
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A guide to integrating immunohistochemistry and chemical imaging
David P.
Bishop†
 ab,
Nerida
Cole†
abc,
Tracy
Zhang
b,
Philip A.
Doble
b and
Dominic J.
Hare
*abd
ab,
Nerida
Cole†
abc,
Tracy
Zhang
b,
Philip A.
Doble
b and
Dominic J.
Hare
*abd
aElemental Bio-imaging Facility, University of Technology Sydney, Broadway, New South Wales, Australia
bAtomic Pathology Laboratory, The Florey Institute of Neuroscience and Mental Health, The University of Melbourne, 30 Royal Parade, Parkville, Victoria 3052, Australia. E-mail: dominic.hare@florey.edu.au; Tel: +61 3 9035 9549
cARC Training Centre in Biodevices, Faculty of Science, Engineering and Technology, Swinburne University of Technology, Hawthorn, Victoria, Australia
dDepartment of Pathology, The University of Melbourne, Parkville, Victoria, Australia
First published on 15th March 2018
Abstract
Chemical imaging provides new insight into the fundamental atomic, molecular, and biochemical composition of tissue and how they are interrelated in normal physiology. Visualising and quantifying products of pathogenic reactions long before structural changes become apparent also adds a new dimension to understanding disease pathogenesis. While chemical imaging in isolation is somewhat limited by the nature of information it can provide (e.g. peptides, metals, lipids, or functional groups), integrating immunohistochemistry allows simultaneous, targeted imaging of biomolecules while also mapping tissue composition. Together, this approach can provide invaluable information on the inner workings of the cell and the molecular basis of diseases.
 David P. Bishop | David Bishop completed his BSc with 1st class Honours and PhD in Chemistry at the University of Technology Sydney. He is a current recipient of the Australian Research Council's Discovery Early Career Researcher Award, and his Fellowship is examining the translational utility of immunohistochemistry and chemical imaging in cell biology. He is a former Fullbright Scholar, spending part of 2016 at the University of California, Los Angeles (UCLA) Center for Duchenne Muscular Dystrophy (CDMD) working with Dr Jonathan Wanagat developing new methods for visualising disease biochemistry. |
 Nerida Cole | Nerida Cole received a BSc (Hons, 1st class) in Chemistry and MSc in Biochemistry from the University of Sydney, prior to completing a PhD in Optometry and Veterinary Science from the University of New South Wales. She has worked in research, teaching and research administration roles, and spent substantial time working on commercial research projects centred on the host-response to implanted biodevices. Now part of the Swinburne University of Technology's Australian Research Council Training Centre in Biodevices, her breadth of knowledge across all facets of contemporary research are being used to accelerate translation of new discoveries to real-world solutions. |
 Tracy Zhang | Tracy Zhang completed her BSc with 1st class Honours in 2017 at the Florey Institute of Neuroscience and Mental Health and The University of Melbourne in Behavioural Neuroscience, and was recently awarded a Parkinson's Victoria PhD scholarship to study the effects of brain iron overexposure throughout lifespan at the Florey with Drs Dominic Hare, Emma Burrow and Scott Kolbe, using a range of in and ex vivo chemical imaging techniques. |
 Philip A. Doble | Philip Doble is Professor of Analytical Chemistry at the University of Technology Sydney and head of the Elemental Bio-imaging Facility. He completed his PhD in Chemistry in 1998 at the University of Tasmania studying the separation and detection of anions using capillary electrophoresis. Philip's main research interests involve the further development of laser ablation-inductively coupled plasma-mass spectrometry as an imaging tool for biologists and medical researchers, as well as the translation of advanced analytical chemistry technology into clinical research. |
 Dominic J. Hare | Dominic Hare completed a BSc in Applied Chemistry with 1st class Honours and a PhD in Chemistry at the University of Technology Sydney, under the supervision of Prof. Philip Doble. He was a co-founder of the Elemental Bio-imaging Facility, before relocating to the Florey Institute of Neuroscience and Mental Health at The University of Melbourne, where he currently heads the Atomic Pathology Laboratory. In 2016 he was awarded a prestigious National Health and Medical Research Council Career Development Fellowship to establish a research program probing the role of iron as a possible causative feature of Parkinson's disease. He is a Fellow of the Royal Society of Chemistry and Advisory Board member of Metallomics. |
Key learning points(1) Chemical imaging must be redefined to encompass all analytical techniques that can spatially map the chemical composition of a sample.(2) Immunohistochemistry has many applications in chemical imaging, from co-localising individual proteins with chemical components to multiplexed experiments for profiling biochemical pathways and cell hierarchies. (3) The significance of an immunohistochemistry and chemical imaging approach is not necessarily based in technology, but how it is applied to answer a primary research question. (4) Immunostaining may inadvertently perturb the native biochemistry of the system being imaged, and the effects of each potentially confounding step should be considered when validating a method. (5) Combining immunohistochemistry with novel applications of chemical imaging presents new opportunities and technical challenges that should be considered research priorities to allow further growth in the field. |
1. Introduction
The complexity of a biological system is reflected by the dynamic microchemical environment found within cells and tissues. Imaging technology has advanced to a point where specific biochemicals can be targeted and visualised, potentially in real time. Traditional anatomical pathology, for which visualising microstructure was once the primary outcome, has greatly benefited from advances in analytical technology and their application to chemical imaging. Biologists now have a range of chemical imaging tools at their disposal that complement, and in some cases even supplant, conventional methods for spatially profiling the structure and biochemical makeup of cells, organs, and even whole organisms.Histology and histochemistry are the standard approaches for visually assessing gross tissue morphology, cell type, subcellular structure, and the presence of endogenous chemicals that react to form coloured compounds with certain dyes and stains. While having an important place in both analytical biochemistry and diagnostics, these methods are primarily qualitative and have limited specificity at the molecular level. Many chemical components of biological systems fall outside the scope of traditional histochemistry and have necessitated the development of alternative approaches to reconstruct their spatial distribution. Simply identifying a biomolecule presents only a partial picture; visualising spatial distribution and associating it with structural or other biochemical features is crucial to fully understand involvement in a defined biological mechanism or relevance to disease.
In the latter case, changes to the chemical properties and compartmentalisation of one or more disease-associated species often precede the appearance of cytopathology. Pathology may manifest from altered concentrations or chemical modifications that impart toxicity either independently or via aberrant interactions with other biological components, though simple redistribution of a biochemical without overt changes to chemical state or structure can also precipitate systemic dysfunction. Identifying when, and most importantly where, normal physiological conditions are disturbed is crucial for elucidating the molecular basis of disease. Accurately measuring the degree of change and where it occurs opens new avenues of investigation for targeted therapies and biomarker discovery.
The use of antibody labelling with a chemical marker was first described with fluorescent β-anthryl-carbamide conjugated to anti-pneumococcus in 1941.1 Biomolecules that were previously undetectable using histochemical staining and light microscopy suddenly became ‘visible’. Three-quarters of a century later, novel uses of technology (some by design, others through creative repurposing) and innovations across all sub-disciplines of chemistry have made available a diverse chemical imaging toolbox, spanning whole organs to single molecules and atoms.
Immunohistochemistry (IHC) has now become routine, although experimental protocols, reagents, detection techniques, and image analysis methods are constantly extending upon what information can be extracted. However, aside from a handful of clinically-approved protocols and the gradual uptake of automation outside high-throughput laboratories, it is also a somewhat imprecise science. Sample preparation requires substantial handling, chemical treatment and, when developing new assays, a degree of trial-and-error. When properly executed, contemporary IHC is enormously powerful for analytical biochemistry; be it as a cell-specific marker for cytometry or probe sensitive only to the subtlest disease-associated chemical change occurring from single point genetic mutations. Coupling IHC with chemical imaging, in all its forms, expands the scope even further—now direct associations between antibody targets and other biological components can be made in single-origin samples. That being said, integrating IHC with chemical imaging is not trivial—considerable planning and preparation, paired with a good understanding of the fundamental principles that underpin both IHC and chemical imaging as separate disciplines is a necessity.
In this Tutorial Review we provide a guide for designing IHC imaging experiments that capitalise on the latest developments in chemical imaging using analytical spectroscopy and mass spectrometry. Integrated IHC and chemical imaging as a research tool falls into two categories with substantial overlap in terms of possible applications: (i) routine IHC protocols are applied in sequence or in parallel with chemical imaging for interrogating spatial associations and relationships between labelled antibodies and endogenous chemicals; and (ii) novel applications of IHC with diverse and exotic chemical reporters that extend beyond the capabilities of routine methods. We summarise the imaging technology available with a focus on the abilities and shortcomings intrinsic to each. We then discuss where sample preparation steps both align with and deviate from standard conventions for IHC, and how the necessary steps to prepare samples for IHC may impact endogenous chemicals of interest. Using the lessons learned from traditional IHC and the common chemical imaging methods employed, we outline how future validation studies should be designed for potential clinical translation. Finally, we examine a selection of novel examples of IHC-chemical imaging, highlighting the achievements that have been made, outline how specific research questions are driving method development, and speculate on where the future potential of IHC-chemical imaging lies.
