
Shedding light on tau protein aggregation: the progress in developing highly selective fluorophores
Peter
Verwilst
 a,
Hyeong Seok
Kim
a,
Soobin
Kim
a,
Chulhun
Kang
*b and
Jong Seung
Kim
*a
a,
Hyeong Seok
Kim
a,
Soobin
Kim
a,
Chulhun
Kang
*b and
Jong Seung
Kim
*a
aDepartment of Chemistry, Korea University, Seoul 02841, Korea. E-mail: jongskim@korea.ac.kr
bThe School of East-West Medical Science, Kyung Hee University, Yongin 17104, Korea. E-mail: kangch@khu.ac.kr
First published on 27th February 2018
Abstract
Historically, in Alzheimer's disease research, a lot of attention has been paid to the development of highly selective fluorophores for beta amyloid plaques. With a shift in the understanding of the disease and the importance of a network of cross-talk interactions, the development of small-molecule fluorescent dyes with high selectivity for (hyperphosphorylated) tau protein aggregates in neurofibrillary tangles has been gaining increasing attention. Fluorescent dyes for the selective labelling of tau aggregates in histological AD brain sections have been described, spanning the entire visible range of the electromagnetic spectrum. Despite the relatively early stages of the development of the field, a large diversity in probe architectures has been reported. Importantly, a handful of near-infrared-emissive dyes have been described as well, and some of these have exhibited good pharmacological profiles, with a significant blood–brain-barrier permeability, and a demonstrated ability to label tau tangles in vivo in small-animal models of Alzheimer's disease and other tauopathies. The developments summarized in the current work are expected to aid the unravelling of the diverse set of players in the etiology of Alzheimer's disease. In this tutorial review, we seek to provide the reader with an overview of the most important recent developments and hope to provide some guidelines for the design of future probes.
 Left to right, top: Peter Verwilst, Soobin Kim and Heong Seok Kim; bottom: Jong Seung Kim and Chulhun Kang | Peter Verwilst received his PhD from the University of Leuven, Belgium under the supervision of Prof. Wim Dehaen and Prof. Wim M. De Borggraeve in 2011. After a postdoctoral position with Dr Nathan D. McClenaghan at the Univeristé Bordeaux 1/CNRS, France, he joined the lab of Prof. Jong Seung Kim as a research professor. He has co-authored 35 scientific publications to date and his research interests include theranostic targeted drug delivery and bio-(in)organic sensors. |
Hyeong Seok Kim received his Bachelor's degree from the Department of Chemistry at the Catholic University of Korea in 2015. He has since worked toward his PhD degree under the guidance of Prof. Jong Seung Kim at Korea university. His current research interests are fluorescent probes for the detection of biomolecules as well as drug delivery systems for cancer therapy. |
Soobin Kim received her Bachelor's degree from the Department of Chemistry at Korea University in 2017. She is currently working towards a master's degree under the guidance of Prof. Jong Seung Kim. Her current research interests are focused on fluorescent probes for Alzheimer's disease as well as cancer therapy. |
Chulhun Kang received an MS Degree in Organic Chemistry from the Department of Chemistry at Seoul National University and a PhD in Biochemistry from the Department of Biochemistry and Biophysics at Iowa State University. Since 1997, he has been a faculty member at Kyung Hee University, where he is currently a professor in the Department of Medical Science. His research record includes 77 scientific publications and 10 domestic and international patents in the fields of organic chemistry, protein chemistry, and biology. |
Jong Seung Kim received his PhD from the Department of Chemistry and Biochemistry at Texas Tech University in 1993. He had one year of research experience at the University of Houston as a post-doc. Currently he is a full professor in the Department of Chemistry at Korea University in Seoul. To date, his research records 394 scientific publications (H-index 75) and 80 domestic and international patents. He has been a member of the Korean Academy of Science and Technology since 2014. |
Key learning points1. What is the current understanding and what are current hypotheses related to the role of tau protein in neurodegenerative diseases?2. What are the structural characteristics of tau selective dyes? 3. Limitations and future directions |
Introduction to tau protein biology and biochemistry
1. Tau protein and physiological function
Tau proteins exist in 6 isoforms in human tissues, originating from the alternative splicing of the MAPT (microtubule-associated tau protein) gene. As shown in Fig. 1, the isoforms range from 352 to 441 amino acids and four distinct regions are identified as the N-terminal projection domain (N), the proline rich domain (PRD), the repeat region (R) and the C-terminal end. Tau protein isoforms are differentiated depending on the number of N-terminal inserts (0, 1 or 2) and the number of repeats in the repeat region (3 or 4), and hence a common naming convention for tau proteins is based on the difference in the N and R length, with 0N3R for the shortest and 2N4R for the longest isoform. Interestingly, the expression of these isoforms is age-dependent; whereas the 0N3R isoform is the only isoform expressed in foetal brains, the proportion of 3R and 4R proteins is roughly equal in adult brains. Furthermore, varying expression ratios of tau protein isoforms are found in different brain structures and even within a single neuron the subcellular localization of the isoforms is distinct, implying that each tau isoform might serve different functions.1,2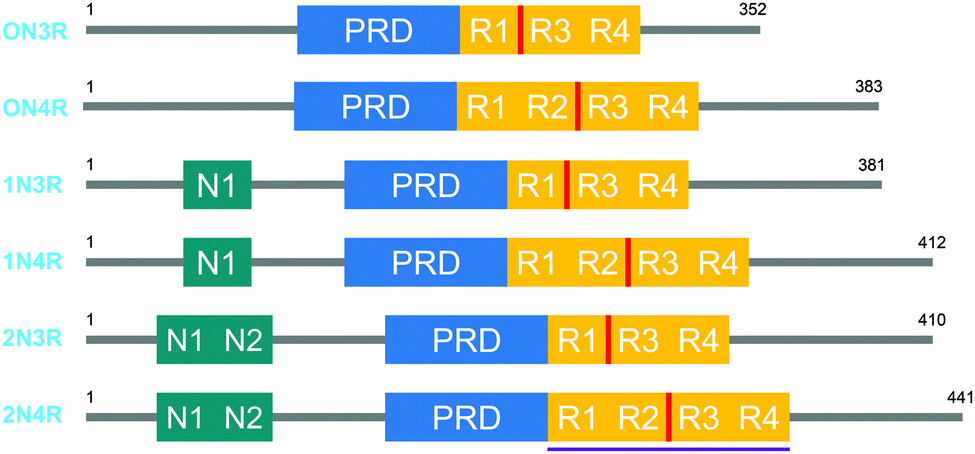 | ||
| Fig. 1 The 6 human tau protein isoforms, resulting from alternative splicing. The location of the crucial PHF6 fragment, and the K18 protein sequence is shown in red and purple, respectively. | ||
As their name suggests, in the axon of neurons, tau proteins are mostly associated with microtubules, the major constituent of the cytoskeleton, composed of a dynamic tubulin polymer. In solution, tau proteins are highly unfolded and disordered, adopting a global hairpin-like structure, where the C-terminal part is sandwiched between the N-terminal tail and the repeat region.3 Upon binding microtubules, the repeat region directly interacts with tubulin and the 4R isoforms exhibit a much stronger affinity than the 3R isoforms. In the microtubule bound state, the C-terminal region is projected away from the microtubule core, potentially governed by coulombic repulsion, contributing to maintaining the proper interstrand distance between microtubules in filaments. However, microtubules are not just a key part of the cell's cytoskeleton; they are also transport highways for vesicles, organelles and proteins. A static interaction between tau and microtubules would significantly interfere with these transportation processes. Recent experiments have shown that the average residence time of tau proteins on microtubules may be as little as 40 ms, even though more than 80% of the available tau proteins will be bound to microtubules at any given moment, demonstrating the highly dynamic nature of this process and revealing why tau protein does not interfere with axonal transport.1
The function of tau protein under physiological conditions is regulated by a plethora of post-translational modifications, with phosphorylation being one of the most important. The 2N4R isoform exhibits up to 85 potential phosphorylation sites (Ser, Thr and Tyr), and with the exception of Thr 50, all observed phosphorylations weaken the interaction with microtubules. Thus, the dynamic phosphorylation by kinases and dephosphorylation by phosphatases allows the cell to have rapid control over the degree of tau proteins bound to microtubules.1,2
Apart from its interaction with microtubules, tau protein also plays important roles in signalling via the proline rich domain, as well as via its post-translational modifications. Intriguingly, the repeat domain of tau proteins has also been shown to exhibit affinity for other macromolecules, including actin (another cytoskeletal component), RNA and DNA. Furthermore, some fractions of tau proteins have also been found in non-axonal compartments, such as the dendritic spines, near synapses and in the nucleus of neurons. Together, this suggests that tau protein plays a much broader role than merely the presumed regulation of microtubule stability. For a more in-depth discussion of the physiological role of tau protein and the implications of non-axonal tau, we refer the reader to recent reviews.1,2
2. Tau protein self-assembles into neurofibrillary tangles (NFTs)
Whereas the existence of so-called neurofibrillary tangles (NFTs) was recognised very early on as one of the major proteinaceous deposits in the brains of Alzheimer's disease (AD) patients, it wasn’t until the 1960s that the paired helical and straight filaments of NFTs were identified as aggregated tau protein (also known as tau tangles). NFTs were discovered later on not just to be a hallmark of AD, but also of other diseases such as Pick's disease, Down syndrome, frontotemporal dementia and Parkinsonism linked to chromosome 17 and progressive supranuclear palsy and other diseases, which are commonly grouped together as tauopathies. In this tutorial review, we will focus the discussion on NFTs in AD, as the vast majority of the fluorescent probes described below have been synthesized and applied in the framework of AD research.In Alzheimer's disease, the other, more abundant, protein deposits forming extracellular senile plaques consist of beta amyloid (Aβ) protein. In the current tutorial review we will focus on the role of tau protein in AD, but as tau protein and Aβ have been demonstrated to exhibit significant cross-talk, as well as exhibiting some common physicochemical properties, we would encourage the reader to consult two excellent reviews on Aβ.4,5
The formation of NFTs is dependent on abnormal post-translational modifications of tau proteins. In particular, tau proteins isolated from NFTs exhibit a greater degree of phosphorylation. Whereas the native protein in the human brain on average bears two phosphate groups, tau in NFTs has been found to have a degree of phosphorylation of up to 8 residues. Although the exact mechanism for this hyperphosphorylation is poorly understood, it is highly likely to originate from a perturbation of phosphoprotein homeostasis by phosphatases and kinases in the affected cells. Indeed PP2A, the most potent phosphatase for tau proteins, has been found to have a significantly reduced activity and/or expression level in AD. Other post-translational modifications such as truncation, N-glycosylation, O-GlcNAcylation and nitration have been found to differ in their extent between soluble and insoluble tau proteins. In particular, truncation is believed to result in an enhanced propensity for self-assembly as it removes the native hairpin conformation; and N-glycosylation has been found to stabilize the tangle state as well as to promote the hyperphosphorylation of neighbouring sites.1,2 It is important to point out that the inherent structural diversity of tau protein, with different isoforms and many different post-translational modifications, complicates studies probing tau protein interactions with their environment either in a physiological or pathological state.
The crucial question in tauopathy is how the hyperphosphorylation and the subsequent self-assembly of tau proteins results in neurodegradation. Current hypotheses either stress a potential gain of toxicity, or a loss of normal function, or a combination of both upon aggregation. Here, we will briefly outline some of the most important observations and their implications for either of these hypotheses.
During development, tau proteins are found throughout the neuron, whereas tau protein is mainly found in the axonal stem in the adult brain. This process may be regulated by development-dependent changes in isoform expression, active transport of tau to the axon, different rates of protein degradation in the soma and the axon, and a diffusional barrier at the axonal base. However, upon hyperphosphorylation, soluble phosphorylated tau proteins, also known as pre-fibrils, are found to relocate to the neuronal body (known as the somatodendritic compartment). Upon aggregation, tau proteins are mainly found in this somatodendritic compartment, with some aggregation along the axis as well. Upon neuronal death, tau protein aggregates from these somatodendritic compartments are left behind under the form of flame-like structures, known as ghost tangles. This somatodendritic accumulation of tau tangles has prompted researchers to theorise that the neuronal depletion of functional tau protein results in destabilized microtubules, significantly hindering axonal transport. As axon structures can be several cm long, the continuous transport of organelles, ribosomes, vesicles and other vital cellular maintenance components is of primordial importance to maintain neuronal health. The interruption of this transport system would explain the degraded neuronal synapses observed in AD.
However, some evidence against this tau tangle-induced transport inhibition exists as well. In tau knockout animal models, neuronal transport appears to not be hindered even without tau protein, suggesting that other redundant microtubule stabilisation proteins may take over the role of tau protein in these animals. Another hypothesis proposes that the accumulation of tau protein tangles induces severe interferences with vital cell processes, yet, neurons in AD are known to function normally for up to a decade, despite significant cellular tau deposits.1,2 Furthermore, only a minority of dead neurons have been found to exhibit tau deposits. In analogy with Aβ,5 it has therefore been proposed that the real toxic species in tauopathies is not the tangled structure, but the soluble oligomeric pre-fibril.1,6 According to this hypothesis the self-assembly into tau tangles would represent a cellular defence mechanism, in order to sequester the toxic oligomeric species as less toxic tangles, until this defence mechanism fails and ultimately cell death follows. Given the putative involvement of tau proteins in other essential functions, for example cell signalling and their low yet potentially crucial expression in other subcellular compartments, it is not unimaginable that oligomers at these locations may in fact be major contributors to tau toxicity as well.
3. Structure of tau protein aggregates
Tau proteins self-aggregate to form loosely intertwined paired helical and more tightly wrapped straight filaments, as identified by electron microscopy, with a diameter of 10–20 nm and a helical periodicity of 80 nm for the paired helical filaments (Fig. 2, 3a and b). The tau tangles consist of a core of stacked beta sheets, likely originating from the repeat domain of tau protein. Surrounding this well-organized dense core, the other protein domains form a flexible polyelectrolyte brush, known as a fuzzy coat. In ghost tangles in the remnant of neurons however, the fuzzy coat has been largely lost. As a result of the inherent flexibility of these polyelectrolyte coats, as well as the polymorphous nature of tau proteins, structural characterisation is significantly hampered, and no structures have been determined at an atomic level.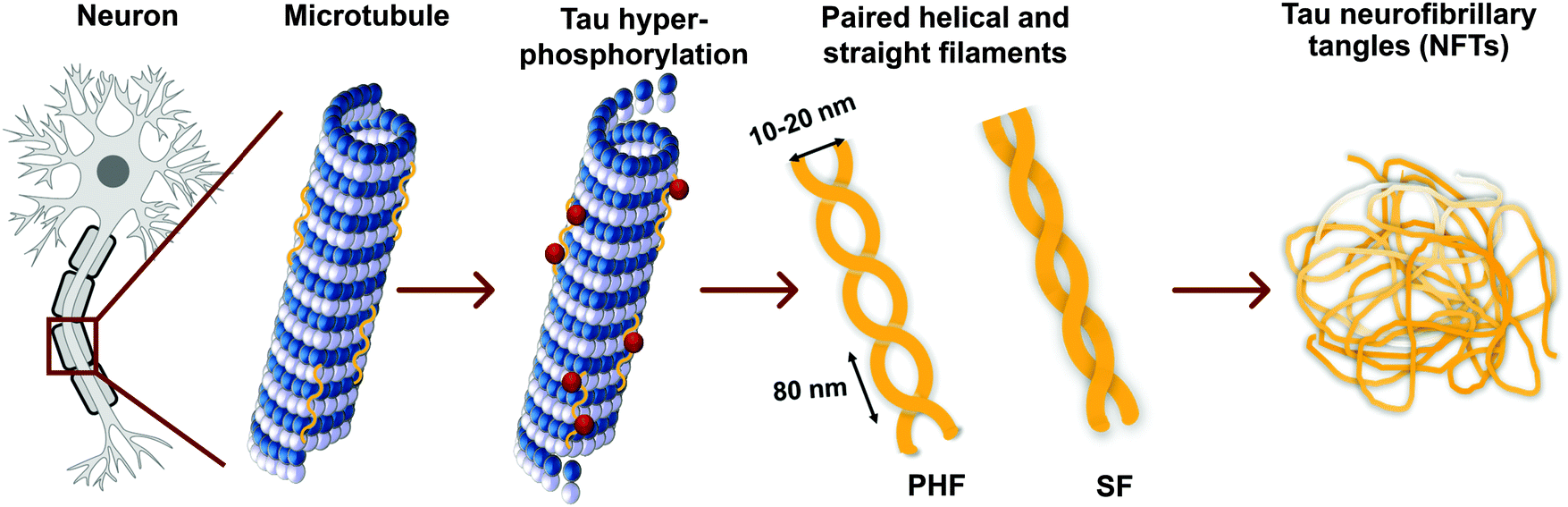 | ||
| Fig. 2 Tau hyperphosphorylation leads to the formation of paired helical and straight filaments resulting in NTFs. See Fig. 3a and b for a TEM image of a paired helical and straight filament, respectively. | ||
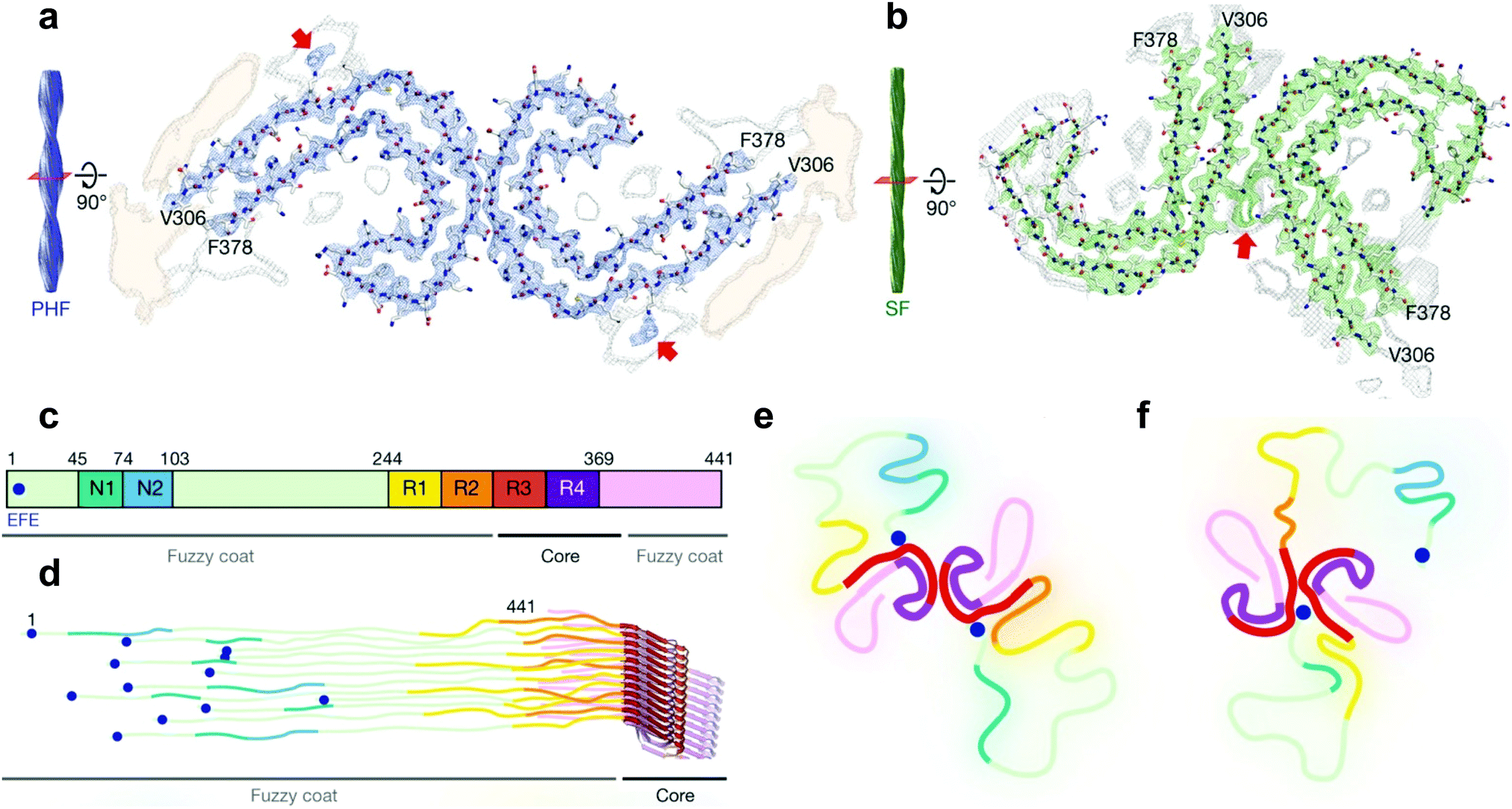 | ||
| Fig. 3 Cryo-EM structural assignments of paired helical and straight filaments. (a and b) Cross section and fitted protein sequence of paired helical filaments (a) and straight filaments (b). (c) 2N4R tau sequence and ultrastructures in tau filaments (7EFE9 is represented by a blue dot). (d) Schematic model of a tau tangle consisting of all 6 tau isoforms. (e and f) Schematic representation of the cross section of paired helical filaments (e) and straight filaments (f) with the hypothesized 7EFE9 “backbiting” interaction, colour scheme following (c), beta sheet region in bold. Reprinted by permission from Macmillan Publishers Ltd: Nature, ref. 7, copyright 2017. | ||
Very recently however, a major breakthrough was disclosed by Fitzpatrick et al. who determined the structure of paired helical and straight filaments, extracted and purified from the brain of an AD patient, at a resolution of 3.4–3.5 Å using cryo-EM,7 an atomic microscopy technique that was awarded the 2017 Nobel Prize in Chemistry. In the work of Fitzpatrick et al., the core of paired helical and straight filaments was determined to consist of two C-shaped protein fragments, with a different lateral contact leading to the two polymorphs. Analysis and modelling of the cryo-EM densities revealed that the β-sheet rich C-shaped cores consisted of R3, R4 and the 10 following C-terminal amino-acids (Fig. 3). Some additional densities around residue 313 were hypothesized to be interactions with the 7EFE9 N-terminal residues (Fig. 3e and f), which is in harmony with the fact that the anti-7EFE9-313VDLSKVTSKC322 antibody is specific to tau tangle structures.7 Virtually all tau tangle-selective fluorescent sensors in this work were developed prior to the determination of this structure and thus did not yet benefit from this knowledge. Novel tau tangle sensors designed using the core model structures are anticipated in the near future.
4. Extracellular tau and its involvement in tau seeding and spreading
One of the most puzzling observations in tauopathies is the fact that different tau pathologies originate in distinct sections of the brain, and exhibit either only 3R (e.g. Pick's disease) or 4R (e.g. progressive supranuclear palsy) or both splicing variants in the aggregated forms (e.g. Alzheimer's disease).1,2 Furthermore, the progression of the diseases in the brain follows a predictable pathway, with different sections becoming affected at different stages of the disease. This observation forms the basis for the Braak staging in AD (see below).8Whereas pathway-specific spreading of tau pathology could be ascribed to an inherent difference in neuronal sensitivity to some of the changes involved during disease progression, the majority of scientists currently support a different mechanism.
Tau protein aggregation in solution has been shown to occur via a seed-based mechanism, where the presence of a small aggregated seed induces protein misfolding in the bulk of the protein solution. This self-catalysed aggregation could explain the presence of either form of tau protein isoform aggregates, with a different type of seed being responsible for either 3R, 4R or mixed 3R/4R pathologies. Current hypotheses build on this observation and confer a prion-type behaviour to tau oligomers.1,2,6 The soluble oligomers, as seeds for tau pathology, would be secreted by afflicted cells through diverse mechanisms, such as via microsomes or synaptic vesicles, or simply as soluble oligomeric proteins. The uptake of these tau oligomers into neighbouring cells would in turn induce tau pathology in those cells.1,2,6
The supplementation of tau seeds to neuronal cultures, or intracranial injection of human AD patients’ brain homogenates into transgenic animals has been shown to induce intraneuronal tauopathy. Furthermore, the observation that tau pathology seems to follow neuronal connectivity networks in AD patients does support the spreading of tau pathology via a prion-type mechanism through synaptic contacts.1,2
It has also been observed that different types of cells excrete small amounts of tau protein under physiological conditions, for example by neurons upon stimulation. This suggests that extracellular tau, apart from acting as a pathological seed when misfolded, could also possess a physiological role, although this function is currently not clear.2
5. Significance of NFT imaging
Finally, whereas over the course of the last decades, increased knowledge of the importance of both tau and Aβ for the progression and neurodegeneration of AD has been gained, their mutual relationship is still shrouded in mystery. For example, Aβ excitotoxicity (cellular damage originating from overstimulation of nerve endings) needs the presence of tau, yet the exact mechanism is currently not fully understood.1,2 The consensus among the majority of researchers is that Aβ precedes tau pathology in AD patients, as clear signs of Aβ-rich plaques are apparent prior to significant amounts of tau pathology in the brains of both AD patients and animal models. The presence and the burden of Aβ-fibril containing plaques in patients do however not correlate well with the progression and clinical manifestation of the disease. Autopsies have even revealed cases with relatively high amounts of senile plaques without any apparent signs of cognitive decline. These facts make Aβ containing plaques a less than ideal biomarker for AD. Several studies by Braak et al. have clearly demonstrated a much more rigorous correlation between the NFT burden and disease progession.8 As such, tau tangles have become an attractive target in the development of clinical diagnostic probes, mainly based on the PET (positron emission tomography) imaging modality. Although there is some overlap between tau probes for fluorescence imaging and PET, the latter one will not be included in the scope of the current manuscript and we direct the readers to a recent review for further information.9As is apparent from the above discussion, the pathogenesis of AD disease is currently still uncertain. It is likely that to understand the true etiology of the disease, a comprehensive study of the ensemble of potential causes and their interplay, such as tau protein, Aβ, ROS, chaperones, mitochondrial dysfunction, metal homeostasis and even genetic factors, would be required. To study this, there is a need for cost-effective, non-invasive, ideally non-disruptive probes to visualize these events at a (sub)cellular and tissue level. In cell biology, pathology and small animal research, the method of choice for this is fluorescence imaging, as this technique combines a relatively low cost with high spatial and temporal control. Despite the challenges, the irrefutable need for the development of specific probes has prompted various researchers to investigate strategies to overcome these limitations, and their strategies as well as design guidelines will be discussed below.
Fluorescent sensors for tau tangles
1. Challenges and key characteristics
Aβ plaques and tau tangles share some striking similarities, for example: both exhibit beta sheet formation and stacking at the core of their structure and represent an environment that is significantly less hydrophilic than the surrounding medium. As we will show below, because of this reason there is a significant overlap between the characteristics and architectures of chemical probes for both these protein aggregates, as well as others, such as α-synuclein contained in Lewy bodies in Parkinson's disease.Depending on the application, the challenges and the desired characteristics differ, but a high affinity for tau in the presence of competitive species such as Aβ, combined with low cyto- and systemic toxicity is a condicio sine qua non for in vitro and in vivo applications. In the case of in vivo imaging, the emission wavelength should be shifted to the red and near-infrared (NIR) regions of the electromagnetic spectrum, as relatively high degrees of tissue penetration can be reached in this region (known as the first NIR window). The combination of low background autofluorescence, low degrees of light scattering and minimized tissue absorption all contribute to the possibility of visualizing events in intact living animals, thus significantly lowering the risk of perturbations of the natural state of events and analytes of interest. Here, an extra important feature is efficient diffusion or translocation across the blood–brain barrier (BBB), a tightly controlled boundary between circulating blood and the extracellular fluid of the central nervous system.
In the case of fluorescent agents for labelling tau tangles for histological purposes or fluorescently imaging cells in vitro, the wavelength requirements are less stringent, but would ideally be in a region devoid of significant background fluorescence. Particularly applied to the brain, the autofluorescence from lipofuscin, endogenous pigment granules with a peak emission in the yellow region, accumulating in lysosomes as a result of aging, suggests that this wavelength region would better be avoided. Furthermore, due to the cytotoxicity of ultraviolet light, the excitation of compounds would ideally be in the visible or NIR region (for example via two photon excitation).
2. Non-selective dyes
One of the major differences between the native tau protein and the aggregated tau species is the secondary protein structure. Upon aggregation, the native random coil protein is transformed into stacked β-sheets. Therefore, it was recognized early on that some of the dyes that exhibited clear staining of Aβ plaques also identified NFTs and other amyloid protein aggregates. Initially, staining of amyloids in brain sections for pathological post-mortem analysis was carried out using traditional tissue staining dyes such as the triarylmethane dyes, for example: Crystal Violet, Hoffman's Violet and Methyl green (Fig. 4). Other dyes used for this purpose are the phenothiazines, in particular Toluidine Blue (Fig. 4). These dyes display a colour change when bound to amyloid proteins, as compared to their staining of the surrounding tissue, allowing their visualization.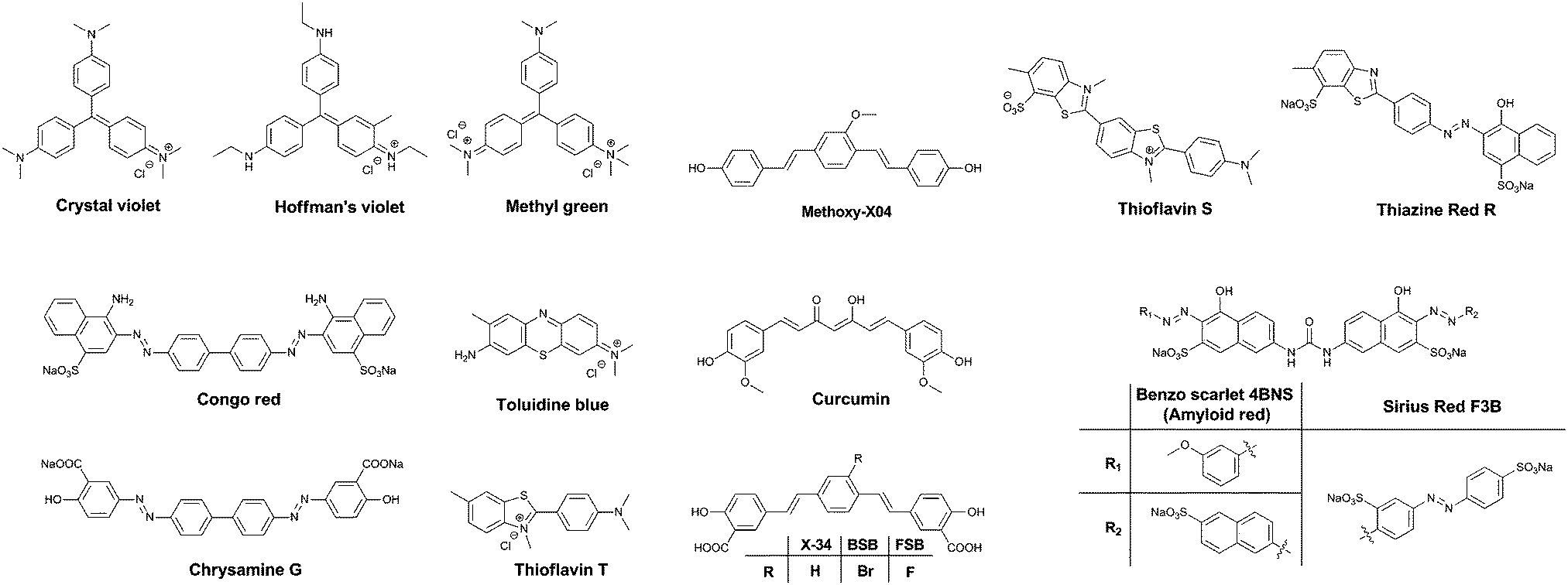 | ||
| Fig. 4 Chemical structures of the non-selective dyes. | ||
Soon, Congo Red (Fig. 4) became the dye of choice for the labelling of amyloid-type protein aggregates. Under polarized light, Congo Red displays birefringence with a greenish colour when bound to diverse types of amyloid deposits, allowing for a higher degree of certainty in assigning the identity of stained spots in tissues. Other dyes showing this type of behaviour are Sirius Red F3B and Benzo Scarlet 4BNS (Fig. 4, also known as Amyloid Red), which are often preferred as they show more intense staining of amyloid deposits. Chrysamine G and Thiazine Red R (Fig. 4), structural analogues to Congo Red, have been employed as well. Sharing a π-conjugated backbone of similar length, curcumin (Fig. 4), a natural dye extracted from turmeric, has been shown to fluorescently stain various protein aggregates too.
Thioflavin T and S (ThT and ThS, Fig. 4) are prototypical molecular rotors, showing a dramatic increase in fluorescence when their rotational freedom is restricted. This restriction is apparent in high viscosity solvents, as well as confined spaces within proteins. Owing to their flat structure, these dyes are particularly well suited to bind to β-sheet rich proteins. Therefore, they show a very strong affinity for both Aβ-rich senile plaques and tau protein aggregate-containing NFTs. In fact, thioflavin staining, demonstrating senile plaques and NFTs, has become the gold standard in the post-mortem diagnosis of AD.
Drawing upon the experiences using the first generation of cross-beta sheet binding dyes, a series of dyes were developed to increase the fluorescence brightness and to optimize the pharmacokinetic profile of the dyes. The first step in the development of tau protein specific compounds was represented by the related dyes X-34,10BSB11 and the more fluorescent analogue FSB (Fig. 4).12 These compounds represent a more lipophilic version of curcumin/Chrysamine G, in order to increase the potential for BBB penetration. The compounds were clearly able to stain NFTs in histological sections from mice and AD patients, as well as label amyloids in vivo. The substitution of a bromine atom with a fluorine resulted in a nearly 2-fold enhancement of the fluorescence, and as a result higher signal over background ratios (SBRs) in histological samples. However, neither of these compounds is selective for tau protein, rather staining all of the proteinaceous aggregates in the brain.
Klunk et al. used a similar approach with Methoxy-X04 (Fig. 4), where a further deviation from the base structure, by removing all carboxylic acid groups, resulted in efficient BBB penetration and in vivo staining of the brain amyloid content.13
3. Cross β-sheet binders
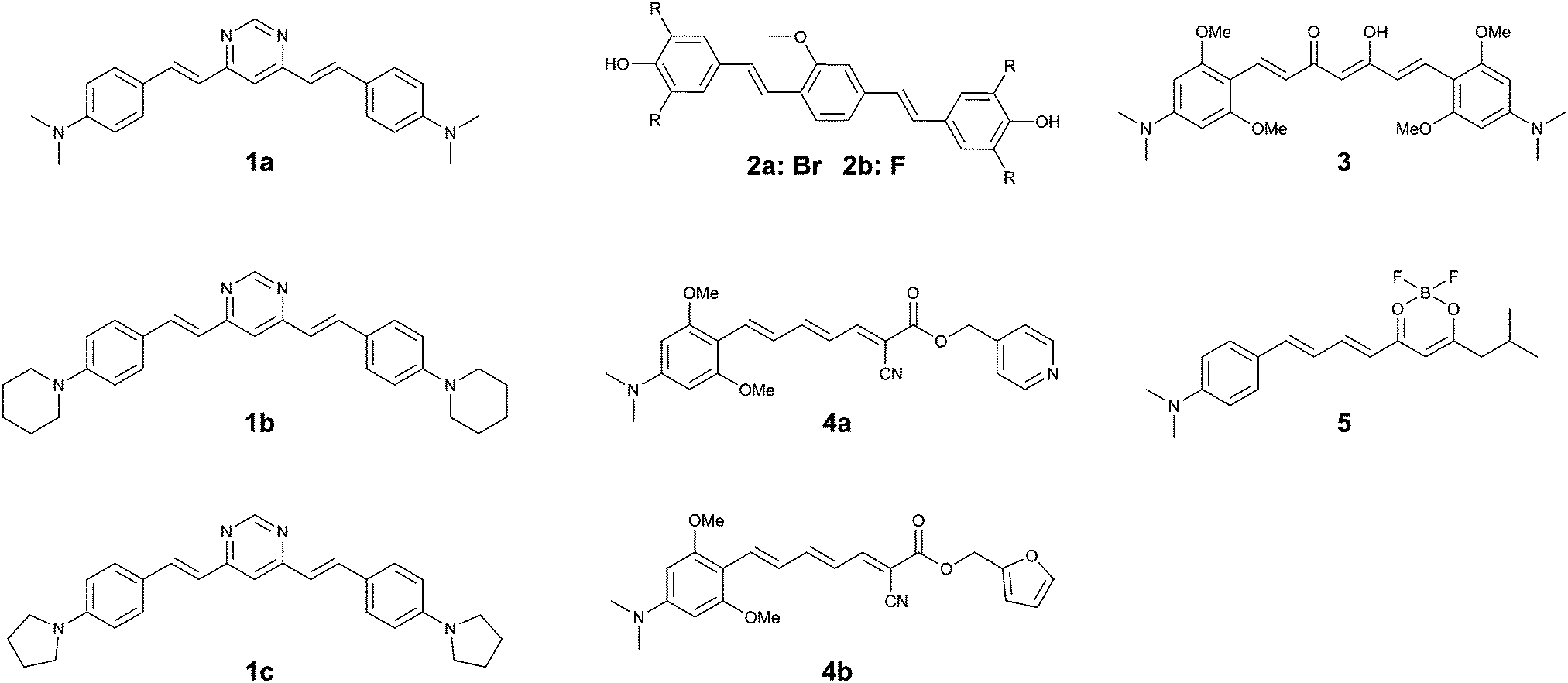 | ||
| Fig. 5 Chemical structures of curcumin- or Congo Red-based fluorophores. | ||
The same group returned to the curcumin-analogue Methoxy-X04, where a series of analogues revealed that a bis-dibrominated (2a) and bis-difluorinated (2b) analogue (Fig. 5) exhibited an affinity that was 24- and 3.5-fold higher for tau tangles than Aβ plaques, although the overall affinity was lower than the previous series of compounds.152a showed efficient staining in post mortem brain slices of AD patients, although some more diffuse staining of Aβ plaques could be observed as well.
The substitution of a dimethylaniline structure on the curcumin scaffold served as a lead compound in the development of fluorophores with an increase in fluorescence intensity upon incubation with aggregated K18 protein, encompassing the 4-repeat domain (Fig. 1).16 Among the compounds studied, 3 (Fig. 5) showed a fluorescence turn-on of 22.9-fold in solution and was able to stain aggregates of the tau 2N4R-GFP (green fluorescent protein) conjugate in vitro. As a result of the electron-donating dimethylaniline groups, and the resulting increased D–A–D intramolecular charge transfer (ICT) efficiency, the emission wavelength of 3 was red-shifted to 620 nm. The β-diketone acceptor core however is likely to preclude efficient BBB penetration, as this is believed to be one of the major contributors to the poor BBB penetrability of the parent compound, curcumin. Neither in vivo or tissue staining experiments, nor competition experiments with Aβ were performed.
Using the same donor pendant, the same group reported a series of D–A dyes, showing an impressive 49- to 108-fold increase in fluorescence upon binding (4a–b) (Fig. 5), while also maintaining a fluorescence intensity selectivity of 5.7- to 3.8-fold higher for tau protein than Aβ, for 4a and 4b, respectively. Whereas for both compounds the binding affinity is higher for Aβ than for tau fibrils, the significantly higher fluorescence quantum yield in the presence of tau protein relative to Aβ is able to overcome this limitation (5.1% and 3.8%; 16.5% and 2.5% for tau and Aβ for 4a and 4b, respectively).17 Due to the strong D–A strength in the extended π-conjugated system, the fluorescence emission wavelength approaches the NIR window cut-off (>650 nm). Both probes demonstrated a clear correlation between the solvent polarity and the fluorescence intensity, typical for molecular rotors. Whereas the probes showed a clear co-localization with GFP-labelled tau aggregates in vitro, staining of human brain sections did reveal the absence of fluorescence in normal brains, whereas in the case of AD pathology bright fluorescence was observed. While 4a–b did label some of the typical tau protein aggregate structures in the slices, the fluorescence of 4a and 4b did not fully overlap with the emission from a fluorescently labelled antibody for hyperphosphorylated tau protein.
Finally, by aromatizing the β-diketonate of the curcumin structure with difluoroboron, a more lipophilic and brighter fluorescent probe (5) (Fig. 5) was constructed, following a strategy that has previously been proven to be successful in the CRANAD series Aβ-specific fluorophores. The D–A curcumin-like compound 5 combines a high turn-on ratio in the presence of tau fibrils with a 14-fold selectivity for tau over Aβ and an emission maximum in the NIR at 660 nm. The molecule was demonstrated to act as a molecular rotor, and showed an excellent overlap with a fluorescently labelled antibody for hyperphosphorylated tau protein in human AD brain sections, whilst only showing a relatively low staining of Aβ plaques at the core of the most dense plaques.18
The group of Jeff Kuret studied a large library of small molecules and determined their affinity for tau protein aggregates, as well as Aβ and α-synuclein filaments using a ThS competition assay. One of the 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 455-member library (6) (Fig. 6) showed very strong affinity for tau protein aggregates whilst maintaining a very high selectivity as compared to α-synuclein, and also showing a 1.8-fold higher binding affinity as compared to Aβ.19 Structurally speaking, it is remarkable to see that the inclusion of a diazo linker between the donor and acceptor part of a thioflavin-like scaffold is able to shift the selectivity towards tau protein.
455-member library (6) (Fig. 6) showed very strong affinity for tau protein aggregates whilst maintaining a very high selectivity as compared to α-synuclein, and also showing a 1.8-fold higher binding affinity as compared to Aβ.19 Structurally speaking, it is remarkable to see that the inclusion of a diazo linker between the donor and acceptor part of a thioflavin-like scaffold is able to shift the selectivity towards tau protein.
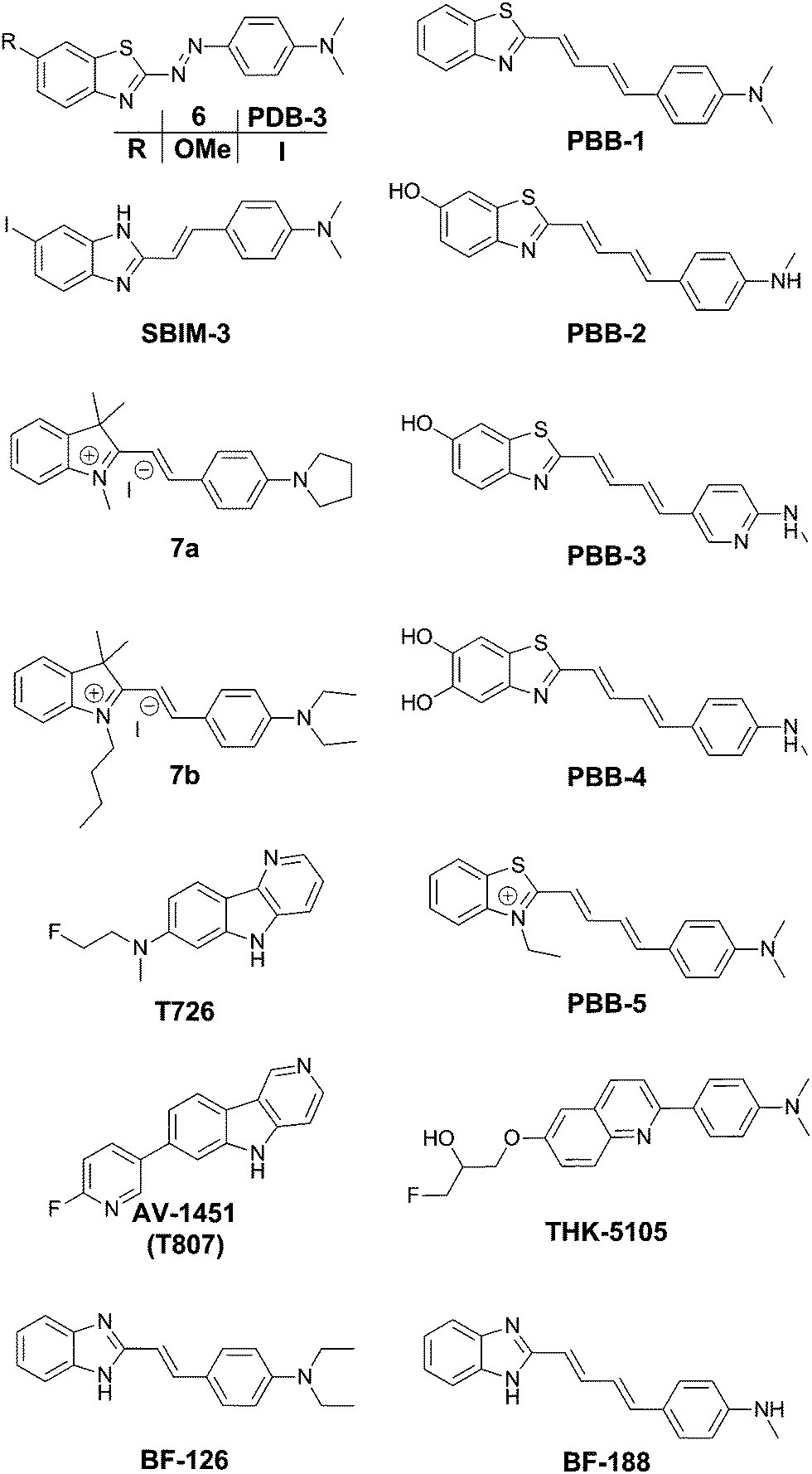 | ||
| Fig. 6 Chemical structures of thioflavin-based fluorophores. | ||
In the quest for an efficient PET probe, Matsumura et al. followed up by replacing the methoxy group of 6 with an iodine, resulting in PDB-3 (Fig. 6).20 Intriguingly PDB-3 demonstrated a significantly enhanced selectivity for tau protein over Aβ (17.2 fold in a ThS competition assay), while the affinity for Aβ was independent of the N-donor substituents (NH2, NHMe or NMe2), the compounds showed a 20-fold increase in affinity for tau tangles in this series. Despite the iodination, PDB-3 was bright enough to clearly demonstrate an excellent overlap with the fluorescence of a fluorescent antibody directed towards hyperphosphorylated tau protein in AD brain sections. However, a radiolabelled PDB-3 analogue revealed a relatively low degree of brain uptake, suggesting that the compound has poor BBB penetrability.
In order to overcome this issue, the same authors made further modifications, changing the benzothiozole acceptor to a benzimidazole ring and replacing the diazo linker with a styryl moiety, resulting in SBIM-3 (Fig. 6).21 Whereas these modifications resulted in a significantly blue-shifted fluorescence and reduced the selectivity of tau/Aβ down to 2.73-fold, the compound showed a 350% increase in brain uptake 2 minutes post injection and a much faster clearance.
Two 2-styrylindolium dyes (7a and 7b, Fig. 6) revealed clear fluorescence staining of NFTs in AD brain sections while only demonstrating faint fluorescence in the presence of Aβ-containing senile plaques.22 The compounds showed a relatively low toxicity in a zebrafish assay, however the presence of the indolium cation would be expected to hinder efficient BBB penetration. The authors have reported many more compounds with varying degrees of tau selectivity based on the thioflavin D–A or the curcumin/Congo Red structure, as well as unrelated scaffolds, in a doctoral thesis.23
Maruyama et al. proposed the hypothesis that not only should probes consist of a narrow π-conjugated D–A molecule, but also the length of the conjugation should ideally be larger than 13 Å. Thus, they described a family of π-extended thioflavin analogues: PBB1–5 (Fig. 6).24 Within this series some interesting aspects were observed. For example, whereas all probes revealed a propensity to fluorescently stain tau aggregates, the affinity for Aβ decreased with decreased probe lipophilicity, and reciprocally associated therewith, the selectivity for tau protein aggregates increased. The most hydrophilic and red-shifted fluorophore PBB5 exhibited the largest selectivity for tau protein, demonstrating NIR fluorescence emission at 685 nm. The probe was demonstrated to enable in vivo discrimination between a transgenic AD mouse model and a normal control, but unfortunately the presence of the benzothiazolium group resulted in metabolic instability and a reduced BBB penetrability as compared with the other PBB dyes. Further analysis identified PBB3 as the most promising candidate for in vivo tau imaging with an emission peak in the 500–550 nm range, combined with efficient two-photon excitation, although it should be noted that the spinal cord in a tauopathy mouse model was exposed and covered with a glass plate to allow for adequate amounts of fluorescent light to be detected. Interestingly, in this time course experiment, following intravenous injection of PBB3, the diffusion through the blood vessels and subsequent infiltration in the surrounding tissues and labelling of tau tangles could clearly be seen (Fig. 7). Finally, a 11C-labeled version of PBB3 was demonstrated to reveal tauopathy and disease progression in both animal models and the brains of living AD patients as compared to healthy controls.
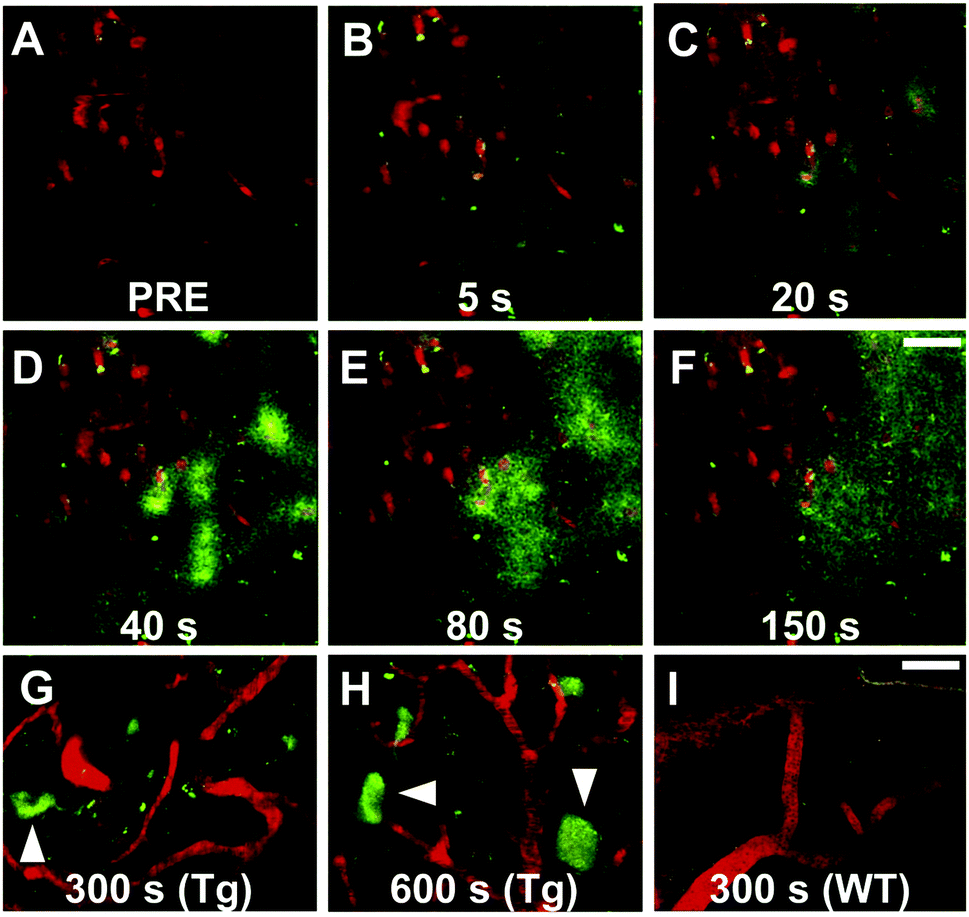 | ||
| Fig. 7 Tracking of PBB3 after intravenous injection in PS19 mice. Panel A: Pre-PBB3 injection. Panels B–F: Diffusion from blood vessels and distribution throughout the surrounding tissues. Panel G and H: Labelling of the somatodendritic compartment of NFT-bearing neurons and background clearance from surrounding tissues. Panel I: Non-Tg mouse control. Red: sulforhodamine 101 as a blood vessel marker. Green: PBB3. Reproduced from ref. 24, Copyright 2013, with permission from Elsevier. | ||
The fluorescent probe T726 (Fig. 6) and the significantly hypo-intense blue-shifted analogue AV-1451 (Fig. 6) (also known as T807) showed a good overlap with tau protein tangles in AD brain sections, while showing a relatively low affinity for Aβ-plaques.25 A comparative follow-up study by Ono et al. revealed that both PBB3 and AV-1451 show similar staining patterns in fluorescence imaging of AD brain sections. Both dyes demonstrated a very intense labelling in the case of ghost tangles and non-ghost tangles, with a relatively high degree of binding to dense-core Aβ-plaques as well. Interestingly, whereas PBB3 was able to visualize both AD tau aggregates, consisting of all six tau isoforms, as well as only 3R isoforms (e.g. Pick's disease) and exclusively 4R tauopathies (e.g. progressive supranuclear palsy), AV-1451 only labelled AD tau pathology. This opens the possibility to differentiate one type of pathology from the others as well as more in-depth studies related to how tau pathologies spread throughout the brain.26 Currently however, there are no hypotheses providing a rationale for these observations.
TK5105 (Fig. 6) represents a further abstraction from the thioflavin building block, bearing a quinoline group. This probe exhibits fluorescence in the blue region of the electromagnetic spectrum and exhibits efficient labelling of tau in AD brain sections. A competitive binding assay revealed a 25-fold higher binding affinity for aggregates made from the 4 repeat K18 domain (with a deletion of the 280 lysine residue) as compared to Aβ fibrils.27
The group of Yukitsuka Kudo disclosed a series of D–A type dyes showing a multichromic response to either Aβ or tau aggregates. Whereas these dyes only demonstrate a relatively small difference in binding affinity for either of these protein aggregates, the different binding modes or protein interactions result in different photophysical properties when staining AD brain sections. The first member of this family to show this remarkable effect was BF-126 (Fig. 6), showing a maximum emission intensity at 490 nm in senile plaques and a maximum emission intensity at 540 nm in NFTs.28 The π-extended analogue BF-188 resulted in an even larger difference with maxima of emission at 520 nm and 600 nm for senile plaques and NFT, respectively.29 Notably, this effect completely disappears when the benzimidazole group is replaced by the analogous benzoxazole and benzothiazole rings, suggesting the involvement of H-bond interactions in the protein binding modes. The use of a single fluorescent agent demonstrating fluorescence labelling and concomitant discrimination between both protein species is particularly useful for fluorescence histopathological imaging, as can be seen in Fig. 8.
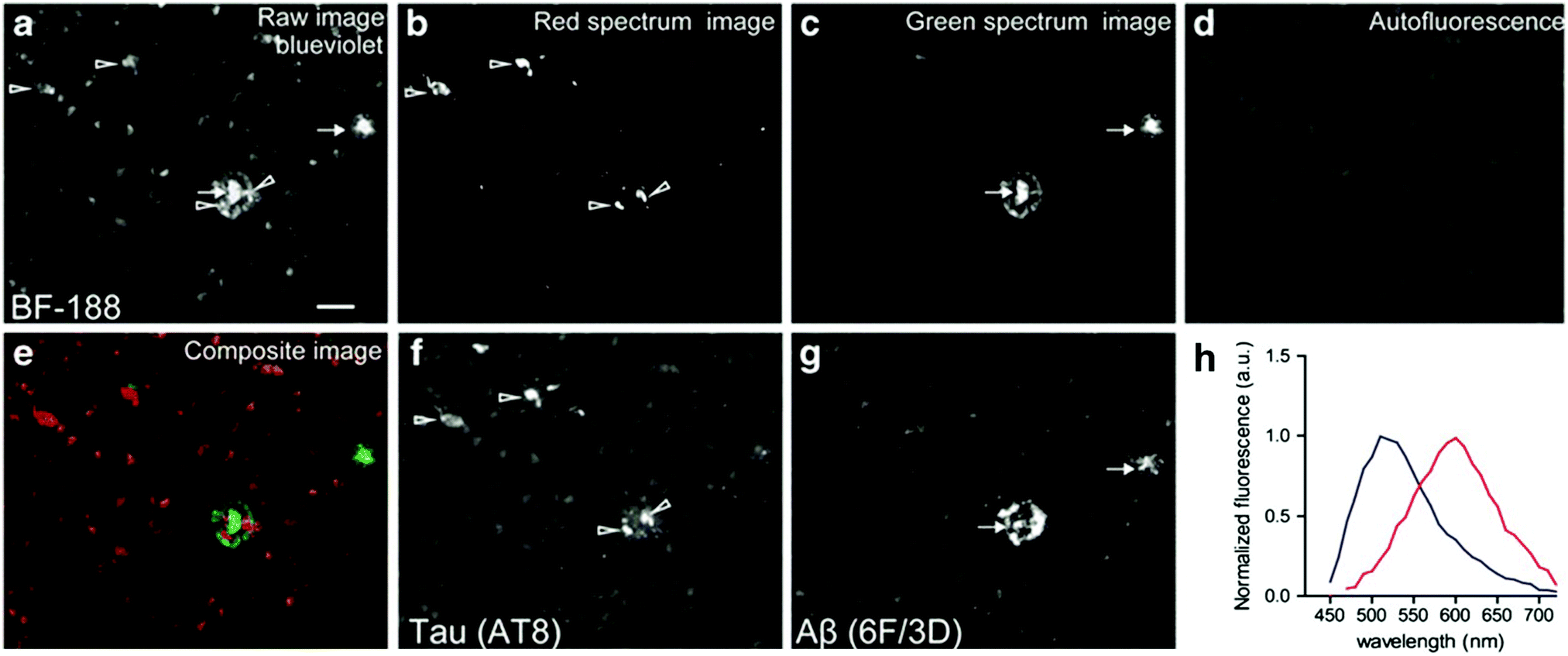 | ||
| Fig. 8 BF-188 multispectral imaging of brain slices using different filter settings. Arrows: senile plaques. Arrowheads: NFTs. (a) Blue-violet excitation shows both species, (b) the calculated red emission reveals NFTs, (c) the green channel correlates with the location of senile plaques, (d) sample autofluorescence, (e) re-coloured image showing both NFTs and senile plaques in red and green, respectively, (f and g) the location of the fluorescence was confirmed to correlate with NFTs and senile plaques using a fluorescently labelled antibody for phosphorylated tau and Aβ fibrils, respectively. (h) Extracted fluorescence profiles of BF-188 in NFTs (red) and senile plaques (blue). Reproduced from ref. 29, Copyright World Molecular Imaging Society 2013, with permission from Springer. | ||
 | ||
| Fig. 9 Chemical structures of BODIPY-based dyes. | ||
Lim et al. presented BD-tau (Fig. 9), obtained from a 1000 fluorophore high-throughput screening. This compound showed a fluorescence enhancement of up to 5.5 times upon the addition of tau protein aggregates in PBS with an emission maximum at 590 nm.31BD-tau shows good selectivity over tau pre-aggregates and bovine serum albumin, however in the presence of Aβ or insulin fibrils a 3.5- to 4-fold fluorescence enhancement was observed as well. BD-tau did reveal a clear co-localisation in tau protein expressing cells and brain slices of a tauopathy mouse model.
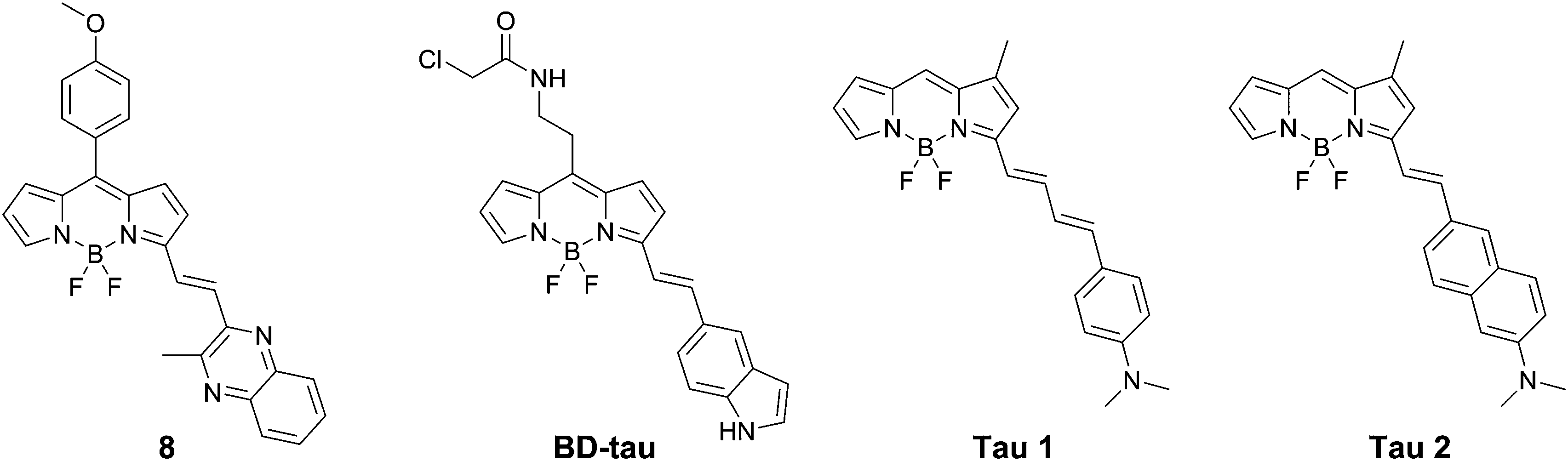
Verwilst et al. demonstrated the rational design of Tau 1 and Tau 2 (Fig. 9), through the π-elongation of a known Aβ-selective BODIPY dye.32 Using the crystal structure of a tau hexapeptide fragment (PHF6) known to nucleate tau protein aggregation, the compounds were subjected to docking studies as an in silico pre-screen, revealing a tight fit in tunnel structures in the crystal (Fig. 10a and b). Solution studies revealed a fluorescence enhancement based on the surrounding solvent polarity, rather than the viscosity. Solution tests in the presence of tau protein aggregates or Aβ fibrils showed a fluorescence enhancement of 6.4- and 9.3-fold around 650 nm for Tau 1 and Tau 2, respectively, whereas virtually no enhancement was observed in the case of Aβ-fibrils. In vitro experiments mirrored the findings of solution experiments and Tau 1, exhibiting the highest solubility, demonstrated the efficient labelling of NFTs in both acute and transgenic mouse models for tauopathy and AD, with minimal labelling of the core of dense core senile plaques (Fig. 10b and c). Finally, the authors demonstrated the BBB crossing of Tau 1 and the ability for in vivo tau aggregation status discrimination in a transgenic mouse model compared to a control (Fig. 10d).
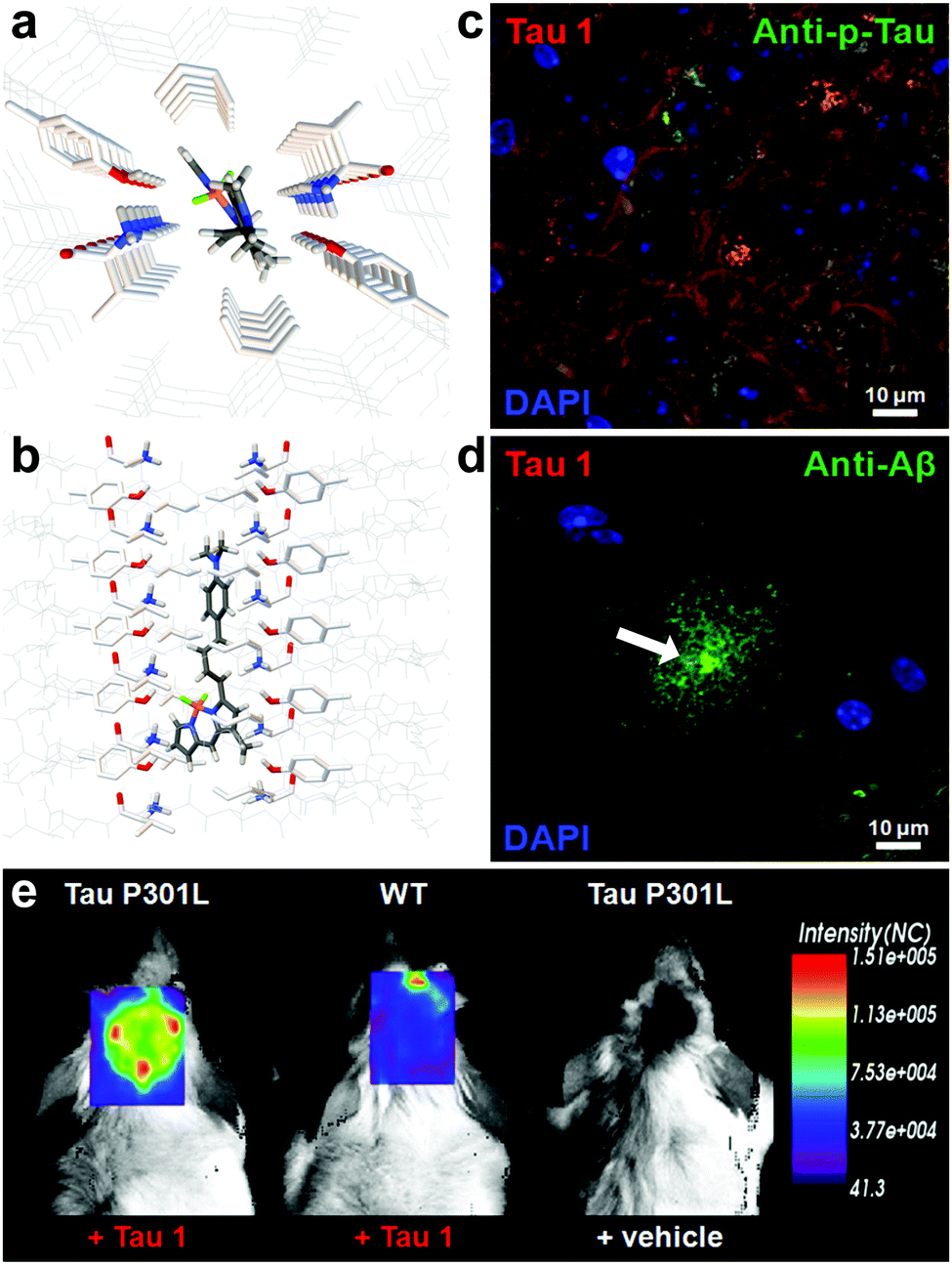 | ||
| Fig. 10 (a and b) Top and side-view of the docking result of Tau 1 in the tyrosine channel of the PHF6 hexapeptide. (c and d) Confocal imaging of brain slices of a 3xTg mouse, showing a NFT-rich and Aβ-containing senile plaque slice. Minimal Tau-1 fluorescence was observed at the plaque's core (arrow). (e) In vivo imaging of tau aggregates with Tau 1 as compared to two controls. Adapted with permission from ref. 32. Copyright 2017 American Chemical Society. | ||
Interestingly, be it by rational design or as a result of library screening, all of these BODIPYs have a few common characteristics, being one styryl-group substituted at the 3-position, bearing an N-heterocyclic or dimethylaniline pendant arm.
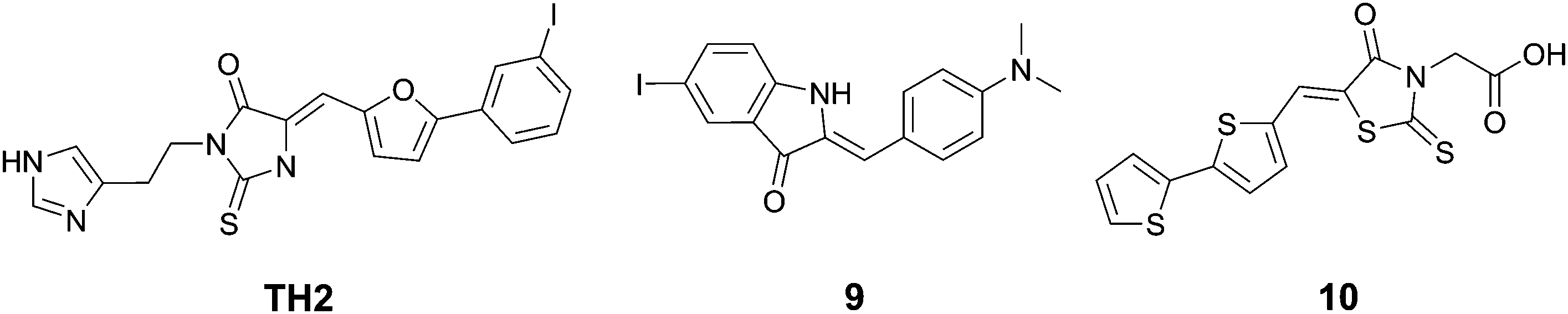 | ||
| Fig. 11 Chemical structures of rhodanines and related dyes. | ||
Wanatabe et al. described a series of oxindole derivatives as potential tau tangle imaging agents.34 Among these 9 (Fig. 11) showed a 1.77-fold higher binding to tau aggregates versus Aβ fibrils. This compound showed an approximately 1.63-fold higher brain uptake 2 minutes post injection than TH2, with a comparable brain clearance rate. The fluorescence imaging of 9 (emission range 600–660) demonstrated a good overlap with thioflavin staining in an entorhinal cortex AD brain section.
Anumala et al. presented a series of fluorescent rhodanine-3-acetic acid derivatives, showing varying degrees of selectivity of aggregated tau protein over Aβ aggregates in a Thiazine Red R displacement assay.35 Among the derivatives, 10 (Fig. 11) showed the highest selectivity for tau tangles (6.8-fold), combined with a high affinity for tau tangles and an emission maximum at 525 nm. Fluorescence imaging of hippocampal AD brain sections clearly showed the flame-like NFTs when stained with 10.
Notably, some of the derivatives described above share a significant structural likeness to known tau aggregation inhibitors,36 opening the path to self-reporting theranostic tau tangle inhibitors.
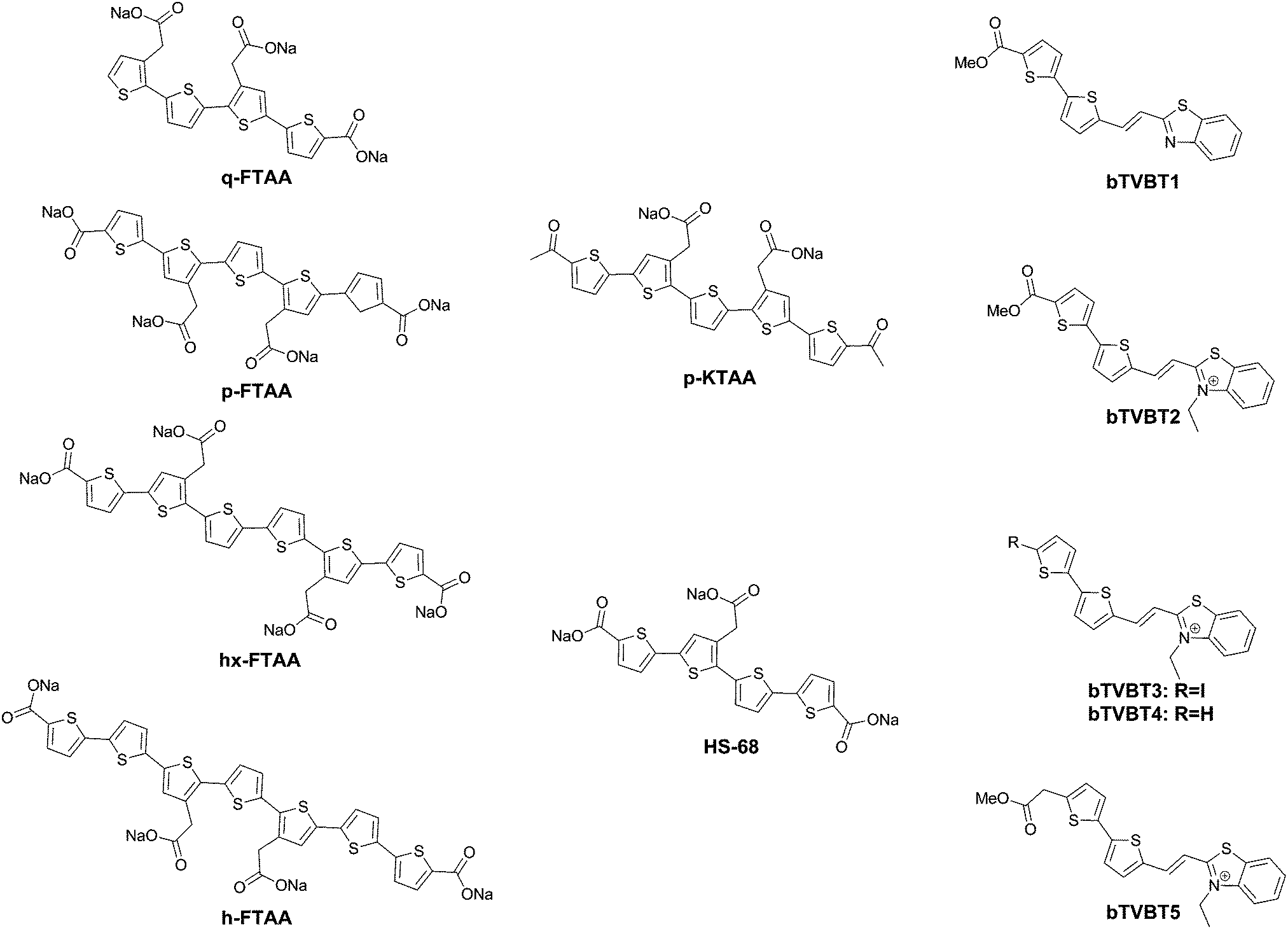 | ||
| Fig. 12 Chemical structure of oligothiophene dyes. | ||
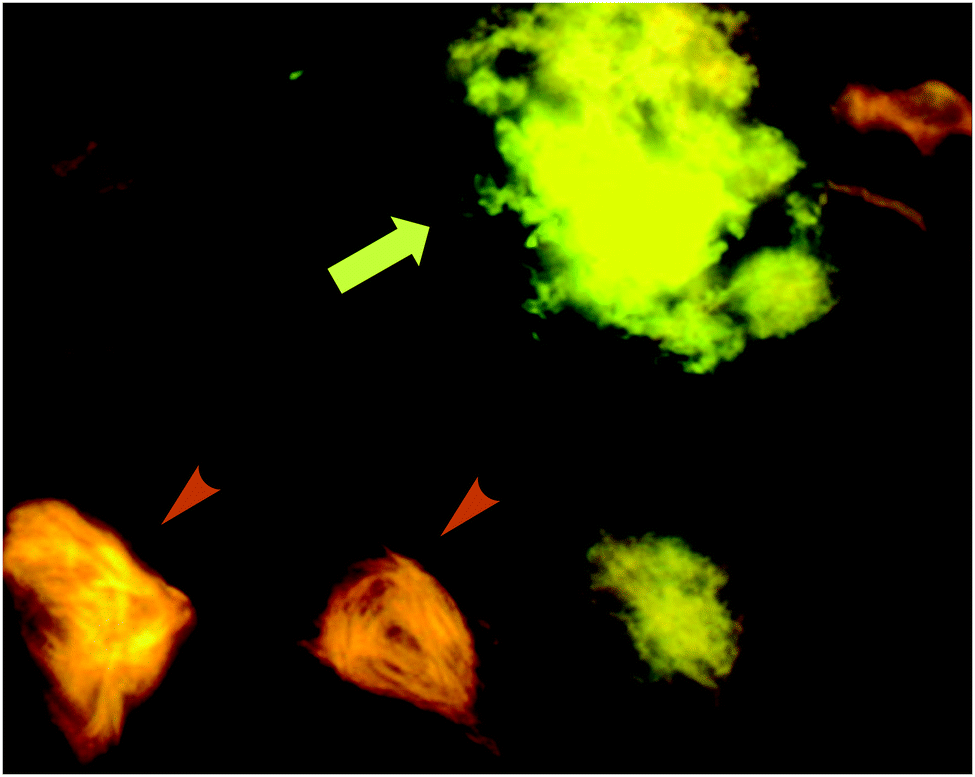 | ||
| Fig. 13 p-FTAA allows for the spectral discrimination between flame-shaped NFTs containing tau proteins (orange arrowheads) and senile plaques with Aβ (green-yellow arrow). Adapted with permission from ref. 37. Copyright 2009 American Chemical Society. | ||
Further work by the same group compared tetra-, penta-, hexa- and heptathiophenes (q-FTAA, p-FTAA, hx-FTAA and h-FTAA, respectively Fig. 12),38 again confirming the crucial importance of the terminal carboxylic acids. Within the series, the tetramer was found to be unable to spectrally resolve both peptidic aggregates, whereas all others were to a degree, although the pentamer clearly outperformed the other compounds.
When analysing the probes, the authors found that p-FTAA differed from the other probes in its solvatochromic behaviour.39 Among all the probes analysed, it showed the highest dependence on both the solvent polarity and viscosity. Whereas the Stokes shift showed an increase with increasing polarity, a decreasing Stokes shift was observed with increasing viscosity, as a result of the probes’ emission from non-relaxed excited state conformations. As such, the results could be interpreted as p-FTAA showing much stronger conformational restrictions on binding Aβ, as attested by the presence of the vibronic peaks as well as a reduced Stokes shift, whereas the binding site on tau fibrils would be less conformationally restricted and more polar. As such, the function of the terminal carboxylates would seem to be to extend the electronic conjugation in planar conformations of the oligothiophenes. Their replacement with acetyl groups (p-KTAA, Fig. 12) leads to a 3.75-fold stronger dependence on the solvent polarity and a larger difference between the emission wavelength in the presence of senile plaques or NFTs, with emission maxima of 570 and 595 nm, respectively, although it should be remarked that no vibronic peaks in the case of senile plaque binding were observed for this fluorophore.39
Interestingly, HS-68 (Fig. 12), while showing large variabilities in spectral results when bound to tau tangles, precluding its use in the differentiation of Aβ and tau, was further investigated, revealing an age-dependence on the emission wavelength of the probe, with blue-shifted emissions in NFTs of older mice, with similar observations in senile plaques as well (Fig. 14).40
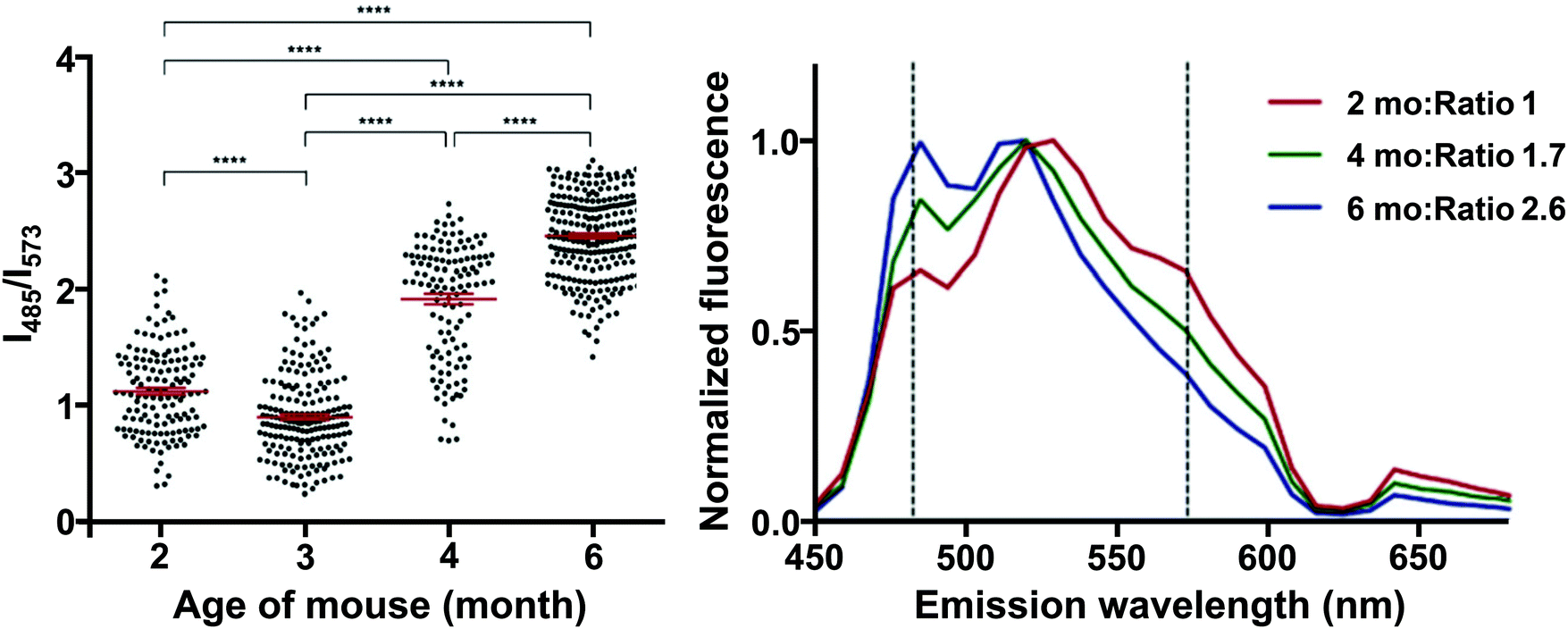 | ||
| Fig. 14 Left: Ratio of the fluorescence intensity of HS-68 at 485 and 573 nm in P301S mouse brain slices. Dots represent individual measurements, the group mean and SEM are depicted in red, significance: **** = p < 0.0001. Right: Normalized fluorescence of HS-68 bound to NFTs in P301S mouse brain slices of various ages (in months). Dashed lines represent the wavelengths used for the ratio determination in the left panel. Reproduced from ref. 40. Copyright 2015 Klingstedt et al., with permission from Wiley-VCH. As the mouse ages, a blue-shifted fluorescence was observed, presumably because of a physicochemical change in the tau aggregates. The results clearly indicate the presence of two distinct states in these tau aggregate populations, in which a switch takes place between 3 and 4 months of age. For further reading, we direct the readers to ref. 40. | ||
Further explorations into the chromophore let to the oligothiophene–PBB3 hybrids bTVBT1–5 (Fig. 12).41 Here, the compounds do not exhibit dual staining for NFTs and senile plaques, but rather bTVBT1–5 show a very high selectivity for tau aggregates. bTVBT1–5 demonstrated emission maxima in the 600–630 nm range with tails reaching well into the NIR region. Intriguingly, the replacement of the benzothiophene ring of bTVBT1 by a benzimidazole completely reversed the selectivity, resulting in high affinity senile plaque binding. The styrene bond was also determined to be highly important, as replacement with a thiophene ring either removed the dyes’ affinities for both NFTs and senile plaques or made the dye non-selective.
 | ||
| Fig. 15 Chemical structure of other classes of tau-selective dyes. | ||
Trimethine dyes have long been used in biological imaging experiments, owing to their generally high brightness with emission at around 570 nm, and relatively low toxicity. 11a–f (Fig. 15) all show intense labelling of tau tangles in hippocampal AD brain sections, while not staining senile plaques contained in the same tissues. The reason for this observation however is not fully understood, as those compounds not showing spectral overlap with Thiazine Red R displayed a much higher affinity to amyloid aggregates in a displacement assay.43 One potential explanation could be that as tau aggregation assays usually only use one isoform of tau protein (often 2N4R), the human brain contains all 6 isoforms as well as many posttranslational modifications in NFTs.
Rejc et al. recently published a small library of long-range D–A dyes showing good affinity to tau tangles (12a–c, Fig. 15). Whereas the dyes all showed some degree of binding to Aβ, 12b showed the lowest affinity to Aβ in a competition assay.44 Similarly to Tau 1,32 the library of compounds was pre-screened in a PHF6 docking assay, revealing a tight fit for all compounds in the tyrosine channel. 12b shows an emission maximum at 654 nm in solution but demonstrates a significant amount of solvatochromism and reveals a maximum emission at 580 and 595 nm for senile plaques and NFTs, respectively, allowing for a visual confirmation of the aggregates’ identity in tissue slices (green versus orange, respectively).
In contrast to the above organic dyes, Gao et al. presented a fluorescent inorganic ruthenium complex (13, Fig. 15). This compound was tested on aggregates of the R3 repeat of tau protein, including the crucial PHF6 fragment. The compound showed a very large increase in fluorescence intensity in the presence of protein aggregates with a virtually fully quenched emission in water.45 The compound, however was not assessed using the full tau protein, nor in the presence of Aβ, and previously had been described to show a similar dramatic turn-on in fluorescence in the presence of G-quadruplex DNA. In the case of this compound, the binding interaction as a cross β-sheet binding species is unclear, and the authors suggested another binding mode in a hinge of a peptide consisting of the R3 region of tau protein, using a software-predicted model.
4. Detecting protein phosphorylation patterns
The group of Itaru Hamachi adopted a radically different approach for sensing tau pathology by focussing on the degree of protein hyperphosphorylation. The use of two dipicolylamines appended to the 2- and 5-positions of a BODIPY scaffold resulted in sensor 14 (Fig. 16). This compound, in the presence of Zn2+ ions revealed a particular sensitivity to bisphosphorylated peptide with a i, i + 4 substitution pattern as assessed in a number of short tau protein derived peptides with an increase in fluorescence intensity at 547 nm.46 This selectivity was rationalized by the rigid nature of the fluorophore separating the two Zn2+ centres. The probe also demonstrated a significantly increased sensitivity for hyperphosphorylated full-length tau protein, with a binding affinity of 8.9- and 72-fold stronger than for non-phosphorylated tau and Aβ fibrils, respectively, which enabled the selective labelling of NFTs in AD brain sections. | ||
| Fig. 16 Chemical structure of dyes recognizing phosphorylation patterns. | ||
The group reported a second fluorescent reporter (15, Fig. 16), demonstrating a very high selectivity for i, i + 1 diphosphorylated peptides by the incorporation of a much shorter rigid linker between the two dipicolylamines.47 Upon the addition of i, i + 1 diphosphorylated tau derived peptides, a 4-fold ratiometric fluorescence change occurred (427 nm/378 nm), with a more than 10-fold selectivity for i, i + 1 diphosphorylated peptides over longer substitution patterns. However, the potential interference by biologically relevant di- and triphosphates, or the applicability to full length tau protein or tissue sections was not reported.
A similar approach was followed by Kim et al. demonstrating the sensitivity of the near-infrared emissive fluorophore CyDPA2 (Fig. 16) for hyperphosphorylated tau protein over non-phosphorylated tau in solution as well as in AD brain sections.48 Interestingly, CyDPA2 shows a ratiometric response to the phosphorylation status of tau protein, with non-phosphorylated tau protein resulting in a shorter absorbance wavelength of the dye (smaller Abs810/Abs750 nm ratio), as well as a decreased fluorescence intesity at 825 nm, whereas the addition of hyperphosphorylated tau protein demonstrated the opposite effect. Similar observations were seen in the brain tissues of normal controls versus AD patients and P301L mice. The interference of Aβ was not tested, however fluorescence imaging of AD brain sections with CyDPA2 did not reveal obvious Aβ pathology. The linker in this bis-Zn2+-dipicolylamine receptor showed a significant amount of flexibility and the i, i + n pattern selectivity was not determined, but is likely not restricted to one specific phosphorylation pattern.
The selective recognition of i, i + 1 phosphorylation patterns was extended to inorganic complexes as well by Wang et al.49 The terbium(III) complex (TC, Fig. 16), bearing two 8-aminoquinoline units, showed a higher than 10-fold fluorescence enhancement upon the addition of bisphosphorylated tau derived short peptides in the presence of Zn2+. Various other bisphosphate species showed virtually no differences in fluorescence, with the exception of pyrophosphate, showing a four-fold increase in fluorescence intensity. The mechanism for the fluorescence enhancement is believed to be a combination of multiple processes, such as the inhibition of PET quenching upon the addition of Zn, followed by templating of the 8-aminoquinolines by the peptide resulting in favourable energy transfer via the antenna effect. The probe was however not applied to full length tau protein or to tissue samples.
None of these probes have been assessed in vitro or in vivo, and the highly charged nature of the compounds might interfere with efficient BBB crossing. Further studies will be required to assess the applicability of these types of probe in animal models of AD.
Potential design guidelines
Currently, despite the progress in the field, no clear structure–activity relationship has been demonstrated, with a wide variety of scaffolds showing a high affinity and selectivity for tau aggregates. One explanation for this complication could be that tau protein likely exhibits a variety of binding pockets, binding different types of compounds with different affinities, as well as inherently being a heterogeneous target. However, most of these compounds have some common characteristics that could aid the design of future tau tangle selective dyes. We will outline some of these features below to formulate guidelines (for cross-β-sheet binding type dyes). Whereas each individual recommendation falls short of explaining all the observations in the probes listed above, taken together they could significantly ease the design of future probes.1. Flat π-conjugated scaffolds with a length exceeding 13 Å
Upon analysing a library of compounds, Maruyama et al. concluded that 13 Å was a threshold value tipping the balance in favour of tau tangles versus Aβ-fibers.24 The authors also remarked that the binding mode of the compounds very likely involved a cross-β-sheet binding mode, with some degree of confinement, with a preference for flat narrow compounds. Furthermore, this type of compound is also expected to show higher degrees of BBB penetration via passive diffusion.2. Pre-screening ligands via docking studies
The prerequisite of a narrow architecture could be rationalized, providing a narrow cross-β-sheet docking site would be an important ligand binding pocket. PHF6, the short peptide sequence nucleating the pathological transformation of random coil tau protein into paired helical filaments was demonstrated to exhibit this architecture in crystals of this peptide. A tyrosine channel was found to demonstrate the tight binding of a large variety of known tau tangle fluorophores and PET agents in silico, and docking studies demonstrated a tight fit for Tau 1 and Tau 2,32 as well as 12a–c.44 Although none of the fluorophores described in this work have been validated on models of the tau core structures identified by cryo-EM,7 it stands to reason that the docking results of these models would likely provide a much better insight into the binding pockets on tau aggregates for various fluorophores. As such the importance of this core structure model cannot be understated. Pre-screening of compounds using docking studies on PHF6 and/or the cryo-EM models could eliminate some unlikely candidates, significantly reducing the costs associated with screening large libraries of ligands on tau tangles or AD brain slides.3. Polarisable ligands
Jensen et al. tested a wide variety of compound scaffolds and their relative affinity for brain-derived paired helical filaments as well as aggregates of recombinant tau protein. A clear trend could be seen. Within the same scaffolds, the more polarisable compounds showed a higher affinity.50 This observation rationalizes the (electron-rich) D–A and D–A–D structures in the vast majority of the compounds described above, as these compounds show the largest electron redistribution in the presence of external electrical fields or environments with large dielectric constants. Furthermore, multichromic compounds showing solvatochromism, such as BF-126,28BF-188,29p-FTAA,37p-KTAA39 and 12b,44 invariably show a bathochromic shift in the emission wavelength when associated with tau, as compared to Aβ fibres or senile plaques. Furthermore, within the same series of scaffolds, it was also observed that the tau/Aβ discrimination in the PBB1–5 series increased with decreasing lipophilicity.24 Taken together, this clearly suggests that the binding pocket in tau tangles has a higher dielectric constant, in other words is less hydrophobic, than the binding site in Aβ fibres.Conclusions and future perspectives
Over the course of the last decade, significant progress in the field of small molecule fluorescent sensors for aggregated tau protein has been made. However, due to complications related to the convoluted nature of (hyperphosphorylated) tau tangles, a clear structure–activity relationship has not been identified to date, yet the use of the recently described core structure models is likely to result in many more rationally designed fluorescent probes. Notwithstanding, some common characteristics such as a slender long π-conjugated backbone, intermediate lipophilicity, large polarizability and affinity for the PHF6 fragment in docking studies have been identified in the absence of the knowledge of the tau core structure.A number of dyes have been reported with excellent signal to background ratios for staining histological AD brain sections. These dyes could present several advantages over fluorescence immunostaining, as they have been demonstrated to have a robust nature, staining histological sections prepared under various fixating conditions, as well as exhibiting the inherent uniform and discrete nature and relatively low production cost of small molecules. The number of dyes with a demonstrated applicability in the field of small animal in vivo imaging is currently still limited, as true NIR emitting dyes are still rare. Yet, their application could be very useful for monitoring the disease progression in AD animal models with minimal invasiveness.
Some recent evidence has been presented, demonstrating a contrasting response to different types of tau aggregates, for example as found in young and old mice, or dyes staining uniquely the 3R, 4R or 3R/4R isoforms of tau aggregates. Over the next years it is expected that more dyes with a selective profile will be developed. Undoubtedly, the progress in these dyes as well as structural models with increasing resolution will result in a much better understanding of the nature and diversity of tau protein aggregates. Some impressive results in the field of multichromic dyes have demonstrated a good differential response for tau tangles or Aβ aggregates and are highly relevant and could find widespread application. Yet their true potential, being the differential labelling of not just these two types of aggregates but also other proteinaceous deposits, such as α-synuclein, prions, amylin and huntingtin in a protocol consisting of one or a combination of several multichromic dyes, has not been reached, but would significantly improve the ease of identification of the disease state in histological samples. Finally, the development of dyes that can selectively label different tau protein pre-aggregates/oligomers is urgently needed, to elucidate the nature and mechanism of cell-to-cell tau pathology spreading.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) (2017R1A2A2A05069805, CK), funded by the Ministry of Science and ICT (CRI project no. 2009-0081566, JSK), and the Basic Science Research Program (NRF-2017R1D1A1B03032561, PV) funded by the Ministry of Education, as well as the Korea Research Fellowship Program funded by the Ministry of Science and ICT through the National Research Foundation of Korea (2016H1D3A1938052, PV).Notes and references
- T. Arendt, J. T. Stieler and M. Holzer, Brain Res. Bull., 2016, 126, 238–292 CrossRef CAS PubMed and references therein.
- Y. Wang and E. Mandelkow, Nat. Rev. Neurosci., 2016, 17, 5–21 CrossRef PubMed and references therein.
- S. Jeganathan, M. von Bergen, H. Brutlach, H.-J. Steinhoff and E. Mandelkow, Biochemistry, 2006, 45, 2283–2293 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC and references therein.
- S. S. Shafiei, M. J. Guerrero-Muñoz and D. L. Castillo-Carranza, Front. Aging Neurosci., 2017, 9, 83 Search PubMed and references therein.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185–190 CrossRef CAS PubMed.
- H. Braak and E. Braak, Acta Neuropathol., 1991, 82, 239–259 CrossRef CAS PubMed.
- H. C. Kolb and J. I. Andrés, Cold Spring Harbor Perspect. Biol., 2017, 9, a023721 CrossRef PubMed.
- S. D. Styren, R. L. Hamilton, G. C. Styren and W. E. Klunk, J. Histochem. Cytochem., 2000, 48, 1223–1232 CrossRef CAS PubMed.
- D. M. Skovronsky, B. Zhang, M.-P. Kung, H. F. Kung, J. Q. Trojanovski and V. M.-Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7609–7614 CrossRef CAS.
- K. Sato, M. Higuchi, N. Iwata, T. C. Saido and K. Sasamoto, Eur. J. Med. Chem., 2004, 39, 573–578 CrossRef CAS PubMed.
- W. E. Klunk, B. J. Bacskai, C. A. Mathis, S. T. Kajdasz, M. E. McLellan, M. P. Frosch, M. L. Debnath, D. P. Holt, Y. Wang and B. T. Hyman, J. Neuropathol. Exp. Neurol., 2002, 61, 797–805 CrossRef CAS PubMed.
- A. Boländer, D. Kieser, C. Voss, S. Bauer, C. Schön, S. Burgold, T. Bittnerm, J. Hölzer, R. Heyny-vonHaußen, G. Mall, V. Goetschy, C. Czech, H. Knust, R. Berger, J. Herms, I. Hilger and B. Schmidt, J. Med. Chem., 2012, 55, 9170–9180 CrossRef PubMed.
- A. Boländer, D. Kieser, C. Scholz, R. Heyny-vonHaußen, G. Mall, V. Goetschy, C. Czech and B. Schmidt, Neurodegener. Dis., 2014, 13, 209–213 CrossRef PubMed.
- K.-s. Park, Y. Seo, M. K. Kim, K. Kim, Y. K. Kim, H. Choo and Y. Chong, Org. Biomol. Chem., 2015, 13, 11194–11199 CAS.
- Y. Seo, K.-s. Park, T. Ha, M. K. Kim, Y. J. Hwang, J. Lee, H. Ryu, H. Choo and Y. Chong, ACS Chem. Neurosci., 2016, 7, 1474–1481 CrossRef CAS PubMed.
- K.-s. Park, M. K. Kim, Y. Seo, T. Ha, K. Yoo, S. J. Hyeon, Y. J. Hwang, J. Lee, H. Ryu, H. Choo and Y. Chong, ACS Chem. Neurosci., 2017, 8, 2124–2131 CrossRef CAS PubMed.
- N. S. Honson, R. L. Johnson, W. Huang, J. Inglese, C. P. Austin and J. Kuret, Neurobiol. Dis., 2007, 28, 251–260 CrossRef CAS PubMed.
- K. Matsumura, M. Ono, S. Hayashi, H. Kimura, Y. Okamoto, M. Ihara, R. Takahashi, H. Mori and H. Saji, Med. Chem. Commun., 2011, 2, 596–600 RSC.
- K. Matsamura, M. Ono, M. Yoshimura, H. Kimura, H. Watanabe, Y. Okamoto, M. Ihari, R. Takahashi and H. Saji, Bioorg. Med. Chem., 2013, 21, 3356–3362 CrossRef PubMed.
- J. Gu, U. R. Anumala, F. Lo Monte, T. Kramer, R. Heyny von Haußen, J. Hölzer, V. Goetschy-Meyer, G. Mall, I. Hilger, C. Czech and B. Schmidt, Bioorg. Med. Chem., 2012, 22, 7667–7671 CrossRef CAS PubMed.
- J. Gu, PhD thesis, Technischen Universität Darmstadt, 2013, http://tuprints.ulb.tu-darmstadt.de/3319/, accessed February 2018.
- M. Maruyama, H. Shimada, T. Suhara, M. Shinotoh, B. Ji, J. Maeda, M.-R. Zhang, J. Q. Trojanowski, V. M.-Y. Lee, M. Ono, K. Masamoto, H. Takano, N. Sahara, N. Iwata, N. Okamura, S. Furumoto, Y. Kudo, Q. Chang, T. C. Saido, A. Takashima, J. Lewis, M.-K. Jang, I. Aoki, H. Ito and M. Higuchi, Neuron, 2013, 79, 1094–1108 CrossRef CAS PubMed.
- C.-F. Xia, J. Arteaga, G. Chen, U. Gangadharmath, L. F. Gomez, D. Kasi, C. Lam, Q. Liang, C. Liu, V. P. Mocharla, F. Mu, A. Sinha, H. Su, K. Szardenings, J. C. Walsh, E. Wang, C. Yu, W. Zhang, T. Zhao and H. C. Kolb, Alzheimer's Dementia, 2013, 9, 666–676 CrossRef PubMed.
- M. Ono, N. Sahara, K. Kumata, B. Ji, R. Ni, S. Koga, D. W. Dickson, J. Q. Trojanowski, V. M.-Y. Lee, M. Yoshida, I. Hozumi, Y. Yoshiyama, J. C. van Swieten, A. Nordberg, T. Suhara, M.-R. Zhang and M. Higuchi, Brain, 2017, 140, 764–780 Search PubMed.
- N. Okamura, S. Furumoto, R. Harada, T. Tago, T. Yoshikawa, M. Fodero-Tavoletti, R. S. Mulligan, V. L. Villemagne, H. Akatsu, T. Yamamoto, H. Arai, R. Iwata, K. Yanai and Y. Kudo, J. Nucl. Med., 2013, 54, 1420–1427 CrossRef CAS PubMed.
- N. Okamura, T. Suemoto, S. Furumoto, M. Suzuki, H. Shimadzu, H. Akatsu, T. Yamamoto, H. Fujiwara, M. Nemoto, M. Maruyama, H. Arai, K. Yanai, T. Sawada and Y. Kudo, J. Neurosci., 2005, 25, 10857–10862 CrossRef CAS PubMed.
- R. Harada, N. Okamura, S. Furumoto, T. Yoshikawa, H. Arai, K. Yanai and Y. Kudo, Mol. Imaging Biol., 2014, 16, 19–27 CrossRef PubMed.
- M. Vedemalai, V. G. Krishnakumar, S. Gupta, S. Mori and I. Gupta, Sens. Actuators, B, 2017, 244, 673–683 CrossRef.
- S. Lim, Md. M. Haque, D. Su, D. Kim, J.-S. Lee, Y. T. Chang and Y. K. Kim, Chem. Commun., 2017, 53, 1607–1610 RSC.
- P. Verwilst, H.-R. Kim, J. Seo, N.-W. Sohn, S.-Y. Cha, Y. Kim, S. Maeng, J.-W. Shin, J. H. Kwak, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2017, 139, 13393–13403 CrossRef CAS PubMed.
- M. Ono, S. Hayashi, K. Matsumura, H. Kimura, Y. Okamoto, M. Ihara, R. Takahashi, H. Mori and H. Saji, ACS Chem. Neurosci., 2011, 2, 269–275 CrossRef CAS PubMed.
- H. Wanatabe, M. Ono, H. Kimura, K. Matsumura, M. Yoshimura, Y. Okamoto, M. Ihara, R. Takahashi and H. Saji, Bioorg. Med. Chem. Lett., 2012, 22, 5700–5703 CrossRef PubMed.
- U. R. Anumala, J. Gu, F. Lo Monte, T. Kramer, R. Heyny-von Haußen, J. Hölzer, V. Goetschy-Meyer, C. Schön, G. Mall, I. Hilger, C. Czech, J. Herms and B. Schmidt, Bioorg. Med. Chem., 2013, 21, 5139–5144 CrossRef CAS PubMed.
- B. Bulic, M. Pickhardt, I. Khlistunova, J. Biernat, E.-M. Mandelkow, E. Mandelkow and H. Waldmann, Angew. Chem., Int. Ed., 2007, 46, 9215–9219 CrossRef PubMed.
- A. Åslund, C. J. Sigurdson, T. Klingstedt, S. Grathwohl, T. Bolmont, D. L. Dickstein, E. Glimsdal, S. Prokop, M. Lindgren, P. Konradsson, D. M. Holtzman, P. R. Hof, F. L. Heppner, S. Gandy, M. Jucker, A. Aguzzi, P. Hammarström and K. P. R. Nilsson, ACS Chem. Biol., 2009, 4, 673–684 CrossRef PubMed.
- T. Klingstedt, A. Åslund, R. A. Simon, L. B. G. Johansson, J. J. Mason, S. Nyström, P. Hammarström and K. P. R. Nilsson, Org. Biomol. Chem., 2011, 9, 8356–8370 CAS.
- R. A. Simon, H. Shirani, K. O. A. Åslund, M. Bäck, V. Haroutunian, S. Gandy and K. P. R. Nilsson, Chem. – Eur. J., 2014, 20, 12537–12543 CrossRef CAS PubMed.
- T. Klingstedt, H. Shirani, J. Mahler, B. M. Wegenast-Braun, S. Nyström, M. Goedert, M. Jucker and K. P. R. Nilsson, Chem. – Eur. J., 2015, 21, 9072–9082 CrossRef CAS PubMed.
- H. Shirani, H. Appelqvist, M. Bäck, T. Klingstedt, N. J. Cairns and K. P. R. Nilsson, Chem. – Eur. J., 2017, 23, 17127–17135 CrossRef CAS PubMed.
- A. Taghavi, S. Nasir, M. Pickhardt, R. Heyny-von Haußen, G. Mall, E. Mandelkow, E.-M. Mandelkow and B. Schmidt, J. Alzheimer's Dis., 2011, 27, 835–843 CAS.
- J. Gu, U. R. Anumala, R. Heyny-von Haußen, J. Hölzer, V. Goetschy-Meyer, G. Mall, I. Hilger, C. Czech and B. Schmidt, ChemMedChem, 2013, 8, 891–897 CrossRef CAS PubMed.
- L. Rejc, L. Šmid, V. Kepe, Č. Podlipnik, A. Golobič, M. Bresjanac, J. R. Barrio, A. Petrič and J. Košmrlj, J. Med. Chem., 2017, 60, 8741–8757 CrossRef CAS PubMed.
- X. Gao, L. Wang, H.-L. Huang, L.-L. Wang, J.-L. Yao, S. Shi and T.-M. Yao, Analyst, 2015, 140, 7513–7517 RSC.
- A. Ojida, T. Sakamoto, M.-a. Inoue, S.-h. Fujishima, G. Lippens and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 6543–6548 CrossRef CAS PubMed.
- Y. Ishida, M.-a. Inoue, T. Inoue, A. Ojida and I. Hamachi, Chem. Commun., 2009, 2848–2850 RSC.
- H.-Y. Kim, U. Sengupta, P. Shao, M. J. Guerrero-Muñoz, R. Kayed and M. Bai, Am. J. Nucl. Med. Mol. Imaging, 2013, 3, 102–117 CAS.
- X. Wang, T. Yang, J. Luo, L. Yang and C. Yao, Chem. Commun., 2015, 51, 8185–8188 RSC.
- J. R. Jensen, K. Cisek, N. S. Honson and J. Kurett, Bioorg. Med. Chem., 2011, 19, 5147–5154 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |