOX complex. According to this, the TON was calculated to be 184 moles cyclopropanes per mol Cu–BOX.178,179 Some improvements were also observed for the PYBOX derivatives. In this case, the catalysts obtained by grafting displayed a similar activity as the homogeneous unmodified catalyst, but up to a twofold increase in catalytic activity was observed for some of the systems prepared by polymerisation.184
Immobilization on an appropriate support can be used to suppress site–site interactions, affording a great effect on activity. Although eventually this can be achieved in microreticular polymers, particularly for low catalyst loadings, this effect can be facilitated by the use of macroporous, highly crosslinked polymers. An interesting example is provided by ruthenium porphyrin catalysts immobilized in a highly crosslinked matrix, where catalyst deactivation by intermolecular reactions was strongly decreased.186 The incorporation of a Ru(CO)(porphyrin) complex in such a matrix was carried out by suspension polymerization of the corresponding vinyl-substituted meso-tetraarylporphyrin complex with ethylene glycol dimethacrylate (EGDMA) in the presence of the appropriate porogenic mixture. The activity of this polymeric catalyst in epoxidation reactions was slightly higher than that of the homogeneous vinyl-substituted meso-tetraarylporphyrin complex (initial TOF value of 40 h−1vs. 27 h−1 for the homogeneous system). The differences in activity were significantly much higher for the catalytic oxidation of aromatic alkanes or secondary alcohols to the corresponding ketones. In some instances, initial TOF values for the supported system were up to one order of magnitude higher than those of the homogeneous counterpart. A further increase in activity by a factor of up to 16 was achieved by preparing the corresponding imprinted polymer using diphenylaminomethane or 1-aminoadamantane as the template.187 This suggested that the presence of the appropriate pseudosubstrate coordinated to the vinylic Ru(CO)(porphyrin) complex can generate an imprint (substrate pocket) in close proximity to the active site. In agreement with this hypothesis, the rate enhancement observed was strongly dependent on the template. An alternative strategy for improving the activity of those supported Ru(CO)(porphyrin) complexes was the use of perfluoromethylcyclohexane as a cosolvent.188 The presence of the fluorous solvent facilitated a favourable partitioning of substrates and oxidant into the polymeric matrix,189,190 leading to an increased local concentration of substrates and reagent in the vicinity of the catalytic site.
Porous organic polymers as supports
In the search of more efficient predesigned polymeric frameworks for the immobilisation of chiral ligands, a bottom-up approach, in which different building blocks are assembled into hierarchical porous polymeric materials with well-defined and tuneable physicochemical properties, is gaining momentum.61–75 These materials exhibit unique properties such as large BET surface areas and pore volumes, enhanced accessibility of catalytically active sites, and decreased diffusional limitations. Therefore, they can lead to highly active heterogeneous catalysts able to surpass the activity found for the homogeneous counterparts. This is illustrated, for example, by the 2D metalloporphyrin polymeric frameworks obtained by cross-coupling polycondensation that displayed impressive TON (6.7 × 107) and TOF (9300 min−1) values for the epoxidation of olefins with superior performance than the non-immobilised catalyst.191,192Among the different approaches for the preparation of POPs and related systems, copolymerization via free-radical polymerization of mono or poly-vinyl-functionalised chiral ligands with additional crosslinked monomers, especially DVB, under solvothermal conditions has led to the synthesis of highly crosslinked and functional polymers.193,194 Based on this methodology the heterogeneous phosphoramidite ligand 120 was prepared by copolymerization of the corresponding di-vinyl-MonoPhos ligand with DVB (1:4 by weight; Fig. 24).195 The corresponding Rh complex was at least three times more active than the homogeneous control. The highly crosslinked framework can inhibit the self-quenching found in solution via multimolecular deactivation. Besides, the excess of chiral ligand in the polymeric network seems to favour the stability of the catalytic complex against deactivation through decomposition into metallic Rh nanoparticles.
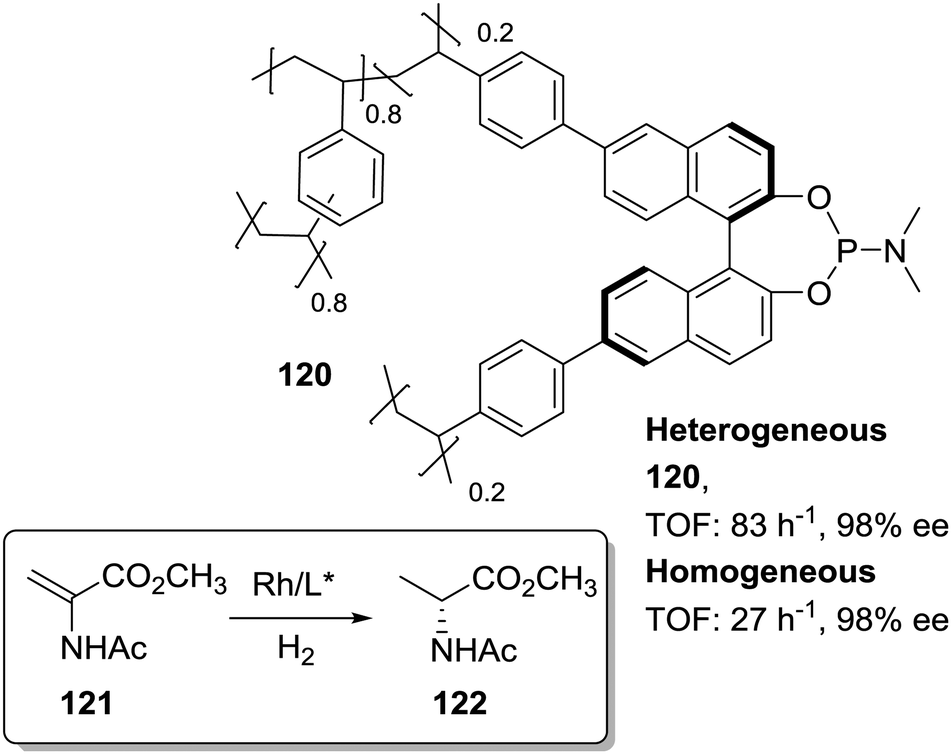 | ||
| Fig. 24 General structure of the POP containing chiral phophoramidite fragments and used for the Rh-catalysed asymmetric hydrogenation. | ||
The position and type of polymerisable moieties introduced on a given chiral ligand are the factors that should be carefully considered. The rigid polymeric organic porous framework is built up through condensation of these functional groups by themselves or in the presence of an additional building block. Thus, the position, number and type of linker can play a decisive role in defining the catalytic activity. For instance, Wang et al. have demonstrated that (S)-5,5′-divinyl-BINAP–DVB (123) prepared by solvothermal copolymerisation of a modified BINAP with DVB (9:81 by weight) produced a mesoporous material with a large surface area (1058–1070 m2 g−1), which was 2.5 times more active for the Ru-catalysed hydrogenation of methyl acetoacetate than the analogous copolymer obtained by copolymerisation of (S)-4,4′-divinyl-BINAP and DVB (124; Fig. 25) for a substrate/catalyst ratio of 1000.196 The derivatisation at the 4,4′ positions, closer to the catalytic sites, is likely to reduce the flexibility of the resulting polymer around the BINAP fragments making the sites less accessible to the substrates. A similar material was prepared with a BINAP bearing at 5,5′-positions acrylate groups instead of the vinylic residues.197 The material obtained by polymerisation with DVB (125, 20% DVB by weight) in spite of its lower surface area (524 m2 g−1) was able to quantitatively convert methyl acetoacetate with good enantioselectivity (90% ee) and at very high substrate/catalyst ratios (up to 5000), being within the more active systems reported for this reaction.
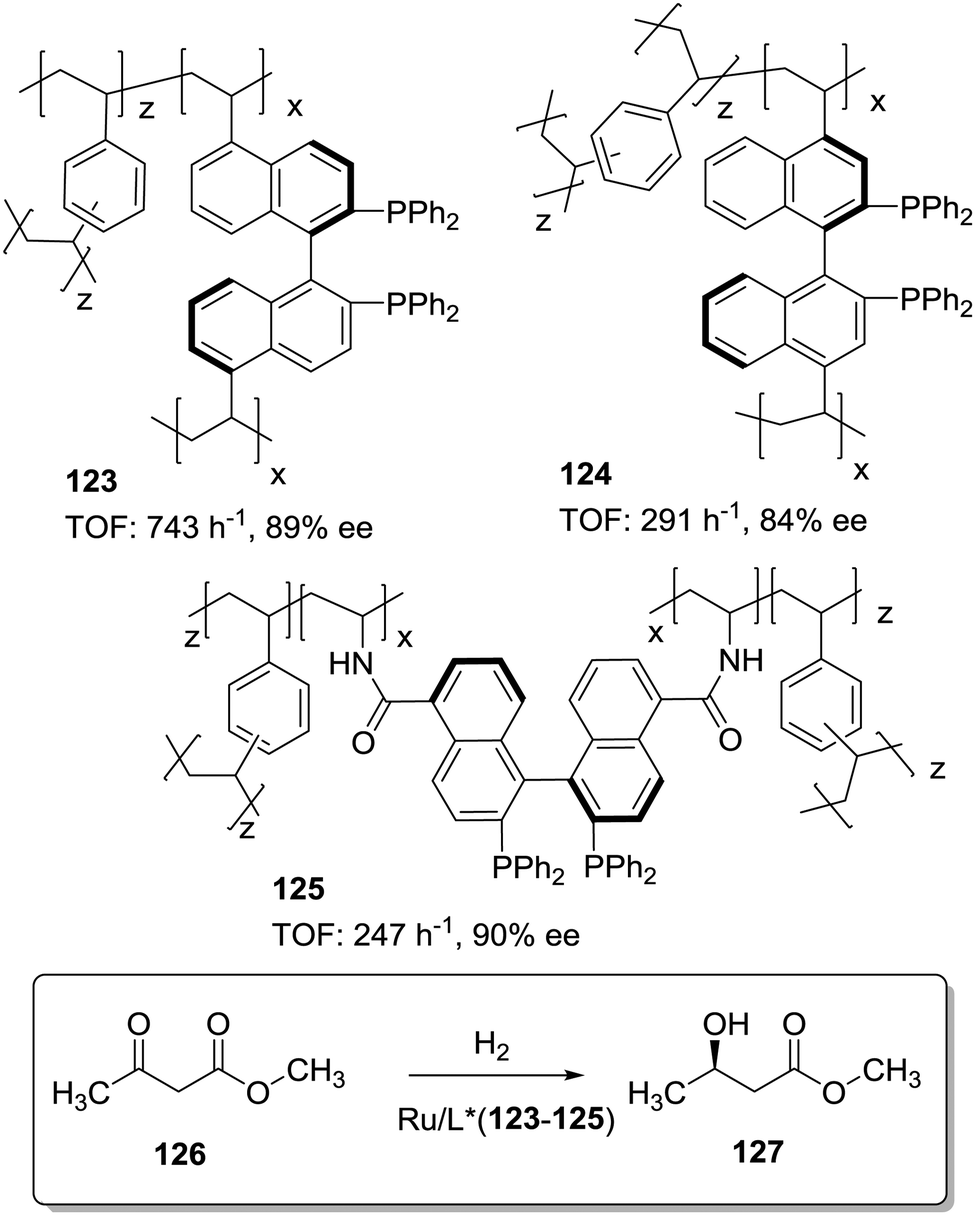 | ||
| Fig. 25 General structures of POPs containing BINAP moieties obtained by radical copolymerization with DVB and used for Ru-catalysed hydrogenations. | ||
In the knitting strategy developed by Tan and coworkers, the very reactive formaldehyde dimethyl acetal (FDA) is employed to crosslink, through rigid methylene bridges, a variety of simple aromatic building blocks like benzene or biphenyl in a FeCl3 catalysed Friedel–Crafts reaction. The resulting polymeric networks have predominant microporosity and a high surface area.198 In this way BINAP has been immobilised in a variety of hypercrosslinked polymeric aromatic networks without requiring the previous preparation of modified BINAP structures (128, Fig. 26).199 These materials are insoluble and porous, with large surface areas of 1100–1200 m2 g−1. The optimised catalyst demonstrated a satisfactory activity for the hydrogenation of 126 at very high substrate/catalyst molar ratios (S/C = 6000) for 10 hours with similar enantioselectivity and activity as those prepared by polymerisation.
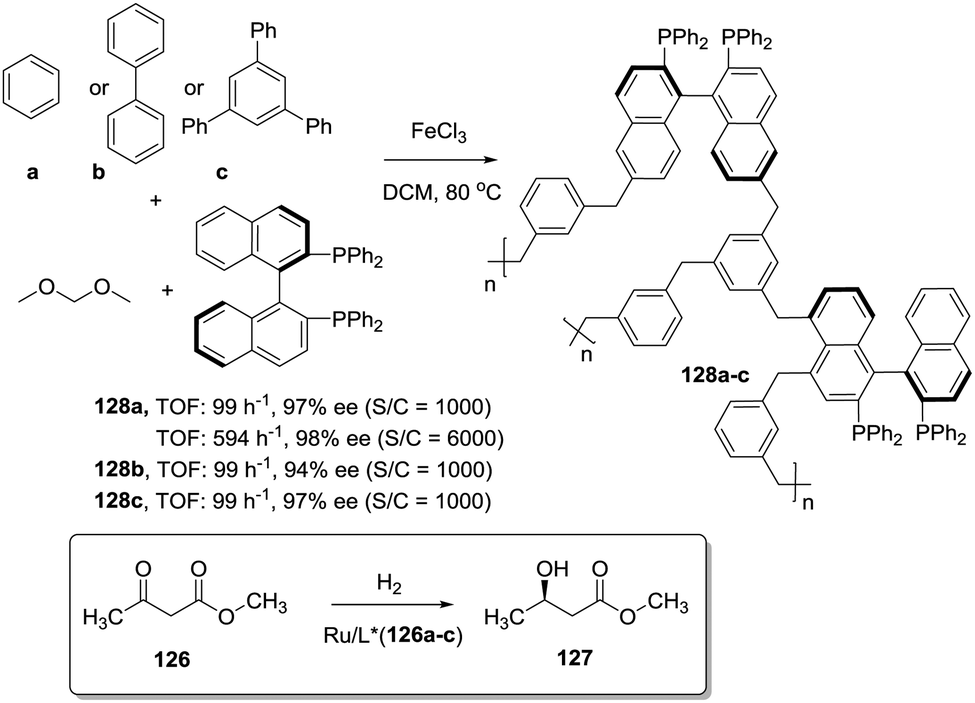 | ||
| Fig. 26 Preparation of a polymeric network containing BINAP fragments not requiring the preparation of polymerisable BINAP derivatives (knitting strategy). | ||
BINAP has been also immobilised in chiral conjugated microporous polymers (CMPs). Apart from the advantages of common POPs, such as large surface areas and high stability, the porous structure and surface area of CMPs can be finely controlled at a molecular level through the rigid node–strut topology of the monomer structure. CMPs functionalized with catalytic units can be regarded as heterogeneous catalysts.72,73
The preparation of BINAPO-CMPs was carried out through the Sonogashira–Hagihara reaction of (R)-4,4′-dibromo BINAPO (129) with alkynes of different lengths (Fig. 27).200 The BET surface areas ranged from 391 m2 g−1 to 509 m2 g−1 depending on the alkyne used. The corresponding Ru-complexes were active for the asymmetric hydrogenation of β-keto esters. More interestingly, a significant activity enhancement was found when their Ir-complexes were used for the asymmetric hydrogenation of quinolines.201 The catalyst 130 prepared by polycondensation of 129 with 1,3,5,7-tetrakis(4-ethynylphenyl) adamantine was four times more active than the homogeneous catalyst providing the same enantioselectivity. This effect was associated with the spatial site isolation of the BINAP catalytic sites that were evenly distributed within the rigid CMP framework preventing the formation of dimers.
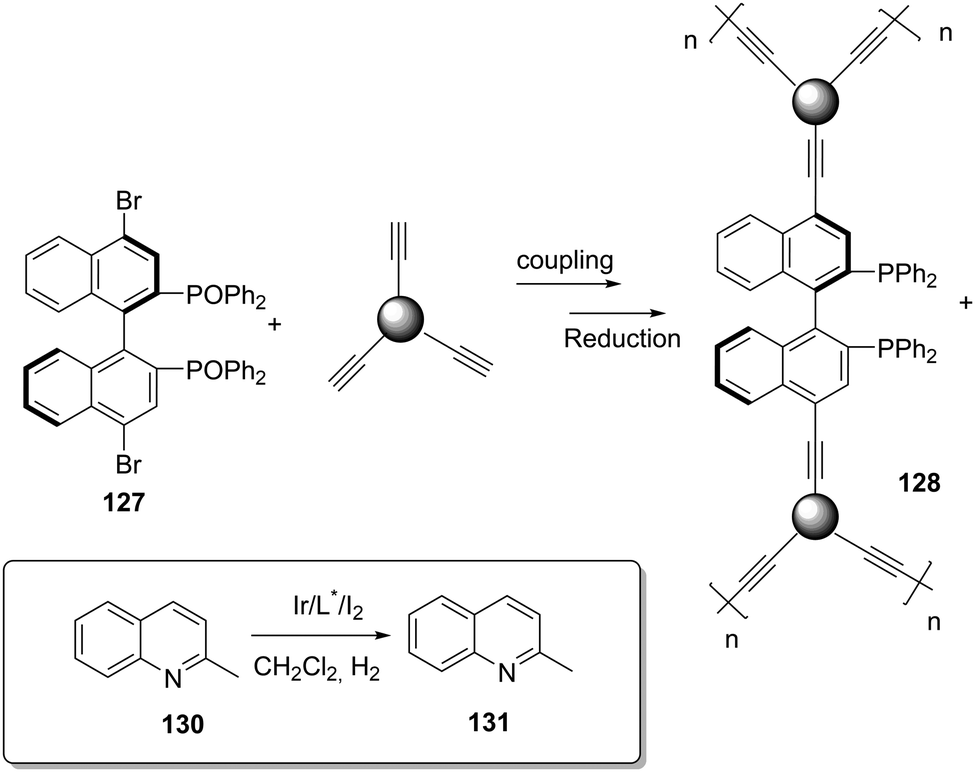 | ||
| Fig. 27 Preparation of BINAPO–CMPs used for the Ir-catalysed hydrogenation of quinaldine. | ||
As in the case of dendrimers discussed above, site isolation is not the only way by which the crosslinked matrix can contribute to increase the activity of supported systems. In fact, the opposite mechanism can also be found. Different catalytic systems involve the participation of more than one catalytic unit. In those cases immobilization with high loading degrees favours the presence of high local concentrations (pseudoconcentration effect) and this can contribute to facilitate the formation of the active species.202
A nice demonstration of the former principle was provided in the case of polymer-supported chiral Co–salen complexes.203 Both silica (133) and polystyrene-bound (134) complexes were used for the hydrolytic kinetic resolution of terminal epoxides (Fig. 28). A clear correlation between the loading of the heterogeneous surface and the rate was observed in the case of silica-supported catalysts, with the presence of a minimum level of loading for the reaction to proceed. The polymeric backbone used for the preparation of 134 contained a very low degree of crosslinking (2%) and this seemed to provide enough flexibility as to favour inter-complex interactions.
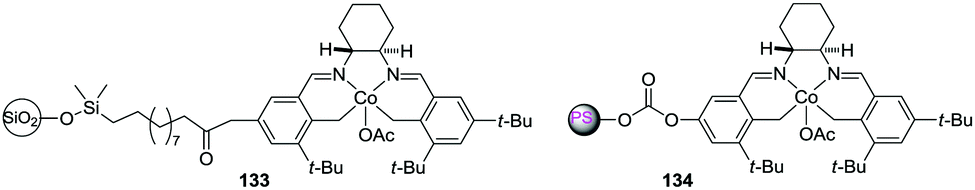 | ||
| Fig. 28 Silica and PS supported chiral Co–salen complexes displaying positive pseudoconcentration effects in the kinetic resolution of terminal epoxides. | ||
The structure of the POPs can be also adjusted considering the mechanistic requirements of the catalyst to be immobilized, by properly selecting the nature of the building blocks to favour site–site interactions. Thus, Zhong et al. have designed flexible porous organic frameworks integrating high loading of Co–salen as the active sites, achieving higher activities than the homogeneous counterpart in the hydration of propylene epoxide (TOF: 3300 vs. 2670 h−1, calculated at a conversion <30%).204 The polymeric porous structure was prepared by poly-condensation of cyclohexanediamine with 1,3,5-tris(3′-tert-butyl-4′-hydroxy-5′-formylphenyl) benzene (TBHFPB) and the 3D-flexible salen network was considered to enhance the cooperation of nearby Co–salen units. Thus, the polycondensation of different building blocks together with the chiral ligand and/or complex by a bottom-up approach offers a wide range of new exciting possibilities.
Sun et al. reported the immobilization of a Noyori–Ikariya asymmetric catalyst onto a superhydrophobic mesoporous polymer prepared by copolymerization of N-p-styrenesulfonyl-1,2-diphenylethylenediamine with DVB under solvothermal conditions (Fig. 29). This Ru catalyst (137) presented a higher activity than the homogeneous analogue for the asymmetric transfer hydrogenation of acetophenone.205 A bottom-up approach allowed the design of a material with the appropriate porous structure (pore size distribution of ca. 11.8 nm, BET surface area of 481 m2 g−1 and pore volume of 0.55 cm3 g−1) and superhydrophobic character that provided a suitable mass transfer of reagents and product to and from the catalytic sites. Due to the wettability properties of the material, acetophenone was absorbed preferentially and the catalytic sites within the porous polymer were enriched with the reactants, leading to a pseudo-concentration effect. The superhydrophobicity also favoured an efficient transfer of the more polar product from the polymer network to the water phase. Noteworthily, when an analogous polymer without nanoporosity was used instead of 137, low levels of conversion were observed (<5% yield). This is in good agreement with the above mentioned results reported by Itsuno using gel-type resins (Fig. 6).115–120
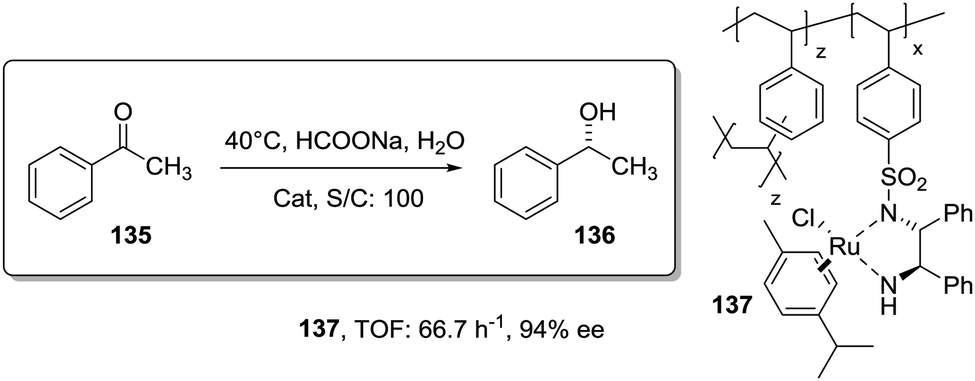 | ||
| Fig. 29 Superhydrophobic mesoporous polymer prepared under solvothermal conditions containing Ru–TsDPEN for the asymmetric transfer hydrogenation of ketones. | ||
Thus the immobilisation strategy determines again the activity of the resulting immobilised catalyst. This can be further highlighted by the immobilisation of BINOL-derived phosphoric acids (Fig. 30). This chiral organocatalyst has been supported by polymerisation of the corresponding monomeric derivatives with styryl substituents in either the 3,3′- or 6,6′-positions of the BINOL skeleton with styrene and DVB.206
 | ||
| Fig. 30 General structures of polymer immobilized BINOL-derived phosphoric acids prepared through different approaches and used for the asymmetric transfer hydrogenation. | ||
Alternatively, a microporous material with a BET surface area of 386 m2g−1 was obtained by self-condensation of chiral 1,1′-binaphthalene-2,2′-diyl hydrogenphosphate (BNPPA) with 9-anthracenyl groups in 3,3′-positions and a 3-thiophenyl group at the 10-position of the anthracene units. These fragments facilitate the condensation reaction by mild oxidative coupling reaction in the presence of FeCl3.207
Using 5 mol% of these polymer-supported catalysts 138 and 139, a 20–24 h reaction time was reported to achieve full conversion of the imine 141 into the corresponding 3-phenyl-2H-1,4-benzoxazine (142) with 64% ee and 94% ee, respectively. However, for the catalyst obtained by self-condensation through oxidative coupling (140), the same reaction was complete within 2 hours with this catalyst being as fast as the homogeneous catalyst. The surface area and pore volume of the microporous polymer networks could be further tuned by co-polymerisation of the chiral building block in the presence of 1,3,5-tris(2-thienyl)benzene. The resulting reaction rate for the same reaction was enhanced by increasing the accessible surface area and pore volume of the microporous polymer network. These copolymers were more active than the corresponding homopolymer 140.208
Jiang and co-workers have exploited the post-modification of the walls of an achiral COF to immobilise in the open channels chiral organocatalytic moieties while retaining the crystallinity and porosity of the framework (Fig. 31).209,210 The mesoporous imine-linked COFs were prepared by condensation of different di-aldehydes with triamines or tetramines. The presence of ethynyl groups on the walls of the pores allowed their further click reaction with pyrrolidine azide to quantitatively yield the corresponding post-functionalised COF. The materials 143 and 144 have been tested for asymmetric Michael reactions in water, displaying better activities than the homogeneous analogue, while retaining enantioselectivity. The results suggest that the crystallinity and porosity of COFs play a vital role in determining their catalytic activities. Analogous amorphous and nonporous polymeric catalytic systems required significantly longer times to complete the reaction (43 and 65 h vs. 1–2 h for the COF). It is worth mentioning that the catalytic activity depends upon the density of the active sites on the pore walls. A high density of pyrrolidine units in the pores induced a steric congestion impeding the mass transport through the channels.
 | ||
| Fig. 31 General structures of imine-linked COFs postfunctionalized with pyrrolidine units on the walls of the porous framework. | ||
Davies and co-workers have reported the immobilisation of dirhodium tetracarboxylate complexes derived from N-arylsulfonyl prolines (147 and 151) and their use as catalysts for the asymmetric cyclopropanation of alkenes using methyl aryldiazoacetates.211–214 For that purpose, they developed a pyridine containing resin in order to axially coordinate the metal centers in the active complexes (Fig. 32). The resulting supported Rh species based on 149 and 153 showed a higher activity than their homogeneous analogue (derived from 148 and 152, respectively) even considering that donor groups such as pyridine tend to deactivate dirhodium tetracarboxylates.215 Some data, in particular the fact that a supported active complex could be also obtained from a resin containing no pyridine moieties (150 and 154), suggested that immobilisation could take place due to a microencapsulation effect.216,217 The differences in activity, in this case, were attributed either to a pseudodilution effect or to the microenvironment inside the polymer.
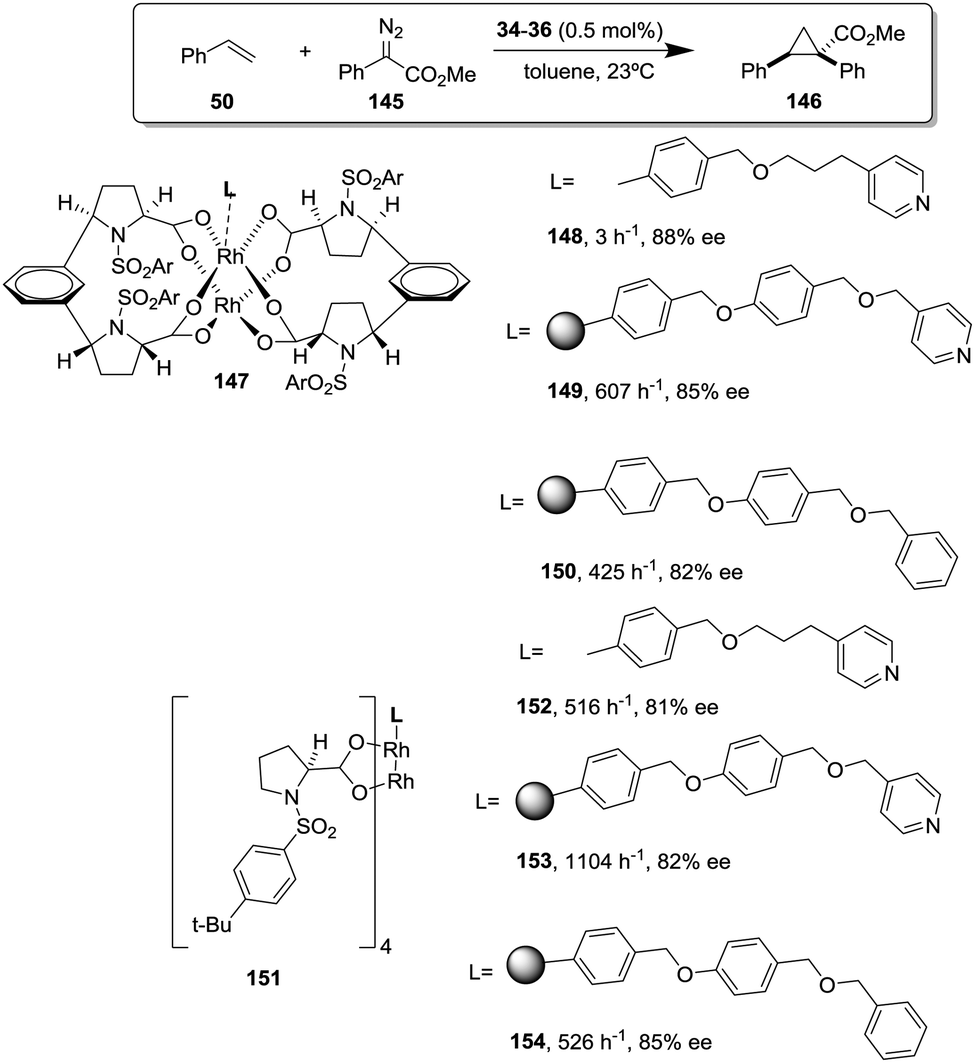 | ||
| Fig. 32 Dirhodium tetracarboxylate complexes derived from N-arylsulfonyl prolines immobilised on resins containing pyridine. | ||
The “microencapsulated (MC) catalysts” and “polymer incarcerated (PI) method” can be considered as a limiting case in between soluble and insoluble supports.217 In MC, catalysts are physically entrapped by a polymeric network (polystyrene derivatives in many cases) and at the same time immobilized by the interaction between the π electrons of the benzene rings of polystyrene and vacant orbitals of the catalysts (metal compounds). In the PI method, a catalyst is first physically microencapsulated in a dynamic system and then the microcapsules formed are crosslinked to afford the desired heterogeneous system. In these systems diffusion through the walls of the “capsule” is a key factor to determine the catalytic activity. In some particular cases, as pointed out before, the microencapsulated catalyst can be more active than the homogeneous counterpart, as is the case of microencapsulated Sc(OTf)3. Kinetic data revealed that its activity was higher than that of monomeric Sc(OTf)3 for aldimine-selective reactions, most likely because of the higher stability of aldimine–Lewis acid complexes using polymer-supported acids.218,219
Soluble polymers as supports
Taking into account some of the previous considerations, it seems reasonable that examples of supported species being more active than the homogeneous ones can be also found for soluble polymeric supports. For soluble polymers diffusion issues encountered in crosslinked resin can be minimised.43–45,220 Soluble polymers, especially when prepared by controlled polymerisation, provide a facile control on the incorporation of catalytic sites either as pendent groups or at the main polymeric chain and an easy adjustment of the nature of the polymer matrix and the catalyst loading, just by using the suitable polymerization technique and monomeric mixture.Thus, for instance, Fan et al. studied the soluble polymer 155 containing BINAP subunits for the Ru-catalyzed asymmetric hydrogenation of 2-(6′-methoxy-2′-naphthyl)acrylic acid (157) to form naproxen (158, Fig. 33).221 A complete conversion was obtained with 155 after ca. 4 hours, while 8–9 hours were required with the use of the non-polymeric analogue 156 and 48 hours for (S)-BINAP itself. Similar increases in activity have been found for other related systems in which the (2S,4S)-pentanediol component was substituted by polyethyleneglycol chains.222,223
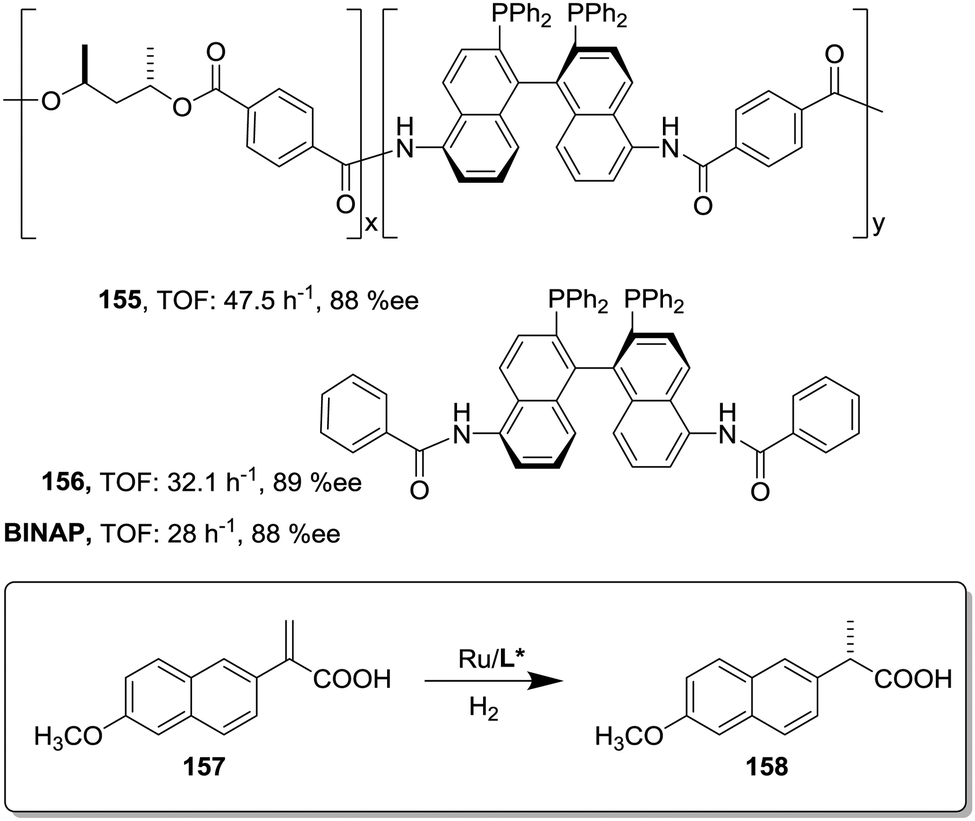 | ||
| Fig. 33 General structure of soluble polymers containing BINAP fragments in the main chain studied for asymmetric Ru-catalysed hydrogenations. | ||
Soluble polymer-supported chiral phosphoramides (159 and 160) have been reported to be significantly more efficient than the corresponding homogeneous analogue (161) for the organocatalytic allylation of benzaldehyde with allyltrichlorosilane (Fig. 34).224
 | ||
| Fig. 34 General structure of soluble polymers containing chiral phosphoramides as pendant groups for asymmetric organocatalytic allylations. | ||
It is important to bear in mind that upon immobilization of a given catalyst, significant changes in the mechanism may occur, including changes in the molecularity of the active complexes, as shown, for instance, for supported Sharpless alkene epoxidation catalysts.225 For these supported chiral phosphoramides, the loading of the catalytic sites on the polymer is rather high (DF = 37–100% of the aromatic rings functionalised). Hence, the pseudo-concentration on the polymeric matrix facilitates site–site interactions.
The 2.4 times increase in activity observed for phosphoramide chiral Lewis bases 159 supported on soluble polystyrene can be assigned to an increase of the local concentration of the phosphoramide units around the polymer chain, as it has been shown that from the two possible transition states detected in solution for the allylation of aromatic and unsaturated aldehydes with allyltrichlorosilanes, the one involving two phosphoramides presents higher reactivity.226–228
The increased local concentration on the support facilitates faster catalytic processes involving bimetallic pathways with lower substrate to catalyst ratios. This has been exploited to improve the activity of metal–salen catalysts by varying the density of catalytic sites along a polymeric backbone.36,229–232
Based on this approach, Weck and coworkers developed highly active polymeric Al–salen complexes for the asymmetric addition of cyanide to α,β-unsaturated imides with high yields and enantioselectivities (Fig. 35).233 The catalyst 166 was designed by incorporating a flexible poly(norbornene) backbone and a long linker to facilitate the bimetallic pathway and four quaternary carbon atoms at the periphery of the catalytic site to improve selectivity. With this rational design the reaction progressed significantly faster with the supported catalyst than with its small molecule analogue. The catalytic transformation using 15 mol% of 166 was complete within 6 h while the same catalyst loading for the non-supported analogue resulted in less than 50% conversions. At lower catalytic loadings (5 mol%), a similar activity was maintained while for the homogeneous analogue less than 5% of the product was found.
 | ||
| Fig. 35 General design of polymeric Al–salen complexes for the asymmetric addition of cyanide to α,β-unsaturated imides involving bimetallic pathways. | ||
The incorporation of different monomeric units in the polymeric framework may lead to cooperativity. Xie et al. have synthesised a pH-responsive soluble polymer by copolymerization of dimethyl aminopropyl acrylamide and N-p-styrenesulfonyl-1,2-diphenylethylenediamine.234 The N-alkyl moieties appeared to play a cooperative role accelerating the Ru-catalysed asymmetric transfer hydrogenation of ketones in water, whereas the mono-N-tosylated derivative moiety played a pivotal role in chiral induction (Fig. 36). The polymer 167 exhibited excellent pH-induced phase-separation behaviour in water. It could be dissolved in water when the pH of the solution was lower than 6.5 and precipitated completely from water when the pH was above 8.5. Furthermore, the catalyst shows an enhanced activity in comparison with the soluble counterpart.
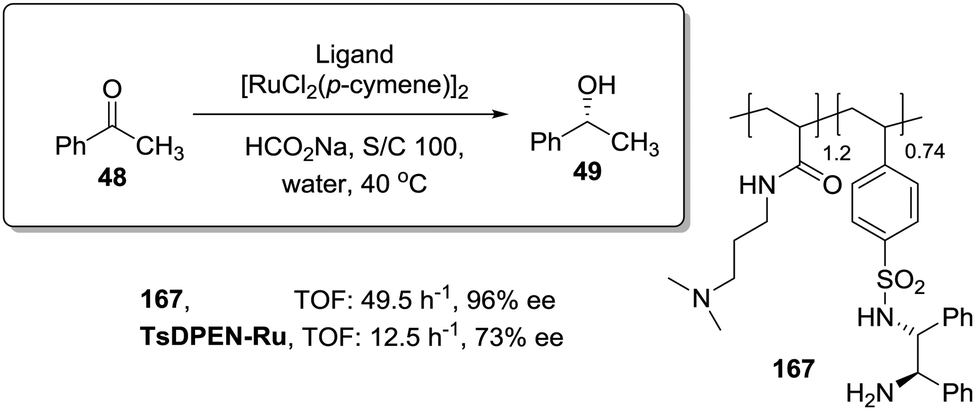 | ||
| Fig. 36 General structure of the pH-responsive soluble linear polymer containing TsDPEN units and dimethyl aminopropyl fragments providing a cooperative effect to increase the activity of the Ru-catalysed asymmetric transfer hydrogenation. | ||
The polymer architecture can be designed using different controlled radical polymerisation (CRP) techniques to influence and tune the reactivity of catalysts in interesting ways. For instance, linear amphiphilic block copolymers, obtained by CRP, can self-assemble in water into nanomicelles, which may provide a hydrophobic environment to lower the interfacial energy of the reaction system and promote organic reactions in water.46 In this regard, O’Reilly and co-workers have covalently attached to the hydrophobic core of a polymeric micelle 4-(dimethylamino)pyridine (DMAP) catalytic moieties for acylation reactions in water employing non-water-soluble substrates. The reactivity of the tethered organocatalyst within the nanostructure was found to be extremely high, improving in some cases the acylation rates up to 100 times compared to those for unsupported DMAP in organic solvents.235,236 Based in those principles, they developed a series of efficient immobilised chiral organocatalysts. For instance, L-proline tethered to amphiphilic block copolymers (169), synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerisation, self-assembled in water leading to highly active catalytic nanoreactors providing an effective concentration of the hydrophobic reagents, thus increasing their effective molarity, based on the difference in core–shell polarity (Fig. 37).237 Low catalyst loadings (1 mol%) of this micellar system catalysed the aldol reaction in water more efficiently than the unsupported L-proline in organic solvents at 10% loading. The modularity of the CRP also allowed the introduction of thermoresponsive polymeric block units in the immobilised systems enabling the control of solubility and activity by temperature.238 Noteworthily other stimuli such as the ion concentration could also be used to tune the catalytic activity of related block polymeric systems.239
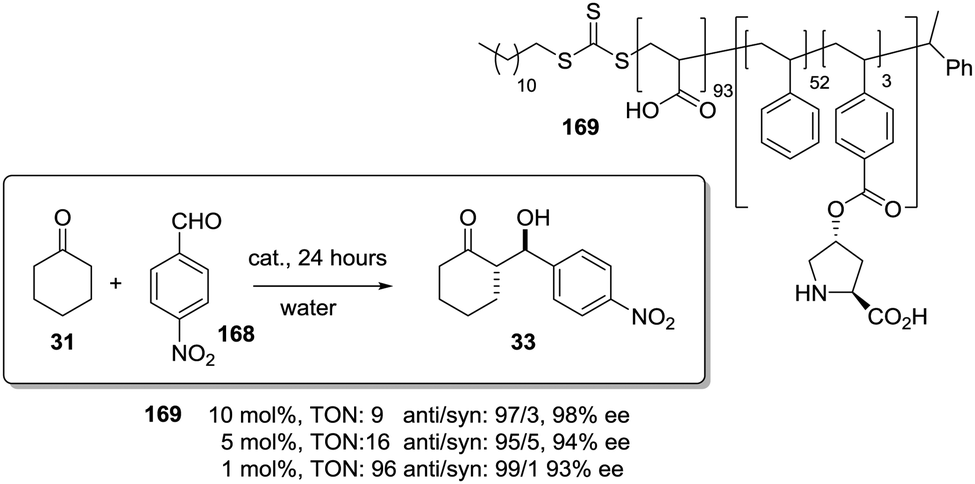 | ||
| Fig. 37 Amphiphilic linear polymer with proline organocatalytic units able to form nanomicelles in water catalysing aldol reactions. | ||
Zhang et al. have reported the immobilisation of a Mn–salen complex onto soluble “smart” poly-N-(isopropylacrylamide) (PNIPAAm) by axially coordinating a supported amine group to the metal center. The resulting macromolecule (170) containing hydrophobic and hydrophilic blocks spontaneously self-assembled in water to form micelles with a hydrophobic catalytic core and a hydrophilic surface.240 A significant acceleration in the reaction rate for the epoxidation of olefins was observed reaching an unprecedented TOF value (2.48 × 103 h−1) in the aqueous epoxidation of styrene, being up to 6 times more active than the homogeneous Mn–salen neat complex (Fig. 38).
 | ||
| Fig. 38 Amphiphilic linear polymer containing Mn–salen fragments able to form micellar structures in water and catalyse the epoxidation of alkenes. | ||
Again, a “pseudo-concentration effect” at the core of the aggregate was used to explain the activity enhancement. The hydrophobic core provides a microenvironment with a high local concentration of catalytic sites along with the concentrated hydrophobic substrates, while the hydrophilic surface guarantees water solubility and the self-assembly of the polymer in the corresponding micelles. The advantages of this approach are further developed with the introduction of a thermo-responsive polymeric block in the polymeric framework.241 The amphiphilic nature of the polymeric catalyst allowed self-assembly in a micellar system while the thermo-sensitive properties facilitated its recovery. Thus, a chiral Ti–salen complex tethered to a hydrophobic block of a thermo-responsive amphiphilic copolymer (poly(N-isopropylacrylamide-co-N,N-dimethyl acrylamide), poly-(NIPAAM-co-DMAAM)) displayed an outstanding catalytic activity and selectivity in the asymmetric sulfoxidation in water for a wide range of methyl aryl sulfides.
A similar catalytic enhancement in water has been reported for the cyclopropanation reaction catalysed by copper–bis(oxazoline) complexes immobilised on the hydrophobic domain of a polymersome membrane (173). These crosslinked polymersomes were obtained by a click reaction of a well-defined soluble block polymer poly(ethyleneglycol)-b-poly-(styrene-co-4-vinylbenzyl-azide) with a C2-BOX modified with an alkyne functionality (Fig. 39).242 The conversion obtained with the catalytic polymersomes was comparable with the conversion of the parent catalyst in CH2Cl2 and, more importantly, surpassed the activity found for the homogeneous Cu–BOX in water. The catalyst inside the polymersomes is surrounded by a hydrophobic environment from which water is excluded, favoring the desired process since water affects both conversion and enantioselectivity of the asymmetric cyclopropanation reaction. This also produced substrate selectivity. Hydrophobic substrates were readily converted into the corresponding cyclopropane products, while hydrophilic substrates did not undergo the reaction. The same strategy has been used, with similar positive effects, for the asymmetric aldol reaction in water catalysed by immobilised L-proline.243
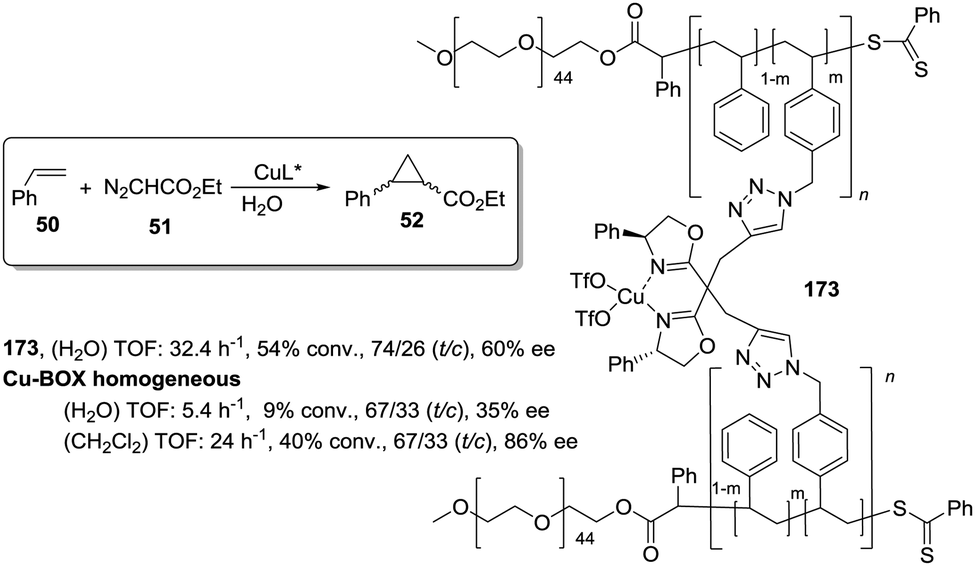 | ||
| Fig. 39 General structure of polymersomes containing Cu–BOX subunits used for the enantioselective cyclopropanation of styrene. | ||
Meijer and coworkers prepared random terpolymers, by controlled RAFT polymerization, bearing in their structure a catalytic unit, a block polymer defining the solubility and an additional block containing chiral benzene-1,3,5-tricarboxamide groups for self-assembly (Fig. 40).244,245 This additional block defines the folding of the polymer in water to form a confined reaction space, mimicking the specific folding of proteins.246 In the case of the polymer 174 containing L-proline as the catalytic unit, the polymer behaved as an enzyme mimic, as its catalytic activity was only expressed in its folded conformation. The structure of the polymer could be fine-tuned to obtain an exceedingly active catalyst.
 | ||
| Fig. 40 General structure of a responsive terpolymer prepared by RAFT polymerization and containing proline catalytic units active for the aldol reaction. | ||
Chiral monomeric imidazolium salts have been used to build up polymers with a well organised secondary structure (Fig. 41).247 The right self-assembly of the monomeric units generated a highly ordered macromolecular structure with a complex hierarchical architecture, which can provide an adequate microenvironment for an efficient catalytic activity. A highly active and selective system for the aldol reaction was obtained for reactions both in water and in the presence of water. Thus, for instance, in acetone containing some water (H2O–acetone 1:4) the polymeric catalysts obtained were up to ca. 6.6 × 103 times more active than their corresponding monomeric counterpart, while in water the only active catalyst was the polymeric one. This enhancement in catalytic activity was related to some degree of chirality transfer to the main polymeric chain, producing a more properly organised polymeric structure and, therefore, a more efficient catalyst. A better transfer of the chirality was obtained for the polymers prepared by RAFT polymerization (177). The catalysts prepared by RAFT polymerization of the chiral monomer were more active and enantioselective than the ones prepared by ATRP (atom transfer radical polymerisation) or grafting.
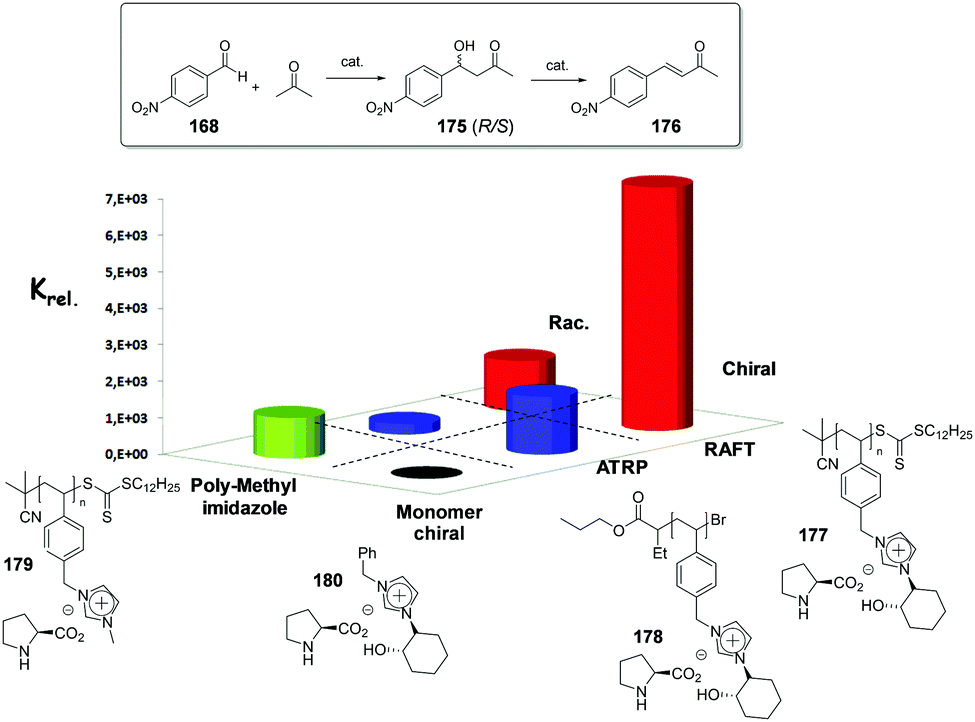 | ||
| Fig. 41 Influence of the polymerization protocol on the activity of proline incorporated into polymeric ionic liquids. | ||
Similar trends have been also observed for a range of well-defined copolymers of styrene and styryl functionalized L-proline prepared by RAFT polymerisation.248 The self-assembly of these functionalized copolymers into well-defined aggregates in DMF/water provided a unique microenvironment for L-proline positively influencing the catalysis of the aldol reaction.
Multi-catalytic systems. Wolf and Lamb approaches
Finally, the preparation of active multi-catalytic systems based on the use of mutually incompatible individual catalysts constitutes a special case of catalytic activity enhancement using polymeric immobilised catalysts.249 The combination in solution of two non-compatible catalysts produces an inactive dual catalyst system. On the contrary, different tools have been developed for the immobilization of those catalysts on polymeric phases so that site isolation is achieved to produce an active dual catalytic system.250,251 Novel approaches to exploit this possibility include the use of sol–gel and star polymers,252,253 as well as polymeric microcapsules.254,255One of such examples is shown in Fig. 42256 The reaction of nitromethane with an alkylaldehyde is catalysed by polyethylenimine (PEI) 181, which may also catalyse the addition of a second molecule of nitromethane. On the other hand, the Ni-based Lewis acid 182 catalyses the addition of dimethylmalonate to β-nitroalkane to give the corresponding Michael adduct.257 When both catalysts were mixed in solution, free PEI strongly coordinated with Ni, making it inactive and only low yields of the Michael adduct (<5%) were obtained.
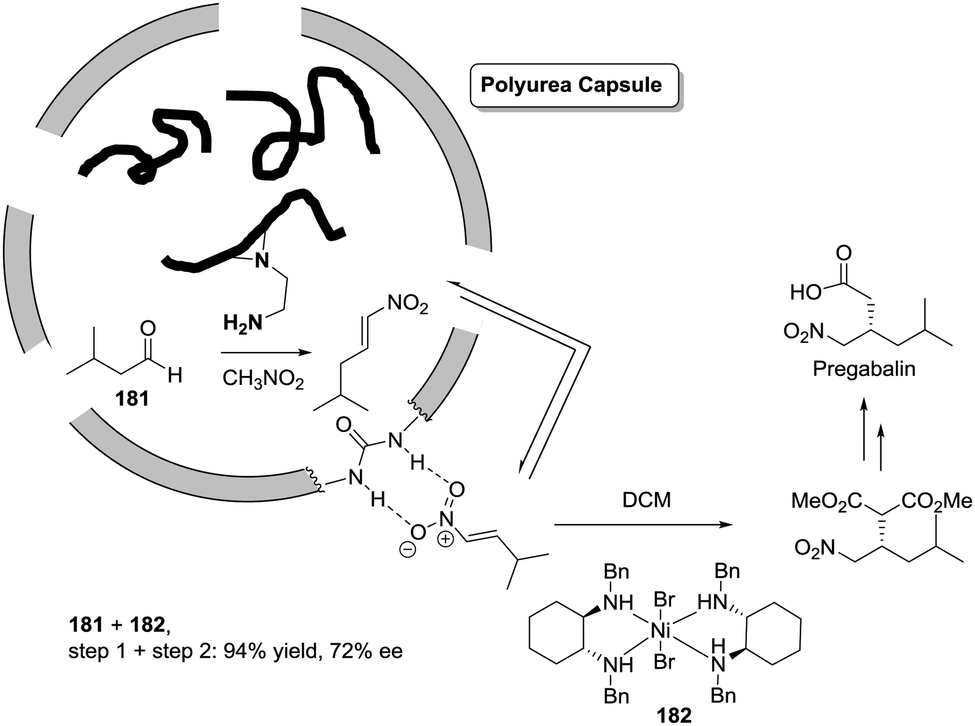 | ||
| Fig. 42 Multicatalytic system based on the encapsulation of PEI in a polyurea capsule. | ||
The microencapsulation of PEI, via interfacial polymerization using polyethylenimine (PEI) and a diisocyanate, prevents interaction between the polyamine and the nickel catalyst. By this isolation of the two catalytic sites, the Michael adduct was obtained in good yield and enantioselectivity. It should be noted that not only did the polymeric shell allowed a right isolation but the ureas present in the shell of the PEI-microcapsules also contributed to accelerate the nickel-catalysed Michael addition.258
The anchoring of two incompatible catalytic functionalities (acidic and basic) to two different solid matrices also allows achievement of one-pot multistep reactions. It has been shown, for instance, how a sulfonic acid resin could be used as the acidic catalyst, while a superparamagnetic spinel ferrite nanoparticle functionalized at the surface with amino groups was the basic catalyst.259 The very different nature of the solid supports provided the development of simple separation protocols for the individual catalysts.
Shell cross-linked micelles (SCMs) can also be used to compartmentalise two incompatible catalysts (e.g. an acid catalyst in the shell and a base in the core). The immobilisation of a dual-catalyst was achieved by using an amphiphilic triblock copolymer of poly(2-oxazoline) with orthogonal functional groups on the side-chain that could covalently cross-link the micelle and separate two incompatible catalysts in two isolated domains of a single micelle (183). This bifunctional SCM was used in the acid–base catalysed one-pot tandem deacetalization-nitroaldol reaction showing excellent catalytic activity,260 and also for the compartmentalisation of stereoincompatible catalysts for one-pot asymmetric cascade reactions.261 The Co-catalysed hydration of an alkyne proceeded in the hydrophobic core, while the Rh-catalysed asymmetric transfer hydrogenation of the intermediate ketone into a chiral alcohol took place in the hydrophilic shell (Fig. 43). While the tandem reaction also worked when the two catalysts were immobilized on different micelles, the multicompartmentalized micelle containing both catalysts gave significantly better results.
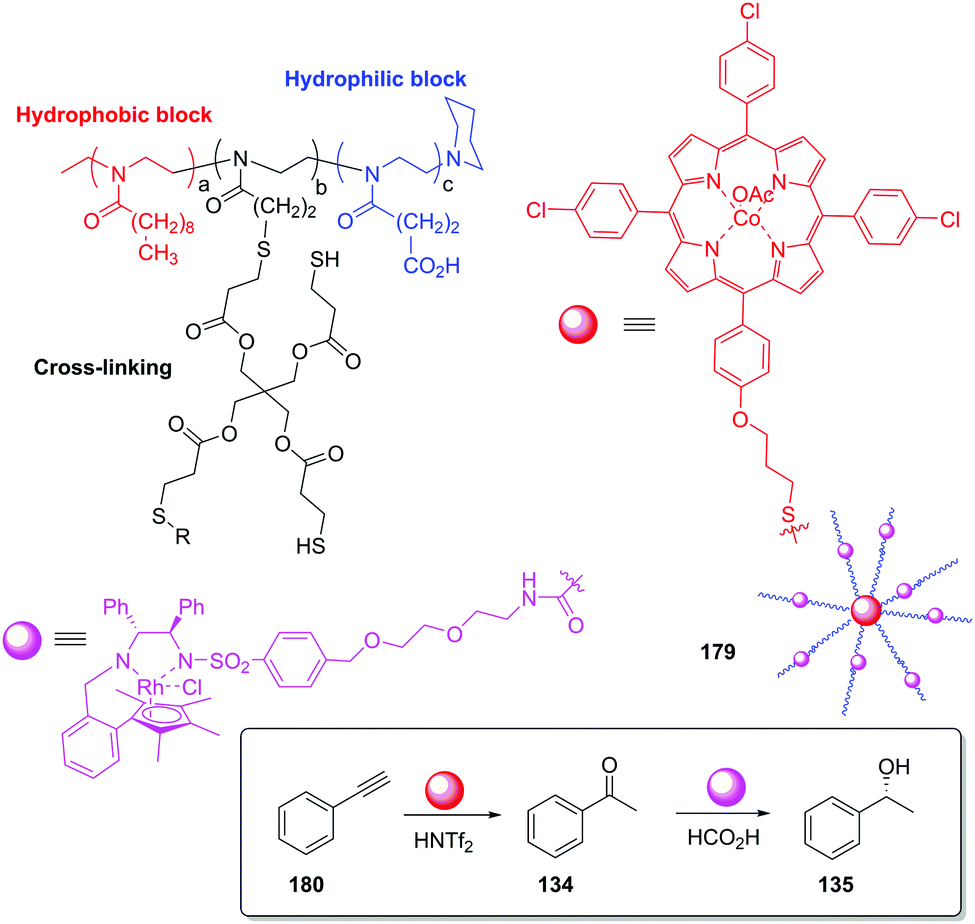 | ||
| Fig. 43 Compartmentalisation of stereoincompatible catalysts at the core and shell of SCMs for one-pot asymmetric cascade processes. | ||
The site isolation properties of non-interpenetrating star polymers have been exploited for encapsulation of multiple, otherwise incompatible, catalysts for asymmetric cascade reactions that involve iminium, enamine, and H-bonding catalysis (Fig. 44). In the case of the encapsulation in the core of soluble star polymers, an efficient one-pot multi-component asymmetric cascade reaction could be carried out with the participation of three different substrates and a catalytic combination involving four species, two of them polymer-bound (185 and 186) and two small molecules (187 and 188). For this system, the multistep reaction took place with good yield and enantioselectivity, while the use of a combination of the corresponding small molecules and/or linear polymer analogues led to little or no cascade reaction.262
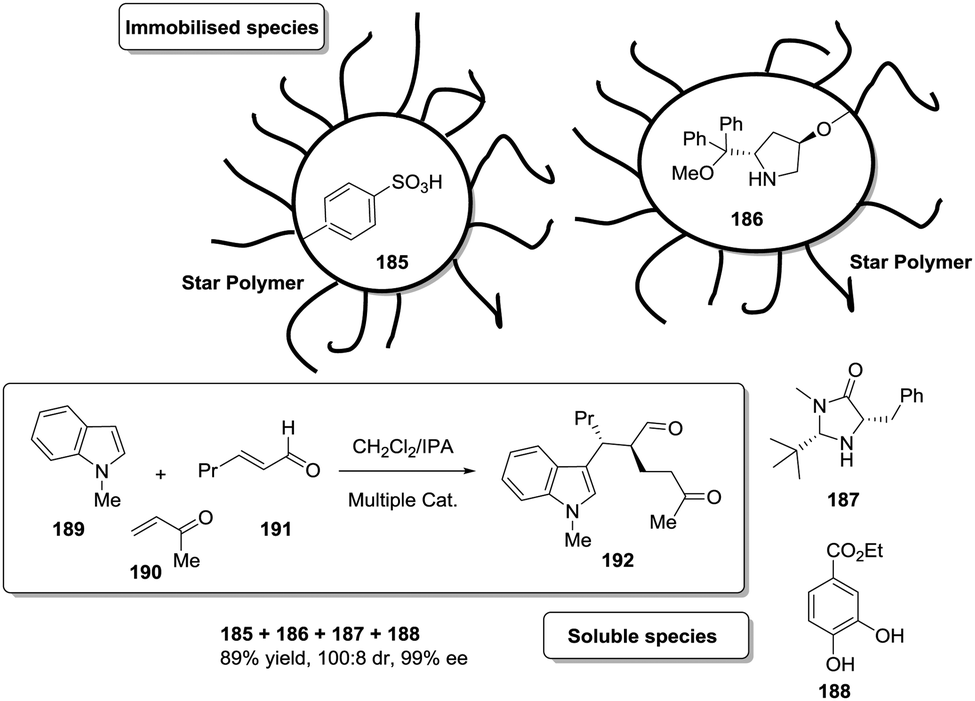 | ||
| Fig. 44 Encapsulation of incompatible catalytic species at the core of star polymers. | ||
Stability
As mentioned, facilitation of the reuse and recycling of supported chiral catalysts represents one of the most important advantages associated with them. This, along with some of the intrinsic properties of the corresponding support, can contribute significantly to enhance their stability and increase their lifetime. In this context, it is necessary to consider both the possibility of reusing the catalyst in successive runs (as many as achievable) and/or for long periods of time and the possibility of its regeneration once the system is not efficient anymore, which is particularly relevant for chiral catalysts where the chiral component and the functional support itself can be the most costly components of the system.Number of runs
The general idea that immobilization onto a support contributes to greatly improve the stability of reagents and catalysts is well established and has been confirmed by data from diverse laboratories. Since the initial reports by pioneering researchers in the field, the use of supported chiral catalysts needs to demonstrate their capacity to be used for several runs.263Although many reports just describe the results for a limited number of reuses (4–5 cycles), examples can be found on systems reused, in batch experiments, for 15 or more runs.78,211,264 The incorporation of cinchona-derived fragments into polymeric matrices has been exploited by different groups active in the field of organocatalysis often with excellent results.265 The immobilization of mono- and bis-cinchona alkaloid derivatives on polystyrene resins using click chemistry for the attachment to the support has allowed the preparation of very stable organocatalysts.266 Some polymer-supported dimeric cinchona systems (193, Fig. 45) were able to efficiently catalyze the asymmetric amination of 2-oxindoles for 100 runs, with more than 5300 h of active life, maintaining the activity (>99% yield) and the enantioselectivity of the final product (>90% ee).267
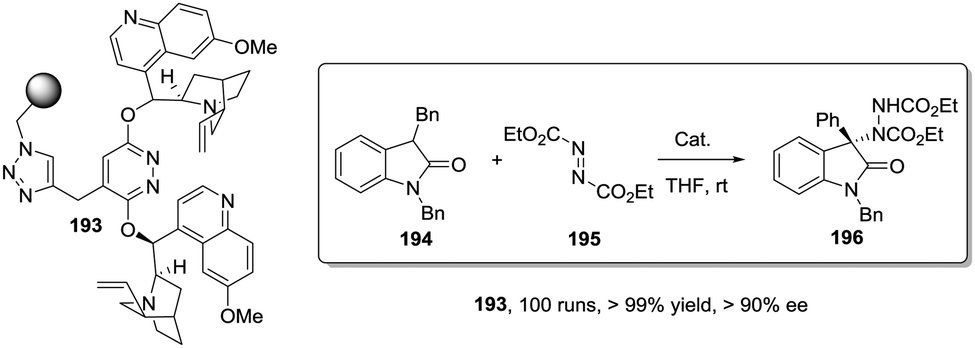 | ||
| Fig. 45 Polymer supported dimeric cinchona derivatives used for the asymmetric amination of oxindoles. | ||
Textiles have been recently reported as excellent polymeric supports for the immobilization of a variety of organocatalytic systems using photochemical approaches for their functionalization.268 The incorporation of a chiral cinchona derivative provided an efficient chiral organocatalyst for the alcoholytic desymmetrization of a cyclic anhydride that could be reused for 300 runs.269 The activity and the corresponding enantioselectivity were maintained essentially constant for the first 200 cycles and the further deactivation could be compensated, at least partly, by an increase in the catalyst loading.
It is worth mentioning that in some instances the reuse of the catalyst is carried out not on the same process but for alternative processes.270 An example is the use of polystyrene-supported TRIP phosphoric acid catalyst (197) for the asymmetric allylboration of aldehydes (Fig. 46).271 This catalyst was used under batch conditions for 18 reuses with different substrates without detecting any deactivation and with excellent yields and enantioselectivities in most cases.
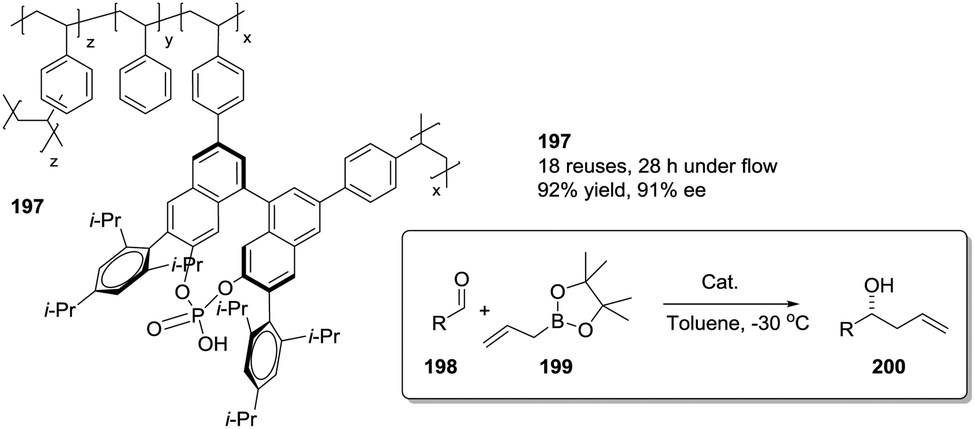 | ||
| Fig. 46 Polymer supported TRIP used for the asymmetric allylboration of aldehydes. | ||
In different instances, some deactivation of the supported catalysts is observed for the first run(s) up to achieving a constant level of activity. In this case, it is assumed that the most accessible sites are more easily deactivated. Very often, in particular in the case of organometallic catalysts, those accessible sites correspond with metal species that are not linked to the supported ligands but just physically entrapped. Although the room temperature solventless octane hydrosilylation catalysed by supported Pt complexes reported by Drake et al. does not involve chiral catalysis, it illustrates very well these issues as much as an extensive analysis of leaching and recycling.273 The activity of the supported-Pt catalysts decreased with the first uses up to reaching an activity plateau, but isomerization decreased for the reused systems, being much lower than that observed for the homogeneous catalyst. The long-term stability of one of the supported catalysts was evaluated after 17 months of its synthesis (stored in contact with air) and 14 months after the first extensive recycling test. The activity for the hydrosilylation process was slightly reduced initially, but fell off with further recycling. However, the level of isomerization was very low and remained so through the reuse. The authors explained this reduction in activity and the concurrent improvement in selectivity based on the oxidation of unbound or weakly bound Pt located on or near the resin surface to PtO2. As isomerization has been associated with the presence of unbound metal in solution, the former oxidation process would lead to a reduction in both activity and isomerization.
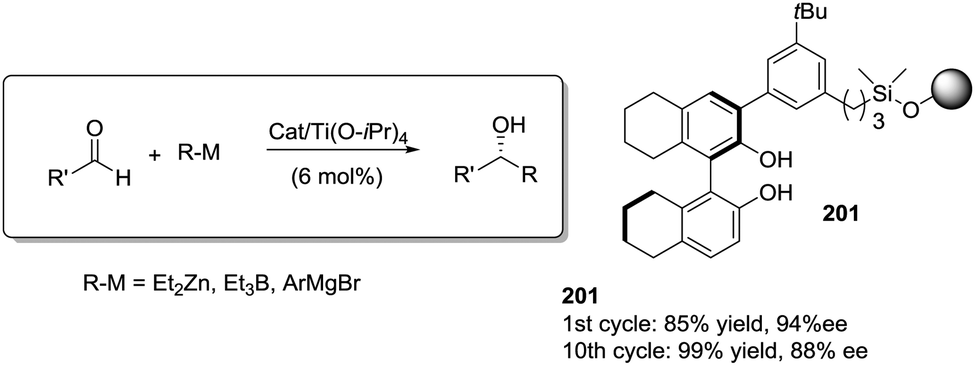 | ||
| Fig. 47 Silica supported BINOL used for the Ti-catalysed alkylation and arylation of aldehydes. | ||
Air–moisture
In general, the stabilisation effect is particularly marked when very reactive, water and air sensitive species are involved. In the case of PS–DVB and related resins, the hydrophobicity of the matrix is likely to contribute to avoid decomposition of such systems. The formation of hydrophobic cavities around the active sites can improve the stability of air/water sensitive species. The introduction of air or moisture in the polymeric matrix was associated with a very small decrease in activity observed for polymer-supported Rh–phosphine–phosphite catalysts in asymmetric hydrogenations (<3% after 11 runs, 97% ee for all runs).275 The small Rh leaching detected in the first cycles did not affect the efficiency of reuse and this leaching was attributed to physically adsorbed Rh species. In the self-supported Mn–salen chiral system reported by Roy et al. for the epoxidation of non-functionalized olefins, the oxidative degradation of the complex was considered to be responsible of the deactivation.276 After modification of the oxidant and optimization of the recycling process the system could be used up to 8 times maintaining the yield and enantioselectivity, but at the expense of an increase in the reaction time. Bissesar et al. have described the preparation of a self-supported Ni(II) metallopolymer capable of acting as an efficient catalyst for the Michael reaction.277 They reported that the self-supported system presented a high air and moisture stability and no change was observed after being maintained in air for two weeks, while the related mononuclear Ni(II) complex decomposed after two days’ exposure to air. In some of the reactions studied, the catalyst could be reused for up to 11 runs maintaining the activity and enantioselectivity (85–83% yield, 99–97% ee). In order to design stable supported species, two factors must be kept in mind: (i) the existence of a robust and appropriate link between the ligand and the support and (ii) the chemical stability of the catalytic entities being immobilised. Even though these points seem to be quite obvious, they should be carefully considered.Deactivation through the metal
In many organometallic complexes deactivation of supported catalysts is generally related to the leaching of the resin-bound catalytic metal or to the possibility of forming deactivated metal species that remain in the polymeric matrix.273When supported catalysts derived from tartaric acid based ligands (Fig. 48) were assayed for the Diels–Alder reaction between methacrolein and cyclopentadiene,278 a sharp contrast was observed for the Al and Ti derivatives of the polymer-supported ester of tartaric acid (202). When the corresponding catalysts were assayed for the Diels–Alder reaction, a rapid decrease in the performance (for both activity and selectivity) was detected for 204. A different situation was found, however, for the Ti catalyst 203 based on a stronger chelate complex. In this case a decrease in activity was observed for the first runs but after the fourth run the activity remained essentially constant for a long period. A significant metal leaching was not detected either for Al or for Ti species, which suggests that in these polymer-supported catalytic systems decomposition of the catalytic site can be accompanied by the formation, inside the polymeric matrix, of insoluble species (i.e. metal oxides). The blockage of the porous structure is a common deactivation mechanism for catalysts based on microporous or mesoporous supports, in particular those of inorganic nature.279
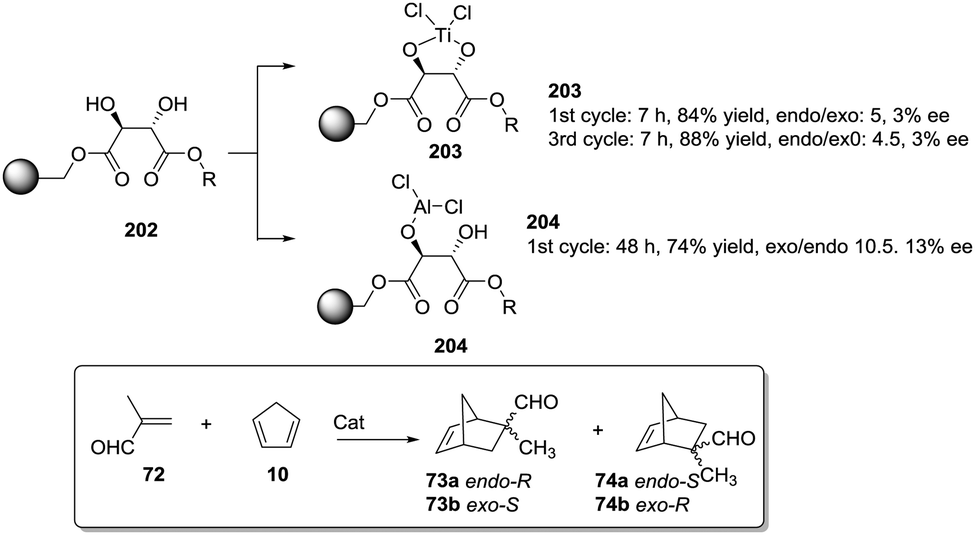 | ||
| Fig. 48 Polymer supported Ti and Al complexes of immobilized tartrate esters used as Lewis acid catalysts for the Diels–Alder reaction of methacrolein and cyclopentadiene. | ||
In the case of polymer-supported TADDOLates prepared by Seebach, the kinetic studies revealed, in general, a decrease in activity for successive runs.136,137,167–169 Only the functional polymer prepared from dendrimer 99 (Fig. 20) retained the original rates. It is interesting to note that this polymer was the only one to maintain the original swelling properties. This suggests that the deactivation mechanism can follow a similar pattern with decomposition of some catalytic sites accompanied by precipitation, inside the polymer, of inorganic particles, which decreases swelling and accessibility to the remaining catalytic centers.
The enantioselective epoxidation of olefins using chiral Mn(III)–salen catalysts is a useful synthetic transformation that suffers from deactivation through the formation of inactive μ-oxo-Mn(IV) dimeric species, and a variety of supports have been studied in order to minimize this deactivation process and facilitate recovery and reuse.280 Huang et al. have reported on the use of a variety of hybrid organic–inorganic supports for the immobilization of Mn–salen catalytic species and found that leaching and formation of μ-oxo-Mn(IV) species were the main deactivation mechanisms.281 Although the catalysts were reused for 9 cycles, some of the optimal systems revealed that the conversion decreased from 99% to 67% and the enantioselectivity from >99% to 62% ee, while the Mn content decreased from 0.73 to 0.45 mmol g−1.282
Non-covalent immobilisation and leaching
Of course, leaching and the associated deactivation are often found in chiral catalysts supported through non-covalent interactions.39,283 This is a common situation when inorganic supports are employed.284 However, the commercially available Rh-(S,S)-EthylDuphos has been anchored efficiently via ionic interactions on phospho tungstic acid dispersed on the surface of aluminum oxide and the stability could be improved by combining a continuous flow process with an appropriate reactor design, allowing for the preparation of an active pharmaceutical ingredient in kilogram scale with excellent enantioselectivity.285 The reactor set-up used suggests that the activation of release and catch strategies can be essential in this regard to limit the leaching and maintain the activity. Developing release and catch strategies is also important to implement the stability in systems based on MNPs (in particular PdNPs) supported on functional polymeric surfaces.270,286 When cellulose was used as a support for RhNPs and the system applied for the enantioselective 1,4-addition of arylboronic acids to enones and enoates in the presence of chiral diene ligands, no Rh leaching was detected in the reaction mixture.287 In the case of a Cu(II) exchanged zeolite modified by direct adsorption of cinchonidine and applied to the asymmetric Henry reaction, the authors studied in detail the deactivation mechanism and observed that after the reaction, the cinchonidine fragments were no longer attached to copper but hydrogen bonded to surface silanols.288 The immobilization of the Noyori catalyst Ru-(R-BINAP) by encapsulation in silica nanocages has been used for the enantioselective hydrogenation of β-ketoesters showing a very good stability, with around 1% Ru leaching after the first cycle, but only ca. 0.3% leaching for the second run.289 The reaction conditions can be very important. Chiral Rh phosphine–phosphite catalysts have been immobilized on sulfonated polystyrene resins and studied for the enantioselective hydrogenation of olefins.290 When the reaction was carried out in MeOH, a significant Rh leaching was detected (9–18%) for each run, while the leaching was much lower when the solvent was changed to water (<1%). Leaching is also a problem associated with chiral organocatalysts based on amines or polyamines immobilized on acidic resins through protonation and electrostatic interactions and some strategies have been devised for its minimization.291,292Detachment-transformation of the ligand
The importance of the nature and chemical stability of the chiral ligand for long-term stability was clearly illustrated by Hodge and co-workers.293 When the supported gel-type camphor derivative 205 (Fig. 49) was used as the catalyst in the reaction of aldehydes with Et2Zn, the first catalytic cycle afforded a chemical yield >95% with >94% ee. However, after being used for ca. 275 h the chemical yields and ee values dropped to 50–60% and to 81–84%, respectively (a decrease of ca. 2.5% in chemical yield and ca. 1.0% in ee per run).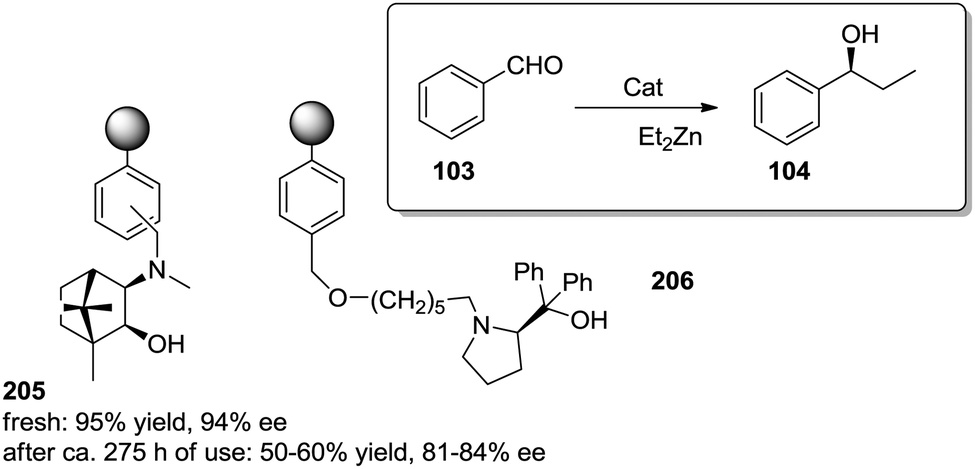 | ||
| Fig. 49 Examples of polymer supported amino alcohols studied for the ethylation of aldehydes with Et2Zn. | ||
It is worth mentioning that the study was carried out in a continuous flow system in order to avoid physical abrasion of the beads through their extensive use and for easy recycling. Data suggested that the secondary β-amino alcohol would be gradually transformed into a β-amino ketone losing its catalytic properties. In the light of this, the same authors designed a more robust and chemically inert supported chiral moiety (206) based on prolinol.294 The corresponding polymers could be successfully used in batch at least nine times without any significant loss of activity or enantioselectivity, showing the importance of choosing the right arrangement of linker, ligand, matrix and reaction conditions in order to achieve a good long-term stability. In the case of the iridium catalysts 2–3 presented in Fig. 1, the authors reported that a good stability was only achieved when the ligand was attached to the resin through an amide bond, while the related system containing an ester bond was much less stable, which was attributed to the more robust amide linkage.78 This is also illustrated in the case of L-proline derivatives supported as the corresponding zirconium phosphates (207) or phosphonates (208) (Fig. 50).295 While the phosphate link was shown to be relatively unstable, the phosphonate bond was stable allowing the recovery of the catalyst by centrifugation (>92%) and its reuse for 6 successive runs.
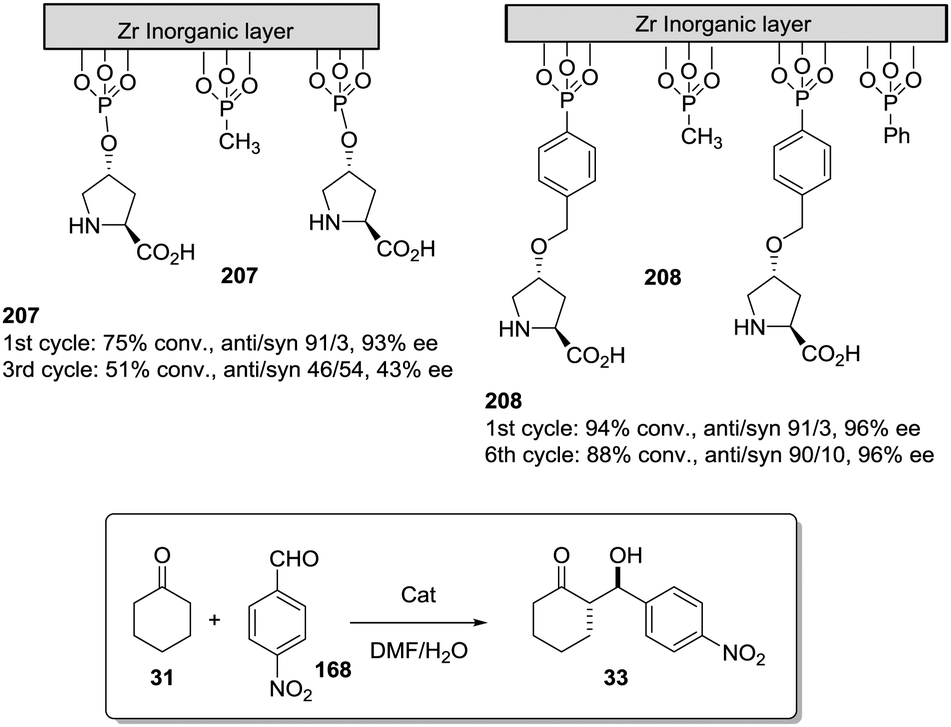 | ||
| Fig. 50 Proline supported on zirconium through phosphate or phosphonate bonds and studied as an organocatalyst for aldol reactions. | ||
Another factor to be considered is the limited mechanical stability of lightly crosslinked polymers, which may affect negatively their long-term use in particular under mechanical stress (i.e. magnetic stirring). The polymer 209 (Fig. 51) containing a fluorinated organocatalytic fragment has been prepared by suspension polymerization and assayed for the enantioselective Michael addition of propanal and (E)-β-nitrostyrene.296
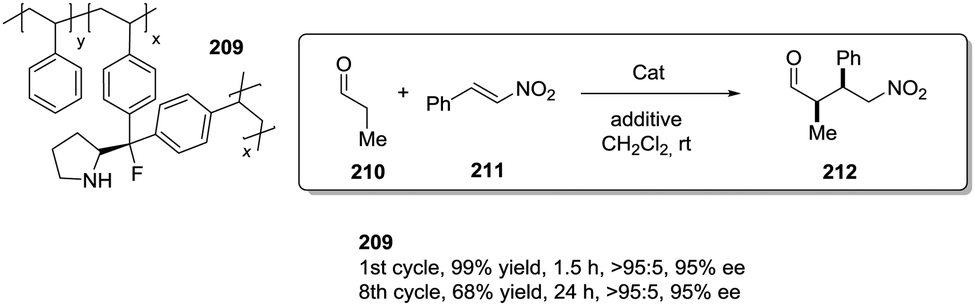 | ||
| Fig. 51 Polymer supported fluorinated organocatalyst derived from proline and studied for the enantioselective Michael addition. | ||
Recycling experiments showed that a full conversion maintaining a high enantioselectivity (>95% ee) could be obtained for 7–8 runs, but at the expense of increasing the reaction time from 1.5 to 24 h. This loss of activity was partly attributed to the mechanical degradation of the polymer. The use of continuous flow systems, as will be discussed below, has been reported to improve the stability of supported systems by reducing this mechanical degradation,263,293 and this was also exploited in the case of the supported organocatalyst 209, allowing its continuous use for 13 h with a single substrate to obtain multigram amounts of the desired product or to sequentially use this flow setup with 13 different substrates for a total of 18.5 h of use.
Mechanical stability can also be achieved by the use of more rigid polymeric matrices. On the other hand, the higher mobility of the functional groups in resins with a low degree of crosslinking can also affect negatively the stability of the resulting species, in particular when the catalytic centers can be deactivated, or lead to diminished selectivity, through site–site interactions.36,297,298
The use of macroporous polymers with a high and adjustable crosslinking degree and the desired porosity would limit those deactivation processes and increase the turnover and the lifetime of the polymer.54,55,181,206,299 As a matter of fact, monolithic macroporous materials can be some of the most suitable materials for the preparation of flow systems due to their porosity properties.170,171,300,301 The morphology of these macroporous materials, when appropriately tailored, avoids diffusional problems and allows them to act as stable and easily recoverable microreactors. However, the properties of the corresponding polymeric support need to be properly optimized for each specific application.
In a recent report on the use of polymer-supported diarylprolinols as organocatalysts for the continuous flow cyclopropanation reaction, it has been shown that the best performance was obtained with catalysts 213 based on a microporous resin and 214 of monolithic nature (Fig. 52).302 Under flow conditions the activity of the monolith remained constant for 24 h and then became rapidly deactivated, which was assigned to the collapse of the macroporous structure. The catalyst based on the microporous resin could be used for the same transformation for 48 h without a significant reduction in the performance. Besides, this catalyst could be used then for several other transformations up to a total of 76 h working in flow.
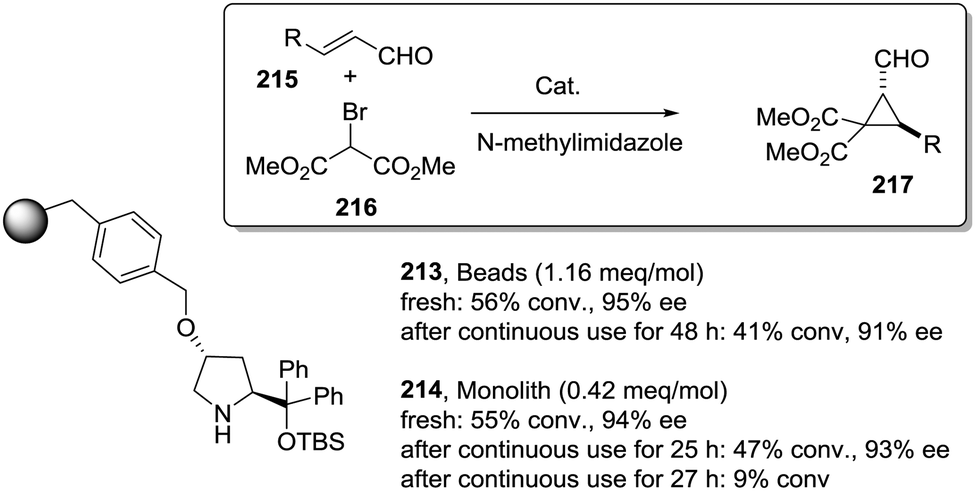 | ||
| Fig. 52 General structure of supported diarylprolinol derivatives studied as organocatalysts for the cyclopropanation reaction. | ||
Recycling protocol
The protocol for recycling is also an essential factor to be considered. This usually involves, for an insoluble polymer, filtering and washing. All these steps need to be carried out under conditions that do not affect the reactive species. The use of monolithic polymeric sticks has been shown to facilitate these recycling protocols. For a variety of polymer supported BINOL phosphates acting as chiral Bronsted acids in asymmetric organocatalytic transfer hydrogenation more than 10 runs could be applied without appreciable loss in activity and enantioselectivity.206 Even small losses of material in the work-up or due to the mechanical breakdown of the resin preclude a long-term use of the supported species. We have to bear in mind that just a loss of about 10% of the material used for each recycle would reduce our catalyst to about 30% of the initial amount after 10 cycles. For insoluble resins, minimisation of losses is better achieved with an experimental design in which the solvent is filtered out of the resin, keeping this in the reaction vessel. Thus, Takeda et al. have used a commercial parallel synthesizer for easy catalyst separation and recycling and were able to carry out the rhodium(II)-catalysed enantioselective intramolecular C–H insertion of a diazo-β-ketoester for 100 cycles with the observation of the same yield (88%) and enantioselectivity (92% ee) as in the first run and without the need to increase the reaction time (20 min for all cycles).303Continuous flow
Clearly the work under flow conditions represents an optimal setup achieving long-term stability as it minimizes the operations that could lead to deactivation of the catalytic sites. This has been highlighted by the recent achievements in the field of flow chemistry, including flow chemistry processes for the preparation of enantiopure compounds through chiral catalysis.315–318 A pioneering and clear exemplification of the advantages of the long-term stability of the supported species for flow processes was reported by Lectka and coworkers in the continuous asymmetric synthesis of different β-lactams (223) (Fig. 53).319 They assembled several columns packed with suitable supported reagents, catalysts and scavengers (220–222) required for the different synthetic and purification steps. The system could be reused sixty times with no significant loss in selectivity or yield (90% ee and 62% yield for the 60th run, in comparison with 99% ee and 65% yield for the first cycle) after following the proper protocols of washing and regeneration.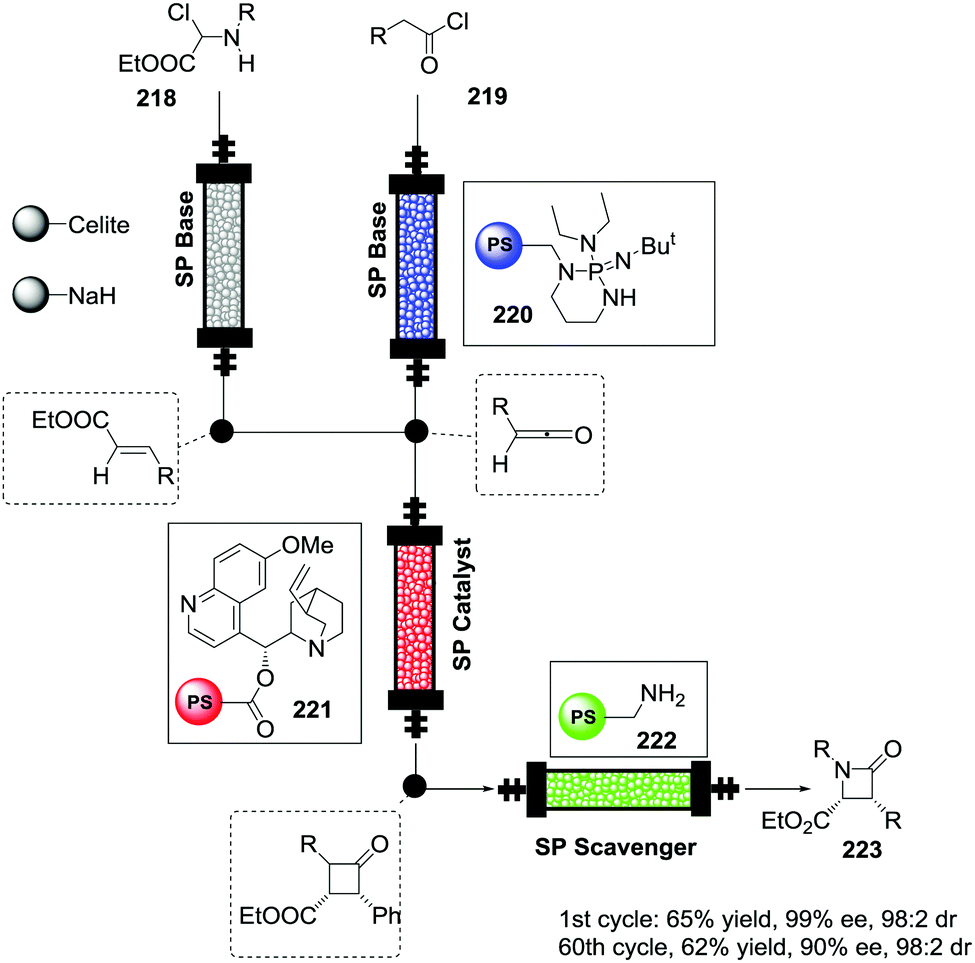 | ||
| Fig. 53 Multistep flow process for the preparation of β-lactams. | ||
This concept has been proved by the design of a continuous flow system based on monolithic catalytic columns containing a bicyclic chiral amino alcohol fragment (224) and its application to the addition reaction of Et2Zn to aldehydes (Fig. 54).320 The monolithic column 224 allowed the design of an efficient flow-system with an excellent activity and selectivity. Besides, this catalytic system could be reused several times with no loss of its catalytic efficiency, in terms of conversion, chemoselectivity and enantioselectivity.
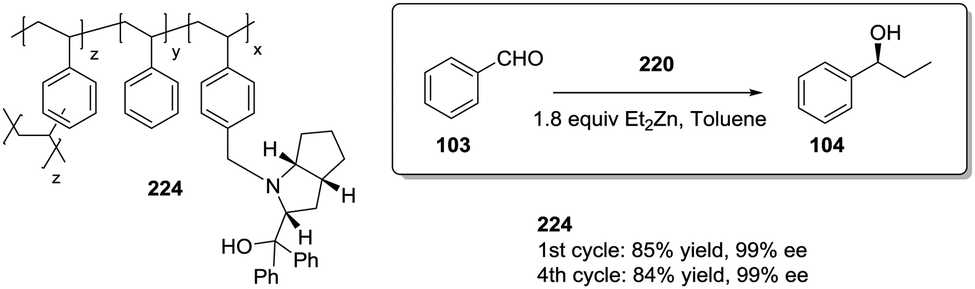 | ||
| Fig. 54 General structure of the monolithic polymer containing chiral bicyclic amino alcohol units studied for the ethylation of aldehydes. | ||
In the case of polymer-supported Ti-TADDOLates prepared as monolithic columns (225), an extraordinary long-term stability has been observed.132,321 Ti species derived from resins such as 225 were assayed as catalysts for the Diels–Alder reaction between cyclopentadiene and N-acryloyl-3-oxazolidinone. In general those supported Ti-TADDOLates maintained their integral performance for several months without the need of any special care except those mentioned for the work-up protocols. In the case of the derivative containing 3,5-dimethylphenyl groups at the α-position the activity remained even when, after the sixth month of use, it was left open in air for an additional month. In this case, however, some loss in selectivity was found (Fig. 55). Similar results were obtained for other monolithic columns containing polymer-supported Ti TADDOLates related to 225 but with different substitution patterns on the aromatic rings; the corresponding catalytic systems were active, in some cases, for more than one year (Fig. 55).322
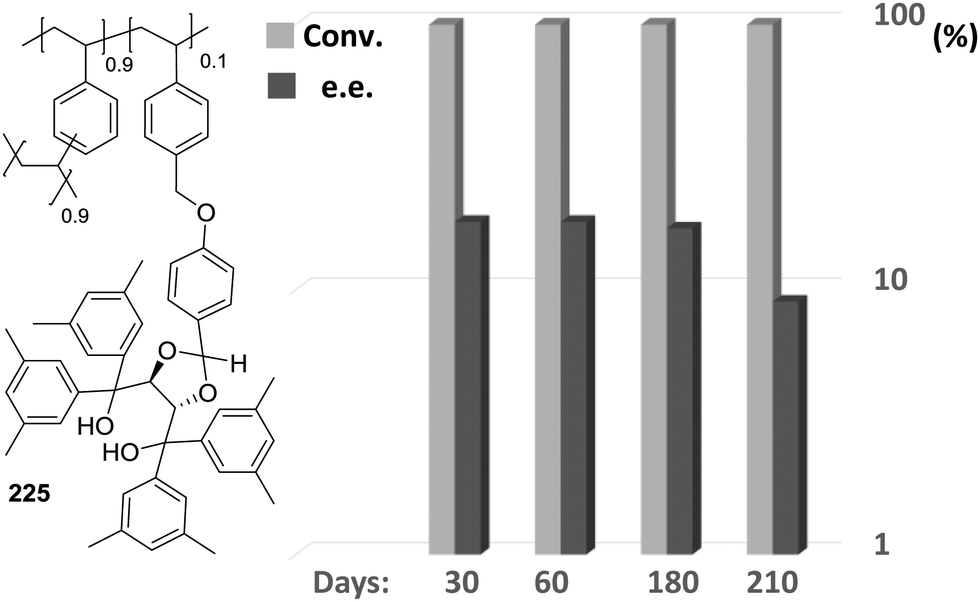 | ||
| Fig. 55 General structure of monolithic TADDOLs assayed under flow conditions for long-term stability. | ||
After screening a series of resin-bound di- and tri-peptides directly prepared on the resin by solid phase peptide synthesis (SPPS) techniques, Ötvös et al. found that a simple supported dipeptide containing an N-terminal proline and acid side chain at the C-terminus provided the best results as an organocatalyst for the asymmetric α-amination of aldehydes with dibenzyl azodicarboxylate (DBAD).323 This supported peptide was used to prepare a packed-bed flow system that was operating for 20 h of continuous use with constant yields in the range of 84–91% and enantioselectivities in the 88–92% ee range to provide the product in gram scale. Stable flow processes with more than 24 h on stream are frequent currently.111 A strategic control over the residence time was demonstrated to be a key parameter. As has been mentioned above, flow setups have allowed the preparation of the desired final products in multigram296 or even kilogram scale.285 Takeda et al. have shown that a polymer-supported dirhodium(II) complex could be applied under flow conditions for the enantioselective intermolecular cycloaddition of a diazodiketoester with styrene retaining the activity (78% yield) and enantioselectivity (99% ee) for 60 h on stream with only a small Rh leaching detected in the solution (2.1 ppm).324 Some of the processes studied could be easily upscaled to produce 22 g of the desired product with a turnover number of 11700. In the case of the continuous-flow asymmetric hydrogenation of an enol ester, the use of an Rh catalyst based on a phosphine–phosphoramidite ligand immobilized on a supported ionic liquid and the use of supercritical carbon dioxide as the mobile phase allowed maintenance of the performance of the process during 233 h on stream, achieving a final turnover number of 70400.325
The use of a monolithic reactor obtained by copolymerization of divinylbenzene and a vinylic imidazolidinone derivative (226, Fig. 56) inside a stainless steel column allowed carrying out the Diels Alder cycloaddition of cyclopentadiene and three different α,β-unsaturated aldehydes for around 150 h on stream achieving excellent reproducibility of the results with the same aldehyde at the beginning and at the end of this period.300 Finally, a similar catalytic column was used sequentially for three different reactions continuously for a total of more than 300 h on stream. Using two coupled catalytic columns packed with a mixture of polystyrene-supported Pybox, CaCl2·2H2O, and Celite, the asymmetric 1,4-addition of 1,3-dicarbonyl compounds to nitroalkenes was carried out in flow for more than 200 h, without loss of activity (95% yield for the last period) and enantioselectivity (91% ee).326 The work under continuous flow was demonstrated, besides, to solve the issue of product inhibition detected in these processes.
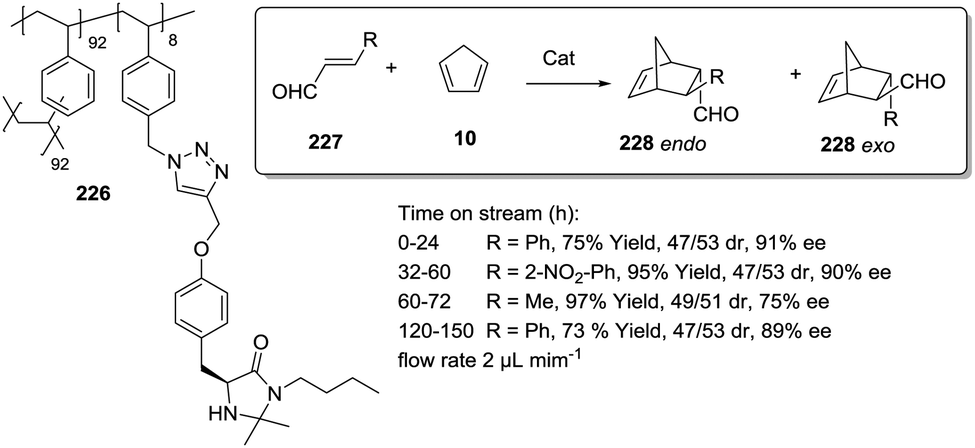 | ||
| Fig. 56 Chemical structure of the organocatalytic monolith used for the Diels–Alder reactions of cyclopentadiene under flow conditions. | ||
Although biotransformations have not been considered in detail in this review, it is worth mentioning that stability issues (i.e. thermal and long term stability) can represent one of the bottlenecks in their practical use for some applications and this has also been approached through the immobilization of the corresponding biocatalysts on an appropriate support. Thus, in the same way that ionic liquids have been demonstrated to be able to maintain the stability of proteins, including enzymes, and are an excellent medium for biocatalysis, supported ionic liquids have also been shown to contribute to the stability of immobilized catalytic enzymes including the development of efficient flow processes.327–329 Thus, the use of polystyrene monoliths modified with imidazolium groups (supported ionic liquid-like phases)330 for the immobilization of CALB provided distinct advantages over other supports for esterification processes.331 Optimum performance and stability was obtained with the most hydrophobic monolith that provided an excellent turnover number (35.8 × 104) working at 80 °C and 10 MP (supercritical CO2). This allowed developing the dynamic kinetic resolution of rac-1-phenylethanol by flow processes using scCO2 and an additional acid zeolite-ionic liquid catalyst, achieving excellent results in terms of yield (92%) and enantioselectivity (>99.9% ee).332 The combination of CALB immobilized on covalently supported ionic liquids with microwave irradiation led to a 28 times activity improvement and operational stability towards reuse for 12 cycles.333
Regeneration and reuse
When regeneration of supported species is involved, the former considerations for the work-up protocols are even more important. In this case, for supported organometallic catalysts, those protocols involve one or several additional steps to ensure the complete removal of metallic species from the polymers. This is not always easy and can require treatment under relatively strong conditions (acids, bases, etc.), followed by a complete washing and then treatment with the corresponding metallic species to regenerate the active sites. Although the protocols for reusing a given immobilized catalyst sometimes include adding some extra amounts of the catalytic metal in the reaction mixture, the regeneration process has been studied only in a limited number of cases.Thus, for instance, even for the case of the supported TADDOLs, which contain a very reactive ketal functionality, Seebach has shown that a complete regeneration of the activity of the Ti catalysts for the addition reaction of dialkyl zinc to aldehydes can be achieved after washing the used functional polymer 101 (Fig. 20) with 3 M HCl followed by thorough washing and reloading with Ti(OPri)2Cl2.137 When Ti catalysts are used for the addition of dialkylzincs to benzaldehyde one equivalent of titanium is consumed at the end of the reaction, and, accordingly, it is essential to reload the resin with TiX4 species after each run. In this regard, when the polymeric Ti-TADDOLate catalyst derived from dendrimer 99 was used for the addition of Et2Zn to PhCHO, a constant performance with respect to enantioselectivity as well as the reaction rate was found during 20 consecutive runs. However, all polymers prepared with non-dendritic TADDOL crosslinkers showed a continuous decrease in reaction kinetics and enantioselectivities, reflecting again how the long-term stability of supported catalysts and reagents is greatly influenced by the linkage used between the chiral ligand and the polymeric matrix.
In the case of Ti-TADDOLates related to 225 used as catalysts for the Diels–Alder reaction between cyclopentadiene and N-acryloyl-3-oxazolidinone, initial attempts for regeneration were discouraging.322 Regeneration of the catalyst afforded species with a reasonable activity but with a lower selectivity. Additionally, loss of performance in successive runs was much sharper than for the original systems. However, the regenerated monolith catalysed the addition of Et2Zn to benzaldehyde with the same performance as a fresh column. Any attempt to rationalise those discrepancies requires an analysis of the differences between the two reactions under study. An important feature of the Et2Zn addition is that it is a ligand-accelerated reaction. This feature is absent in the Diels–Alder reaction. Thus, the presence of metallic particles/species physically entrapped on the polymeric matrix, in a non-chiral microenvironment, could be partially responsible for the activity in the catalysis of the Diels–Alder reaction, giving rise to a decrease in the selectivity. Those particles could also catalyse the polymerisation of cyclopentadiene, a factor that has been considered to contribute to deactivate the polymeric catalysts by decreasing the porosity of the resin via physical entrapping or copolymerisation with residual vinylic groups. In highly crosslinked PS–DVB a significant percentage of vinylic groups can remain unreacted according to FT-Raman and NMR studies.334–336 In the case of the Et2Zn addition only the recovered chiral active sites catalyse the process. Polymer supported α-amino amides assayed also for the enantioselective addition of Et2Zn to aldehydes presented some deactivation after some cycles, but the activity and selectivity could be fully recovered after an appropriate washing protocol that included a washing with 1 M HCl.146
In the case of the silica-supported BINOL derivative (201) considered in Fig. 47, the regeneration of the Ti-catalyst used for 10 successive runs for the ethylation of 1-naphthaldehyde with Et3B was studied revealing that the regenerated catalyst achieved the same performance for four additional cycles.274 Lunden et al. reported that the polymer-supported pyridine-bis(oxazoline) ligand developed for the ytterbium-catalyzed silylcyanation of benzaldehyde could be recovered from the reaction mixture and regenerated for at least 30 times.264 In their study of silica-supported organocatalysts for stereoselective Diels–Alder reactions under flow conditions, Porta et al. analysed the regeneration of the organocatalytic column after 200 h of use and found that in this way the life of the reaction was prolonged to 300 h in total, but the performance of this regenerated catalytic column was slightly lower than that of the fresh system.337
Selectivity
One of the main aims of the use of a catalyst is, besides the lowering of activation energies, to increase the selectivity of the process under consideration. From a theoretical and practical point of view, enantioselectivity, and in general stereoselectivity, is the most important goal to be achieved. The very small energy differences at the transition states that determine the stereochemical course of the reaction, in particular for enantiotopic transition states, make this an always difficult target. Minor changes in the reaction conditions/environment can interfere as to drastically modify those differences and, accordingly, alter the selectivity of a given reaction. This explains the key role that the polymer matrix can play in determining the selectivity in polymer-supported catalysts and reagents. The polymeric framework is a major factor in determining the actual microenvironment of the active site.It is often considered, as a general rule, that the selectivity observed for a given chiral catalyst tends to be reduced when heterogenisation is carried out. This is not necessarily true, however, and very different trends can be found. Most of the targeted homogeneous catalysts to be immobilised are selected from those that perform, after long and costly optimisation, with excellent (enantio)selectivities (>95% ee). Hence, upon immobilisation, in the best scenario the levels of selectivity will be maintained, obscuring such cases in which the matrix induces a positive effect and highlighting, on the contrary, those in which a negative effect is found. Therefore, a careful examination of the different aspects of the immobilisation process should be taken into consideration to shed light on the effect of the support regarding selectivity.
In order to really assess the effect of the matrix, the first point to be considered is to ensure that proper comparisons are being made. Quite often, the structural changes made in the chiral ligand in order to provide the required features for the immobilisation are responsible for the changes observed and not the immobilisation itself. Therefore, as mentioned, it is essential to compare results obtained for homogeneous and supported ligands having the same structure.
This is clearly illustrated in the case of the immobilised Cu–BOX used as catalyst for the cyclopropanation reaction of styrene in the presence of ethylazodicarboxylate (Fig. 57). As outlined before, an efficient bisoxazoline immobilization (polymer 109) can be carried out either by homo-polymerisation of the derivative 103 containing two vinylbenzyl groups at the methylene bridge (see Fig. 21) or by copolymerisation in the presence of styrene and DVB.172–175 When the initial Cu loaded catalyst (109d) was used for the cyclopropanation reaction, a trans/cis selectivity of 60:40 and an enantioselectivity of 46% ee (trans) and 42% ee (cis) were obtained (Fig. 57). These results were far away from the ones obtained for the homogeneous derivative 230 and suggested a negative effect of the immobilisation. This is not true, however, as the decrease in the selectivity is not associated with the heterogenisation process itself, but with the change in the nature of the substitution at the methylene bridge. Thus, when the appropriate homogeneous bisoxazolines, containing two benzyl groups at the methylene bridge, 230 and 231 were synthesised, the results obtained under the same conditions for the polymeric species prepared by polymerisation (109b–f and 228) were essentially identical or even slightly better than those found for the homogeneous counterpart.
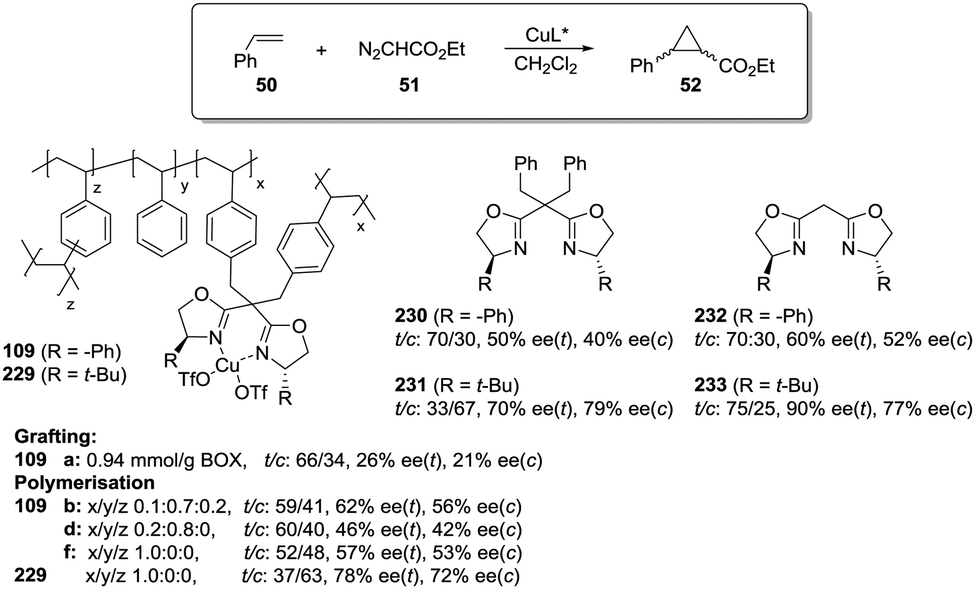 | ||
| Fig. 57 Comparison of selectivity for different polymer supported Cu–BOX catalysts with that of related soluble analogues for the cyclopropanation of styrene. | ||
It is worth mentioning here that the enantioselectivity observed for the related catalyst prepared by grafting onto a Merrifield resin (109a) was much lower (t/c: 71/29; ee 26% (trans), 21% (cis)). This represents a situation that is not uncommon: very often similar polymer-bound chiral ligands can be prepared both by grafting onto a preformed resin or by polymerisation of the corresponding functional monomer containing the chiral fragment. In many instances, selectivities found for catalysts prepared using the second strategy are higher, in particular when high degrees of crosslinking are involved. It is important to mention that the supported catalyst is prepared by polymerization of the corresponding di-vinylic chiral monomer (105, Fig. 21) which provides additional crosslinking. During the polymerisation the chiral moieties are almost randomly distributed throughout the polymeric matrix and the polymer itself may become chiral, to some extent, creating many different chiral microenvironments. The exact microenvironment distribution depends on a complex series of factors, such as solvents, polarity of monomers, kinetics of polymerization of each monomer, etc.80,82 Chiral moieties will be incorporated as cross linkers but can also be integrated in the linear chain (with unreacted alkene groups left). This can lead to catalytic sites with different selectivity profiles that can largely affect the enantioselectivity depending on their distribution and accessibility. These effects can be enhanced in flow processes, as illustrated by the results with the monolith 109b when used under continuous flow conditions.338 At low flow rates (2 mL min−1) the immobilised catalyst led to better enantioselectivities than the homogeneous catalyst under batch conditions (ee: 71% (trans), 50% (cis) vs. 50% (trans), 40% (cis)). The flow forces the reactants to pass-through the active sites, minimising diffusional problems, and also allows an increase in the instant S/C ratio. The former results suggest that immobilisation of the catalyst can produce positive effects on selectivity after the appropriate design of the whole set of experimental conditions. Similar effects have been also observed, for the same reaction, with the chiral Ru–PYBOX monolith 119d (Fig. 23).339 The immobilised catalyst provided, under flow conditions, better enantioselectivities (trans/cis: 85:15, 82% ee (trans), 45% ee (cis) in CH2Cl2 or trans/cis: 85:15, 89% ee (trans), 56% ee (cis) in scCO2) than those found for the homogeneous catalyst in batch (trans/cis: 87:13, 77% ee (trans), 48% ee (cis) in CH2Cl2).
It can be expected that these effects could be minimised when attaching the catalytic sites to the polymer through a long spacer. This strategy was very successful for supporting Cu–BOX catalysts 114 and 115 from BOX ligands, prepared by grafting or polymerisation respectively, lacking the C2 symmetry and tethered to the polymeric network through a long aliphatic linker (Fig. 22).182,183 In this case, no polymeric effect was found, with both heterogeneous catalysts showing comparable selectivities (114: trans/cis: 67/33, 92% ee (trans), 90% ee (cis) and 115: trans/cis: 67/33, 93% ee (trans), 90% ee (cis)) to those achieved for the homogeneous counterpart.
On the contrary the positive role played by the proximity of the polymeric backbone to the catalytic site was highlighted for the case of supported pyridineoxazoline (PYOX) (Fig. 58) for which a remarkable enhancement in enantioselectivity was reported.340 In contrast with the almost no-enantioselection obtained with both homogeneous catalysts 234 and 236 in solution, the analogous supported catalysts led to a significant asymmetric induction, 46% ee (cis) and 40% ee (trans) for 235 and 65% ee (cis) and 56% ee (trans) for 237. The steric hindrance provided by the rigid highly crosslinked polymeric backbone around the position 6 of pyridine can cooperate with the bulky substituent at the oxazoline ring to enhance the differences in energy for the different possible transition states, enhancing the chirality transfer and accordingly the enantioselectivity. To check the existence of this positive “polymeric effect”, a catalyst 238 with a phenoxy spacer was also synthesized. The introduction of the spacer led to a reduction of the enantioselectivity relative to 237, supporting the positive role played by the polymeric network in the sevenfold improvement in enantioselectivity observed for the supported Cu–PYOX catalyst 237.
 | ||
| Fig. 58 General structures of the polymer supported and homogeneous Cu–PYOX catalysts studied for the cyclopropanation of styrene. | ||
Similar effects have been described for the immobilized Cu–PYOX catalyst 239 (Fig. 59) prepared from the linear soluble poly[(−)-(S)-4-tert-butyl-2-(3-vinylpyridin-2-yl)-oxazoline] in the Diels–Alder reaction between 2-alkenoyl pyridine N-oxide (241) and cyclopentadiene (10).341 The polymeric catalyst (239) showed an enhancement of both activity (fivefold increase) and enantioselectivity (2.5 times increase) in comparison with the unsupported homogeneous analogue (240). It was considered that the high population of Cu(II)–PYOX groups around the polymer backbone brings about a high local concentration of the catalytic sites providing a constrained environment for the Diels–Alder reaction, which favours the stereoselectivity.
 | ||
| Fig. 59 Homogeneous and supported Cu–PYOX complexes as catalysts for the Diels–Alder reaction of 2-alkenoyl pyridine N-oxide and cyclopentadiene. | ||
In the light of these results, the proximity of the catalytic site to the polymeric framework and their relative spatial disposition, which can be tuned by the right selection of the tethering point and the use or not of spacer, seem to be key parameters in the design of polymer-supported catalysts with a positive polymeric effect. This can define, among other parameters, site-isolation and accessibility, but also the steric hindrance in the proximity of the catalytic center, which are essential to fine-tune the catalyst performance. Thus, for instance, in the case of BINOL Yang et al. reported a positive polymeric effect when BINOL was anchored to the polymer at 3,3′-position of the BINOL subunit. The polymeric body was designed as a bulky substituent to improve the steric control of the reaction. The C2-symmetric supported catalyst 244 provided much higher enantioselectivities than its homogeneous counterpart (243) for the diethylzinc addition to benzaldehyde (see Fig. 60).342 Noteworthily, the BINOL supported derivative (246) lacking the C2 symmetry showed only a slightly better performance than the homogeneous analogue (245). Both the higher hindrance and rigidity of the supported BINOL 244 may be at the origin of this positive effect. To some extent, the polymer backbone can freeze the proper conformation of the naphthyl rings making their twisting more difficult. This effect would be more important in the case of the 3,3′-C2-BINOL supported derivative (244) attached to two polymeric chains, greatly restricting the conformational flexibility of the two naphthyl rings, than in the case of the supported ligand 246 with only one polymeric arm.
 | ||
| Fig. 60 Supported and homogeneous 3,3′-substituted BINOL derivatives for the Ti-catalysed Et2Zn addition to aldehydes. | ||
In an alternative approach, Sellner et al. (Fig. 61) selected the attachment via the 6,6′-positions using long spacers (247) and dendrimeric spacers (248–249). The corresponding supported BINOL derivatives were prepared by copolymerisation of different 6,6′-branched C2-BINOL crosslinking monomers with styrene.343 The distance from the crosslinking points to the catalytic centre was large enough in resins 247–249 as to provide a good accessibility to the active sites and as to afford an appreciable degree of flexibility, and no polymer effect, neither positive nor negative, was detected. Selectivities achieved were comparable to those obtained in solution, even considering that the catalytic active sites were placed in the crosslinked region of the polymer.
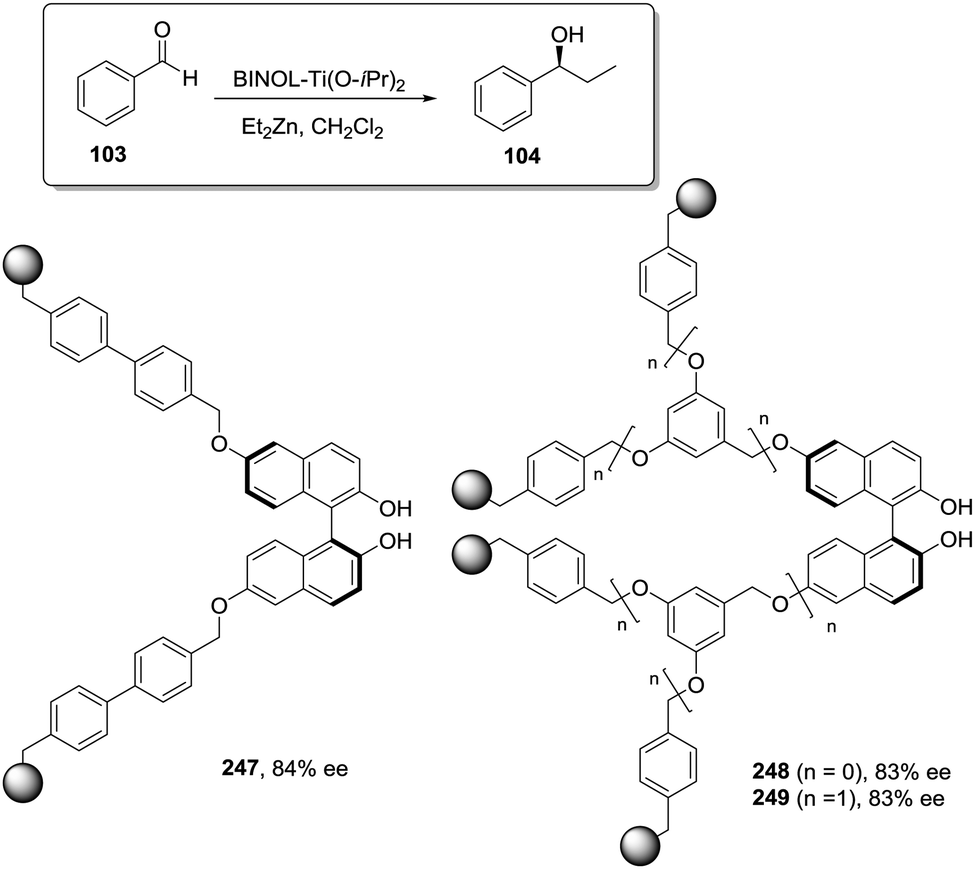 | ||
| Fig. 61 Supported 6,6′-substituted BINOL derivatives incorporating long spacers for the Ti-catalysed Et2Zn addition to aldehydes. | ||
However, when a similar approach was investigated using much more rigid 3-3′ and 6-6′ C2-BINOL crosslinking agents to obtain supported BINOLs 250 and 251, a negative matrix effect was found (Fig. 62).344 The enantioselectivities observed were in all cases lower than in solution, being strongly affected by the substitution pattern of the supported BINOL. In this sense, the polymers containing 6,6′-disubstituted BINOLs showed higher selectivities than those with the more hindered 3,3′-disubstituted-BINOLs (78% ee for 251vs. 20% for 250).
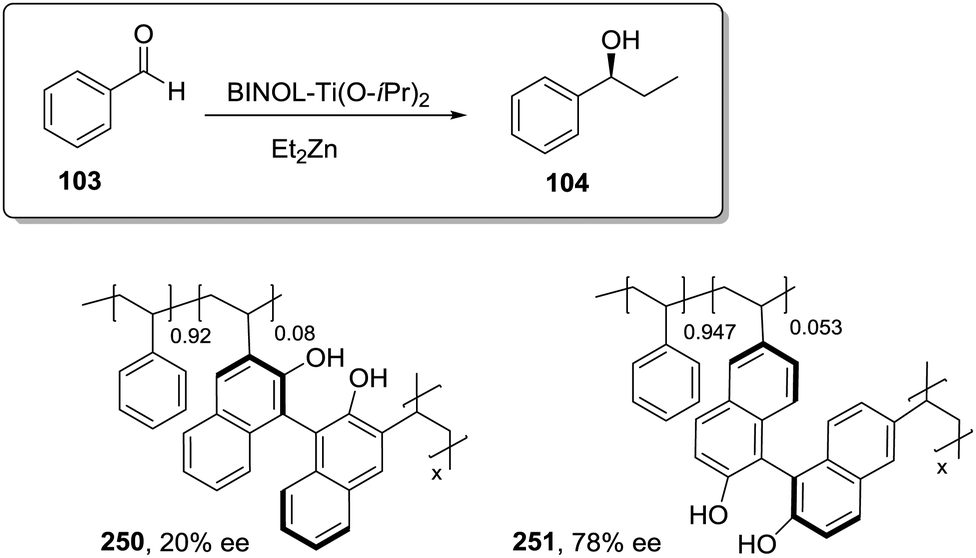 | ||
| Fig. 62 Supported 3,3′ and 6,6′-substituted BINOL derivatives fusing very short spacers for the Ti-catalysed Et2Zn addition to aldehydes. | ||
The very rigid framework achieved in these polymers and the steric interference of the polymeric matrix, particularly in 250, can prevent the diol moiety from achieving a proper conformation and limit the accessibility to the active sites. This is in good agreement with the work of Hodge and coworkers using supported aminoalcohols, who demonstrated that in those cases where the aldehyde and Et2Zn cannot diffuse freely into the polymer to reach the catalytic sites, a non-catalysed reaction is possible giving a racemate and so reducing the stereochemical performance.293,294,345
A related effect was described, for instance, by Kamahori et al. in their study of boron species, prepared from polymer supported chiral N-sulphonyl amino acids, for the catalysis of the Diels–Alder reaction between cyclopentadiene and methacrolein (Fig. 63).346 The catalyst 254a, prepared by grafting, from resin 252, was essentially unselective, whilst a 57% ee was found for 254b prepared by polymerisation of the N-vinylbenzsulfonyl amino acid 253. A possible explanation for this behaviour was suggested by the solvent effects observed in homogeneous solution. The selectivity observed in THF was much higher (86% ee) than that in CH2Cl2 (20% ee), suggesting that the non-donor solvent CH2Cl2 favours the presence of non-selective aggregates. Immobilisation through grafting does not destroy such a possibility, as functionalisation occurs in the more accessible sites of the resin in a very flexible matrix, allowing for site-to-site interactions. Those are much more difficult when the functional groups are introduced by polymerisation in a highly crosslinked matrix. According to this, the use of a CH2Cl2/THF mixture with the supported catalyst 254b had a minor effect on the selectivity. It was also observed that an increase in loading was accompanied by up to a 4 times decrease in enantioselectivity.
 | ||
| Fig. 63 Preparation of chiral boron supported catalysts for the Diels–Alder reaction between cyclopentadiene and methacrolein. | ||
Similar improvements in asymmetric induction for the immobilised catalyst were also found for the asymmetric transfer hydrogenation of aromatic ketones in water. The supported catalyst 44a (Fig. 7) provided higher enantioselectivities than the homogeneous analogue (water-soluble modified TsDPEN)347 for the Ru and Ir-catalysed reduction of aromatic ketones (e.g.: C6H5COCH3 98% vs. 94% ee, C6H5COCH2CH3 96 vs. 86% ee, p-Cl-C6H4COCH3 99% vs. 91% ee, NaptCOCH3 87% vs. 87% ee).119,348
The effect of loading is crucial for the performance of many supported reagents and catalysts. In the case of the enantioselective reduction of acetophenone with a LAH derivative of the resin-bound ephedrine, enantioselectivity values ranged from 55% for a resin containing 2.1 mmol g−1 to 79% for a loading of 0.7 mmol g−1.349 The same phenomenon has been reported by Seebach for supported Ti-TADDOLate catalysts, where better enantioselectivities were observed for the polymers with lower loadings.167 In other cases, however, high loadings have been found to be associated with better enantioselectivities. Thus, the enantioselectivities achieved for the asymmetric borane reduction of acetophenones (257) catalysed by polymer-supported chiral sulphonamides (255) showed a steady improvement when the functionalisation degree was slightly increased (63, 75 and 85% ee, respectively for loadings of 2.23 mmol g−1, 2.26 mmol g−1 and 2.29 mmol g−1). Additionally the enantioselectivity obtained in this case by the supported catalyst was higher than that by the homogeneous analogue 256 (Fig. 64).350,351
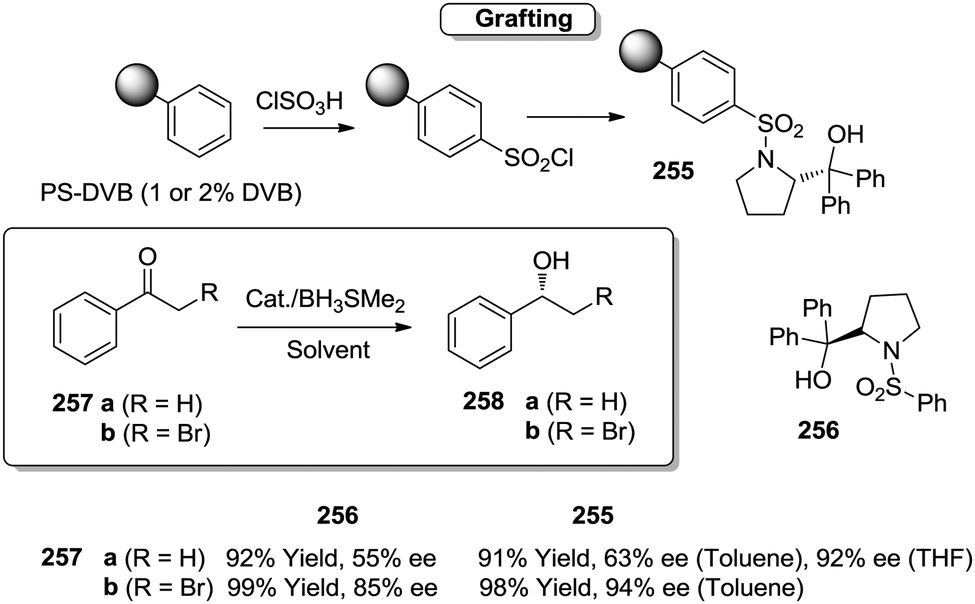 | ||
| Fig. 64 Polymer supported chiral sulfonamides for the enantioselective reduction of acetophenones. | ||
Itsuno et al. have reported another remarkable example highlighting a positive matrix effect on both reactivity and selectivity (Fig. 65).352 This enhancement may well be related to pseudodilution/isolation effects related to immobilization. Thus, after the adequate optimization of the polymeric catalyst, the supported oxazaborolidine derived from 259b showed a better performance in terms of activity and selectivity than the low molecular counterpart (260). When used at room temperature for the borane reduction of the acetophenone O-methoxy oxime (261), 259b led to 65% yield and 96% ee. These results were even better at 0 °C (70% yield and 99% ee). However, the process with the homogeneous 260 did not occur under the same experimental conditions. The temperature should be increased to 50 °C to achieve 61% yield and 84% ee. This may be related to the formation of inactive/nonselective aggregates in solution at room temperature. An increase in temperature may favour the breakdown of such aggregates liberating the active forms of the catalyst. In the case of the supported catalyst, the formation of aggregates will not be favoured, allowing the involvement of the active catalytic species even at lower temperatures.
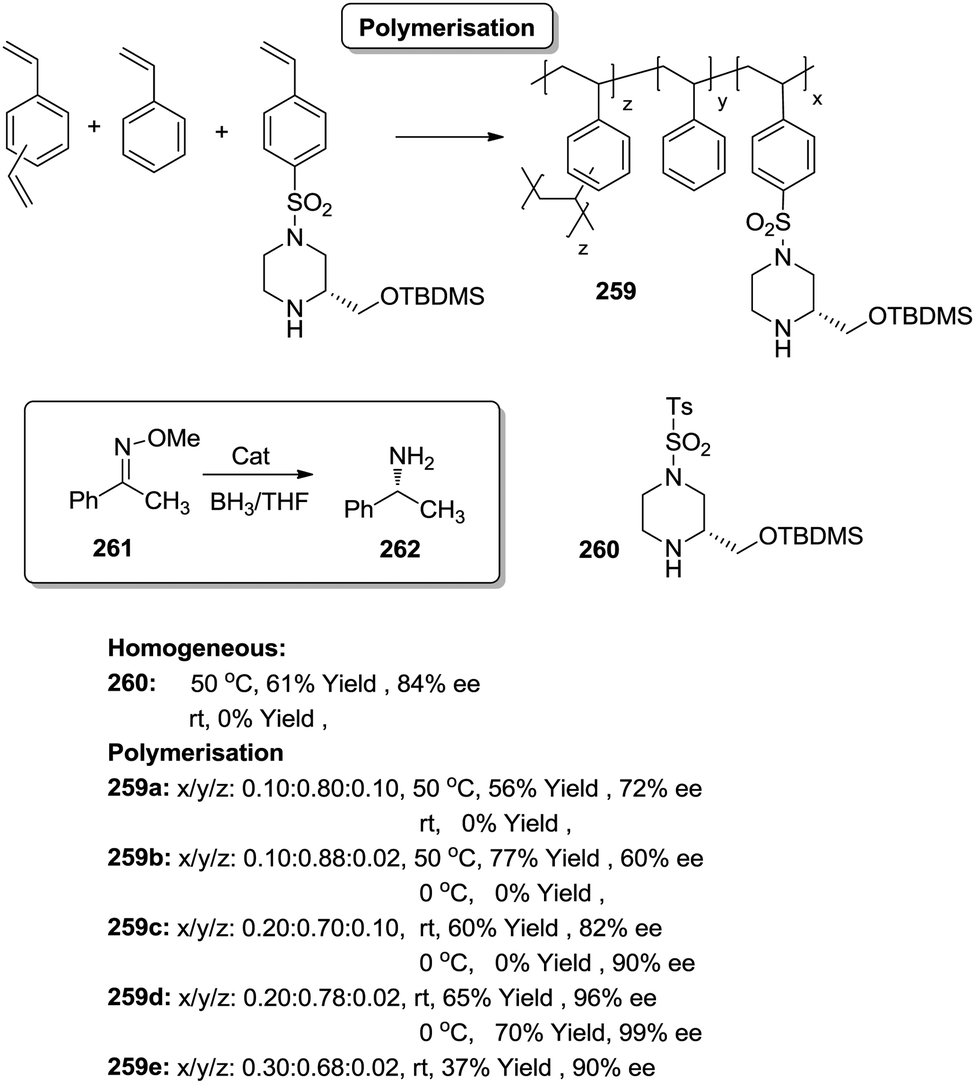 | ||
| Fig. 65 Polymer supported piperazine methanol ligands for the catalytic borane reduction of O-methoxy oximes. | ||
A similar effect can be accounted for in the case of the supported amino-alcohol 224 (Fig. 54) obtained as a polymeric monolith and used as a catalyst for the Et2Zn addition to benzaldehyde under flow conditions.320 The polymer prepared by polymerisation showed an improved selectivity (99% ee) in comparison with those found for the related homogeneous catalyst (87% ee) or the one immobilised onto a Merrifield resin by grafting protocols (89% ee).
Sundararajan et al. prepared a polymeric analogue of chiral C2-symmetric (R,R)-3-aza-3-benzyl-1,5-dihydroxy-1,5-diphenylpentane (266) by copolymerization of the corresponding vinylic derivative with styrene and DVB (Fig. 66).353 The corresponding supported Al complex (265) catalysed the Michael additions of nitromethane and the addition of thiophenols to unsaturated cycloalkenones. In the case of the Michael addition between nitromethane and chalcone 267, the supported catalyst 265a led to higher asymmetric induction (51% ee) than the related homogeneous analogue (266, 5% ee). However, for the polymer 265b, with lower crosslinking content and higher styrene incorporation, the enantioselectivity significantly dropped (11% ee). The crosslinking degree can define the rigidity of the ligand, modifying to some extent the steric restrictions around the catalytic site and leading to more efficient heterobimetallic catalysts inducing a better chiral transfer.
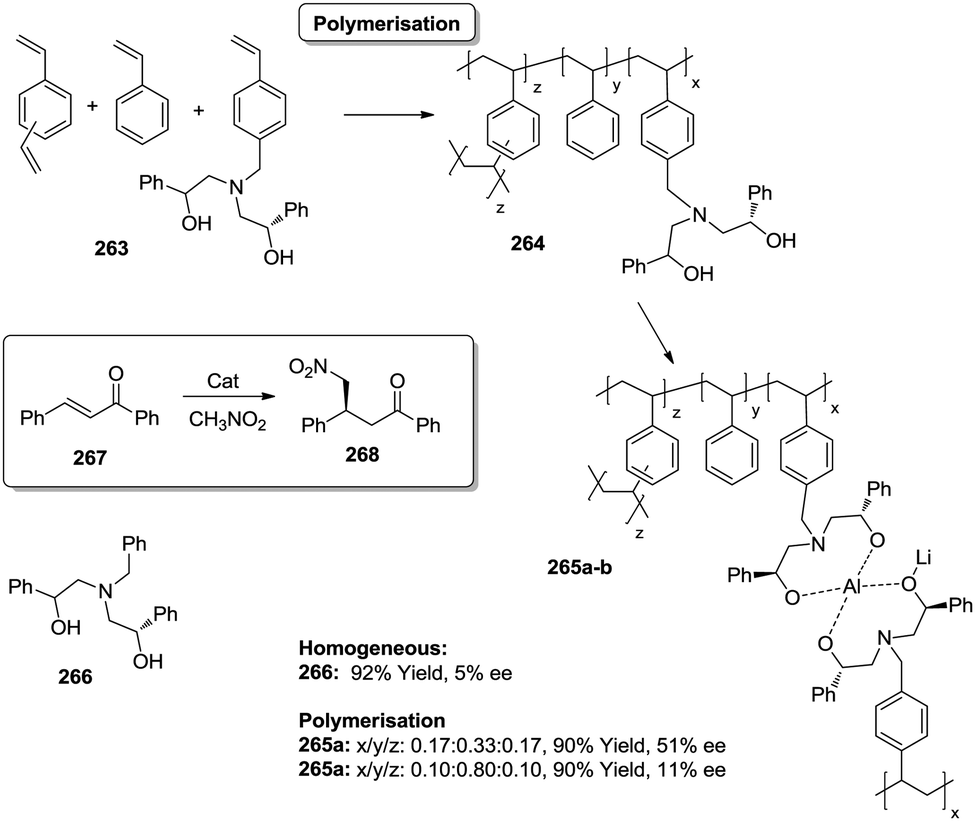 | ||
| Fig. 66 Polymer supported Al complexes for the asymmetric Michael addition. | ||
The formation of different active species as a consequence of different polymerisation procedures has been proved for the Rh-catalysed asymmetric hydroformylation of styrene involving two polymer supported (R,S)-BINAPHOS ligands (271, Fig. 67).354,355 When the polymerisation of 269 was performed in the absence of the metal, the ligand adopted a conformation that apparently was not suitable to coordinate the Rh. Thus the resulting polymeric catalyst 271a gave rise to a modest selectivity of 68% ee (method A). However, the conformation of the ligand could be fixed in a suitable disposition when the polymerisation was performed directly with the corresponding chiral Rh-monomeric complex (270) (method B). Indeed, a better selectivity was obtained under the same conditions (85% ee).
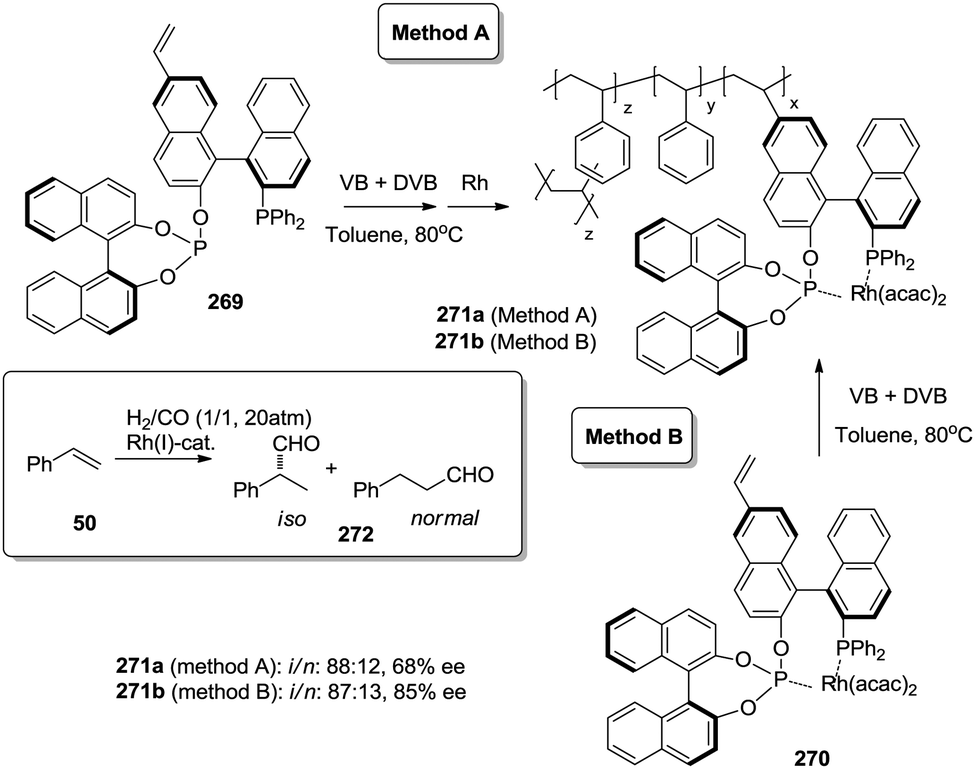 | ||
| Fig. 67 Polymer supported (R,S)-BINAPHOS prepared by two alternative polymerisation approaches. | ||
Another important factor to be considered is the crosslinking. When the immobilised catalysts are prepared by a bottom-up approach by direct polymerisation of the chiral functional monomers, different crosslinking agents can lead to different microenvironments, which, in turn, can modify, even enhance, the asymmetric induction of a given reaction. This was the case of the Diels–Alder reaction between methacrolein and cyclopentadiene catalysed by polymeric chiral oxazaborolidines obtained by polymerisation in the presence of different crosslinking agents.92 The selectivities found were significantly influenced by the crosslinkers (Fig. 68). The polymeric catalyst 275a derived from DVB catalysed the reaction with moderate selectivity (65% ee), while the polymeric catalyst prepared with the more flexible crosslinking agent 276 gave an enhanced enantioselectivity (275b, 84% ee). This was comparable to the selectivity obtained from using the unsupported catalyst in solution as reported by Helmchen (86% ee).356 Finally, the polymeric catalyst with a longer oxyethylene crosslinker gave enantioselectivities as high as 95% ee (275e), results that are superior to those obtained with the related homogeneous catalyst in solution.
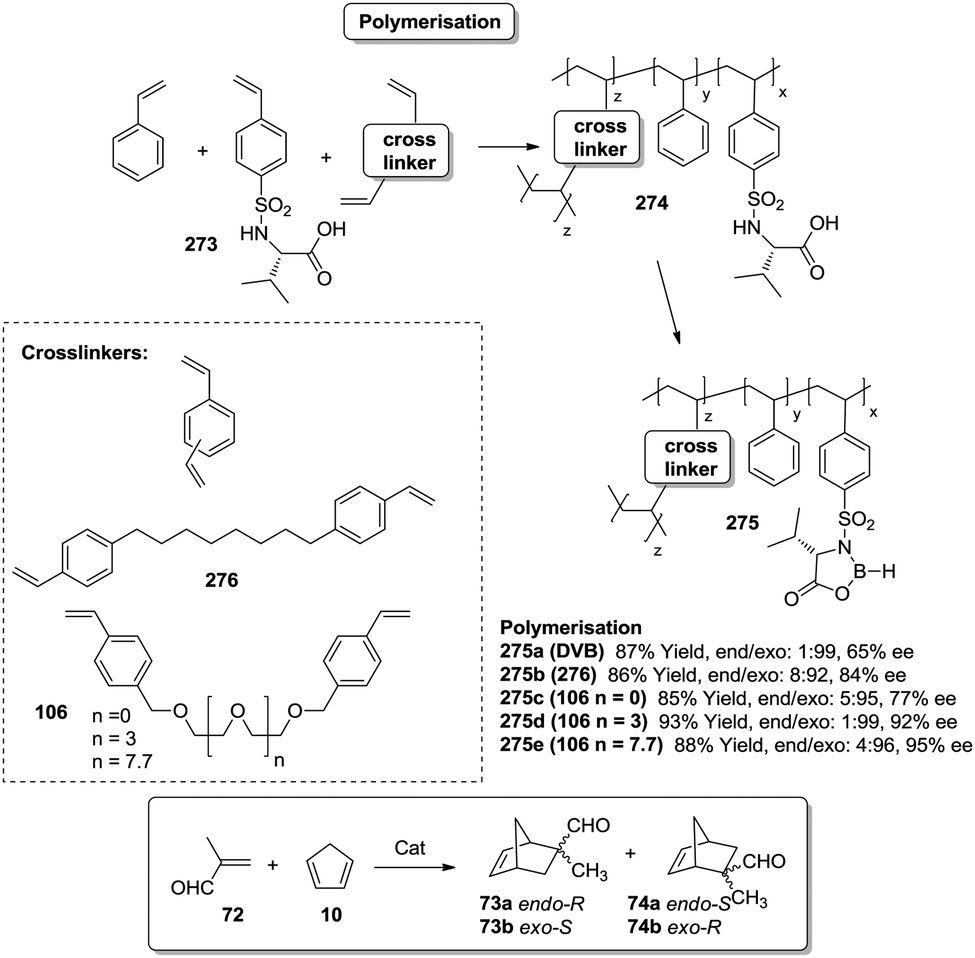 | ||
| Fig. 68 Preparation of supported oxazaborolidines by polymerization involving different crosslinkers. | ||
When supported Al catalysts derived from polymeric prolinols 65–66 (Fig. 10) were used for the same Diels–Alder reaction, selectivities found were low or moderate, being similar for the functional polymers prepared by grafting or by polymerisation (suspension or “bulk” polymerisation) using low crosslinking degrees (up to 20%), but a twofold increase in ee was found when the crosslinking (DVB) was increased to 90%.357
The polymer-supported Rh catalysts obtained through the copolymerization of (S)-5,5′-divinyl-BINAP (277) also exhibited better enantioselectivity than the corresponding homogeneous complex (Fig. 69).358 The BINAP immobilisation was performed by copolymerisation of 277 with different crosslinking agents (DVB, 1,3,5-tri(4-vinylphenyl)benzene (278) and EGDMA) and the corresponding Rh-complexes were assayed in the asymmetric hydroformylation of styrene. The results showed that the pore structures had little effect on the yields, but had a clear effect on the regioselectivity and enantioselectivity. Catalysts 279a and 279b exhibited higher enantioselectivity (59% and 45% ee, respectively) than the related homogeneous system (35% ee). However, 279c containing ethylene glycol dimethacrylate as crosslinker provided only a modest enantioselectivity of 30% ee. These results highlight once again that the nature of the crosslinkers can be used to modify/improve the selectivity.
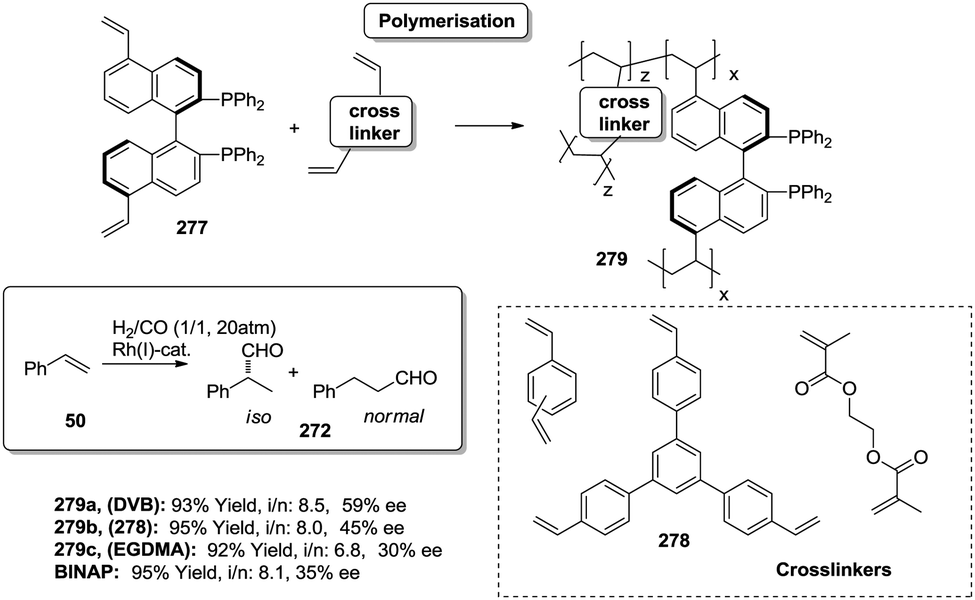 | ||
| Fig. 69 Polymerisation of a vinylic BINAP derivative in combination with different crosslinkers for the Rh-catalysed hydroformylation. | ||
A very particular case regarding the influence of polymerization on enantioselectivity is given by the preparation of polymers displaying intrinsic main chain chiral organization, in particular through the adoption of helical structures.29,30 In many cases, the reported asymmetric inductions are rather limited.247 In the last few years, however, different groups have reported examples in which the helical organization of the polymeric chain provides a significant contribution towards the enantioselectivity for a variety of catalytic reactions. In some instances it has been reported that the presence of helicity can increase the enantioselectivity from 18% ee for the non-helical support to 94% ee for the helical one.359 In some extreme cases, even attachment of achiral catalyst groups on the helical polymer matrix, whose helix sense is switchable, allows selective production of both enantiomers with enantioselectivities higher than 90% ee in several mechanistically different palladium-catalyzed and organocatalytic asymmetric reactions.360–362
Cinchona-alkaloid derived quaternary ammonium salts have been immobilized through ionic interactions to polymers possessing sulfonate groups and were used as supported organocatalysts in the asymmetric alkylation of N-diphenylmethylene glycine tert-butyl ester (283, Fig. 70).363 Their synthesis was approached either by polymerization of a chiral quaternary ammonium sulfonate monomer or by the immobilization of a chiral quaternary ammonium salt onto a sulfonated polymer through an ion exchange reaction. In this particular case, both approaches provided similar results. The enantioselectivity obtained by using the polymeric catalyst 281a (78% ee) was higher than that obtained from the unsupported salt 282a (68% ee). The 9-anthracenemethyl derivative of the cinchonidine-based quaternary ammonium salt gave better enantioselectivities both in solution (282b, 93% ee) and with the supported derivative (281b, 93%ee; 281c, 95% ee).
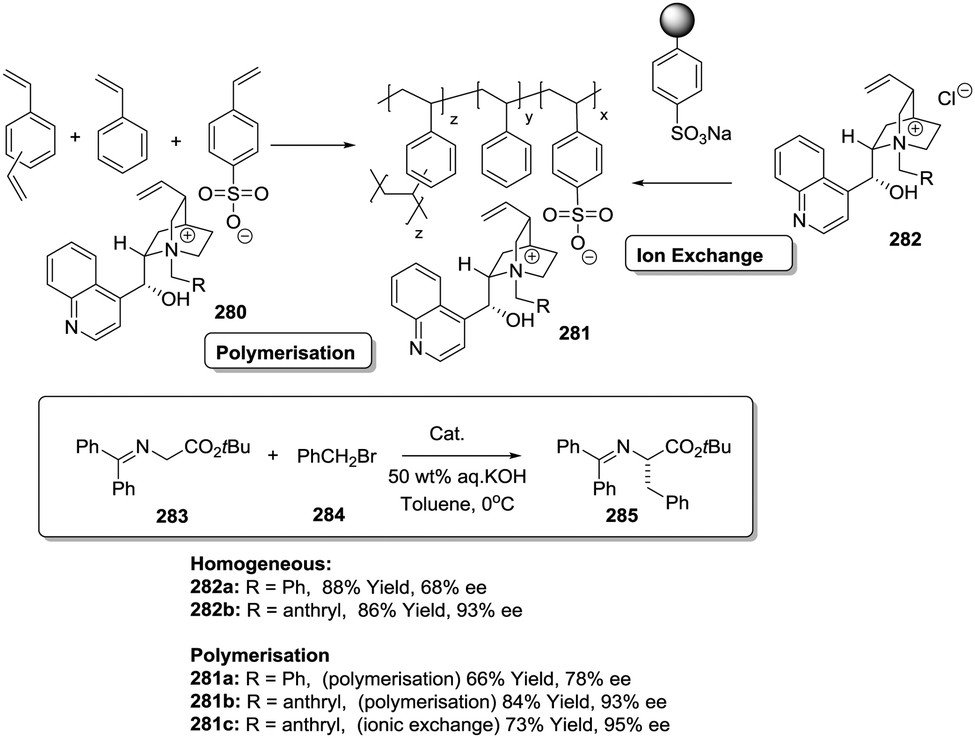 | ||
| Fig. 70 Preparation of polymer supported quaternary ammonium salts derived from cinchona alkaloids by polymerization and ion exchange as catalysts for asymmetric alkylations. | ||
Based on the immobilisation through ionic bonding and due to the exceptional stability of the quaternary ammonium sulfonate salts, the same authors have developed a very versatile strategy to immobilise different chiral salts in the main polymeric chain.364 This strategy is based on bringing together different modular building blocks (monomeric or dimeric chiral salts and sulfonate salts) to create a chiral ionic polymer allowing fine-tuning of the catalytic efficacy from a bottom-up approach. In this way by tuning the structure of these building blocks, the polymer main chain can be adjusted to positively affect the conformation of the catalytic sites enhancing the catalytic efficiency.365Fig. 71 schematically represents some of the strategies developed using cinchona-alkaloid-derived quaternary ammonium salts as chiral catalysts for the asymmetric benzylation of N-diphenylmethylene glycine tert-butyl ester. Main chain supported chiral salts immobilised by completely ionic interaction, combining both ionic and covalent bonds or just solely based on covalent immobilisation have been successfully obtained.366–370 A wide range of structural building blocks have been evaluated in order to optimise the supported catalyst. In most of the cases, it has been possible to adjust the polymer structure to obtain catalysts (286–289) which are often more selective than the homogeneous analogues.
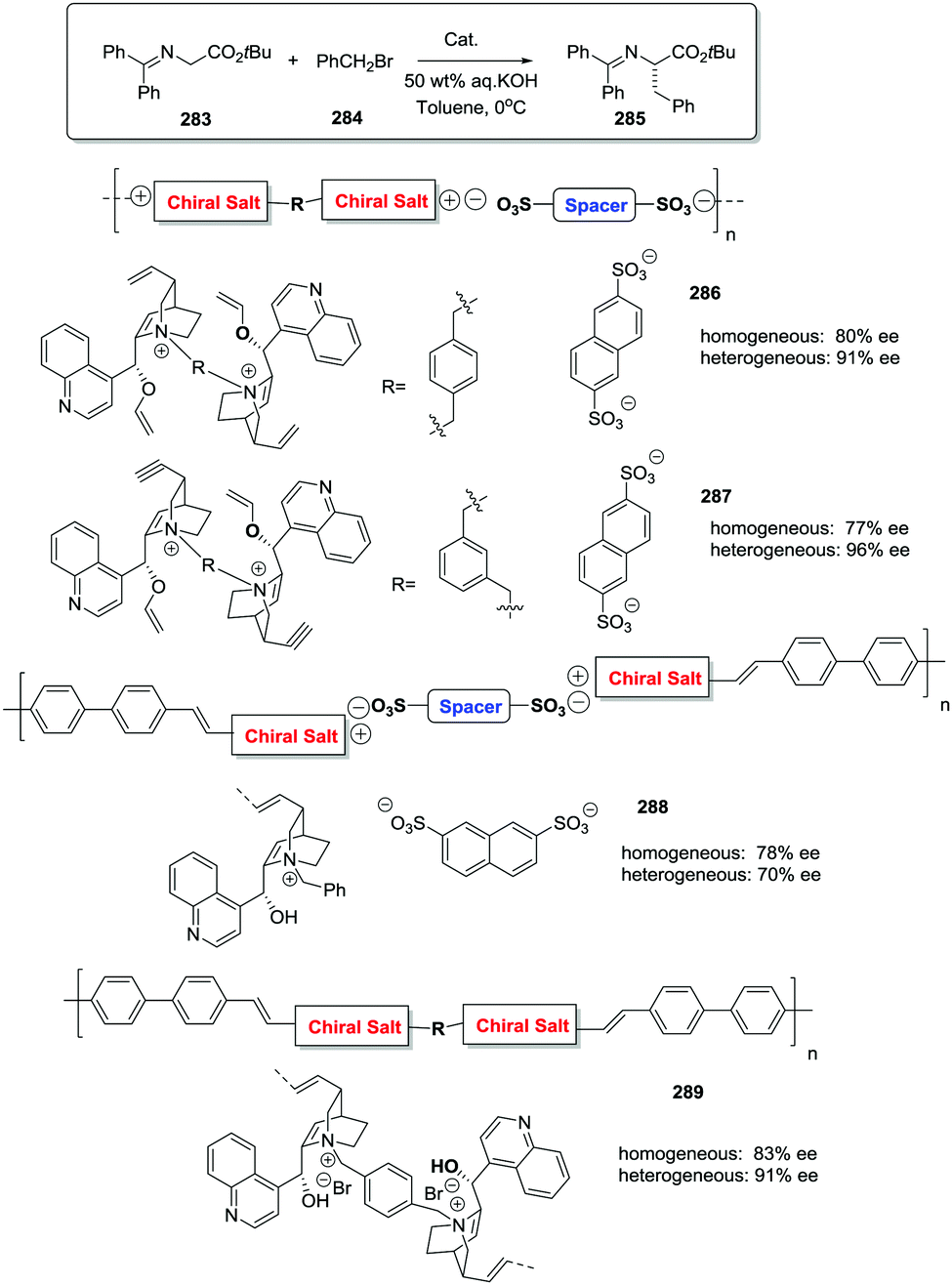 | ||
| Fig. 71 Preparation of polymer supported quaternary ammonium salts derived from cinchona alkaloids by polymerization and ion exchange as catalysts for asymmetric alkylations. | ||
The great modularity of the approach has also allowed extending this methodology to other types of organocatalysts opening avenues to a wide range of asymmetric transformations.371,372 For instance, main chain polymers 290–291 derived from chiral imidazolidinone have been reported as efficient organocatalysts for the Diels–Alder reaction (Fig. 72).373–375 By using the right combination of building blocks, the immobilized systems displayed an equivalent or even better selectivity than the homogeneous counterpart.
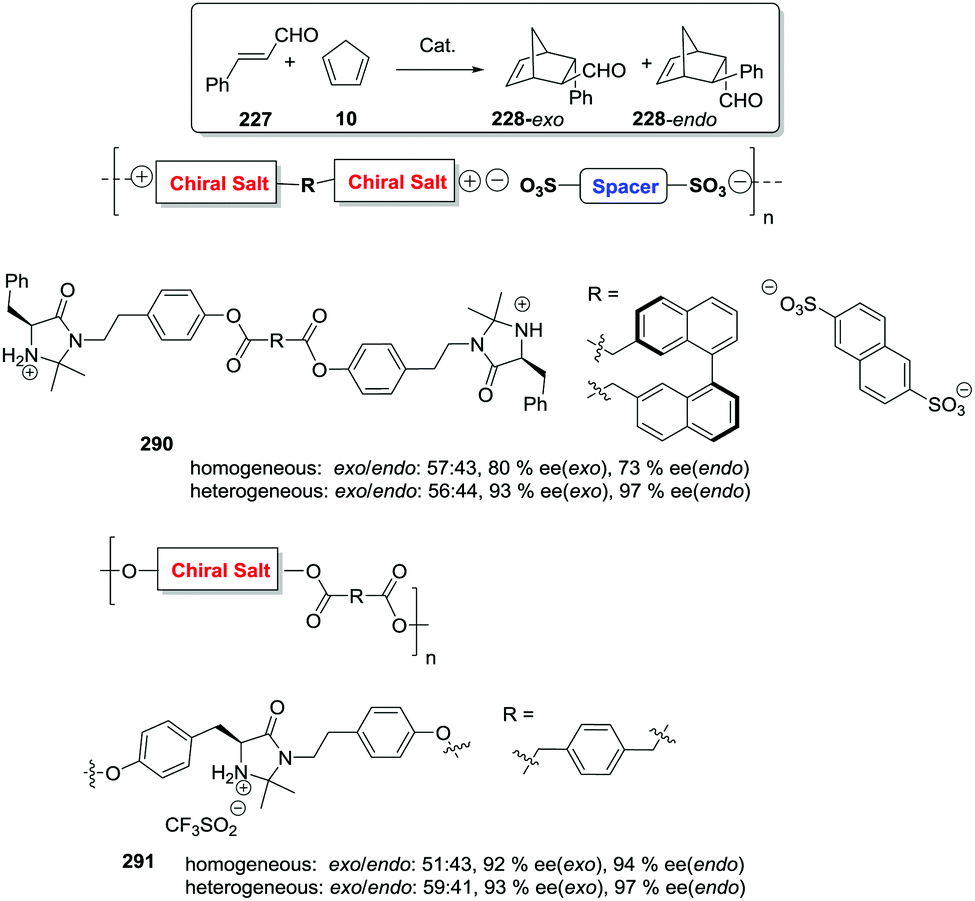 | ||
| Fig. 72 Preparation of polymer supported ammonium salts derived from MacMillan Imidazolidinone by polymerization and ion exchange as catalysts for Diels–Alder reaction. | ||
The ionic complexation has been also employed by Hu, Li and co-workers to obtain a nanoparticle-supported catalyst by in situ ionic complexation between an imidazolium-based polymer ionic liquid (PIL) and poly(L-prolinamide-co-MAA) (Fig. 73).376 The structure and hydrophobicity–hydrophilicity balance of the nanoparticles can be fine-tuned by varying the PIL reaction ratio and by applying different anions.
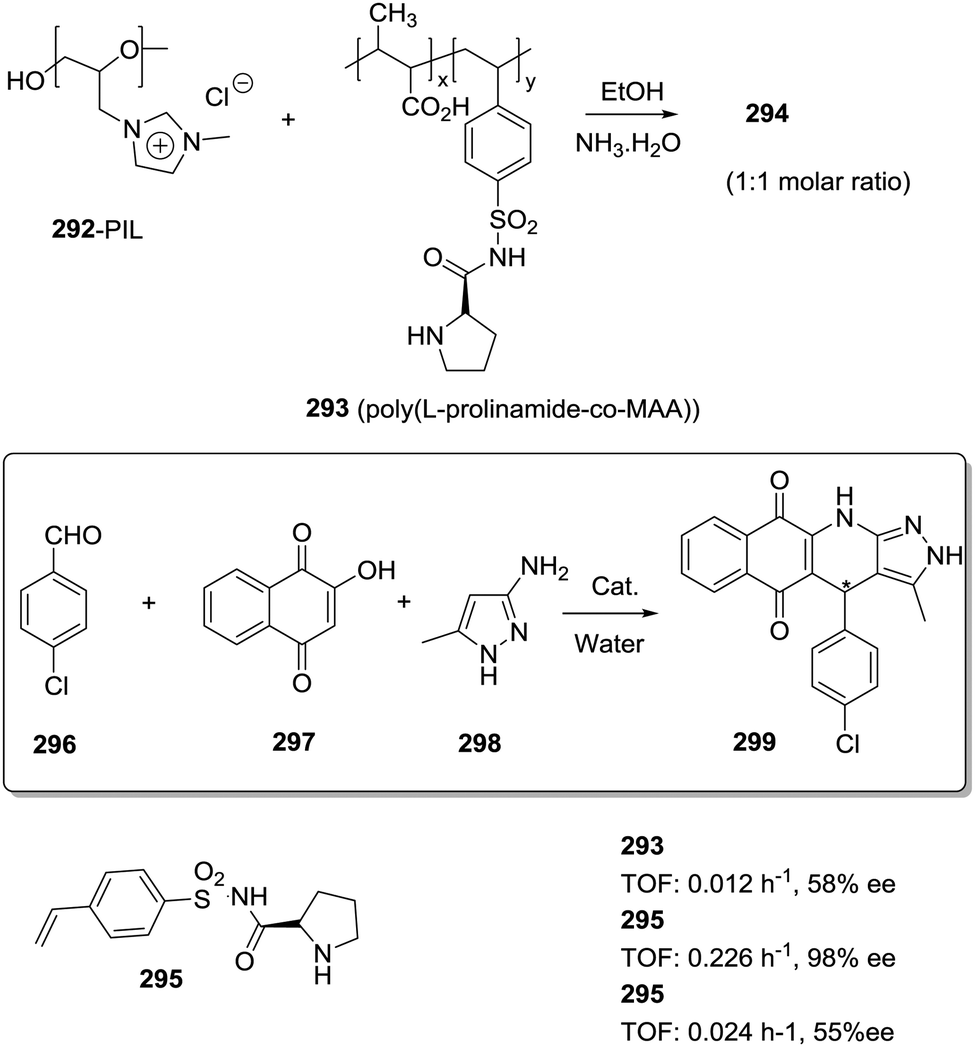 | ||
| Fig. 73 Preparation of a nanoparticle-supported catalyst by in situ ionic complexation between an imidazolium-based polymer ionic liquid (PIL) and poly(L-prolinamide-co-MAA) for the three component reaction. | ||
The chiral solid efficiently catalysed both Aldol and multicomponent reactions (MCRs) in water exhibiting much better catalytic activity and enantioselectivity than its homogeneous catalytic analogue. For instance, the poly(L-prolinamide-co-MAA) copolymer (293) and the corresponding monomer (295) gave modest activity and enantioselectivity for the three component reaction between 4-chlorobenzaldehyde (296), 2-hydroxy-1,4-naphthoquinone (297), and 3-amino-5-4-methylpyrazole (298) at 80 °C in water. However, the activity and enantioselectivity found for the nanoparticle-supported catalyst (294) clearly surpassed those values. The combination of both polymeric elements creates a polymeric nanoparticle with a PIL outer structure that facilitates the mass transfer of reaction substrates into the nanoparticle and creates the required micro-environment for the induction of chirality.
Similar selectivity enhancement can be also found when soluble polymers are carefully designed. For instance, the immobilization of [Mn(salen)] complexes on smart polymers can render catalytic systems which are not only more active but also more selective than the homogeneous counterpart (Fig. 74). The introduction of adequate building blocks in the polymer structure contributes to tune and enhance the asymmetric induction by favoring the polymer self-assembly in a micellar well-organized structure. Thus, the polymeric catalyst 302 led to quantitative conversion (>99%) of methyl phenyl sulfide (301) with up to 96% chemoselectivity and 95% enantioselectivity, whereas the neat complex was far less efficient (9% conversion with 72% chemoselectivity and 79% enantioselectivity).241 In a similar way, amphiphilic block copolymer 301 based on the N-isopropylacrylamide and vinylimidazolium ionic liquid modified chiral salen TiIV complex (poly(NIPAAm-co-PIL-[Ti(salen)])) efficiently catalyzed the enantioselective oxidation of aryl sulfide in water. The optimized supported catalyst showed, for the oxidation of methyl phenyl sulfide in water, better activity and selectivity (almost quantitative yield (95%), 95% of chemoselectivity and with up to 98% ee) than the related neat monomeric catalyst (48% of conversion, 65% of chemoselectivity and 76% of ee).377 In this case, the stable structure of the self-folded single-chain polymeric nanoparticles (SCPNs) and the presence of the ionic liquid moiety in the confined SCPNs provide a suitable micro-environment to favor the concentration of the reactants and to induce a better enantioselection.
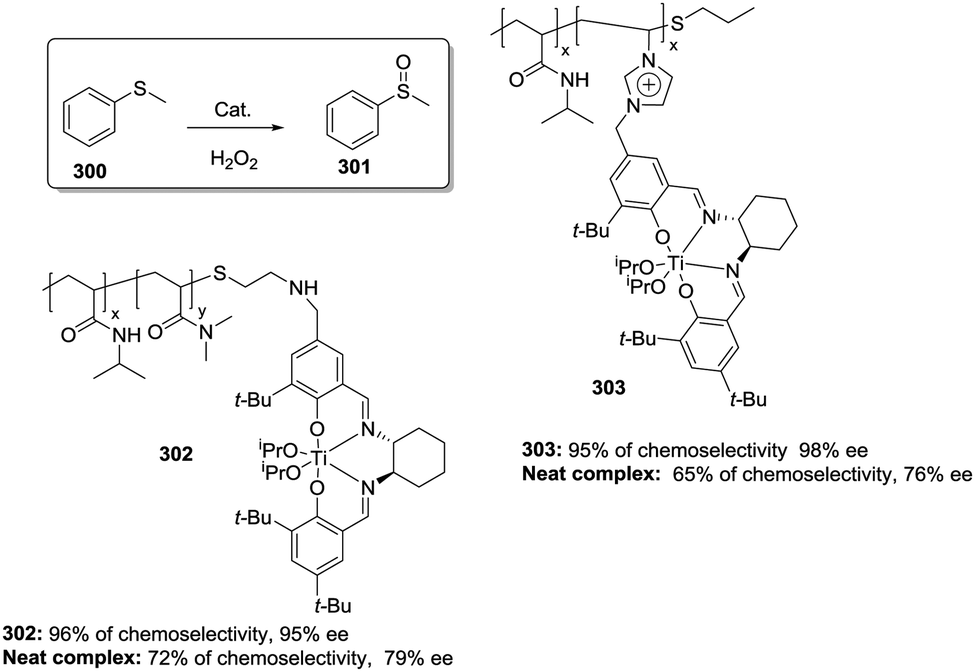 | ||
| Fig. 74 Soluble chiral Ti–Salen complexes as catalysts for highly enantioselective sulfoxidation in water. | ||
Dendrimers
In the same way that catalytic activity can be enhanced by the “positive dendrimer effect”, under certain circumstances an enhancement in the selectivity, especially the chiral induction, can be achieved upon catalyst immobilization on a dendritic support. In this regard, Ribourdouille et al. have demonstrated that Pd–PYRPHOS catalytic units immobilized onto polyamidoamine dendrimers (PAMAM) can surpass the selectivity found for the monomeric reference.378 The higher generation dendrimers decorated with a chiral Pd-complex (PAMAM(PYRPHOS-PdCl2)64, 304) rendered a better chiral induction (69% ee) in the allylic amination than the monomeric model (305, 9% ee). The improvement was related to the changes in the conformational flexibility and preferential orientation of the aryl substituents of the diphosphine by increased crowdedness at the periphery of the dendrimer upon increasing the dendrimer wedges (Fig. 75).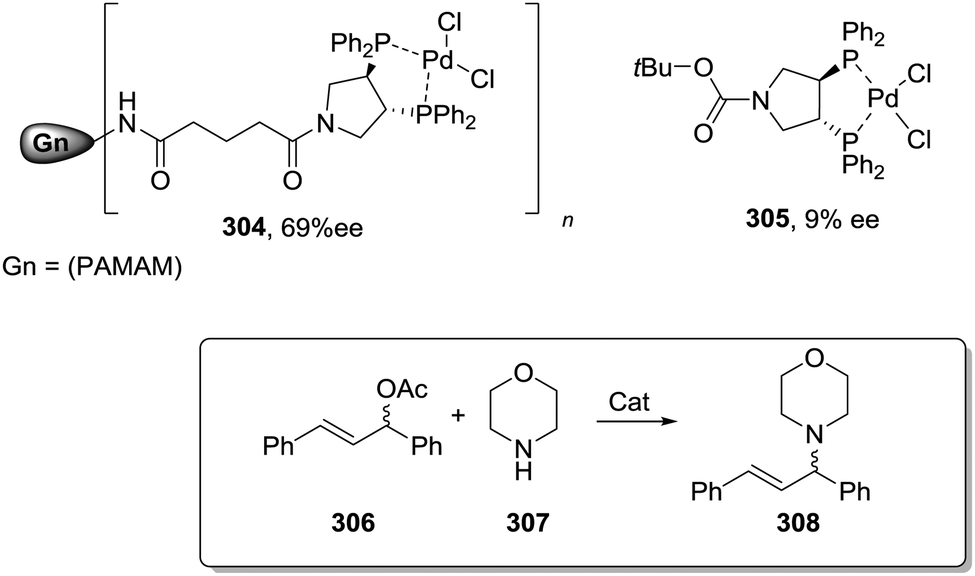 | ||
| Fig. 75 Dendrimer supported Pd–PYPHOS catalysts for the allylic amination displaying an enhanced chirality transfer than the unsupported catalyst. | ||
Similar positive effects have been reported by Zhang et al. for the asymmetric hydrogenation of α-dehydroamino acid esters and enamides catalyzed by Rh–chiral dendritic monodentate phosphoramidite complexes (Fig. 76).379 The corresponding chiral dendritic ligands (309) were obtained through substitution of the dimethylamino moiety in the original monodentate phosphoramidite by Fréchet-type polyaryl ether dendritic wedges. In this case, the catalytic unit is located at the core and not at the periphery. As an illustrative example, the enantioselectivity for the hydrogenation of N-(1-(p-tolyl)vinyl)acetamide (310) increased from 47% ee for the monomeric catalyst to 92% ee for the third dendrimer generation. However, a negative dendrimer effect on the catalytic activity was observed. This suggests an effective encapsulation of the catalytic unit within the dendritic wedges. The increasing steric repulsion between dendritic wedges might influence the chiral pocket of the catalyst where the hydrogenation reaction occurs enhancing the selectivity in comparison with the monomeric catalyst, but at the expense of reducing the rate of the reaction.
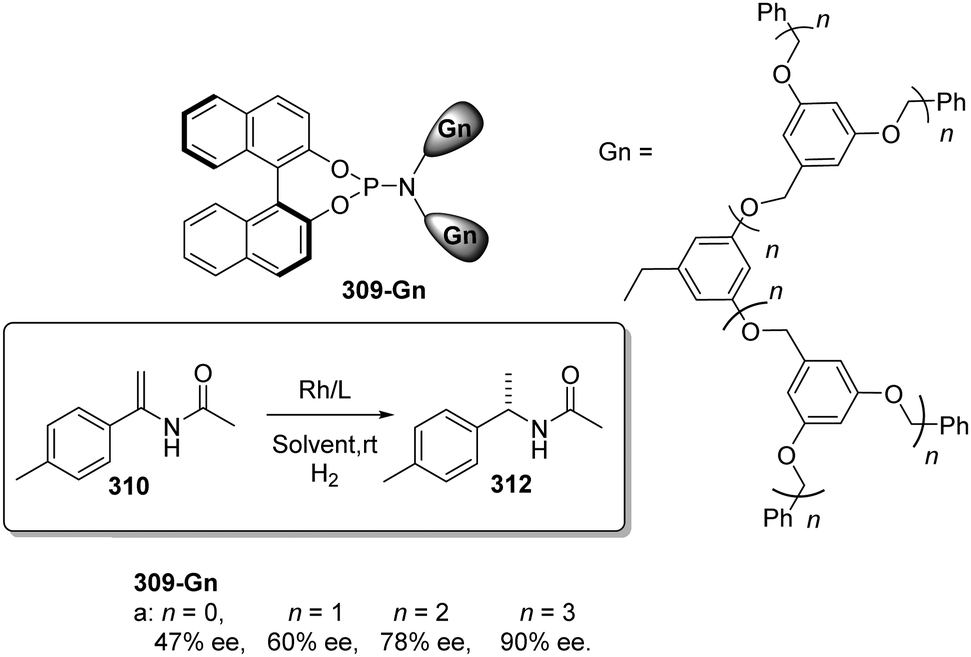 | ||
| Fig. 76 Dendritic chiral monodentate phosphoramidite ligands developed for enantioselective Rh-catalysed hydrogenations. | ||
Different approaches for the preparation of BINAP systems immobilized on dendrimers have been reported. The existence of a “dendrimer effect” seems to depend on the immobilization strategy applied. The BINAP containing dendritic wedges at the 5,5′-position of the binaphthyl backbone linked by amide groups (Fig. 13) induced a positive dendrimer effect regarding the catalytic activity for the hydrogenation of β-keto esters; however no improvement in selectivity was observed. In this case, the dendritic units are far away from the chiral center.150 However, when the dendritic units are directly attached to the diphenylphosphine chelating units of the BINAP (313-Gn) significant dendrimer effects can be found for the ruthenium-catalysed asymmetric hydrogenation of β-keto esters (314) or α-ketoesters (316), as well as α-ketoamides (318) (Fig. 77).380,381 Indeed, the high-generation catalysts exhibited excellent enantioselectivities, which were superior to those obtained from the related small molecular catalysts. The attachment of bulky dendritic wedges on the four phenyl rings at the P atoms was able to fine-tune the steric environment around the metal center. The increasing steric repulsion between the dendritic wedges might be responsible for the different arrangement of the four phenyl rings, and consequently modulate the catalytic behaviour of the [Ru(Gn-BINAP)] complexes.
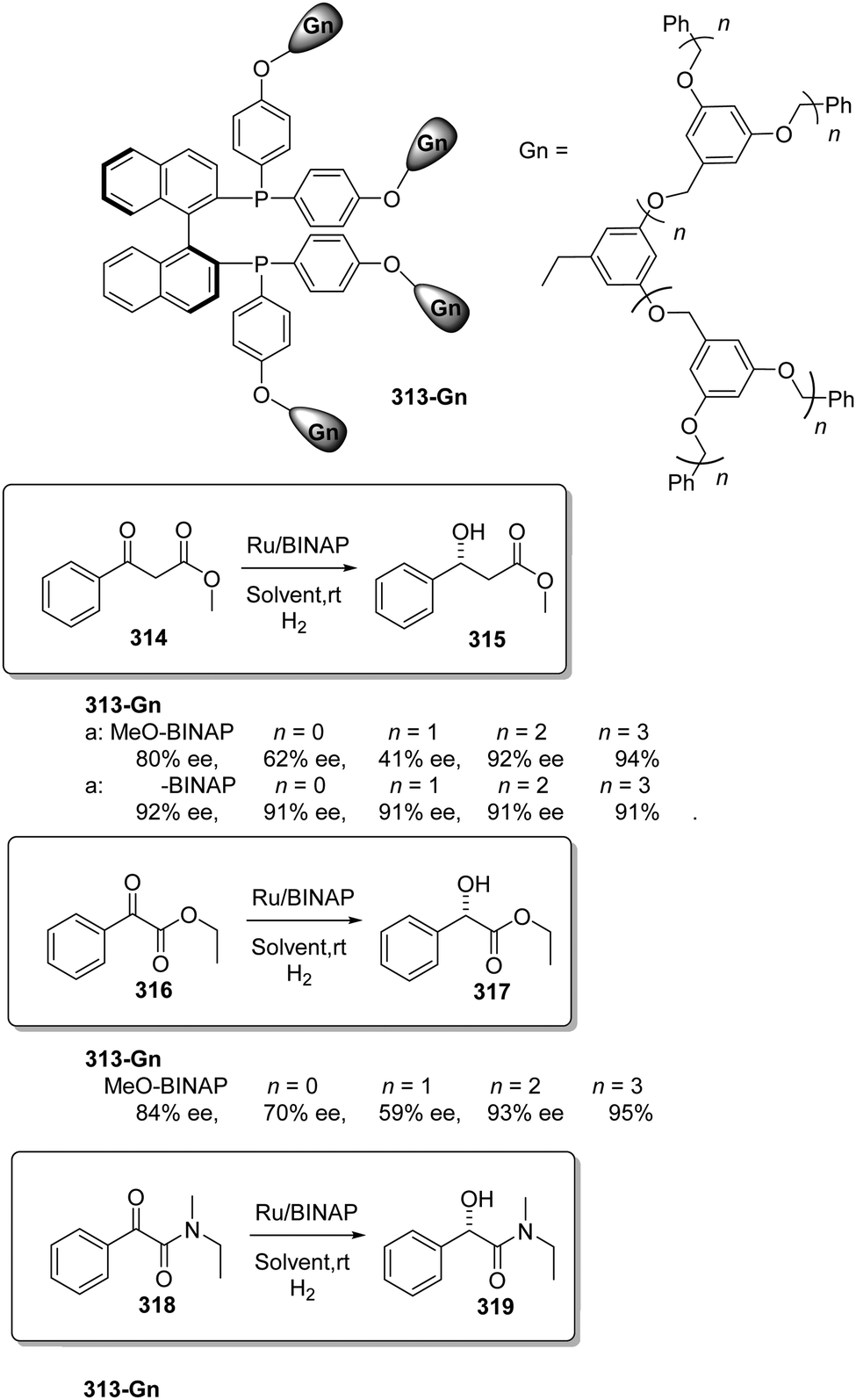 | ||
| Fig. 77 BINAP systems with dendrimeric wedges attached to the four phenyl rings on the P atoms, used for the Ru-catalysed asymmetric hydrogenation. | ||
Chen et al. have designed chiral Ti–salen complexes on the periphery of a polyamidoamine (PAMAM) dendrimer capable of providing enhanced activity, chemoselectivity and enantioselectivity in comparison with the unsupported catalyst. The peripheral free amine groups of PAMAM provide a suitable grafting point for the Ti–salen complex (Fig. 78).382 However, in the initial design, the dense packing of active sites at the periphery of PAMAM increased the difficulty for all complexes to adopt their preferred conformation, which is detrimental to catalysis. This was overcome by the introduction of IL-like units as spacers (320-Gn). The role of this spacer was multiple. It located the chiral complexes away from the dendrimer backbone, ensured a high local concentration of the catalyst and allowed achieving the preferred conformation of the active sites, favouring the intramolecular cooperation between the catalytic sites and therefore enhancing the selectivity. Furthermore, the IL-like units modified the hydrophilic/hydrophobic balance of the support making it more compatible with organic sulfides and aqueous H2O2, facilitating the mass transfer of the reagents and therefore enhancing the activity. The performance of the catalyst 320-Gn (conv. 91%, chemoselectivity 90% and 85% ee) was enhanced relative to that of the analogue 321 (conv. 93%, chemoselectivity 74% and 74% ee) and was even better than that of the homogeneous analogue 322 (conv. 78%, chemoselectivity 64% and 68% ee).
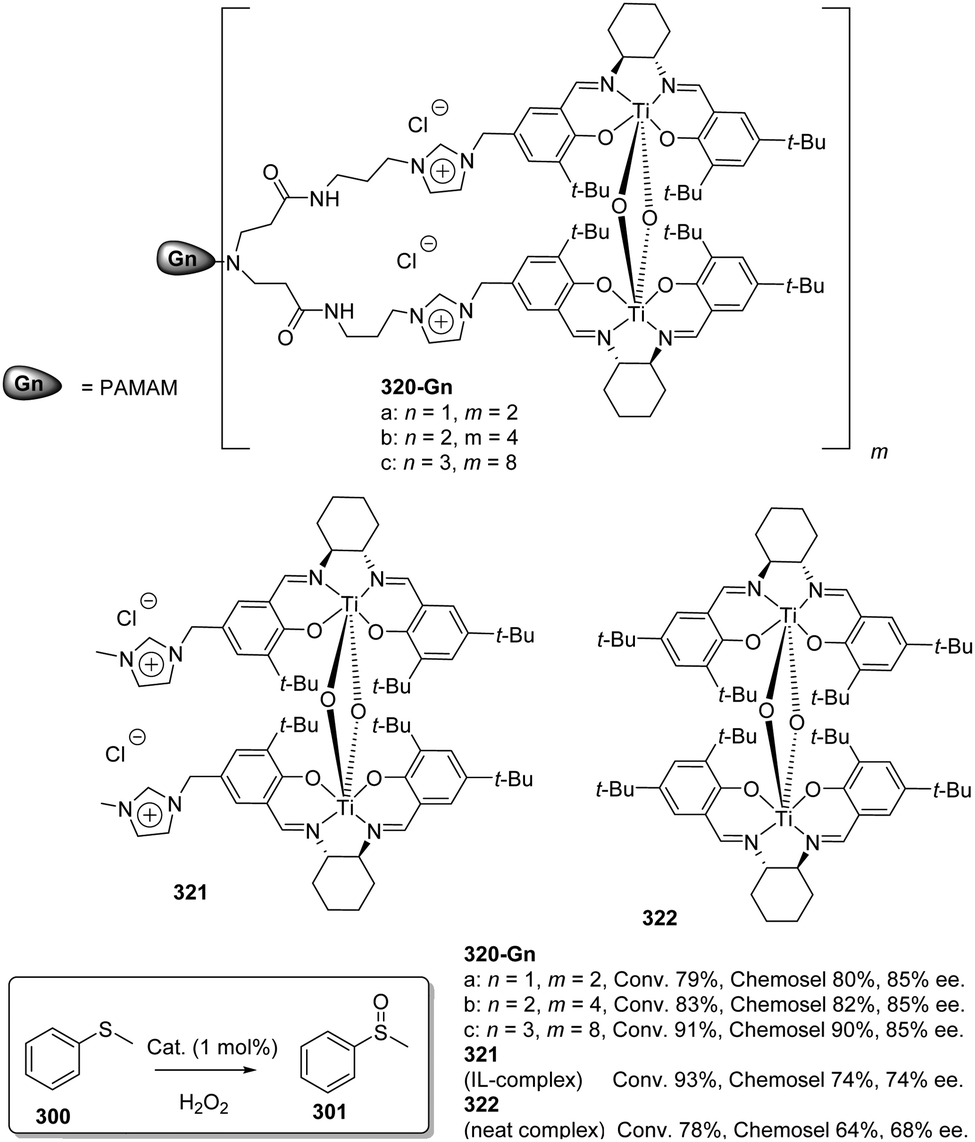 | ||
| Fig. 78 Ti–salen complexes attached to PAMAM dendrimers through an imidazolium linker used for the asymmetric oxidation of sulfides. | ||
Gissibl et al. have immobilized azabis(oxazoline) ligands onto the terminal groups of phosphorus-containing dendrimers (polyphosphorhydrazone, PPH) (327-Gn). The corresponding copper(II) complexes were used as immobilised catalysts for the asymmetric benzoylation of diols (Fig. 79).383 Interestingly, an excellent enantioselectivity was observed for the first and the second generation, but there was a large detrimental effect with the third generation. The dendritic catalysts afforded compound 324b with better selectivities than the monomeric counterpart (328) or those found for the azabis(oxazolines) supported onto soluble polymers such as MEOPEG (329) or polystyrene (330).384 The lack of selectivity found for these non-dendritic systems was associated with the ability of the achiral triazole moiety, used as a linker for the attachment, to coordinate with copper.385 In the case of the dendrimers the triazole moieties were suggested to be soaked in the globular structure of the dendrimer and only the chiral azabis(oxazoline) fragments were located at the periphery of the dendrimer and exposed enough to coordinate to copper, avoiding non- or less selective reaction pathways.
 | ||
| Fig. 79 General structure of azabis(oxazoline) ligands attached to PPH dendrimers and their homogeneous, PEG and PS supported analogues assayed for the asymmetric benzoylation of diols. | ||
A very impressive enhancement in selectivity related to a positive dendrimer effect has been reported by García et al. for the Rh-catalysed [2+2+2] cycloaddition reactions between N-tosyl-1,6-diyne and 2-methoxynaphthalene alkynyl derivatives (Fig. 80).386 The phosphoramidite ligands for Rh complexation were also immobilized onto the peripheral groups of PPH dendrimers (331-Gn). An enhancement in both yield and enantioselectivity through a positive dendrimer effect was observed. The monomer (332) almost induced no enantiomeric excess, whereas all the generations of the dendrimers, from 331-G1 to 331-G3, afforded a very high enantioselection. It should be noted that the dimeric homogeneous system (333) was not more efficient than the monomer 332, excluding simple cooperative site–site effects. These data pointed out to an effect associated with a large number of chiral entities in close proximity.
 | ||
| Fig. 80 General structures of dendritic and non-dendritic phosphoramidite ligands studied for Rh-catalysed [2+2+2] cycloaddition reactions. | ||
Topicity inversion
The effect of the polymer can be so important that the morphology of the polymeric backbone can even alter the topicity of the resulting products. The literature contains a limited number of examples of polymer-bound enantioselective catalysts for which the topicity of the resulting products can be controlled through the morphology of the support with the potential for achieving a dual stereocontrol.387Two of such examples have been reported for catalysts derived from immobilized tartatic acid esters and used for the enantioselective Sharpless asymmetric epoxidation reaction (Fig. 81). Reed et al. found that in the epoxidation of 2-hexen-1-ol when using poly(ethylene glycol) trans-esterified chiral tartrates (337–338), the enantioselection observed could be reversed depending on the PEG chain length.388 A clear change in topicity was found to occur when the molecular weight of the PEG chain acting as the support was around 800 or higher. Similarly, García et al. have also reported that when using for this chiral epoxidation a supported tartrate derivative 340, immobilised onto polyhedral oligomeric silsesquioxanes (POSS), a reversed enantioselectivity was observed with regard to the use of the related non-supported ligand 339.389 In both cases, the change in enantioselectivity was proposed to occur as a result of the coexistence of two Ti–ligand complexes which differ in the molecularity of the ligand, one with a monomeric ligand and the other with a dimeric ligand. The POSS support and the longer PEG chains seem to favor 2:1 instead of 2:2 Ti:ligand complexes.
 | ||
| Fig. 81 Examples of supported tartrate ligands providing topicity reversal of the major enantiomer associated with the nature of the achiral polymeric support. | ||
The possibility of site isolation or preconcentration of the catalytic sites as a function of polymer structure and morphology has also been exploited to achieve a dual stereocontrol in the case of supported amino amides that are able to provide the two enantiomers for the Ni(II) catalysed Et2Zn addition to aldehydes depending on the ligand:metal ratio.390,391
Schurig and coworkers had also reported a reversal in enantioselectivity upon immobilization for the hetero-Diels–Alder reaction between trans-1-methoxy-3-trimethylsiloxy-1,3-butadiene (343) and benzaldehyde catalysed by an oxovanadium complex (Fig. 82). The (1R)-(+)-oxovanadium(IV) bis[3-heptafluorobutanoylcamphorate] attached onto a soluble polymeric dimethylsiloxane support (341) provided the corresponding adduct with a similar enantioselection but with an opposite configuration than the one found for the unsupported catalyst 342.392
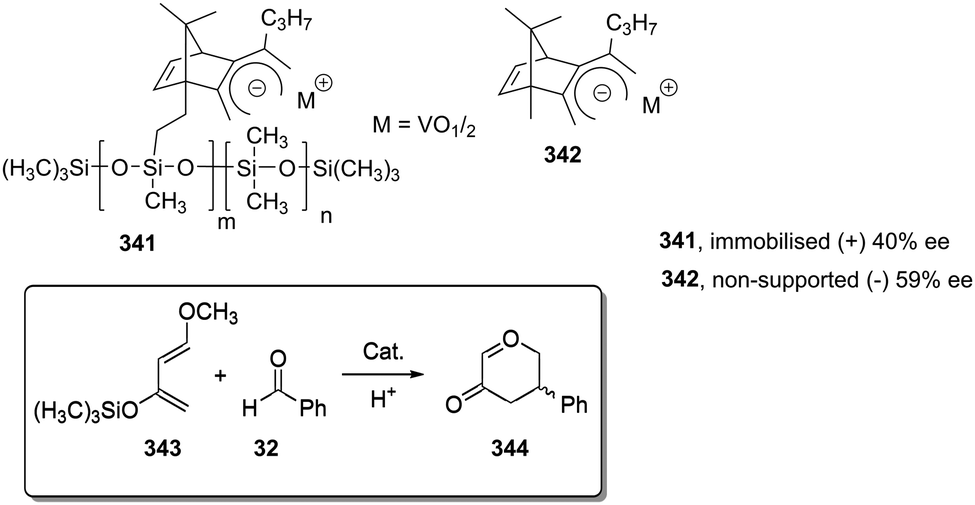 | ||
| Fig. 82 Reversal of enantioselectivity upon immobilization of an oxovanadium complex in a Diels–Alder reaction. | ||
Another example clearly illustrating the importance of the immobilization strategy is given by immobilised Ti-TADDOLates as catalysts for the Diels–Alder reaction between cyclopentadiene and 3-crotonoyl-1,3-oxazolidin-2-one (Fig. 83).132,321,322 The systems prepared by polymerization providing a highly crosslinked matrix showed, in some cases, a better selectivity than the analogues prepared by grafting and even better than the homogeneous counterpart. For instance, the enantioselectivity obtained at 0 °C for the monolithic Ti-TADDOLate from 349b (43% ee) was significantly higher than that observed for the homogeneous (from 345, 20% ee) and grafted (from 349a, 13% ee) analogues, under the same conditions. A very remarkable result, supporting the effect of the polymer, was obtained with the monolithic derivative 225 (Ar = –3,5-(CH3)2–C6H3–). In this case, the major endo enantiomer obtained with the monolithic ligand 225b was the 2S,3R isomer instead of the 2R,3S isomer obtained with the homogeneous analogue 346 or with the supported catalyst 225a prepared by grafting. The steric interactions between the structural components of the TADDOL structure can be modified during the polymerization to form part of a very rigid crosslinked matrix. This effect can be so dramatic that it leads to a different kind of asymmetric induction.
 | ||
| Fig. 83 Effect of the immobilisation strategy of TADDOLs on the use of Ti-TADDOLates as catalysts in Diels–Alder reactions. | ||
The aromatic subunits at the groups bound to the C-2 position of the dioxolane ring are important, as it has been shown that π–π interactions with the α-substituent play a key role in determining both the nature of the most stable conformer and the relative stability of the possible transition states.393,394 In this regard, polymerization of the corresponding vinylic TADDOL in a very rigid, highly crosslinked matrix might modify or even preclude some of those π–π interactions between the aromatic group at C-2 and one of the 3,5-dimethylpenyl substituents at the α-positions, modifying the relative energies of the participating transition states, and favouring in this way the formation of the endo-2S,3R adduct. The generation of main-chain chirality/chiral cavities derived from the polymerization of a chiral vinylic monomer should not be excluded.
As mentioned above (Fig. 30) BINOL phosphoric acids (BNPPA) modified at the 3- and 3′-positions with fragments containing thienyl groups can be immobilized by direct oxidative coupling of the thiophenes to build up a polymeric network with intrinsic beneficial properties, such as high surface area and distinctive microporosity.207,208,395 Remarkably, the corresponding immobilized organocatalyst (354) was not only able to surpass the enantioselectivity of the related monomer (353) in solution but was also able to invert the absolute configuration of the product obtained (56% ee (S) for 354vs. 34% ee (R) for 353) (Fig. 84). In this case, the crosslinking close to the reactive center resulted in a significantly bigger steric hindrance, which seems to benefit the conformation more favourably for the formation of the S-enantiomer in opposition to the more flexible system (353). In good agreement with this, the introduction of more bulky anthracen-9-yl substituents at the 3,3′-positions of the BINOL led to higher enantioselectivities for the monomeric and the polymeric organocatalysts (99% ee (S) for 140).207 In this case, both the monomeric and the polymeric systems provided the S-enantiomer confirming the importance of the steric hindrance.
 | ||
| Fig. 84 Microporous polymeric BINOL phosphoric acids displaying reversed enantioselection compared to the monomeric unit. | ||
Conclusions
Although many additional examples of polymer supported chiral catalytic systems can be found in the literature, the examples presented here illustrate with clarity how the support is not an innocent partner and plays an essential role in defining the performance of these systems. The support needs to be considered as an essential design factor for the preparation of chiral immobilized catalysts. Through an appropriate design and selection of the physico-chemical properties of the polymeric support, it is possible to contribute to enhance the desired properties of the corresponding catalytic system in terms of its activity, stability and selectivity, including the enantioselectivity in the case of chiral systems considered here. Still, however, there is a need for further studies aiming to properly understand the molecular basis of many of the positive support effects discussed here. This will allow in the future a rational approach to the development of new and improved polymer supported chiral catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from MINECO (CTQ2015-68429-R) and GV (PROMETEO/2016/071) is acknowledged.Notes and references
- H.-J. Federsel, Nat. Rev. Drug Discovery, 2005, 4, 685–697 CrossRef CAS PubMed.
- C. Challener, Pharm. Technol., 2016, 40, 28–29 Search PubMed.
- S. Wendeborn, E. Godineau, R. Mondière, T. Smejkal and H. Smits, Chirality in Agrochemicals, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, vol. I, pp. 120–166 Search PubMed.
- C. Lamberth, S. Jeanmart, T. Luksch and A. Plant, Science, 2013, 341, 742–746 CrossRef PubMed.
- G. M. R. Tombo and D. Belluš, Angew. Chem., Int. Ed. Engl., 1991, 30, 1193–1215 CrossRef.
- C. Wang, D. Lu, J. Yang, Y. Xu, C. Gong and Z. Li, Curr. Protein Pept. Sci., 2017, 18, 15–21 CrossRef CAS PubMed.
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer-Verlag, Berlin, 1999 Search PubMed.
- H. U. Blaser and E. Schmidt, Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, John Wiley & Sons Inc., New York, 2003 Search PubMed.
- Q.-L. Zhou, Privileged Chiral Ligands and Catalysts, Wiley-VCH, Weinheim, 2011, ISBN: 978-352732704-1 Search PubMed.
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998–2007 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS PubMed.
- K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2024–2032 CrossRef CAS PubMed.
- B. List, Chem. Rev., 2007, 107, 5413–5415 CrossRef CAS.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994 Search PubMed.
- Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385–3466 CrossRef CAS PubMed.
- A. M. P. Salvo, F. Giacalone and M. Gruttadauria, Molecules, 2016, 21, 1288 CrossRef PubMed.
- T. Frenzel, W. Solodenko and A. Kirschning, in Polymeric Materials in Organic Synthesis and Catalysis, ed. M. R. Buchmeiser, Wiley-VCH, Weinheim, 2003, ch. 4 Search PubMed.
- T. J. Dickerson, N. N. Reed and K. D. Janda, in Polymeric Materials in Organic Synthesis and Catalysis, ed. M. R. Buchmeiser, Wiley-VCH, Weinheim, 2003, ch. 5 Search PubMed.
- A. Kirschning, H. Monenschein and R. Wittenberg, Angew. Chem., Int. Ed., 2001, 40, 650–679 CrossRef CAS PubMed.
- S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 3815–4195 RSC.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- S. Itsuno and M. Hassan, RSC Adv., 2014, 4, 52023–52043 RSC.
- S. Hîbner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- C. Yu and J. He, Chem. Commun., 2012, 48, 4933–4940 RSC.
- J. M. Notestein and A. Katz, Chem. – Eur. J., 2006, 12, 3954–3956 CrossRef CAS PubMed.
- C. E. Song, D. H. Kim and D. S. Choi, Eur. J. Inorg. Chem., 2006, 2927–2935 CrossRef CAS.
- J. M. Thomas and R. Raja, Acc. Chem. Res., 2008, 41, 708–720 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- R. P. Megens and G. Roelfes, Chem. – Eur. J., 2011, 17, 8514–8523 CrossRef CAS PubMed.
- (a) A. Ikeda, K. Terada, M. Shiotsuki and F. Sanda, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3783–3796 CrossRef CAS; (b) K. Maeda, K. Tanaka, K. Morino and E. Yashima, Macromolecules, 2007, 40, 6783–6785 CrossRef CAS; (c) H. Zhang, W. Yang and J. Deng, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1816–1823 CrossRef CAS.
- (a) M. Reggelin, M. Schultz and M. Holbach, Angew. Chem., Int. Ed., 2002, 41, 1614–1617 CrossRef CAS; (b) M. Reggelin, S. Doerr, M. Klussmann, M. Schultz and M. Holbach, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5461–5466 CrossRef CAS PubMed.
- (a) T. Yamamoto, Y. Akai, Y. Nagata and M. Suginome, Angew. Chem., Int. Ed., 2011, 50, 8844–8847 CrossRef CAS PubMed; (b) Y. Akai, T. Yamamoto, Y. Nagata, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2012, 134, 11092–11095 CrossRef CAS PubMed; (c) T. Yamamoto, R. Murakami and M. Suginome, J. Am. Chem. Soc., 2017, 139, 2557–2560 CrossRef CAS PubMed.
- A. Warshawsky, Isr. J. Chem., 1979, 18, 318–324 CrossRef CAS.
- B. Altava, M. I. Burguete, E. Garcia-Verdugo, S. V. Luis, M. J. Vicent and J. A. Mayoral, React. Funct. Polym., 2001, 48, 25–35 CrossRef CAS.
- N. Madhavan, C. W. Jones and M. Weck, Acc. Chem. Res., 2008, 41, 1153–1165 CrossRef CAS PubMed.
- E. S. Choong, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2005, 101, 143–173 RSC.
- Chiral Catalyst Immobilization and Recycling, ed. D. E. de Vos, I. F. J. Vankelecom and P. A. Jacobs, Wiley-VCH, New York, 2000 Search PubMed.
- J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS PubMed.
- P. Barbaro and F. Liguori, Chem. Rev., 2009, 109, 515–529 CrossRef CAS PubMed.
- E. Bayer and W. Schumann, J. Chem. Soc., Chem. Commun., 1986, 949–952 RSC.
- P. Wentworth and K. D. Janda, Chem. Commun., 1999, 1917–1924 CAS.
- D. E. Bergbreiter, Polymeric reagents and catalysts, ACS Symp. Ser., 1986, 308, 17–41 CrossRef CAS.
- D. E. Bergbreiter, J. Tian and C. Hongfa, Chem. Rev., 2009, 109, 530–582 CrossRef CAS PubMed.
- D. E. Bergbreiter, ACS Macro Lett., 2014, 3, 260–265 CrossRef CAS.
- A. Lu and R. K. O'Reilly, Curr. Opin. Biotechnol., 2013, 24, 639–645 CrossRef CAS PubMed.
- R. van Heerbeek, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Rev., 2002, 102, 3717–3756 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Coord. Chem. Rev., 2013, 257, 2317–2334 CrossRef CAS.
- E. de Jesús and Juan C. Flores, Ind. Eng. Chem. Res., 2008, 47, 7968–7981 CrossRef.
- A. Akelah and D. C. Sherrington, Chem. Rev., 1981, 81, 557–587 CrossRef CAS.
- J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815–838 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- M. Benaglia, A. Puglisi and F. Cozzi, Chem. Rev., 2003, 103, 3401–3430 CrossRef CAS PubMed.
- H. P. Hentze and M. Antonietti, Rev. Mol. Biotechnol., 2002, 90, 27–53 CrossRef CAS PubMed.
- T. Gokmen and F. E. Du Prez, Prog. Polym. Sci., 2012, 37, 365–405 CrossRef.
- M. Gericke, J. Trygg and P. Fardim, Chem. Rev., 2013, 113, 4812–4836 CrossRef CAS PubMed.
- D. Hudson, J. Comb. Chem., 1999, 1, 403–457 CrossRef CAS PubMed.
- P. H. Toy and K. D. Janda, Tetrahedron Lett., 1999, 40, 6329–6332 CrossRef CAS.
- D. Lumpi, C. Braunshier, E. Horkel, C. Hametner and J. Fröhlich, ACS Comb. Sci., 2014, 16, 367–374 CrossRef CAS PubMed.
- P. H. Toy, T. S. Reger and K. D. Janda, Aldrichimica Acta, 2000, 33, 87–93 CAS.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- M. Rose, ChemCatChem, 2014, 6, 1166–1182 CAS.
- Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC.
- X. Zou, H. Rena and G. Zhu, Chem. Commun., 2013, 49, 3925–3936 RSC.
- J. Huang and S. R. Turner, Polym. Rev., 2017, 6, 1–41 Search PubMed.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- Y. Zhang and J. Y. Ying, ACS Catal., 2015, 5, 2681–2691 CrossRef CAS.
- U. Díaz and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef.
- Q. Sun, Z. Dai, X. Meng, L. Wang and F.-S. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17–26 RSC.
- Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, Weinheim, 2nd edn, 2005 Search PubMed.
- Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, ed. H. U. Blaser and H.-J. Federsel, Wilei-VCH, Weinheim, 2nd edn, 2010 Search PubMed.
- V. A. Larionov, T. Cruchter, T. Mietke and E. Meggers, Organometallics, 2017, 36, 1457–1460 CrossRef CAS.
- B. Corain, M. Zecca and K. Jeràbek, J. Mol. Catal. A: Chem., 2001, 177, 3–20 CrossRef CAS.
- P. Hodge, Chem. Soc. Rev., 1997, 26, 417–424 RSC.
- D. Walsh, D. Wu and Y.-T. Chang, Curr. Opin. Chem. Biol., 2003, 7, 353–361 CrossRef CAS PubMed.
- D. C. Sherrington, Chem. Commun., 1998, 2275–2286 RSC.
- A. Deratani, G. D. Darling, D. Horak and J. M. J. Frechet, Macromolecules, 1987, 20, 767–772 CrossRef CAS.
- R. Santini, M. C. Griffith and M. Qi, Tetrahedron Lett., 1998, 39, 8951–8954 CrossRef CAS.
- B. P. Santora, M. R. Gagné, K. G. Moloy and N. S. Radu, Macromolecules, 2001, 34, 658–661 CrossRef CAS.
- A. R. Vaino and K. D. Janda, J. Comb. Chem., 2000, 2, 579–596 CrossRef CAS PubMed.
- S. W. Gerritz, Curr. Opin. Chem. Biol., 2001, 5, 264–268 CrossRef CAS PubMed.
- B. S. Lee, S. Mahajan and K. D. Janda, Tetrahedron Lett., 2005, 46, 807–810 CrossRef CAS.
- A. Guyot, P. Hodge, D. C. Sherrington and H. Widdecke, React. Polym., 1992, 16, 233–259 CrossRef CAS.
- M. Watanabe and K. Soai, J. Chem. Soc., Perkin Trans. 1, 1994, 837–842 RSC.
- K. Soai and M. Watanabe, Tetrahedron: Asymmetry, 1991, 2, 97–100 CrossRef CAS.
- K. Kamahori, K. Ito and S. Itsuno, J. Org. Chem., 1996, 61, 8321–8324 CrossRef CAS PubMed.
- A. Marrocchi, P. Adriaensens, E. Bartollini, B. Barkakaty, R. Carleer, J. Chen, D. K. Hensley, C. Petrucci, M. Tassi and L. Vaccaro, Eur. Polym. J., 2015, 73, 391–401 CrossRef CAS.
- F. Montanari and P. Tundo, J. Org. Chem., 1981, 46, 2125–2130 CrossRef CAS.
- P. L. Anelli, F. Montanari and S. Quici, J. Chem. Soc., Perkin Trans. 2, 1983, 1827–1830 RSC.
- P. L. Anelli, B. Czech, F. Montanari and S. Quici, J. Am. Chem. Soc., 1984, 106, 861–869 CrossRef CAS.
- P. W. Dyer, S. Handa, T. B. Reeve and S. Suhard, Tetrahedron Lett., 2005, 46, 4753–4756 CrossRef CAS.
- A. Seki, F. Ishiwata, Y. Takizawa and M. Asami, Tetrahedron, 2004, 60, 5001–5011 CrossRef CAS.
- L.-J. Zhao, H. S. He, M. Shi and P. H. Toy, J. Comb. Chem., 2004, 6, 680–683 CrossRef CAS PubMed.
- L.-J. Zhao, C. K.-W. Kwong, M. Shi and P. H. Toy, Tetrahedron, 2005, 61, 12026–12032 CrossRef CAS.
- R. Pedrosa, J. M. Andrés, D. P. Ávila, M. Ceballos and R. Pindado, Green Chem., 2015, 17, 2217–2225 RSC.
- J. M. Andres, N. de La Cruz, M. Valle and R. Pedrosa, ChemPlusChem, 2016, 81, 86–92 CrossRef CAS.
- L. Wang and F.-S. Xiao, ChemCatChem, 2014, 6, 3048–3052 CrossRef CAS.
- S. Cañellas, C. Ayats, A. H. Henseler and M. A. Pericàs, ACS Catal., 2017, 7, 1383–1391 CrossRef.
- B. Thierry, J.-C. Plaquevent and D. Cahard, Tetrahedron: Asymmetry, 2001, 12, 983–986 CrossRef CAS.
- K. Hallman and C. Moberg, Tetrahedron: Asymmetry, 2001, 12, 1475–1478 CrossRef CAS.
- L. Canali, J. K. Karjalainen, D. C. Sherrington and O. Hormi, Chem. Commun., 1997, 123–124 RSC.
- S. D. Alexandratos and D. H. J. Miller, Macromolecules, 1996, 29, 8025–8029 CrossRef CAS.
- D. Font, S. Sayalero, A. Bastero, C. Jimeno and M. A. Pericas, Org. Lett., 2008, 10, 337–340 CrossRef CAS PubMed.
- D. Font, C. Jimeno and M. A. Pericas, Org. Lett., 2006, 8, 4653–4655 CrossRef CAS PubMed.
- C. Ayats, A. H. Henseler and M. A. Pericàs, ChemSusChem, 2012, 5, 320–325 CrossRef CAS PubMed.
- E. Alza, X. C. Cambeiro, C. Jimeno and M. A. Pericàs, Org. Lett., 2007, 9, 3717–3720 CrossRef CAS PubMed.
- S. D. Alexandratos and D. H. J. Miller, Macromolecules, 2000, 33, 2011–2015 CrossRef CAS.
- C.-W. Kwong, R. Huang, M. Zhang, M. Shi and P. H. Toy, Chem. – Eur. J., 2007, 13, 2369–2376 CrossRef CAS PubMed.
- S. Itsuno, J. Synth. Org. Chem., Jpn., 2009, 67, 1025–1031 CrossRef CAS.
- S. Hashiguchi, A. Fujita, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- H. Sugie, Y. Hashimoto, N. Haraguchi and S. Itsuno, J. Organomet. Chem., 2014, 751, 711–716 CrossRef CAS.
- N. Haraguchi, K. Tsuru, Y. Arakawa and S. Itsuno, Org. Biomol. Chem., 2009, 7, 69–75 CAS.
- Y. Arakawa, N. Haraguchi and S. Itsuno, Tetrahedron Lett., 2006, 47, 3239–3243 CrossRef CAS.
- Y. Arakawa, A. Chiba, N. Haraguchi and S. Itsuno, Adv. Synth. Catal., 2008, 350, 2295–2304 CrossRef CAS.
- F. Guendouz, R. Jacquier and J. Verducci, Tetrahedron, 1988, 44, 7095–7108 CrossRef CAS.
- S. V. Luis and I. Alfonso, Acc. Chem. Res., 2014, 47, 112–124 CrossRef CAS PubMed.
- M. I. Burguete, J. M. J. Frechet, E. Garcia-Verdugo, M. Janco, S. V. Luis, F. Svec, M. J. Vicent and M. Xu, Polym. Bull., 2002, 48, 9–15 CrossRef CAS.
- F. M. Adrian, B. Altava, M. I. Burguete, S. V. Luis, R. V. Salvador and E. Garcia-España, Tetrahedron, 1998, 54, 3581–3588 CrossRef CAS.
- B. Altava, M. I. Burguete, J. C. Frias, E. Garcia-España, S. V. Luis and J. F. Miravet, Ind. Eng. Chem. Res., 2000, 39, 3589–3595 CrossRef CAS.
- F. Adrian, M. I. Burguete, J. M. Fraile, J. I. Garcia, J. Garcia, E. Garcia-España, S. V. Luis, A. J. Royo and M. C. Sanchez, Eur. J. Inorg. Chem., 1999, 2347–2354 CrossRef CAS.
- G. Nováková, P. Drabina, B. Frumarová and M. Sedlák, Adv. Synth. Catal., 2016, 358, 2541–2552 CrossRef.
- M. Sedlák, P. Drabina, R. Keder, J. Hanusek, I. Císařová and A. Růžička, J. Organomet. Chem., 2006, 691, 2623–2630 CrossRef.
- J. Irurre, A. Fernandez-Serrat, M. Altayo and M. Riera, Enantiomer, 1998, 3, 103–120 CAS.
- J. Irurre, A. Fernández-Serrat and F. Rosanas, Chirality, 1997, 9, 191–197 CrossRef CAS.
- D. Seebach, A. K. Beck and A. Heckel, Angew. Chem., Int. Ed., 2001, 40, 92–138 CrossRef CAS PubMed.
- B. Altava, M. I. Burguete, B. Escuder, S. V. Luis, R. V. Salvador, J. M. Fraile, J. A. Mayoral and A. J. Royo, J. Org. Chem., 1997, 62, 3126–3134 CrossRef CAS PubMed.
- S. Degni, C.-E. Wilén and R. Leino, Org. Lett., 2001, 3, 2551–2554 CrossRef CAS PubMed.
- W.-K. An, M.-Y. Han, C.-A. Wang, S.-M. Yu, Y. Zhang, S. Bai and W. Wang, Chem. – Eur. J., 2014, 20, 11019–11028 CrossRef CAS PubMed.
- S. Degni, S. Strandman, P. Laari, M. Nuopponen, C.-E. Wilen, H. Tenhu and A. Rosling, React. Funct. Polym., 2005, 62, 231–240 CrossRef CAS.
- A. Heckel and D. Seebach, Chem. – Eur. J., 2002, 8, 560–572 CrossRef.
- A. Heckel and D. Seebach, Angew. Chem., Int. Ed., 2000, 39, 163–165 CrossRef CAS PubMed.
- X. Wang, J. Zhang, Y. Liu and Y. Cui, Bull. Chem. Soc. Jpn., 2014, 87, 435–440 CrossRef CAS.
- S. Legrand, H. Heikkinen, I. A. Nicholls, A. Root, J. Svenson and C. R. Unelius, Tetrahedron Lett., 2010, 51, 2258–2261 CrossRef CAS.
- B. Altava, M. I. Burguete, J. I. Garcia, S. V. Luis, J. A. Mayoral and M. J. Vicent, Tetrahedron: Asymmetry, 2001, 12, 1829–1835 CrossRef CAS.
- J. M. Fraile, J. A. Mayoral, A. J. Royo, R. V. Salvador, B. Altava, S. V. Luis and M. I. Burguete, Tetrahedron, 1996, 52, 9853–9862 CrossRef CAS.
- M. I. Burguete, M. Collado, E. Garcia-Verdugo, M. J. Vicent, S. V. Luis, N. G. Von Keyserling and J. Martens, Tetrahedron, 2003, 59, 1797–1804 CrossRef.
- B. Altava, M. I. Burguete, E. Garcia-Verdugo, S. V. Luis, O. Pozo and R. V. Salvador, Eur. J. Org. Chem., 1999, 2263–2267 CrossRef CAS.
- B. Altava, M. I. Burguete, M. Collado, E. Garcia-Verdugo, S. V. Luis, R. V. Salvador and M. J. Vicent, Tetrahedron Lett., 2001, 42, 1673–1675 CrossRef CAS.
- M. I. Burguete, M. Collado, J. Escorihuela, F. Galindo, E. García-Verdugo, S. V. Luis and M. J. Vicent, Tetrahedron Lett., 2003, 44, 6891–6894 CrossRef CAS.
- J. Escorihuela, L. González, B. Altava, M. I. Burguete and S. V. Luis, Appl. Catal., A, 2013, 462–463, 23–30 CrossRef CAS.
- A.-M. Caminade, P. Servin, R. Laurent and J.-P. Majoral, Chem. Soc. Rev., 2008, 37, 56–67 RSC.
- A.-M. Caminade, A. Ouali, R. Laurent, C.-O. Turrin and J.-P. Majoral, Chem. Soc. Rev., 2015, 44, 3890–3899 RSC.
- A.-M. Caminade, A. Ouali, R. Laurent, C. O. Turrin and J.-P. Majoral, Coord. Chem. Rev., 2016, 308, 478–497 CrossRef CAS.
- Y.-M. He, Y. Feng and Q.-H. Fan, Acc. Chem. Res., 2014, 47, 2894–2906 CrossRef CAS PubMed.
- A. John, F. M. Nachtigall and L. S. Santos, Curr. Org. Chem., 2012, 16, 1776–1787 CrossRef CAS.
- B. Helms and J. M. J. Fréchet, Adv. Synth. Catal., 2006, 348, 1125–1148 CrossRef CAS.
- A. Ouali, R. Laurent, A.-M. Caminade, J.-P. Majoral and M. Taillefer, J. Am. Chem. Soc., 2006, 128, 15990–15991 CrossRef CAS PubMed.
- E. Delort, T. Darbre and J.-L. Reymond, J. Am. Chem. Soc., 2004, 126, 15642–15643 CrossRef CAS PubMed.
- Z.-J. Wang, G.-J. Deng, Y. Li, Y.-M. He, W.-J. Tang and Q.-H. Fan, Org. Lett., 2007, 9, 1243–1246 CrossRef CAS PubMed.
- Q.-H. Fan, Y.-M. Chen, X.-M. Chen, D.-Z. Jiang, F. Xi and A. S. C. Chan, Chem. Commun., 2000, 789–790 RSC.
- G.-J. Deng, Q.-H. Fan and X.-M. Chen, Chin. J. Chem., 2002, 20, 1139–1141 CrossRef CAS.
- B. Ma, G. Deng, J. Liu, Y. He and Q.-H. Fan, Acta Chim. Sin., 2013, 71, 528–534 CrossRef CAS.
- B. Ma, Z. Ding, J. Liu, Y. He and Q.-H. Fan, Chem. – Asian J., 2013, 8, 1101–1104 CrossRef CAS PubMed.
- S. P. Smidt, A. Pfaltz, E. Martinez-Viviente, P. S. Pregosin and A. Albinati, Organometallics, 2003, 22, 1000–1009 CrossRef CAS.
- B. Helms, C. O. Liang, C. J. Hawker and J. M. J. Fréchet, Macromolecules, 2005, 38, 5411–5415 CrossRef CAS.
- J. Liu, Y. Feng, B. D. Ma, Y. M. He and Q. H. Fan, Eur. J. Org. Chem., 2012, 6737–6744 CrossRef CAS.
- R. Breinbauer and E. N. Jacobsen, Angew. Chem., Int. Ed., 2000, 39, 3604–3607 CrossRef CAS PubMed.
- K. B. Hansen, J. L. Leighton and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 10924–10925 CrossRef CAS.
- A.-M. Abu-Elfotoh, K. Phomkeona, K. Shibatomi and S. Iwasa, Angew. Chem., Int. Ed., 2010, 49, 8439–8443 CrossRef CAS PubMed.
- A. Petri, D. Pini, S. Rapaccini and P. Salvadori, Chirality, 1999, 11, 745–751 CrossRef CAS PubMed.
- H. Sellner, P. B. Rheiner and D. Seebach, Helv. Chim. Acta, 2002, 85, 352–387 CrossRef CAS.
- P. B. Rheiner and D. Seebach, Chem. – Eur. J., 1999, 5, 3221–3236 CrossRef CAS.
- H. Sellner and D. Seebach, Angew. Chem., Int. Ed., 1999, 38, 1918–1920 CrossRef CAS.
- F. Svec, J. Chromatogr. A, 2010, 1217, 902–924 CrossRef CAS PubMed.
- M. R. Buchmeiser, Polymer, 2007, 48, 2187–2198 CrossRef CAS.
- M. I. Burguete, J. M. Fraile, J. I. García, E. García-Verdugo, S. V. Luis and J. A. Mayoral, Org. Lett., 2000, 2, 3905–3908 CrossRef CAS PubMed.
- M. J. Fernández, J. M. Fraile, J. I. García, J. A. Mayoral, M. I. Burguete, E. García-Verdugo, S. V. Luis and M. A. Harmer, Top. Catal., 2000, 13, 303–309 CrossRef.
- M. I. Burguete, J. A. Fraile, J. I. García, E. García-Verdugo, C. I. Herrerías, S. V. Luis and J. A. Mayoral, J. Org. Chem., 2001, 66, 8893–8901 CrossRef CAS PubMed.
- M. I. Burguete, J. M. Fraile, E. Garcia-Verdugo, S. V. Luis, V. Martinez-Merino and J. A. Mayoral, Ind. Eng. Chem. Res., 2005, 44, 8580–8587 CrossRef CAS.
- H. Nogami, S. Matsunaga, M. Kanai and M. Shibasaki, Tetrahedron Lett., 2001, 42, 279–283 CrossRef CAS.
- W. Li and B. Yan, J. Org. Chem., 1998, 63, 4092–4097 CrossRef CAS.
- M. I. Burguete, E. Diez-Barra, J. M. Fraile, J. I. Garcia, E. Garcia-Verdugo, R. Gonzalez, C. I. Herrerias, S. V. Luis and J. I. Mayoral, Bioorg. Med. Chem. Lett., 2002, 12, 1821–1824 CrossRef CAS PubMed.
- E. Diez-Barra, J. M. Fraile, J. I. Garcia, E. Garcia-Verdugo, C. I. Herrerias, S. V. Luis, J. A. Mayoral, P. Sánchez-Verdú and J. Tolosa, Tetrahedron: Asymmetry, 2003, 14, 773–778 CrossRef CAS.
- S. Xie, R. W. Allington, J. M. J. Frechet and F. Svec, Adv. Biochem. Eng./Biotechnol., 2002, 76, 87–125 CrossRef CAS PubMed.
- F. Svec and J. M. J. Frechet, Rigid Macroporous Organic Polymer Monoliths Prepared by Free Radical Polymerization, in Monolithic materials, ed. F. Svec, T. B. Tennikova and Z. Deyl, Elsevier, Amsterdam, 2003, ch. 2, pp. 19–50 Search PubMed.
- A. Mandoli, S. Orlandi, D. Pini and P. Salvadori, Chem. Commun., 2003, 2466–2467 RSC.
- A. Mandoli, R. Garzelli, S. Orlandi, D. Pini, M. Lessi and P. Salvadori, Catal. Today, 2009, 140, 51–57 CrossRef CAS.
- A. Cornejo, J. M. Fraile, J. I. García, M. J. Gil, G. Legarreta, S. V. Luis, V. Martínez-Merino and J. A. Mayoral, Org. Lett., 2002, 4, 3927–3930 CrossRef CAS PubMed.
- A. Cornejo, J. M. Fraile, J. I. García, M. J. Gil, S. V. Luis, V. Martínez-Merino and J. A. Mayoral, J. Org. Chem., 2005, 70, 5536–5544 CrossRef CAS PubMed.
- O. Nestler and K. Severin, Org. Lett., 2001, 3, 3907–3909 CrossRef CAS PubMed.
- E. Burri, M. Öhm, C. Daguenet and K. Severin, Chem. – Eur. J., 2005, 11, 5055–5061 CrossRef CAS PubMed.
- E. Burri, S. M. Leeder, K. Severin and M. R. Gagné, Adv. Synth. Catal., 2006, 348, 1640–1644 CrossRef CAS.
- S. M. Leeder and M. R. Gagné, J. Am. Chem. Soc., 2003, 125, 9048–9054 CrossRef CAS PubMed.
- S. L. Vinson and M. R. Gagné, Chem. Commun., 2001, 1130–1132 RSC.
- L. Chen, Y. Yang, Z. Guo and D. Jiang, Adv. Mater., 2011, 23, 3149–3154 CrossRef CAS PubMed.
- L. Chen, Y. Yang and D. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- Q. Sun, M. Jiang, Z. Shen, Y. Jin, S. Pan, L. Wang, X. Meng, W. Chen, Y. Ding, J. Lib and F.-S. Xiao, Chem. Commun., 2014, 50, 11844–11847 RSC.
- Q. Sun, Z. Dai, X. Liu, N. Sheng, F. Deng, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2015, 137, 5204–5209 CrossRef CAS PubMed.
- Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Mater., 2017, 29, 5720–5726 CrossRef CAS.
- T. Wang, Y. Lyu, K. Xiong, W. Wang, H. Zhang, Z. Zhan, Z. Jiang and Y. Ding, Chin. J. Catal., 2017, 38, 890–897 CrossRef CAS.
- Q. Sun, X. Meng, X. Liu, X. Zhang, Y. Yang, Q. Yang and F.-S. Xia, Chem. Commun., 2012, 48, 10505–10507 RSC.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- T. Wang, Y. Lyu, X. Chen, C. Li, M. Jiang, X. Song and Y. Ding, RSC Adv., 2016, 6, 28447–28450 RSC.
- X. Wang, S.-M. Lu, J. Li, Y. Liu and C. Li, Catal. Sci. Technol., 2015, 5, 2585–2589 CAS.
- X. Wang, J. Li, S. Lu, Y. Liu and C. Li, Chin. J. Catal., 2015, 36, 1170–1174 CrossRef.
- R. Marchetto, E. M. Cilli, G. N. Jubilut, S. Schreier and C. R. Nakaie, J. Org. Chem., 2005, 70, 4561–4568 CrossRef CAS PubMed.
- D. A. Annis and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 4147–4154 CrossRef CAS.
- M. M. Zhong, H. Li, J. Chen, L. Tao, C. Li and Q. H. Yang, Chem. – Eur. J., 2017, 23, 11504–11508 CrossRef CAS PubMed.
- Q. Sun, Y. Jin, L. Zhu, L. Wang, X. Meng and F.-S. Xiao, Nano Today, 2013, 8, 342–350 CrossRef CAS.
- M. Rueping, E. Sugiono, A. Steck and T. Theissmann, Adv. Synth. Catal., 2010, 352, 281–287 CrossRef CAS.
- D. S. Kundu, J. Schmidt, C. Bleschke, A. Thomas and S. Blechert, Angew. Chem., Int. Ed., 2012, 51, 5456–5459 CrossRef CAS PubMed.
- J. Schmidt, D. S. Kundu, S. Blechert and A. Thomas, Chem. Commun., 2014, 50, 3347–3349 RSC.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- H. Xu, X. Chen, J. Gao, J. Lin, M. Addicoat, S. Irle and D. Jiang, Chem. Commun., 2014, 50, 1292–1294 RSC.
- T. Nagashima and H. M. L. Davies, Org. Lett., 2002, 4, 1989–1992 CrossRef CAS PubMed.
- H. M. L. Davies and A. M. Walji, Org. Lett., 2003, 5, 479–482 CrossRef CAS PubMed.
- H. M. L. Davies, A. M. Walji and T. Nagashima, J. Am. Chem. Soc., 2004, 126, 4271–4280 CrossRef CAS PubMed.
- H. M. L. Davies and A. M. Walji, Org. Lett., 2005, 7, 2941–2944 CrossRef CAS PubMed.
- F. A. Cotton and R. A. Walton, Multiple Bonds between Metal Atoms, Clarendon Press, Oxford, UK, 2nd edn, 1993 Search PubMed.
- S. Kobayashi and R. Akiyamaa, Chem. Commun., 2003, 449–460 RSC.
- R. Akiyama and S. Kobayashi, Chem. Rev., 2009, 109, 594–642 CrossRef CAS PubMed.
- S. Kobayashi and S. Nagayama, J. Am. Chem. Soc., 1998, 120, 2985–2986 CrossRef CAS.
- S. Kobayashi and S. Nagayama, Synlett, 1997, 653–654 CrossRef CAS.
- P. H. Toy and K. D. Janda, Acc. Chem. Res., 2000, 33, 546–554 CrossRef CAS PubMed.
- Q.-H. Fan, C.-Y. Ren, C.-H. Yeung, W.-H. Hu and A. S. C. Chan, J. Am. Chem. Soc., 1999, 121, 7407–7408 CrossRef CAS.
- Q.-H. Fan, G.-J. Deng, C.-C. Lin and A. S. C. Chan, Tetrahedron: Asymmetry, 2001, 12, 1241–1247 CrossRef CAS.
- Q.-H. Fan, G.-J. Deng, X.-M. Chen, W.-C. Xie, D.-Z. Jiang, D.-S. Liu and A. S. C. Chan, J. Mol. Catal. A: Chem., 2000, 159, 37–43 CrossRef CAS.
- T. Oyama, H. Yoshioka and M. Tomoi, Chem. Commun., 2005, 1857–1859 RSC.
- J. K. Karjalainen, O. E. O. Hormi and D. C. Sherrington, Tetrahedron: Asymmetry, 1998, 9, 1563–1575 CrossRef CAS.
- S. E. Denmark and T. Wynn, J. Am. Chem. Soc., 2001, 123, 6199–6200 CrossRef CAS PubMed.
- S. E. Denmark and J. Fu, J. Am. Chem. Soc., 2001, 123, 9488–9489 CrossRef CAS PubMed.
- S. E. Denmark, D. M. Coe, N. E. Pratt and B. D. Griedel, J. Org. Chem., 1994, 59, 6161–6163 CrossRef CAS.
- X. Zheng, C. W. Jones and M. Weck, J. Am. Chem. Soc., 2007, 129, 1105–1112 CrossRef CAS PubMed.
- M. Holbach and M. Weck, J. Org. Chem., 2006, 71, 1825–1836 CrossRef CAS PubMed.
- P. Goyal, X. Zheng and M. Weck, Adv. Synth. Catal., 2008, 350, 1816–1822 CrossRef CAS.
- X. Zheng, C. W. Jones and M. Weck, Chem. – Eur. J., 2006, 12, 576–583 CrossRef PubMed.
- N. Madhavan and M. Weck, Adv. Synth. Catal., 2008, 350, 419–425 CrossRef CAS.
- Y. Xie, M. Wang, X. Wu, C. Chen, W. Ma, Q. Dong, M. Yuan and Z. Hou, ChemPlusChem, 2016, 81, 541–549 CrossRef CAS.
- P. Cotanda, A. Lu, J. P. Patterson, N. Petzetakis and R. K. O’Reilly, Macromolecules, 2012, 45, 2377–2384 CrossRef CAS.
- P. Cotanda and R. K. O'Reilly, Chem. Commun., 2012, 48, 10280–10282 RSC.
- A. Lu, P. Cotanda, J. P. Patterson, D. A. Longbottom and R. K. O’Reilly, Chem. Commun., 2012, 48, 9699–9701 RSC.
- H. A. Zayas, A. Lu, D. Valade, F. Amir, Z. Jia, R. K. O’Reilly and M. J. Monteiro, ACS Macro Lett., 2013, 2, 327–331 CrossRef CAS.
- Y. Xu, Z. Hua, J. Zhang, J. Yang, Z. Cao, D. Zhang, L. He, V. S. J. Craig, G. Zhang and G. Liu, Chem. Commun., 2016, 52, 3392–3395 RSC.
- Y. Zhang, R. Tan, G. Zhao, X. Luo and D. Yin, Catal. Sci. Technol., 2016, 6, 488–496 CAS.
- Y. Zhang, R. Tan, G. Zhao, X. Luo, C. Xing and D. Yin, J. Catal., 2016, 335, 62–71 CrossRef CAS.
- M. C. M. van Oers, L. K. E. A. Abdelmohsen, F. P. J. T. Rutjes and J. C. M. van Hest, Chem. Commun., 2014, 50, 4040–4043 RSC.
- M. C. M. van Oers, W. S. Veldmate, J. C. M. van Hest and F. P. J. T. Rutjes, Polym. Chem., 2015, 6, 5358–5361 RSC.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2013, 52, 2906–2910 CrossRef CAS PubMed.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- P. J. M. Stals, C.-Y. Cheng, L. van Beek, A. C. Wauters, A. R. A. Palmans, S. Han and E. W. Meijer, Chem. Sci., 2016, 7, 2011–2015 RSC.
- E. Karjalainen, D. F. Izquierdo, V. Martí-Centelles, S. V. Luis, H. Tenhu and E. García-Verdugo, Polym. Chem., 2014, 5, 1437–1446 RSC.
- A. Lu, T. P. Smart, T. H. Epps, D. A. Longbottom and R. K. O’Reilly, Macromolecules, 2011, 44, 7233–7241 CrossRef CAS PubMed.
- M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. Van Hest, Curr. Opin. Biotechnol., 2014, 28, 10–16 CrossRef CAS PubMed.
- B. J. Cohen, M. A. Kraus and A. Patchornik, J. Am. Chem. Soc., 1981, 103, 7620–7629 CrossRef CAS.
- B. Voit, Angew. Chem., Int. Ed., 2006, 45, 4238–4240 CrossRef CAS PubMed.
- F. Gelman, J. Blum and D. Avnir, Angew. Chem., Int. Ed., 2001, 40, 3647–3649 CrossRef CAS PubMed.
- B. Helms, S. J. Guillaudeu, Y. Xie, M. McMurdo, C. J. Hawker and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2005, 44, 6384–6387 CrossRef CAS PubMed.
- K. E. Price, B. P. Mason, A. R. Bogdan, S. J. Broadwater, J. L. Steinbacher and D. T. McQuade, J. Am. Chem. Soc., 2006, 128, 10376–10377 CrossRef CAS PubMed.
- B. P. Mason, A. R. Bogdan, A. Goswami and D. T. McQuade, Org. Lett., 2007, 9, 3449–3451 CrossRef CAS PubMed.
- S. L. Poe, M. Kobašlija and D. T. McQuade, J. Am. Chem. Soc., 2006, 128, 15586–15587 CrossRef CAS PubMed.
- D. A. Evans and D. Seidel, J. Am. Chem. Soc., 2005, 127, 9958–9959 CrossRef CAS PubMed.
- S. L. Poe, M. Kobašlija and D. T. McQuade, J. Am. Chem. Soc., 2007, 129, 9216–9221 CrossRef CAS PubMed.
- N. T. S. Phan, C. S. Gill, J. V. Nguyen, Z. J. Zhang and C. W. Jones, Angew. Chem., Int. Ed., 2006, 45, 2209–2212 CrossRef CAS PubMed.
- L.-C. Lee, J. Lu, M. Weck and C. W. Jones, ACS Catal., 2016, 6, 784–787 CrossRef CAS.
- J. Lu, J. Dimroth and M. Weck, J. Am. Chem. Soc., 2015, 137, 12984–12989 CrossRef CAS PubMed.
- Y. Chi, S. T. Scroggins and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 6322–6323 CrossRef CAS PubMed.
- S. Itsuno, Y. Sakurai, K. Ito, T. Maruyama, S. Nakahama and J. M. J. Frechet, J. Org. Chem., 1990, 55, 304–310 CrossRef CAS.
- S. Lundgren, S. Lutsenko, C. Johnsson and C. Moberg, Org. Lett., 2003, 5, 3663–3665 CrossRef CAS PubMed.
- S. V. Luis, M. I. Burguete and B. Altava, Polymer Supported Organocatalysts, in The Power of Functional Resins in Organic Synthesis, ed. F. Albericio and J. Tulla-Puche, Wiley-WCH, Weinheim, 2008 Search PubMed.
- R. P. Jumde, A. Di Pietro, A. Manariti and A. Mandoli, Chem. – Asian J., 2015, 10, 397–404 CrossRef CAS PubMed.
- R. P. Jumde and A. Mandoli, ACS Catal., 2016, 6, 4281–4285 CrossRef CAS.
- T. Mayer-Gall, J.-W. Lee, K. Opwis, B. List and J. S. Gutmann, ChemCatChem, 2016, 8, 1428–1436 CrossRef CAS.
- J.-W. Lee, T. Mayer-Gall, K. Opwis, C. E. Song, J. S. Gutmann and B. List, Science, 2013, 341, 1225–1229 CrossRef CAS PubMed.
- V. Sans, F. Gelat, N. Karbass, M. I. Burguete, E. García-Verdugo and S. V. Luis, Adv. Synth. Catal., 2010, 352, 3013–3021 CrossRef CAS.
- L. Clot-Almenara, C. Rodríguez-Escrich, L. Osorio-Planes and M. A. Pericàs, ACS Catal., 2016, 6, 7647–7651 CrossRef CAS.
- K. R. Corbin and S. H. Bergens, Organometallics, 2007, 26, 1571–1574 CrossRef.
- R. Drake, D. C. Sherrington and S. J. Thomson, J. Chem. Soc., Perkin Trans. 1, 2002, 1523–1534 RSC.
- J. Akai, S. Watanabe, K. Michikawa and T. Harada, Org. Lett., 2017, 19, 3632–3635 CrossRef CAS PubMed.
- F. J. L. Heutz and P. C. J. Kamer, Dalton Trans., 2016, 45, 2116–2123 RSC.
- T. Roy, R. I. Dureshy, N. H. Khan, S. H. R. Abdi and H. C. Bajaj, ChemPlusChem, 2015, 80, 1038–1044 CrossRef CAS.
- D. Bissessar, T. Achard and S. Bellemin-Laponnaz, Adv. Synth. Catal., 2016, 358, 1982–1988 CrossRef CAS.
- B. Altava, M. I. Burguete, S. V. Luis and J. A. Mayoral, Tetrahedron, 1994, 50, 7535–7542 CrossRef CAS.
- T. B. Zid, M. Fadhli, I. Khedher and J. M. Fraile, Microporous Mesoporous Mater., 2017, 332, 38–47 Search PubMed.
- Z.-H. Shi, N.-G. Li, Q.-P. Shi, Y.-P. Tang, H. Tang, M. Z. Shen and J.-A. Duan, Curr. Org. Synth., 2014, 11, 204–243 CrossRef CAS.
- J. Huang, J. Cai, C. M. Li and X. Fu, Inorg. Chem. Commun., 2014, 44, 20–24 CrossRef CAS.
- J. Huang, L. Yuan, J. Cai, X. Chen and D. Qi, Inorg. Chem. Commun., 2016, 65, 4–8 CrossRef CAS.
- L. Zhang, S. Z. Luo and J. P. Cheng, Catal. Sci. Technol., 2011, 1, 507–515 CAS.
- T. Cheng, Q. Zhao, D. Zhang and G. Liu, Green Chem., 2015, 17, 2100–2122 RSC.
- Z. Amara, M. Poliakoff, R. Duque, D. Geier, G. Francio, C. M. Gordon, R. E. Meadows, R. Woodward and W. Leitner, Org. Process Res. Dev., 2016, 20, 1321–1327 CrossRef CAS.
- M. I. Burguete, E. García-Verdugo, I. García-Villar, F. Gelat, P. Licence, S. V. Luis and V. Sans, J. Catal., 2010, 269, 150–160 CrossRef CAS.
- T. Yasukawa, H. Miyamura and S. Kobayashi, Chem. Sci., 2015, 6, 6224–6229 RSC.
- J. Deka, L. Satyanarayana, G. V. Karunakar, P. K. Bhattacharyya and K. K. Bania, Dalton Trans., 2015, 44, 20949–20963 RSC.
- J. Peng, X. Wang, X. Zhang, S. Bai, Y. Zhao, C. Li and Q. Yang, Catal. Sci. Technol., 2015, 5, 666–672 CAS.
- P. Kleman, P. Barbaro and A. Pizzano, Green Chem., 2015, 17, 3826–3836 RSC.
- S. Z. Luo, J. Y. Li, L. Zhang, H. Xu and J. P. Cheng, Chem. – Eur. J., 2008, 14, 1273–1281 CrossRef CAS PubMed.
- R. Zhang, G. Yin, Y. Li, X. Yan and L. Chen, RSC Adv., 2015, 5, 3461–3464 RSC.
- P. Hodge, D. W. L. Sung and P. W. Stratford, J. Chem. Soc., Perkin Trans. 1, 1999, 2335–2342 RSC.
- P. Hodge, R. J. Kell, J. B. Ma and H. Morris, Aust. J. Chem., 1999, 52, 1041–1046 CrossRef CAS.
- M. Angeloni, O. Piermatti, F. Pizzo and L. Vaccaro, Eur. J. Org. Chem., 2014, 1716–1726 CrossRef CAS.
- I. Sagamanova, C. Rodríguez-Escrich, I. G. Molnár, S. Sayalero, R. Gilmour and M. A. Pericàs, ACS Catal., 2015, 5, 6241-–6248 CrossRef CAS.
- L. T. Scott, J. Rebek, L. Ovsyanko and C. L. Sims, J. Am. Chem. Soc., 1977, 99, 625–626 CrossRef CAS.
- F. Gaviña, S. V. Luis, A. M. Costero, M. I. Burguete and J. Rebek, J. Am. Chem. Soc., 1988, 110, 7140–7143 CrossRef.
- F. Svec and J. M. J. Fréchet, Science, 1996, 273, 205–211 CAS.
- V. Chiroli, M. Benaglia, A. Puglisi, R. Porta, R. P. Jumde and A. Mandoli, Green Chem., 2014, 16, 2798–2806 RSC.
- N. Karbass, V. Sans, E. García-Verdugo, M. I. Burguete and S. V. Luis, Chem. Commun., 2006, 3095–3097 RSC.
- P. Llanes, C. Rodríguez-Escrich, S. Sayalero and M. A. Pericàs, Org. Lett., 2016, 18, 6292–6295 CrossRef CAS PubMed.
- K. Takeda, T. Oohara, M. Anada, H. Nambu and S. Hashimoto, Angew. Chem., Int. Ed., 2010, 49, 6979–6983 CrossRef CAS PubMed.
- J. I. García, J. Gracia, C. I. Herrerías, J. A. Mayoral, A. C. Miñana and C. Sáenz, Eur. J. Org. Chem., 2014, 1531–1540 CrossRef.
- W.-X. Zhao, N. Liu, G.-W. Li, D.-L. Chen, A.-A. Zhang, M.-C. Wang and L. Liu, Green Chem., 2015, 17, 2924–2930 RSC.
- M. Chen, Z.-M. Zhang, Z. Yu, H. Qiu, B. Ma, H.-H. Wu and J. Zhang, ACS Catal., 2015, 5, 7488–7492 CrossRef CAS.
- Y. Zhou, G. Yang, C. Lu, J. Nie, Z. Chen and J. Ren, Catal. Commun., 2016, 75, 23–27 CrossRef CAS.
- X. Li, B. Yang, X. Jia, M. Chen and Z. Hu, RSC Adv., 2015, 5, 89149–89156 RSC.
- X. Li, M. Chen, B. Yang, S. Zhang, X. Jia and Z. Hu, RSC Adv., 2014, 4, 43278–43285 RSC.
- R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Coord. Chem. Rev., 2015, 287, 137–156 CrossRef.
- X. Li, S. Zhang, B. Yang, C. Lv, X. Jia and Z. Hu, RSC Adv., 2016, 6, 86531–86539 RSC.
- S. Pagoti, T. Ghosh and J. Dash, ChemistrySelect, 2016, 1, 4386–4391 CrossRef CAS.
- S. Ranjbar, P. Riente, C. Rodríguez-Escrich, J. Yadav, K. Ramineni and M. A. Pericàs, Org. Lett., 2016, 18, 1602–1605 CrossRef CAS PubMed.
- A. Rostami and B. Atashkar, J. Mol. Catal. A: Chem., 2015, 398, 170–176 CrossRef CAS.
- D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS.
- S. Kobayashi, Chem. – Asian J., 2016, 11, 425–436 CrossRef CAS PubMed.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- H. Ishitani, Y. Saito and S. Kobayashi, Top. Organomet. Chem., 2016, 57, 213–248 CrossRef CAS.
- A. M. Hafez, A. E. Taggi and T. Lectka, Chem. – Eur. J., 2002, 8, 4115–4119 CrossRef.
- M. I. Burguete, E. García-Verdugo, M. J. Vicent, S. V. Luis, H. N. Pennemann, N. G. Keyserling and J. Martens, Org. Lett., 2002, 4, 3947–3950 CrossRef CAS PubMed.
- B. Altava, M. I. Burguete, J. M. Fraile, J. I. García, S. V. Luis, J. A. Mayoral and M. J. Vicent, Angew. Chem., Int. Ed., 2000, 39, 1503–1506 CrossRef CAS PubMed.
- B. Altava, M. I. Burguete, E. García-Verdugo, S. V. Luis and M. J. Vicent, Green Chem., 2006, 8, 717–726 RSC.
- S. B. Ötvös, A. Szloszár, I. M. Mándity and F. Fülöp, Adv. Synth. Catal., 2015, 357, 3671–3680 CrossRef.
- K. Takeda, T. Oohara, N. Shimada, H. Nambu and S. Hashimoto, Chem. – Eur. J., 2011, 17, 13992–13998 CrossRef CAS PubMed.
- Z. Zhang, G. Francio and W. Leitner, ChemCatChem, 2015, 7, 1961–1965 CrossRef CAS.
- T. Tsubogo, Y. Yamashita and S. Kobayashi, Chem. – Eur. J., 2012, 18, 13624–13628 CrossRef CAS PubMed.
- P. Lozano, J. M. Bernal, S. Nieto, C. Gomez, E. Garcia-Verdugo and S. V. Luis, Chem. Commun., 2015, 51, 17361–17374 RSC.
- E. Garcia-Verdugo, B. Altava, M. I. Burguete, P. Lozano and S. V. Luis, Green Chem., 2015, 17, 2693–2713 RSC.
- P. Lozano, S. Nieto, J. L. Serrano, J. Perez, G. Sanchez-Gomez, E. Garcia-Verdugo and S. V. Luis, Mini-Rev. Org. Chem., 2017, 14, 65–74 CrossRef CAS.
- V. Sans, N. Karbass, M. I. Burguete, V. Compañ, E. García-Verdugo, S. V. Luis and M. Pawlak, Chem. – Eur. J., 2011, 17, 1894–1906 CrossRef CAS PubMed.
- P. Lozano, E. García-Verdugo, R. Piamtongkama, N. Karbass, T. De Diego, M. I. Burguete, S. V. Luis and J. L. Iborra, Adv. Synth. Catal., 2007, 349, 1077–1084 CrossRef CAS.
- P. Lozano, M. I. Burguete, N. Karbass, K. Montague, T. de Diego and S. V. Luis, Green Chem., 2010, 12, 1803–1810 RSC.
- D. F. Izquierdo, J. M. Bernal, M. I. Burguete, E. Garcia-Verdugo, P. Lozano and S. V. Luis, RSC Adv., 2013, 3, 13123–13126 RSC.
- R. V. Law, D. C. Sherrington and C. E. Snape, Macromolecules, 1996, 29, 6284–6293 CrossRef CAS.
- R. V. Law, D. C. Sherrington and C. E. Snape, Macromolecules, 1997, 30, 2868–2875 CrossRef CAS.
- B. Altava, M. I. Burguete, E. García-Verdugo, S. V. Luis and M. J. Vicent, Tetrahedron, 2001, 57, 8675–8683 CrossRef CAS.
- R. Porta, M. Benaglia, V. Chiroli, F. Coccia and A. Puglisi, Isr. J. Chem., 2014, 54, 381–394 CrossRef CAS.
- M. I. Burguete, A. Cornejo, E. Garcıa-Verdugo, J. Garcıa, M. J. Gil, S. V. Luis, V. Martínez-Merino, J. A. Mayoral and M. Sokolova, Green Chem., 2007, 9, 1091–1096 RSC.
- M. I. Burguete, A. Cornejo, E. Garcia-Verdugo, M. J. Gil, S. V. Luis, J. A. Mayoral, V. Martinez-Merino and M. Sokolova, J. Org. Chem., 2007, 72, 4344–4350 CrossRef CAS PubMed.
- C. Aranda, A. Cornejo, J. M. Fraile, E. García-Verdugo, M. J. Gil, S. V. Luis, J. A. Mayoral, V. Martinez-Merino and Z. Ochoa, Green Chem., 2011, 13, 983–990 RSC.
- H. Wang, N. Li, Z. Yan, J. Zhang and X. Wan, RSC Adv., 2015, 5, 2882–2890 RSC.
- X. Yang, W. Su, D. Liu, H. Wang, J. Shen, C. Da, R. Wang and A. S. C. Chan, Tetrahedron, 2000, 56, 3511–3516 CrossRef CAS.
- H. Sellner, C. Faber, P. B. Rheiner and D. Seebach, Chem. – Eur. J., 2006, 20, 3692–3705 Search PubMed.
- S. Herres, P. Hesemann and J. J. E. Moreau, Eur. J. Org. Chem., 2003, 99–105 CrossRef CAS.
- D. W. L. Sung, P. Hodge and P. W. Stratford, J. Chem. Soc., Perkin Trans. 1, 1999, 1463–1472 RSC.
- K. Kamahori, S. Tada, K. Lto and S. Itsuno, Tetrahedron: Asymmetry, 1995, 6, 2547–2555 CrossRef CAS.
- J. Wu, F. Wang, Y. Ma, X. Cui, L. Cun, J. Zhu, J. Deng and B. Yu, Chem. Commun., 2006, 1766–1768 RSC.
- S. Itsuno, Y. Hashimoto and N. Haraguchi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3037–3044 CrossRef CAS.
- J. M. Frechet, E. Bald and P. Lecavalier, J. Org. Chem., 1986, 51, 3462–3467 CrossRef CAS.
- J.-B. Hu, G. Zhao, G.-S. Yang and Z.-D. Ding, J. Org. Chem., 2001, 66, 303–304 CrossRef CAS PubMed.
- J.-B. Hu, G. Zhao and Z.-D. Ding, Angew. Chem., Int. Ed., 2001, 40, 1109–1111 CrossRef CAS.
- S. Itsuno, T. Matsumoto, D. Sato and T. Inoue, J. Org. Chem., 2000, 65, 5879–5881 CrossRef CAS PubMed.
- G. Sundararajan and N. Prabagaran, Org. Lett., 2001, 3, 389–392 CrossRef CAS PubMed.
- K. Nozaki, Y. Itoi, F. Shibahara, E. Shirakawa, T. Ohta, H. Takaya and T. Hiyama, J. Am. Chem. Soc., 1998, 120, 4051–4052 CrossRef CAS.
- S. Kinoshita, F. Shibahara and K. Nozaki, Green Chem., 2005, 7, 256–258 RSC.
- D. Sartor, J. Saffrich and G. Helmchen, Synlett, 1990, 197–198 CrossRef CAS.
- B. Altava, M. I. Burguete, E. Garcia-Verdugo, S. V. Luis, R. V. Salvador and M. J. Vicent, Tetrahedron, 1999, 55, 12897–12906 CrossRef CAS.
- T. Wang, W. Wang, Y. Lyu, K. Xiong, C. Li, H. Zhang, Z. Zhan, Z. Jiang and Y. Ding, Chin. J. Catal., 2017, 38, 691–698 CrossRef CAS.
- Z. Tang, H. Iida, H.-Y. Hu and E. Yashima, ACS Macro Lett., 2012, 1, 261–265 CrossRef CAS.
- T. Yamamoto and M. Suginome, Angew. Chem., Int. Ed., 2009, 48, 539–542 CrossRef CAS PubMed.
- T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899–7901 CrossRef CAS PubMed.
- Y. Yoshinaga, T. Yamamoto and M. Suginome, ACS Macro Lett., 2017, 6, 705–710 CrossRef CAS.
- Y. Arakawa, N. Haraguchi and S. Itsuno, Angew. Chem., Int. Ed., 2008, 47, 8232–8235 CrossRef CAS PubMed.
- S. Itsuno, D. K. Paul, M. A. Salam and N. Haraguchi, J. Am. Chem. Soc., 2010, 132, 2864–2865 CrossRef CAS PubMed.
- M. M. Parvez, N. Haraguchi and S. Itsuno, Org. Biomol. Chem., 2012, 10, 2870–2877 CAS.
- N. Haraguchi, P. Ahamed, M. Parvez and S. Itsuno, Molecules, 2012, 17, 7569–7583 CrossRef CAS PubMed.
- P. Ahamed, M. A. Haque, M. Ishimoto, M. M. Parvez, N. Haraguchi and S. Itsuno, Tetrahedron, 2013, 69, 3978–3983 CrossRef CAS.
- M. M. Parvez, N. Haraguchi and S. Itsuno, Macromolecules, 2014, 47, 1922–1928 CrossRef CAS.
- M. R. Islam, P. Ahamed, N. Haraguchi and S. Itsuno, Tetrahedron: Asymmetry, 2014, 25, 1309–1315 CrossRef CAS.
- M. M. Hassan, N. Haraguchi and S. Itsuno, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 621–627 CrossRef CAS.
- S. Takata, Y. Endo, M. Shahid Ullah and S. Itsuno, RSC Adv., 2016, 6, 72300–72305 RSC.
- M. S. Ullah and S. Itsuno, Mol. Catal., 2017, 438, 239–244 CrossRef CAS.
- N. Haraguchi, H. Kiyono, Y. Takemura and S. Itsuno, Chem. Commun., 2012, 48, 4011–4013 RSC.
- S. Itsuno, T. Oonami, N. Takenaka and N. Haraguchi, Adv. Synth. Catal., 2015, 357, 3995–4002 CrossRef CAS.
- N. Haraguchi, T. L. Nguyen and S. Itsuno, ChemCatChem, 2017, 9, 3786–3794 CrossRef CAS.
- X. Li, C. Lv, X. Jia, M. Cheng, K. Wang and Z. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 827–835 CAS.
- Y. Zhang, R. Tan, M. Gao, P. Hao and D. Yin, Green Chem., 2017, 19, 1182–1193 RSC.
- Y. Ribourdouille, G. D. Engel, M. Richard-Plouetbc and L. H. Gade, Chem. Commun., 2003, 1228–1229 RSC.
- F. Zhang, Y. Li, Z.-W. Li, Y.-M. He, S.-F. Zhu, Q.-H. Fan and Q.-L. Zhou, Chem. Commun., 2008, 6048–6050 RSC.
- W.-J. Tang, Y.-Y. Huang, Y.-M. He and Q.-H. Fan, Tetrahedron: Asymmetry, 2006, 17, 536–543 CrossRef CAS.
- B. Ma, T. Miao, Y. Sun, Y. He, J. Liu, Y. Feng, H. Chenn and Q.-H. Fan, Chem. – Eur. J., 2014, 20, 9969–9978 CrossRef CAS PubMed.
- Y. Chen, R. Tan, Y. Zhang, G. Zhao and D. Yin, ChemCatChem, 2015, 7, 4066–4075 CrossRef CAS.
- A. Gissibl, C. Padie, M. Hager, F. Jaroschik, R. Rasappan, E. Cuevas-Yañez, C.-O. Turrin, A.-M. Caminade, J.-P. Majoral and O. Reiser, Org. Lett., 2007, 9, 2895–2898 CrossRef CAS PubMed.
- A. Gissibl, M. G. Finn and O. Reiser, Org. Lett., 2005, 7, 2325–2328 CrossRef CAS PubMed.
- R. Rasappan, M. Hager, A. Gissibl and O. Reiser, Org. Lett., 2006, 8, 6099–6102 CrossRef CAS PubMed.
- L. Garcia, A. Roglans, R. Laurent, J. P. Majoral, A. Pla-Quintana and A. M. Caminade, Chem. Commun., 2012, 48, 9248–9250 RSC.
- J. Escorihuela, M. I. Burguete and S. V. Luis, Chem. Soc. Rev., 2013, 42, 5595–5617 RSC.
- N. N. Reed, T. J. Dickerson, G. E. Boldt and K. D. Janda, J. Org. Chem., 2005, 70, 1728–1731 CrossRef CAS PubMed.
- R. A. García, R. Van Grieken, J. Iglesias, D. C. Sherrington and C. L. Gibson, Chirality, 2010, 22, 675–683 Search PubMed.
- M. I. Burguete, M. Collado, J. Escorihuela and S. V. Luis, Angew. Chem., Int. Ed., 2007, 46, 9002–9005 CrossRef CAS PubMed.
- J. Escorihuela, B. Altava, M. I. Burguete and S. V. Luis, RSC Adv., 2015, 5, 14653–14662 RSC.
- F. Keller, H. Weinmann and V. Schurig, Chem. Ber., 1997, 130, 879–885 CrossRef CAS.
- B. Altava, M. I. Burguete, J. M. Fraile, J. I. Garcia, S. V. Luis, J. A. Mayoral, A. J. Royo and M. J. Vicent, Tetrahedron: Asymmetry, 1997, 8, 2561–2570 CrossRef CAS.
- B. Altava, M. I. Burguete, E. Garcıa-Verdugo, S. V. Luis, J. F. Miravet and M. J. Vicent, Tetrahedron: Asymmetry, 2000, 11, 4885–4893 CrossRef CAS.
- C. Bleschke, J. Schmidt, D. S. Kundu, S. Blechert and A. Thomas, Adv. Synth. Catal., 2011, 353, 3101–3106 CrossRef CAS.
Footnote |
| † TOF from literature data or calculated from data reported as: (mol of substrate/mol catalyst) × (yield (%)/100) × (1/time (h)). |
| This journal is © The Royal Society of Chemistry 2018 |