2. Redefining chemical imaging
The current definition of chemical imaging describes the use of spectroscopy to measure unique interactions between photons and matter in two, three, and occasionally four dimensions, though the emergence of imaging mass spectrometry (MS) in recent years justifies extension to include direct spatial profiling of chemical composition. This expanded definition shifts the focus to the shared ability of spectroscopy and spectrometry to collect multiple spectra reflecting specific chemical features present in a defined area, and to use this information to construct spatial measures of chemical distribution. This broader description of chemical imaging, when used in sequence and combined with IHC, emphasises that nearly every class of biomolecule is amenable to spatial analysis at resolutions extending from the mesoscale into the subcellular domain, depending on the method used. Rather than present an in-depth discussion of technologies available, we have summarised key features and considerations in the context of IHC-chemical imaging in Table 1 and direct the reader to key technical reviews and texts.| Spectroscopic detection | ||||||
|---|---|---|---|---|---|---|
| Analyte type | Target biochemicals and markers | Typical image resolution range | Advantages | Limitations | Key technical references | |
| Light microscopy | • Chromogenic dyes and stains | • Structural/organelle-specific markers | To 200–250 nm | • Rapid staining | • Diffraction limited | Thorn2 |
| • Enzyme-catalysed chromogens | • Dye-specific substrates (proteins, carbohydrates, nucleic acids, etc.) | • Digital image capture | • Relatively non-specific (dyes and stains) | |||
| • Chromogenic substrate-tagged antibodies | • Wide image resolution range | • Inconsistent staining and variable contrast | ||||
| • Capacity for high volume staining and image capture | • Quantitative analysis limited by observational scoring and interobserver bias | |||||
| • Automation | ||||||
| • Well suited to archived samples on standard glass mounts | ||||||
| Epifluorescence/confocal microscopy | • Fluorophores | • Fluorophore-tagged antibodies | To 200–250 nm | • High specificity | • Diffraction limited | Kubitscheck;3 Thorn2 |
| • Structural/organelle-specific markers | • Diverse range of targets | • Autofluorescence interference | ||||
| • Small molecules | • Live cell imaging | • Photobleaching | ||||
| • Fluorescent proteins | • Three-dimensional imaging (confocal) | • Phototoxicity (live-cell imaging) | ||||
| • Potential for quantitative analysis | • Spectral overlap limits multiplexing to <10 analytes | |||||
| Super-resolution microscopy | • Fluorophores | • Fluorophore-tagged antibodies | 250–20 nm | • Nanoscale imaging | • Requires bright fluorophores | Kubitscheck;3 Thorn;2 Vogler et al.4 |
| • Structural/organelle-specific markers | • High specificity | • Emerging technology with limited turnkey options | ||||
| • Small molecules | • Diverse range of targets | • Large field of views restricted by scanning speeds and high resolution | ||||
| • Fluorescent proteins | • Live cell imaging | • Resolution and multiplexing limited for large antigen-1°–2°-antibody complexes | ||||
| • Fast confocal imaging | ||||||
| • Reduced phototoxicity | ||||||
| Electron microscopy (EM) | • Structural components | • Heavy-metal contrast-enhanced ultrastructure | 200 nm–50 pm | • Molecular-level resolution | • Complex sample preparation and analysis | Fernandez-Leiro and Scheres5 |
| • Nanoparticles | • Gold nanoparticle-tagged antibodies | • Multiplexing possible with gold nanoparticle conjugates of differing diameters | • Requires 50–100 nm sections | |||
| • Simultaneous elemental analysis by energy-dispersive X-ray spectroscopy (EDX) | • Vacuum required for non-ESEM | |||||
| • Environmental scanning (ES)EM for hydrated specimens | • Potential radiation effects on hydrated samples in ESEM | |||||
| • Specialised sample mounts | ||||||
| X-ray emission/fluorescence spectroscopy | • Structural components | • Ultrastructure | 1 μm–10 nm | • Density and structural information from untreated samples | • Requires synchrotron radiation for high resolution imaging | McRae et al;6 Pushie et al.7 |
| • Elements | • Metal-labelled antibodies | • High specificity and multiplexing using metal-labelled antibodies | • Overlapping K-shell emission lines reduce sensitivity for many bioelements | |||
| • Spatial mapping of element oxidation state | • Time-consuming spectral deconvolution for imaging | |||||
| • No sample preparation when using cryostreams | • Photoreduction and radiation-induced observer effects | |||||
| • Potential for quantitative analysis | • Specialised sample mounts | |||||
| Infrared and vibrational spectroscopy | • Organic compounds | • Proteins, carbohydrates, nucleic acids, etc. | 10–4 μm (FTIR); 1 μm–400 nm (Raman) | • Rapid and simultaneous collection of over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 spectra using FPAs 000 spectra using FPAs |
• Detection of functional groups only is relatively non-specific | Vogler et al.4 |
| • Nanoparticles (for SERS) | • Vibrational tagged antibodies (for Raman spectroscopy) | • Live-cell imaging (Raman) | • General low sensitivity | |||
| • Gold nanoparticle conjugated antibodies (for SERS) | • Combined FTIR and Raman microscopes available | • Long spectral collection times | ||||
| • Selection of specific wavenumbers increases sensitivity | • Specialised sample mounts | |||||
| • Quantitative volumetric imaging | ||||||
| Atomic emission spectroscopy | • Elements | • Endogenous elements | 200–10 μm | • Simultaneous detection using CCD cameras | • Few reported applications in bioimaging | Kaiser et al.8 |
| • Metal-labelled antibodies | • Low-volume sampling with minimal sample damage | • Low sensitivity for many endogenous elements | ||||
| • Potential for quantitative analysis | • Low spatial resolution | |||||
| • Simultaneous LIBS and Raman imaging using common laser source | ||||||
| Spectrometric detection | ||||||
| Molecular mass spectrometry | • Organic compounds | • Proteins, carbohydrates, lipids, etc. | 200–1 μm | • Direct detection of high-abundance species | • Limited sensitivity for high mass compounds and low abundance biomolecules | Bodzon-Kulakowska and Suder;9 Buchberger et al.10 |
| • Metabolites and small molecules | • IHC and on-tissue for antigen-specific Fab and V fragments | • Low spatial resolution | ||||
| • Photocleavable mass tags | • Selective and sensitive photocleavable mass tags suitable for highly-multiplexed imaging | • Specialised sample mounts | ||||
| • Potential for quantitative analysis | ||||||
| • Wide range of application-specific ambient ionisation sources | ||||||
| Elemental mass spectrometry | • Elements | • Endogenous elements | 200–1 μm (LA-ICP-MS); 1 μm–100 nm (SIMS) | • High sensitivity | • Substantial matrix effects | McRae et al.6 |
| • Metal-labelled antibodies | • Simultaneous detection (for TOF-MS and SIMS) | • Low spatial resolution (for ICP-MS) | ||||
| • Small molecules (for SIMS) | • Potential for quantitative analysis | |||||
| • Commercial systems for highly-multiplexed imaging (MaxPar® reagents and LA-CyTOF-MS) | ||||||
In most cases, combining IHC and chemical imaging does not necessarily supersede histochemistry; rather, it offers an opportunity to gather more detailed information about the presence, quantity, and chemical state of specific biomolecules. Each chemical imaging technique normally excels at one particular task, such as how Fourier-transform infrared (FTIR) and Raman spectroscopy can plot spectroscopic qualities of the predominant chemical environment while mass spectrometry imaging (MSI) uses a more targeted approach to isolate and map specific chemicals by mass. While investigation of a specific target may be optimally achieved with single imaging technique, the true power is in spatial profiling of multiple chemicals and using this data to decipher the complex interplay occurring between them. The utility of chemical imaging in biology is further enhanced by the capacity to simultaneously measure and spatially associate biomolecules identified with exogenous tags and unlabelled chemical species.
3. A beginner's guide to immunohistochemistry for chemical imaging
Antibodies are glycoproteins with a specific affinity to a portion of a target biomolecule, known as an antigen. The antigen-binding site, or paratope, of an antibody is sensitive to the epitope of the antigen, which may be as small as a specific amino acid sequence (linear epitopes) or as large as part of the antigen tertiary structure (conformational epitopes). Antibodies are part of an organism's innate immune response and can be produced with specificity for nearly all biomolecules, including misfolded or mutated variants involved in specific disease states. However, even the most robustly-validated and clinical-approved IHC assays are prone to analytical error and interference, and the number of research-only applications dwarfs those that have been approved for clinical use. Prior to explicit guidelines for IHC analytical validation issued by the US College of American Pathologists (CAP) Pathology and Laboratory Quality Center in 2014 (discussed in detail in Section 3.4),11 clinical pathology laboratories had no set instructions to follow, and instead either replicated prior examples of validated and Food and Drug Administration (FDA)-approved predictive immunoassays (such as the oncogene-product human epidermal growth factor 2 [HER2]), or developed internal method validation protocols.Considering that IHC-chemical imaging is still in relative infancy, now is a critical time to establish consensus-driven guidelines for method development and eventual clinical translation. Instituting such standards cannot be the responsibility of one laboratory or research group, though we can identify key considerations in designing chemical imaging experiments that employ antigen-specific detection for both immediate research use, and as parameters for future multi-site concordance studies. It is also important to recognise that, in a rapidly growing field, new confounding factors are constantly being identified, and designing future experiments should encompass these as they are reported.
At this point it is also important to make a clear distinction in the nomenclature used throughout this Tutorial Review: when referring to an analyte, we are describing the species that is directly detected using chemical imaging, whereas antigen is used to describe the associated biomolecule (Fig. 1).
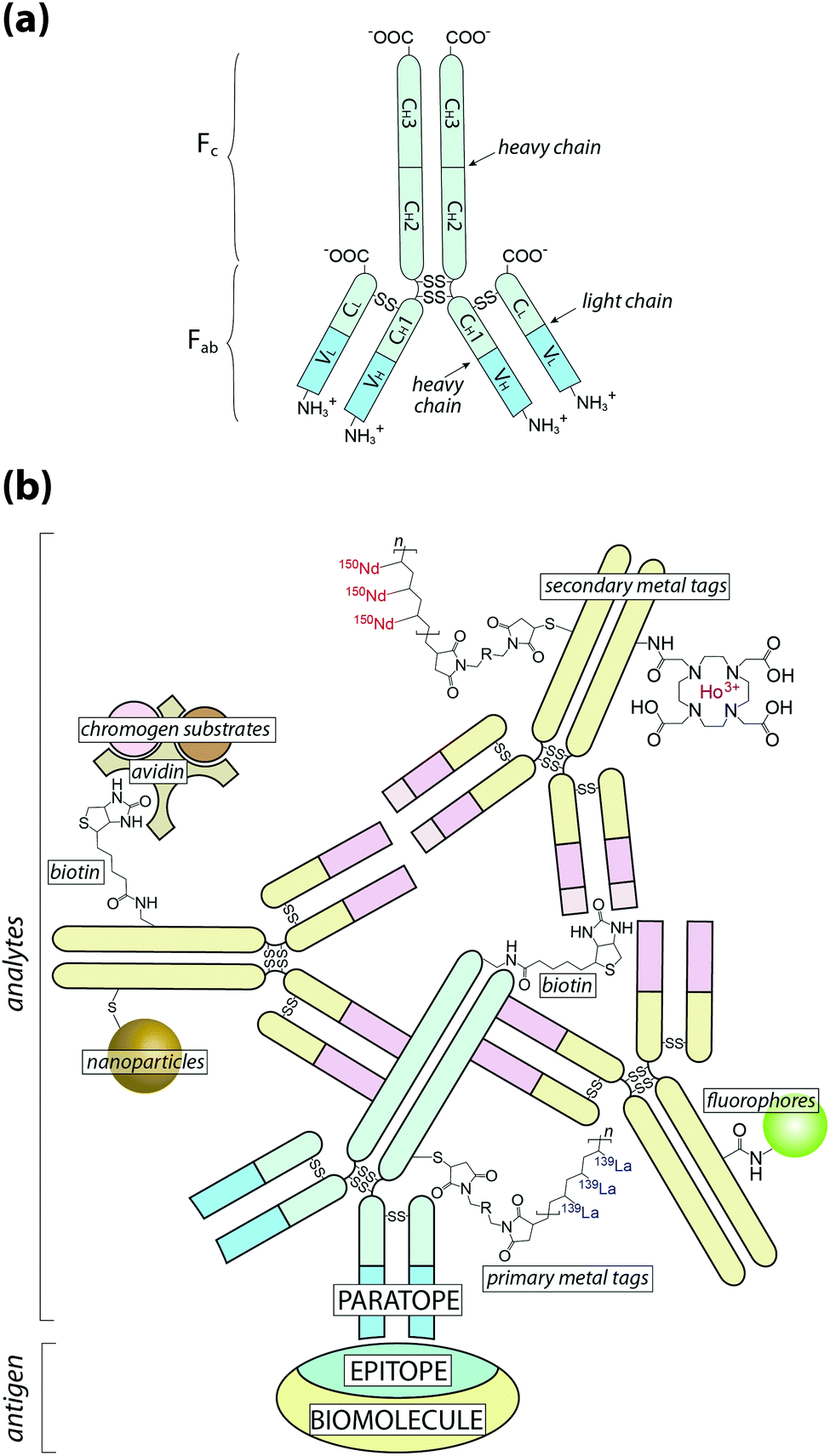 | ||
| Fig. 1 Schematic of antigen–antibody binding for detecting biomolecules. (a) Antibodies are large glycoprotein immunoglobulins with a ‘Y’-shaped monomeric structure. The two ∼50 kDa heavy chains consists of three constant domains (CH1–3) and a N-terminus variable domain (VH) that forms one half of the antigen-binding site. The two light chains (∼25 kDa) contain a constant domain (CL) at the C-terminus and a corresponding variable domain (VL) to complete the antigen-binding paratope. Interchain disulfide bonds link the heavy and light chains within the constant domains, with two additional disulfide bridges at the hinge region between CH1 and CH2. The Fab region contains the paratope, whereas the Fc region is the site of secondary antibody binding. (b) Primary antibodies recognise specific amino acid sequences or structural features of the antigen, referred to as the epitope, at 1:1 stoichiometry. Signal amplification is typically achieved by secondary antibodies labelled with a fluorophore or chromogen, though metal tags have specific applications. Multiple binding sites in the Fc region increases the number of chemical analytes that can be associated with a single antigen (see Fig. 2). | ||
The class of analyte suitable for IHC-chemical imaging is limited only by what can form a stable and specific association with either the primary or secondary antibody. Traditional IHC relies on spectroscopic properties of the analyte, such as propensity to form a coloured precipitate with specific stains or fluoresce when excited. Manipulating photon–matter interactions and improving how they are detected drives development of new technology. Fluorescence microscopy can therefore be considered as both routine—it is one of the most widely used laboratory techniques in the world—and intersecting the cutting-edge of physics, chemistry and biology. Recent technical advances, such as overcoming the light diffraction limit to allow super-resolution microscopy certainly offer unique opportunities, though ‘routine’ fluorescence microscopy is still central to many IHC-chemical imaging applications. This highlights a key learning point: the significance of an imaging approach is not necessarily based in technology, but how it is applied to answer a primary research question. The development of a new analyte, its integration into an existing IHC protocol, and use in aiding visualisation of the surrounding microchemical environment is as important as emergence of new imaging techniques. In this Tutorial Review, we focus on selecting (or developing) the analyte rather than imaging technique, acknowledging that certain analyte classes are applicable to multiple analytical methods.
As most protocols involve labelling an antibody with an analyte prior to incubation with a sample, standard protocols for IHC are generally applicable. Depending on the analyte, an IHC-chemical imaging experiment may use primary antibodies pre-labelled with a chemical reporter, or apply labelled secondary antibodies as an amplification step (Fig. 2). Both approaches have intrinsic advantages and limitations related to the choice of analyte and imaging technique. In this section, we provide a guide to developing an IHC-chemical imaging experiment, emphasising the importance of good experimental design based on the principles of analytical method validation.
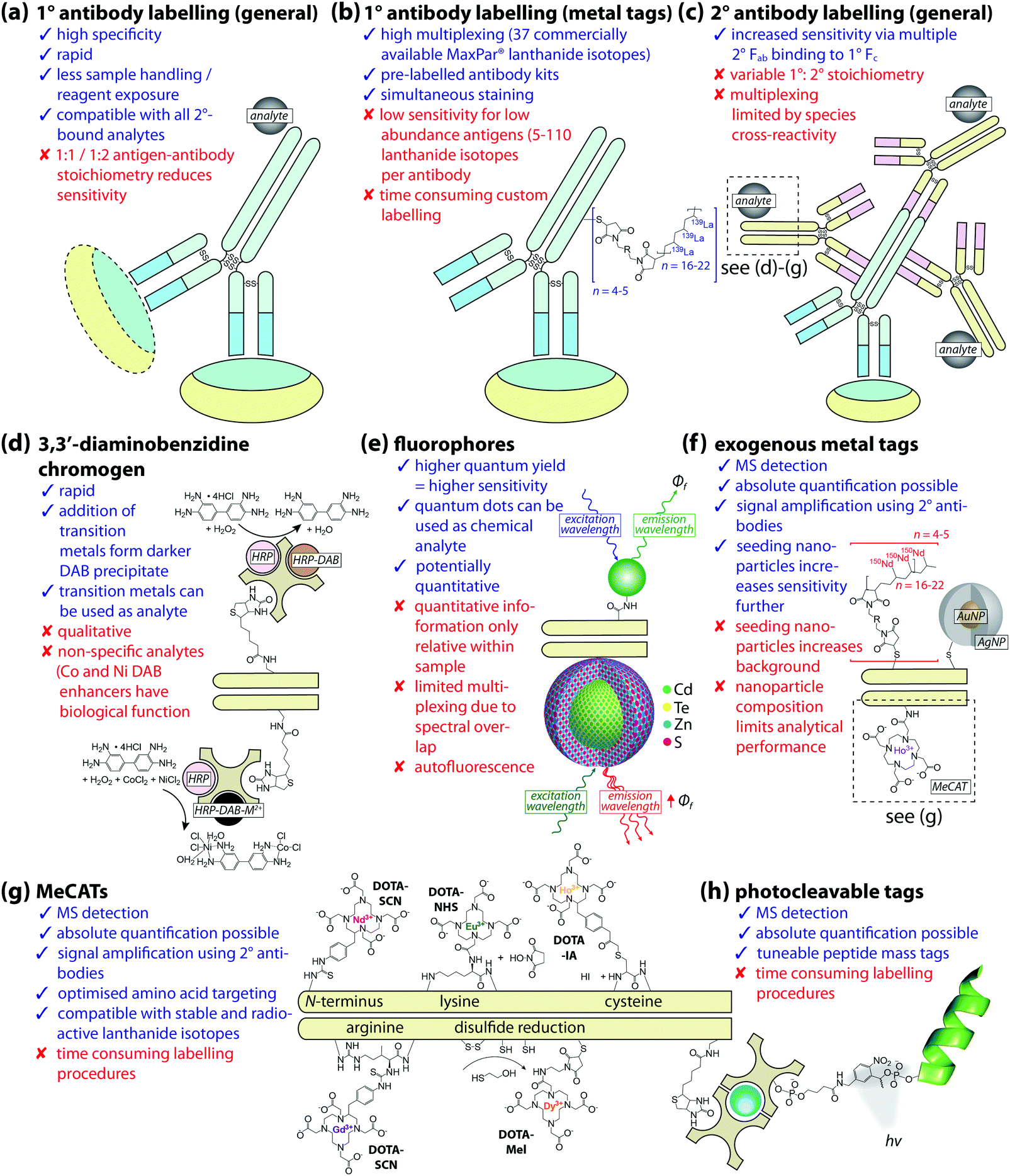 | ||
| Fig. 2 Antibodies and analytes for IHC-chemical imaging. (a) Primary (1°) antibodies provide the highest specificity and greatest potential for multiplexing using unique chemical reporters (such as isotopically-enriched lanthanides); (b), though have lower sensitivity. (c) Secondary (2°) antibodies raised against the primary host organism are therefore used to amplify signal and improve detection limits, coming with their own limitations. (d) Light microscopy employs chromogens, normally linked to the 2° IgG via a biotin–streptavidin complex with a peroxidase (shown here as horseradish peroxidase, or HRP) substrate for 3,3′-diaminobenzadine (DAB) oxidation. Addition of divalent metal ions (e.g. Ni2+, Co2+) forms a darker precipitate, and while these could be used as a proxy for imaging by LA-ICP-MS, caution should be taken due to potential interference from physiological sources. (e) Fluorophores use an excitation source to induce emission of Stokes-shifted photons with brightness proportional to the product of the fluorophore quantum yield (ϕf) and extinction coefficient. Quantum dots are photon-emitting nanomaterials with cores comprised of group II–VI, group III–V, or group IV–VI binary compounds and zinc sulfide shells that produce stable, bright, and narrow emission spectra which can be tuned by manipulating particle size. Quantum dots can be conjugated to antibodies via several methods, most commonly using –COOH, –NH2 and –SH functional groups on the particle surface to crosslink with amino acids. In addition to superior ϕf, near-infrared properties of quantum dots can be exploited with IR imaging, and the exogenous metal/metalloid composition with atomic spectrometry. (f) Gold and silver nanoparticles (Au/Ag NP) are particularly useful in MS imaging for amplifying signal attributable to low-abundance antigens, or as a substrate for SERS; while MeCATs use 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) macrocycles to chelate lanthanide ions, including radionuclides. (g) Addition of prosthetic reactive groups permits selective binding of the DOTA chelate to specific amino acid residues on the IgG carrier (isothiocyanate, SCN; N-hydroxysuccinimide, NHS; maleimide, Mel; iodoacetamide, IA). (h) Photocleavable tags are also used in MS imaging, and typically consist of a peptide of known mass and structure linked to a photosensitive moiety and biotin–avidin complex, represented here with an avidin-coated glass bead. | ||
3.1 Designing an IHC-chemical imaging experiment
The first question an analyst should ask when embarking on experiments that integrate IHC with chemical imaging is: can the intended outcome provide information not currently achievable with existing protocols and technology? Developing a method using a novel chemical analyte that produces a similar result to that which could be obtained using traditional methods may be of interest to a subset of researchers, though its potential impact within the wider field of anatomical pathology will be limited. Where it is clear that traditional IHC approaches are insufficient to answer the question at hand and that chemical imaging offers capabilities that overcome such limitations, the next step is to identify what properties of the sample in question are subject to analysis. These include, but are not limited to, quantifying the amount of endogenous chemicals present, spatial assessment of how multiple antigens and biochemicals interact, high-resolution imaging of subcellular compartmentalisation, or even providing complementary information for traditional IHC methods. Here we provide here a guide based on known quantities that various chemical imaging methods can inform. The reader should not consider this list exhaustive as the discipline is rapidly evolving, and the emergence of new technologies will see the scope of IHC-chemical imaging broaden.Immunohistochemical detection of an antigen can be divided into two categories: (i) direct detection, where the primary antibody is conjugated to a suitable analyte in the Fc domain; or (ii) indirect detection, where an immunoglobulin (typically IgG) raised against the primary host species (e.g. rabbit anti-mouse IgG for a murine-derived primary antibody) with a conjugated analyte is introduced. Direct detection requires fewer sample processing steps and has a high capacity for multiplexing by eliminating species cross-reactivity of secondary antibodies, though in the majority of cases antibody-to-antigen stoichiometry is 1:1 which can limit sensitivity relative to indirect detection. The signal intensity attributable to the analyte is proportional to its concentration—which is technically an indirect measure of total antigen recognition by the primary antibody, though we use terms specific to IHC protocols in this Tutorial Review—and as multiple secondary antibodies can bind to the Fc region of a primary immunoglobulin, signal amplification is achieved by increasing the number of analytes associated with a single antigen.
Sensitivity is also determined by the detection technique. Spectroscopic chemical imaging requires the ability to detect and spatially resolve subtle changes in energy resulting from interactions between photons and matter, while mass spectrometry-based imaging depends on efficient physical transfer, separation, and detection of material directly removed from the sample surface. For a technique such as matrix assisted laser desorption/ionisation (MALDI) MS, this can be as little as 1–10 attomoles of total material per pulse (depending on laser and matrix conditions),12 within which the total analyte may be present at levels several orders of magnitude lower.
The amount of detectable signal that can be attributed to a defined area determines both the sensitivity and achievable spatial resolution (see Section 3.1.3) of an imaging technique. Technical limits such as focal point, laser pulse diameter and the dynamic range of the detector are nominally fixed, making control of variables in sample preparation and IHC protocols the major determinant of analytical sensitivity. A successful IHC protocol applied to chemical imaging should demonstrate reproducible measurements of analyte areal density with high signal-to-noise.
Equal attention should also be given to the practical detection limits for endogenous chemicals of interest. Factors to consider include whether the dynamic range of the detector is compatible with the different concentration ranges of the analytes and associated biochemicals, if further chemical derivatisation is necessary, and whether an additional mode of detection should be used in sequence. It is not uncommon for individual factors affecting sensitivity of either the analyte or endogenous chemicals to impose a compromise in imaging parameters used.
With the exception of secondary-ion mass spectrometry (SIMS), MS imaging techniques are typically limited to mesoscale (>1 μm) resolution. This is primarily due to the same determinants of sensitivity, and resolution will improve as MS technology does.
3.2 Developing an IHC-chemical imaging experiment
The next question to be addressed when designing an experiment is: how can the desired outcome be met? This requires an understanding of the variables that influence analytical validity and knowledge of the strengths and limitations of the imaging technique used. There is no universal protocol or checklist to follow for integrating IHC with chemical imaging—antibodies and biological systems are a far cry from the standard reference materials an analytical chemist would typically use to develop and validate a new method. Acknowledging that combined IHC and chemical imaging is an emerging field where potential measures and experimental outcomes are still being actively developed, we provide a general guide that can be adapted to suit the experiment to be performed.Each step exerts some influence on the microchemical environment, dependent on preparation method and the class of chemical affected (Fig. 3). A true depiction of an undisturbed biological system is inherently unobtainable; even live-cell imaging is subject to an ‘observer effect’, a concept that acknowledges chemical alterations can result from the imaging technique itself.14 As such, there is no ‘standard’ of a completely undisturbed system against which an image can be compared, and the few studies that have specifically measured chemical alterations resulting from sample preparation indicate the effects are wide-ranging and cover every class of biochemical. When developing an IHC protocol intended to assess spatial relationships between antibody-bound antigens and native species, the aim should be to both minimise ex vivo perturbations to natural chemical distribution and, while recognising that some degree of modification is unavoidable, ensure all samples undergo identical preparation procedures.
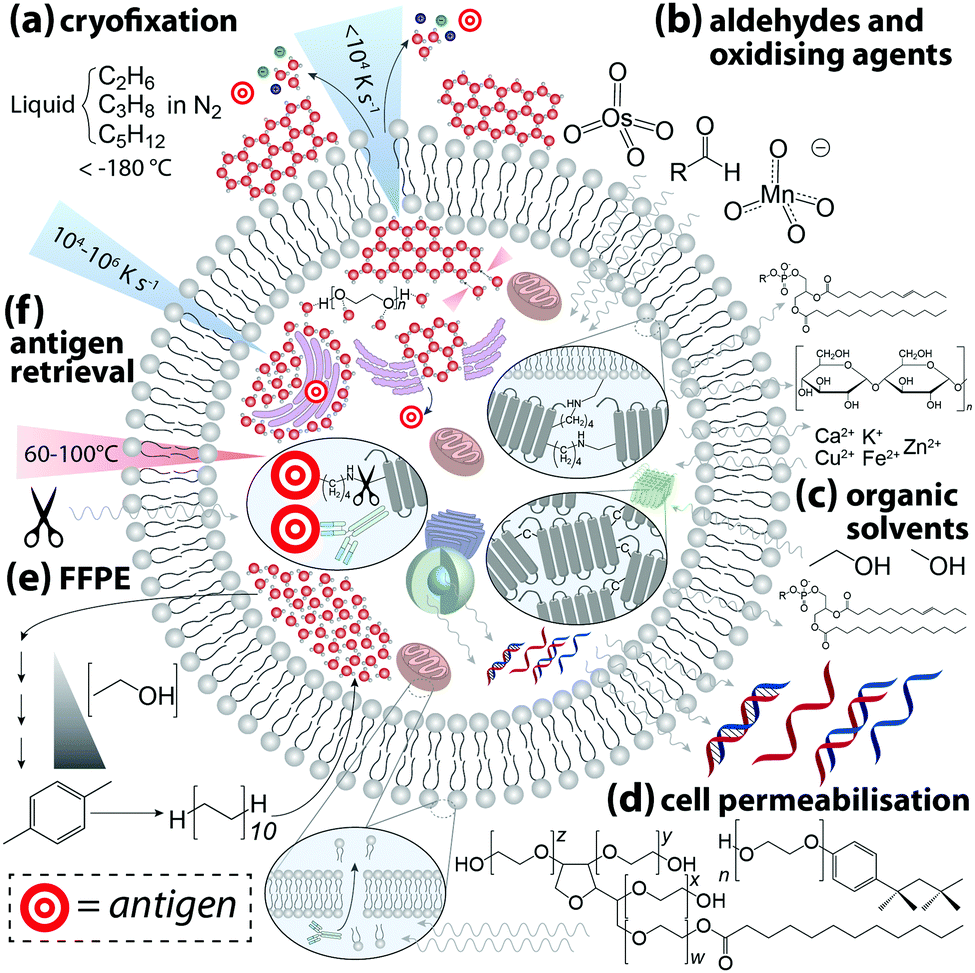 | ||
| Fig. 3 Sample processing steps for IHC and associated effects on native cell biochemistry. (a) The aim of cryofixation is to cool the cell at a rate that prevents H2O molecules forming an ordered crystalline structure in favour of an amorphous glass-like disordered arrangement of supercooled H2O molecules that retains mechanical properties of a liquid. If the rate of cooling is too slow spontaneous nucleation and growth of ice crystals can rupture membranes, damaging cellular microstructures and causing both intra- (e.g. release of immature proteins from inside ribosomes on rough endoplasmic reticulum into the cytoplasm) and intercellular (e.g. interstitial exchange of molecules and ions through ruptured cell membranes as a result of changing concentration gradients) chemical redistribution. Cryoprotectants (e.g. polyethylene glycol) can hinder ice crystal formation by temporarily stabilising liquid H2O via hydrogen bonding. (b) Aldehydes and oxidising agents diffuse across the cell membrane and covalently crosslink lysine residues with an aliphatic bridge, disrupting tertiary protein structure while also anchoring proteins to cell membranes. Proteins, phospholipids and carbohydrates are partially lost to aldehyde fixation, and labile metals undergo two-way exchange. (c) Alcohols and organic solvents rapidly denature proteins to induce aggregation while simultaneously permeabilising membranes. This causes a loss of phospholipids and leaching of nucleic acids into the cytosol. (d) Non-ionic surfactants (e.g. polysorbate-20, Triton X-100) permeate bilipid membranes and allow antibodies to penetrate cells and organelles, which undoubtedly causes some degree of chemical redistribution. (e) Long-term archiving of biological samples at room temperature requires protein stabilisation, dehydration, and substitution of H2O with aliphatic hydrocarbons via formalin fixation and paraffin embedding (FFPE). (f) Fixation and preservation may mask the target epitope, thus heat or proteolytic (e.g. trypsin) cleavage of crosslinks, known as antigen retrieval, may be necessary to expose antigens to the antibody paratope. | ||
Cryofixation is the least damaging form of sample preservation and is the preferred method for the majority of chemical imaging techniques, though it is also the most technically challenging. Rapid uniform cryogenic freezing vitrifies a sample, transforming liquid H2O into molecularly-disordered amorphous ice. By preventing H2O molecules from assembling into a hexagonal lattice structure, damage to cell morphology resulting from ice crystal growth is minimised. Cryoprotectants including polyethylene glycol and disaccharides can also be added to reduce crystal formation. Water is then removed by either addition of cooled organic solvents (freeze-substitution) or sublimation via low pressure lyophilisation, after which IHC can be performed on resin-embedded sections at standard laboratory conditions. Optimal cryofixation methods vary depending on sample type, and detailed protocols can be found in the book by Wolkers and Oldenhorf.15 Excellent preservation of cell ultrastructure makes cryofixation particularly attractive for electron microscopy, and subsequent immunostaining with gold–nanoparticle conjugated antibodies has been used to demonstrate superior labelling efficiency of insoluble proteins compared to chemically-fixed samples.16 Rapid freezing of whole tissue samples followed by cryosectioning and chemical fixation of cut tissue is an alternative approach for imaging techniques where damage to cell ultrastructure is beyond the practical image resolution obtained.
Aldehydes, oxidising agents, and organic solvents are the most commonly-used chemical fixatives. Aldehydes and oxidants forms covalent cross-links between amino acids, stabilising protein structure and anchoring proteins to cell membranes. Organic solvents denature proteins and induce the formation of insoluble aggregates. Fixation efficiency is determined by adequate penetration of a cell; therefore, exposure time, sample thickness, and temperature are variables that may produce inconsistencies. Organic solvents have the advantage of simultaneously permeabilising the cell, while aldehyde fixation is normally followed by immersion in a buffer containing a non-ionic surfactant. By removing lipids from the outer cell membrane antibodies are able to penetrate the cell, though removing such barriers allows soluble chemicals to diffuse beyond their native cellular compartment.
Hackett et al.17 applied a multimodal imaging approach using both FTIR and PIXE imaging to assess effects of formalin fixation on the native microchemistry of neurological tissue. Using cryofixed and freeze-substituted tissue as a reference, FTIR spectra demonstrated marked decreases in vibrational intensities assigned to amides, olefinic carbon–hydrogen bonds, covalent carbon–oxygen bonds, and phosphates, indicating loss of proteins, carbohydrates, lipids, and lipid oxidation products. Even simple immersion in phosphate buffered saline had measurable effects on organic content. Hobro and Smith18 capitalised on the ability of Raman spectroscopy to image live cells to evaluate the effects of immersion in aldehydes, organic solvents, and permeabilising surfactants. All reagents produced measurable changes in Raman spectra indicative of chemical redistribution and structural modification. This study is particularly informative as it not only demonstrates the differential effects of each reagent, but also how ambient conditions and order of chemical exposure can be optimised to preserve a specific class of biomolecule.
Unsurprisingly, both studies found that cytoplasmic proteins are particularly susceptible to leaching and redistribution. The same can also be said for labile and weakly-bound inorganics; quantitative elemental mapping of formalin-fixed brain tissue sections using PIXE showed near-total loss of intracellular potassium and reduction in chlorine. Conversely, calcium and the major biologically-relevant transition metals (iron, copper and zinc) all showed increased concentrations within cells. Similar observations were made by Chwiej et al.,19 who used X-ray fluorescence spectroscopy to show differential influx and efflux of biologically-relevant elements in various areas of rat brain sections following formalin or paraformaldehyde fixation, with paraffin-embedded counterparts showing a marked decrease in nearly all measured elements. External contamination is clearly a point of consideration; when high-purity reagents are used, aldehyde fixation and sucrose cryoprotection resulted in loss of most biologically-relevant metals.20 The degree of metal loss is not uniform, and species-specific effects can be attributed to variable affinities of metals to protein ligands, per the Irving–Williams series:21
| Mg2+ < Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ ≈ Zn2+ |
Finally, cross-linked proteins often mask an antigenic site and antigen retrieval may be necessary to expose sufficient numbers of epitopes prior to antibody incubation. The two most common methods used are: (i) heat-induced, where the sample is incubated in buffers containing surfactants and EDTA at high temperature and pressure; and (ii) protease-induced, where protein cross-links are enzymatically cleaved. Both approaches undoubtedly cause modifications to other biomolecules, and it is fitting that the authors of the first heat-induced antigen retrieval protocol commented on the need for standardisation in IHC, particularly regarding chemical fixation.24 Over a quarter of a century later, many of the cautions raised have still not been addressed.
Chemical alterations to the sample during preparation for IHC are ultimately unavoidable. Therefore, steps taken should be sufficient to produce adequate (and equivalent) immunostaining using conditions that are both compatible with the antibody used and have minimal effects on the native biochemicals being analysed.
Non-specific binding below the ordinary threshold of detection for routine microscopy may cause significant interference when more sensitive chemical imaging modalities are used. Conversely, higher antibody concentrations may be necessary for detecting certain analytes. When a new antibody is being tested, a titration experiment should be performed specific to the detection method used where antibody concentration is the only variable. This is also the ideal point to prepare an isotype control, where the primary antibody is substituted with an otherwise-identical immunoglobulin raised against an antigen not present in the sample to determine levels of non-specific binding (Fig. 4). These should also be conducted in parallel with conventional immunohistochemical detection by optical microscopy.
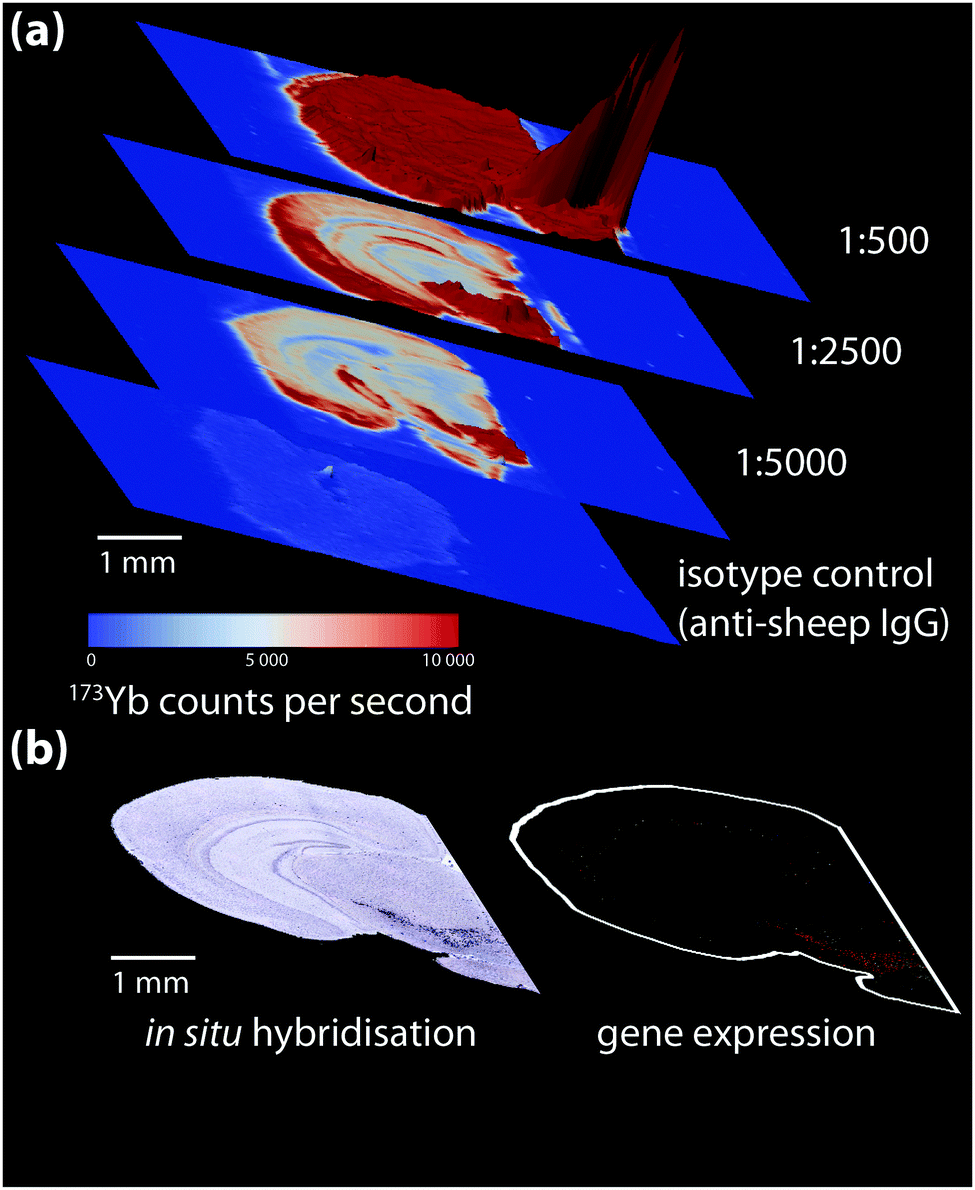 | ||
| Fig. 4 (a) Isotype control and titration experiment for a 173Yb-labelled sheep anti-mouse tyrosine hydroxylase (TH) primary antibody-stained coronal mouse brain section, imaged using LA-ICP-MS. 173Yb-labelled sheep IgG, which should have no specific reactivity to mouse tissue, shows a uniform pattern of non-specific binding slightly above background. Increasing antibody concentration increases background to a point where non-specific binding decreases signal-to-noise to a point that obscures antigen-specific analyte signal. (b) In situ hybridisation and gene expression maps from the Allen Brain Atlas25 can be used as additional reference for wild-type protein distribution; here, sections taken at the same anatomical coordinates shows TH expression is mainly in the lower right quadrant of the brain hemisphere containing the basal ganglia formation. | ||
Multiplexing is dependent on the ability of an imaging technique to resolve signals from each chemical reporter. For fluorescence microscopy, brightness and areal density of the fluorophore determine intensity of emission. Peak broadening proportional to fluorophore density can produce spectral overlap and potential false-positive measurements. A typical fluorescence IHC assay uses emission from three sources (e.g. two fluorophore-labelled secondary antibodies and a dye such as 4′,6-diamidino-2-phenylindole [DAPI], that fluoresces when bound to a specific structural marker, in this case DNA), though some commercial autostainer and microscopy systems have been optimised for up to seven specific fluorophores. Quantum dots (QDs) offer several advantages over traditional fluorophores. Fluorescent emission of QDs is proportional to size, with larger diameter dots emitting into the near-IR region. QDs are resistant to photobleaching and brighter than conventional fluorophores, and surface chemistry can target specific ligands on whole antibodies or antibody fragments, as well as single-chain variant fragment fusion proteins. A detailed overview and protocol for designing multiplexed IHC experiments with QDs can be found in Xing et al.26
Even greater flexibility is provided by MS for imaging metal-tagged antibodies, which shifts the limiting factor from spectral overlap to the number of unique combinations of primary and secondary antibody host species used for indirect detection (Fig. 5a), or the number of antibodies to which unique photocleavable mass tags can be conjugated for molecular mass spectrometry (see below). The true power of elemental MS imaging for multiplexed IHC is evident when applied to direct detection, which is limited only by the number of separate analytes that can be associated with each primary antibody (Fig. 5b). This was demonstrated by Giesen et al.,27 who used commercial MaxPar® monoisotopic lanthanide tagging kits to label and simultaneously image 32 antibodies at 1 μm resolution (Fig. 5c) with a laser ablation system coupled to a CyTOF-MS (an ICP-MS design optimised for mass cytometry). This particularly elegant study not only demonstrated a near-five-fold improvement on the maximum number of antigens that can be detected using fluorescence microscopy; it also described an image processing algorithm using lanthanide-labelled structural features to segment and extract data from single cells which subsequently underwent spanning-tree progression analysis of density-normalised events (SPADE) to characterise tumour heterogeneity in breast cancer tissue microarrays. Designed for CyTOF mass cytometry, SPADE examines multidimensional datasets to characterise cell–cell interactions and transitions to construct hierarchical trees of diverse cell populations, it has enormous potential for examining spatial interactions between antigens and associated endogenous chemicals. Angelo et al.28 used a similar IHC protocol with nanoSIMS detection and their own cell-segmentation algorithm to image 10 analytes at 200–300 nm resolution, and used both signal intensity and categorical expression (i.e. positive or negative) to build composite images representing immunohistochemical and phenotypic features of malignant cells (Fig. 5d). Both studies used classical light and fluorescence microscopy to demonstrate specificity and sensitivity (Fig. 5e and f). The LA-CyTOF-MS system originally described by Giesen et al.27 was recently commercialised as the Helios™ by Fluidigm, from which the first applications are now emerging.29 By design, the Helios™ system is optimised for high-mass lanthanide detection, thus simultaneous imaging of biologically-relevant elements, most of which fall below the 80 amu threshold, is hardware-limited. New ICP-TOF-MS systems capable of concurrently measuring both low- and high-mass elements, such as the TOFWERK design, will eventually be used for correlative LA-ICP-MS imaging of multiplexed IHC with endogenous metals and heteroatoms.
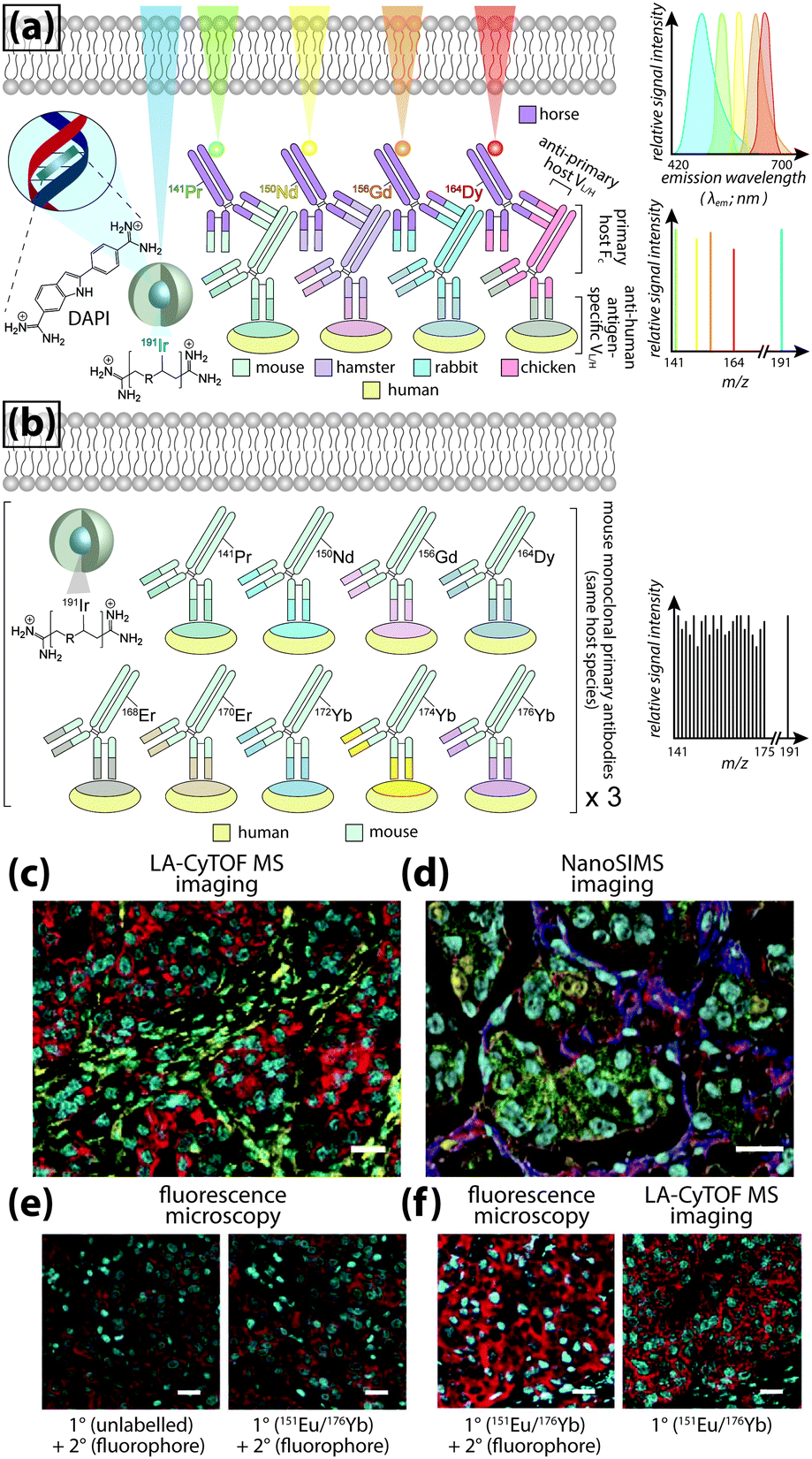 | ||
| Fig. 5 Multiplexed IHC using fluorescence microscopy and MS imaging. (a) Simplified conceptual schematic of quintuplexed IHC using four secondary antibodies and a DNA-specific probe. Primary antibodies for human antigens raised in four different species are indirectly detected by secondary antibodies with conjugated fluorophores. Resolving signal associated with each antigen requires complex optics and fluorophore emission spectra with minimal overlap. An alternative to overcome spectral overlap uses secondary antibodies labelled with monoisotopic lanthanides (coloured here to match fluorescent tags) and an iridium-containing DNA intercalator, followed by MS imaging. (b) Direct detection of primary antibodies capitalising on the resolving power of MS can be used to substantially increase the number of target antigens. (c) Three-colour LA-CyTOF-MS image of 174Yb-tagged cytokeratin 8/18 (red), 176Yb-tagged H3 (cyan) and 162Dy-tagged vimentin (yellow) from a 32-plexed human breast cancer tissue microarray.27 (d) Composite nanoSIMS image of mass intensities for cytoplasmic 168Er-tagged actin (red), 158Gd-tagged E-cadherin (green) and 154Sm-tagged vimentin (blue) with nuclei categorically classified as either 139La-ERα + 145Nd-PR positive (aqua) or 139La-ERα + 145Nd-PR + 150Nd-Ki67 positive (yellow).28 (e) Addition of lanthanide tags to primary antibodies (151Eu-tagged HER2 [red] and 176Yb-tagged H3 [cyan]) does not affect binding of fluorophore-conjugated secondary antibodies, and (f) MS imaging reproduces immunofluorescence specificity.27 Panels (c), (e) and (f) reproduced with permission from Giesen et al.;27 panel (d) reproduced with permission from Angelo et al.28 (both Nature Publishing Group, Copyright 2014). | ||
Photocleavable mass tags can be used in a similar approach for multiplexed IHC experiments using molecular MS imaging, allowing detection of intact proteins normally outside the mass range of TOF mass analysers. Here, the analyte consists of a molecule or peptide with a known mass linked to the antibody via an organic linker (either directly or as part of an avidin–biotin complex) with a photo-sensitive region that releases the mass tag when exposed a specific wavelength, normally that of the MALDI ionisation source. By not applying a matrix prior to analysis, signal-to-noise ratios for the applied mass tags allow for highly sensitive targeted multiplexed detection using both primary antibodies30 and enzyme activity probes.31 Subsequent matrix application and traditional MALDI-MS imaging can then be used to examine spatial associations between mass-tagged antigens and endogenous biomolecules.
Most examples of IHC-chemical imaging using vibrational spectroscopy take advantage of stimulated Raman scattering, which produces vibrational transitions around 150 times narrower than fluorescence emission spectra. Wei et al.32 synthesised a panel of 14 π-conjugated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C or CN-containing dyes with isotopic 13C and 15N substitution to supplement six commercial Raman-scattering dyes and four fluorophores to simultaneously resolve 24 analytes in multiplexed IHC-stained cultured cells and tissue sections. Functionalisation of antibodies with materials such as gold and silver nanomaterials can be used with surface enhanced Raman scattering (SERS) and surface plasmon resonance spectroscopy for multiplexed imaging, where the size and shape of conjugated particles can be manipulated to produce a unique, resolvable signal.33
C or CN-containing dyes with isotopic 13C and 15N substitution to supplement six commercial Raman-scattering dyes and four fluorophores to simultaneously resolve 24 analytes in multiplexed IHC-stained cultured cells and tissue sections. Functionalisation of antibodies with materials such as gold and silver nanomaterials can be used with surface enhanced Raman scattering (SERS) and surface plasmon resonance spectroscopy for multiplexed imaging, where the size and shape of conjugated particles can be manipulated to produce a unique, resolvable signal.33
3.3 Validating IHC-chemical imaging experiments
To date, most applications of IHC using chemical imaging have been proof-of-concept and focused primarily on the analytical technique used, as opposed to the full suite of analytical validation steps that would ordinarily be taken for more routine experiments where a known target is isolated, identified, and quantified. Fitzgibbons et al.'s Principles of Analytic Validation of Immunohistochemical Assays11 is required reading when developing any IHC-based chemical imaging experiment. Although the 14 guideline statements are geared toward eventual FDA approval of a new IHC method, they are conceptually analogous to standard validation steps common to analytical chemistry. When setting the rubric, the CAP approached the issue with the overarching question of “[w]hat is needed for initial analytic assay validation before placing any IHC test into clinical service and what are the revalidation requirements?”.11 Rather than dissect the 14 guideline statements, we will examine the five scope questions on which Fitzgibbons et al. based their recommendations, and use a hypothetical experiment studying copper metabolism in the mouse brain using LA-ICP-MS (Fig. 6a) to illustrate how analytical validation can be applied to any research-based IHC chemical imaging scenario.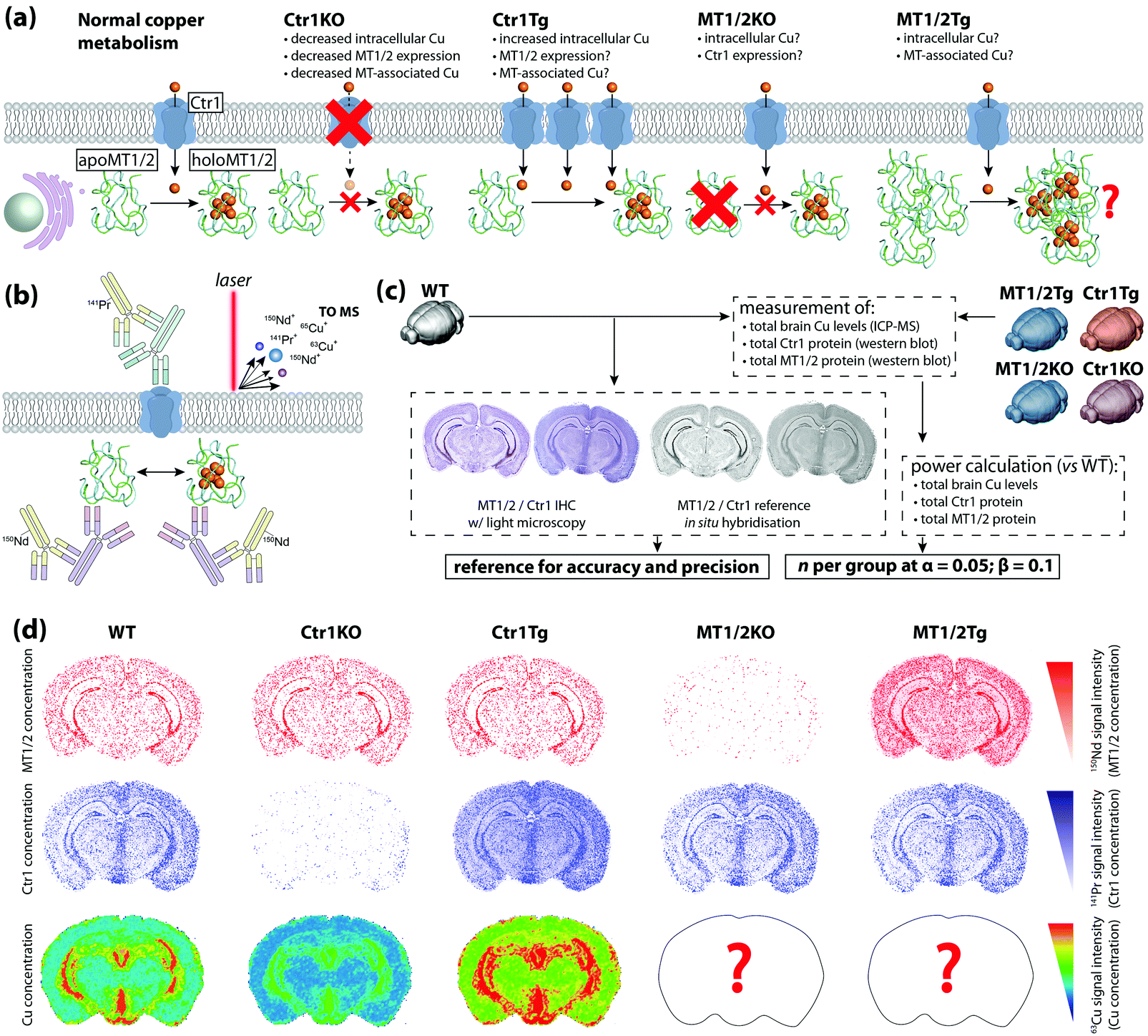 | ||
| Fig. 6 Hypothetical experimental design and validation using IHC-chemical imaging. (a) Two major regulators of cellular copper homeostasis are the intramembrane protein copper transporter 1 (Ctr1) and cytosolic metallothionein (MT) family, of which MT1 and 2 isoforms are most abundant. Knock-out (KO) of Ctr1 decreases cellular concentration, whereas transgenic overexpression (Tg) increases copper content. The aim of this hypothetical experiment is to compare Ctr1 modulation with MT1/2 KO and Tg mice with respect to brain copper concentration and distribution. (b) Ctr1 and MT1/2 are detected using modified IHC protocols with lanthanide-tagged secondary antibodies, and distribution of lanthanides and endogenous copper imaging using LA-ICP-MS. (c) Several routine techniques including standard IHC with light microscopy detection, use of reference in situ hybridisation databases,25 total tissue copper assay by ICP-MS, and western blotting for relative protein concentration are used to determine accuracy, precision and sample size. (d) When applied to a fully developed IHC-LA-ICP-MS method, spatial copper distribution in the brain can be compared and contrasted with wild type and Ctr1 KO/Tg animal. | ||
In our hypothetical experiment, a multiplexed approach using lanthanide-tagged secondary antibodies was developed and applied to image two major copper regulatory proteins, copper transporter-1 (Ctr1) and metallothionein (MT) in sections from fresh-frozen, paraformaldehyde-fixed wild-type and transgenic murine brains using modified IHC protocols, while simultaneously monitoring how regional copper levels are affected by ablation and overexpression of either protein (Fig. 6b). The animals used were the wild-type background strain (hereafter WT), heterozygous Ctr1 mice (Ctr1KO)34 and a hypothetical overexpressor (Ctr1Tg; overexpression of Ctr1 and resulting copper accumulation has been demonstrated in vivo34), combined MT1/2-null (MT1/2KO) and transgenic overexpressors (MT1/2Tg).35 Analytical sensitivity and specificity were assessed from the outset using antibody titrations and isotype controls to experimentally determine the optimal antibody concentrations.
Two questions regarding specificity can be raised. Firstly, can MT1 be distinguished from MT2, and could cross-reactivity with MT3 produce false-positive data? Most commercial antibodies target N-terminus epitopes in the β-domain that share 100% homology between MT1 and 2; differentiation of MT1 and 2 would require raising monocloncal antibodies specific to one isoform and confirmation by ELISA.36 Within the context of this experiment, where MT1 and 2 perform an equivalent function and manipulation of protein expression was universal, differentiation of isoforms was not considered necessary. Regarding MT3, the β-domain epitope shares only three common residues and cross-reactivity is absent36—this could be experimentally demonstrated by the presence of MT3 staining in MT1/2KO animals.
The second is whether MT and non-MT copper can be distinguished. In this case, only spatial association between MT1/2 and copper can be inferred from LA-ICP-MS images as the analyte marking MT1/2 spatial distribution is indicative of the protein only, whereas copper images represent all chemical forms in the sample. Thus, appraising the direct relationship between MT1/2 and associated copper levels would require additional biochemical analysis and subsequent loss of spatial information.
Determining accuracy and precision requires a reference standard against which chemical images can be compared (Fig. 6c). Where a relatively low spatial resolution is being used comparisons against light microscopy images may be sufficient, though using a non-IHC assay (i.e. in situ hybridisation in either the source material11 or from open-access databases25) increases confidence. The proportional response to concentration adds an additional dimension when assessing precision. Inter-experiment and inter-operator precision is assessed as reproducible localisation of tagged Ctr1 and MT1/2, and a consistent signal intensity in repeated measures of WT animals where relative protein expression levels have been externally confirmed (i.e. by western blot). A minimum concordance rate of 90% is the recommended standard for IHC.11
4. Opportunities and future challenges
Potential applications for IHC-chemical imaging are vast and varied, and primarily depend on two variables: the chemical nature of the analyte serving as a fiducial proxy for the antigen of interest; and other biomolecules with which they are associated, which may be either endogenous or labelled with different chemical reporter. In the following section, we provide a brief summary of where IHC-chemical imaging can provide new insight into functional biochemistry and discuss the challenges that will arise as the field continues to evolve.4.1 Multimodal imaging
Multimodal imaging describes the use of different detection techniques used in sequence (asynchronous) or simultaneous collection of signals from multiple analyte classes (synchronous). Both approaches have inherent technical challenges; asynchronous imaging requires registration of sequential images using appropriate fiducial markers, while synchronous imaging often involves complex, multistep sample preparation. Asynchronous imaging should ideally have similar spatial resolution. As an example on the extreme end, super resolution microscopy can achieve spatial resolutions at or below the spatial dimensions of a typical IgG molecule, and identification of a biomolecule is typically achieved through recognition of a specific peptide or amino acid sequence by a small molecule fluorophore attached by either direct conjugation or enzymatic delivery.39 When coupled with electron microscopy and a common detectable analyte, such as fluorescent tetramethylrhodamine which mediates photooxidation and induces polymerisation of DAB, correlative multimodal images of genetically-encoded tetramethylrhodamine-sensitive proteins and cell ultrastructure can be generated.40Que et al.41 applied a particularly innovative asynchronous multimodal approach using a Zn2+-sensitive fluorescent sensor and live-cell imaging (Fig. 7a) with subsequent scanning transmission electron microscopy with energy-dispersive spectroscopy, and synchrotron-based XFM and three-dimensional elemental tomography to characterise zinc flux during oocyte maturation. This method demonstrated that multimodal imaging need not necessarily be performed on the same sample; information gleaned from one technique was built upon by unique capabilities of following imaging modalities to quantitatively asses how ‘zinc sparks’ necessary for transition from egg-to-embryo form and change subcellular compartmentalisation.
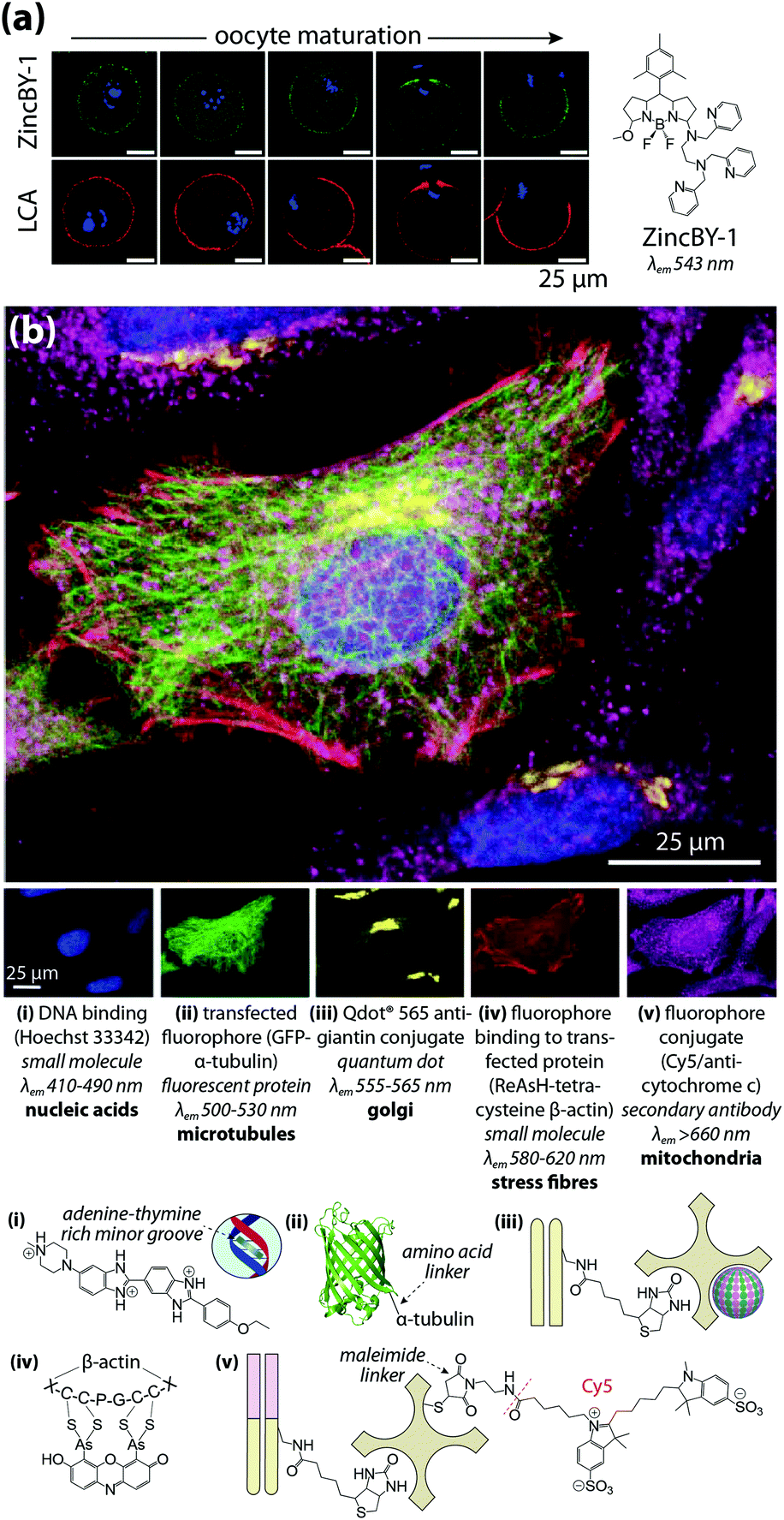 | ||
| Fig. 7 Asynchronous and synchronous multimodal imaging (a) sequential fluorescence imaging of ‘zinc sparks’ in maturing mammalian oocytes. Murine cells at five distinct stages of meiosis were labelled with the Zn2+ sensor ZincBY-1 (green) and counterstained with a DNA-binding fluorophore (blue). Oocytes were then fixed with paraformaldehyde and stained with fluorescent Lens culinaris agglutinin (LCA; red) and nucleic acid-marker DAPI (blue). LCA selectively binds to lectins on the surface of cortical granules, and the similar pattern of ZincBY-1 fluorescence suggests these vesicles are laden with Zn2+. Combined with XFM imaging, this method was able to quantify the total zinc content of a single oocyte (6 × 1010 atoms). Reproduced with permission from Que et al. (Nature Publishing Group, Copyright 2014).41 (b) Merged (top) and false-colour (bottom panels) images of parallel microscopy of HeLa cell structural features using five distinct fluorescent analytes: (i) bisbenzimide (Hoechst 33342) binding to adenine–thymine-rich DNA minor grooves; (ii) transfection and expression of green fluorescent protein (GFP) bound to α-tubulin via a six-amino acid linker; (iii) secondary antibodies with quantum dot conjugates; (iv) 4′,5′-bis(arsenoso)resorufin (ReAsH) binding to mutant β-actin containing a tetracysteine (Cys-Cys-Pro-Gly-Cys-Cys) motif; and (v) traditional ICH with a Cy5–streptavadin–biotin fluorophore bound to the Fab region of a secondary IgG antibody. Reproduced with permission from Giepmans et al. (American Association for the Advancement of Science, Copyright 2006).42 | ||
Capabilities of diffraction-limited fluorescence microscopy are constantly being expanded by application of fluorophores with greater quantum yields, and the development of novel fluorescent sensors for endogenous chemicals, recently reviewed by Kolanowski et al.43 Synchronous fluorescence microscopy combining IHC with inducible fluorescence in transfected cell lines, organelle markers, and small molecule sensors have produced subcellular images with remarkable levels of detail (Fig. 7b).
4.2 Small molecule sensors and temporal imaging
Over the last several decades a catalogue of cell-permeable and highly-specific compounds that fluoresce in the presence of small molecules have become commercially available, and the synthesis of novel chemical sensors is a vibrant field of research.44 Fluorophores specific to by-products of cellular respiration (such as reactive nitrogen, oxygen, and sulfur species) at nanomolar levels have provided new insight into cellular redox state and subcellular effects of oxidative stress, and can be multiplexed to spatially profile biochemical interactions.43 Specificity to a particular redox state permits snapshots of dynamic biochemical equilibria, and reversible probes with stable quantum yields over multiple redox cycles can be used to temporally profile changes to cellular oxidative state as a result of normal cellular function or chemical stimuli used as a disease mimetic. While these probes can theoretically be coupled with antibody-conjugated fluorophores in permeabilised cells, unavoidable perturbations to native cellular redox state arising from IHC protocols limits applications in live cell imaging.4.3 Intact systems
Several methods for optical clearing, such as 3DISCO and CLARITY, are allowing interrogation of structural and molecular networks in intact biological systems using three-dimensional microscopy such as confocal and light-sheet methods. CLARITY is particularly appealing as impregnation of tissue with a fixative and hydrogel monomer forms a polymerised ‘mesh’ support to which proteins are bound.45 Addition of sodium dodecyl sulfate then allows electrophoretic clearing of optically-opaque lipids, leaving a transparent sample amenable to repeated IHC staining, imaging, and antibody clearing using a detergent. Sequential staining cycles produce diminishing returns, as approximately 8% of hydrogel-anchored proteins are lost during each surfactant clearing step. The ideal scenario for CLARITY would be single clearing step followed by multiplexed IHC with several fluorophores covering a pre-determined panel of target biomolecules. Superior optical properties of QDs would appear to be best suited, though QD-conjugated antibodies are unable to match traditional fluorophores with respect to tissue penetration depth. Nevertheless, CLARITY and associated methods are relatively new tools that will undoubtedly improve over the coming years.4.4 In situ quantification
Traditional IHC assesses sensitivity and quantification in a somewhat arbitrary and subjective manner based on visual inspection of sections using light microscopy. A pathologist may assign a binary positive/negative score with reference to a positive control, use a narrow numerical range to rank staining intensity (i.e. 1–4), or apply other grading systems that assess both staining intensity and percentage of positive cells (e.g. H-scoring, quick scoring or the Allred method46). Each method is manual, prone to interobserver bias, and chromogen development is difficult to standardise. Recent advances in digital image analysis, such as the IHC Profiler plugin for ImageJ automate spectral deconvolution of 3,3′-diaminobenzidine (DAB)-enhanced IHC-labelled cancer biopsies to segment and score staining intensity on a pixel-by-pixel basis, reducing observer bias,47 though the resultant 8-bit colour image is still used to stratify data according to four-point grading. The availability of more comprehensive image analysis algorithms for ImageJ, such as those in the Fiji (Fiji Is Just ImageJ) package,48 have dramatically expanded the capabilities of this public-domain resource, and are applicable to all imaging modalities. Additionally, by making algorithms used for cell segmentation and data analysis, such as that used by Giesen et al.,27 publicly accessible and in either open-access or commonly-used proprietary programing languages, templates for custom-designed image analysis software suited to the method used are available.Absolute quantification of an antigen using chemical imaging is, at least in theory, achievable, though considering the multiple variables in existing IHC protocols that necessitate re-validation it is unlikely that reproducible in situ quantification would ever become mainstream. The ‘antibody binding capacity’, or ABC, of a sample can be determined according to the following:49
| ABC = n1θ1λ1 | (1) |
| Ψ = n2θ2λ2 | (2) |
5. Conclusions
Integrating IHC with chemical imaging represents the forefront of transdisciplinary chemistry. As outlined in this Tutorial Review, designing, executing, and validating an imaging experiment using antibodies and novel analytes requires knowledge spanning the fundamental principles of IHC to the technical intricacies of advanced analytical methods. However, with a sound and methodical approach to conducting IHC-chemical imaging experiments, the potential insight into complex biochemical processes that stands to be gained is, metaphorically speaking, almost immeasurable.Conflicts of interest
P. A. D. and D. J. H. receive research and material support from Agilent Technologies and ESI Ltd through the Australian Research Council Linkage Projects scheme. D. J. H. also receives research and material support from Agilent Technologies through the National Health and Medical Research Council Career Development Fellowship (Industry) program.Acknowledgements
DPB is supported by an Australian Research Council Discovery Early Career Researcher Award (DE180100194). PAD is the recipient of an Australian Research Council Discovery Project (DP170100036). TZ is supported by a Parkinson's Victoria PhD Scholarship. DJH is supported by an Australian National Health and Medical Research Council Industry Career Development Fellowship (1122981) in partnership with Agilent Technologies. Due to space limitations, this Tutorial Review could not give appropriate credit to all research in this field, and we wish to acknowledge to the authors of the many great works that were not cited directly.References
- A. Coons, H. Creech and R. Jones, Exp. Biol. Med., 1941, 47, 200–202 CrossRef CAS.
- K. Thorn, Mol. Biol. Cell, 2016, 27, 219–222 CrossRef CAS PubMed.
- U. Kubitscheck, Fluorescence Microscopy: From Principles to Biological Applications, John Wiley & Sons, 2nd edn, 2017 Search PubMed.
- N. Vogler, S. Heuke, T. W. Bocklitz, M. Schmitt and J. Popp, Annu. Rev. Anal. Chem., 2015, 8, 1–29 CrossRef PubMed.
- R. Fernandez-Leiro and S. H. W. Scheres, Nature, 2016, 537, 339–346 CrossRef CAS PubMed.
- R. McRae, P. Bagchi, S. Sumalekshmy and C. J. Fahrni, Chem. Rev., 2009, 109, 4780–4827 CrossRef CAS PubMed.
- J. M. Pushie, I. J. Pickering, M. Korbas, M. J. Hackett and G. N. George, Chem. Rev., 2014, 114, 8499–8541 CrossRef PubMed.
- J. Kaiser, K. Novotný, M. Z. Martin, A. Hrdlička, R. Malina, M. Hartl, V. Adam and R. Kizek, Surf. Sci. Rep., 2012, 67, 233–243 CrossRef CAS.
- A. Bodzon-Kulakowska and P. Suder, Mass Spectrom. Rev., 2016, 35, 147–169 CrossRef CAS PubMed.
- A. R. Buchberger, K. DeLaney, J. Johnson and L. Li, Anal. Chem., 2017, 90, 240–265 CrossRef PubMed.
- P. L. Fitzgibbons, L. A. Bradley, L. A. Fatheree, R. Alsabeh, R. S. Fulton, J. D. Goldsmith, T. S. Haas, R. G. Karabakhtsian, P. A. Loykasek, M. J. Marolt, S. S. Shen, A. T. Smith and P. E. Swanson, Arch. Pathol. Lab. Med., 2014, 138, 1432–1443 CrossRef PubMed.
- K. Dreisewerd, Chem. Rev., 2003, 103, 395–426 CrossRef CAS PubMed.
- Y. Chen, J. Cai, Q. Xu and Z. W. Chen, Mol. Immunol., 2004, 41, 1247–1252 CrossRef CAS PubMed.
- D. J. Hare, E. J. New, M. D. de Jonge and G. McColl, Chem. Soc. Rev., 2015, 44, 5941–5958 RSC.
- W. F. Wolkers and H. Oldenhorf, Cryopreservation and Freeze-Drying Protocols, Springer-Verlag, New York, 2015 Search PubMed.
- A. G. Bittermann, G. Knoll, A. Németh and H. Plattner, Histochemistry, 1992, 97, 421–429 CAS.
- M. J. Hackett, J. A. McQuillan, F. El-Assaad, J. B. Aitken, A. Levina, D. D. Cohen, R. Siegele, E. A. Carter, G. E. Grau, N. H. Hunt and P. A. Lay, Analyst, 2011, 136, 2941–2952 RSC.
- A. J. Hobro and N. I. Smith, Vib. Spectrosc., 2017, 91, 31–45 CrossRef CAS.
- J. Chwiej, M. Szczerbowska-Boruchowska, M. Lankosz, S. Wojcik, G. Falkenberg, Z. Stegowski and Z. Setkowicz, Spectrochim. Acta, Part B, 2005, 60, 1531–1537 CrossRef.
- D. J. Hare, J. L. George, L. Bray, I. Volitakis, A. Vais, T. M. Ryan, R. A. Cherny, A. I. Bush, C. L. Masters, P. A. Adlard, P. A. Doble and D. I. Finkelstein, J. Anal. At. Spectrom., 2013, 29, 565–570 RSC.
- A. W. Foster, D. Osman and N. J. Robinson, J. Biol. Chem., 2014, 289, 28095–28103 CrossRef CAS PubMed.
- P. B. Bass, K. B. Engel, S. R. Greytak and H. M. Moore, Arch. Pathol. Lab. Med., 2014, 138, 1520–1530 CrossRef PubMed.
- J. A. Ramos-Vara, J. D. Webster, D. DuSold and M. A. Miller, Vet. Pathol., 2014, 51, 102–109 CrossRef CAS PubMed.
- S. R. Shi, M. E. Key and K. L. Kalra, J. Histochem. Cytochem., 1991, 39, 741–748 CrossRef CAS PubMed.
- E. S. Lein, M. J. Hawrylycz, N. Ao, M. Ayres, A. Bensinger, A. Bernard, A. F. Boe, M. S. Boguski, K. S. Brockway, E. J. Byrnes, L. Chen, L. Chen, T.-M. Chen, M. Chin, J. Chong, B. E. Crook, A. Czaplinska, C. N. Dang, S. Datta, N. R. Dee, A. L. Desaki, T. Desta, E. Diep, T. A. Dolbeare, M. J. Donelan, H.-W. Dong, J. G. Dougherty, B. J. Duncan, A. J. Ebbert, G. Eichele, L. K. Estin, C. Faber, B. A. Facer, R. Fields, S. R. Fischer, T. P. Fliss, C. Frensley, S. N. Gates, K. J. Glattfelder, K. R. Halverson, M. R. Hart, J. G. Hohmann, M. P. Howell, D. P. Jeung, R. A. Johnson, P. T. Karr, R. Kawal, J. M. Kidney, R. H. Knapik, C. L. Kuan, J. H. Lake, A. R. Laramee, K. D. Larsen, C. Lau, T. A. Lemon, A. J. Liang, Y. Liu, L. T. Luong, J. Michaels, J. J. Morgan, R. J. Morgan, M. T. Mortrud, N. F. Mosqueda, L. L. Ng, R. Ng, G. J. Orta, C. C. Overly, T. H. Pak, S. E. Parry, S. D. Pathak, O. C. Pearson, R. B. Puchalski, Z. L. Riley, H. R. Rockett, S. A. Rowland, J. J. Royall, M. J. Ruiz, N. R. Sarno, K. Schaffnit, N. V. Shapovalova, T. Sivisay, C. R. Slaughterbeck, S. C. Smith, K. A. Smith, B. I. Smith, A. J. Sodt, N. N. Stewart, K.-R. Stumpf, S. M. Sunkin, M. Sutram, A. Tam, C. D. Teemer, C. Thaller, C. L. Thompson, L. R. Varnam, A. Visel, R. M. Whitlock, P. E. Wohnoutka, C. K. Wolkey, V. Y. Wong, M. Wood, M. B. Yaylaoglu, R. C. Young, B. L. Youngstrom, X. Yuan, B. Zhang, T. A. Zwingman and A. R. Jones, Nature, 2007, 445, 168–176 CrossRef CAS PubMed.
- Y. Xing, Q. Chaudry, C. Shen, K. Kong, H. E. Zhau, L. W. Chung, J. A. Petros, R. M. O'Regan, M. V. Yezhelyev, J. W. Simons, M. D. Wang and S. Nie, Nat. Protoc., 2007, 2, 1152–1165 CrossRef CAS PubMed.
- C. Giesen, H. A. O. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schüffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Günther and B. Bodenmiller, Nat. Methods, 2014, 11, 417–422 CrossRef CAS PubMed.
- M. Angelo, S. C. Bendall, R. Finck, M. B. Hale, C. Hitzman, A. D. Borowsky, R. M. Levenson, J. B. Lowe, S. D. Liu, S. Zhao, Y. Natkunam and G. P. Nolan, Nat. Med., 2014, 20, 436–442 CrossRef CAS PubMed.
- D. Schulz, V. Zanotelli, J. Fischer, D. Schapiro, S. Engler, X.-K. Lun, H. Jackson and B. Bodenmiller, Cell Syst., 2018, 6, 25–36 CrossRef CAS PubMed.
- G. Thiery, M. S. Shchepinov, E. M. Southern, A. Audebourg, V. Audard, B. Terris and L. G. Gut, Rapid Commun. Mass Spectrom., 2007, 21, 823–829 CrossRef CAS PubMed.
- J. Yang, P. Chaurand, J. L. Norris, N. A. Porter and R. M. Caprioli, Anal. Chem., 2012, 84, 3689–3695 CrossRef CAS PubMed.
- L. Wei, Z. Chen, L. Shi, R. Long, A. V. Anzalone, L. Zhang, F. Hu, R. Yuste, V. W. Cornish and W. Min, Nature, 2017, 544, 465–470 CrossRef CAS PubMed.
- H. Xu, Q. Li, L. Wang, Y. He, J. Shi, B. Tang and C. Fan, Chem. Soc. Rev., 2014, 43, 2650–2661 RSC.
- J. Lee, M. J. Petris and D. J. Thiele, J. Biol. Chem., 2002, 277, 40253–40259 CrossRef CAS PubMed.
- S.-i. Ono, D. J. Koropatnick and M. G. Cherian, Toxicology, 1997, 124, 1–10 CrossRef CAS PubMed.
- G. Emri, E. Emri, L. Beke, G. Boros, C. Hegedűs, E. Janka, E. Gellén, G. Méhes and É. Remenyik, J. Met. Nano, 2015, 3, 33–42 Search PubMed.
- E. J. New, V. C. Wimmer and D. J. Hare, Cell Chem. Biol., 2018, 25, 7–18 CrossRef CAS PubMed.
- S. Tricot, M. Meyrand, C. Sammicheli, J. Elhmouzi-Younes, A. Corneau, S. Bertholet, M. Malissen, R. Grand, S. Nuti, H. Luche and A. Cosma, Cytometry, Part A, 2015, 87, 357–368 CrossRef CAS PubMed.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef PubMed.
- V. Liss, B. Barlag, M. Nietschke and M. Hensel, Sci. Rep., 2015, 5, 17740 CrossRef CAS PubMed.
- E. L. Que, R. Bleher, F. E. Duncan, B. Y. Kong, S. C. Gleber, S. Vogt, S. Chen, S. A. Garwin, A. R. Bayer, V. P. Dravid, T. K. Woodruff and T. V. O'Halloran, Nat. Chem., 2014, 7, 130–139 CrossRef PubMed.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- J. L. Kolanowski, F. Liu and E. J. New, Chem. Soc. Rev., 2018, 47, 195–208 RSC.
- E. J. New, ACS Sens., 2016, 1, 328–333 CrossRef CAS.
- K. Chung, J. Wallace, S.-Y. Kim, S. Kalyanasundaram, A. S. Andalman, T. J. Davidson, J. J. Mirzabekov, K. A. Zalocusky, J. Mattis, A. K. Denisin, S. Pak, H. Bernstein, C. Ramakrishnan, L. Grosenick, V. Gradinaru and K. Deisseroth, Nature, 2013, 497, 332–337 CrossRef CAS PubMed.
- D. C. Allred, J. M. Harvey, M. Berardo and G. M. Clark, Mod. Pathol., 1998, 11, 155–168 CAS.
- F. Varghese, A. B. Bukhari, R. Malhotra and A. De, PLoS One, 2014, 9, e96801 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- R. J. Zagursky, D. Sharp, K. A. Solomon and A. Schwartz, Biotechniques, 1995, 18, 504–509 CAS.
- K. E. McCloskey, K. Comella, J. J. Chalmers, S. Margel and M. Zborowski, Biotechnol. Bioeng., 2001, 75, 642–655 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |