
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical methods for mapping cysteine oxidation
Lisa J.
Alcock
 ,
Michael V.
Perkins
and
Justin M.
Chalker
*
,
Michael V.
Perkins
and
Justin M.
Chalker
*
College of Science and Engineering, Flinders University, Bedford Park, South Australia, Australia. E-mail: justin.chalker@flinders.edu.au
First published on 15th December 2017
Abstract
Cysteine residues in proteins are subject to diverse redox chemistry. Oxidation of cysteine to S-nitrosocysteine, cysteine sulfenic and sulfinic acids, disulfides and persulfides are a few prominent examples of these oxidative post-translational modifications. In living organisms, these modifications often play key roles in cell signalling and protein function, but a full account of this biochemistry is far from complete. It is therefore an important goal in chemical biology to identify what proteins are subjected to these modifications and understand their physiological function. This review provides an overview of these modifications, how they can be detected and quantified using chemical probes, and how this information provides insight into their role in biology. This survey also highlights future opportunities in the study of cysteine redox chemistry, the challenges that await chemists and biologists in this area of study, and how meeting such challenges might reveal valuable information for biomedical science.
 Lisa J. Alcock | Lisa J. Alcock obtained her BSc (Honours) in Organic Chemistry from Flinders University in 2015. Her Honours research was carried out under the supervision of Assoc. Prof. Michael Perkins where she completed the total synthesis of a marine natural product. Currently, Lisa is a PhD candidate at Flinders University working in the area of chemical biology, under the supervision of Dr Justin Chalker and Assoc. Prof. Michael Perkins. Her research is focused on developing new probes to understand cysteine redox chemistry in cells. In 2017, Lisa was selected as an Australian delegate for the SciFinder Future Leaders programme. Lisa was also awarded a 2018 Endeavour Postgraduate Scholarship. |
 Michael V. Perkins | Michael V. Perkins earned a PhD from the University of Queensland in 1990 under the guidance of William Kitching. He carried out post-doctoral research on natural product synthesis with Ian Paterson at the University of Cambridge and then with Lew Mander at the Australian National University. He joined the Faculty of Flinders University in 1995, where he is currently an Associate Professor. His primary research interests are in the area of natural product synthesis and medicinal chemistry. |
 Justin M. Chalker | Justin M. Chalker earned a BS in Chemistry and a BA in the History and Philosophy of Science at The University of Pittsburgh in 2006. At Pittsburgh, he contributed to the total synthesis of several natural products under the direction of Theodore Cohen. As a Rhodes Scholar, Justin completed his DPhil at the University of Oxford under the supervision of Benjamin Davis where he developed several tools for protein modification. In 2012, Justin started his independent career at The University of Tulsa and then moved to Flinders University in 2015 where he is currently a Senior Lecturer. Justin's research interests are diverse with current projects spanning protein chemistry to new materials for environmental protection. Justin has been recognised with several awards including the ARC Discovery Early Career Research Award and the 2016 Tall Poppy of the Year for South Australia in recognition of his achievements in research, teaching and science communication. |
1.1 Introduction
The amino acid cysteine (1) exhibits diverse redox chemistry in proteins. For instance, the thiol group of the cysteine side chain is subject to a variety of oxidative post-translational modifications (oxPTMs) and can be converted to S-nitrosothiols 2, sulfenic acids 3, sulfinic acids 4, sulfonic acids 5, sulfenamides 6, persulfides 7, and various disulfides (8–9) including intramolecular disulfide bridges and intermolecular disulfides with small molecules such as glutathione (9) (Fig. 1). Each oxidation state of cysteine exhibits different reactivity which, in turn, may determine function. These differences in reactivity may be subtle, presenting an interesting challenge in developing chemoselective probes to detect these modifications. The formation of each of these oxPTMs is often in response to various stimuli within cells, such as the generation of reactive oxygen species (ROS), reactive nitrogen species (RNS) and reactive sulfur species (RSS).1 Since cysteine can react with these species and adopt a variety of oxidation states, cysteine residues in proteins are key regulators of redox homeostasis and signalling.2 Interestingly, only certain cysteine residues are susceptible to oxidation due to variations in their protein microenvironment such as solvent accessibility, pKa, and polarity of neighbouring residues. The type of oxidant (ROS, RNS, RSS) may also influence which cysteine residue is modified. Understanding these issues of selectivity is important for understanding the biological roles of cysteine oxidation.3–5 Therefore, methods to determine the site of a specific oxidative modification of cysteine are critical in biology. More generally, this information may allow us to understand the mechanisms of signalling, the role of oxidative regulation in cells, and how these pathways and processes might be associated with diseases linked to oxidative stress.6–8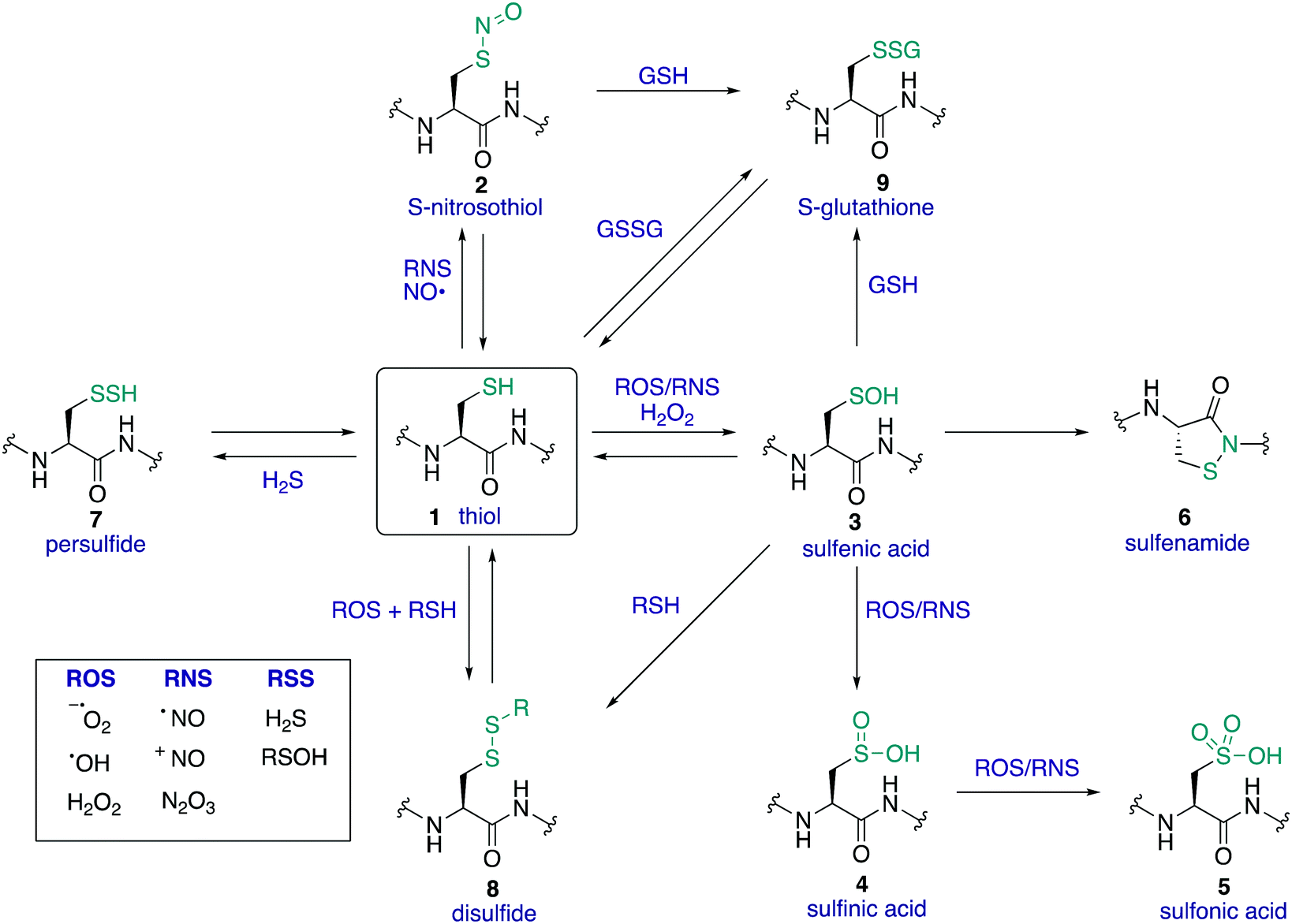 | ||
| Fig. 1 A selection of biologically relevant oxidative post-translational modifications of cysteine. | ||
As highlighted above, determination of the site and type of oxidative modification of cysteine is required for an understanding of its biological role. This is a very challenging task given the similarities between several of the cysteine oxidation states in Fig. 1 (1–9). However, as we will see, there are often subtle differences in reactivity between these modifications that allow selective detection, even in cells. Another challenge is the short lifetime of some of these oxidative modifications of cysteine, necessitating methods of detection that operate on a similar time scale as the non-persistent OxPTMs. In this review, a brief overview of cysteine redox chemistry will first be provided that highlights some of the key considerations that govern cysteine chemistry and its oxidative modification in proteins. Then, the focus of the review will turn primarily to the current chemical methods for mapping cysteine oxidation states, with selected examples of what biochemistry was learned in these studies. Finally, an outlook on future challenges will be provided to highlight the opportunities for developing selective chemical probes that will help identify hitherto unknown cysteine modifications and how this new information might advance biology and medicine.
1.2 Overview of cysteine chemistry
Cysteine is found in relatively low abundance in proteins (∼2% of all residues are cysteine),9 but its versatile chemistry makes it a critical determinant of protein structure and function. The cysteine sulfur atom can adopt a variety of oxidation states (oxidation numbers can range from –2 to +6), with diverse chemistry observed across this series.10 For example, the cysteine thiol can behave as a potent nucleophile or reducing agent, while its corresponding disulfide might behave as an electrophile or oxidising agent. The sulfur of cysteine can also exist as a thiyl radical, opening entirely different modes of reactivity. The specific reactivity of each cysteine thiol is governed by its microenvironment in the protein, with its pKa and redox potential influenced by the local polarity and interactions with neighbouring residues. For example, most thiols have a pKa around 8–9 making them almost fully protonated at physiological pH and therefore less susceptible to oxidation or reaction with electrophiles in those scenarios. However, the pKa of protein thiols can vary dramatically, and values ranging from 2.5 to 12 have been reported.11–14 This diverse chemistry and array of oxidation states (and the ability to interconvert between several of these oxidation states in vivo) is one of the reasons cysteine was selected by nature for diverse roles in cellular redox regulation and signalling.1.3 Mapping the function of cysteine
Within a given protein, not all cysteine residues are equal. Due to subtle changes in the protein microenvironment, different cysteine residues will exhibit unique reactivity, including the susceptibility towards oxidation by reactive oxygen, nitrogen, and sulfur species. In many cases, these differences in reactivity may be the basis for cysteine redox signalling. Therefore, it is imperative to understand the impact of these subtle differences by locating oxidisable cysteine residues in proteins. The selectivity for oxidation of particular cysteines is not well understood or easily predicted, although several studies have looked at factors that may influence selectivity.8,15–17Understanding the type and level of oxidant is also important in cysteine redox chemistry. Some cysteine oxPTMs occur at the basal levels of oxidants, suggesting that oxidative modification of certain cysteines is expected, perhaps playing an essential regulating role in healthy cells. These oxidative modifications increase in number and type, however, during oxidative stress. In such events, an influx of ROS, RNS, and/or RSS may lead to cysteine modifications that are harmful to the cell. Understanding the different oxPTM profiles that occur in healthy and diseased states in humans is one reason why profiling cysteine chemistry is important, but redox regulation is important in a variety of species. The most biologically relevant cysteine oxidation states are shown in Fig. 1. Many of these cysteine oxidation states have been implicated in some form of redox-based regulation in proteins.18–20 Several modes of cysteine redox regulation have been identified that influence signal transduction, often by way of structural changes that influence enzymatic activity. For example, protein tyrosine phosphatase activity,3,21 EGFR signalling,22,23 and thioredoxin activity24,25 are all influenced by cysteine oxidation. It is also important to note that oxPTMs at cysteine can be deactivating (e.g. reduced structural stability and activity of M. tuberculosis tyrosine phosphatase A upon S-nitrosylation of a non-catalytic cysteine),26 activating (e.g. oxidative activation of OxyR transcription factor in E. coli by disulfide formation),27,28 or part of a normal catalytic cycle (e.g. the active site of thioredoxins features interconversion of thiols and disulfides in the active site).24,29,30
Locating sites of cysteine oxidation is important for understanding these functions and meeting this goal is highly challenging. For instance, many early studies used exogenous sources of oxidants or performed experiments with purified proteins which may not fully represent the redox process in vivo. For studying protein oxidation in cells, mapping of the cysteine proteome can occur through indirect or direct methods. Indirect methods generally involve conversion of the modification of interest into a detectible group, such as one labelled with biotin or a fluorophore. These methods often require masking the reactivity of all other cysteines (for instance by alkylation), followed by selective reduction and labelling of the cysteine bearing the oxPTM of interest. In contrast, direct methods for detecting oxPTMS feature chemistry that is highly selective for the modification of interest, ligating the reporter group directly to the modified cysteine (and no other residues). Direct methods are attractive because, in principle, they can be applied to living cells. Several of these methods will be discussed in the next sections for each type of cysteine oxPTM.
1.4 Dual roles of reactive oxygen species (ROS) and reactive nitrogen species (RNS)
Reactive oxygen and reactive nitrogen species—such as hydrogen peroxide, superoxide, nitric oxide, and peroxynitrite2—have been viewed traditionally as destructive oxidants that compromise the structure and activity of proteins and genetic material. However, while elevated levels of ROS and RNS may be associated with disease, it is becoming increasingly apparent that these species also act as second messengers in healthy cells. For example, hydrogen peroxide has been shown to have a proliferative effect on cells and can promote wound healing.19 There are many sources of ROS and RNS within cells. For example, production of H2O2 occurs in the mitochondria as part of the electron transport chain, NADPH oxidase produces superoxide in the neutrophil-based defence against bacteria, and the production of nitric oxide by nitric oxide synthase is also critical as a defence mechanism.5,19,31These reactive species can be categorised into two main types, radical and non-radical, and this reactivity profile may influence which cysteines are oxidised for a given protein. For non-radical or two electron reactions, cysteine thiolates may act as nucleophiles, as in the case of the direct reaction of the thiolate with hydrogen peroxide to form a sulfenic acid. The rate and site of such modifications often depends on the steric hindrance and pKa of the cysteine thiol.5 What is important to note is that not all cysteine residues are susceptible to oxidation. Some ROS/RNS/RSS react far faster with thiolates (the deprotonated thiol), and more slowly with the thiol in its neutral, protonated form—often the predominant form at physiological pH. This differential reactivity of ROS and RNS with cysteines is often the basis for selective redox-regulation. Another factor that can be considered is the proximity of a protein to the ROS/RNS/RSS source, where ROS/RNS/RSS can exist in concentration gradients within cellular compartments. Therefore, proteins in closer proximity to ROS/RNS/RSS sources through various inter-protein interactions are more likely to become oxidised.
Antioxidant molecules are also present in the intracellular environment that contribute to maintenance of the overall redox homeostasis. These antioxidants include glutathione, superoxide dismutase, peroxiredoxins, thioredoxins, and glutaredoxins.32 These antioxidant molecules assist in redox-regulation presumably as a defence against overoxidation or as a way to restore pre-oxidation function, making them active players in redox signalling.
Overall, the important message here is that cysteine oxidation is integral to signalling in healthy cells, but also a hallmark of diseases associated with oxidative stress. Understanding the delicate balance between these two seemingly opposing functions motivates the development of tools that help detect the proteins involved in these processes.
1.5 Detecting oxidative modifications at cysteine: challenges and limitations
Since it has been established that oxidation of certain protein thiols is involved in redox-regulation events, it is important to develop strategies for detecting the level and site of these oxidations in vivo. This is not a simple task. In order to detect specific oxidation states of cysteine, chemical probes must react selectively (or better yet, specifically) with the oxPTM of interest and not any other oxidation state. Furthermore, the probe must be compatible with the hundreds of other reactive groups within a protein and cellular environment. Most cysteine oxidation states are themselves reactive and may react further to higher oxidation states. In cases where a modification of interest is short-lived, the probe must react rapidly. To date, a wide array of detection methods have been reported for mapping oxPTMs at cysteine in isolated proteins and in cells. Chemical methods are the focus of this review.When testing a chemical probe for a particular oxidation state of cysteine in cells, it is critical to use conditions relevant to the cellular redox environment. For instance, subjecting cells to extreme oxidative stress could result in false positives or overoxidation of key sites that are not normally present in these states. Another important consideration is the source of the ROS/RNS/RSS used in experiments. Since not all cysteines can be oxidised equally by different ROS/RNS/RSS, this should be taken into consideration. Additionally, it is critical to consider if cell lysis is required, as this process will immediately change the redox environment of key proteins and perhaps lead to a different outcome than if the probe acted inside the cell. Finally, many of the oxPTMs of cysteine react similarly with common reagents (sulfenic acids, nitrosothiols and disulfides can all react with strong reducing agents, for instance) so it is critical to evaluate the outcomes of mapping experiments with these considerations in mind. In the discussion of specific methods that follow, these challenges in selectivity will be discussed where specific issues have been identified in the literature.
2.1 S-Nitrosothiols (RSNO)
The S-nitrosothiol post-translational modification of cysteine (2, Fig. 2) has gained attention as a regulatory mechanism over the last 30 years thanks to foundational work by Stamler which first suggested that proteins can be carriers of nitric oxide, through the formation of S-nitrosylated cysteines.33,34 The studies by Stamler documented S-nitrosothiol formation from endogenously derived ˙NO using the Saville method35 in which the interaction of RSNO with Hg2+ generates +NO, which in turn can be trapped by an aniline and naphthalene derivative to form an intensely coloured azo dye. Other more direct spectrometric techniques such as 15N-NMR spectroscopy or UV-Vis spectroscopy have also been used to observe bovine serum albumin bearing nitrosylation at its single cysteine. S-Nitrosoproteins in these studies were found to exhibit endothelium-derived relaxing factor-like properties, suggesting a mechanism to preserve the biological activity of ˙NO, and an additional regulatory mechanism.33,34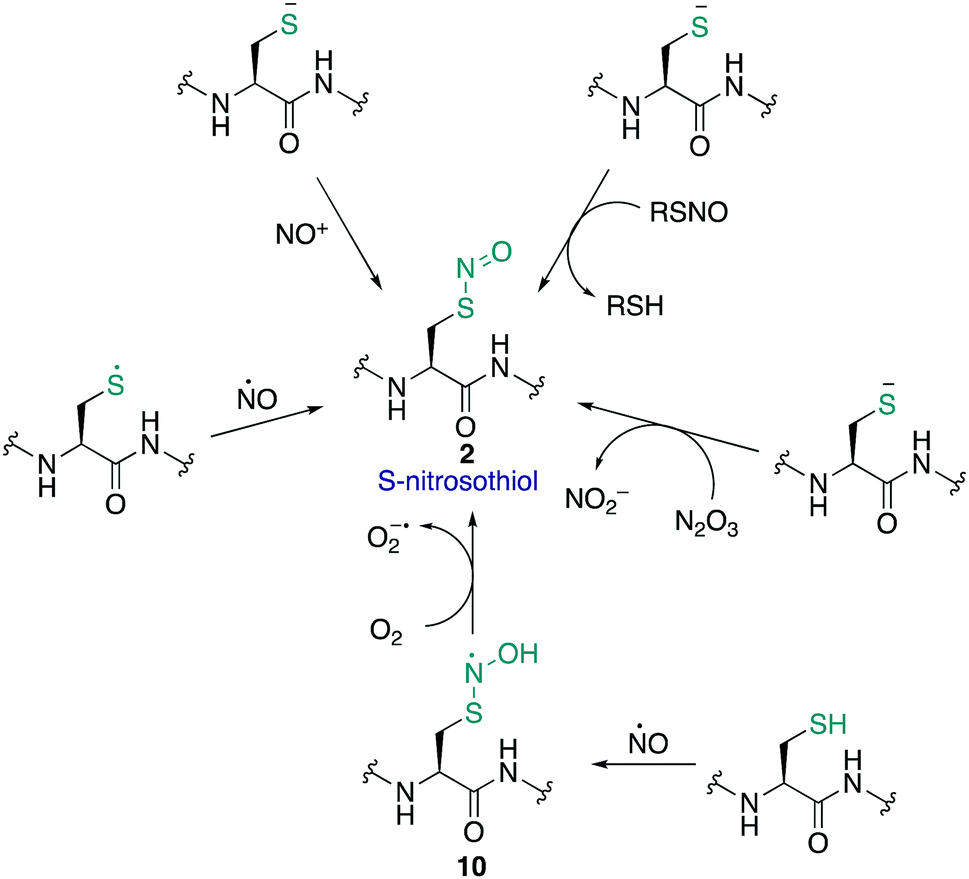 | ||
| Fig. 2 The formation of S-nitrosothiols in proteins. | ||
˙NO itself serves as a signalling molecule involved in many physiological processes and is produced by a family of enzymes known as nitric oxide synthases (NOS). Protein-SNO can be formed through several pathways (Fig. 2). The formation of protein-SNO has not been well characterised but several mechanisms have been proposed. Thiyl radicals, for instance, can directly combine with ˙NO giving the corresponding protein-SNO. This was demonstrated in a study by Goldstein and co-workers through generation of thiyl radicals in the presence of ˙NO. Formation of the S-nitrosothiol was monitored by following the characteristic SNO absorption maxima. LC-MS provided additional evidence for the expected nitrosylation of model compounds. Rate constants for the reactions of nitric oxide with thiyl radicals derived from cysteine were on the order of 109 M−1 s−1.36,37 Evidence for the intermediate role of ˙NO2 in formation of RSNO through radical combination of RS˙ and ˙NO has also been explored.37 Intermediate nitrosylating agents such as N2O3 may be formed from oxidation of ˙NO, which in turn can form nitrosothiols. Studies by Goldstein using stopped-flow kinetic measurements monitored formation of S-nitroso compounds using characteristic absorption maxima (330–340 nm) found N2O3 can directly nitrosylate thiols and amines with rate constants exceeding 6 × 107 M−1 s−1 for low molecular weight compounds.38 Similar studies gave a rate constant on the order of 105 M−1 s−1 for low molecular weight thiols such as glutathione (GSH) demonstrating this reaction is competitive with the hydrolysis of N2O3, and comparable to the rate of nitrosylation for human serum albumin (rate constant = 3 × 104 M−1 s−1).39 It is also possible that NO groups may be transferred from the iron of heme groups to cysteine thiols. Using human methemoglobin, Singel and co-workers demonstrated that protein-SNO could be produced through a heme-Fe(III)NO intermediate with formal loss of NO+.40 The direct addition of ˙NO to a thiol can occur but generates the radical intermediate 10. This radical species 10 can be oxidised by oxygen or some other one-electron acceptor to yield the RSNO.41 Additionally, redox-active metal ions and metalloproteins may assist in these mechanisms.42 Transnitrosylation is another mechanism for protein-SNO formation, for instance through reaction of protein thiols and nitrosylated glutathione (GSNO). Evidence for this pathway was revealed by Hogg through monitoring the transnitrosylation events by HPLC as a function of time.43 However, these transnitrosylation reactions were shown to be slow, with the largest rate constants for the reaction between S-nitrosocysteine and glutathione (GSH) found to be on the order of 102 M−1 s−1. Of course, different protein microenvironments will influence the rate of reaction of these mechanisms on proteins, but the comparison to GSH is important because of its high concentration in cells making it an active competitor. Indeed, evidence for the presence of GSNO was detected in mitochondria, where GSNO may be considered a storage and transport molecule of ˙NO and facilitate nitrosylation of target proteins.44,45
Cysteine nitrosylation occurs on diverse classes of proteins, but it is common to modify cysteine thiols with a low pKa and those in an exposed environment.46 The specificity of certain cysteines for S-nitrosylation may also be influenced by neighbouring amino acids which could direct nitrosylating and denitrosylating agents to the cysteine.15,16,47S-Nitrosylation is also relatively unstable and can, for instance, undergo photolytic decomposition to form ˙NO and a thiyl radical. The short lifetime of certain nitrosylated residues therefore complicates detection of this oxPTM by conventional methods. And yet this detection is important because ˙NO has been shown to mediate a number of important processes such as neuronal survival, synaptic plasticity, gene expression and caspase activity.15,46,48
Increased levels of ˙NO, however, can be pathogenic with S-nitrosylation of proteins associated with Alzheimer's disease, Parkinson's disease, neuronal degeneration and Huntingson's disease.6,15,49–51 Lipton and co-workers discovered pathologically relevant amounts of S-nitrosylated dynamin-related protein 1 (Drp1) in the brains of patients with Alzheimer's disease.52S-Nitrosylation of Drp1 caused dimer formation which then increased GTPase activity, accelerated mitochondrial fission, and contributed to neuronal synaptic damage or cell death. Preventing nitrosylation of Drp1 by a cysteine mutation diminished these neurotoxic events. More recently, they also found that an increase in ˙NO levels provoked S-nitrosylation of the insulin-degrading enzyme (IDE) and once again Drp1, which in turn inhibited insulin and oligomeric β-amyloid peptide (Aβ) catabolism, resulting in elevated levels, which also caused hyperactivity in mitochondrial fission.53 Increased levels of glucose were found to result in an increase of neuronal RNS, and eventually synaptic dysfunction and loss. Overall, this lead to the conclusion that nitrosylation of specific brain proteins (IDE and Drp1) leads to cognitive dysfunction in metabolic syndrome, type 2 diabetes mellitus, and Alzheimer's disease, which was shown to be linked with high levels of glucose and Aβ by loss of function of these enzymes.
Lipton and co-workers also undertook a similar study on S-nitrosylation of protein-disulfide isomerase (PDI).54 They were able to show that in brains manifesting sporadic Parkinson's or Alzheimer's disease, the PDI was S-nitrosylated to excessive levels. This modification inhibits the ability of PDI to catalyse disulfide exchange reactions, leads to the accumulation of polyubiquitinated proteins, and activates the unfolded protein response. PDI also acts to reduce neuronal cell death under normal function, preventing neurotoxicity associated with endoplasmic reticulum stress and protein misfolding. However, production of ˙NO blocks this protective effect in neurodegenerative disorders through S-nitrosylation of PDI.
Other proteins such as S-nitrosoglutathione reductase (GSNOR) have also been shown to be enzymatically inhibited by nitrosylation at non-zinc-coordinating cysteines. GSNOR monitors accumulation of GSNO, where nitrosylation of GSNOR may permit GSNO accumulation through ˙NO signalling.55 Since GSNO may further cause nitrosylation of other protein cysteine targets, this may present an additional regulatory mechanism.
These studies highlight the growing amount of evidence that S-nitrosylation is a hallmark of certain diseases. Therefore, it is clear that reliable methods to detect such modifications are critical in understanding the mechanism of these pathologies. These methods are discussed in the following section.
2.2 Chemical methods for detecting RSNO
Due to the relatively unstable nature of the RSNO modification, its efficient and reliable detection has been an on-going challenge in chemical biology. As highlighted above, proteins are known to contain this RSNO modification, but some uncertainty still exists as several S-nitrosylated proteins were identified using exogenous ˙NO donors that may not reflect endogenous ˙NO sources.56 Likewise, indirect methods of detection may not accurately capture the physiologically relevant form of the proteins of interest.The biotin switch technique
The most reported method for detection of protein S-nitrosylation is the so-called biotin switch technique (BST), first described by Jaffrey and co-workers to convert nitrosylated cysteines into biotinylated cysteines for detection by western blot.57,58 This method follows three general steps (Fig. 3a): first, free thiols are converted to disulfides by reaction with methylmethanethiosulfonate (MMTS, 11). Second, nitrosothiols are selectively reduced with ascorbate to form thiols. The disulfides formed from reaction with MMTS are not reduced by ascorbate in this step. Third, the newly formed thiols (from cysteines that previously were nitrosylated) are treated with disulfide 12—a thiol specific biotinylating reagent (Fig. 3a).59 Using this method, Jaffrey and co-workers were able to establish protein-SNO as a physiological cell signalling mechanism. While it is true that the BST has been used on numerous occasions to locate and infer involvement of many protein-SNO sites, limitations with this technique have been identified. For example, this is an indirect detection method which requires initial blocking steps that require time and manipulations that might result in the loss of more labile RSNO species. There have also been reports of non-specific binding or endogenously biotinylated background.60 The specificity of the technique relies on the assumptions that free thiols are completely blocked in the first step, and that ascorbate will convert RSNO to free thiols without reducing other cysteine oxidation states. Several studies have shown that in fact, ascorbate is not a very efficient reducing agent for S-nitrosothiols. In cases where the reduction of nitrosylated cysteines is slow, there is the possibility of not detecting certain RSNO modifications.61 Furthermore, ascorbate was shown to reduce disulfide bonds at higher concentrations which could lead to false positives when searching for RSNO sites.62,63 The role of copper in the ascorbate mediated reduction of nitrosylated cysteines has also been explored, and Gladwin et al. have suggested that addition of Cu(II) promotes the direct reduction of protein-SNO, rather than ascorbate.64 In such cases, the decomposition of SNO likely occurs through the one-electron reduction by Cu(I) to give the free thiol, ˙NO, and Cu(II). Ascorbate can then reduce the Cu(II) to Cu(I) and complete the catalytic cycle.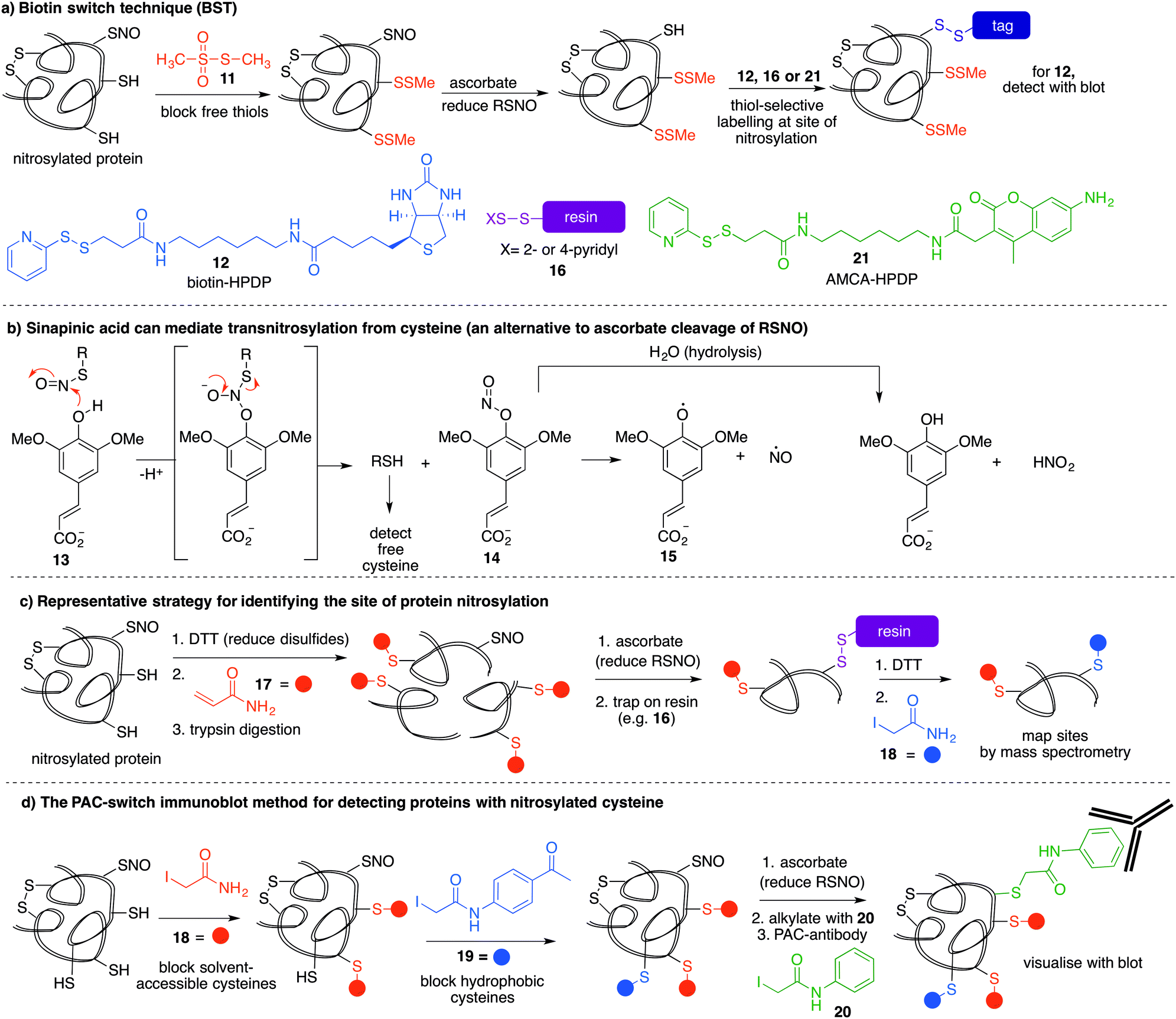 | ||
| Fig. 3 (a) The biotin switch technique can be used to detect protein-SNO. Free thiols are blocked and then the nitrosylated cysteine is reduced. The liberated thiol is then labelled for detection with a biotin tag (12). (b) Sinapinic acid (13) can reduce nitrosylated cysteines. (c) Blocking both thiols and disulfides before uniquely labelling nitrosylated cysteine residues. Note that DTT may reduce RSNO. Mass spectrometry can be used to identify the sites of nitrosylation after affinity enrichment of the key peptide fragments. (d) The PAC-switch technique uses two thiol blocking reagents tailored to solvent accessible and hydrophobic cysteines. Ensuring complete thiol blocking minimises false positives. Protein-SNOs are reduced with ascorbate and labelled with 20 which can be detected using an anti-PAC antibody. | ||
Mutus et al. demonstrated that ascorbate can be replaced with sinapinic acid (13) in the S–NO cleavage step of the biotin switch technique.65 Sinapinic acid is thought to behave as a nucleophile, producing O-nitrosinapinic acid (14) and cysteine in the transnitrosylation mechanism shown in Fig. 3b. The O-nitrosinapinic acid is likely unstable and may decompose by homolytic cleavage to produce ˙NO and the sinapinic radical (15) or by hydrolysis (Fig. 3b).
Many modifications of the BST have since been implemented to improve sensitivity and provide additional information about the site of nitrosylation.59 Stamler et al. used thiol-reactive resin 16 to trap the key cysteine residue after reduction of the nitrocysteine by ascorbate (Fig. 3a).66 Using this strategy, Lu et al. demonstrated that phosphodiesterase type 5 (PDE5) activity and expression were regulated by S-nitrosylation at Cys-220.67 This study demonstrated for the first time that S-nitrosylation of PDE5 resulted in reduced activity and degradation via the ubiquitin–proteasome system.
Other variations of the trapping technique include the use of resins and blocking sequences that trap either nitrosylated or oxidised cysteines.68 The BST has also been modified to include a proteolytic digestion before avidin capture in order to identify the site of nitrosylation.69 In this strategy, false positives may arise from detection of disulfide bonds in the final reductive removal of the peptide fragment from the resin.70 To overcome this problem, more stable linkages to biotin can be used, such as maleimide derivatives.
Ding and co-workers developed a so-called site-specific high-throughput identification of protein S-nitrosylation (SHIPS) method to identify sites of cysteine nitrosylation (Fig. 3c).71 First, DTT was added to reduce all disulfide bonds, all free thiols were blocked using acrylamide (17), and then the protein was digested using trypsin. Apparently under these conditions DTT does not reduce the nitrosyl cysteine, but this should be considered when deploying this method. Then, ascorbate mediated reduction of SNO-peptides provided the free thiol which was captured on a resin. Peptides were then eluted with DTT and alkylated with iodoacetamide (18) to differentiate between free thiols in the native protein and SNO-derived thiols. These peptide fragments were then analysed by mass spectrometry and reported to identify sites of nitrosylation.
A more recent strategy to detect cysteine nitrosylation used a phenylacetamidyl group as the detectable epitope in an immunoblotting protocol (Fig. 3d).72 This study demonstrated that by using iodoacetamide (18) to alkylate solvent accessible and hydrophilic regions, and N-(4-acetylphenyl)-2-iodoacetamide (19) to alkylate cysteines in hydrophobic environments, false positives were largely avoided. After ascorbate reduction of the remaining nitrosocysteines, the resultant thiols were labelled with iodoacetanilide (20) and detected by the anti-phenylacetamidyl antibody (anti-PAC) (Fig. 3d).
Several methods have been reported that replace the biotin tag with a fluorescent counterpart. This tactic allows direct visualisation of the protein-SNO without the need for the immunoblotting step. For example, Chen et al. used thiol-reactive fluorophore 21 as a biotin substitute (Fig. 3a).73 Likewise, Berkowitz et al. used CyDyes 22 and 23 for detection, identification and quantification of protein-SNO (Fig. 4a). This selective fluorescent labelling of S-nitrosothiols (S-FLOS) technique benefitted from low background, high signal to noise, and compatibility with in situ tissue staining.60 Both stimulated and unstimulated controls were conducted, labelling with 22 and 23, respectively. Comparison of fluorescent intensities on the same 2D gel allows relative quantification of S-nitrosylation. The S-FLOS method was also shown to detect changes in S-nitrosylation by signal increase with the same time dependency as increase in total protein S-nitrosylation. Hogg et al. also incorporated the use of the CyDyes 22 and 23 as a direct in-gel fluorescence method using difference in gel electrophoresis (DIGE).74 However, some modifications were made to the original BST procedure by replacing 11 with N-methylmaleimide (NEM, 25) since the thioether formed by blocking with 25 is more stable to reducing conditions than the disulfide formed with 11. The DIGE method allows examination of the difference in modification before and after exposure to the nitrosylation stimulus because the control and experimental samples are labelled with different cyanine dyes. This is advantageous as any thiol groups that are not blocked in the first step will be labelled by both samples and differentiated from SNO. Similarly, Murphy and co-workers used this technique for detection of mitochondrial proteins that are targets of S-nitrosylation.75 In this study, nitrosylation of mitochondrial proteins was stimulated by the treatment of cells with a lipophilic phosphonium cation bearing a nitrosylated thiol.
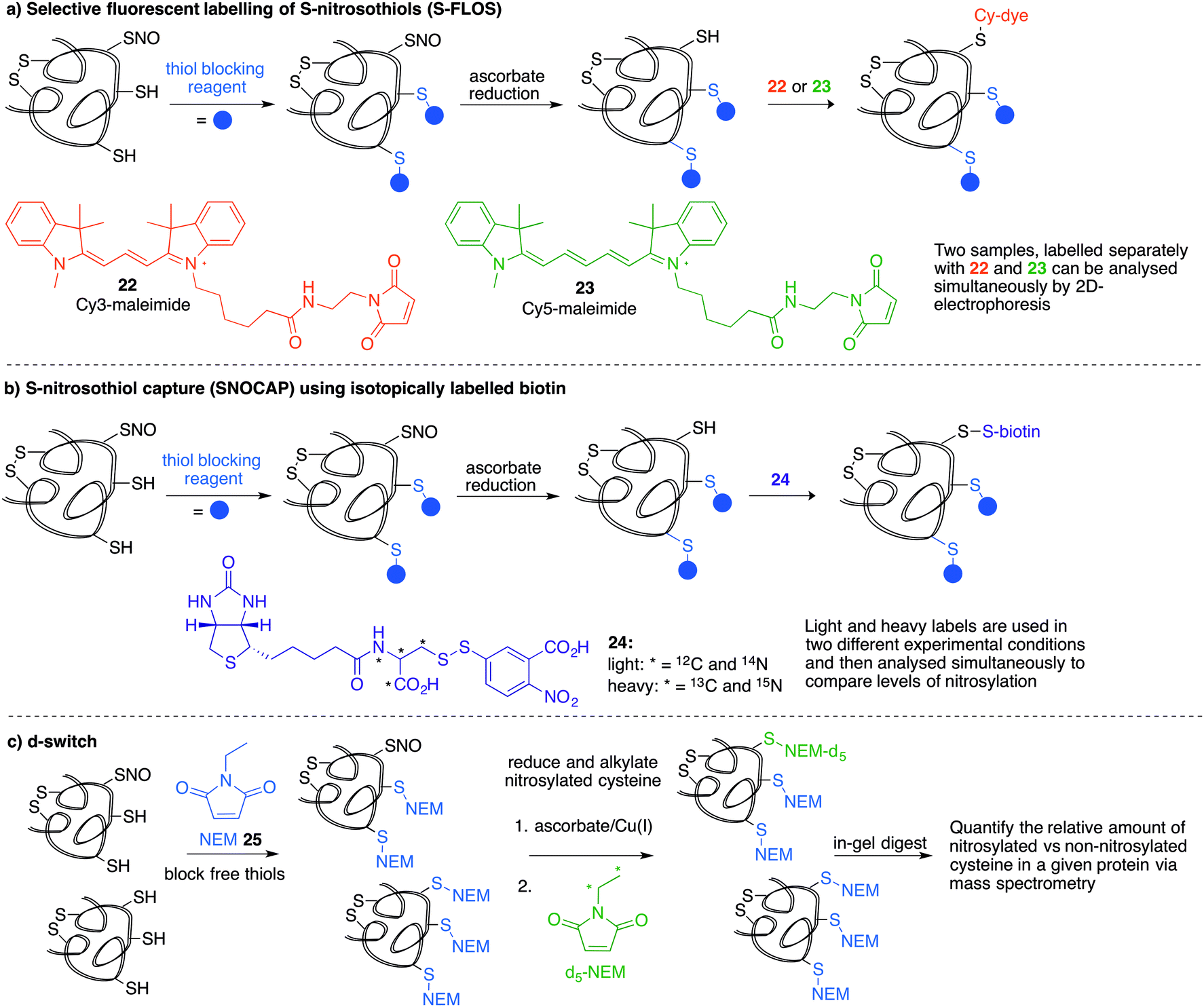 | ||
| Fig. 4 (a) The selective fluorescent labelling of S-nitrosothiols (S-FLOS) uses 2 CyDyes (22 and 23) to label both control and stimulated samples where the relative fluorescence between samples can be compared. (b) The SNOCAP method uses an isotopically labelled biotin reagent 24 to label nitrosylated cysteines after reduction. Comparison of the ratio of light to heavy isotopes allows quantification of two different samples by mass spectrometry. (c) The d-switch technique uses NEM (25) to block free thiols followed by reduction of nitrosylated thiols with an ascorbate/Cu(I) system. The liberated cysteines are then labelled with d5-NEM. Mass spectrometric analysis allows determination of the levels of nitrosylation for a given protein at a specific site by comparison of the MS peak intensities for the heavy and light NEM groups at a particular cysteine. | ||
A limitation of several of the methods discussed thus far is the inability to precisely determine the levels of protein-SNO and identify the site of the labelled protein. Jaffrey and co-workers designed a method coined S-nitrosothiol capture (SNOCAP) using the reagent 24 which consists of a biotin affinity tag containing an isotope-coded section adapted from the ICAT technique (see Disulfide section).7624 was synthesised as both the light and heavy forms. The method uses a similar workflow to BST including blocking free thiols with 11, reduction of R-SNO with ascorbate, and finally reaction of the liberated thiols with 24. The proteins, now biotinylated at the site of nitrosylation, were then trypsinised and the key peptides isolated by using avidin affinity enrichment for further analysis by mass spectrometry. Additionally, two samples can be quantitatively compared if the light SNOCAP reagent is used in one and the heavy SNOCAP reagent in the other. Quantitative information about protein-SNO can then be inferred by mass spectrometry (Fig. 4b). Other variations such as the d-switch developed by Thatcher also enable a quantitative proteomics approach by using N-ethylmaleimide (NEM, 25) to alkylate free thiols and a deuterated version (d5-NEM) to alkylate the thiols liberated after reduction of the R-SNO groups (Fig. 4c). This technique allows the ratio of nitrosylated versus non-nitrosylated cysteines for a particular site to be assessed by mass spectrometry.77 Recently, Murphy and co-workers devised an improved strategy to detect nitrosylated cysteines coined S-nitrosothiol redox isotope-coded affinity tag (SNOxICAT).78 This technique is based on the isotope-coded affinity tag labelling technique (OxICAT) commonly applied in the analysis of reversibly oxidised protein thiols. Samples were stabilised by solubilisation in 20% TCA in the presence of 0.5% sulfanilamide to both preserve endogenous S-nitrosothiols and prevent artefactual nitrosylation during work-up. Samples can then be neutralised and unmodified cysteine residues labelled with light ICAT (26). S-Nitrosothiols can then be reduced selectively with Cu(I) (ascorbic acid/CuSO4) and labelled with heavy ICAT (26) (Fig. 5a). Samples are then digested and enriched by affinity purification using the biotin tag on 26, and analysed by LC-MS. Since both ICAT reagents are chemically identical, the extent of modification could be quantified and the proteins identified by MS/MS. Application of SNOxICAT allowed both the identification of cysteine residues that are S-nitrosylated and the quantification of the extent of S-nitrosylation. The impact of NO2− on mouse heart was studied in vivo, where it was suggested that the combination of NO2− and ischemia were necessary for extensive S-nitrosylation.
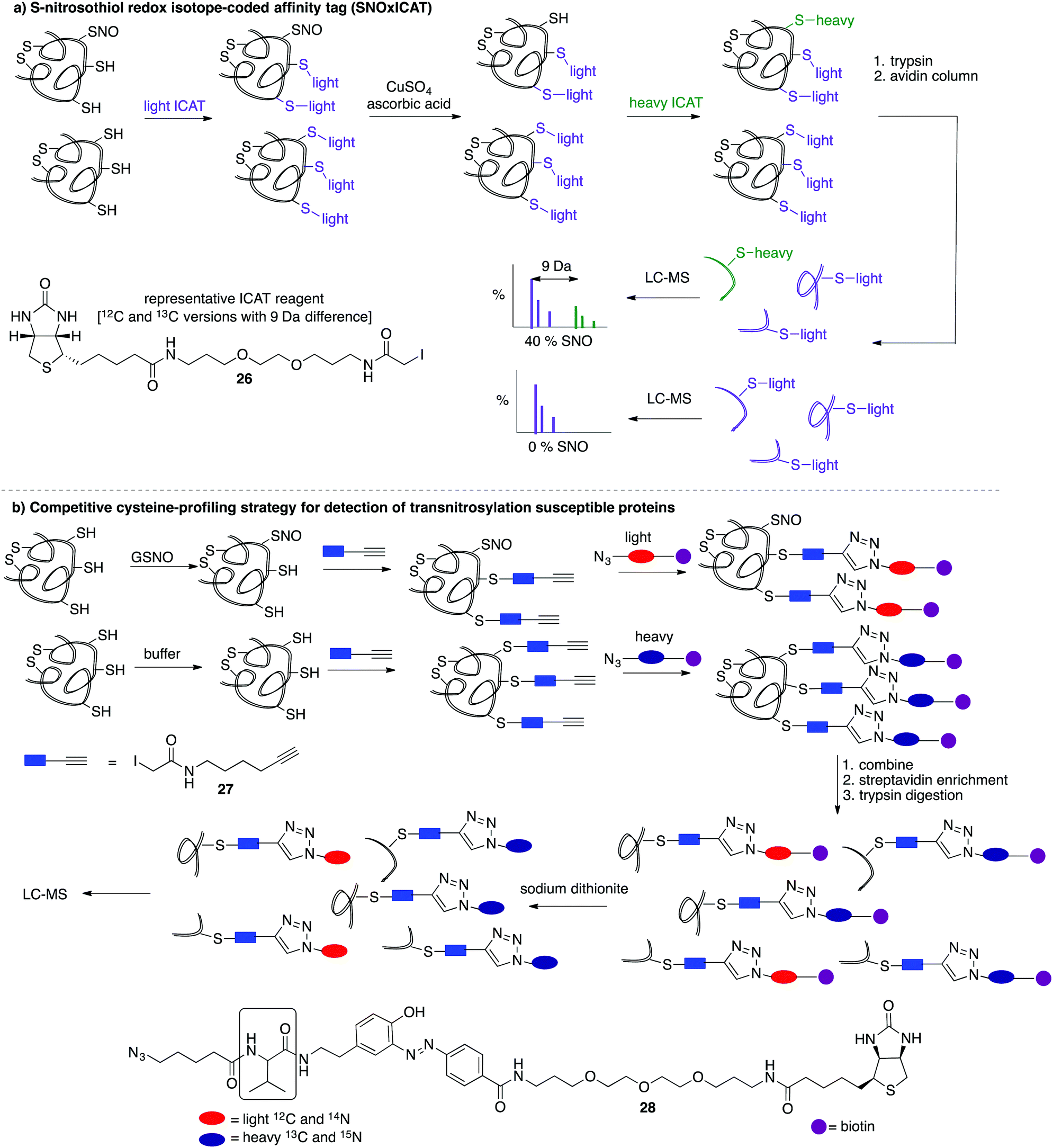 | ||
| Fig. 5 (a) S-Nitrosothiol redox isotope-coded affinity tag (SNOxICAT) for the quantitative detection of protein S-nitrosothiols. Free cysteines are reacted with light ICAT (26), followed by reductions with CuSO4/ascorbic acid to reduce SNO which are subsequently labelled with heavy ICAT (26). This allows the extent of S-nitrosylation to be quantified. (b) A competitive cysteine-profiling strategy to detect proteins which are susceptible to transnitrosylation by GSNO. Two samples are treated with either GSNO or buffer (a negative control) and then free cysteine thiols are labelled with 27. An azo-biotin tag 28 is then covalently bound by CuAAC with light (for GSNO samples) and heavy (buffer control samples) isotopic labels. Samples are combined, enriched by streptavidin, and digested. The biotin tag is then cleaved by sodium dithionite and analysed by MS. | ||
A method was also developed to monitor transnitrosylation events by the endogenous donor S-nitrosoglutathione (GSNO) to determine which cysteine residues were most sensitive to GSNO-mediated transnitrosylation.79 This method was adapted from a similar approach developed within the Cravatt group termed isotopic tandem orthogonal proteolysis-activity-based protein profiling (isoTOP-ABPP) for detection of reactive cysteines.80,81 Cell lysates are exposed to GSNO or a negative control and then treated with the cysteine reactive iodoacetamide-alkyne (27) probe, bearing a terminal alkyne for further functionalisation through copper(I)-catalysed azide–alkyne cycloaddition reactions (CuAAC). The 27-labelled proteins are then covalently linked to light (GSNO-treated) or heavy (buffer control) biotin-azide tags (28) containing an azobenzene linker for later reductive cleavage by sodium dithionite. Both samples are combined and enriched by streptavidin, trypsin digested, cleaved, then analysed by MS (Fig. 5b). A loss in cysteine reactivity—read out as a lower intensity signal in the mass spectrum—indicates a transnitrosylation event (from GSNO) or other cysteine modification.
Gold nanoparticle (AuNP) probes for cysteine nitrosylation
Another property of thiols that can be exploited for biochemical analysis is their affinity for soft metals such as gold. Gold nanoparticles (AuNPs) have been shown to react with SNO-proteins to give ˙NO and AuNP–protein conjugates (Fig. 6a).82 After first alkylating free thiols with iodoacetamide (18), proteins were proteolysed and then treated with AuNPs.83 SNO containing peptides are designed to react with the AuNPs to release ˙NO and form peptides with a AuNP label at the site of nitrosylation. The AuNP-bound peptides can be harvested by centrifugation and the Au–S bond cleaved with excess DTT. The released peptide fragments can then be analysed by mass spectrometry. It is important to note that disulfides and thioethers also have affinity for AuNPs, so issues of selectivity should be considered when interpreting results. Additionally, the final DTT reduction step will likely cleave any disulfides present, which could complicate MS analysis. Still, it is notable that the peptides with high affinity for gold can be enriched, analysed and relevant hits can then be validated by independent assays.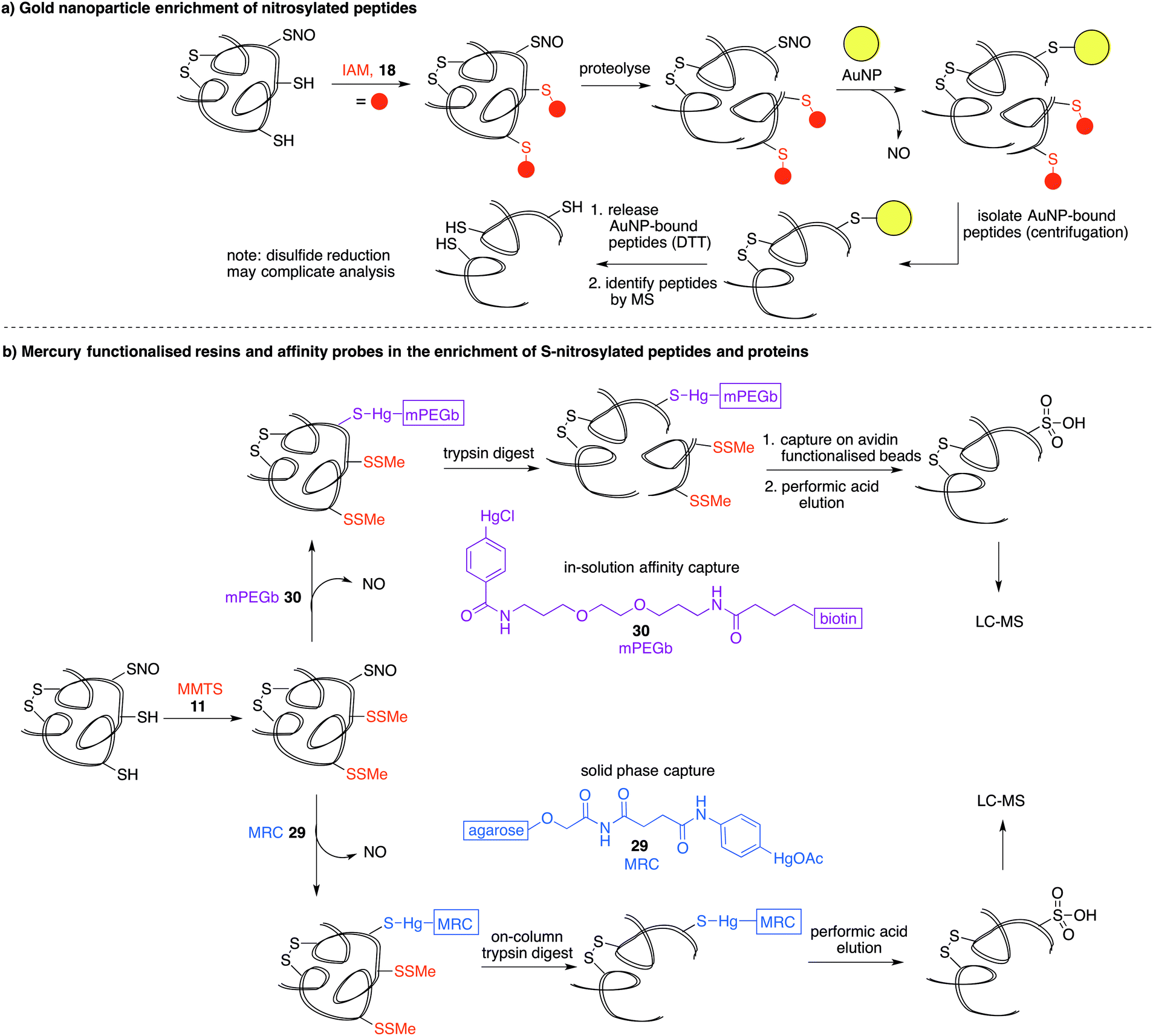 | ||
| Fig. 6 (a) Gold nanoparticle (AuNP) capture of protein-SNO. Free thiols are blocked by iodoacetamide (IAM, 18), proteolysed, and trapped on AuNPs with release of NO. The AuNP-bound peptides are isolated and released by DTT reduction. (b) Organomercury capture of protein-SNO using either affinity capture (mPEGb, 30) or mercury resin bound capture (MRC, 29). Both methods require blocking of free thiols (by MMTS 11, for instance), with performic acid elution to remove the captured peptides for analysis by mass spectrometry. | ||
Phenylmercury probes for cysteine nitrosylation
Phenylmercury compounds also react with S-nitrosothiols to give a relatively stable thiol–mercury bond. Leveraging this property, a method was developed whereby reduced cysteine residues are first blocked with MMTS (11) and then S-nitrosylated proteins or peptides are captured by an organomercury functionalised resin or affinity probe. Oxidation of the S-Hg group to the cysteine sulfonic acid provides the diagnostic group detected by LC-MS analysis (Fig. 6b).16 Organomercury resin (MRC) 29 and phenylmercury-polyethyleneglycol-biotin (mPEGb) 30 are representative mercury-containing reagents used to capture the S-nitrosylated proteins and peptides. The proteins or peptides were released from the mercury by treatment with performic acid which results in concomitant oxidation of the cysteine of interest to its corresponding sulfonic acid, which can be detected by mass spectrometry. Using this method, the identification of protein targets for S-nitrosylation in murine livers was achieved.16,84 More recently, mercury resin capture has been used to study S-nitrosylation in the wild-type mouse brain, where several S-nitrosylated proteins identified were implicated in glutamate metabolism.85 This method has also implicated S-nitrosylation in the regulation of β-oxidation of fatty acids in mitochondria.86 These discoveries notwithstanding, it is worth noting that both inorganic and organic mercury are known to bind strongly to polysulfides,87,88 so non-selective binding to disulfides should be considered when validating hits. It is also worth noting the inherent toxicity of organomercury compounds may limit their general applicability in chemical biology.Organophosphine probes for cysteine nitrosylation
In the methods discussed thus far for the detection of nitrosylated cysteine residues, several chemical steps are required to functionalise the putative site. These methods are therefore indirect and may be confounded by lack of chemoselectivity in each step and the need for sequential manipulation before final analysis. It is therefore valuable to consider methods that react directly, in the first step, with the nitrosylated cysteine. Early work by Xian et al. showed the utility of a novel reductive ligation reaction of nitrosylated cysteines with functionalised phosphines.89 Reminiscent of the Bertozzi–Staudinger ligation, triarylphosphines (31) bearing an ester in the ortho position of one of the aryl groups, were shown to react with small molecule nitrosylated thiols, forming sulfenamide 35 in good yield and high rates of reaction (Fig. 7a). This reaction constituted the first method to directly convert unstable nitrosylated thiols to isolable conjugates. The mechanism is likely similar to that of the Bertozzi–Staudinger ligation of such phosphines to azides. The RSNO reacts with 2 equivalents of phosphine reagent 31 to produce the phosphine oxide 32 and aza-ylide 33. An intramolecular reaction between the ester and aza-ylide provides intermediate 34, which undergoes hydrolysis to give the isolable sulfenamide product 35.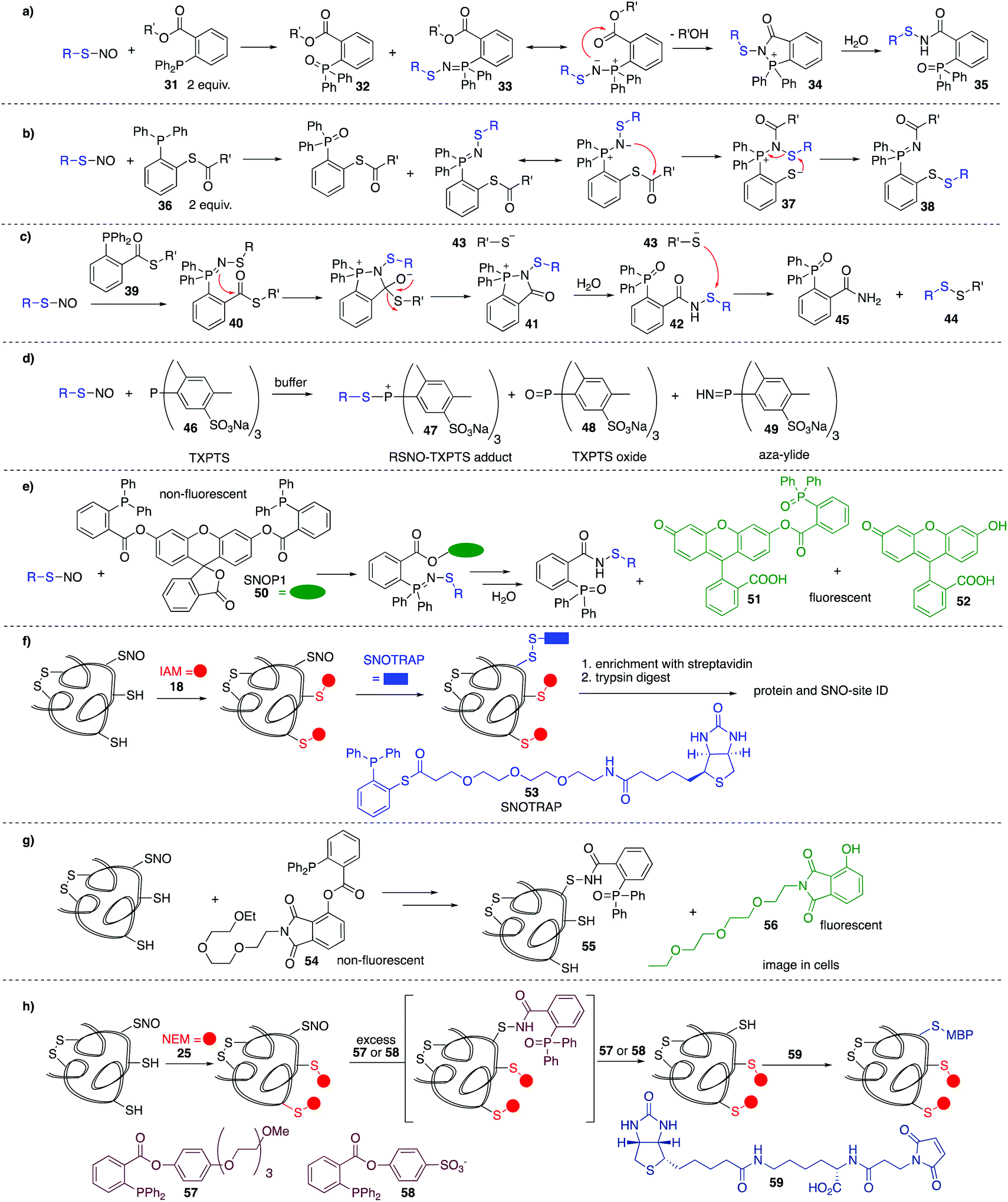 | ||
| Fig. 7 (a and b) Reductive ligation of triarylphosphine derivatives with small molecule S-nitrosothiols. (c) Reductive ligation of triarylphosphine derivatives with S-nitrosothiols, followed by in situ conversion to an isolable disulfide. (d) Water soluble phosphine tris(4,6-dimethyl-3-sulfonatophenyl) phosphine trisodium salt (TXPTS) reacts with nitrosylated thiols. (e) Reductive ligation of triarylphosphine derivatives and nitrosylated thiols that generates a fluorescent product. (f) Reductive ligation of triarylphosphine derivatives with nitrosylated cysteines, leading to biotinylation at the key cysteine. (g) Cell-compatible reductive ligation of triarylphosphine derivatives and nitrosylated thiols that generates a fluorescent product. (h) A variation of the biotin switch method in which nitrosylated cysteines are selectively reduced with a phosphine-based reagent. | ||
Variations on this reductive ligation include the transformation shown in Fig. 7b. In this reaction, phosphine reagent 36 reacts with nitrosylated cysteines to produce the isolable disulfide-iminophosphorane 38.90,91 In another variation, reagent 39 reacts with nitrosylated thiols to produce disulfide 44 and phosphine oxide 45. In this transformation, the reductive ligation proceeds as in Fig. 7a, but the final sulfenamide reacts with the thiol (43) generated in the ligation. Ultimately, the nitrosylated thiol is isolated as a disulfide (Fig. 7c).92
King et al. have shown that the water soluble phosphine tris(4,6-dimethyl-3-sulfonatophenyl)phosphine trisodium salt (TXPTS, 46) reacts with biological RSNOs.93 The formation of various products was observed, including an S-alkylphosphonium adduct 47, TXPTS oxide 48, and TXPTS-derived ylide 49 which were monitored using 13P NMR (Fig. 7d). Presumably adduct 47 is slow to hydrolyse due to the bulk of TXPTS. Some reactivity was also observed with certain disulfides. Notably, this was the first covalent labelling of an S-nitrosylated protein in buffer.
King et al. revisited the reductive ligation of phosphines and nitrosylated cysteines, this time employing the fluorogenic probe SNOP1 (50) (Fig. 7e).94 Upon reaction of 50 with RSNO and subsequent ligation, fluorescent products 51 or 52 are liberated, providing a convenient means for monitoring protein S-nitrosylation events in real time.
Tannenbaum et al. merged the reductive ligation techniques with the biotin switch strategy, a proteomic approach termed SNOTRAP 53 (Fig. 7f).15 Phosphine-based probe 53 contains a thioester linked with a polyethylenegylcol (PEG) spacer to biotin, allowing for subsequent affinity capture of labelled proteins. Free thiols are first blocked and then 53 is used to react directly at nitrosylated cysteine residues. This method allowed the study of early stages of neurodegeneration.
Fluorogenic probe 54 was developed by Li et al. for selective in situ imaging of protein S-nitrosylation in live cells (Fig. 7g).9554 acts through phosphine-mediated reductive ligation at nitrosylated cysteine residues. Much like King's SNOP1 probe 50, this phosphine probe is non-fluorescent, but liberates a fluorescent product 56 upon ligation to RSNOs. This probe was used to image S-nitrosylated GAPDH in live cells in real time.
Based on the phosphine-mediated reductive ligation work by Xian, Whorton et al. thought to use this as an alternative reducing agent in a BST modification. For some phosphine substrates such as 57 or 58, the sulfenamide intermediate 35 can be over-reduced in the presence of excess phosphine to give the free thiol. The free thiol could be labelled with biotinylated maleimide derivative 59, for detection or isolation by standard affinity methods (Fig. 7h).96
This last example in Fig. 7h highlights potential selectivity issues in the reductive ligation, as sulfenamide derivatives such as 35, 42, and 55 can apparently be reduced with phosphines or thiols. Additionally, phosphine reagents are known to react with disulfides and they can also be oxidised under aerobic conditions to the phosphine oxide. These potential complications should be considered when selecting and deploying a phosphine based detection method in biological systems.
Benzenesulfinate probes for cysteine nitrosylation
Based on work related to the conversion of S-nitrosoglutathione (GSNO) to S-phenylsulfonylglutathione (GSSO2Ph) by reaction with benzenesulfinic acid, Grieco and co-workers explored this reaction as a method to trap nitrosylated cysteine residues on proteins.97 Under acidic conditions, the SNO moiety can be protonated and activated for nucleophilic attack by benzenesulfinate 60. The resulting adduct (61) can react with a second equivalent of benzenesulfinate, forming products 62 and 63 (Fig. 8a). However, 62 is prone to hydrolysis at physiological pH. To overcome this, a variation termed the thiosulfonate switch was developed which displaces the S-phenylthiosulfonates (such as 62 and 66) with a thiol containing fluorescent probe 65 to give a stable disulfide linkage.98 First, free thiols are blocked by S-phenylsulfonyl cysteine (SPSC, 64), generating the benzenesulfinate 60 as a co-product. Addition of more 60 converts S-nitrosothiols to the S-phenylthiosulfinates 66. Finally, addition of a water-soluble rhodamine based probe containing a reactive thiol (Rhod-SH, 65) reacts with 66 giving a mixed disulfide 67 of the probe and protein-SNO (Fig. 8b). This transformation requires an acidic pH (4) which may limit its use in live cells.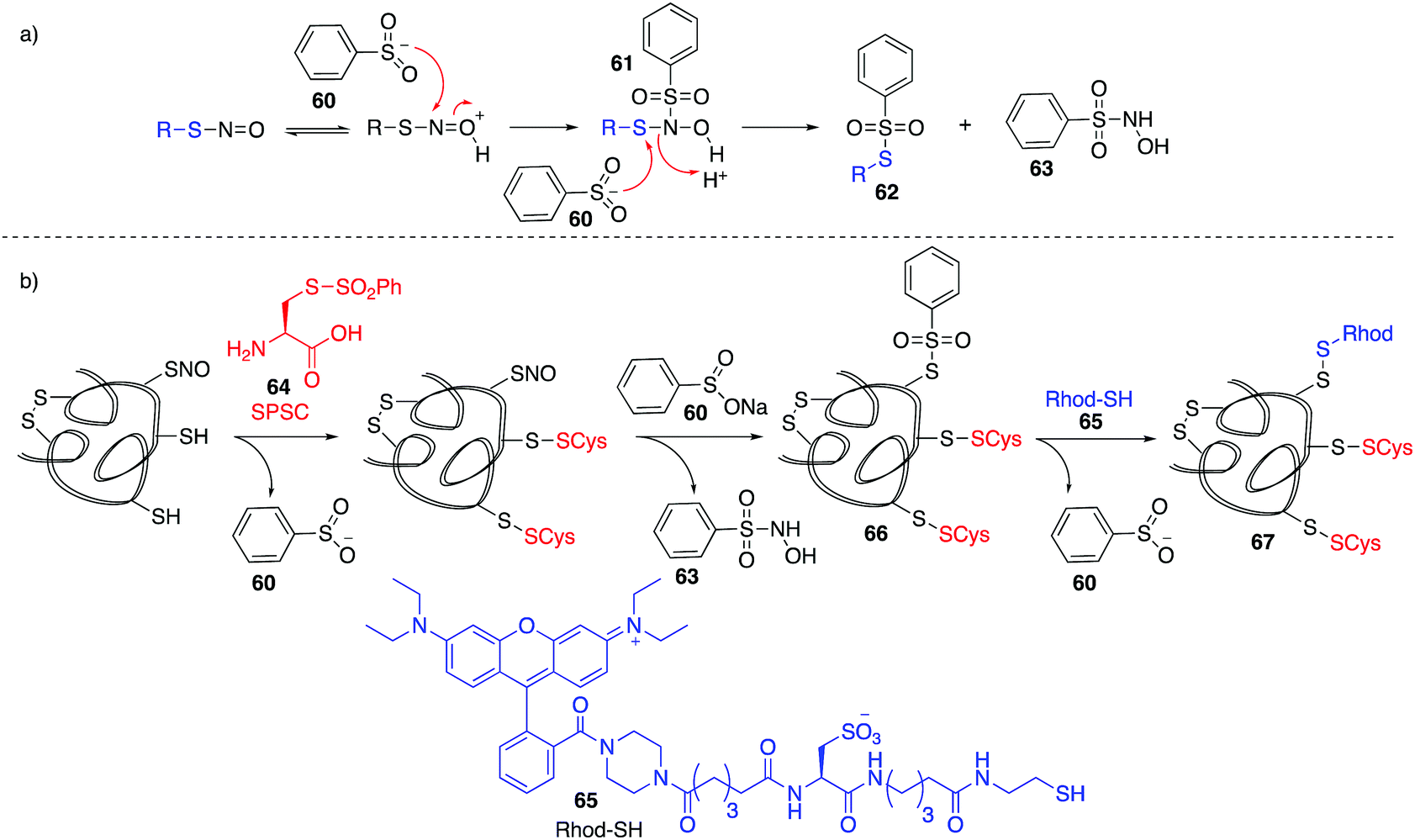 | ||
| Fig. 8 (a) Proposed mechanism for the reaction of benzenesulfinate (60) with nitrosothiols. (b) Reaction of benzenesulfinate (60) with nitrosylated proteins, followed by addition of a thiol-conjugated with rhodamine (65) for detection. | ||
Detection of cysteine nitrosylation in proteins is challenging due to the potential for cross-reactivity with other cysteine oxidation states, particularly when switch techniques are involved. There is therefore opportunity for development in direct methods to trap nitrosylated cysteines in which there are side reactions with other groups such as thiols, disulfides, or sulfenic acids. These selectivity issues notwithstanding, the existing methods for studying cysteine nitrosylation strongly suggest that this protein modification is a critical determinant of redox regulation with important physiological implications.
3.1 Sulfenic acids (RSOH)
Sulfenic acids (3) have garnered increasing attention since Allison and Benitez trapped the sulfenic acid form of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) with dimedone (68) and various alkenes, thereby inhibiting the enzyme.99,100 Poole and Claiborne also provided evidence for a stabilised cysteine sulfenic acid in the redox-centre of the streptococcal NADH peroxidase,101 and Denu and Tanner revealed the reversible inactivation of protein tyrosine phosphatases by cysteine sulfenic acid formation several years later.21 Together these studies indicated that cysteine sulfenic acid is not merely an intermediate oxidation state of cysteine (though it can be) but rather it is a key regulator of protein function, catalysis and signalling. Protein sulfenic acids are also intriguing because they are often an early oxidation product of the reaction of cysteine with ROS and RNS. For this reason, cysteine sulfenic acids may also be a biomarker for oxidative stress and play a key role in redox-regulation. Commonly, sulfenic acids are formed in proteins through oxidation of the thiolate side chain of cysteine by H2O2. Acidic cysteine residues are particularly reactive, as the nucleophilic thiolate (RS−) can attack H2O2 directly via an SN2 reaction.102 The pKa of cysteine residues is therefore an important factor in assessing its susceptibility to oxidation. Most protein thiols have a pKa of 8–9, making them largely protonated at physiological pH and hence less reactive.17 However, the pKa is not the only determinant and the protein microenvironment (varying in polarity, charge and steric congestion) can modulate reactivity. For example, the rate constant determined for the reaction of the active site cysteine of peroxiredoxin 2 with H2O2 was 1.3 × 107 M−1 s−1 (pKa 5–6),103 compared to tyrosine phosphatases (PTPs) which was only 10–20 M−1 s−1 (pKa 5.4).21H2O2 has been described as both a second messenger and a toxic by-product of metabolic processes. The mitochondrial electron transport chain104 and NADPH oxidases105 generate superoxide as a by-product of oxygen reduction. Superoxide (O2˙−) can undergo dismutation to H2O2 and O2 with a rate constant of ≈105 M−1 s−1, or when catalysed by superoxide dismutase, ≈109 M−1 s−1.106,107 Other ROS and RNS can form sulfenic acids by reaction with thiolates such as organic hydroperoxides (ROOH),108 peroxynitrous acid (ONOOH), and the peroxynitrite anion (ONOO−).109 Comparative rate constants for the oxidation of cysteine and BSA by H2O2 and ONOO− showed the latter to be a stronger oxidant at pH 7.4. The reported rate constants for ONOO− oxidation of cysteine and BSA were 5.9 × 103 and 2.7 × 103 M−1 s−1 respectively, with H2O2 oxidation proceeding with rate constants of 4.69 and 1.14 M−1 s−1 respectively.109 It was also found that cysteine can react with ONOO− as either the thiol or thiolate.
Sulfenic acids can exist in two tautomeric forms, with the proton bound to either the oxygen or the sulfur (Fig. 9). Early spectroscopic work by Bruice showed that the tautomer with the proton bound to the oxygen is favoured almost exclusively—likely due to the stronger O–H bond versus the weaker S–H bond.110 The sulfur atom in sulfenic acids exhibits both electrophilic and weak nucleophilic character. This is evident in the self-condensation of sulfenic acids to form thiosulfinates. While this frequently occurs in small molecule sulfenic acids, the occurrence of thiosulfinates in proteins has not been reported. The sulfenic acid oxidation state is generally unstable and short lived, and depending on the protein microenvironment, will undergo further reactions with neighbouring groups such as thiols and amides to form disulfides or cyclic sulfenamides, respectively. Due to its reactive nature, sulfenic acids are a challenge to detect and isolate and the mechanisms by which they are trapped are often unclear.8,30,111,112
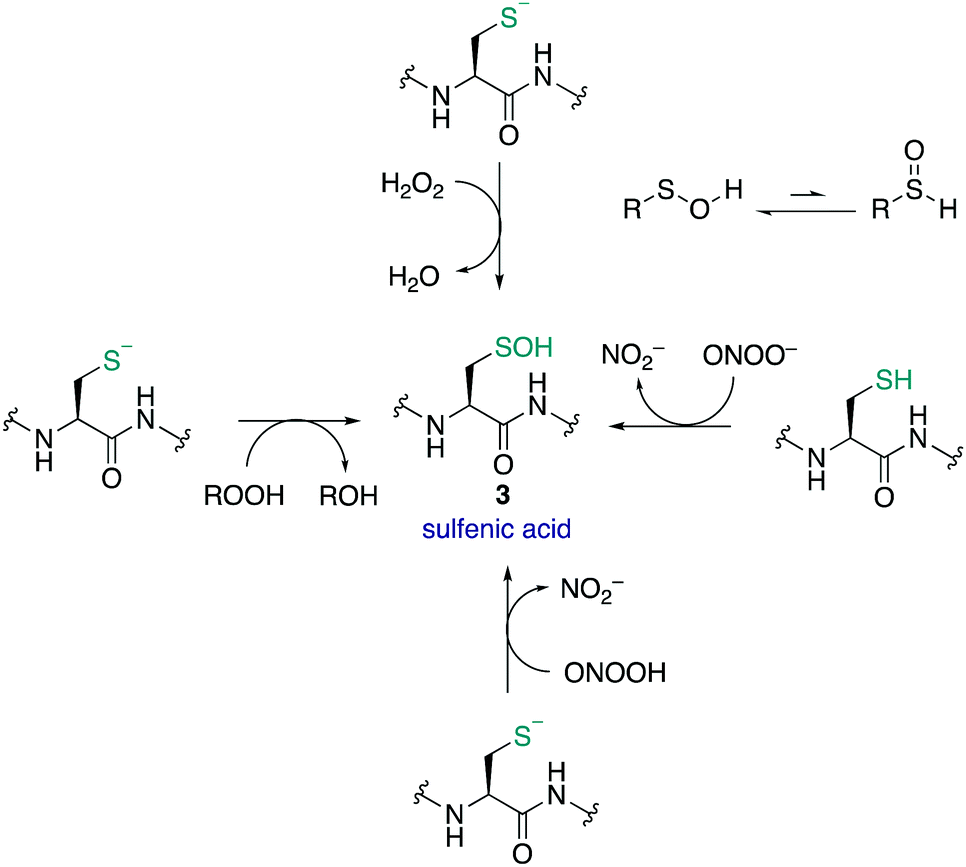 | ||
| Fig. 9 The formation of cysteine sulfenic acids in proteins. | ||
Cysteine sulfenic acids have been suggested to play key roles in the immune response, stem cell development, pathogenesis of cancers, neurodegeneration, and growth factor signalling.113 Sulfenic acids are involved in the protein tyrosine phosphatase (PTP) signal transduction pathways,21 activation and function of T-cells (cell growth, calcium flux, proliferation and phosphorylation),114 redox-switching to regulate kinase activity (formation of the sulfenic acid represses or enhances certain kinase activity),115−117 and regulation of intracellular signalling of the EGFR by oxidation of a key cysteine residue.22 With such ranging function attributed to this intriguing oxidation state of cysteine, it is critical to understand its incidence and mechanism in biology.
Because cysteine sulfenic acids are most often the first oxidation product formed during oxidative stress events, they likely play an intermediate role in downstream redox signalling. Because of its potential role in diverse pathways, the oxidation of cysteine to its sulfenic acid is sometimes compared to (and also linked) to universal signalling mechanisms such as protein phosphorylation. For example, protein tyrosine phosphatases (PTP), a family of about 80 proteins, are inactivated by oxidation of the cysteine to its sulfenic acid.21,118 This reversible oxidation to the sulfenic acid can promote phosphorylation-dependent signalling cascades as seen in the epidermal growth factor receptor pathway (EGFR).23 The resultant sulfenic acid may also react with a backbone amide to form a cyclic sulfenamide,29 or react with an adjacent thiol to form an intramolecular disulfide.119 This reversible conversion prevents overoxidation to the sulfinic and sulfonic acids.
Using A431 cell lines, Carroll and co-workers investigated epidermal growth factor (EGF)-induced protein sulfenylaton.23 Binding of epidermal growth factor (EGF) to the epidermal growth factor receptor (EGFR) induces the production of H2O2 through the enzyme Nox2. The H2O2 generated can directly oxidise the EGFR Cys-797 to the sulfenic acid, which enhances the tyrosine kinase activity up to 5-fold. H2O2 may also oxidise and deactivate PTPs, which regulate the level of EGFR phosphorylation.22 Together these events lead to an increase in receptor autophosphorylation which promotes downstream signalling pathways related to proliferation, differentiation, growth, migration and survival. Using a histochemical approach, Carroll and Seo found that EGFR was overexpressed in Her2 breast cancer cell lines that exhibited increased levels of H2O2 and protein sulfenylation.120 More recently, further work on EGFR from A431 cell lines has confirmed the Cys-797 as a functional redox switch that stimulates EGFR autophosphorylation.121 Using computational modelling, oxidation of Cys-797 to the sulfenic acid may influence the catalytic loop through stabilisation, stimulating kinase activity.
The monoacylglycerol lipase (MGL) is a serine hydrolase that can deactivate the endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG). Two regulatory cysteines Cys-201 and Cys-208 control entry of substrates into the active site. Studies by Piomelli and co-workers explored the effect of H2O2 induced oxidation of these two cysteine residues using site-directed mutagenesis and dimedone 68 (a chemoselective probe for cysteine sulfenic acids).122 They found that H2O2 inhibited the enzyme MGL through an allosteric mechanism involving the two redox-sensitive cysteines Cys-201 and Cys-208. This in turn regulates 2-AG-mediated signalling in neurons, which may serve as a presynaptic control point.
Recent work by the Toledao and Rahuel-Clermont groups have also demonstrated the intricate ways in which sulfenic acid signalling can be mediated.123 In this study it was shown that a key cysteine sulfenic acid residue in the peroxidase Orp1 is protected through binding to the Yap1-binding protein (Ybp1). These two proteins then form a ternary complex with Yap1. This ternary complex leads to a directed and rate-enhanced formation of disulfide on Yap1 through reaction with the Orp1 cysteine sulfenic acid. Without Ybp1, this process is slow and unselective. These results indicate that Yap1-binding protein operates as a sulfenic acid chaperone and facilitates this redox signalling.
With growing evidence for the intermediate role of sulfenic acids in diverse signalling pathways, the importance of defining these pathways has become clear. Chemical tools for such endeavours are discussed next.
3.2 Chemical methods for detecting RSOH
Many cysteine sulfenic acid residues are thought to be short-lived species. Therefore, prolonged manipulation may lead to degradation of the sulfenic acid (by over oxidation, self-condensation, or reaction with a nucleophile, for instance). Therefore, direct and rapid methods are required to detect this oxidation state of cysteine—a significant challenge in chemical biology.Modified biotin switch technique for detecting cysteine sulfenic acids
A method similar to the biotin switch technique initially developed for S-nitrosothiols was used to detect sulfenic acids, exploiting the selective reduction of the sulfenic acid by arsenite (Fig. 11a).124 Free thiols are first blocked with maleimide in the presence of SDS, followed by reduction of SOH by sodium arsenite in the presence of biotin-maleimide. However, much like the biotin switch technique used to study S-nitrosothiols, several limitations restrict the overall efficiency of this indirect approach, particularly the long time scale over which short-lived sulfenic acids will not persist. The method is also performed under denaturing conditions, which could compromise sulfenic acids that rely on the protein microenvironment for stability. The selectivity of the reduction by sodium arsenate has also been debated and because arsenic is toxic, the disruption to normal cellular function must also be considered. Nevertheless, this study provided motivation for further study of cysteine sulfenylation.1,3-Diketone chemical probes for cysteine sulfenic acids
Detection of the sulfenic acid modification has become increasingly more versatile due to a large set of chemical probes that contain an enolisable 1,3-diketone which can react with sulfenic acids directly. The most commonly used sulfenic acid trap is dimedone 68 (5,5-dimethyl-1,3-cyclohexanedione) and its derivatives. The reaction of dimedone with cysteine sulfenic acids was first discovered by Allison during studies of glyceraldehyde 3-phosphate dehydrogenase (GAPDH)—a discovery that paved the way for the majority of current sulfenic acid detection methods.99 The exact mechanism for the reaction of 68 with sulfenic acids has not been established but several hypotheses have been advanced to account for the formation of the putative adduct 70 including hydrogen-bond assisted substitution (via the enol 69a) or direct substitution (via the enolate 69b) (Fig. 10a). Other mechanisms in which dimedone acts as an electrophile have also been proposed.11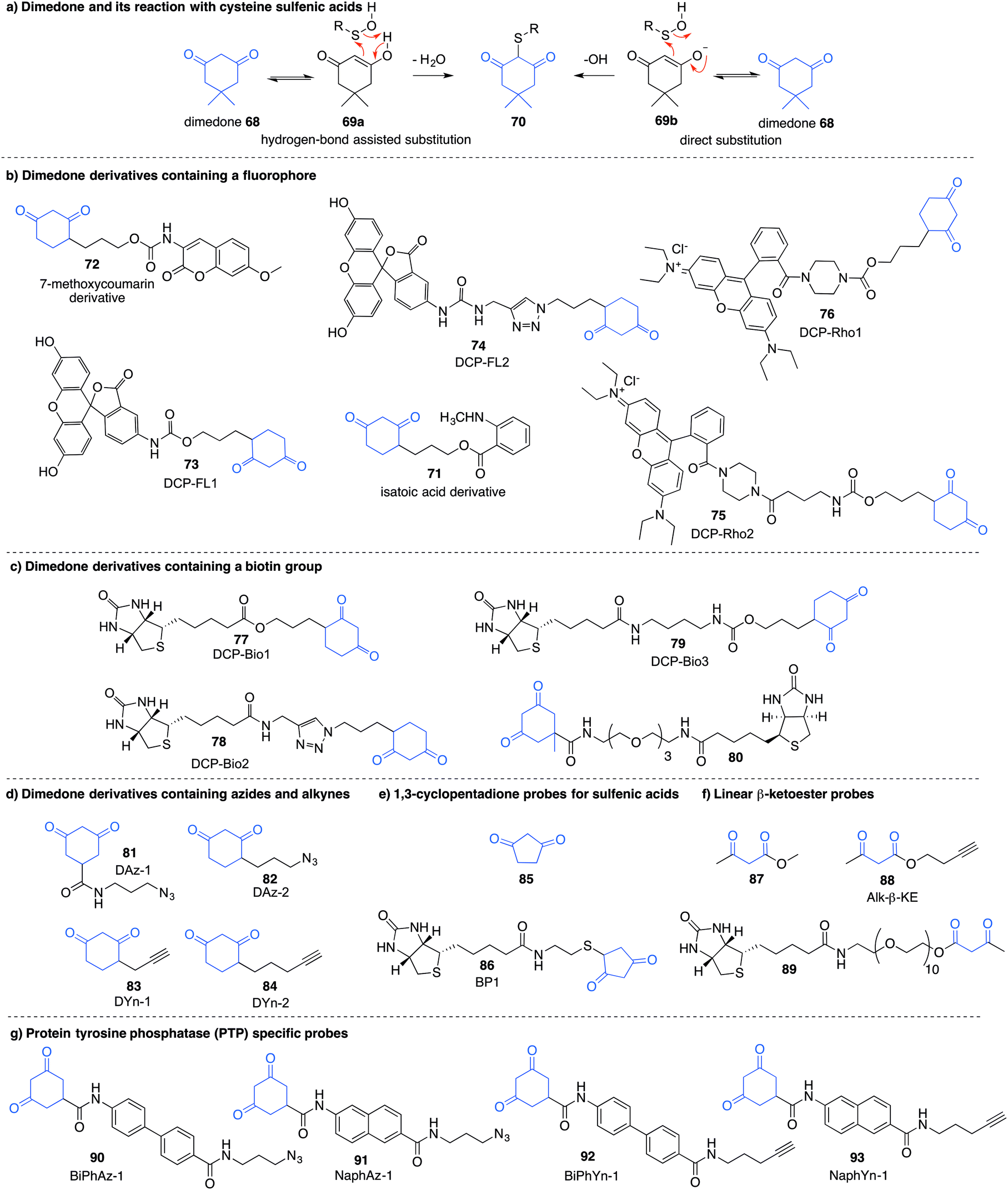 | ||
| Fig. 10 Sulfenic acid probes containing 1,3-diketone functionalities with various detectable tags. | ||
Because dimedone does not contain a detectable handle, many derivatives have been synthesised that contain fluorophores, affinity probes, or functionalisable groups such as azides and alkynes (Fig. 10). King et al. first attached fluorescent labels in the form of isatoic acid derivative 71 and 7-methoxy coumarin derivative 72 (Fig. 10b).125 Testing these probes on a cysteine-dependent peroxidase enzyme from Salmonella typhimurium, small changes in the excitation and emission wavelength maxima were observed upon protein sulfenic acid adduct formation. But, due to bleaching of the fluorophore, monitoring the protein reaction was technically difficult. King and co-workers then disclosed a new library of dimedone probes containing fluorescein (73–74), rhodamine (75–76) and biotin (77–79).126 Charles et al also disclosed a similar biotin probe (80) to study oxidation in rat hearts (Fig. 10b and c).127 Both 76 and 77 were further utilised to study sulfenic acid formation during lysophosphatidic acid-mediated cell signalling.128 The probe 77 has been used in several recent studies focussed on different proteins such as, endoplasmic reticulum (ER) transmembrane protein IRE-1129 and ERK-1/2 kinase.116
While these probes offered a convenient means for detection through fluorescence analysis or affinity enrichment using the biotin tag, the steric bulk of the probes might limit the detection of more hindered cysteine sulfenic acids. Smaller probes were therefore prepared to address this potential issue. Accordingly, azide and alkyne handles were explored as they can be imaged through azide–alkyne cycloadditions and Staudinger ligation reactions that install a biotin group or fluorophore after the dimedone derivative has trapped the sulfenic acid.130 Using this strategy, Carroll et al. devised a dimedone probe DAz-1 (81) containing an azide chemical handle (Fig. 10d).131,132 Using this probe, a new method for detecting sulfenic acids directly in un-manipulated, intact cells was achieved using Jurkat cells as a model. This study also revealed the important differences between labelling cell lysates and live cells, with significantly more oxidation occurring during the lysis process. However, 81 was less reactive relative to dimedone (68), and the amide linkage was thought to reduce cell permeability and accessibility making the probe non-optimal for global proteomic studies. Improving upon this probe, Carroll et al. synthesised derivative DAz-2 (82) which lead to increased labelling in vitro and in live cell studies, providing the first global analysis of the sulfenome (Fig. 11b).133,134 These studies on tumour cell lines validated DAz-2 as a general sulfenic acid probe demonstrating membrane permeability and reaction with cysteine sulfenic acids in cells. Importantly, 82 was not toxic and did not appear to trigger oxidative stress or cell death.
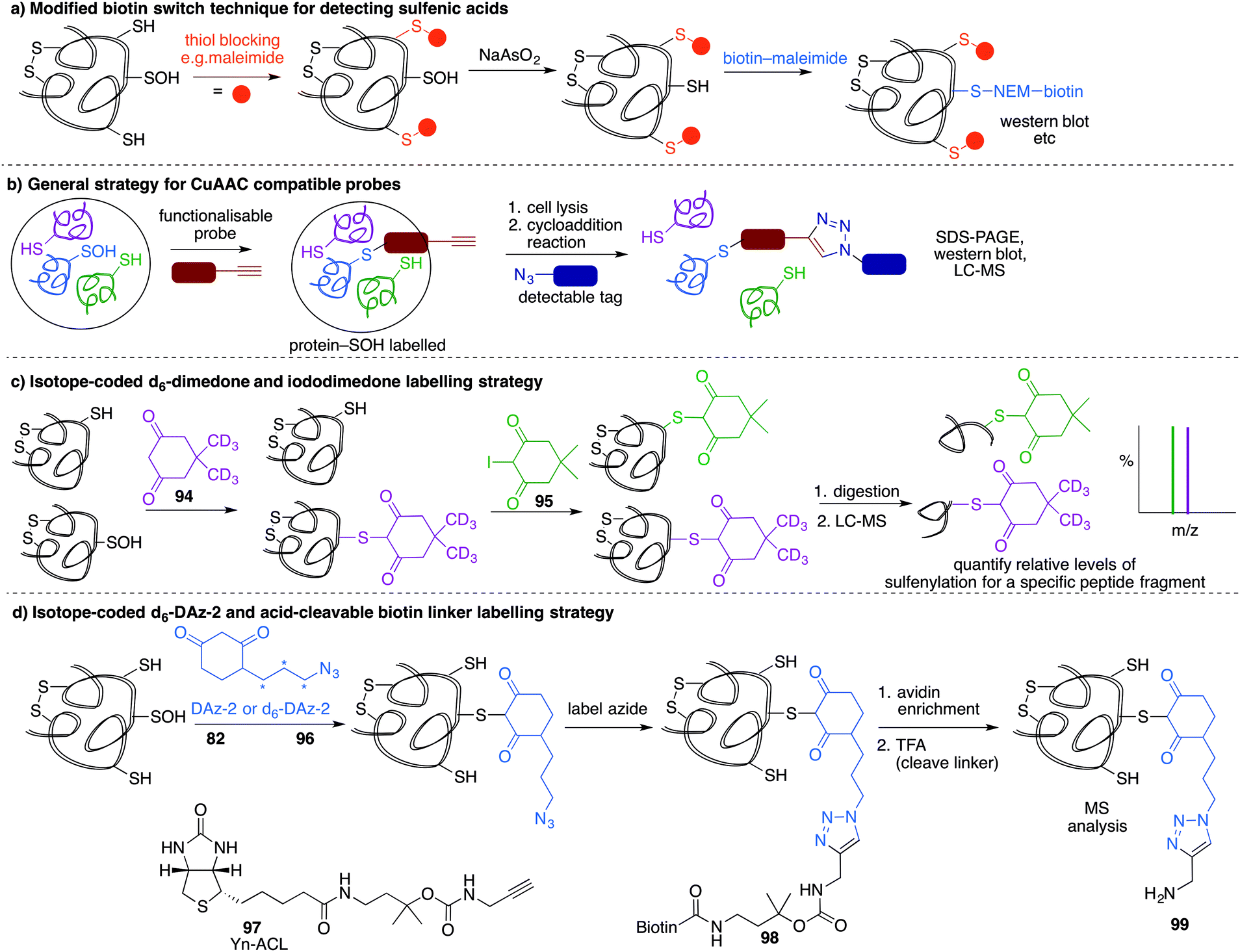 | ||
| Fig. 11 (a) Modified biotin switch technique for sulfenic acid detection. Free thiols are blocked, then remaining SOH reduced with arsenate to the thiol and detected with biotin-maleimide. (b) General strategy for using alkyne-labelled sulfenic acid probes. After ligation to the sulfenic acid, probes are visualised through the Cu-catalysed azide–alkyne cycloaddition reaction which can attach a fluorophore or affinity tag. (c) Isotope labelling method using d6-dimedone (94). Sulfenic acids are labelled with 94 then thiols are labelled with iododimedone (95). Proteolytic digestion and mass spectrometric analysis can reveal the ratio of thiol and sulfenic acid for a specific cysteine. (d) DAz-2 (82) (and its isotopically labelled derivative, 96) can trap sulfenic acids and then be isolated after conjugation to a biotin affinity tag. Cleavage of the biotin tag facilitates analysis by mass spectrometry. | ||
In a related approach using an alkyne handle on dimedone, probes DYn-1 (83) and DYn-2 (84) were found to offer superior sensitivity (Fig. 10d).23 Detection of sulfenic acids was also dependent on probe dose and incubation time with no loss of cell viability and redox-balance was maintained for cells treated with 84. This probe was integral in the discovery that a key cysteine residue in epidermal growth factor receptor, when oxidised to its sulfenic acid, enhances the kinase activity of the protein. Development of this direct detection method revealed the molecular details of at least one way in which hydrogen peroxide can act as a signalling molecule,135 and how oxidation of cysteine can be regarded as a method of signalling akin to phosphorylation.
Furdui et al. developed 1,3-cyclopentadione sulfenic acid probes (85 and 86, Fig. 10e). These probes exhibited reasonable reactivity however some pH-dependence was seen, where lowering the pH increased reactivity (suggesting the enol form as the reactive species).136 Additionally, linear β-ketoesters (87–89) were also shown to trap sulfenic acids with improved reactivity at physiological pH (Fig. 10f).137 The β-ketoester is cell permeable and does not cause cell death. An advantage of 88 and 89 is their ability to react with NH2OH, cleaving the ester and removing the affinity tag, ultimately generating a 3-methyl-5-isoxazolone at the key cysteine—a modification readily integrated with mass spectrometry. However, these probes still possess modest reaction rates, a key issue with most sulfenic acid probes to date.
In order to further study protein tyrosine phosphatases (PTPs) specifically, Carroll and co-workers designed several probes (90–91) for direct detection of PTP oxidation (Fig. 10g).138 Oxidative inhibition of PTPs have been shown to promote phosphorylation-dependent signalling cascades, but low cellular abundance has hindered further investigation. The trifunctional probes consisted of a cyclic 1,3-diketone group to form the covalent adduct with the PTP sulfenic acids, a module that directs binding to target the PTP (such as naphthalene and biphenyl), and a reporter tag used for detection. Compounds containing the binding module showed increase in labelling for PTPs compared to DAz-1 (81). Related probes were also prepared so that they contained an alkyne handle (92–93). The key cycloaddition reaction used to visualise the alkyne appeared to be enhanced in comparison to the azide analogue.139 The study examined protein tyrosine phosphatase 1B (PTP1B)—a negative regulator in the insulin signalling pathway, which acts by dephosphorylating tyrosine residues on the insulin receptor. Probes 92 and 93 were able to bind with oxidised and inactivated PTP, which prolonged signalling by preventing this de-phosphorylation.
Based on the cysteine-specific acid-cleavable isotope-coded affinity tag (ICAT) approach used to quantify oxidant-sensitive thiols,140,141 Carroll et al. developed a similar dimedone-based strategy for detection and quantification of sulfenic acids in proteins.142 The isotope-coded dimedone (94) and iododimedone (95) were used to quantify the extent of sulfenic acid modification for a given residue (Fig. 11c). In the event, sulfenic acids are first labelled with 94 and then free thiols are labelled with 95. The protein samples are then digested and analysed by LC-MS. The extent of cysteine oxidation for a specific site on a protein is then determined by the relative peak intensities of the heavy and light dimedone labels in the mass spectrum. Using isotope-coded dimedone/iododimedone-labelling strategies, other proteins such as the heme-thiolate cysteine (Cys-457) of cytochrome P450 4A11 was found to be selectively oxidised to the sulfenic acid, leading to loss of activity.143
A complementary strategy couples an isotope-coded DAz-2 derivative 96 with an acid-cleavable biotinylated tag (97).144 In this method, protein sulfenic acids are trapped by 82 or 96 and then labelled with Yn-ACL 97 to give the triazole 98 (Fig. 11d). Biotinylated proteins are enriched on avidin beads and then cleaved from biotin by treatment with acid to give 99. The advantage of this method is that after enrichment of the proteins of interest, the biotin label can be removed. This is important as biotin can, in some circumstances, complicate protein MS analysis. Additionally, the isotope pair 82 and 96 can be used to monitor relative changes in protein oxidation. DYn-2 (84) has also been used in development of proteomic methods for global identification and quantitation of sulfenic acid modified sites using light and heavy DYn-2 derivatives.145
Alternative chemical probes
Other chemical probes for detection of cysteine sulfenic acids that are complementary to dimedone (68) have been explored. The reagent 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl, 100) was studied by Poole et al. as an electrophilic sulfenic acid trap, which is complementary to the nucleophilic dimedone (Fig. 12a). 100 had previously been shown to react with thiols, the phenolic group of tyrosine, and amino groups under basic conditions, which can present challenges with selectivity. However, each product had a unique absorbance maximum (420, 382 and 480 nm respectively) which can provide a way to potentially distinguish products.146100 was also found to react with sulfenic acids with a characteristic absorbance maximum at 347 nm allowing it to be distinguished spectroscopically from other adducts. Importantly, the reaction of 100 with cysteine gives a product with a different mass than when it reacts with cysteine sulfenic acid. With that said, there is some uncertainty as to whether sulfoxide 101 or sulfenate ester 102 (or both) are formed when 100 reacts with a sulfenic acid. In any event, 100 has been used in several studies including the reversible inactivation of protein tyrosine phosphatases,21 OhrR,108 and the Orp1 protein.147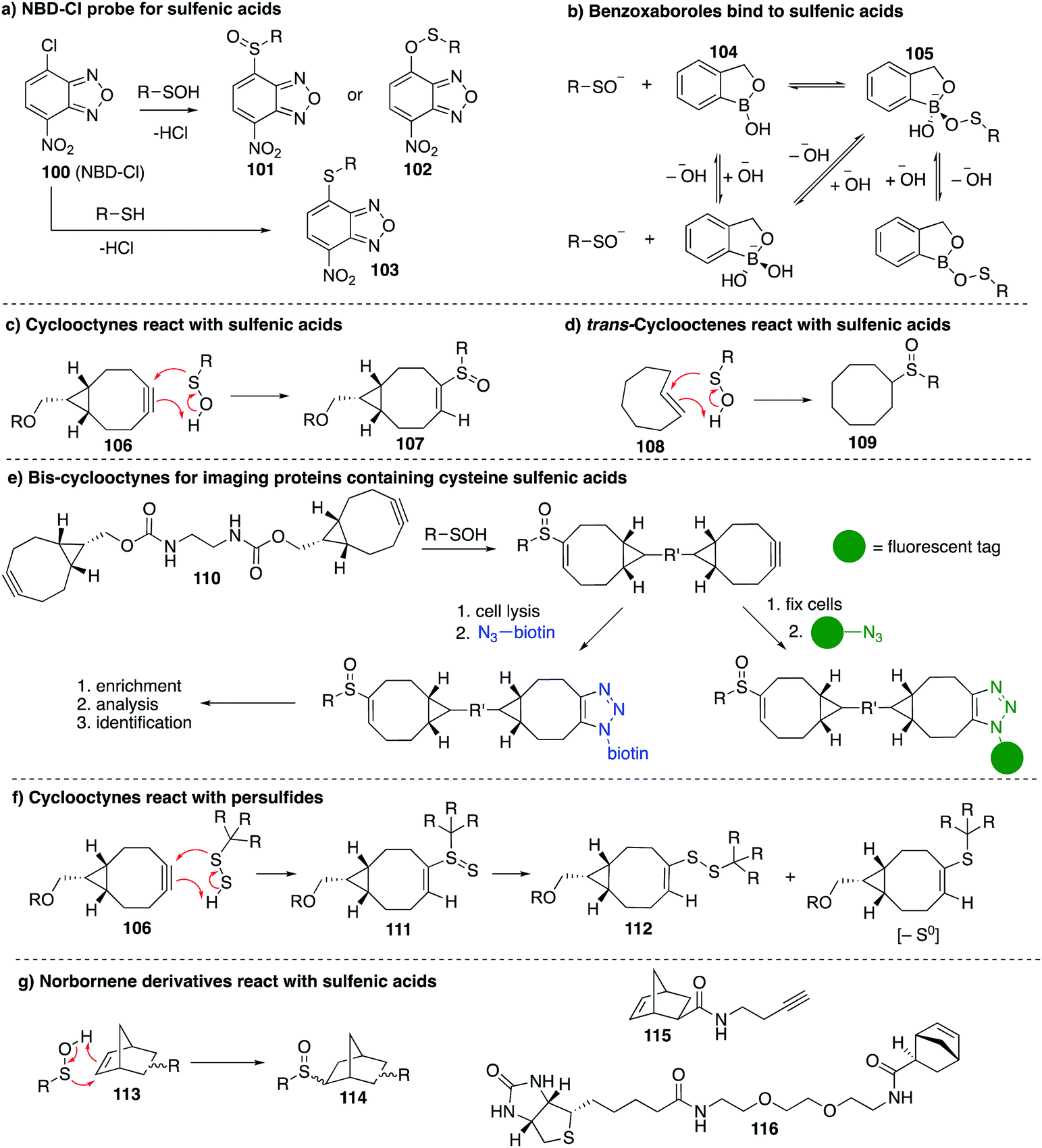 | ||
| Fig. 12 (a) NBD-Cl (100) is an electrophile that reacts with cysteine sulfenic acid. (b) Boronic acids and benzoxaboroles such as 104 bind reversibly to sulfenic acids. (c and d) Strained alkynes and alkenes react with sulfenic acids. (e) A biscyclooctyne probe 110 can react with protein sulfenic acids. The second alkyne can be used to ligate a reporter group for affinity purification or imaging. (f) In addition to sulfenic acids, strained alkynes also react with persulfides. (g) Norbornene derivatives react with short-lived sulfenic acids. Two norbornene probes for cysteine sulfenic acid detection are shown, one labelled with an alkyne (115) and the other with biotin (116). | ||
Benkovic et al. explored boronic acids as sulfenic acid traps (Fig. 12b).148 Arylboronic acids and cyclic benzoxaboroles such as 104 can reversibly form adducts such as 105 with sulfenic acids under aqueous conditions. The benzoxaborole 104 was the most effective probe for reversibly binding to sulfenic acids. While this may not be an effective sulfenic acid trap for proteomics analysis, boronic acids may be useful for reversible inhibition of proteins that rely on cysteine sulfenic acid formation for function.
The main limitation of most sulfenic acid probes is the slow rate of reaction, possibly preventing the detection of shorter-lived sulfenic acids. As a consequence, there are likely protein sulfenic acids that remain undiscovered. Sulfenic acids are known to react with alkenes and alkynes via a concerted mechanism to give sulfoxide adducts.99,100 Early work by Allison found that the olefins 3-cyclohexene-1-carboxylate, dihydropyran and tetrahydrophthalimide inactivated GAPDH, presumably by covalent inhibition due to the cycloaddition of the sulfenic acid and the alkene. King et al. revisited this concept in their study of strained alkenes and alkynes and their reaction with cysteine sulfenic acids. The key advance was to increase the rate of reaction by virtue of strain relief in the cycloaddition.149 Using bicyclo[6.1.0]nonyne derivatives (106), they demonstrated rapid reaction with small molecule sulfenic acids as well as sulfenic acids on proteins. The sulfoxide adduct 107 was formed with reaction rates up to 2 orders of magnitude greater than dimedone (Fig. 12c). The strained alkene trans-cyclooctene (108) was also tested and verified to form sulfoxide 109 after reaction with sulfenic acids, though the rate was less than that of the cyclooctyne (Fig. 12d). Biscyclooctyne 110 has also been explored recently in mapping sulfenic acids. After the sulfenic acid reacts with one alkyne in 110, the other alkyne can be labelled through a cycloaddition with an azide (Fig. 12e).150
While cyclooctynes have been shown to react rapidly with cysteine sulfenic acids, this highly strained system can also react with other groups such as thiols and persulfides. For example 106 reacts rapidly with synthetic persulfides—a functional group that may have biological importance (Fig. 12f).151 The reaction of 106 with persulfides proceeds analogously to the reaction with sulfenic acids, providing adduct 111, which can undergo subsequent rearrangement to disulfide 112 or the corresponding thioether after extrusion of sulfur (Fig. 12f). The thiol–yne reaction is another well-documented reaction between an alkyne and thiyl radical that should be considered when using cyclooctynes in a biological context. Indeed the direct reaction of strained alkynes and cysteines has been demonstrated for peptides and proteins in live cells,152 which prompts an issue of caution when using cyclooctynes to map sulfenic acids. In such cases, clear mass spectrometric analysis and hit validation is required to eliminate such off-target reactions.
Our group has recently explored other strained alkenes as cysteine sulfenic acid traps.153 It was demonstrated that easily accessible norbornene probes (113) could trap the short-lived sulfenic acid formed by oxidation of N-acetylcysteine with H2O2 through a cycloaddition mechanism to form sulfoxide adduct 114 (Fig. 12g). This work was inspired by the early work of Allison and Benitez using water soluble cyclohexene derivatives to trap cysteine sulfenic acids and later Barton and co-workers who specifically used norbornene derivatives to trap the sulfenic acid formed during thermally induced syn-elimination of the sulfoxides of penicillin.99,154–156 The strain of the norbornene was sufficient to lead to rapid reaction with the unstable sulfenic acid. Dimedone was far less reactive and unable to trap the same short-lived cysteine sulfenic acid. The use of moderately strained alkenes, as opposed to highly strained alkynes, was designed to help overcome off-target reactions such as direct reaction with the cysteine thiol. Some thiol–ene chemistry was still observed with norbornene, but the rapid reaction with short-lived sulfenic acids is a promising and complementary feature to the chemistry of dimedone. The preparation of the probes 115 and 116 in Fig. 12g153 encourages future studies on proteins and live cells.
Other carbon-centred nucleophiles
Despite the relatively low reactivity, dimedone-derived probes currently appear to offer the best selectivity in studying cysteine sulfenic acids in cells. Carroll et al. recently conducted a wide ranging study on various carbon-centred nucleophiles (inspired by dimedone) to improve their rate of reaction with sulfenic acids (Fig. 13).157 Using pseudo first-order conditions with a dipeptide-based sulfenic acid model 117 (Fig. 13a), rate constants were compared across a wide range of potential probes. It was found that increasing the ring size from a 6 to 7-membered ring such as 118 increased the rate of reaction slightly (Fig. 13b). Rate enhancements relative to dimedone were also seen for linear 1,3-diketone 119. Substitution at the C-4 or C-5 position of 6-membered ring 1,3-carbonyls (120) resulted in relatively small effects, but N-alkyl analogues such as 121 proved remarkably reactive with a 100-fold increase in reaction rate relative to dimedone. Other heteroatom substitutions such as 122 and 123 also resulted in increased rates in comparison to dimedone. Replacement of one of the carbonyls with a sulfonamide moiety (124 and 125) gave dramatic rate enhancements up to 2000-fold greater than dimedone. The increased nucleophilicity of the anion derived from deprotonation of 124 and 125 is thought to arise from the half-boat conformation of the heterocyclic ring that decreases resonance stabilisation, making the anion more reactive.158 | ||
| Fig. 13 (a) A dipeptide-based sulfenic acid model. (b) Various 1,3-diketones and related derivatives that react with sulfenic acid/sulfenamide probe 117 (kobs shown for pseudo first-order conditions with >10-fold excess of nucleophile). (c) Linear carbon-centred nucleophiles that can react with sulfenic acid/sulfenamide probe 117 (kobs shown for pseudo first-order conditions with >10-fold excess of nucleophile). (d) Selection of probes with the highest reaction rates with cysteine sulfenic acids (second order rate constants shown for the reaction with 117). These five probes were used to identify cysteine sulfenic acids in RKO colon adenocarcinoma cells. Only nine of the 761 proteins identified in the screen were detected by all five nucleophilic probes. | ||
A range of sulfenic acid probes were also designed based on linear C-nucleophiles (Fig. 13c).159 Rather than the 1,3-dicarbonyl groups, these probes contain the electron withdrawing sulfone and nitrile or nitro groups that render the α-carbon acidic and nucleophilic (126–128). Analogues of these probes were also prepared with the reactive α-carbon, and an alkyne handle for detection (129–130). It was also found that under reducing conditions (addition of DTT, GSH or TCEP), the thioether adduct of some linear C-nucleophiles such as 130 were unstable. Other comparative studies have also identified next generation probes which aim to improve the slow reaction kinetics of current probes.160
Extending on these discoveries, Carroll et al. developed additional C-nucleophiles containing an alkyne reporter group (131–134, Fig. 13d) and compared them with 84.161 Interestingly, when all five probes were used to map the sulfenic acid sites in RKO cells, only nine of the 761 proteins detected were labelled by all five probes. This result suggests that each sulfenic acid has a unique reactivity and it also highlights how different probes can display complementary reactivity towards protein sulfenic acids. On one hand, this means that it might actually be challenging to generate a general probe for cysteine sulfenic acids. On the other hand, this discovery opens the opportunity for selective manipulation of proteins containing sulfenic acids—a useful capability in chemical biology and perhaps medicinal chemistry. Future research will clarify the parameters that match a particular probe with its protein target.
Very recently, a report on the reactivity and selectivity of dimedone (68) was published by Ursini et al. in which the authors provide evidence that dimedone can react directly with certain sulfenamides.162 For example, dimedone was shown to cleave the S–N bond of the sulfenamide 2-methyl-4-isothiazolin-3-one and provide the dimedone adduct in which a C–S bond is formed. This result means that dimedone derivatives might, in some cases, react with sulfenamides of the type found in 117. The product of the reaction of dimedone with the sulfenamide or the sulfenic acid is the same, which makes it challenging to distinguish whether the probe reacted with the sulfenamide or sulfenic acid. In cases where the site of oxidation is the primary interest, this distinction might not matter as the sulfenamide likely is derived reversibly from the sulfenic acid. However when this distinction is important, the validation by mechanistically distinct probes (e.g. cycloaddition of the putative sulfenic acid with a strained alkyne or alkene vs. no reaction with the sulfenamide) may prove useful.
Chemical methods for detecting cysteine sulfenic acids have made remarkable strides over the last decade. In particular, cell-compatible probes have revealed the importance of this oxidative modification in cellular signalling and redox regulation. With suggestions that cysteine sulfenic acids may play as critical a role as phosphorylation, there are clearly exciting opportunities ahead in mapping and understanding its precise role in living organisms.
4.1 Sulfinic and sulfonic acids (RSO2H and RSO3H)
Cysteine sulfinic (4) and sulfonic (5) acids are often considered overoxidation products of cysteine. The formation of these oxidation states has been regarded as an artefact of protein handling, but it has since been revealed that these higher oxidation states of cysteine form in vivo and may even be involved in redox-regulation.163 Sulfenic acids are oxidised to sulfinic or sulfonic acids through reaction with H2O2 and other ROS.102 Compared to the initial oxidation of the cysteine thiolate by H2O2, further oxidation of the sulfenic acid by H2O2 typically occurs more slowly. For example, human serum albumin reacts with H2O2 with a rate constant of 2.7 ± 0.7 M−1 s−1, while the sulfenic acid reacts with a rate constant of 0.4 ± 0.2 M−1 s−1.164 Typically, sulfinic acids will have a pKa of about 2 so they are fully deprotonated at physiological pH and persist as the sulfinate anion—a soft nucleophile.7 Sulfonic acids are the most highly oxidised form of cysteine. The pKa is usually <2 so cysteine sulfonic acids exist as the negatively charged sulfonate anions.Historically, it was not clear if these higher oxidation states of cysteine were anything more than the terminal products of oxidation (a consequence of oxidative stress, for instance). For a more intricate role in signalling, enzymatic reduction of cysteine sulfinic and sulfonic acids back to lower oxidation states would likely be necessary. Indeed, with discovery of a sulfinic acid reductase (sulfiredoxin), the regulatory role of this protein modification in biology is now the subject of great interest.165,166 Such discoveries have motivated further investigation into the function of cysteine sulfinates, such as their roles in the regulation of peroxiredoxin activity167 and metal binding.168 The association of this oxidation with certain pathologies is also of interest, as cancer tissues generally have higher levels of cysteine sulfinic acid compared to normal tissue.113,169,170
Notable examples of cysteine sulfinic acid function include its formation in peroxiredoxin enzymes where it plays a reversible, deactivating role.171 Other proteins such as matrilysin are regulated by formation of a cysteine sulfinic acid that disrupts Zn2+ coordination.172 Likewise, nitrile hydratases are known that contain a sulfenic and sulfinic acid in the active site for metal ion coordination of Co(III) or Fe(III).173 Another example is DJ-1174 which is a protein associated with Parkinson's disease that protects neurons from oxidative damage.7 A study by Wilson and co-workers provided evidence that the formation of a sulfinic acid in DJ-1 was responsible for its protective function.174 Interestingly, the conversion of the key cysteine to its sulfinic acid is not merely the product of ROS scavenging, but also a potential mechanism for localising DJ-1 in mitochondria. Cysteine oxidation is also important in the copper-zinc superoxide dismutase (SOD1), which catalyses the conversion of superoxide anion (O2−) into O2 and H2O2, protecting cells against oxidative stress. Studies by Fujiwara showed that the Cys-111 of SOD1 can readily be oxidised to the sulfonic acid even by air oxidation and it has been speculated that this hyperoxidative damage may be linked to diseases like familial amyotrophic lateral sclerosis.175
All of these examples highlight the potential roles of the historically overlooked cysteine sulfinic and sulfonic acids as redox-regulators that are critical for cellular health. Further investigation into these roles is needed to better understand the exact role of these oxidative modifications.
4.2 Chemical methods for detecting RSO2H and RSO3H
To date, there are only scattered reports of the selective detection of cysteine sulfinic and sulfonic acids in cells. Higher oxidation states of sulfur exhibit decreased reactivity in comparison to lower oxidation states, so designing chemical probes to react specifically with this modification is difficult. Antibodies do exist that are selective in their binding to both cysteine sulfinic and sulfonic acids, but their effectiveness and suitability for global profiling are still limited.176 Enrichment protocols have been reported for sulfopeptides. For example, Wu et al. explored the possibility of trapping peptidic sulfonic acids using polyarginine (PA) covalently conjugated to a solid support such as nanodiamonds (135, Fig. 14a)—a strategy inspired by related work in phosphopetide analysis.177 Strong interactions occur between the guanidinium groups of PA and the negatively charged cysteine sulfonate, enabling enrichment of sulfopeptides for MS analysis. Studies by Cordwell and co-workers explored the use of other enrichment strategies by the electrostatic repulsion of sulfinic and sulfonic acid containing peptides first from cationic resins, followed by further selection using hydrophilic interaction liquid chromatography. This method was used to study rat myocardial peptides with irreversible sulfinylation and sulfonylation.178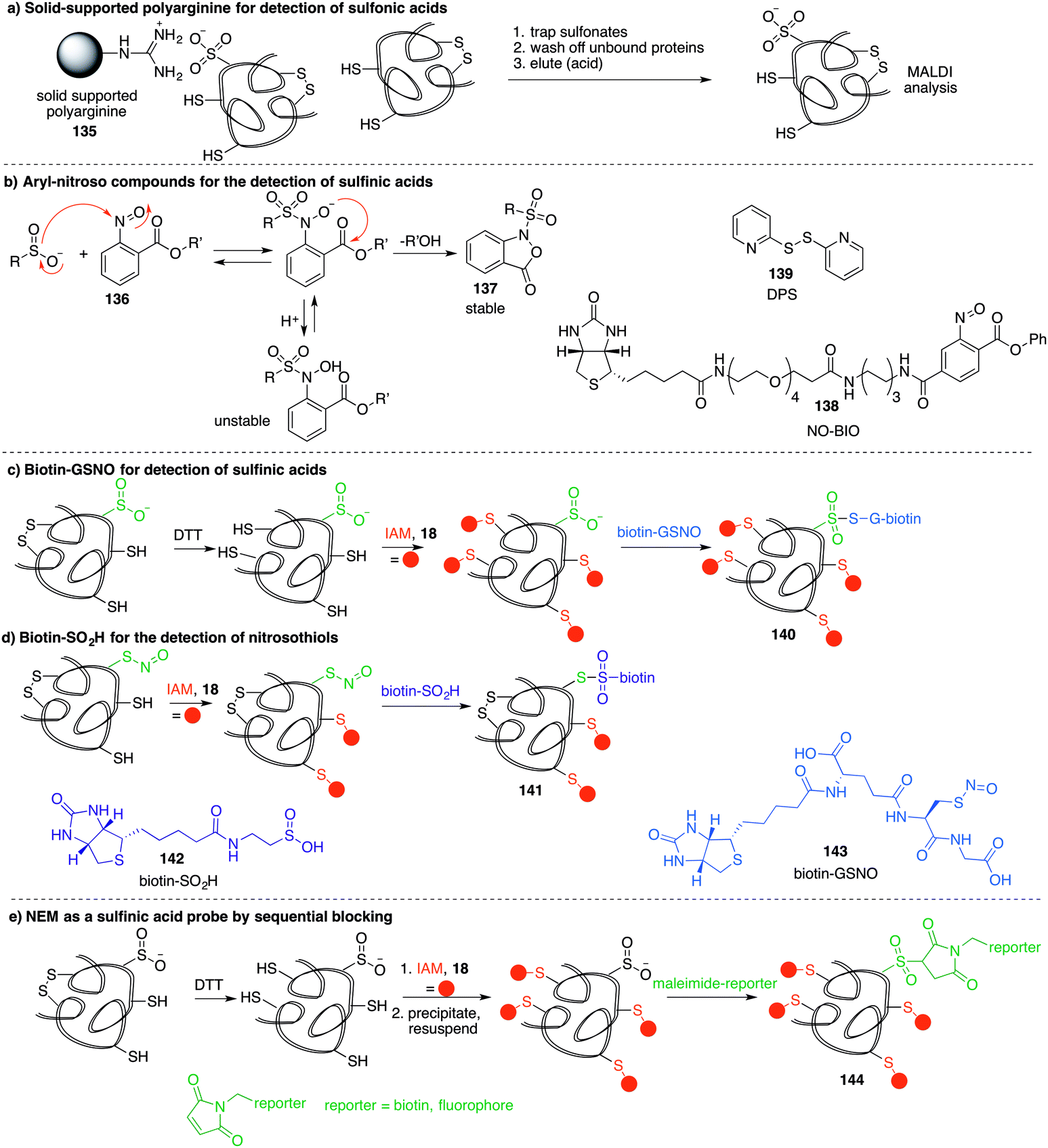 | ||
| Fig. 14 (a) Proteins and peptides containing cysteine sulfonic acids can be enriched by their affinity for solid-supported polyarginine (135). (b) Aryl nitroso compounds bearing an ester in the ortho position can react with and trap cysteine sulfinates. (c) Biotin-GSNO (143) can be used to detect protein sulfinic acids by forming the stable thiosulfonate 140. Samples are first reduced with DTT to prevent disulfide exchange with residual GSNO, followed by blocking free thiols with IAM (18). (d) Using the reverse reaction of (c), biotin-SO2H (142) can be used to detect protein-SNO by first blocking free thiols with IAM (18), and treating with 142 to give the thiosulfonate. (e) NEM (25) can be used as a sulfinic acid probe by sequential reaction, first with DTT to reduce disulfide bonds, followed by IAM (18) to block free thiols. NEM containing a reporter group can then react with sulfinic acids for detection. | ||
To detect sulfinic acids directly, Carroll et al. focused on aryl nitroso compounds such as 136 as potential sulfinic acid traps (Fig. 14b).179 The product of the reaction between the aryl nitroso compounds and sulfinic acids (the N-sulfonyl hydroxylamine) is prone to hydrolysis, but the addition of an ester group in the ortho position favours intramolecular rearrangement to N-sulfonylbenzisoxazolone 137 (Fig. 14b). Translating this reaction to protein analysis, Carroll et al. prepared biotin-labelled 138 (NO-Bio).170 Because the aryl nitroso compounds react with thiols as well as sulfinic acids, free thiols must first be blocked (with 139, for instance) before adding the NO-Bio probe for cysteine sulfinic acid detection. Using this strategy, the NO-Bio assay was validated on both purified proteins and live cells. In the case of lung tumour tissues, higher levels of sulfinylation were detected in comparison to healthy tissues. This study provided, for the first time, a chemical tool for the detection of protein sulfinylation. And while there have been few studies on the more general roles that cysteine sulfinic and sulfonic acids might play, the analytical technologies in Fig. 14 are important enabling strategies for future studies. More recently, the Martin group explored the cross reactivity of S-nitrosothiols and S-sulfinic acids, forming stable thiosulfonates (140–141) (Fig. 14c and d).180 For example, phenylsulfinic acid reacts rapidly with N-acetyl-S-nitroso-cysteine methyl ester to give the thiosulfonate product. They noted the selective reaction of sulfinic acids and S-nitrosothiols, and the former's in lack of reactivity with free thiols, biological disulfides, aldehydes or sulfenamides. However, since the thiosulfonate products are readily exchangeable with thiols, free thiols must first be alkylated with iodoacetamide (18). A biotin or fluorescein-labelled N-hydroxysuccinimide ester was directly coupled to the sulfinic acid metabolite hypotaurine (biotin-SO2H, 142). However, while 142 is stable during the 1 hr labelling process, it is fully oxidised after approximately 5 h in atmospheric oxygen so probe stability is a concern. It was also noted that if the alkylating reagent MMTS (11) is used instead of 18, complete loss of 142 labelling is observed. It was proposed that upon thiol alkylation, 11 releases methyl sulfinic acid which reacts with S-nitrosothiols, highlighting another issue when using 11 as a blocking reagent in various biotin switch assays where fewer S-nitrosothiols will be detected. It was also noted that the product of the reaction with 142 was sensitive to reduction by TCEP, requiring careful consideration during sample preparation and analysis. The reverse process was also explored using biotin-GSNO (143) to detect sulfinic acids (Fig. 14c).170,179 In this assay, the reduced lysates were blocked with 18 and then treated with 143. Using this probe, it was found that DJ-1 reacts with N-acetyl-S-nitroso-cysteine methyl ester or GSNO to form the thiosulfonate at Cys-106. Both methods, while shown to be selective, have unstable probes. To address this issue, the Martin group have recently found that N-ethylmaleimide (NEM, 25) reacts with both aryl and alkyl sulfinic acids in aqueous buffer, forming the sulfonyl-succinimide 144 (Fig. 14e).181 Since 25 is also a thiol-reactive reagent, selective blocking of thiols with 18 at neutral pH is followed by reaction of sulfinic acids with 25 at lowered pH. The sulfonyl-succinimide conjugate (144) is stable for analysis below pH 6, but decomposes at higher pH. This method was applied to sulfinic acid analysis of purified DJ-1 protein and mammalian cell lysates HEK-293T.
5.1 Persulfides (RSSH)
Oxidation of cysteine to the persulfide oxidation state (7) is thought to occur through various reactions with hydrogen sulfide (H2S). Cavallini and co-workers demonstrated early evidence for persulfide formation on proteins through the addition of sodium sulfide to disulfide containing proteins such as insulin and RNase.182 The reaction was monitored using spectrophotometry with characteristic absorption of the persulfide appearing at 335 nm. Treating this product with cyanide resulted in the formation of the reduced cysteine thiol and thiocyanate.H2S does not react with thiols to form persulfides in the reducing cellular environment without an appropriate redox couple. H2S exists predominantly as the hydrosulfide anion (HS−) in cells—an important signalling molecule in many physiological processes.183–186 In mammals, endogenous H2S is produced from cysteine and homocysteine by enzymes such as cystathionine β-synthase (CBS),187 cystathionine γ-lyase (CSE),188 and mercaptopyruvate sulfurtransferase (MST).189 Formation of persulfides is thought to occur through several pathways. Disulfides can react with the hydrosulfide anion to form both a persulfide and thiol.190,191 Other cysteine derivatives such as sulfenic acids (3) and nitrosothiols (2) may also react with hydrosulfide to form persulfides. The cysteine thiol may also react with sulfane sulfurs or other polysulfides to form cysteine persulfide (Fig. 15).192,193
Hydrogen disulfides are unstable in aqueous buffers and can decompose to form thiols, polysulfides and elemental sulfur. The pKa of persulfides is approximately 1–2 pKa units lower than their parent thiols, making them stronger acids, and likely deprotonated at physiological pH. Persulfides can act as both nucleophiles and electrophiles but compared to their parent thiols, persulfides are more nucleophilic due to the alpha effect.192 When considering the electrophilic nature of cysteine persulfides, nucleophiles can attack at either the inner (γ) or outer (δ) sulfur atom. Thiols such as glutathione, for example, can react with cysteine persulfide. If the internal sulfur is attacked, this leads to disulfide formation and generation of H2S. If the outer sulfur atom is attacked, this leads to regeneration of the parent thiol and generation of a new persulfide.191 Persulfides can also undergo one-electron oxidations to give the perthiyl radical (RSS˙), which can add to di- or polysulfides or react with thiols by hydrogen atom abstraction.194
The reversible sulfhydration of cysteine (forming cysteine persulfide) can alter the activity of several proteins: protein tyrosine phosphatases 1B is inhibited,195 ATP-sensitive potassium channels become hyperpolarised,196 actin polymerisation is enhanced,190 and nuclear factor κB (NF-κB) is activated for anti-apoptotic function.197 These functional responses are part of the reason for growing interest in H2S as a signalling molecule, which acts in many cases by converting cysteine to its corresponding persulfide. In the examples of sulfhydration mentioned above, the formation of H2S by cystathionine γ-lyase is critical. For example, Snyder and co-workers have shown that H2S generated enzymatically by cystathionine γ-lyase (CSE) sulfhydrates glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-tubulin, and actin. The sulfhydration enhanced actin polymerisation and augmented GAPDH activity.190 Other studies have also shown the decrease in enzymatic activity of GAPDH,198 which contrasts reports of an increase in GAPDH catalytic activity upon sulfhydration.190 The free Cys-152 is critical for activity, and sulfhydration of Cys-156 and Cys-152 decreased enzymatic activity.198 Other studies involving CSE production of H2S have shown sulfhydration of the peroxisome proliferator activated receptor γ (PPARγ) at Cys-139.199 This was found to increase activity and promote glucose uptake in lipid storage, where mutating the PPARγ at Cys-139 removed sulfhydration, and lead to reduced glucose uptake. Using human hepato-cellular liver carcinoma cell lines (HepG2) and mouse primary hepatocytes from both wild-type and CSE knockout mice, it was found that pyruvate carboxylase (PC) activity could be increased by H2S sulfhydrating Cys-265, where increased activity contributed to gluconeogenesis.200
Similarly, the antiapoptotic activity of nuclear factor κB (NF-κB) is mediated through H2S synthesised by CSE.197 H2S sulfhydrates the p65 subunit of NF-κB at Cys-38, promoting binding of the co-activator ribosomal protein S3 and, in turn, binding to the promoters of antiapoptotic genes. CSE-deficient specimens could not sulfhydrate p65 and showed decreased NF-κB activity. H2S was also found to protect against cellular senescence. In a study by Yang et al. it was shown that mouse embryonic fibroblasts isolated from CSE knockout mice, had increased oxidative stress and cellular aging compared to wild-type mice.201 Incubation of CSE deficient specimens with NaHS (a H2S donor) increased GSH levels and decreased cellular senescence. It was also found that S-sulfhydration of Keap1 (Kelch-like ECH-associated protein 1) at Cys-151 caused dissociation of Nrf2 (nuclear (erythroid-derived 2)-like 2) from Keap1. After dissociation, Nrf2 translocates to the nucleus and activates transcription of multiple antioxidant genes encoding for proteins that protect cells against oxidative stress.202
The Bian group demonstrated that H2S can sulfhydrate p66Shc at Cys-59.203 The effect of H2S on mitochondrial ROS generation was investigated and found that sulfhydration of Cys-59 of p66Shc decreases phosphorylation due to its close proximity to the phosphorylation site serine-36. This in turn results in reduction of p66Shc-mediated mitochondrial ROS production. Like the other cysteine modifications discussed thus far, clearly these diverse biological functions motivate methods to detect and analyse cysteine persulfides and their link to the subtle roles of H2S signalling.
5.2 Chemical methods for detecting RSSH
Because the persulfide group is a strong nucleophile like the native cysteine thiol, developing selective methods for its detection is a challenge. One of the first detection methods used for persulfides was cyanolysis.204 The cyanide anion nucleophilically attacks the terminal sulfur forming thiocyanate, which can then be analysed spectroscopically after a second reaction with ferric nitrate (Fig. 16a). However, many drawbacks exist with this method. Competing attack of the inner sulfur atom releases the sulfide anion and complicates analysis, and the method cannot differentiate between persulfides and polysulfides, making it unsuitable for complex mixtures. | ||
| Fig. 15 The formation of cysteine persulfides in proteins. | ||
Neal and Sawahata used 1-fluoro-2,4-dinitrobenzene (145) as a probe for persulfides.205 The persulfide reacts in a nucleophilic aromatic substitution with 145. The dinitrobenzene disulfide product (146) can then be reduced with DTT to release 2,4-dinitrothiophenol (147), which can be analysed spectroscopically (Fig. 16b). This method can differentiate between persulfides and thiols because the latter reacts with 145 to form a stable thioether that is not cleaved when DTT is added. This assay has largely been applied to purified proteins at present, rather than lysates or live cells.
 | ||
| Fig. 16 (a) Cyanide reacts with persulfides to generate thiocyanate, which can be monitored spectroscopically after reacting further with Fe3+. (b) Persulfides react with 145 in a nucleophilic aromatic substitution. Reduction of the product disulfide 146 generates 147, which can be conveniently monitored by spectrophotometry. | ||
Switch techniques to detect cysteine persulfides
The biotin switch technique can also be adapted to detect the cysteine persulfide oxidation state. Snyder et al. first blocked thiols through reaction with MMTS (11), converting free cysteines to the corresponding methyl disulfides. The persulfides were then labelled with biotin tag 12 for detection (Fig. 17a).190 It is unclear whether 11 will block persulfides as well, so this side reaction should be considered when using this strategy. With this biotin switch technique, it was found that H2S generated from cystathionine β-lyase acts as a physiologic vasorelaxant and sulfhydration was shown to augment GAPDH activity and enhance actin polymerisation. Tonks et al. presented a second variation which employed iodoacetic acid (148) to first label both the persulfide and thiol groups, addressing the selectivity concerns of 11.195 Treatment with DTT was then used to reduce the persulfide adduct to the free thiol which could then be labelled with iodoacetamide-linked biotin (IAP, 149) and detected (Fig. 17b). However, this method relies on selective reduction of the persulfide and other groups that react with DTT (disulfides, sulfenic acids, or nitrosylated cysteine for example) may lead to false positives. Nevertheless, this modified method was used to study the sulfhydration of PTP1B, where conversion to the persulfide at Cys-215 inhibited PTP1B activity, and promoted the activity of protein kinase-like endoplasmic reticulum kinase.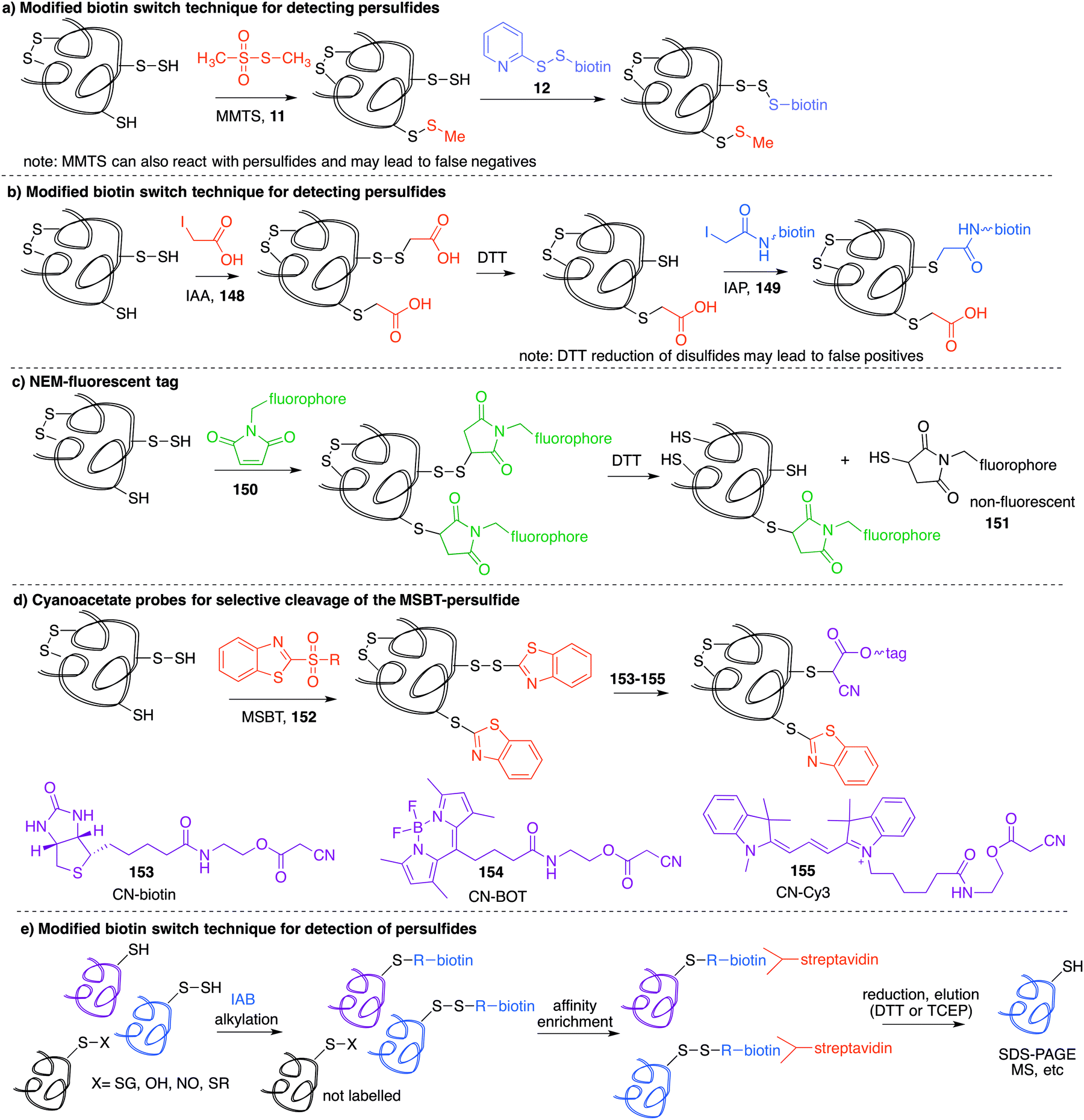 | ||
| Fig. 17 (a and b) A modified biotin switch technique for detecting cysteine persulfides. Note that in the initial blocking step, MMTS (11) may also react with the cysteine persulfide. Iodoacetic acid (IAA, 148) is a more suitable blocking reagent because both thiols and persulfides are alkylated and the latter can be reduced and selectively labelled with biotin. (c) A fluorescent maleimide (150) can react with cysteine thiols and persulfides. Cleaving the disulfide product derived from the persulfide results in a decrease in fluorescence that can be monitored. (d) Reagent 152 reacts with cysteine thiols and persulfides. The disulfide formed from the reaction of cysteine persulfide and 152 can subsequently be tagged by reaction to a nucleophilic cyanoacetate group conjugated to biotin or a fluorescent tag. (e) A variation of the biotin switch technique illustrating how cysteine persulfides can be enriched by binding to immobilised streptavidin. The proteins bound through the persulfide (but not through the non-reducible thioether) can then be cleaved with DTT or TCEP and isolated for analysis. | ||
Investigating the cross reactivity of 11, Carroll examined the reactivity of protein persulfides towards different electrophiles and nucleophiles.206 Using an analogue of 11, S-4-bromobenzyl methanethiosulfonate (BBMTS), it was confirmed that the nucleophilic protein persulfides do indeed react with electrophilic compounds such as MMTS and BBMTS. Snyder et al. also modified their previous method by implementing an Alexa Fluor 680-conjugated maleimide 150 to monitor levels of persulfides.197 No blocking step is required: 150 reacts with both persulfides and thiols but not with other oxidation states such as nitrosothiols. The fluorescence is monitored, and upon reduction with DTT, the disulfide linkage of the original persulfides is cleaved, but the thioether linkage of the original thiols remains intact. Cleavage of the disulfide maleimide tag provides a non-fluorescent product 151, so persulfides can be quantified by measuring the decrease in fluorescence (Fig. 17c).
Xian and Filipovic et al. proposed yet another tag-switch technique to detect persulfides.207 Here, methylsulfonyl benzothiazole (MSBT, 152) reacts with both thiols and persulfides. The disulfide product that forms when persulfides react with 152 can be selectively cleaved with enolate nucleophiles such as dimedone, malonitrile and methyl cyanoacetate. These nucleophiles can be functionalised with reporter groups such as 153–155 (Fig. 17d).208 Because 152 is not cell membrane permeable, this strategy has been applied to cell lysates rather than live cells, presenting a notable limitation.
Nagy et al. reported a protein persulfide detection protocol with high specificity.209 First, protein cysteine thiols and persulfides are blocked with the biotin-labelled alkylating reagent iodoacetyl-PEG2-biotin (IAB). The labelled proteins can then be enriched through binding to streptavidin-coated magnetic beads. The derivatised protein persulfides are selectively cleaved from the beads through reduction (TCEP or DTT) for analysis by SDS-PAGE (Fig. 17e). This method was used to establish that thioredoxin (Trx) and glutathione (GSH) systems catalyse reduction of inorganic polysulfides and protein persulfides. In intact HEK293 cells lacking TrxR1 and TRP14, increased protein persulfide levels were observed demonstrating the role of both Trx and GSH systems in redox homeostasis. A few limitations of this technique are worth noting. For instance, if a protein contains a persulfide as well as a free thiol on the surface, this thiol may become alkylated and prevent release from the beads during the reduction step giving a false negative. If a protein contains intermolecular disulfide bonds as well as a free thiol (but no persulfides), it may be captured during the affinity pull down and the disulfide bond cleaved upon reduction with DTT, resulting in a false positive. As such, careful validation of hits is essential.
Due to the similar reactivities of persulfides and thiols, designing chemical tools that specifically react with persulfides is difficult. The cyclooctyne reaction with persulfides is a notable exception (Fig. 12f). Exploring these new modes of reactivity may lead to more general methods for the detection and analysis of protein persulfides. Such studies are further motivated by the growing evidence that at least some aspects of H2S are directly linked to the formation of cysteine persulfides on proteins.
6.1 Disulfides (RSSR)
Disulfides (8) are important for maintaining the structure of proteins as well as participating in various catalytic and signalling processes. Within a cellular environment, both enzymes and small-molecules are capable of reducing disulfide bonds. The reversible and ubiquitous nature of cysteine disulfides is one reason for their key roles in redox-regulation and signalling. Protein disulfides can form inter- or intramolecularly depending on the accessibility and proximity to other cysteine groups. Formation of disulfides can also facilitate protein folding.210 Disulfides can be formed through several pathways in cells (Fig. 18). These include disulfide exchange reactions with thiols,211 thiyl radical coupling with cysteine, or the reaction of thiols with nitrosylated cysteine212 or sulfenic acids210 (Fig. 18).213 The disulfide exchange reaction in particular plays a key role in cellular function. In this process the thiolate acts as a nucleophile and attacks one of the sulfur atoms of the disulfide bond, forming a disulfide and releasing the other sulfur group as a thiol.211,214 Highly nucleophilic cysteines that are deprotonated are often involved in these disulfide exchange reactions.215 Non-catalysed disulfide exchanges do occur but several enzymes such as thioredoxin (Trx), glutaredoxin (Grx), and protein disulfide isomerase (PDI) can accelerate the process. These enzymes are important in mediating protein functions that are controlled by disulfide formation.216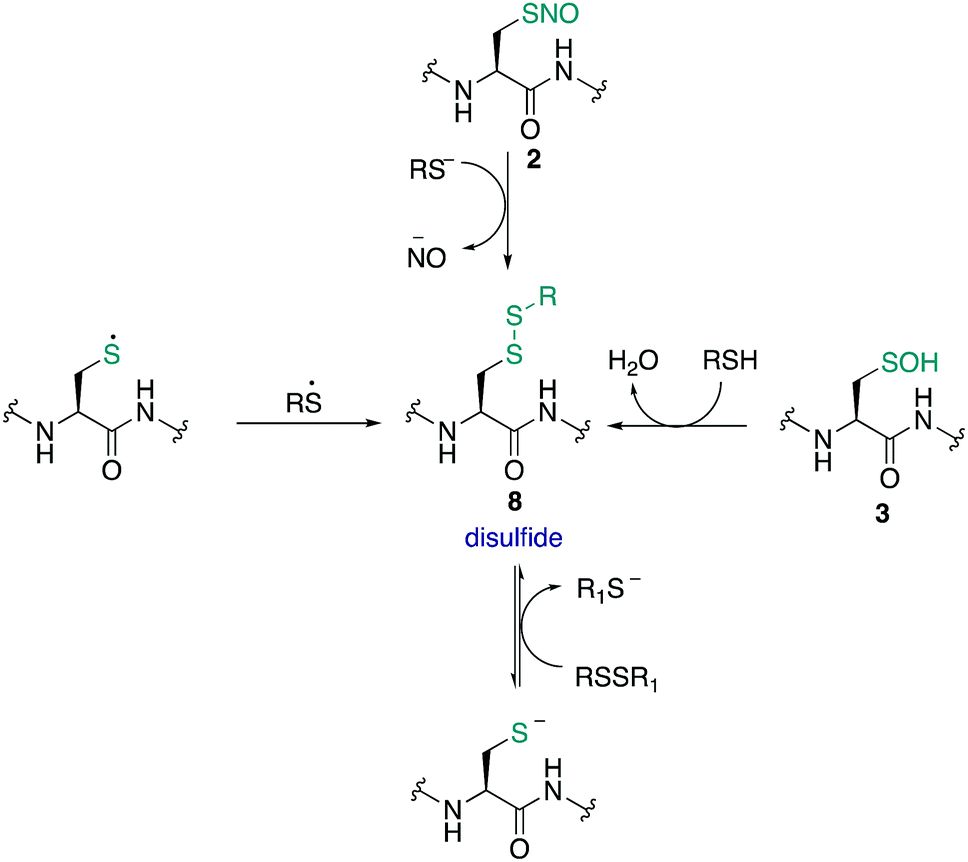 | ||
| Fig. 18 The formation of disulfides in proteins. | ||
The regulation of protein function by disulfide formation is important in a number of diverse processes. For example, the OxyR transcription factor is activated through formation of a disulfide bond in response to elevated levels of H2O2.27,217 OxyR is deactivated through disulfide reduction by Grx1, providing a means of regulation. Cell structure is also dependent on cysteine disulfides, as microtubule assembly is mediated in part by disulfide bonds.218 Protein multimers are also often linked through cysteine disulfides. For instance the ataxia-telangiectasia mutated (ATM) protein kinase forms a dimer linked through a cysteine disulfide,219 and type I protein kinase A forms an interprotein disulfide bond between two regulatory RI subunits.220 While there are many other examples of disulfide formation on proteins, these examples highlight the diverse functions that are associated with this cysteine modification. Dick and colleagues explored the thiol peroxidase peroxiredoxin-2 (Prx2), and its interactions with transcription factor STAT3 to form a redox relay. Prx2 acts as a primary receptor for H2O2 and specifically transmits an oxidative equivalent to STAT3 by performing disulfide exchange intermediates. This generates disulfide linked STAT3 oligomers, decreasing transcriptional activity.221
6.2 Chemical methods for detecting RSSR
The presence of intermolecular disulfide bonds can often be inferred through SDS-PAGE analysis. For example, Sommer and Traut devised a diagonal electrophoresis 2D-gel method using the reagent methyl-4-mercaptobutyrimidate as a protein–protein cross-linking reagent which can subsequently be reduced, revealing proteins which were cross-linked due to different migration in the second dimension of electrophoresis. Any protein that contains an intermolecular disulfide bond will appear off the diagonal in the second dimension. This diagonal electrophoresis has been used to study formation of disulfides in both the cytoplasm and myocardial cells.222–224Bardwell et al. developed a method when studying the heat shock protein Hsp33 which utilised a IAM/AMS (4-acetamido-4′-maleimidylstilbene-2,2′-disulfonate, (156)) trapping method. First free thiols were blocked with IAM (18) and then disulfides reduced with DTT. The newly exposed thiols could then be labelled with AMS 156. AMS adds a molecular mass of ≈500 Da which causes a detectable mobility shift during SDS-PAGE (Fig. 19a and b).225 Similarly, Momand et al. proposed reducing the disulfide bonds then labelling the thiols with methoxypolyethylene glycol-maleimide (MAL-PEG, 157).226 The MAL-PEG-protein conjugate can then be detected by band shifts in gel and western blot analysis.
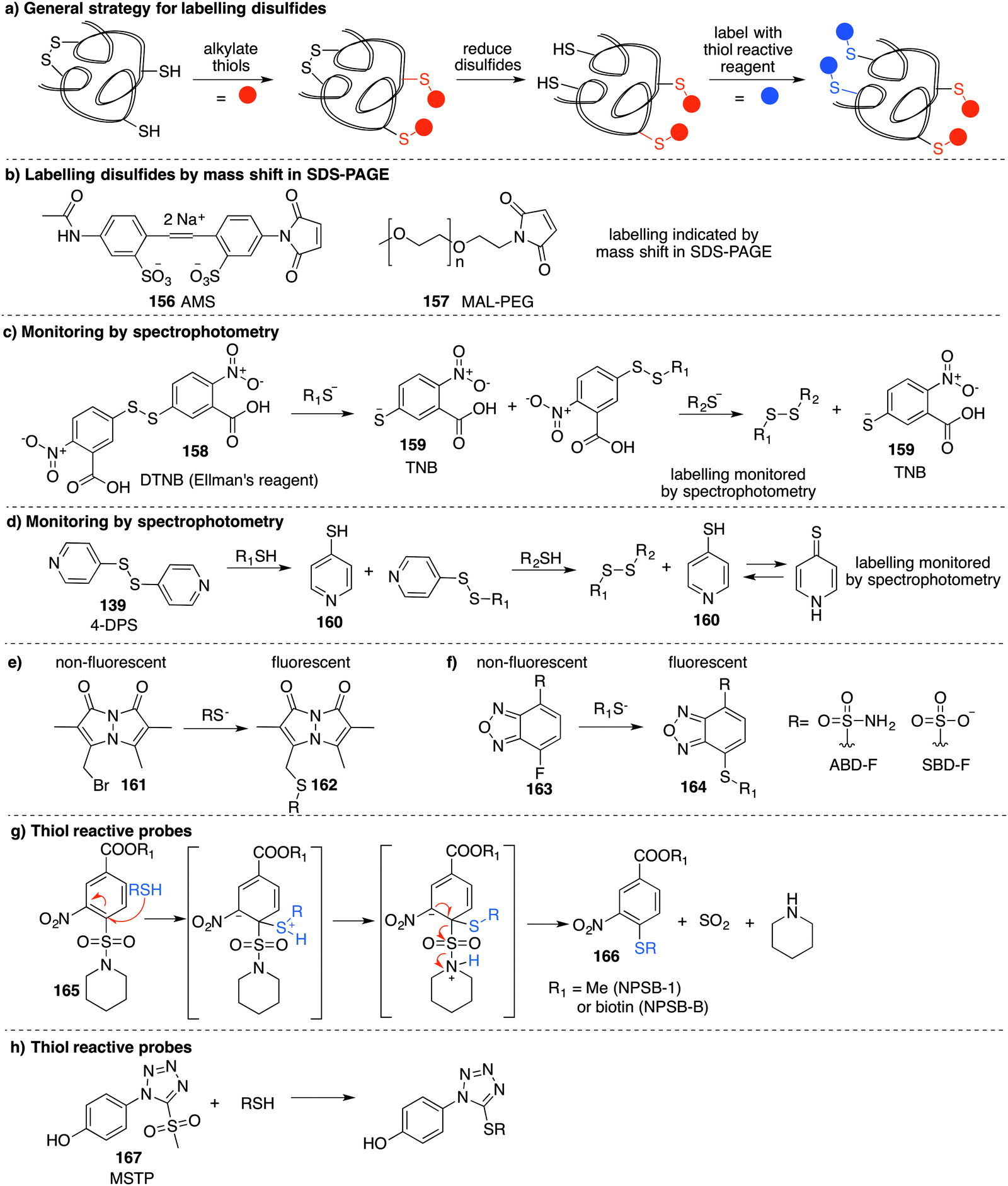 | ||
| Fig. 19 (a) A general strategy for labelling the cysteine residues of disulfides with small molecule probes. Examples of the thiol reactive reagents are shown in (b–f). (b) Labelling thiols with 156 or 157 results in a change in the mobility of the protein during SDS-PAGE. (c and d) Ellman's reagent (158) reacts with thiols and generates a product 159 that is easily monitored by spectrophotometry. Reagent 139 is less sensitive to pH than Ellman's reagent, but is mechanistically similar. (e and f) Fluorogenic probes for detecting free thiols. (g) Methyl-3-nitro-4-(piperidin-1-ylsulfonyl)benzoate (NPSB-1) thiol-selective probe. (h) 4-(5-Methanesulfonyl-[1,2,3,4]tetrazol-1-yl)-phenol (MSTP) thiol-selective probe with enhanced reaction rate compared to 18. | ||
The same strategy can also be used to attach smaller chemical labels or reporter groups at cysteine disulfides. Typically, free thiols are first blocked by alkylation, and then disulfides are reduced to thiols which can then react with the reporter group (Fig. 19a). Common thiol blocking agents include iodoacetamide (18), maleimide, N-ethyl maleimide (25), and iodoacetic acid (148). Common reductants include TCEP, DTT and sodium borohydride.227,228 For thiol detection, the chromogenic 5,5′-dithiobis-(2-nitrobenzoic)acid (DTNB or Ellman's reagent, 158) reacts to produce 5-thio-2-nitrobenzoic acid (TNB, 159) which can be monitored spectroscopically (Fig. 19c).229 However, 158 can undergo hydrolysis at higher pH and the absorption maximum for the product 159 is pH dependent. Similarly, Winther et al. used 4,4′-dithiodipyridine (4-DPS, 139) for quantification of protein thiols and disulfides.228 Upon reaction with thiols, 139 generates a strongly absorbing resonance-stabilised 4-thiopyridone tautomer of 160 which has a pH independent absorption, an advantage over Ellman's assay (Fig. 19d).
Fluorogenic labelling of thiols is also possible.230 Representative reagents include monobromobimane (161)231 and 7-fluoro-2,1,3-benzoxadiazole derivatives such as 163 (Fig. 19e and f).232,233 Initially, 161 and 163 are non-fluorescent but upon reaction with thiol groups, they generate strongly fluorescent products 162 and 164 respectively. It should be noted that while 163 may require elevated temperature and pH for labelling (60 °C, pH 9.5 for 1 h), it has been used to study disulfides in proteins.234 It is also worth noting that 161 is more sensitive than Ellman's reagent and it can pass freely through biological membranes.235,236
Since a key component of many cysteine oxidation detection methods is subsequent reaction or prior blocking of the reduced thiols with detectable reagents, several recent studies have explored the potential cross-reactivity of many thiol-reactive reagents and their use in various strategies. Reactivity of electrophilic thiol-blocking reagents such as IAM (18), NEM (25), and MMTS (11) have been reported with sulfenic and sulfinic acids in addition to thiols, with 11 also reacting with persulfides and nitrosothiols.180,206,237 This presents many concerns for several of the above mentioned strategies for detection of various cysteine oxidation states where cross-reactivity of the initial thiol blocking reagent may be reacting with the oxidation state of interest, preventing detection and an under-representation of that oxidation state. To address these concerns, recent attempts to produce reagents with greater selectivity for thiols have been reported. Using a 2,4-dinitriobenzenesulfonamide(nosyl)group, the thiol selective probe methyl-3-nitro-4-(piperidin-1-ylsulfonyl)benzoate (NPSB-1, 165) was synthesised (Fig. 19g).238 Design of this probe was inspired by deprotection of the nosyl group by thiols, which produces a stable aryl sulfide 166. While 165 was selective for thiols and did not react with sulfenic or sulfinic acids, it had slower reactivity towards thiols than 18, which may hinder its widespread use. More recently, a heteroaromatic alkylsulfone, 4-(5-methanesulfonyl-[1,2,3,4]tetrazol-1-yl)-phenol (MSTP, 167) was investigated by Furdui and co-workers as a selective thiol blocking reagent with comparable blocking ability to that of 25 (Fig. 19h).239 It had a rate enhancement of 160-fold compared to 18, and could provide a more reliable alternative to currently used thiol-blocking reagents which suffer cross-reactivity with several cysteine oxidation states, a key area of concern for many detection strategies.
Other thiol labelling methods can be adapted for disulfide analysis. Reagents such as 3H-IAM,118 5-iodoacetamido-fluorescein,240 isotope coded affinity tag (ICAT) reagents,140,241 and biotin-conjugated iodoacetamide (BIAM)242 are commonly used. Radiolabelling the thiols derived from disulfide reduction allows straightforward identification of these disulfide-containing proteins.243,244 Using isotopically labelled reagents is also useful for quantification by mass spectrometry. For example, the ICAT method can reveal which cysteines react under oxidative stress (Fig. 20a). The ICAT reagents (e.g.168) consist of the cysteine reactive component, an isotopically labelled linker containing light (1H) or heavy (2H) isotopes, and a biotin group which allows affinity purification. Some ICAT variants have cleavable linkers to remove the biotin group before MS analysis. Two separate samples (e.g. cell lysates from a control and lysate from cells subject to oxidative stress) are treated with either the light or heavy ICAT. The two samples are then mixed in equal quantities, digested, and analysed by mass spectrometry to determine which sites are not labelled due to oxidative stress (Fig. 20a). In this experimental design, any reduction in signal relative to the control indicates a cysteine in that peptide fragment was oxidised (for instance to a sulfenic acid). Jakob and co-workers adapted the ICAT method for global analysis of cysteine oxidation to identify, for instance, what cysteines are susceptible to oxidation and form disulfides during oxidative stress (Fig. 20b).245 Such analyses help identify the specific cysteine in proteins that may be involved in redox regulation.
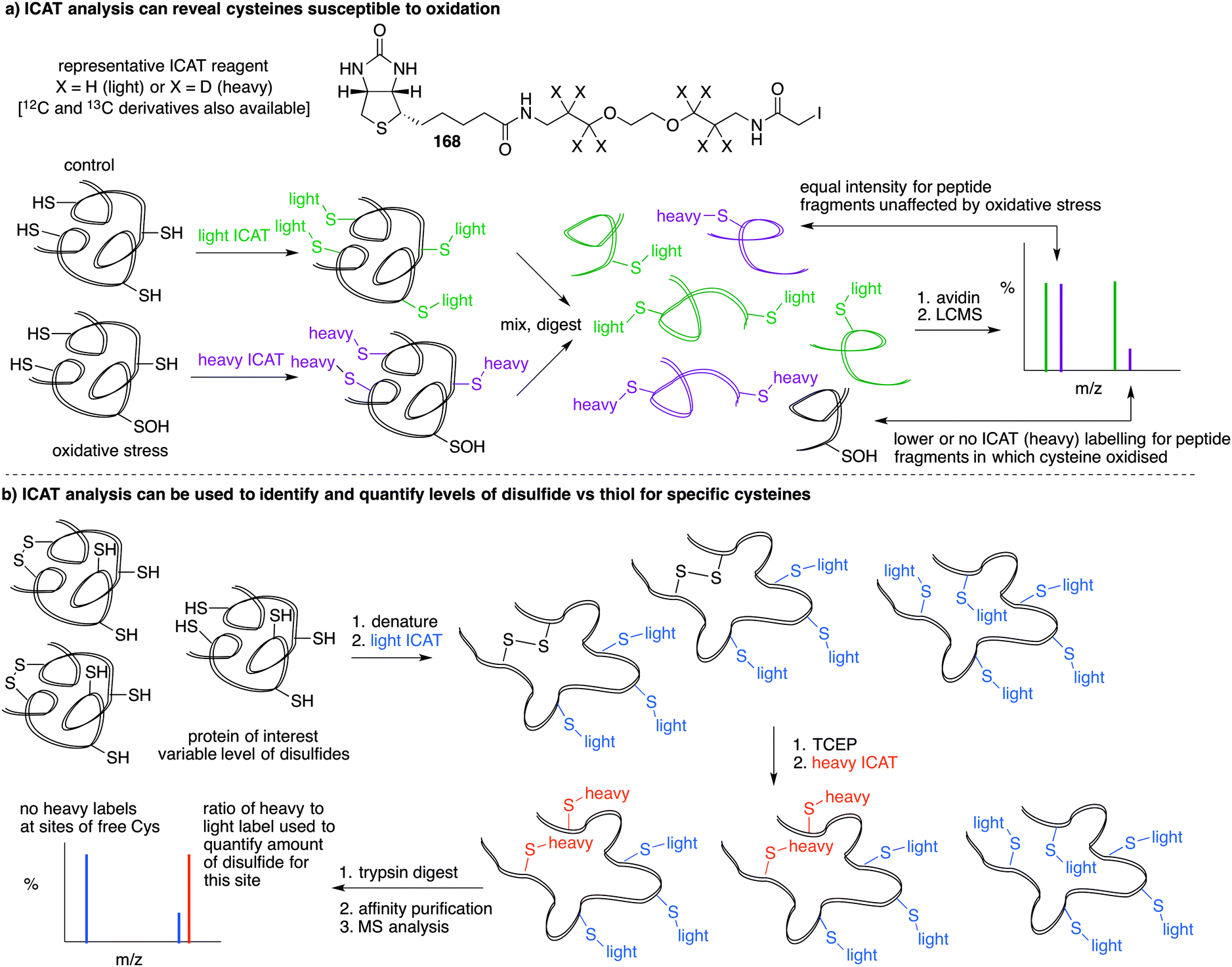 | ||
| Fig. 20 (a) The ICAT method can be used to identify cysteines susceptible to oxidation. (b) A variation of the ICAT method can be used to identify which cysteines exist as disulfides and the relative levels of thiol/disulfide for a particular cysteine residue. | ||
7.1 Protein–glutathiones (RSSG)
An important regulator of cellular redox balance is the tripeptide glutathione (GSH, 169). GSH is synthesised in the cytosol by γ-glutamylcysteine synthetase (GCS) and glutathione synthetase (GS) where GCS activity is monitored by feed-back inhibition of GSH concentrations.246,247 Glutathione is present at millimolar concentrations (2–10 mM) in the intracellular environment and can exist as the thiol (GSH) or its homodimer disulfide (GSSG). GSSG is usually present at much lower concentrations (20–40 μM) due to the activity of glutathione reductase. Therefore, the GSH/GSSG ratio is typically 100–400.248 The mixture of GSH and GSSG acts as a redox buffer where the thiol is a reducing agent and the disulfide an oxidant. Changes in the GSH/GSSG ratio, (for instance due to oxidative stress) can lead to the formation of mixed disulfides with glutathione and proteins.249 Another important role of GSH is its participation in detoxification processes through scavenging of free radicals, reduction of peroxides, or reaction with electrophilic compounds. Many of these processes are mediated by enzymes such as glutathione peroxidases (GPX) and glutathione S-transferases (GST).250 Glutathione reductase is also a critical protein for recycling GSH by the reduction of its disulfide.250Formation of protein–glutathione disulfides can occur through direct reaction of GSH with other cysteine oxidation states such as thiyl radicals (forming intermediate 170), sulfenic acids, and nitrosothiols. Disulfide exchange reactions also lead to protein glutathionylation (Fig. 21).248 Additionally, the cysteine of GSH may be oxidised to various thiol oxidation states (e.g. GSNO, GSOH) and then react with protein thiols. Glutathionylation can either be an intermediate species that ultimately is converted into a protein disulfide bond, or the protein–glutathione conjugate may persist under oxidative conditions. Protein glutathionylation may serve many functions. For instance, it may protect cysteine residues from overoxidation during oxidative stress, and it can alter protein function and intracellular redox signalling. Removal of GSH from proteins can occur through direct disulfide exchange reactions with another molecule of GSH. Additionally, several enzymes are capable of reducing these protein–GSH conjugates including glutaredoxins (GRX), thioredoxins and sulfiredoxins.248,250
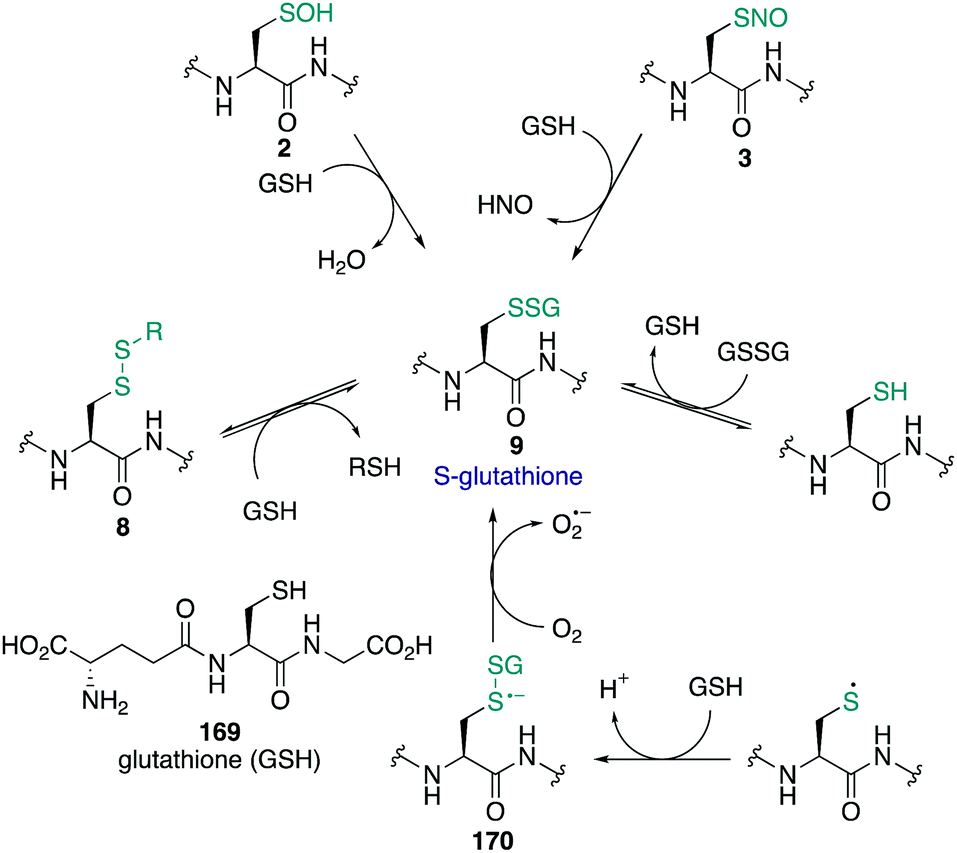 | ||
| Fig. 21 The formation of protein–glutathione conjugates. | ||
Proteins that are known to be modified with glutathione are involved in cell growth, transcription, cell cycle progression, metabolism, and control of redox state in the mitochondria.248,251,252 For example, protein tyrosine phosphatases (PTPs) are inactivated by formation of a mixed disulfide with GSH.253 The mitochondrial carnitine/acylcarnitine carrier (CAC) is also regulated by variations in GSH/GSSG concentrations.251 The oxidised CAC (containing a disulfide) is activated by GSH-dependent reduction. Because the oxidation state of the key CAC cysteine residues depends on the levels of GSH and GSSG, it is thought that the protein may act as a cellular redox sensor. Understanding such processes is important for elucidating signalling pathways and how they respond to oxidative stress. Winterbourn and co-workers explored the oxidation of the active site Cys of Prx2 under oxidative stress. They were able to show that upon the addition of GSH to either the disulfide or oxidised Prx2 (by H2O2), Prx2 formed a stable mixed disulfide with glutathione that could readily be reversed by glutaredoxin 1.254 This could indicate a role of glutathionylation in protecting Prx2 from hyperoxidation during oxidative stress, as it was shown that hyperoxidation was suppressed when GSH was present compared to treatments with H2O2 alone.
The elastic protein titin has also been shown to undergo S-glutathionylation.255 Mechanical unfolding of the titin immunoglobulin (Ig) domains exposes buried cysteines which can be glutathionylated. This causes increased elasticity of titin through mechanical weakening and prevents folding, leading to decrease in the stiffness of cardiomyocytes.
Other studies have looked at the S-glutathionylation of STAT3, a transcription factor that is activated in a variety of cancers that plays a critical role in inhibition of apoptosis and induction of chemoresistance.256 Further work has demonstrated that S-glutathionylation of STAT3 at Cys-238 and Cys-542 can impair phosphorylation, implying redox-regulation of STAT3 in cells.257
Because the GSH/GSSG redox couple is present in high concentrations in cells and responds to changes in the redox balance, developing strategies to better understand this relationship are beneficial.
7.2 Chemical methods for detecting RSSG
Detection of glutathionylation in proteins can be performed using a number of strategies. One common strategy is to detect the release of GSH upon cleavage of the disulfide bond. This first requires separation of proteins, followed by reduction and release of protein-bound GSH for detection and analysis. While these methods allow the quantification of total protein glutathionylation, they do not allow detection of specific proteins that have undergone glutathionylation which is considered more important when trying to determine the role of protein glutathionylation in various biological pathways.Radiochemically, 35S-labelled GSH can either be prepared directly as [35S]GSH258 or biosynthesised from L-[35S]cysteine.259 This 35S labelling method, coupled with proteomic analysis, has been used to identify proteins of T lymphocytes that undergo glutathionylation during oxidative stress.260 Such radiolabelling methods alone, however, do not allow enrichment of modified proteins. To overcome this problem GSH can be labelled with biotin and delivered to cells, with 171 as an example.261,262 This approach (Fig. 22a) was used to discover that protein glutathionylation is important in tumour necrosis factor (TNF-α) signalling.261 A complementary reagent (biotin-GSSG 172) was used to identify proteins that become glutathionylated through disulfide exchange (Fig. 22b).263 Other GSSG reagents include an eosin-labelled glutathione disulfide that was used to study glutaredoxin activity on deglutathionylation.264
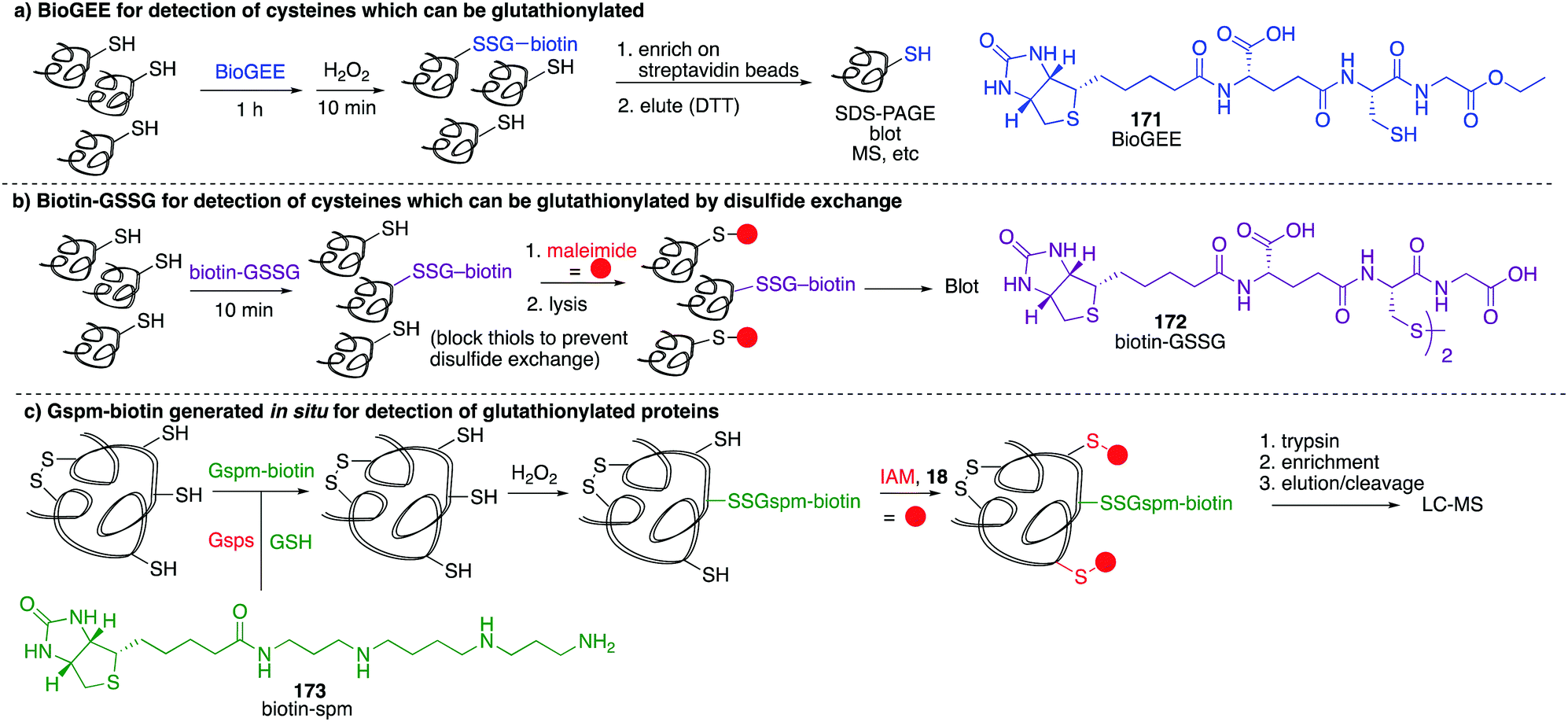 | ||
| Fig. 22 (a and b) Biotin labelled GSH (171) or GSSG (172) can be fed to cells and incorporated into proteins. Affinity enrichment allows the proteins to be isolated and characterised further. (c) Biotin-spm (173) can be incubated with cells and using the enzyme Gsps, can form an amide bond with GSH in situ. This biotin labelled GSH derivative can then react with glutathione reactive cysteines, followed by blocking of free thiols with 18 and subsequent detection. | ||
Khoo, Lin and associates developed a selective and rapid chemoenzymatic method for detecting proteins bearing glutathione. This technique takes advantage of an E. coli glutathionylspermidine synthetase (GspS) that can form an amide bond between GSH and biotin-labelled spermidine (biotin-spm, 173) when GspS is expressed in mammalian cells. The resulting biotin labelled GSH forms disulfides with proteins in the same way as unlabelled GSH (Fig. 22c).265 Glutathionylated proteins were identified by GspS-transfected 293T cell lysates incubated with 173 and then treated with H2O2. Free cysteines were then capped with 18 to prevent interference. Proteins were precipitated, unreacted biotin compounds were removed, and the proteins were digested. Gspm-biotin bound peptides were captured by streptavidin-conjugated resin. After washing, peptides were eluted with Gsp amidase (to remove biotin-spm) and then analysed by mass spectrometry. An advantage of this approach is that no reduction step is required, reducing chances of cross-reactivity of other cysteine oxidation states such as sulfenic acids or nitrosothiols. Additionally, no external addition of GSH is required so the results are more likely to reflect the innate GSH/GSSG ratio.
An enzymatic approach was developed by Cotgreave and co-workers that took advantage of the specific reduction of protein glutathiones by a glutaredoxin-3 (Grx-3) mutant from E. coli which shows preference for reduction of protein–GSH mixed disulfides.266 After blocking free thiols, the protein mixture is reduced with Grx-3, with selective reduction of glutathionylated proteins. The resulting thiols can then be selectively labelled for enrichment and mass spectrometric analysis (Fig. 23a). This strategy was used to monitor human ECV304 cells during enhanced oxidation of GSH by diamide. A similar proteomic approach using resin-capture (174) was developed by Qian and co-workers which relies on initial blocking of free thiols by alkylation, followed by selective reduction of glutathionylated thiols. The liberated thiols are captured with affinity resins, digested and analysed by MS (Fig. 23b).267 This method was validated on RAW 264.7 macrophage cells. Before blocking, ascorbate/Cu(I) was added to reduce SNO to free thiols. All thiols were then blocked with 25. Glutathione proteins were then selectively reduced with Grx3, captured with a thiol-affinity resin, digested, and labelled with iTRAQ for analysis.
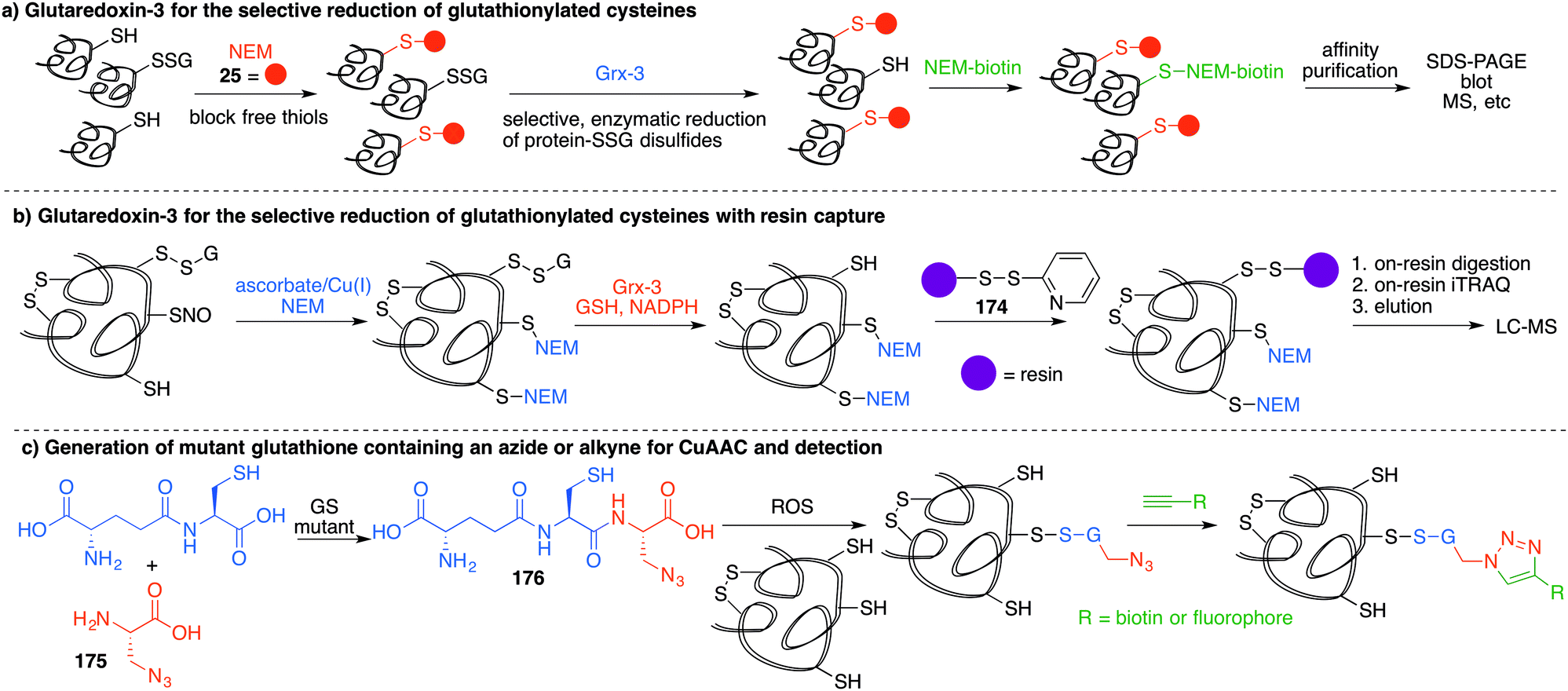 | ||
| Fig. 23 (a and b) Glutaredoxin-3 (Grx-3) can selectively reduce protein-SSG disulfides. After blocking free thiols, Grx-3 can be used to reduce the sites of glutathionylation. The resulting cysteine thiols can then be labelled for affinity enrichment and further analysis. In (b), a resin-capture (174) is used to improve detection of reduced glutathione thiols. (c) Glutathione containing an azide or alkyne (176) incorporated using a mutant GS can react with reactive thiols and later be detected by using an alkyne or azide with a reporter group for analysis. | ||
While these methods provide convenient ways of monitoring glutathionylation, the bulky nature of the reporter tags (e.g. biotin or fluorophores) may hinder incorporation of these probes into the proteins of interest. And, because most of these processes use exogenous glutathione-based probes, the addition of this excess thiol has the potential to alter the redox balance of the cell (namely the GSH/GSSG ratio). This balance is particularly important for biotin-GSSG (172) as the levels of GSSG make up only 1% of all glutathione.265 The biotin-switch inspired techniques also have several concerns with incomplete reduction, and the specificity of Grx-3 for protein–glutathiones. Addressing these issues, the Ahn group engineered a glutathione synthetase (GS) mutant which could incorporate azidoalanine (175) in place of the glycine in GSH (Fig. 23c).268 This technology allows the production of GSH bearing a small azide reporter tag for subsequent detection (176), without increasing the levels of glutathione in the cell. The glutathione synthetase mutant was expressed in cells in the presence of 175, producing glutathione with the azide handle (176). The cells are then treated with H2O2, and the lysates then subject to the copper-catalysed cycloaddition reaction with a biotin- or rhodamine-alkyne and resolved by SDS-PAGE. It was also found that Grx1 can efficiently catalyse the removal of azido-glutathione from proteins by the reduction of the disulfide. Glutathionylation could also be visualised in cells by adding a fluorescent tag using Alexa-Fluor647-alkyne, which gave a strong wide-spread signal in HEK293 cells overexpressing the GS mutant. Ahn and associates later used a similar approach with a terminal alkene derived from allyl-Gly and allyl-Ser. This technique relies on the alkene–tetrazine ligation for attachment of a reporter group. An advantage of using allyl-Glu is that it is smaller than azido-Glu and therefore closer to the native structure of GSH.269
Other groups have incorporated an alkyne into GSH and used these derivatives as probes to study glutathionylation.270 The alkyne-labelled GSH (installed as a propargyl amide at Gly) was first synthesised chemically and then added to cell lysates. Protein glutathionylation was detected by subsequent reaction with an azido-biotin reagent. This technique was used to map glutathionylation and revealed that glutathionylation occurs at a higher frequency on cysteines near negatively charged residues. It should be noted, however, this technique required the addition of exogenous GSH which may perturb the innate redox balance in the cell.
8.1 Conclusions and future opportunities
The oxidation of cysteine in cells features diverse chemistry. The thiol of this unique amino acid residue can be oxidised to the sulfenic, sulfinic and sulfonic acids or converted to a disulfide, persulfide or nitrosylated cysteine. These oxidative modifications are formed in response to oxidative stress. However, these modifications are also integral to several signalling pathways in healthy cells that control enzyme activity, protein structure, and protein–protein interactions. This review has surveyed common methods for mapping these oxidised derivatives of cysteine, with spotlights on a few of the important biological processes that are impacted by these modifications.Rather than reiterate these achievements here, it is worth noting some future opportunities for chemists and biologists in advancing our understanding of cysteine oxidation. For example, probes that can react directly with a specific modification are highly valuable because they obviate the need for blocking steps. Additionally, if these probes are compatible with cells, then the modification of interest can be trapped under native conditions, rather than through analysis of cellular lysates. Such probes must overcome a substantial challenge in chemoselectivity: the probe needs to react with the modification of interest and not with any other biomolecule. For oxidative modifications of cysteines that contain a labile group (especially RSOH, RSSR’, RSNO, and RSSH), this is particularly challenging because each modification is, in principle, susceptible to reduction or reaction with nucleophiles. Identifying probes that react through a unique mechanism with each of these modifications is therefore important. For new probes, it is also critical that selectivity for the modification of interest is demonstrated. For example, if a new probe for nitrosylated cysteine is developed, this probe should be tested under the same conditions against reduced cysteine, a cysteine disulfide, a cysteine sulfenic acid, and a cysteine persulfide. The identification of off-target reactivity will help researchers anticipate false positives and validate hits. Rigorous mechanistic insight can also help guide deployment of new probes in a biological context—especially in anticipating any potential off-target reactions.
Many of the chemical probes discussed in this review demonstrated off-target reactions with cysteine oxidation states other than the one intended. These issues in selectivity were often not addressed in initial studies, and only later found through related studies on other cysteine oxidation states. While this cross-reactivity doesn’t necessarily prevent the use of these probes, it warrants careful consideration when assessing the outcomes of these experiments.
Several indirect detection methods relied on thiol blocking reagents, which have long been thought to be thiol-selective. However, evidence has steadily been put forward which demonstrates that several of these key electrophilic thiol-blocking reagents are not selective for thiols, and can react with sulfenic and sulfinic acids, with some also reacting with persulfides and nitrosothiols. This is obviously a concern when these blocking agents react non-selectively and mask a modification of interest. Since many detection methods rely on thiol-blocking, such as the biotin switch technique and its many variations, development of truly thiol-specific reagents would be greatly beneficial.
Another significant challenge is to develop probes that react rapidly with their target so that short-lived oxidative modifications can be identified. Cells respond rapidly to oxidative stress and second messengers such as NO and H2S. The availability of selective probes that react with cysteine modifications on the same timescale would be highly useful for monitoring how proteins are modified in real-time. Increasing reactivity, however, can often come at the expense of selectivity so this is not a trivial task.
The majority of methods outlined in this review were used to identify proteins that are modified during oxidative stress and other signalling events. In some cases, identifying the site of the protein allowed hypotheses to be generated about how that oxidative modification of cysteine was involved in a specific signalling pathway. Identifying and annotating such signalling networks will continue to be a major aspect of research into cysteine oxidation. With that said, there are many opportunities to translate this fundamental information into useful strategies for chemical biology and medicinal chemistry. For example, if a cysteine modification leads to an unwanted event (uncontrolled cell proliferation, for example), it might be possible to intercept the modification with a selective probe and prevent that unwanted event from occurring. If possible, this could be a potential route to new cancer therapies or treatments for the many other diseases linked to oxidative stress. Considering this possibility further, there is substantial opportunity to identify how these molecular probes can be modified to react not just with a specific cysteine modification, but with that modification on a specific protein. If these probes are to be developed into pharmaceuticals, protein-specific probes will likely be required.
The diverse redox chemistry of cysteine is harnessed by nature in a variety of cellular processes. Understanding these modifications is a critical part of fundamental biology and medicine, so the development of selective probes for each type of modification is important. Ultimately developing such probes is a challenge in chemoselective labelling, which presents a unique opportunity and challenge for the chemical community to contribute to this area.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge generous financial support from the Australian Research Council (DE150101863).References
- C. E. Paulsen and K. S. Carroll, ACS Chem. Biol., 2010, 5, 47–62 CrossRef CAS PubMed.
- H. J. Forman, J. M. Fukuto and M. Torres, Am. J. Physiol.: Cell Physiol., 2004, 287, C246–C256 CrossRef CAS PubMed.
- T. Finkel, FEBS Lett., 2000, 476, 52–54 CrossRef CAS PubMed.
- M. S. B. P. A. M. J. Buttner, Annu. Rev. Genet., 2003, 37, 91 CrossRef PubMed.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- M. W. Foster, T. J. McMahon and J. S. Stamler, Trends Mol. Med., 2003, 9, 160–168 CrossRef CAS PubMed.
- M. Lo Conte and K. S. Carroll, J. Biol. Chem., 2013, 288, 26480–26488 CrossRef CAS PubMed.
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS PubMed.
- A. Miseta and P. Csutora, Mol. Biol. Evol., 2000, 17, 1232–1239 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2008, 4, 630–640 CrossRef PubMed.
- K. G. Reddie and K. S. Carroll, Curr. Opin. Chem. Biol., 2008, 12, 746–754 CrossRef CAS PubMed.
- O. Rudyk and P. Eaton, Redox Biol., 2014, 2, 803–813 CrossRef CAS PubMed.
- S. E. Leonard and K. S. Carroll, Curr. Opin. Chem. Biol., 2011, 15, 88–102 CrossRef CAS PubMed.
- G. Roos, N. Foloppe and J. Messens, Antioxid. Redox Signaling, 2012, 18, 94–127 CrossRef PubMed.
- U. Seneviratne, A. Nott, V. B. Bhat, K. C. Ravindra, J. S. Wishnok, L.-H. Tsai and S. R. Tannenbaum, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4152–4157 CrossRef CAS PubMed.
- P.-T. Doulias, J. L. Greene, T. M. Greco, M. Tenopoulou, S. H. Seeholzer, R. L. Dunbrack and H. Ischiropoulos, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16958–16963 CrossRef CAS PubMed.
- G. Ferrer-Sueta, B. Manta, H. Botti, R. Radi, M. Trujillo and A. Denicola, Chem. Res. Toxicol., 2011, 24, 434–450 CrossRef CAS PubMed.
- Z. A. Wood, E. Schröder, J. Robin Harris and L. B. Poole, Trends Biochem. Sci., 2003, 28, 32–40 CrossRef CAS PubMed.
- H. Miki and Y. Funato, J. Biochem., 2012, 151, 255–261 CrossRef CAS PubMed.
- J. V. C. a. D. J. Templeton, Antioxid. Redox Signaling, 2006, 8, 1819–1827 CrossRef PubMed.
- J. M. Denu and K. G. Tanner, Biochemistry, 1998, 37, 5633–5642 CrossRef CAS PubMed.
- T. H. Truong and K. S. Carroll, Biochemistry, 2012, 51, 9954–9965 CrossRef CAS PubMed.
- C. E. Paulsen, T. H. Truong, F. J. Garcia, A. Homann, V. Gupta, S. E. Leonard and K. S. Carroll, Nat. Chem. Biol., 2012, 8, 57–64 CrossRef CAS PubMed.
- J. Haendeler, J. Hoffmann, V. Tischler, B. C. Berk, A. M. Zeiher and S. Dimmeler, Nat. Cell Biol., 2002, 4, 743–749 CrossRef CAS PubMed.
- K. Hirota, M. Murata, Y. Sachi, H. Nakamura, J. Takeuchi, K. Mori and J. Yodoi, J. Biol. Chem., 1999, 274, 27891–27897 CrossRef CAS PubMed.
- C. Matiollo, G. Ecco, A. C. O. Menegatti, G. Razzera, J. Vernal and H. Terenzi, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 191–196 CrossRef CAS PubMed.
- M. Zheng, F. Aslund and G. Storz, Science, 1998, 279, 1718–1721 CrossRef CAS PubMed.
- H.-J. Choi, S.-J. Kim, P. Mukhopadhyay, S. Cho, J.-R. Woo, G. Storz and S.-E. Ryu, Cell, 2001, 105, 103–113 CrossRef CAS PubMed.
- A. Salmeen, J. N. Andersen, M. P. Myers, T.-C. Meng, J. A. Hinks, N. K. Tonks and D. Barford, Nature, 2003, 423, 769–773 CrossRef CAS PubMed.
- V. Gupta and K. S. Carroll, Biochim. Biophys. Acta, 2013, 1840, 847–875 CrossRef PubMed.
- L. B. Poole and K. J. Nelson, Curr. Opin. Chem. Biol., 2008, 12, 18–24 CrossRef CAS PubMed.
- T. Finkel, J. Cell Biol., 2011, 194, 7–15 CrossRef CAS PubMed.
- J. S. Stamler, D. I. Simon, J. A. Osborne, M. E. Mullins, O. Jaraki, T. Michel, D. J. Singel and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 444–448 CrossRef CAS.
- J. S. Stamler, O. Jaraki, J. Osborne, D. I. Simon, J. Keaney, J. Vita, D. Singel, C. R. Valeri and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7674–7677 CrossRef CAS.
- B. Saville, Analyst, 1958, 83, 670–672 RSC.
- E. Madej, L. K. Folkes, P. Wardman, G. Czapski and S. Goldstein, Free Radical Biol. Med., 2008, 44, 2013–2018 CrossRef CAS PubMed.
- D. Jourd'heuil, F. L. Jourd'heuil and M. Feelisch, J. Biol. Chem., 2003, 278, 15720–15726 CrossRef PubMed.
- S. Goldstein and G. Czapski, J. Am. Chem. Soc., 1996, 118, 3419–3425 CrossRef CAS.
- V. G. Kharitonov, A. R. Sundquist and V. S. Sharma, J. Biol. Chem., 1995, 270, 28158–28164 CrossRef CAS PubMed.
- B. P. Luchsinger, E. N. Rich, A. J. Gow, E. M. Williams, J. S. Stamler and D. J. Singel, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 461–466 CrossRef CAS PubMed.
- A. J. Gow, D. G. Buerk and H. Ischiropoulos, J. Biol. Chem., 1997, 272, 2841–2845 CrossRef CAS PubMed.
- S. Basu, A. Keszler, N. A. Azarova, N. Nwanze, A. Perlegas, S. Shiva, K. A. Broniowska, N. Hogg and D. B. Kim-Shapiro, Free Radical Biol. Med., 2010, 48, 255–263 CrossRef CAS PubMed.
- N. Hogg, Anal. Biochem., 1999, 272, 257–262 CrossRef CAS PubMed.
- M. Steffen, T. M. Sarkela, A. A. Gybina, T. W. Steele, N. J. Trasseth, D. Keuhl and C. Giulivi, Biochem. J., 2001, 356, 395–402 CrossRef CAS PubMed.
- K. A. Broniowska, A. R. Diers and N. Hogg, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3173–3181 CrossRef CAS PubMed.
- T. Nakamura and S. A. Lipton, Neurochem. Res., 2016, 41, 510–514 CrossRef CAS PubMed.
- T. M. Greco, R. Hodara, I. Parastatidis, H. F. G. Heijnen, M. K. Dennehy, D. C. Liebler and H. Ischiropoulos, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7420–7425 CrossRef CAS PubMed.
- J. B. Mannick, A. Hausladen, L. Liu, D. T. Hess, M. Zeng, Q. X. Miao, L. S. Kane, A. J. Gow and J. S. Stamler, Science, 1999, 284, 651–654 CrossRef CAS PubMed.
- S. D. Ryan, N. Dolatabadi, S. F. Chan, X. Zhang, M. W. Akhtar, J. Parker, F. Soldner, C. R. Sunico, S. Nagar, M. Talantova, B. Lee, K. Lopez, A. Nutter, B. Shan, E. Molokanova, Y. Zhang, X. Han, T. Nakamura, E. Masliah, J. R. Yates III, N. Nakanishi, A. Y. Andreyev, S.-I. Okamoto, R. Jaenisch, R. Ambasudhan and S. A. Lipton, Cell, 2013, 155, 1351–1364 CrossRef CAS PubMed.
- L. Gasperini, E. Meneghetti, B. Pastore, F. Benetti and G. Legname, Antioxid. Redox Signaling, 2015, 22, 772–784 CrossRef CAS PubMed.
- S. Zahid, R. Khan, M. Oellerich, N. Ahmed and A. R. Asif, Neuroscience, 2014, 256, 126–136 CrossRef CAS PubMed.
- D.-H. Cho, T. Nakamura, J. Fang, P. Cieplak, A. Godzik, Z. Gu and S. A. Lipton, Science, 2009, 324, 102–105 CrossRef CAS PubMed.
- M. W. Akhtar, S. Sanz-Blasco, N. Dolatabadi, J. Parker, K. Chon, M. S. Lee, W. Soussou, S. R. McKercher, R. Ambasudhan, T. Nakamura and S. A. Lipton, Nat. Commun., 2016, 7, 10242 CrossRef CAS PubMed.
- T. Uehara, T. Nakamura, D. Yao, S. Zhong-Qing, Z. Gu, Y. Ma, E. Masliah, Y. Nomura and S. A. Lipton, Nature, 2006, 441, 513 CrossRef CAS PubMed.
- D. Guerra, K. Ballard, I. Truebridge and E. Vierling, Biochemistry, 2016, 55, 2452–2464 CrossRef CAS PubMed.
- H. Wang and M. Xian, Curr. Opin. Chem. Biol., 2011, 15, 32–37 CrossRef CAS PubMed.
- S. R. Jaffrey, H. Erdjument-Bromage, C. D. Ferris, P. Tempst and S. H. Snyder, Nat. Cell Biol., 2001, 3, 193–197 CrossRef CAS PubMed.
- M. T. Forrester, M. W. Foster and J. S. Stamler, J. Biol. Chem., 2007, 282, 13977–13983 CrossRef CAS PubMed.
- M. T. Forrester, M. W. Foster, M. Benhar and J. S. Stamler, Free Radical Biol. Med., 2009, 46, 119–126 CrossRef CAS PubMed.
- L. Santhanam, M. Gucek, T. R. Brown, M. Mansharamani, S. Ryoo, C. A. Lemmon, L. Romer, A. A. Shoukas, D. E. Berkowitz and R. N. Cole, Nitric Oxide, 2008, 19, 295–302 CrossRef CAS PubMed.
- Y. Zhang, A. Keszler, K. A. Broniowska and N. Hogg, Free Radical Biol. Med., 2005, 38, 874–881 CrossRef CAS PubMed.
- L. M. Landino, M. T. Koumas, C. E. Mason and J. A. Alston, Biochem. Biophys. Res. Commun., 2006, 340, 347–352 CrossRef CAS PubMed.
- B. Huang and C. Chen, Free Radical Biol. Med., 2006, 41, 562–567 CrossRef CAS PubMed.
- X. Wang, N. J. Kettenhofen, S. Shiva, N. Hogg and M. T. Gladwin, Free Radical Biol. Med., 2008, 44, 1362–1372 CrossRef CAS PubMed.
- V. M. Kallakunta, A. Staruch and B. Mutus, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 23–30 CrossRef CAS PubMed.
- M. T. Forrester, J. W. Thompson, M. W. Foster, L. Nogueira, M. A. Moseley and J. S. Stamler, Nat. Biotechnol., 2009, 27, 557–559 CrossRef CAS PubMed.
- Y. Wang, P. Zhang, Z. Xu, W. Yue, Y. Zhuang, Y. Chen and Z. Lu, Free Radical Biol. Med., 2015, 86, 343–351 CrossRef CAS PubMed.
- M. J. Kohr, J. Sun, A. Aponte, G. Wang, M. Gucek, E. Murphy and C. Steenbergen, Circ. Res., 2011, 108, 418–426 CrossRef CAS PubMed.
- G. Hao, B. Derakhshan, L. Shi, F. Campagne and S. S. Gross, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1012–1017 CrossRef CAS PubMed.
- B. Huang and C. Chen, Free Radical Biol. Med., 2010, 49, 447–456 CrossRef CAS PubMed.
- M. Liu, J. Hou, L. Huang, X. Huang, T. H. Heibeck, R. Zhao, L. Pasa-Tolic, R. D. Smith, Y. Li, K. Fu, Z. Zhang, S. H. Hinrichs and S.-J. Ding, Anal. Chem., 2010, 82, 7160–7168 CrossRef CAS PubMed.
- M.-F. Hsu, K.-T. Pan, F.-Y. Chang, K.-H. Khoo, H. Urlaub, C.-F. Cheng, G.-D. Chang, F. G. Haj and T.-C. Meng, Free Radical Biol. Med., 2016, 99, 199–213 CrossRef CAS PubMed.
- P. Han, X. Zhou, B. Huang, X. Zhang and C. Chen, Anal. Biochem., 2008, 377, 150–155 CrossRef CAS PubMed.
- N. J. Kettenhofen, X. Wang, M. T. Gladwin and N. Hogg, Methods Enzymol., 2008, 441, 53–71 CAS.
- E. T. Chouchani, T. R. Hurd, S. M. Nadtochiy, P. S. Brookes, I. M. Fearnley, K. S. Lilley, R. A. J. Smith and M. P. Murphy, Biochem. J., 2010, 430, 49–59 CrossRef CAS PubMed.
- J. S. Paige, G. Xu, B. Stancevic and S. R. Jaffrey, Chem. Biol., 2008, 15, 1307–1316 CrossRef CAS PubMed.
- V. Sinha, G. T. Wijewickrama, R. E. P. Chandrasena, H. Xu, P. D. Edirisinghe, I. T. Schiefer and G. R. J. Thatcher, ACS Chem. Biol., 2010, 5, 667–680 CrossRef CAS PubMed.
- E. T. Chouchani, A. M. James, C. Methner, V. R. Pell, T. A. Prime, B. K. Erickson, M. Forkink, G. Y. Lau, T. P. Bright, K. E. Menger, I. M. Fearnley, T. Krieg and M. P. Murphy, J. Biol. Chem., 2017, 292, 14486–14495 CrossRef CAS PubMed.
- Y. Zhou, S. L. Wynia-Smith, S. M. Couvertier, K. S. Kalous, M. A. Marletta, B. C. Smith and E. Weerapana, Cell Chem. Biol., 2016, 23, 727–737 CrossRef CAS PubMed.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–795 CrossRef CAS PubMed.
- Y. Qian, J. Martell, N. J. Pace, T. E. Ballard, D. S. Johnson and E. Weerapana, ChemBioChem, 2013, 14, 1410–1414 CrossRef CAS PubMed.
- H. Y. Jia, Y. Liu, X. J. Zhang, L. Han, L. B. Du, Q. Tian and Y. C. Xu, J. Am. Chem. Soc., 2009, 131, 40–41 CrossRef CAS PubMed.
- A. Faccenda, C. A. Bonham, P. O. Vacratsis, X. Zhang and B. Mutus, J. Am. Chem. Soc., 2010, 132, 11392–11394 CrossRef CAS PubMed.
- P.-T. Doulias, M. Tenopoulou, K. Raju, L. A. Spruce, S. H. Seeholzer and H. Ischiropoulos, J. Proteomics, 2013, 92, 195–203 CrossRef CAS PubMed.
- K. Raju, P.-T. Doulias, P. Evans, E. N. Krizman, J. G. Jackson, O. Horyn, Y. Daikhin, I. Nissim, M. Yudkoff, I. Nissim, K. A. Sharp, M. B. Robinson and H. Ischiropoulos, Sci. Signaling, 2015, 8, ra68–ra68 CrossRef PubMed.
- P.-T. Doulias, M. Tenopoulou, J. L. Greene, K. Raju and H. Ischiropoulos, Sci. Signaling, 2013, 6, rs1–rs1 CrossRef PubMed.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera, I. S. Albuquerque, C. T. Gibson, A. Sibley, A. D. Slattery, J. A. Campbell, S. F. K. Alboaiji, K. A. Muller, J. Young, N. Adamson, J. R. Gascooke, D. Jampaiah, Y. M. Sabri, S. K. Bhargava, S. J. Ippolito, D. A. Lewis, J. S. Quinton, A. V. Ellis, A. Johs, G. J. L. Bernardes and J. M. Chalker, Chem. – Eur. J., 2017, 23, 16219–16230 CrossRef CAS PubMed.
- H. Wang and M. Xian, Angew. Chem., Int. Ed., 2008, 47, 6598–6601 CrossRef CAS PubMed.
- J. Zhang, H. Wang and M. Xian, J. Am. Chem. Soc., 2009, 131, 3854–3855 CrossRef CAS PubMed.
- J. Zhang, H. Wang and M. Xian, Org. Lett., 2009, 11, 477–480 CrossRef CAS PubMed.
- J. Zhang, S. Li, D. Zhang, H. Wang, A. R. Whorton and M. Xian, Org. Lett., 2010, 12, 4208–4211 CrossRef CAS PubMed.
- E. Bechtold, J. A. Reisz, C. Klomsiri, A. W. Tsang, M. W. Wright, L. B. Poole, C. M. Furdui and S. B. King, ACS Chem. Biol., 2010, 5, 405–414 CrossRef CAS PubMed.
- D. Zhang, W. Chen, Z. Miao, Y. Ye, Y. Zhao, S. B. King and M. Xian, Chem. Commun., 2014, 50, 4806–4809 RSC.
- S. Shao, B. Chen, J. Cheng, C. Wang, Y. Zhang, L. Shao, Y. Hu, Y. Han, F. Han and X. Li, Biosens. Bioelectron., 2017, 94, 162–168 CrossRef CAS PubMed.
- S. Li, H. Wang, M. Xian and A. R. Whorton, Nitric Oxide, 2012, 26, 20–26 CrossRef CAS PubMed.
- B. D. Reeves, J. K. Hilmer, L. Mellmann, M. Hartzheim, K. Poffenberger, K. Johnson, N. Joshi, D. J. Singel and P. A. Grieco, Tetrahedron Lett., 2013, 54, 5707–5710 CrossRef CAS PubMed.
- B. D. Reeves, N. Joshi, G. C. Campanello, J. K. Hilmer, L. Chetia, J. A. Vance, J. N. Reinschmidt, C. G. Miller, D. P. Giedroc, E. A. Dratz, D. J. Singel and P. A. Grieco, Org. Biomol. Chem., 2014, 12, 7942–7956 CAS.
- L. V. Benitez and W. S. Allison, J. Biol. Chem., 1974, 249, 6234–6243 CAS.
- W. S. Allison, Acc. Chem. Res., 1976, 9, 293–299 CrossRef CAS.
- L. B. Poole and A. Claiborne, J. Biol. Chem., 1989, 264, 12330–12338 CAS.
- M. Hugo, L. Turell, B. Manta, H. Botti, G. Monteiro, L. E. S. Netto, B. Alvarez, R. Radi and M. Trujillo, Biochemistry, 2009, 48, 9416–9426 CrossRef CAS PubMed.
- A. V. Peskin, F. M. Low, L. N. Paton, G. J. Maghzal, M. B. Hampton and C. C. Winterbourn, J. Biol. Chem., 2007, 282, 11885–11892 CrossRef CAS PubMed.
- O. Dionisi, T. Galeotti, T. Terranova and A. Azzi, Biochim. Biophys. Acta, Enzymol., 1975, 403, 292–300 CrossRef CAS.
- Y.-A. Suh, R. S. Arnold, B. Lassegue, J. Shi, X. Xu, D. Sorescu, A. B. Chung, K. K. Griendling and J. D. Lambeth, Nature, 1999, 401, 79–82 CrossRef CAS PubMed.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CAS.
- H. J. Forman and I. Fridovich, Arch. Biochem. Biophys., 1973, 158, 396–400 CrossRef CAS PubMed.
- M. Fuangthong and J. D. Helmann, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6690–6695 CrossRef CAS PubMed.
- R. Radi, J. S. Beckman, K. M. Bush and B. A. Freeman, J. Biol. Chem., 1991, 266, 4244–4250 CAS.
- T. C. Bruice and A. B. Sayigh, J. Am. Chem. Soc., 1959, 81, 3416–3420 CrossRef CAS.
- G. Roos and J. Messens, Free Radical Biol. Med., 2011, 51, 314–326 CrossRef CAS PubMed.
- N. J. Kettenhofen and M. J. Wood, Chem. Res. Toxicol., 2010, 23, 1633–1646 CrossRef CAS PubMed.
- T. F. Brewer, F. J. Garcia, C. S. Onak, K. S. Carroll and C. J. Chang, Annu. Rev. Biochem., 2015, 84, 765–790 CrossRef CAS PubMed.
- R. D. Michalek, K. J. Nelson, B. C. Holbrook, S. Y. John, D. Stridiron, L. W. Daniel, J. S. Fetrow, S. B. King, L. B. Poole and J. M. Grayson, J. Immunol., 2007, 179, 6456–6467 CrossRef CAS.
- J. Wu, Z. Cheng, K. Reddie, K. Carroll, L. A. Hammad, J. A. Karty and C. E. Bauer, J. Biol. Chem., 2013, 288, 4755–4762 CrossRef CAS PubMed.
- J. D. Keyes, D. Parsonage, R. D. Yammani, L. C. Rogers, C. Kesty, C. M. Furdui, K. J. Nelson and L. B. Poole, Free Radical Biol. Med., 2017, 112, 534–543 CrossRef CAS PubMed.
- J. Wu, Z. Cheng, K. Reddie, K. Carroll, L. A. Hammad, J. A. Karty and C. E. Bauer, J. Biol. Chem., 2013, 288, 4755–4762 CrossRef CAS PubMed.
- S.-R. Lee, K.-S. Kwon, S.-R. Kim and S. G. Rhee, J. Biol. Chem., 1998, 273, 15366–15372 CrossRef CAS PubMed.
- J. Sohn and J. Rudolph, Biochemistry, 2003, 42, 10060–10070 CrossRef CAS PubMed.
- Y. H. Seo and K. S. Carroll, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16163–16168 CrossRef CAS PubMed.
- T. H. Truong, P. M.-U. Ung, P. B. Palde, C. E. Paulsen, A. Schlessinger and K. S. Carroll, Cell Chem. Biol., 2016, 23, 837–848 CrossRef CAS PubMed.
- E. Y. Dotsey, K.-M. Jung, A. Basit, D. Wei, J. Daglian, F. Vacondio, A. Armirotti, M. Mor and D. Piomelli, Chem. Biol., 2015, 22, 619–628 CrossRef CAS PubMed.
- A. Bersweiler, B. D'Autréaux, H. Mazon, A. Kriznik, G. Belli, A. Delaunay-Moisan, M. B. Toledano and S. Rahuel-Clermont, Nat. Chem. Biol., 2017, 13, 909–915 CrossRef CAS PubMed.
- A. T. Saurin, H. Neubert, J. P. Brennan and P. Eaton, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17982–17987 CrossRef CAS PubMed.
- L. B. Poole, B.-B. Zeng, S. A. Knaggs, M. Yakubu and S. B. King, Bioconjugate Chem., 2005, 16, 1624–1628 CrossRef CAS PubMed.
- L. B. Poole, C. Klomsiri, S. A. Knaggs, C. M. Furdui, K. J. Nelson, M. J. Thomas, J. S. Fetrow, L. W. Daniel and S. B. King, Bioconjugate Chem., 2007, 18, 2004–2017 CrossRef CAS PubMed.
- R. L. Charles, E. Schroder, G. May, P. Free, P. R. J. Gaffney, R. Wait, S. Begum, R. J. Heads and P. Eaton, Mol. Cell. Proteomics, 2007, 6, 1473–1484 CAS.
- C. Klomsiri, L. C. Rogers, L. Soito, A. K. McCauley, S. B. King, K. J. Nelson, L. B. Poole and L. W. Daniel, Free Radical Biol. Med., 2014, 71, 49–60 CrossRef CAS PubMed.
- J. M. Hourihan, L. E. Moronetti Mazzeo, L. P. Fernández-Cárdenas and T. K. Blackwell, Mol. Cell, 2016, 63, 553–566 CrossRef CAS PubMed.
- N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648 CrossRef CAS PubMed.
- K. G. Reddie, Y. H. Seo, W. B. M. Iii, S. E. Leonard and K. S. Carroll, Mol. BioSyst., 2008, 4, 521–531 RSC.
- Y. H. Seo and K. S. Carroll, Bioorg. Med. Chem. Lett., 2009, 19, 356–359 CrossRef CAS PubMed.
- S. E. Leonard, K. G. Reddie and K. S. Carroll, ACS Chem. Biol., 2009, 4, 783–799 CrossRef CAS PubMed.
- S. Akter, J. Huang, N. Bodra, B. De Smet, K. Wahni, D. Rombaut, J. Pauwels, K. Gevaert, K. Carroll, F. Van Breusegem and J. Messens, Mol. Cell. Proteomics, 2015, 14, 1183–1200 CAS.
- M. Sundaresan, Z.-X. Yu, V. J. Ferrans, K. Irani and T. Finkel, Science, 1995, 270, 296–299 CAS.
- J. Qian, C. Klomsiri, M. W. Wright, S. B. King, A. W. Tsang, L. B. Poole and C. M. Furdui, Chem. Commun., 2011, 47, 9203–9205 RSC.
- J. Qian, R. Wani, C. Klomsiri, L. B. Poole, A. W. Tsang and C. M. Furdui, Chem. Commun., 2012, 48, 4091–4093 RSC.
- S. E. Leonard, F. J. Garcia, D. S. Goodsell and K. S. Carroll, Angew. Chem., Int. Ed., 2011, 50, 4423–4427 CrossRef CAS PubMed.
- F. J. Garcia and K. S. Carroll, Eur. J. Med. Chem., 2014, 88, 28–33 CrossRef CAS PubMed.
- M. Sethuraman, M. E. McComb, T. Heibeck, C. E. Costello and R. A. Cohen, Mol. Cell. Proteomics, 2004, 3, 273–278 CAS.
- M. Sethuraman, M. E. McComb, H. Huang, S. Huang, T. Heibeck, C. E. Costello and R. A. Cohen, J. Proteome Res., 2004, 3, 1228–1233 CrossRef CAS PubMed.
- Y. H. Seo and K. S. Carroll, Angew. Chem., Int. Ed., 2011, 50, 1342–1345 CrossRef CAS PubMed.
- M. E. Albertolle, D. Kim, L. D. Nagy, C.-H. Yun, A. Pozzi, Ü. Savas, E. F. Johnson and F. P. Guengerich, J. Biol. Chem., 2017, 292, 11230–11242 CrossRef CAS PubMed.
- T. H. Truong, F. J. Garcia, Y. H. Seo and K. S. Carroll, Bioorg. Med. Chem. Lett., 2011, 21, 5015–5020 CrossRef CAS PubMed.
- J. Yang, V. Gupta, K. S. Carroll and D. C. Liebler, Nat. Commun., 2014, 5, 4776 CrossRef CAS PubMed.
- H. R. Ellis and L. B. Poole, Biochemistry, 1997, 36, 15013–15018 CrossRef CAS PubMed.
- L.-H. Ma, C. L. Takanishi and M. J. Wood, J. Biol. Chem., 2007, 282, 31429–31436 CrossRef CAS PubMed.
- C. T. Liu and S. J. Benkovic, J. Am. Chem. Soc., 2013, 135, 14544–14547 CrossRef CAS PubMed.
- T. H. Poole, J. A. Reisz, W. Zhao, L. B. Poole, C. M. Furdui and S. B. King, J. Am. Chem. Soc., 2014, 136 Search PubMed.
- D. J. McGarry, M. M. Shchepinova, S. Lilla, R. C. Hartley and M. F. Olson, ACS Chem. Biol., 2016, 11, 3300–3304 CrossRef CAS PubMed.
- E. Galardon and D. Padovani, Bioconjugate Chem., 2015, 26, 1013–1016 CrossRef CAS PubMed.
- R. van Geel, G. J. M. Pruijn, F. L. van Delft and W. C. Boelens, Bioconjugate Chem., 2012, 23, 392–398 CrossRef CAS PubMed.
- L. J. Alcock, K. D. Farrell, M. T. Akol, G. H. Jones, M. M. Tierney, H. B. Kramer, T. L. Pukala, G. J. L. Bernardes, M. V. Perkins and J. M. Chalker, Tetrahedron, 2017 DOI:10.1016/j.tet.2017.11.011.
- D. H. R. Barton, D. G. T. Greig, G. Lucente, P. G. Sammes and M. V. Taylor, Chem. Commun., 1970, 1683–1684 RSC.
- I. Ager, D. H. R. Barton, D. G. T. Greig, G. Lucente, P. G. Sammes, M. V. Taylor, G. H. Hewitt, B. E. Looker, A. Mowatt, C. A. Robson and W. G. E. Underwood, J. Chem. Soc., Perkin Trans. 1, 1973, 1187–1196 RSC.
- A. G. M. Barrett, D. H. R. Barton and S. Nagubandi, J. Chem. Soc., Perkin Trans. 1, 1980, 237–239 RSC.
- V. Gupta and K. S. Carroll, Chem. Sci., 2016, 7, 400–415 RSC.
- M. N. Tahir, M. Shafiq, I. U. Khan, W. A. Siddiqui and M. N. Arshad, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o557 CAS.
- V. Gupta and K. S. Carroll, Chem. Commun., 2016, 52, 3414–3417 RSC.
- V. Gupta, H. Paritala and K. S. Carroll, Bioconjugate Chem., 2016, 27, 1411–1418 CrossRef CAS PubMed.
- V. Gupta, J. Yang, D. C. Liebler and K. S. Carroll, J. Am. Chem. Soc., 2017, 139, 5588–5595 CrossRef CAS PubMed.
- H. J. Forman, M. J. Davies, A. C. Krämer, G. Miotto, M. Zaccarin, H. Zhang and F. Ursini, Arch. Biochem. Biophys., 2017, 617, 26–37 CrossRef CAS PubMed.
- T. Rabilloud, M. Heller, F. Gasnier, S. Luche, C. Rey, R. Aebersold, M. Benahmed, P. Louisot and J. Lunardi, J. Biol. Chem., 2002, 277, 19396–19401 CrossRef CAS PubMed.
- L. Turell, H. Botti, S. Carballal, G. Ferrer-Sueta, J. M. Souza, R. Durán, B. A. Freeman, R. Radi and B. Alvarez, Biochemistry, 2008, 47, 358–367 CrossRef CAS PubMed.
- B. Biteau, J. Labarre and M. B. Toledano, Nature, 2003, 425, 980–984 CrossRef CAS PubMed.
- T.-S. Chang, W. Jeong, H. A. Woo, S. M. Lee, S. Park and S. G. Rhee, J. Biol. Chem., 2004, 279, 50994–51001 CrossRef CAS PubMed.
- Z. A. Wood, L. B. Poole and P. A. Karplus, Science, 2003, 300, 650–653 CrossRef CAS PubMed.
- C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742–4758 CrossRef CAS PubMed.
- C. Jacob, A. L. Holme and F. H. Fry, Org. Biomol. Chem., 2004, 2, 1953–1956 CAS.
- M. Lo Conte, J. Lin, M. A. Wilson and K. S. Carroll, ACS Chem. Biol., 2015, 10, 1825–1830 CrossRef CAS PubMed.
- H. A. Woo, H. Z. Chae, S. C. Hwang, K.-S. Yang, S. W. Kang, K. Kim and S. G. Rhee, Science, 2003, 300, 653–656 CrossRef CAS PubMed.
- X. Fu, S. Y. Kassim, W. C. Parks and J. W. Heinecke, J. Biol. Chem., 2001, 276, 41279–41287 CrossRef CAS PubMed.
- T. Murakami, M. Nojiri, H. Nakayama, M. Odaka, M. Yohda, N. Dohmae, K. Takio, T. Nagamune and I. Endo, Protein Sci., 2000, 9, 1024–1030 CrossRef CAS PubMed.
- J. Blackinton, M. Lakshminarasimhan, K. J. Thomas, R. Ahmad, E. Greggio, A. S. Raza, M. R. Cookson and M. A. Wilson, J. Biol. Chem., 2009, 284, 6476–6485 CrossRef CAS PubMed.
- N. Fujiwara, M. Nakano, S. Kato, D. Yoshihara, T. Ookawara, H. Eguchi, N. Taniguchi and K. Suzuki, J. Biol. Chem., 2007, 282, 35933–35944 CrossRef CAS PubMed.
- H. A. Woo, S. Won Kang, H. K. Kim, K.-S. Yang, H. Z. Chae and S. G. Rhee, J. Biol. Chem., 2003, 278, 47361–47364 CrossRef CAS PubMed.
- Y.-C. Chang, C.-N. Huang, C.-H. Lin, H.-C. Chang and C.-C. Wu, Proteomics, 2010, 10, 2961–2971 CrossRef CAS PubMed.
- J. Paulech, K. A. Liddy, K. Engholm-Keller, M. Y. White and S. J. Cordwell, Mol. Cell. Proteomics, 2015, 14, 609–620 CAS.
- M. Lo Conte and K. S. Carroll, Angew. Chem., Int. Ed., 2012, 51, 6502–6505 CrossRef CAS PubMed.
- J. D. Majmudar, A. M. Konopko, K. J. Labby, C. T. M. B. Tom, J. E. Crellin, A. Prakash and B. R. Martin, J. Am. Chem. Soc., 2016, 138, 1852–1859 CrossRef CAS PubMed.
- Y.-H. Kuo, A. M. Konopko, N. B. Borotto, J. D. Majmudar, S. E. Haynes and B. R. Martin, ChemBioChem, 2017, 18, 2028–2032 CrossRef CAS PubMed.
- D. Cavallini, G. Federici and E. Barboni, Eur. J. Biochem., 1970, 14, 169–174 CrossRef CAS PubMed.
- G. K. Kolluru, X. Shen, S. C. Bir and C. G. Kevil, Nitric Oxide, 2013, 35, 5–20 CrossRef CAS PubMed.
- V. S. Lin, W. Chen, M. Xian and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4596–4618 RSC.
- P. Nagy, Z. Pálinkás, A. Nagy, B. Budai, I. Tóth and A. Vasas, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 876–891 CrossRef CAS PubMed.
- E. Cuevasanta, M. N. Möller and B. Alvarez, Arch. Biochem. Biophys., 2017, 617, 9–25 CrossRef CAS PubMed.
- X. Chen, K.-H. Jhee and W. D. Kruger, J. Biol. Chem., 2004, 279, 52082–52086 CrossRef CAS PubMed.
- S. Singh, D. Padovani, R. A. Leslie, T. Chiku and R. Banerjee, J. Biol. Chem., 2009, 284, 22457–22466 CrossRef CAS PubMed.
- A. Meister, P. E. Fraser and S. V. Tice, J. Biol. Chem., 1954, 206, 561–575 CAS.
- A. K. Mustafa, M. M. Gadalla, N. Sen, S. Kim, W. Mu, S. K. Gazi, R. K. Barrow, G. Yang, R. Wang and S. H. Snyder, Sci. Signaling, 2009, 2, ra72–ra72 Search PubMed.
- N. E. Francoleon, S. J. Carrington and J. M. Fukuto, Arch. Biochem. Biophys., 2011, 516, 146–153 CrossRef CAS PubMed.
- E. Cuevasanta, M. Lange, J. Bonanata, E. L. Coitiño, G. Ferrer-Sueta, M. R. Filipovic and B. Alvarez, J. Biol. Chem., 2015, 290, 26866–26880 CrossRef CAS PubMed.
- T. V. Mishanina, M. Libiad and R. Banerjee, Nat. Chem. Biol., 2015, 11, 457–464 CrossRef CAS PubMed.
- S. A. Everett, L. K. Folkes, P. Wardman and K. D. Asmus, Free Radical Res., 1994, 20, 387–400 CrossRef CAS PubMed.
- N. Krishnan, C. Fu, D. J. Pappin and N. K. Tonks, Sci. Signaling, 2011, 4, ra86–ra86 CrossRef PubMed.
- A. K. Mustafa, G. Sikka, S. K. Gazi, J. Steppan, S. M. Jung, A. K. Bhunia, V. M. Barodka, F. K. Gazi, R. K. Barrow, R. Wang, L. M. Amzel, D. E. Berkowitz and S. H. Snyder, Circ. Res., 2011, 109, 1259–1268 CrossRef CAS PubMed.
- N. Sen, B. D. Paul, M. M. Gadalla, A. K. Mustafa, T. Sen, R. Xu, S. Kim and S. H. Snyder, Mol. Cell, 2012, 45, 13–24 CrossRef CAS PubMed.
- A. P. Jarosz, W. Wei, J. W. Gauld, J. Auld, F. Özcan, M. Aslan and B. Mutus, Free Radical Biol. Med., 2015, 89, 512–521 CrossRef CAS PubMed.
- J. Cai, X. Shi, H. Wang, J. Fan, Y. Feng, X. Lin, J. Yang, Q. Cui, C. Tang, G. Xu and B. Geng, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2016, 1861, 419–429 CrossRef CAS PubMed.
- Y. Ju, A. Untereiner, L. Wu and G. Yang, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 2293–2303 CrossRef CAS PubMed.
- G. Yang, K. Zhao, Y. Ju, S. Mani, Q. Cao, S. Puukila, N. Khaper, L. Wu and R. Wang, Antioxid. Redox Signaling, 2012, 18, 1906–1919 CrossRef PubMed.
- A. T. Dinkova-Kostova, W. D. Holtzclaw, R. N. Cole, K. Itoh, N. Wakabayashi, Y. Katoh, M. Yamamoto and P. Talalay, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11908–11913 CrossRef CAS PubMed.
- Z.-Z. Xie, M.-M. Shi, L. Xie, Z.-Y. Wu, G. Li, F. Hua and J.-S. Bian, Antioxid. Redox Signaling, 2014, 21, 2531–2542 CrossRef CAS PubMed.
- U. Branzoli and V. Massey, J. Biol. Chem., 1974, 249, 4346–4349 CAS.
- T. Sawahata and R. A. Neal, Anal. Biochem., 1982, 126, 360–364 CrossRef CAS PubMed.
- J. Pan and K. S. Carroll, ACS Chem. Biol., 2013, 8, 1110–1116 CrossRef CAS PubMed.
- D. Zhang, I. Macinkovic, N. O. Devarie-Baez, J. Pan, C.-M. Park, K. S. Carroll, M. R. Filipovic and M. Xian, Angew. Chem., Int. Ed., 2014, 53, 575–581 CrossRef CAS PubMed.
- R. Wedmann, C. Onderka, S. Wei, I. A. Szijarto, J. L. Miljkovic, A. Mitrovic, M. Lange, S. Savitsky, P. K. Yadav, R. Torregrossa, E. G. Harrer, T. Harrer, I. Ishii, M. Gollasch, M. E. Wood, E. Galardon, M. Xian, M. Whiteman, R. Banerjee and M. R. Filipovic, Chem. Sci., 2016, 7, 3414–3426 RSC.
- É. Dóka, I. Pader, A. Bíró, K. Johansson, Q. Cheng, K. Ballagó, J. R. Prigge, D. Pastor-Flores, T. P. Dick, E. E. Schmidt, E. S. J. Arnér and P. Nagy, Sci. Adv., 2016, 2, e1500968 Search PubMed.
- D. S. Rehder and C. R. Borges, Biochemistry, 2010, 49, 7748–7755 CrossRef CAS PubMed.
- R. D. Bach, O. Dmitrenko and C. Thorpe, J. Org. Chem., 2008, 73, 12–21 CrossRef CAS PubMed.
- D. R. Arnelle and J. S. Stamler, Arch. Biochem. Biophys., 1995, 318, 279–285 CrossRef CAS PubMed.
- P. Eaton, Free Radical Biol. Med., 2006, 40, 1889–1899 CrossRef CAS PubMed.
- P. A. Fernandes and M. J. Ramos, Chem. – Eur. J., 2004, 10, 257–266 CrossRef CAS PubMed.
- A. Fava, A. Iliceto and E. Camera, J. Am. Chem. Soc., 1957, 79, 833–838 CrossRef CAS.
- J. Ying, N. Clavreul, M. Sethuraman, T. Adachi and R. A. Cohen, Free Radical Biol. Med., 2007, 43, 1099–1108 CrossRef CAS PubMed.
- F. Åslund and J. Beckwith, Cell, 1999, 96, 751–753 CrossRef.
- A. R. Chaudhuri, I. A. Khan and R. F. Ludueña, Biochemistry, 2001, 40, 8834–8841 CrossRef CAS PubMed.
- Z. Guo, S. Kozlov, M. F. Lavin, M. D. Person and T. T. Paull, Science, 2010, 330, 517–521 CrossRef CAS PubMed.
- J. P. Brennan, S. C. Bardswell, J. R. Burgoyne, W. Fuller, E. Schröder, R. Wait, S. Begum, J. C. Kentish and P. Eaton, J. Biol. Chem., 2006, 281, 21827–21836 CrossRef CAS PubMed.
- M. C. Sobotta, W. Liou, S. Stöcker, D. Talwar, M. Oehler, T. Ruppert, A. N. D. Scharf and T. P. Dick, Nat. Chem. Biol., 2014, 11, 64 CrossRef PubMed.
- A. Sommer and R. R. Traut, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3946–3950 CrossRef CAS.
- J. P. Brennan, R. Wait, S. Begum, J. R. Bell, M. J. Dunn and P. Eaton, J. Biol. Chem., 2004, 279, 41352–41360 CrossRef CAS PubMed.
- R. C. Cumming, N. L. Andon, P. A. Haynes, M. Park, W. H. Fischer and D. Schubert, J. Biol. Chem., 2004, 279, 21749–21758 CrossRef CAS PubMed.
- U. Jakob, W. Muse, M. Eser and J. C. A. Bardwell, Cell, 1999, 96, 341–352 CrossRef CAS PubMed.
- L. Makmura, M. Hamann, A. Areopagita, S. Furuta, A. Muñoz and J. Momand, Antioxid. Redox Signaling, 2001, 3, 1105–1118 CrossRef CAS PubMed.
- J. R. Winther and C. Thorpe, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 838–846 CrossRef CAS PubMed.
- R. E. Hansen, H. Østergaard, P. Nørgaard and J. R. Winther, Anal. Biochem., 2007, 363, 77–82 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- X. Chen, Y. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2010, 39, 2120–2135 RSC.
- G. L. Newton, R. Dorian and R. C. Fahey, Anal. Biochem., 1981, 114, 383–387 CrossRef CAS PubMed.
- K. Imai, T. Toyo'oka and Y. Watanabe, Anal. Biochem., 1983, 128, 471–473 CrossRef CAS PubMed.
- T. Toyooka and K. Imai, Anal. Chem., 1984, 56, 2461–2464 CrossRef CAS.
- T. L. Kirley, Anal. Biochem., 1989, 180, 231–236 CrossRef CAS PubMed.
- I. A. Cotgreave and P. Moldéus, J. Biochem. Biophys. Methods, 1986, 13, 231–249 CrossRef CAS PubMed.
- B. G. Hill, C. Reily, J.-Y. Oh, M. S. Johnson and A. Landar, Free Radical Biol. Med., 2009, 47, 675–683 CrossRef CAS PubMed.
- J. A. Reisz, E. Bechtold, S. B. King, L. B. Poole and C. M. Furdui, FEBS J., 2013, 280, 6150–6161 CrossRef CAS PubMed.
- J.-J. Lee, S. Ha, H.-J. Kim, H. J. Ha, H.-Y. Lee and K.-J. Lee, ACS Chem. Biol., 2014, 9, 2883–2894 CrossRef CAS PubMed.
- X. Chen, H. Wu, C.-M. Park, T. H. Poole, G. Keceli, N. O. Devarie-Baez, A. W. Tsang, W. T. Lowther, L. B. Poole, S. B. King, M. Xian and C. M. Furdui, ACS Chem. Biol., 2017, 12, 2201–2208 CrossRef CAS PubMed.
- Y. Wu, K.-S. Kwon and S. G. Rhee, FEBS Lett., 1998, 440, 111–115 CrossRef CAS PubMed.
- M. Sethuraman, N. Clavreul, H. Huang, M. E. McComb, C. E. Costello and R. A. Cohen, Free Radical Biol. Med., 2007, 42, 823–829 CrossRef CAS PubMed.
- J.-R. Kim, H. W. Yoon, K.-S. Kwon, S.-R. Lee and S. G. Rhee, Anal. Biochem., 2000, 283, 214–221 CrossRef CAS PubMed.
- C. Gitler, B. Zarmi and E. Kalef, Anal. Biochem., 1997, 252, 48–55 CrossRef CAS PubMed.
- N. Le Moan, G. Clement, S. Le Maout, F. Tacnet and M. B. Toledano, J. Biol. Chem., 2006, 281, 10420–10430 CrossRef CAS PubMed.
- L. I. Leichert, F. Gehrke, H. V. Gudiseva, T. Blackwell, M. Ilbert, A. K. Walker, J. R. Strahler, P. C. Andrews and U. Jakob, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8197–8202 CrossRef CAS PubMed.
- P. G. Richman and A. Meister, J. Biol. Chem., 1975, 250, 1422–1426 CAS.
- A. Meister, J. Biol. Chem., 1988, 263, 17205–17208 CAS.
- I. Dalle-Donne, R. Rossi, D. Giustarini, R. Colombo and A. Milzani, Free Radical Biol. Med., 2007, 43, 883–898 CrossRef CAS PubMed.
- G. Bellomo, F. Mirabelli, D. DiMonte, P. Richelmi, H. Thor, C. Orrenius and S. Orrenius, Biochem. Pharmacol., 1987, 36, 1313–1320 CrossRef CAS PubMed.
- J. D. Hayes and L. I. McLellan, Free Radical Res., 1999, 31, 273–300 CrossRef CAS PubMed.
- N. Giangregorio, F. Palmieri and C. Indiveri, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5299–5304 CrossRef CAS PubMed.
- R. J. Mailloux and J. R. Treberg, Redox Biol., 2016, 8, 110–118 CrossRef CAS PubMed.
- W. C. Barrett, J. P. DeGnore, S. König, H. M. Fales, Y.-F. Keng, Z.-Y. Zhang, M. B. Yim and P. B. Chock, Biochemistry, 1999, 38, 6699–6705 CrossRef CAS PubMed.
- A. V. Peskin, P. E. Pace, J. B. Behring, L. N. Paton, M. Soethoudt, M. M. Bachschmid and C. C. Winterbourn, J. Biol. Chem., 2016, 291, 3053–3062 CrossRef CAS PubMed.
- J. Alegre-Cebollada, P. Kosuri, D. Giganti, E. Eckels, J. A. Rivas-Pardo, N. Hamdani, C. M. Warren, R. J. Solaro, W. A. Linke and J. M. Fernández, Cell, 2014, 156, 1235–1246 CrossRef CAS PubMed.
- E. Butturini, A. Carcereri de Prati, G. Chiavegato, A. Rigo, E. Cavalieri, E. Darra and S. Mariotto, Free Radical Biol. Med., 2013, 65, 1322–1330 CrossRef CAS PubMed.
- E. Butturini, E. Darra, G. Chiavegato, B. Cellini, F. Cozzolino, M. Monti, P. Pucci, D. Dell’Orco and S. Mariotto, ACS Chem. Biol., 2014, 9, 1885–1893 CrossRef CAS PubMed.
- S. M. Beer, E. R. Taylor, S. E. Brown, C. C. Dahm, N. J. Costa, M. J. Runswick and M. P. Murphy, J. Biol. Chem., 2004, 279, 47939–47951 CrossRef CAS PubMed.
- K. Rokutan, J. A. Thomas and R. B. Johnston, J. Immunol., 1991, 147, 260–264 CAS.
- M. Fratelli, H. Demol, M. Puype, S. Casagrande, I. Eberini, M. Salmona, V. Bonetto, M. Mengozzi, F. Duffieux, E. Miclet, A. Bachi, J. Vandekerckhove, E. Gianazza and P. Ghezzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3505–3510 CrossRef CAS PubMed.
- D. M. Sullivan, N. B. Wehr, M. M. Fergusson, R. L. Levine and T. Finkel, Biochemistry, 2000, 39, 11121–11128 CrossRef CAS PubMed.
- A. Pfefferle, R. J. Mailloux, C. N.-K. Adjeitey and M.-E. Harper, Biochim. Biophys. Acta, Mol. Cell Res., 2013, 1833, 80–89 CrossRef CAS PubMed.
- J. P. Brennan, J. I. A. Miller, W. Fuller, R. Wait, S. Begum, M. J. Dunn and P. Eaton, Mol. Cell. Proteomics, 2006, 5, 215–225 CAS.
- L. Coppo, S. J. Montano, A. C. Padilla and A. Holmgren, Anal. Biochem., 2016, 499, 24–33 CrossRef CAS PubMed.
- B.-Y. Chiang, C.-C. Chou, F.-T. Hsieh, S. Gao, J. C.-Y. Lin, S.-H. Lin, T.-C. Chen, K.-H. Khoo and C.-H. Lin, Angew. Chem., Int. Ed., 2012, 51, 5871–5875 CrossRef CAS PubMed.
- C. Lind, R. Gerdes, Y. Hamnell, I. Schuppe-Koistinen, H. B. von Löwenhielm, A. Holmgren and I. A. Cotgreave, Arch. Biochem. Biophys., 2002, 406, 229–240 CrossRef CAS PubMed.
- D. Su, M. J. Gaffrey, J. Guo, K. E. Hatchell, R. K. Chu, T. R. W. Clauss, J. T. Aldrich, S. Wu, S. Purvine, D. G. Camp, R. D. Smith, B. D. Thrall and W.-J. Qian, Free Radical Biol. Med., 2014, 67, 460–470 CrossRef CAS PubMed.
- K. T. G. Samarasinghe, D. N. P. Munkanatta Godage, G. C. VanHecke and Y.-H. Ahn, J. Am. Chem. Soc., 2014, 136, 11566–11569 CrossRef CAS PubMed.
- D. N. Kekulandara, K. T. G. Samarasinghe, D. N. P. Munkanatta Godage and Y.-H. Ahn, Org. Biomol. Chem., 2016, 14, 10886–10893 CAS.
- S. Feng, Y. Chen, F. Yang, L. Zhang, Y. Gong, G. Adilijiang, Y. Gao and H. Deng, Chem. Biol., 2015, 22, 1461–1469 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |