DOI:
10.1039/C7CS00508C
(Tutorial Review)
Chem. Soc. Rev., 2018,
47, 3831-3848
Recent advances in spirocyclization of indole derivatives
Received
4th December 2017
First published on 10th April 2018
Abstract
Spiroindolines and spiroindoles are an important class of spirocyclic compounds present in a wide range of pharmaceuticals and biologically important natural alkaloids. Various spiroindolines and spiroindoles possess versatile reactivity which enables them to act as precursors for other privileged heterocycles. In view of the importance of this scaffold, many researchers focused their efforts to develop facile and mild synthetic methods for spirocyclization of indoles. However, the synthesis of spiroindolines and spiroindoles is known to be difficult due to rapid 1,2-migration to restore aromaticity. This review aims to briefly discuss the latest developments to access highly functionalized spiroindolines and spiroindoles to stimulate further research in the field to find new and efficient methodologies for accessing new spiroindolines and spiroindoles.

Jitender Bariwal
| Jitender Bariwal received his PhD degree in Pharmaceutical Sciences from Saurashtra University, Rajkot, Gujarat, India in 2009 under the supervision of Prof. Kishor S. Jain and Prof. Anamik K. Shah. He worked as a Post Doctorate researcher with Prof. Erik Van der Eycken from 2009 to 2010. Then, he joined the ISF College of Pharmacy, Moga, Punjab, India as Associate Professor. Later in 2015, he moved to Shiva Institute of B. Pharmacy, Bilaspur, Himachal Pradesh, India as a Principal. His current research interest includes synthesis of novel heterocycles and new polymeric nanomaterials for delivery of anticancer drugs. |

Leonid G. Voskressensky
| Leonid G. Voskressensky received his PhD degree in Organic Chemistry from the Peoples Friendship University of Russia in 1999. In 2001 he joined the Prof. Cosimo Altomare (Universita Degli Studi di Bari, Italy) group as a Post-Doc Fellow (Medicinal Chemistry). In 2001 he became Assistant Professor, in 2006, Associate Professor and in 2011, Full Professor of the Organic Chemistry department of the RUDN. Since 2013, he has been the Dean of the Science Faculty of the RUDN. His group scientific interests mainly focus on domino reactions methodology, new MCR reactions as well as medicinal chemistry. |

Erik V. Van der Eycken
| Erik Van der Eycken received his PhD degree (1987) in organic chemistry from the University of Ghent, with Prof. Maurits Vandewalle. From 1988 to 1992, he worked as a scientific researcher at the R&D-laboratories of AGFA-Gevaert, Belgium and moved back to the University of Ghent in 1992. In 1997 he became a Doctoral-Assistant at the Katholieke Universiteit Leuven. He has been a visiting scientist at the University of Graz (C. Oliver Kappe), at The Scripps Research Institute (K. Barry Sharpless), and at Uppsala University (Mats Larhed, Anders Hallberg). He was appointed Professor at the Katholieke Universiteit Leuven in 2007. |
Key learning points
(1) This review will provide a good introduction to the field of spirocyclization reactions of indoles and will serve as a springboard for further reading.
(2) This review highlights the recent and significant advances in the construction of spiroindolines and spiroindoles.
(3) It highlights the recently used ligands and catalysts to achieve diastereoselective and enantioselective synthesis of spiroindolenines.
|
1. Introduction
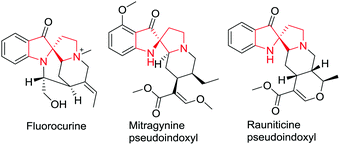
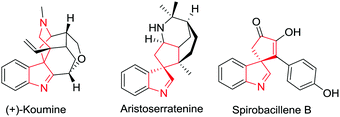
Spirocyclic compounds are unique because of their rigidity and three-dimensional geometries. Spiroindolines and spiroindoles are an important class of spirocyclic compounds present in a wide range of pharmaceuticals and biologically important natural alkaloids.1 There are two classes of spiroindoles based on the position of the spirocycle: C2-spirocyclicindolines and C3-spirocyclicindoles or C3-spirocyclicindolines. These spirocyclic compounds are rich in biological activities and are widespread in nature2 (Fig. 1 and 2). Spiroindolines and spiroindoles possess versatile reactivity which enables them to act as precursors for other privileged heterocycles including indolines, oxindoles, carbazoles, indoles and others.3
 |
| | Fig. 1 Natural alkaloids containing the C2-spirocyclicindole core structure. | |
 |
| | Fig. 2 Natural alkaloids containing the C3-spirocyclicindoline core structure. | |
The spirocyclic core structure was first reported by A. Pictet and T. Spengler in 1911 as an intermediate, which rapidly underwent 1,2-migration to restore aromaticity. The first successful isolation of a spiroindoline was achieved in 2010 by the research group of S.-L. You utilizing an Ir catalyst.4 Since then, many synthetic strategies have been successfully applied for the synthesis of spiroindolines (sometimes referred as spiroindolenines or spirocyclic 3H-indoles) which were reviewed by Unsworth and co-workers a few years back.2 They have described various synthetic strategies for the synthesis of spirocyclic indolenines mainly grouped into three categories: (1) interrupted Fischer indolisations, (2) dearomatization reactions of indoles, and (3) condensation reactions.
In recent years, spirocyclization has attracted great interest of chemists around the world, proved by hundreds of reports in this field appearing within the last two years. Looking at the biological significance and synthetic challenges for the construction of spiroindolines and spiroindoles, there is an urgent need to compile the recent developments in the field. The recently developed methodologies to access diverse spiroindolines and spiroindoles are divided into categories based on their structural complexity as detailed in Fig. 3.
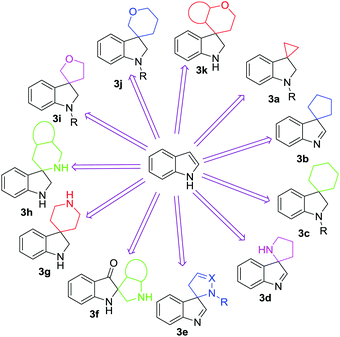 |
| | Fig. 3 Different categories of spiroindolines and spiroindoles. | |
2. Spirocyclization of indoles
Indoles may be spirocyclized via their 2- or 3-position to give variably substituted spirocyclic indolines and spiroindoles as discussed in the following sub-sections.
a. Spirocyclization via the 2-position of the indole skeleton
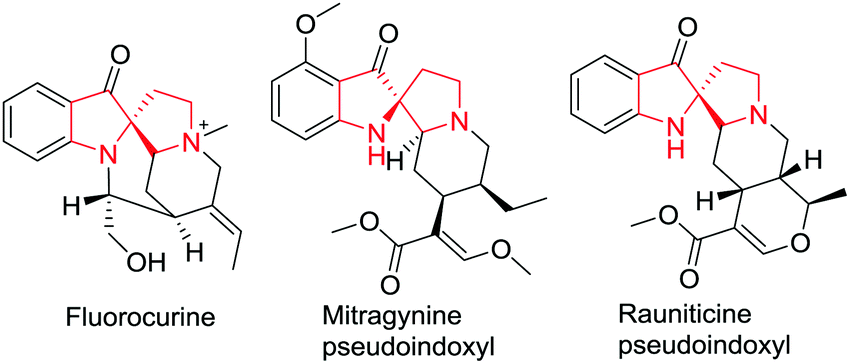
Indoles containing a C2-spirocyclic quaternary carbon center are important structural subunits found in a variety of indole alkaloids such as fluorocurine, mitragynine pseudoindoxyl and rauniticine pseudoindoxyl (Fig. 1). Despite their important biological properties, C2-spirocyclic indolines are scarcely explored as compared to C3-spirocyclic indolines and spiroindoles. This section is devoted to the latest developments in C2-spirocyclic indoline synthesis.
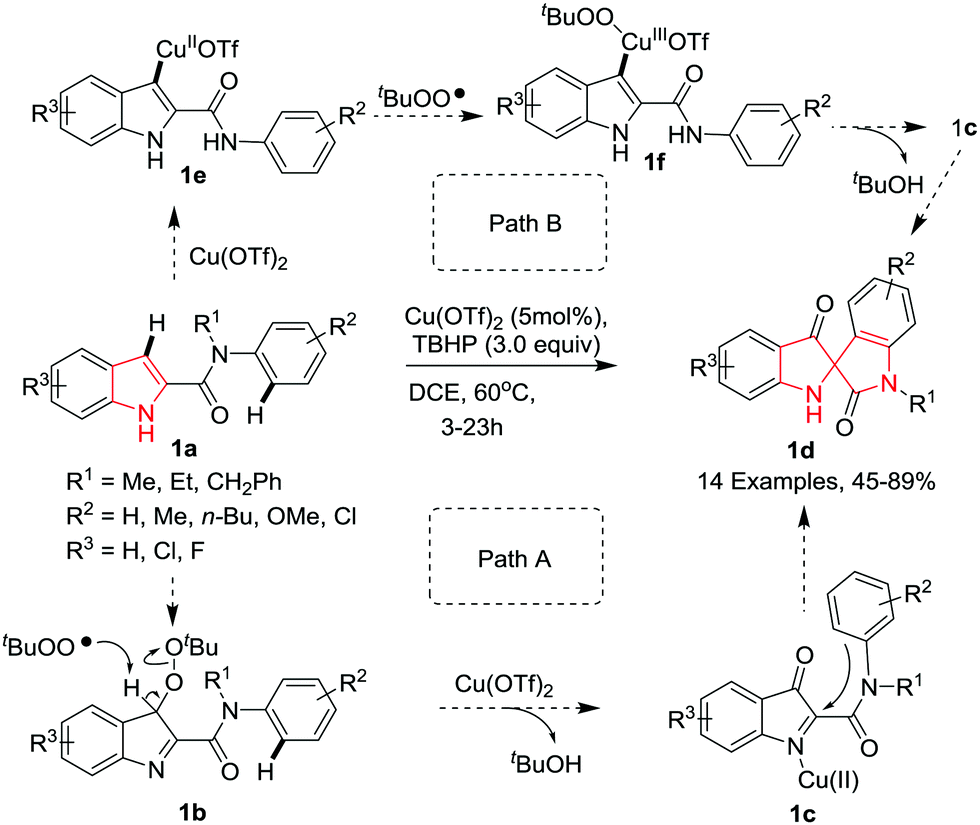
Li and co-workers5 have reported a novel copper-catalyzed oxidative dearomatization/spirocyclization of indole-2-carboxamides 1a using tert-butyl hydroperoxide (TBHP) as oxidant, giving access to C2-spiro-pseudoindoxyls 1d. The reaction proceeds smoothly under optimized conditions where Cu(OTf)2 proved to be the best catalyst among other copper salts in DCE at 60 °C. It was found that a variety of N-alkyl substituents (R1) on the amide group were compatible with the reaction delivering the desired spiro compounds. With various electron-donating groups (R2) at the 4-position of the N-phenyl substituent good yields were obtained. However, electron-withdrawing groups led to low yields due to the decreased nucleophilicity of the N-phenyl ring. Interestingly, when a 2-methyl substituted N-phenyl ring was employed, two isomeric spirocyclized products were obtained in almost equal amount, indicating that both the ortho C–H bonds on the N-phenyl ring could be competitively functionalized. Further, indole-2-carboxamide bearing a tetrahydroquinoline on the amide moiety cyclized readily under the standard reaction conditions to afford the highly strained polycyclic spiroindole. Substitution of chlorine or fluorine on the indole was well tolerated and gave access to the corresponding products in moderate yields. The reaction proceeds probably via either of two pathways where path A involves the formation of a highly reactive 3H-indol-3-one intermediate 1b followed by aromatic electrophilic substitution with the N-aryl ring of the amide moiety to produce the product 1d. Alternatively, path B involves the formation of indolylcopper(II) intermediate 1e followed by reaction with the tert-butylperoxy radical to give the Cu(III) intermediate 1f which undergoes reductive elimination followed by elimination of t-BuOH to deliver the same intermediate 1c as in path A (Scheme 1).
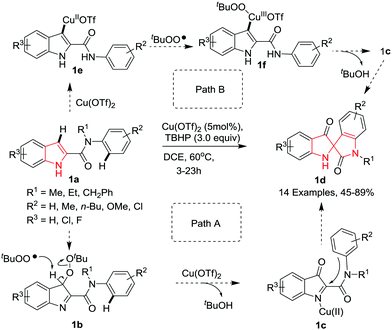 |
| | Scheme 1 Synthesis of C2-spiropseudoindoxyls 1d. | |
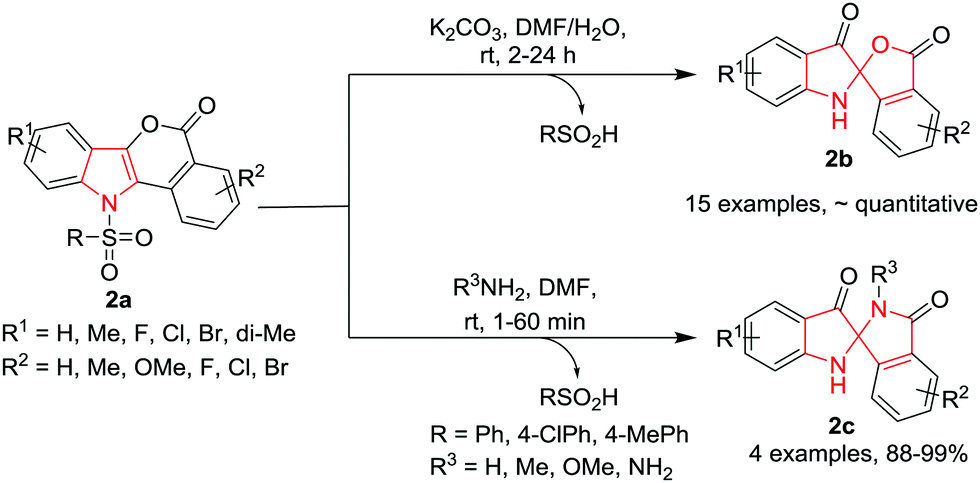
Du, Zhao and co-workers6 have developed a new strategy for the ring contraction of isochromeno[4,3-b]indol-5(11H)-ones 2avia an unusual nucleophile-induced disproportionation/spirocyclization cascade process to gain access to N-unsubstituted spiro[indoline-2,1′-isobenzofuran]-3,3′-diones 2b and spiro-[indoline-2,1′-isoindoline]-3,3′-diones 2c without using any transition-metal catalyst or oxidant. The reaction proceeds smoothly at room temperature using K2CO3 as the base in a mixture of DMF and H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the solvent. The use of strong bases such as NaOH or KOH led to a significant decrease of the yields, although the reactions were completed within short times. Diversely substituted isochromeno-[4,3-b]indol-5(11H)-ones 2a were converted into the corresponding spiro products in almost quantitative yields. It has been observed that substitution (R1 and R2) on the aromatic rings of 2a does not affect the yields of the product. However, an electron-withdrawing halogen as R2 was found to speed up the reactions whereas electron-donating groups slow them down. Further, replacement of the tosyl group with other sulfonyl groups such as 4-ClPhSO2 or PhSO2 gave access to the corresponding product 2b in quantitative yield. However, with a butanesulfonyl group a mixture of the desired product 2b was obtained in a moderate yield, along with the deprotected starting substrate. Interestingly, the scope of other nucleophiles revealed that nitrogen nucleophiles, such as NH3·H2O, MeNH2 in aqueous solution, or MeONH2, gave the corresponding N,N′-(un)substituted ketal 2c in excellent yields within a few minutes. However, sulfur nucleophiles, such as Na2S and (NH4)2S, failed to give the N,S-ketal product but furnished the corresponding deprotected isochromeno[4,3-b]indol-5(11H)-one as the sole product (Scheme 2).
:1) as the solvent. The use of strong bases such as NaOH or KOH led to a significant decrease of the yields, although the reactions were completed within short times. Diversely substituted isochromeno-[4,3-b]indol-5(11H)-ones 2a were converted into the corresponding spiro products in almost quantitative yields. It has been observed that substitution (R1 and R2) on the aromatic rings of 2a does not affect the yields of the product. However, an electron-withdrawing halogen as R2 was found to speed up the reactions whereas electron-donating groups slow them down. Further, replacement of the tosyl group with other sulfonyl groups such as 4-ClPhSO2 or PhSO2 gave access to the corresponding product 2b in quantitative yield. However, with a butanesulfonyl group a mixture of the desired product 2b was obtained in a moderate yield, along with the deprotected starting substrate. Interestingly, the scope of other nucleophiles revealed that nitrogen nucleophiles, such as NH3·H2O, MeNH2 in aqueous solution, or MeONH2, gave the corresponding N,N′-(un)substituted ketal 2c in excellent yields within a few minutes. However, sulfur nucleophiles, such as Na2S and (NH4)2S, failed to give the N,S-ketal product but furnished the corresponding deprotected isochromeno[4,3-b]indol-5(11H)-one as the sole product (Scheme 2).
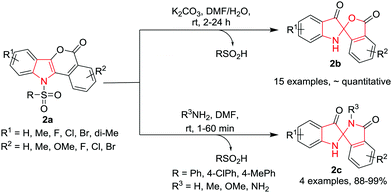 |
| | Scheme 2 Synthesis of N-unsubstituted spiro[indoline-2,1′-isobenzofuran]-3,3′-diones 2b and spiro-[indoline-2,1′-isoindoline]-3,3′-diones 2c. | |
Hu, Xu and co-workers7 have developed a novel approach for the construction of 2,2′-pyrrolidinyl-spirooxindoles 3c by employing a hydrogen-bonding network activation strategy through a catalytic asymmetric [3+2] cycloaddition reaction. The reaction proceeds well by reacting aromatic aldimines 3a with (Z)-1-acetyl-2-benzylideneindolin-3-ones (azaaruone) 3b in the presence of cinchona-thiourea based catalyst 3d in DCE at rt. Under the optimized reaction conditions, a variety of electron-donating and electron-withdrawing substituents on the phenyl ring of the 2-benzylideneindolin (R2) gave access to high yields of the product with excellent diastereoselectivity and enantioselectivity (>20:1 d.r. and 95% e.e.). Replacement of the phenyl ring with a heterocyclic system such as furan or piperonyl aldehyde resulted in good product yields but with lower enantioselectivities. Electron-withdrawing or electron-donating groups at the 5-indolin (R1) were also found to be well tolerated and provided similar results. Control experiments using pure (Z)- and (E)-azaaurone, respectively, revealed that under the optimal reaction conditions, the same product 3c was obtained with similar yields and stereoselectivities (Scheme 3).
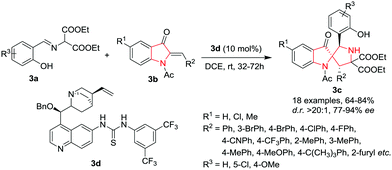 |
| | Scheme 3 Synthesis of 2,2′-pyrrolidinyl-spirooxindoles 3c. | |
b. Spirocyclization via the 3-position of the indole skeleton
The interesting biological activities possessed by many C3-spirocyclic indolines and spiroindoles have attracted the interest of medicinal chemists to extensively explore this class of compounds. This section has been subdivided as per the nature of the C3-spirocyclic ring of the indole moiety for better understanding.
1. Three membered spiro-cyclic compounds.
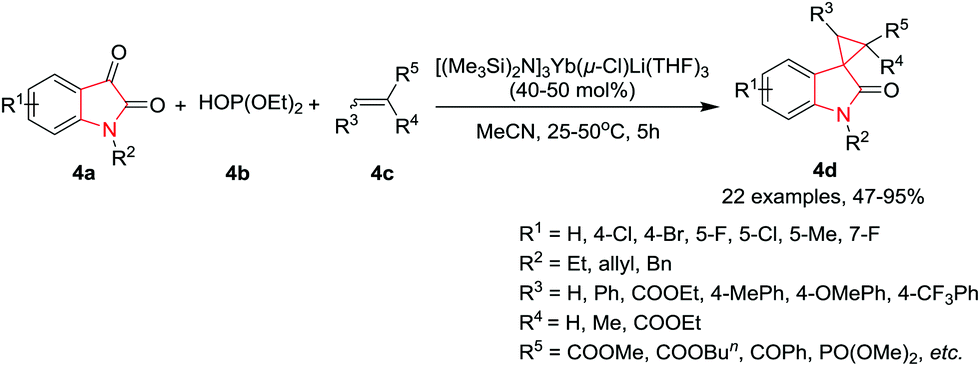
Xue, Xu and co-workers8 have developed a tandem reaction of isatin 4a, dialkyl phosphite 4b, and an activated alkene 4c for the synthesis of spiro[cyclopropan-1,3′-oxindoles] 4d in the presence of a lanthanide amide based catalyst, [(Me3Si)2N]3Yb(μ-Cl)Li(THF)3, in CH3CN at 25 °C. The reaction proceeds via a four-step mechanism involving the Pudovik reaction, a Brook rearrangement, the Michael addition, and an intramolecular nucleophilic substitution. The substituents at the 2- and 3-positions of the cyclopropane unit are obtained in a trans configuration. Under the optimum reaction conditions, activated alkenes such as acrylates gave the corresponding 1′-allylspiro[cyclopropane-1,3′-indolin]-2′-ones in moderate to excellent yields, whereas cinnamates were found to be less reactive and required an enhanced reaction temperature (50 °C) to give high product yields. Interestingly, the reaction with diethyl maleate or diethyl fumarate gave the same main product accompanied by a small amount of its stereoisomer. Among the α,β-unsaturated ketones, 4-phenylbut-3-en-2-one gave only a moderate yield, whereas the use of chalcones resulted in high yields. Chalcones with the aryl at the 3-position, bearing electron-donating groups, afforded the corresponding products in higher yields than those bearing electron-withdrawing groups. The reaction also tolerated heteroaromatics. Further, isatins substituted at the 4-, 5-, or 7-position undergo the reaction with good to high yields, whereas N-ethyl and N-benzyl substituted isatins required an increased catalyst loading of 50 mol% to afford the corresponding spiro compound in good yields (Scheme 4).
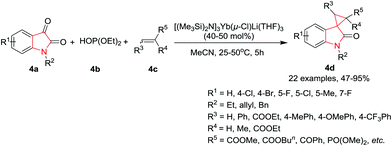 |
| | Scheme 4 Synthesis of spiro[cyclopropan-1,3′-oxindoles] 4d. | |
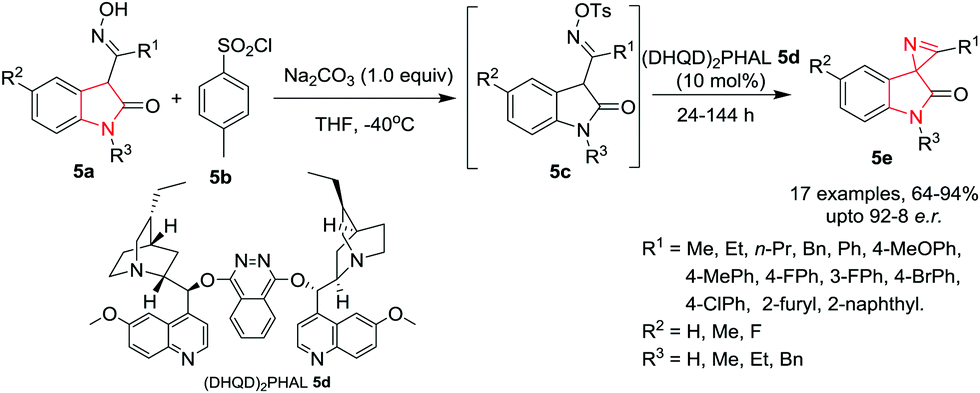
Xu, Yuan and co-workers9 have employed the Neber reaction process for the synthesis of chiral spirooxindole 2H-azirines 5e by reacting 3-O-sulfonyl ketoxime 5c which is in situ generated from isatin ketoxime 5a and sulfonyl chloride 5b. The reaction proceeds smoothly in the presence of hydroquinidine 1,4-phthalazinediyl diether [(DHQD)2PHAL] 5d as catalyst and Na2CO3 as base in THF at −40 °C to give the spiro compound in high yields and high enantiomeric ratio (e.r.) under the optimized conditions. R1 substituents like ethyl, n-propyl, benzyl or phenyl could be used without affecting the yields. Substitution on isatin (R1) with both electron-donating and electron-withdrawing substituents, bulky groups such as a naphthyl group and heteromatic groups was well tolerated. Interestingly, isatin ketoximes with an R1 alkyl substituent exhibited slightly higher reactivity than the aromatic ones. Isatin ketoximes bearing electron-withdrawing or electron-donating R2 groups at the phenyl ring of the core skeleton were compatible and provided their respective products in high yields. R3 substituents at the N1-position of isatin were well tolerated and gave access to the chiral spirooxindole 2H-azirines in high yields with moderate to good enantioselectivities. Further, it has been found that the electronic nature of the phenyl group of the sulfonyl chlorides had little impact on the reactivity. However, it significantly influences the enantioselectivity. The use of electron rich 4-methylbenzene-1-sulfonyl chloride gave high yields and enhanced enantioselectivity whereas electron deficient 2,4-dinitrobenzene-1-sulfonyl chloride afforded a relatively low yield and poor enantioselectivity. Alkyl sulfonyl chlorides were also tolerated and provided similar results as their aromatic counterparts (Scheme 5).
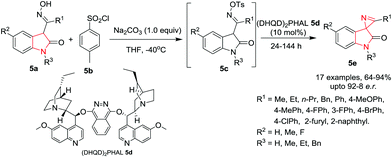 |
| | Scheme 5 Synthesis of spirooxindole 2H-azirines 5e. | |
2. Five membered carbocyclic spiro-ring containing compounds.
Van der Eycken and co-workers4b have described a high yielding Brønsted acid (TFA)-promoted protocol for the synthesis of spiroindolenines 6b from indolyl ynones 6a at rt within a few minutes. TFA promotes the dearomatization of the indole and results in the formation of the spiroindolenine. Interestingly, when the reaction was performed at higher temperature, a rearrangement results in the formation of a quinoline. (Un)substituted aromatic R1-substituents gave excellent yields compared to aliphatic ones. A trimethyl silyl substituent proved to be unstable under the acidic conditions. An electron-donating R2 substituent on the indole core resulted in moderate yields, while an electron-withdrawing group gave excellent yields. Interestingly, replacing the C-2 hydrogen of the indole (R3) with methyl provided the desired product in excellent yield but required an extended reaction time. Further, the R4-benzyl-substituted substrate resulted in the formation of an inseparable mixture of diastereomers in an excellent yield and with a d.r. = 5:4 (Scheme 6).
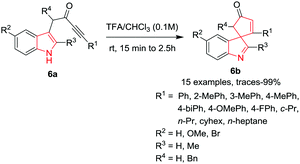 |
| | Scheme 6 Synthesis of spiro-indolenines 6b. | |
Wang and co-workers10 have developed an asymmetric Michael-intramolecular aldol-lactonization cascade of enals 7a with oxindolyl β,γ-unsaturated α-keto esters 7b in the presence of N-heterocyclic carbine (NHC) generated from triazole salt 7c as a catalyst to gain access to the β-propiolactone-fused spiro[cyclopentane-oxindoles] 7d in moderate to high yield with excellent diastereoselectivities and enantioselectivities. The reaction gave best results when 4 Å Molecular Sieves (MS) were used as additive with 10 mol% of NHC at 0 °C. Under the optimized conditions, a variety of cinnamaldehydes bearing electron-releasing and electron-withdrawing substituents performed well to give the desired product in moderate to good yields and high to excellent enantioselectivities. Various oxindolyl β,γ-unsaturated α-keto esters bearing different N-substituents such as benzyl, allyl or Me on the oxindole backbone were found to be well tolerated, affording the desired products in moderate yields and with good enantioselectivities. The steric and electronic properties of the substituents on the oxindole skeleton were found to be crucial and affected the reactivity and stereoselectivity. The substrates bearing electron-donating substituents on the oxindole gave moderate yields of the products whereas electron-withdrawing or halogen substituents gave only moderate yields but with excellent enantioselectivities (Scheme 7).
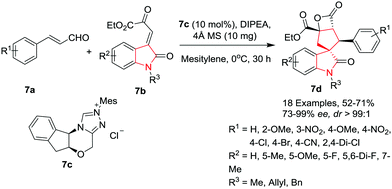 |
| | Scheme 7 Synthesis of β-propiolactone-fused spiro[cyclopentane-oxindoles] 7d. | |
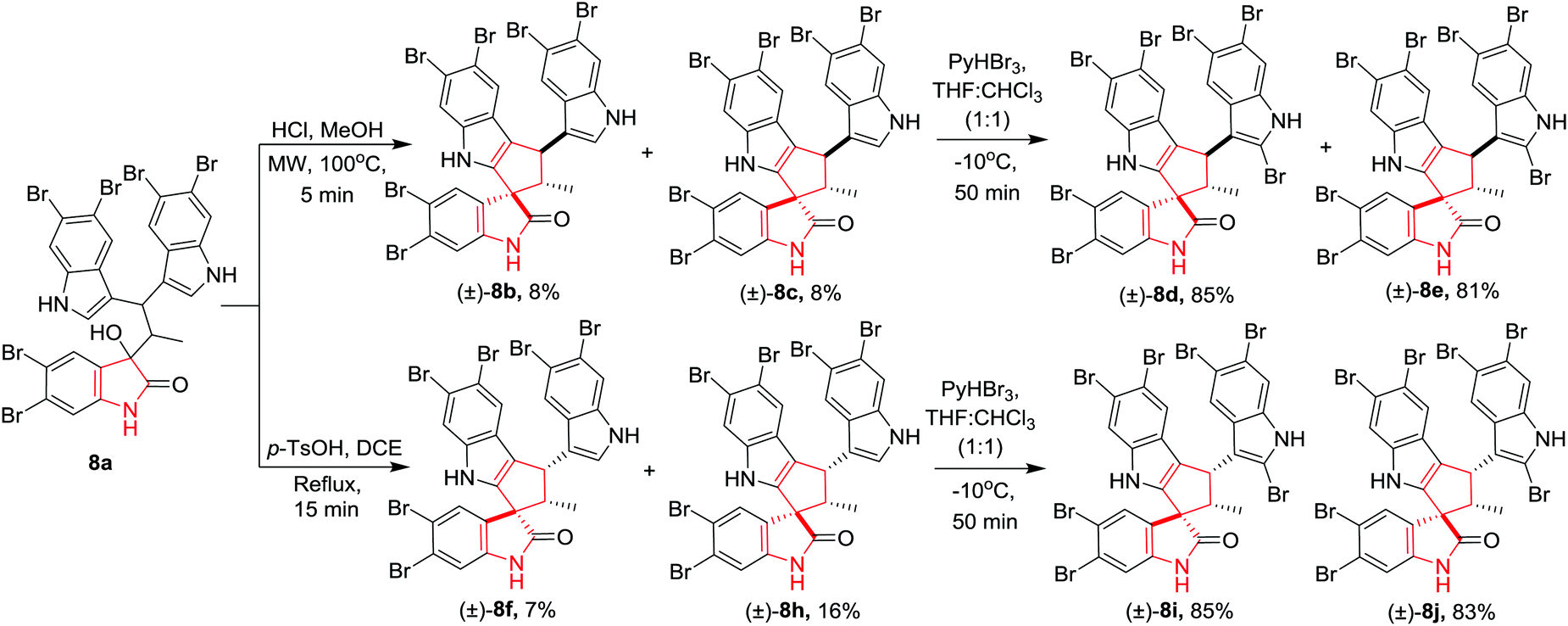
Zhang, Jia and co-workers11 have reported the first total synthesis of polybrominated spiro-trisindole alkaloids, similisines A (+)-8d and B (−)-8d along with their stereoisomers which were first isolated from Laurencia similis. The important intermediate trisindole 8a was synthesised from 5,6-dibromoindole in a few steps. The construction of key spirooxindole 8(b–c) was achieved through acid-promoted intramolecular Friedel–Crafts cyclization. A variety of Lewis acids and Brønsted acids did not afford the desired product whereas upon treatment of trisindole 8a with HCl in MeOH under MW irradiation (50 W, 100 °C, 5 min) provided two racemic isomers of (±)-8b and (±)-8c in very low yields along with an undesired trisindole derivative. Further, treatment of trisindole 8a with p-TsOH in DCE under reflux conditions gave access to racemic isomers (±)-8f and (±)-8h in low yields. Synthesis of similisines A, B (±)-8d and their stereoisomers (±)-8e–(±)-8j was achieved by bromination of (±)-8b–(±)-8h with pyridinium tribromide in THF and CHCl3 (1:1) in high yields. Compound (±)-8d is the racemic mixture of natural similisines A and B (Scheme 8).
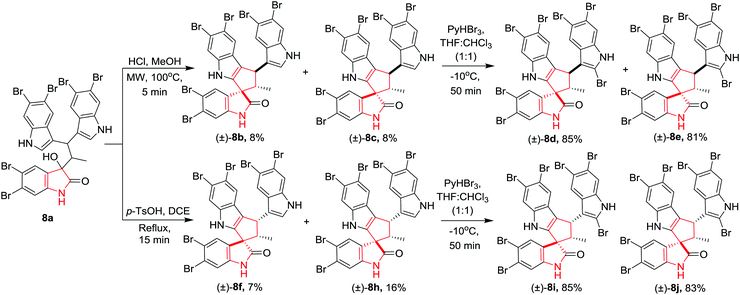 |
| | Scheme 8 Synthesis of similisines A (+)-8d and B (−)-8d along with their stereoisomers (±)-8e- to (±)-8j. | |
3. Six membered carbocyclic spiro-ring containing compounds.
You and co-workers12 have developed a Pd-catalyzed allylic dearomatization reaction of substituted indoles 9a with vinyl oxirane 9b to gain access to diverse spiroindolenines 9c in moderate yields and moderate diastereoselectivities using 1,4-bis(diphenylphosphino)butane (dppb) as ligand, MgSO4 as additive and Et3B as promoter in THF at 50 °C. Under the optimized conditions, substrates bearing an electron-donating group on the indole ring underwent the reaction smoothly, providing spiroindoles in satisfactory yields and moderate diastereoselectivities. The 6-Cl bearing substrate must be reduced from indolenine to indoline for allowing the separation of the two diastereomers (Scheme 9). Interestingly, the substrate bearing a bulky group at the C-2 position of the indole ring provided the allylic alcohol instead of the desired spiroindolenines.
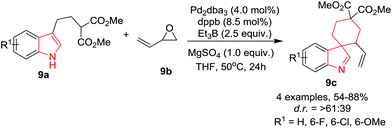 |
| | Scheme 9 Synthesis of Pd catalyzed spiroindolenines 9c. | |
Tanaka and co-workers13 have described a diastereoselective formal (4+1) cycloaddition reaction of enones 10a with cyclic 1,3-diketone 10b to construct spiro[4,5]decanes 10c bearing an oxindole moiety. A subsequent Michael–Henry cascade transformation with nitroalkenes 10d afforded the highly functionalized polycarbocyclic compounds 10f, bearing both the spiro[4,5]decane system and the spirooxindole system, with high diastereo- and enantioselectivities. Spiro[4,5]decanes 10c were converted to polycarbocyclic compounds 10f within a few hours using quinine 10e as catalyst in the presence of 4 Å molecular sieves in benzene at rt. Under the optimized conditions, various nitrostyrenes afforded the spirooxindole polycyclic products. Irrespective of the electronic nature of the R2 substituent on the nitrostyrene, all products were obtained in low yields but with high enantioselectivity. Spiro[4,5]decanes 10c bearing (un)substituted phenyl or alkyl groups (R1) underwent the reaction with low yields of the corresponding polycyclic compounds 10f. Interestingly, the enantiopurity was found to decrease as the yield of 10f increased with prolonged reaction times. Thus, isolation of 10f from the reaction mixture within 5 to 10 hours is necessary for high enantioselectivity (because of the kinetic resolution) (Scheme 10).
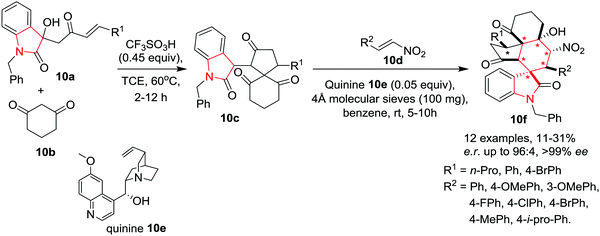 |
| | Scheme 10 Synthesis of functionalized polycarbocyclic compounds 10f. | |
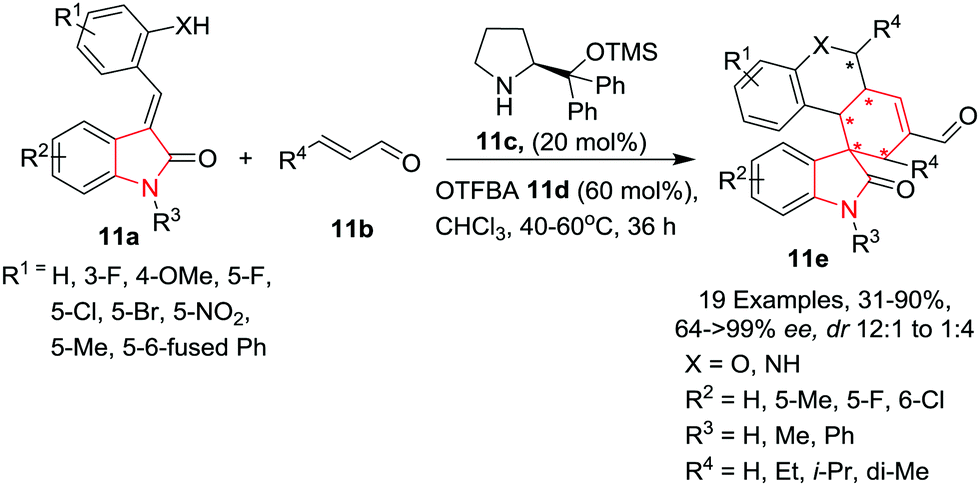
Liu, Zhao, Zhang and co-workers14 have reported the synthesis of polycyclic spiro-fused carbocyclicoxindoles 11e in moderate to high yields, excellent enantioselectivity and good control over the diastereoselectivity by reacting (E)-3-(2-hydroxybenzylidene) oxindole 11a with crotonaldehyde 11b in the presence of α,α-L-diphenylprolinol trimethylsilyl ether 11c and 2-(trifluoromethyl)benzoic acid (OTFBA) 11d in a quadruple-cascade manner at 40–60 °C in chloroform. The substitution pattern on the benzylic part of (E)-3-(2-hydroxybenzylidene) oxindole 11a has a significant influence on the outcome of the reaction as halogens and electron donating substitutions gave high yields with excellent enantioselectivity and good diastereoselectivity, whereas electron withdrawing groups such as NO2 drastically reduced the product yield and diastereoselectivity. Further, fused phenyl rings as R1 resulted in complete inversion of diastereoselectivity. Hydroxyl groups as well as amines are well tolerated on the benzyl ring. Substituents such as halogens and a small alkyl group (Me) on the indole are well tolerated and gave high to excellent yields and enantioselectivities of the products and good control over the diastereoselectivities. Substitution on the indole nitrogen drastically reduced the product yield but provided excellent control over the diastereoselectivities. Substitution on the crotonaldehyde also has a significant influence on the product yields whereas unsubstituted (R4 = H) crotonaldehyde resulted in inversion of diastereoselectivity. Some of the compounds were found to induce apoptosis in HCT116 cells through a cascade-dependent pathway by blocking the MDM2-mediated p53 degradation (Scheme 11).
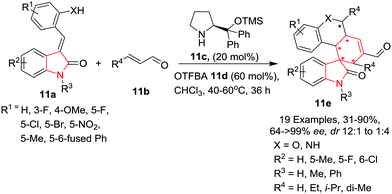 |
| | Scheme 11 Synthesis of polycyclic spiro-fused carbocyclicoxindoles 11e. | |
4. Five membered one nitrogen containing spiro-cyclic compounds.
Wang and co-workers15 have reported a catalytic asymmetric [3+2] cycloaddition reaction of 3-amino oxindole HCl-salt 12a, aldehyde 12b and α,β-enone 12c to gain access to a diverse array of spiro[pyrrolidine-2,3′-oxindoles] 12e with high levels of regio- and enantiocontrol using 3,3′-(9-phenanthryl)-modified phosphoric acid 12d as catalyst under basic conditions in Et2O at 35 °C. Under the optimal conditions, a broad range of aromatic as well as heteroaromatic aldehydes were found to be compatible with this new process, irrespective of the electronic properties of the substituents on the phenyl ring, and provided access to the cycloadducts in good to excellent yields. However, the use of aliphatic aldehydes resulted in very low regio- and enantioselectivity. Electron-donating or electron-withdrawing groups at the 5-position of the oxindole framework are well tolerated. Further, a diverse array of chalcones were found to be suitable dipolarophiles in this cycloaddition reaction. In addition, aliphatic ketones also work well in this reaction (Scheme 12).
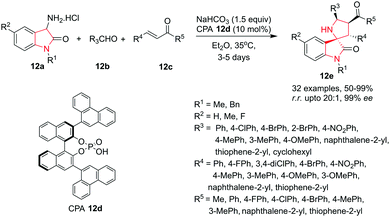 |
| | Scheme 12 Synthesis of spiro[pyrrolidine-2,3′-oxindoles] 12e. | |
Van der Eycken and co-workers16 have developed an efficient process for spirocyclization of 3-substituted indoles 13a and 13c to access 3-spiroindolenines 13b and 13d in moderate to high yield with high to excellent diastereoselectivity by employing novel supported Ag-nanoparticles in ethanol at 28 °C. Under the optimized conditions, the 5-exo-dig process for 3-substituted indoles 13a was found to be efficient and provided high yields of 3-spiroindolenines 13b. Substrates bearing terminal alkynes (R2 = H) showed good to excellent reactivity resulting in high product yields. However, internal alkynes (R2 ≠ H) cyclize slowly and required extended reaction times, high reaction temperature and higher loading of catalyst. Further, in some cases, small amounts of inseparable 6-endo-dig products were also formed. The electronic nature of the substituent at C5-(R3) did not affect the reaction output whereas substitution of chlorine at C6-position resulted in a drastic loss of product yield. By slight alteration in reaction conditions, diversified Ugi adducts 13c were cyclized to the desired spiroindolenines 13d. Various substrates bearing terminal alkynes provided high product yields along with very minor amounts of the undesired tetracyclic product. Substituents at the C5- or C6-position (R4) did not affect the yields; however, a nitro-substituent at the C4-position decreased the product yield drastically and no diastereoselectivity was observed. Further, a less sterically demanding R3-substituent, such as butyl, caused a severe drop in the yield. Furthermore, the performance of the catalyst was found to be unchanged up to 10 catalytic cycles and only a small amount of metal was leached (0.35% leaching of the original silver) during the reaction (Scheme 13).
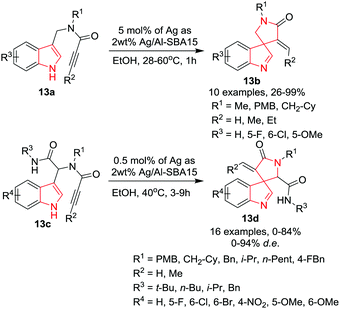 |
| | Scheme 13 Synthesis of 3-spiroindolenines 13b and 13d. | |
Wang and co-workers17 have developed a novel one-pot 1,3-dipolar cycloaddition of indolenines 14c, 3-aminooxindoles 14a, and aldehydes 14b to provide access to indolenine-substituted spiro[pyrrolidin-2,3′-oxindoles] 14d in high yields and excellent diastereoselectivities using 3 Å MS in DCM at rt. Various benzaldehydes bearing electron-donating and electron-withdrawing substituents including naphthaldehyde and heteroaromatic aldehydes were well tolerated. However, electron deficient benzaldehydes afforded moderately lowered yields and exhibited lower reactivity than the neutral or electron-rich derivatives, whereas no difference in diastereoselectivities was observed. Interestingly, ortho substitution on the benzaldehydes decreased diastereoselectivity compared to meta- and para-substitution. Substitutions on the benzene ring of 3-aminooxindole were well tolerated and provided good product yields with excellent diastereoselectivities. Indolenines with electron-withdrawing or electron-donating groups at the C5-position reacted smoothly and afforded desirable products with excellent yields and diastereoselectivities, while a nitro group reduced the activity. To further induce diversification at the C3 and C5 positions of the pyrrolidine ring, preformed 2-alkenylindolenines 14e were employed in the cycloaddition reaction. However, diphenyl phosphate (DPP) must be used to enhance the efficiency and diastereoselectivity of the products. Under these modified conditions, various 2-alkenylindolenines underwent the [3+2] cycloaddition reaction smoothly and gave indolenine substituted spiro[pyrrolidin-2,3′-oxindole] 14f in excellent yields and diastereoselectivities. Interestingly, the use of NaHCO3 as base and DPP as catalyst resulted in switching of the diastereomeric ratio. Under these new conditions, diversely substituted 2-alkenylindolenines underwent the reaction smoothly to give epimer 14h as the major product with high reactivity in high yields and with moderate to good diastereoselectivity. The switch of diastereoselectivity of the product by using NaHCO3 and DPP is still unclear (Scheme 14).
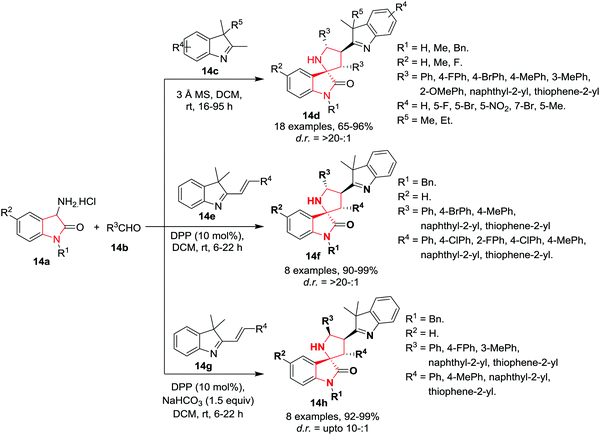 |
| | Scheme 14 Synthesis of indolenine-substituted spiro[pyrrolidin-2,3′-oxindoles] 14d, 14f and 14h. | |
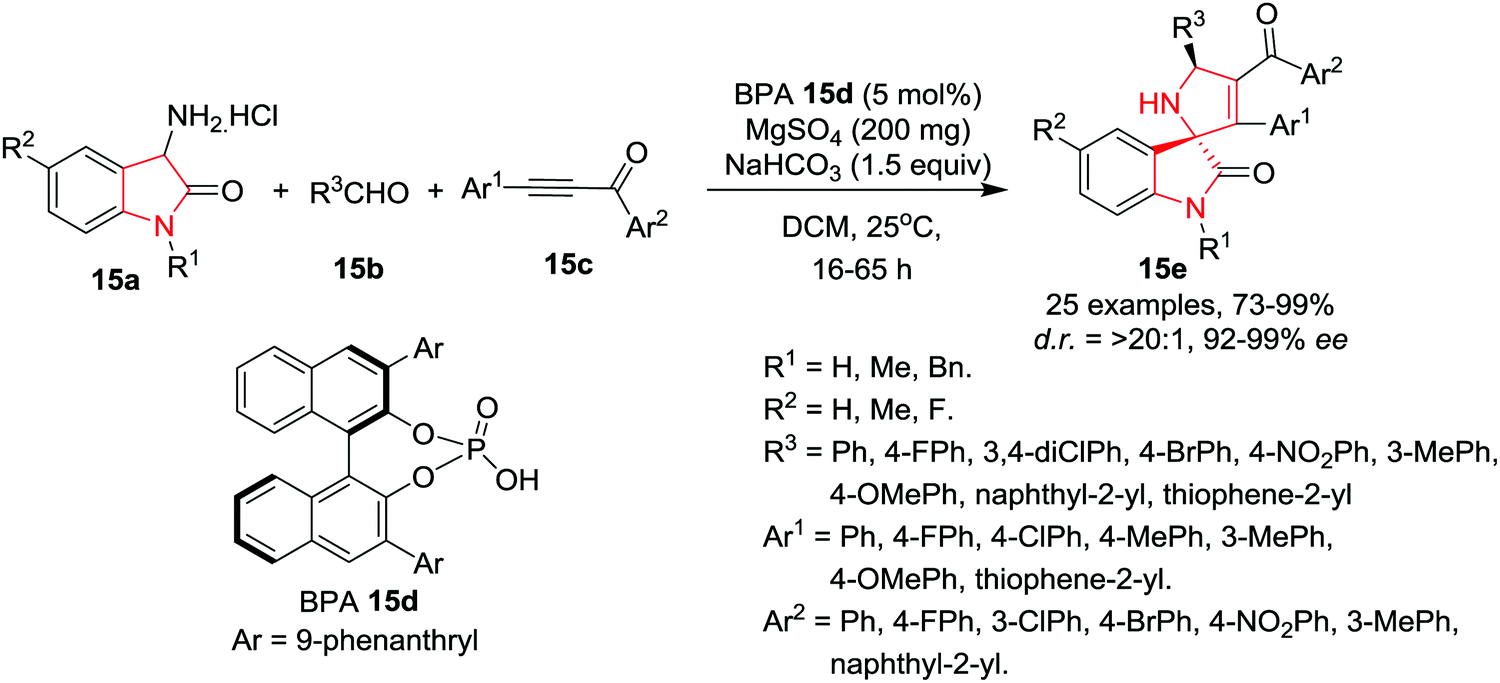
Wang and co-workers18 have developed an organocatalytic asymmetric 1,3-dipolar [3+2] cycloaddition reaction of 3-amino oxindole-based azomethine ylides (obtained by the reaction of 3-amino oxindole 15a and aldehydes 15b) with α,β-ynones 15c in the presence of chiral 3,3′-(9-phenanthryl)binaphthol-phosphoric acid (BPA) 15d as catalyst, MgSO4 as a dehydrating agent and NaHCO3 as base in DCM at rt to afford spiro[dihydropyrrole-2,3′-oxindoles] 15e in high yields and with excellent enantio- and diastereoselectivities. A diverse array of aromatic aldehydes bearing electron-neutral, electron-deficient, or electron-rich groups were found to be suitable in this reaction and afforded the corresponding products in high yields with excellent enantioselectivities and diastereoselectivities. Additionally, heteroaromatic and naphthaldehydes were also found to be suitable and afforded excellent product yields. Substitution at C-5 of the 3-amino oxindole framework is well tolerated. A diverse array of ynones were found to be suitable as dipolarophiles for this cycloaddition reaction as electronically diverse substituents on aromatic rings or heteroaromatic groups at the alkynyl terminus are well amenable to the cycloaddition reaction, delivering enantiopure cycloadducts in excellent yields. Similarly, with respect to the ketone terminus, a variety of aromatic groups are well tolerated. The scaled up reaction of cycloadducts maintained the efficiency and stereoselectivity (Scheme 15).
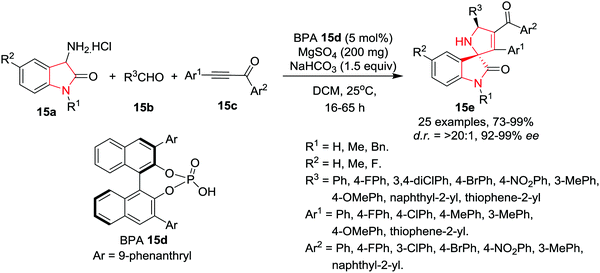 |
| | Scheme 15 Synthesis of spiro[dihydropyrrole-2,3′-oxindoles] 15e. | |
Zhao, Shang and co-workers19 have developed a highly stereoselective 1,3-dipolar cycloaddition reaction of imino esters 16a and methyleneindolinones 16b using a chiral thiourea based quaternary ammonium salt 16c as phase-transfer catalyst at rt in Et2O using K2CO3 as base to provide facile access to chiral spiro[pyrrolin-3,3′-oxindoles] 16d in high yields and with excellent enantio- and diastereoselectivities. Among the imino esters bearing different ester groups, the t-butyl imino ester provided the product with the highest enantioselectivity compared to other esters (i.e. Et, Bn). Further, a wide array of t-butyl imino esters originating from heterocyclic or aromatic aldehydes bearing electron-donating or electron-withdrawing substituents on the aromatic ring provided the spiroindoles with excellent enantioselectivities and good to excellent diastereoselectivities. Interestingly, N-methyl or benzyl protected indolinone provided excellent results whereas N-Boc-protection reduced the enantioselectivities and diastereoselectivities. Further, methyleneindolinones derived from aromatic and aliphatic aldehydes underwent the reaction smoothly and provided excellent product yields. However, reactions with propyl- and phenethyl-substituted methyleneindolinones afforded the products with comparatively lower diastereoselectivities. The indolinone moiety bearing various substituents at the C5 or C6 position were well tolerated and provided excellent results (Scheme 16).
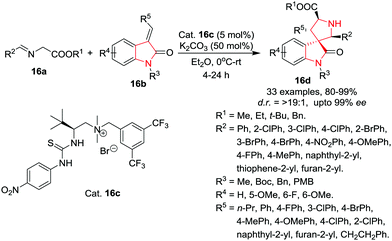 |
| | Scheme 16 Synthesis of chiral spiro[pyrrolin-3,3′-oxindoles] 16d. | |
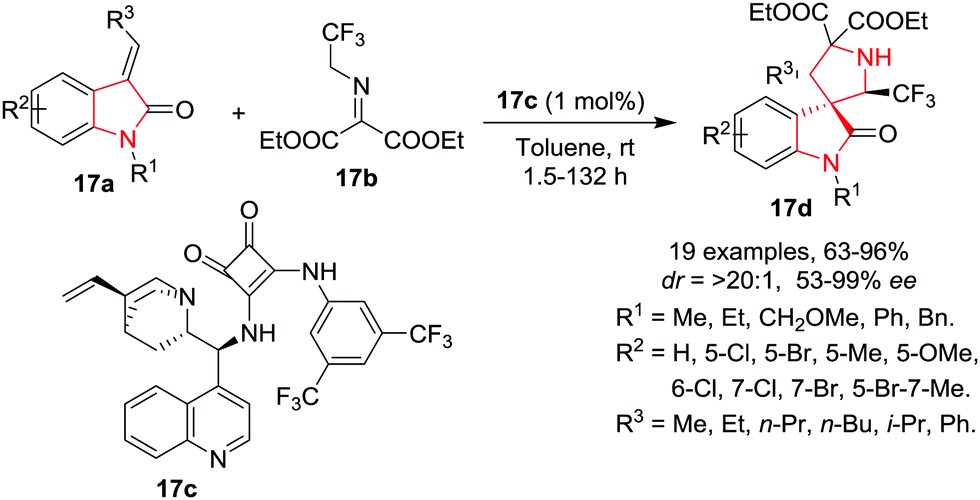
Recently, Yan, K. Wang, R. Wang and co-workers20 have reported a facile 1,3-dipolar cycloaddition reaction of 3-alkenyloxindoles 17a with CF3-containing ketimines 17b in the presence of Cinchona alkaloids-derived thiourea catalyst 17c to access 2′-CF3-spiro-pyrrolidine-3,3′-oxindoles 17d with excellent yields, enantioselectivities and diastereoselectivities using toluene as reaction medium at rt. With the optimized conditions, various 3-alkenyl-oxindoles as dipolarophiles were found to be suitable in this reaction as various substituents such as ethyl, CH2OCH3 or benzyl groups at the N-1 position underwent the reaction smoothly, and gave the cycloadducts with excellent enantioselectivities and diastereoselectivities, although in lower yields and with longer reaction times. Meanwhile, a phenyl group at the N-1 position of the 3-alkenyloxindole resulted in high yield, excellent enantioselectivity but moderate diastereoselectivity. Further, various electron-withdrawing and electron-donating groups at the aromatic ring of the 3-alkenyloxindole were well tolerated, and provided the corresponding adducts in good yields, excellent enantioselectivities as well as diastereoselectivities. Dipolarophiles bearing an electron-withdrawing substituent at the 5-position of 3-alkenyloxindole have higher reactivity. Substituents on the alkenyl moiety played an important role in the reaction as an increase in the length of the substituent from methyl to ethyl, n-propyl, n-butyl or isopropyl resulted in a slower reaction rate, even with increased catalytic loading. Despite the slow reaction rate, the cycloadducts were obtained in high yields with excellent enantioselectivities and diastereoselectivities. Further, the phenyl substituted alkenyl substrate resulted in low product yield and poor enantioselectivity even after prolonged reaction time (Scheme 17).
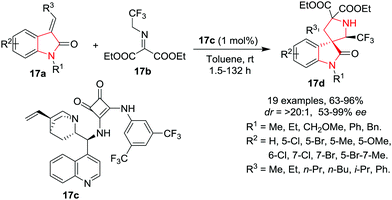 |
| | Scheme 17 Synthesis of 2′-CF3-spiro-pyrrolidine-3,3′-oxindoles 17d. | |
5. Five/six membered spiro-ring with more than one heteroatom.
Chen and co-workers21 have developed a formal [4+1] annulation reaction of 3-bromooxindoles 18a and 1,2-diaza-1,3-dienes 18b to gain access to spiropyrazoline oxindoles 18d in high yields, using Cs2CO3 as base in CH2Cl2 at rt within a few hours. A one-pot, three-component variant of the same reaction also works smoothly to give the desired products in comparable yields. In this reaction, variation of the electronic properties at the 5-position of the phenyl ring of the 3-bromoxindoles has no effect on the reaction efficiency. However, electron-withdrawing groups such as a bromo or trifluoromethyl substituent at the 6 or 7-position of the phenyl ring furnished moderate yields of the products. Further, different protecting groups on the nitrogen atom of 3-bromooxindoles, such as benzyl, phenyl, and 4-methoxylphenyl, are well tolerated in the reaction resulting in good yields. Also the scaling-up of this procedure was elaborated. This reaction is believed to progress through intermediate 18c, which further undergoes an intramolecular nucleophilic SN2-substitution to form the final formal [4+1] annulation product (Scheme 18).
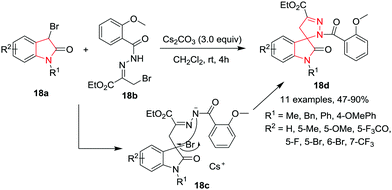 |
| | Scheme 18 Synthesis of spiropyrazoline oxindoles 18d. | |
Nath and co-workers22 have reported an efficient methodology for the synthesis of spiro[indoline-3,2′-thiazolidinones] 19e in good to excellent yields by the sequential reaction of primary amines 19a with various isatins 19b and then the resultant Schiff base 19c was reacted with thioglycolic acid 19d in the presence of p-dodecylbenzenesulfonic acid (DBSA) as Brønsted acid catalyst in aqueous medium at 25 °C. Under the optimized conditions, various aryl amines bearing an electron-donating substituent at the 4-position of the phenyl ring underwent the reaction smoothly and produced the desired spiro-indoles in high yields, whereas substrates with electron-withdrawing substituents were found to be sluggish and afforded the desired product in slightly less yields. Interestingly, the reactions of benzylamine with various isatins and thioglycolic acid proceeded with a faster rate and afforded the desired products in higher yields. Additionally, substituents on the 5-position of isatin or the N-1 nitrogen have a minimal effect on the reaction output and provide high product yields. This synthetic protocol has the advantages of operational simplicity, energy-efficiency, and the use of green solvent to access the desired spiro[indoline-3,2′-thiazolidinones] in high yields (Scheme 19).
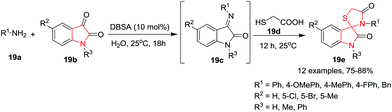 |
| | Scheme 19 Synthesis of spiro[indoline-3,2′-thiazolidinones] 19e. | |
Jeong and co-workers23 have developed a rapid synthetic procedure for spiro[chromeno[4′,3′:4,5]pyrimido[1,2-b]indazole-7,3′-indoline]-2′,6(9H)-diones 20d by one-pot condensation reaction of 4-hydroxy-2H-chromen-2-one 20a, isatin 20b and 1H-indazole-3-amine 20c in the presence of acetic acid in EtOH under refluxing conditions for 2 h. Under the optimized reaction conditions, a variety of substituents on isatin and on 1H-indazole-3-amine, including electron-withdrawing or electron-donating groups, were acceptable and gave high product yields. This protocol has general applicability and can accommodate a variety of substitution patterns. This method has the advantages of operational simplicity and high product yield via a simple work-up procedure as compared to the conventional methods (Scheme 20).
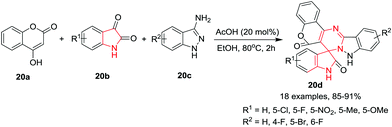 |
| | Scheme 20 Synthesis of spiro[chromeno[4′,3′:4,5]pyrimido[1,2-b]indazole-7,3′-indoline]-2′,6(9H)-diones 20d. | |
6. Six/seven membered nitrogen containing spiro-cyclic compounds.
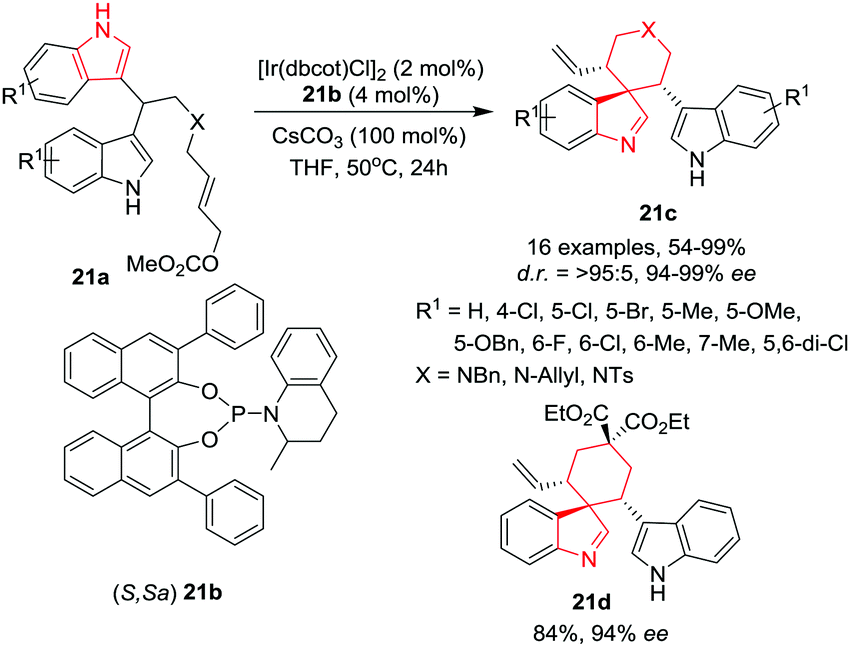
Zheng, You and co-workers24 have developed a highly efficient desymmetrizing allylic dearomatization reaction of bis(indol-3-yl)allylic carbonates 21a to gain access to spiroindolenines 21c in high yields and excellent diastereo- and enantioselectivities utilizing an Ir-based catalyst and phosphoramidite ligand 21b under basic conditions in THF at 50 °C. Under the optimal conditions, substrates bearing electron-donating or electron-withdrawing groups on the nitrogen atom of the linkage underwent the reaction smoothly providing good to excellent product yields and excellent enantioselectivities. Additionally, substrates with different linkages revealed that allylic carbonates linked with a C(CO2Me)2 group underwent the reaction smoothly and provided access to compound 21d whereas substrates with an ester or amide tether were not reactive. A wide range of electron-donating and electron-withdrawing substituents on the indole ring were well tolerated. However, the 5,6-di-chloro bearing indole provided low yield and poor diastereoselectivity. Interestingly, the substrate bearing a one methylene elongated linkage (X = –CH2N(Bn)) did not afford the seven-membered spiroindolenine. Additionally, when an unsymmetrical allylic carbonate was used, both indole rings were involved in the reaction, providing a pair of inseparable constitutional isomers (≈1:1) in a high combined yield and enantioselectivities. The reaction was found to be robust and can be performed on a gram scale, providing high yields with excellent enantioselectivities (Scheme 21).
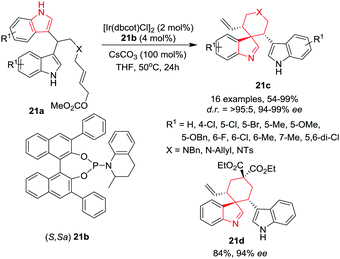 |
| | Scheme 21 Ir-Catalyzed synthesis of chiral spiroindolenines 21c. | |
Voituriez, Guinchard and co-workers25 have reported a gold-catalyzed protocol to cyclize 2-bromo-N-propargyltryptamines 22a to gain access to spiro[indoline-3,4′-piperidin]-2-ones 22b in excellent yields by utilizing Gagosz catalyst (Ph3PAuNTf2) in toluene at 50 °C. 2-Bromotryptamines gave superior yields compared to their chlorinated counterparts. Similarly, the nitrobenzenesulfonyl (Ns) protected amines proved superior to tosyl-protected analogs. The reaction of indoles bearing electron-donating or electron-withdrawing groups afforded good product yields. Further, the use of L-tryptophan esters in this cyclization afforded the corresponding spirocyclic compounds in excellent yields and diastereoselectivities. Additionally, the scope of this cyclization was extended by installing an allene function on the 2-bromotryptamine to give tryptamines 22c which underwent smoothly a gold-catalyzed cyclization/hydrolysis sequence to furnish the spirocyclic 22d in good yields as a mixture of diastereomers (1:1). Interestingly, replacement of the 2-propanediyl unit by a cyclohexanediyl unit on the allene function increased the diastereomeric ratio to 78/22 (Scheme 22).
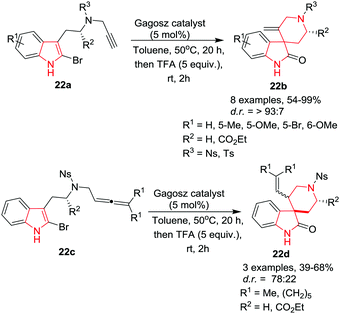 |
| | Scheme 22 Synthesis of spiro[indoline-3,4′-piperidin]-2-ones 22b and 22d. | |
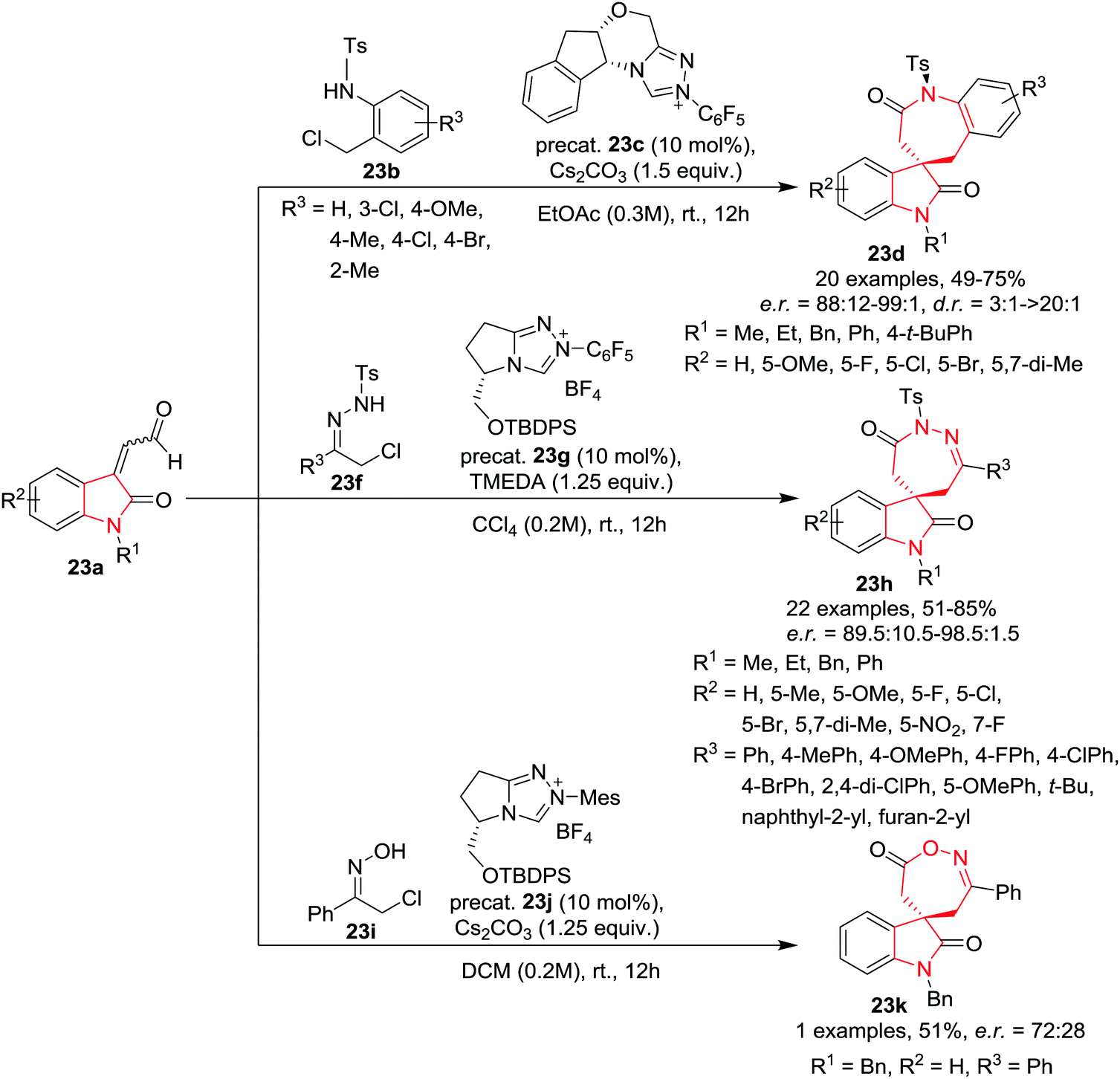
Enders and co-workers26 have developed an asymmetric, N-heterocyclic carbene (NHC) catalyzed formal [3+4] cycloaddition reaction of isatin-derived enals 23a (3C component) with in situ formed aza-o-quinone methides and azoalkenes or nitrosoalkenes (4 atom components) to synthesize spirobenzazepinones 23d, spiro-1,2-diazepinones 23h and spiro-1,2-oxazepinones 23k in good yield and with very good to excellent enantioselectivities by slight alteration in the reaction conditions. The atropo- and enantioselective synthesis of spiro-benzazepinones 23d was achieved by the reaction of isatin derived enals 23a with N-(ortho-chloromethyl)aryl amides 23b using a chiral triazolium salt based precatalyst 23c and Cs2CO3 as base in EtOAc at rt. Various electron-withdrawing and electron-donating substituents on the benzene ring of the enal provided the corresponding spirobenzazepinones in good yields, high enantiomeric ratio and with good diastereoselectivities. Variation of the N-substituents of the enal resulted in good to excellent enantioselectivities of the product. A wide range of electron-rich and electron-deficient substituents on the N-(ortho-chloromethyl)aryl amides were found to be compatible to afford the respective spiroheterocycles in good yields, high enantiomeric ratios but with moderate diastereoselectivities. The use of 6-Cl-substituted amides resulted in reduced enantioselectivity. Interestingly, N-(ortho-chloromethyl)aryl amides bearing a methyl group at the ortho-position afforded excellent diastereoselectivities probably due to the fact that substituents at this position of the benzene ring of seven-membered benzolactams increase the conformational barrier of the corresponding atropisomers. This methodology was extended to achieve the synthesis of spiro-1,2-diazepinones 23h by reacting α-halogenohydrazones 23f with isatin-derived enals 23a using chiral triazolium salt based precatalyst 23g as precatalyst and TMEDA as base in CCl4. A broad range of enal substrates bearing electron-neutral, -releasing, and -withdrawing substituents on the aromatic ring provided the corresponding spiro-1,2-diazepinone products with excellent enantioselectivities and moderate to very good yields. Enal substrates with varied N-substituents when employed in this annulation procedure provided excellent yields and high enantioselectivities. Various aromatic, aliphatic and heterocyclic α-chloro N-tosyl hydrazones were also found to be suitable as azoalkene precursors, yielding the desired spiro-1,2-diazepinones with excellent yields and high enantioselectivities in most cases. Additionally, polycyclic spiro-1,2-diazepinones were obtained in good yields and excellent diastereo- and enantioselectivities utilizing the standard NHC catalysis conditions by using cyclic hydrazones. Interestingly, seven-membered hydrazine under the standard catalytic conditions provided the corresponding polycyclic spiro-1,2-diazepinone with two fused seven-membered rings in good yield and with an enantiomeric ratio of 91.5:8.5. Further, spiro-1,2-oxazepinones 23k were obtained with good yields and moderate enantiomeric ratio utilizing a similar NHC strategy where α-chloro oximes 23i were used as substrates and utilizing precatalyst 23j and Cs2CO3 as base in DCM (Scheme 23).
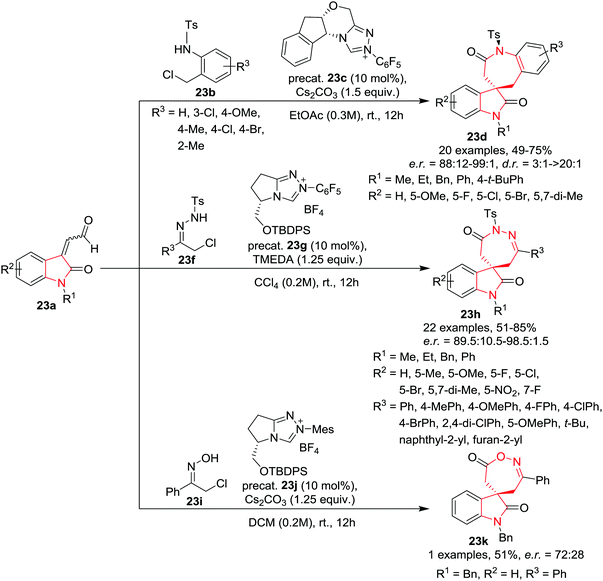 |
| | Scheme 23 Synthesis of spirobenzazepinones 23d, spiro-1,2-diazepinones 23h and spiro-1,2-oxazepinones 23k. | |
7. Fused nitrogen-heterocycle containing spiro-cyclic compounds.
Boitsov, Stepakov and co-workers27 have developed a highly efficient stereoselective synthesis of a novel class of substituted 3-spiro[cyclopropa[a]pyrrolizine] 24d, 24e and 3-spiro[3-azabicyclo[3.1.0]hexane]-oxindoles 24g, 24i and 24kvia a one-pot three-component 1,3-dipolar cycloaddition reaction of cyclopropenes 24a with azomethine ylides generated from isatins 24b and α-amino acids 24c and with azomethine ylides generated from isatins and benzylamine 24h, dipeptide 24f or Gly–Gly 24j under refluxing conditions in MeOH alone or in a mixture with water or with benzene. A variety of cyclopropenes were found to be suitable for this reaction to gain access to the corresponding spirocyclic-3,3′-oxindoles 24d, 24e in moderate to high yields and with moderate to excellent stereoselectivities. However, cyclopropenes bearing methyl substituents at the double bond or tetra-substituted cyclopropene did not undergo this reaction. Further, a number of primary α-amino acids 24f or dipeptides 24j were also found to be suitable substrates for this reaction by slightly modifying the solvent to a mixture of MeOH/EtOH and water (3:1) to gain access to the 3-spiro[3-azabicyclo[3.1.0]hexane]oxindoles 24g, 24k in good to moderate yields and with high diastereoselectivity. Surprisingly, glycine and DL-alanine were found to be unsuitable for this reaction and produced only trace amounts of the desired products along with an unidentified mixture of compounds. Additionally, the application of benzylamine 24h as an amine component in a modified solvent mixture of MeOH and benzene (3:1) under reflux conditions provided the spirooxindoles 24i in excellent yields irrespective of the substitution pattern of the reactants (Scheme 24).
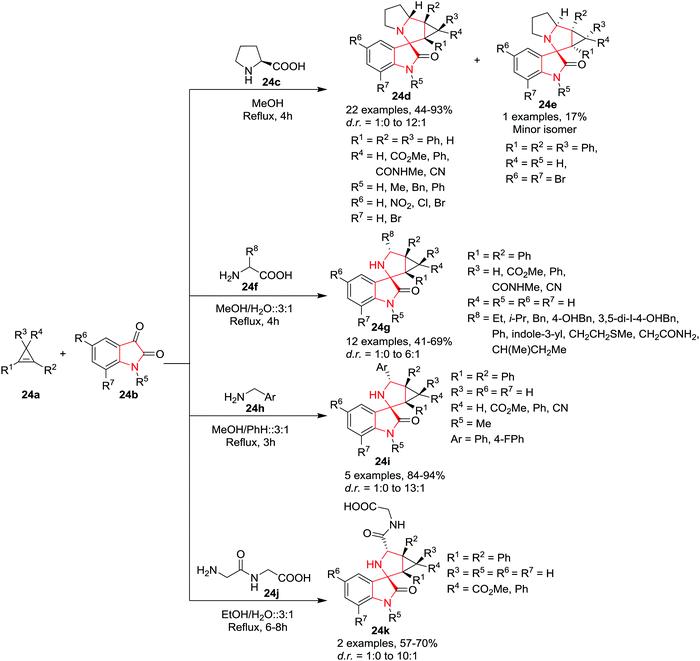 |
| | Scheme 24 Synthesis of 3-spiro[cyclopropa[α]pyrrolizine] 24d, 24e and 3-spiro[3-azabicyclo[3.1.0]hexane]-oxindoles 24g, 24i and 24k. | |
Wang and co-workers28 have developed a cascade asymmetric Friedel–Crafts alkylation/N-hemiketalization/Friedel–Crafts alkylation reaction of 3-alkylindoles 25a and oxindolyl β,γ-unsaturated α-ketoesters 25b to give spiro-polycyclic indoles 25d in high yields with excellent enantioselectivities and diastereoselectivities. This cascade sequence is catalyzed by a combination of Pd(PPh3)2Cl2 and AgSbF6 using (R)-DM-SegPhos 25c as ligand in PhCF3. Under the optimized conditions, the various N-(un)protected oxindolyl β,γ-unsaturated α-ketoesters 25b underwent the reaction smoothly and provided the desired products in very good yields and with excellent enantioselectivities. The oxindolyl ring bearing either electron-donating or electron-withdrawing groups at the 5-position are well tolerated giving the desired products in excellent yields and enantioselectivities with >20:1 d.r. However, substrates bearing a methyl group at the 5-position of the oxindolyl ring gave the desired products in comparatively low yield and low enantioselectivity. Various substituents on the 3-substituted indoles at the 5- or 6-position are well tolerated under the reaction conditions. Interestingly, a 4-Br substituent gave a low product yield with moderate enantioselectivity. At the 3-position, small or large alkyl chains are well tolerated whereas sterically hindered groups such as isopropyl were found to be unsuitable in this reaction and provided very poor yields (Scheme 25).
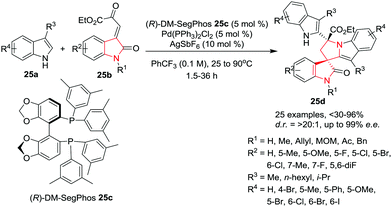 |
| | Scheme 25 Synthesis of spiro-polycyclic indoles 25d. | |
Van der Eycken and co-workers29 have reported an efficient reaction which converted the Ugi-adduct 26a to the corresponding polycyclic spirocyclic indoles 26b by using a catalytic system of Au(PPh3)SbF6 in CDCl3 at rt in a short time. Under these conditions, various substituents on the alkyne, isonitrile, indole, and amine groups were tolerated and provided good product yields. However, bulky substituents on the alkyne yielded low product yields. Noticeably, methyl substitution at the 2-position of the indole core completely inhibited the cyclization. Further, the tosyl group on the indole nitrogen was found to be unfavorable for cyclization (Scheme 26).
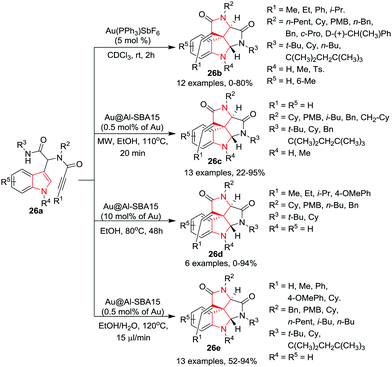 |
| | Scheme 26 Synthesis of polycyclic spirocyclic indoles 26b–26e. | |
Further, Van der Eycken and co-workers30 have applied a MW-assisted protocol to the Ugi-adduct 26a to access the corresponding polycyclic spirocyclic indoles 26c in good to excellent yields using Au nanoparticles supported on Al-SBA15. For substrates bearing terminal alkynes, the reaction proceeds smoothly and provides good to excellent product yields within 20 min in ethanol at 110 °C. However, bulky substituents at the amide nitrogen (R3) provided poor product yields which is attributed to the interference in the trapping of the iminium moiety by the amide in the spirointermediate. Interestingly, these reaction conditions were found to be unsuitable for substrates bearing substituents on the alkynes, as even increased temperature and longer reaction times fail to give satisfactory product yields. However, under conventional heating these substrates were spirocyclized to give polycyclic indoles 26d. Noticeably, with the increase of the bulkiness of the alkyne substituent, the product yield dropped sharply. For example, changing the substituent from a methyl-group to an ethyl-group resulted in a drop of yield by 76% whereas the change with a t-butyl group resulted in complete failure of the reaction (Scheme 26).
Further, Noël, Van der Eycken and co-workers31 have developed an efficient protocol for the cycloisomerization of Ugi-adduct 26a using a heterogeneous gold based catalyst Au@Al-SBA15 (0.5 mol% of Au as 2.0 wt% Au@Al-SBA15) in a mixture of EtOH and water to yield polycyclic spirocyclic indoles 26e under microflow conditions (15 μL min−1) in good to excellent yields. Under the optimized conditions (120 °C, Res. time 5.5 min), several aliphatic, aromatic or unsubstituted alkynes underwent the spiro-cyclization reaction to give good to excellent product yields. Similarly, various aliphatic substitutions at the R2 position were tolerated and provided the product in good yields. Interestingly, bulky aliphatic groups at the R3 position gave relatively low yields compared to other aliphatic groups. Noticeably, this reaction did not show any spiro-cyclization conversion under conventional batch conditions probably because of the very low catalyst/reactant ratio as compared to the packed bed reactor. In addition, leaching of the gold particles form the Au@Al-SBA15 catalytic bed was not observed. However, under continuous-flow conditions the activity of the catalyst usually decreased after eight injections of each 50 μmol (Scheme 26).
Zheng, You and co-workers32 have recently reported a highly efficient synthesis of enantio-enriched spiroindolines 27dvia a sequential isomerization/spirocyclization/transfer hydrogenation of indolyl dihydropyridine 27a by employing a chiral phosphoric acid 27b as a catalyst, Hantzsch ester 27c as a hydrogen transfer agent and 3 Å molecular sieves as an additive in EtOAc at 40 °C. Under the optimized conditions, electron-donating groups or halogens on the phenyl ring of the indole ring provided good to high yields of spiroindolines with excellent enantioselectivities. However, substrates with an electron-withdrawing group such as methoxycarbonyl did not undergo the reaction. The 1,4-dihydropyridines bearing various substituents such as aliphatic, cyclic, aryl, or heteroaryl ketone underwent the reaction smoothly to provide spiroindolines in good yields and with excellent enantioselectivities. Electron-withdrawing substituents on the C3′-position of the 1,4-dihydropyridine such as ester, cyano, and sulfonyl or a bulky group such as naphthalene underwent the reaction smoothly, although providing moderate yields but with good to excellent enantioselectivities. Notably, all the spiroindolines were obtained as single diastereoisomers, except the substrate bearing 5-chloro or 5-methyl as a substituent on the indole ring. The reaction proceeds through the enamine isomerization of indolyl dihydropyridine 27a to generate iminium intermediate 27e followed by cyclization at the C3 position of the indole ring, leading to intermediate 27f, followed by transfer hydrogenation of intermediate 27f to spiroindole 27d (Scheme 27).
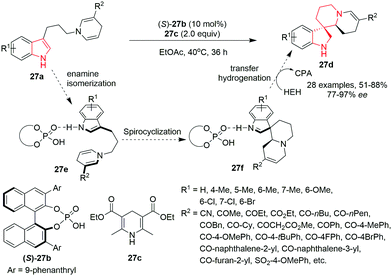 |
| | Scheme 27 Synthesis of tetrahydrospiro[indoline-3.1′-quinolizine] 27d. | |
8. Five membered oxygen containing spiro-cyclic compounds.
Hajra and co-workers33 have reported an efficient Lewis acid catalyzed formal [3+2]-annulation reaction of spiro-epoxyoxindoles 28a and allyltrimethylsilane 28b for the synthesis of spiro-furanoindolines 28c in DCE at 0 °C. This reaction proceeds smoothly with differently substituted (electron-donating as well as electron-withdrawing groups) spiro-epoxyoxindoles. Interestingly, it has been observed that Sc(OTf)3 (10 mol%) is optimal for all N-methyl and N-allyl spiro-epoxyoxindoles, whereas 10 mol% of BF3·OEt2 is best for all N-benzyl spiro-epoxyoxindoles. These spirocyclic oxindoles were obtained as an equal diastereomeric mixture (1:1). Noticeably, in this reaction, a polar solvent facilitated the C–Si bond cleavage, whereas the β-silicon effect was predominant in a nonpolar solvent (Scheme 28).
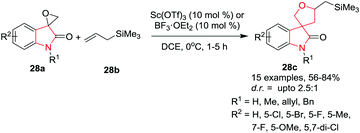 |
| | Scheme 28 Synthesis of spiro-furanoindolines 28c. | |
Further, Kumar and co-workers34 have extended this methodology to get access to substituted spirocyclic oxindoles as a diastereomeric mixture in appreciable yields (60–78%) with substituted allylsilanes using a similar methodology except using DCM as reaction solvent.
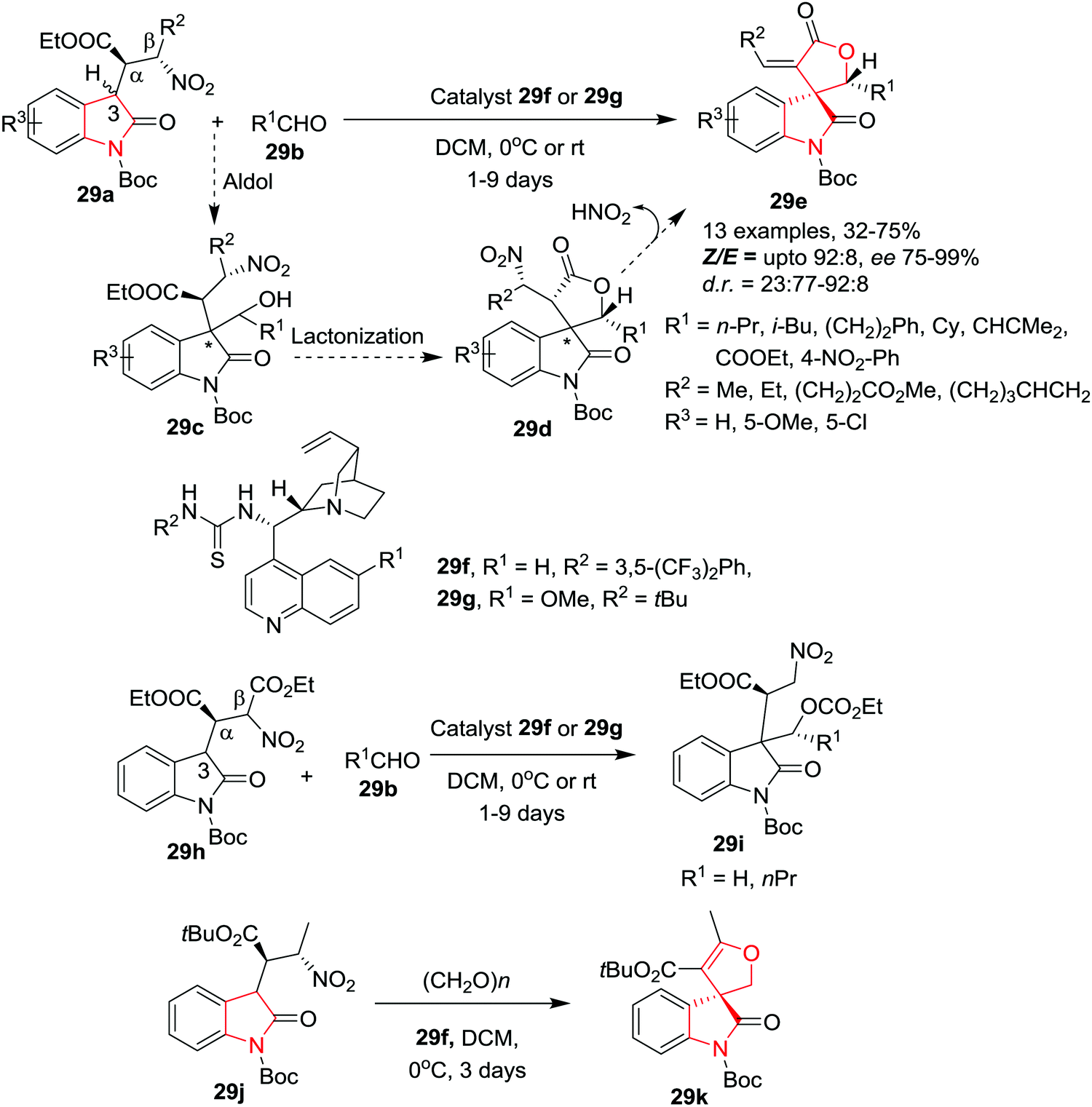
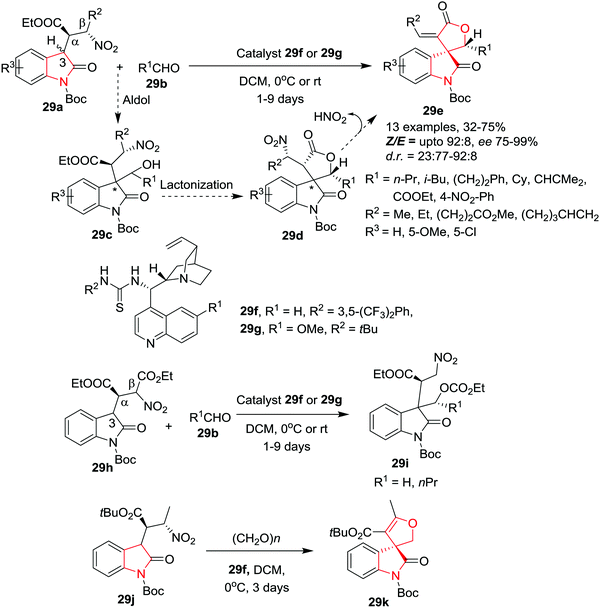
Quintavalla and co-workers35 have developed a new methodology to access pharmaceutically important 3-spiro-α-alkylidene-γ-butyrolactone oxindoles 29e with excellent enantioselectivity and high E/Z ratio by reacting β-nitro oxindoles 29a and aldehydes 29b, in the presence of a bifunctional chiral thiourea based catalyst 29f or 29g in DCM at 0 °C or rt. Under the optimized conditions, several aliphatic aldehydes provided the corresponding spirolactone oxindoles in acceptable yields, good d.r. values, Z/E ratios and with high e.e. However, sterically hindered aldehydes required longer reaction times. Interestingly, when ethyl glyoxylate was subjected to this reaction at rt, significantly lower diastereoselectivity and decreased e.e. for the major isomer (86% e.e.) were observed, whereas the minor isomer was produced with excellent enantio-control (96% e.e.). Further, the same reaction at low temperature (0 °C) unexpectedly resulted in a reversed diastereomeric ratio and with high enantioselectivity of the major isomer. The use of aromatic aldehydes led to good enantiomeric excess at rt which can be improved by carrying out the reaction at 0 °C. However, this required longer reaction times and gave only low product yields. The spirolactones, variably substituted on the exocyclic double bond with alkyl groups, were obtained with good stereoselectivities and yields. Further, an electron-donating or electron-withdrawing group on the oxindole aromatic ring is well tolerated. The reaction proceeds with the proposed aldol/lactonization/elimination domino sequence. Interestingly, nitroester-derived oxindoles 29h gave uncyclized product 29i as a mixture of four inseparable diastereoisomers which is generated through an unexpected intramolecular 1,5 C → O migration of the ethyl ester from the Cβ to the hydroxyl group. Further, the β-nitro oxindoles 29j bearing a tert-butyl ester at Cα (instead of ethyl ester) gave access to only 3-spiro dihydrofuran oxindole 29k in low yield (15%) but with excellent enantiomeric excess (Scheme 29).
 |
| | Scheme 29 Synthesis of 3-spiro-α-alkylidene-γ-butyrolactone oxindoles 29e. | |
9. Six membered oxygen containing spiro-cyclic compounds.
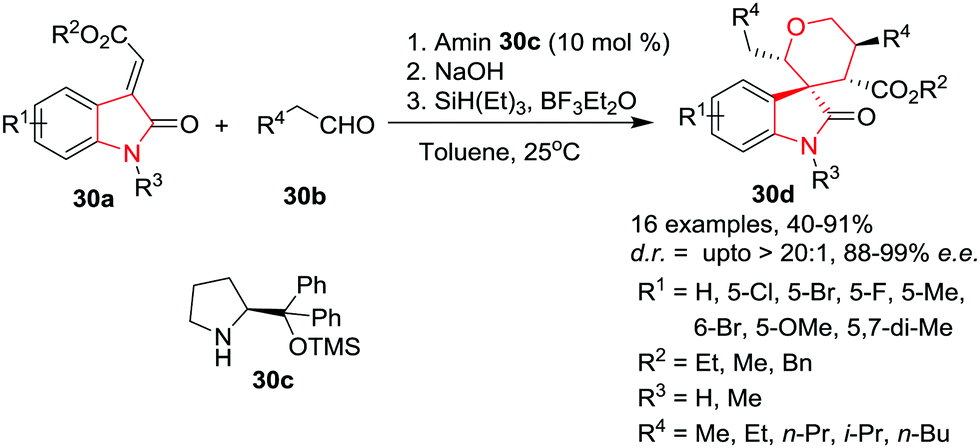
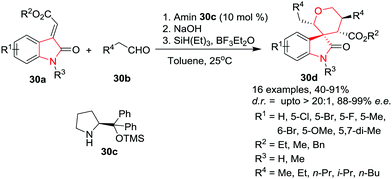
Zeng, Zhong and co-workers36 have developed an efficient organocatalytic Michael/aldol/hemiacetalization cascade reaction of electron-deficient oxindole olefins 30a and aliphatic aldehydes 30b to get access to highly functionalized 5′,6′-dihydro-2′H,4′H-spiro[indoline-3,3′-pyran]-2-ones 30d in good yields and with high stereoselectivities. This one pot protocol was catalyzed by a chiral amine 30c under basic conditions in toluene at rt. It has been found that the reaction could accommodate a broad range of substituents (irrespective of the electronic nature) on the 3-ylideneoxindoles, and provided the products in moderate to good yields with good to excellent diastereo- and enantioselectivities. The 3-ylideneoxindoles with methyl and benzyl esters were also good substrates for the reaction and provided the desired products in moderate to good yields and diastereoselectivities with excellent enantioselectivities. Notably, changing the propionaldehyde to other aliphatic aldehydes resulted in slightly lower yields with good to excellent stereoselectivities. Furthermore, N-methyl protected oxindole olefin also gave the desired product in good yield and with excellent diastereoselectivity, although the e.e. was lowered to 85% (Scheme 30).
 |
| | Scheme 30 Synthesis of 5′,6′-dihydro-2′H,4′H-spiro[indoline-3,3′-pyran]-2-ones 30d. | |
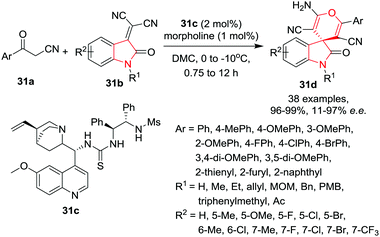
Wu and co-workers37 have reported the first organocatalytic enantioselective Michael/cyclization cascade reaction of α-cyano ketones 31a and isatylidene malononitriles 31b to get access to new spiro[4H-pyran-oxindoles] 31d in the presence of quinidine-derived organo-catalyst 31c and morpholine as additive in DCM at 0 °C in quantitative yields with good to excellent enantioselectivity within 2 h. Under optimized conditions, differently substituted isatylidene malononitriles provided excellent yields irrespective of the electronic nature of the substituents and with good to excellent enantioselectivities. Interestingly the 6-methyl substituted isatylidene malononitriles required extended reaction times to give the desired product with excellent yield and enantioselectivity. Substituents on the nitrogen atom have a significant impact on the stereoselectivity of the reaction. The low steric hindrance of an N-alkyl substituent increased the enantioselectivity of the product whereas N-acetyl-substituted substrate provided inferior enantioselectivity. Isatylidene malononitrile with an unsubstituted NH group underwent the reaction smoothly but gave inferior enantioselectivity. Interestingly, when performing the same reaction at −10 °C, the enantioselectivity was improved significantly. The α-cyano ketones bearing an electron-rich substituent at the 4-position of the phenyl ring provided better enantioselectivities than those with an electron-withdrawing group. Further, substituents at the 3- and 2-position of the phenyl ring provided lower enantioselectivities likely due to the different stereoelectronic effects. Heterocyclic and polycyclic α-cyano ketones also provided excellent yields and enantioselectivities (Scheme 31).
 |
| | Scheme 31 Synthesis of spiro[4H-pyran-oxindoles] 31d. | |
10. Fused oxygen-heterocycle containing spiro-cyclic compounds.
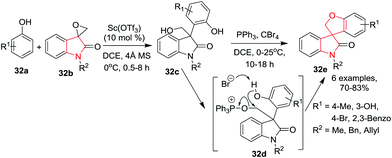
Hajra and co-workers38 have developed a metal triflate-catalyzed intermolecular Friedel–Crafts reaction of arenes 32a and spiroepoxyoxindoles 32b to achieve the regioselective synthesis of 3-(hydroxymethyl)-3-(2-hydroxyaryl)indolin-2-ones 32c in good yields. These were subsequently utilized for the synthesis of 2H-spiro[benzofuran]-3,3′-oxindoles 32e. The intermediate 3-aryl-(3-hydroxymethyl)oxindoles 32c were subjected to Ph3P and CBr4 under Appel reaction conditions. This intramolecular etherification reaction was found to be general with a number of 3-(hydroxymethyl)-3-(2-hydroxyaryl)indolin-2-ones and proceeded smoothly to provide very high yields of 2H-spiro[benzofuran]-3,3′-oxindoles. The reaction is believed to proceed through the intermediate alkoxyphosphonium salt 32d which underwent intramolecular SN2 cyclization with the phenolic OH, leading to the formation of the desired spiro[benzofuran]-3,3′-oxindoles (Scheme 32).
 |
| | Scheme 32 Synthesis of 2H-spiro[benzofuran]-3,3′-oxindoles 32e. | |
Wei and co-workers39 have developed an efficient synthesis of 3-(3-indolyl)-oxindole-3-methanols 33c through an Fe(III)-catalyzed, regioselective ring-opening/Friedel–Crafts-type arylation of spiro-epoxyoxindoles 33a with phenols 33b. Subsequently the generated 3-(3-indolyl)-oxindole-3-methanol 33c has been utilized for the concise synthesis of novel 2H-spiro[benzofuran-3,3′-indolin]-2′-ones 33d. Under the standard Mitsunobu reaction conditions, irrespective of the electronic nature of the substituents on the 3-(3-indolyl)-oxindole-3-methanols 33c, 2H-spiro[benzofuran-3,3′-indolin]-2′-ones 33d were synthesized in excellent yields. Interestingly, spiro-epoxyoxindoles underwent tandem Friedel–Crafts type arylation and O-cyclization with naphthols 33e and 33g in the presence of a catalytic amount of FeCl3·6H2O in DCM to yield novel naphthofuranyl-spirooxindoles 33f and 33h in high regioselectivity and excellent yields. The electronic nature of the substituent on the oxindole rings or on the naphthols was found to have a minimal effect on the reaction outcome and provided the addition/O-cyclization products in high to excellent yields. Besides N-methyl or benzyl-protected spiroepoxyoxindoles, N-unsubstituted spiro-epoxy-oxindoles also underwent this reaction smoothly, albeit with slightly lower yield (Scheme 33).
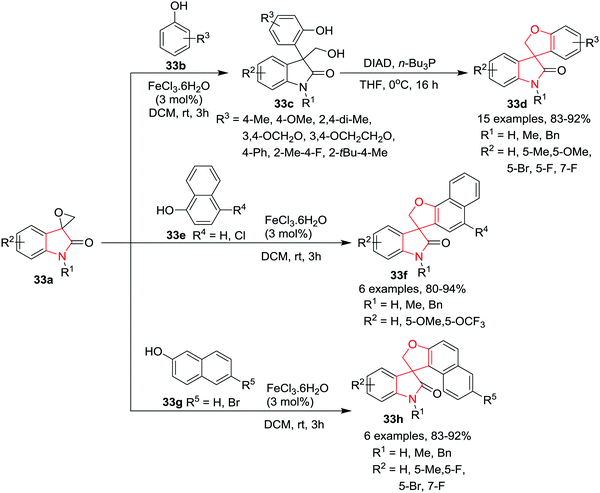 |
| | Scheme 33 Synthesis of 2H-spiro[benzofuran-3,3′-indolin]-2′-ones 33d and naphthofuranyl-spirooxindoles 33f and 33h. | |
Hu and co-workers40 have reported a simple, mild and efficient rhodium-catalyzed reaction of α-phenoxy ketones 34a and 3-diazooxindoles 34b for the synthesis of functionalized spiro[chroman-4,3′-oxindoles] 34c in very high yields and stereoselectivities. The use of Rh2(OAc)4 in DCE at 25 °C in the presence of 4 Å molecular sieves was found to be optimal to achieve excellent product yields and diastereoselectivities. The optimized transformation protocol tolerates differently substituted aryl groups (electron-donating or electron-withdrawing as well as heterocyclic) on the α-phenoxyarylethanones and provides access to the desired products in very good yields and excellent diastereoselectivities, even with bulky aryl groups such as naphthyl substituted substrates. Notably, 4-CF3-phenyl-substitution provided the product with low diastereoselectivity probably due to partial isomerization of the syn-product under the standard reaction conditions. Importantly, small alkyl groups (methyl or ethyl) were also compatible whereas bulkier groups such as n-butyl afforded the corresponding spiro[chroman-4,3′-oxindole] with relatively low diastereoselectivity. Further, various electron-donating as well as electron-withdrawing substituents at the C-5 and C-6 positions of the 3-diazooxindoles gave access to the desired products in good yields with high diastereoselectivities. Importantly, replacement of the N-benzyl substituent of the 3-diazooxindole by N-Cbz or with N-Me slightly reduces the yields and diastereoselectivities. The synthetic efficiency of this process was proved by a gram-scale reaction that gave the desired product in 86% yield (1.09 g) with 95:5 diastereoselectivity (Scheme 34).
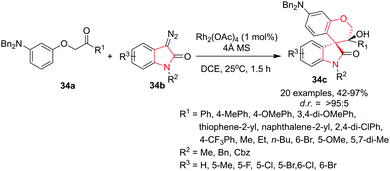 |
| | Scheme 34 Synthesis of functionalized spiro[chroman-4,3′-oxindoles] 34c. | |
3. Outlook and conclusion
In this tutorial review, we have highlighted some recent advances in the construction of spiroindolines and spiroindoles which are the centre of attraction among medicinal chemists, due to their rigidity and unique three-dimensional geometries. Spirocyclization of indoles via the 2- or 3-position has been achieved successfully in high yields by utilizing various inorganic or organic catalytic systems. We have witnessed the use of various cinchona/quinidine based chiral catalysts for efficient spirocyclization in excellent yields, enantioselectivities and diastereoselectivities. The use of phosphoric acid based chiral ligands proved to be helpful for reaching a high level of regio- and enantiocontrol in spirocyclization reactions. Among these reactions, [3+2] or [3+4] cycloaddition, and [4+1] annulation reactions have proved to be a hallmark over the last few years. Further, the cascade Friedel–Crafts alkylation reaction has been applied successfully for the spirocyclization of indoles. Another interesting approach utilizes the cycloisomerization of appropriately substituted indoles. Our own contribution in this field utilized this approach where post Ugi-adducts were cycloisomerized, yielding highly functionalised polycyclic spirocyclic indoles. The application of MW-assisted and continuous-flow conditions in such processes opened a new avenue for spirocyclic reactions by employing a high catalyst/reactant ratio. It is likely that over the time, many more powerful and mild methodologies will emerge in this field.11,21,27 Overall, we trust that this review will serve to update the researchers focused on the synthesis of spiroindolines/spiroindoles, and will serve as an anchor to encourage further growth in this field.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge the support from Shiva Institute of B. Pharmacy and RUDN University Program 5-100.
References
- J. Reymond and M. Awale, ACS Chem. Neurosci., 2012, 3, 649–657 CrossRef PubMed.
- M. J. James, P. O’Brien, R. J. K. Taylor and W. P. Unsworth, Chem. – Eur. J., 2016, 22, 2856–2881 CrossRef PubMed.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef PubMed.
-
(a) Q. F. Wu, H. He, W. B. Liu and S. L. You, J. Am. Chem. Soc., 2010, 132, 11418–11419 CrossRef PubMed;
(b) P. D. Fedoseev and E. V. Van der Eycken, Chem. Commun., 2017, 53, 7732–7735 RSC.
- L. Kong, M. Wang, F. Zhang, M. Xu and Y. Li, Org. Lett., 2016, 18, 6124–6127 CrossRef PubMed.
- X. Zhang, G. Zhou, Y. Zhang, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2016, 81, 11397–11403 CrossRef PubMed.
- L. Zhang, Y. Wang, X. Hu and P. Xu, Chem. – Asian J., 2016, 11, 834–838 CrossRef PubMed.
- C. Peng, J. Zhai, M. Xue and F. Xu, Org. Biomol. Chem., 2017, 15, 3968–3974 Search PubMed.
- J. Zhao, D. Yue, X. Zhang, X. Xu and W. Yuan, Org. Biomol. Chem., 2016, 14, 10946–10952 Search PubMed.
- J. Zhang, N. Li, S. Yin, B. Sun, W. Fan and X. Wang, Adv. Synth. Catal., 2017, 359, 1541–1551 CrossRef.
- L. Shi, L. Li, J. Wang, B. Huang, K. Zeng, H. Jin, Q. Zhang and Y. Jia, Tetrahedron Lett., 2017, 58, 1934–1938 CrossRef.
- R. D. Gao, Q. L. Xu, L. X. Dai and S. L. You, Org. Biomol. Chem., 2016, 14, 8044–8046 Search PubMed.
- J. Huang, M. Sohail, T. Taniguchi, K. Monde and F. Tanaka, Angew. Chem., 2017, 56, 5853–5857 CrossRef PubMed.
- L. Zhang, W. Ren, X. Wang, J. Zhang, J. Liu, L. Zhao and X. Zhang, Eur. J. Med. Chem., 2017, 126, 1071–1082 CrossRef PubMed.
- G. Zhu, Q. Wei, H. Chen, Y. Zhang, W. Shen, J. Qu and B. Wang, Org. Lett., 2017, 19, 1862–1865 CrossRef PubMed.
- F. Schröder, U. K. Sharma, M. Mertens, F. Devred, D. P. Debecker, R. Luque and E. V. Van der Eycken, ACS Catal., 2016, 6, 8156–8161 CrossRef.
- G. Zhu, S. Liu, S. Wu, L. Peng, J. Qu and B. Wang, J. Org. Chem., 2017, 82, 4317–4327 CrossRef PubMed.
- G. Zhu, S. Wu, X. Bao, L. Cui, Y. Zhang, J. Qu, H. Chen and B. Wang, Chem. Commun., 2017, 53, 4714–4717 RSC.
- J. Zhang, H. Wang, Q. Jin, C. Zheng, G. Zhao and Y. Shang, Org. Lett., 2016, 18, 4774–4777 CrossRef PubMed.
- J. Su, Z. Ma, X. Li, L. Lin, Z. Shen, P. Yang, Y. Li, H. Wang, W. Yan, K. Wang and R. Wang, Adv. Synth. Catal., 2016, 358, 3777–3785 CrossRef.
- D. Chen, W. Xiaoa and J. Chen, Org. Chem. Front., 2017, 4, 1289–1293 RSC.
- A. Preetam and M. Nath, Tetrahedron Lett., 2016, 57, 1502–1506 CrossRef.
- A. M. Jadhav, S. G. Balwe, K. T. Lim and Y. T. Jeong, Tetrahedron, 2017, 73, 2806–2813 CrossRef.
- Y. Wang, C. Zheng and S. L. You, Angew. Chem., Int. Ed., 2017, 56, 15093–15097 CrossRef PubMed.
- V. Magne, F. Blanchard, A. Marinetti, A. Voituriez and X. Guinchard, Adv. Synth. Catal., 2016, 358, 3355–3361 CrossRef.
- L. Wang, S. Li, M. Blumel, A. R. Philipps, A. Wang, R. Puttreddy, K. Rissanen and D. Enders, Angew. Chem., 2016, 55, 11110–11114 CrossRef PubMed.
- A. S. Filatov, N. A. Knyazev, A. P. Molchanov, T. L. Panikorovsky, R. R. Kostikov, A. G. Larina, V. M. Boitsov and A. V. Stepakov, J. Org. Chem., 2017, 82, 959–975 CrossRef PubMed.
- N. Li, J. Zhang, B. Sun, H. Li and X. Wang, Org. Lett., 2017, 19, 1954–1957 CrossRef PubMed.
- S. G. Modha, A. Kumar, D. D. Vachhani, J. Jacobs, S. K. Sharma, V. S. Parmar, L. V. Meervelt and E. V. Van der Eycken, Angew. Chem., Int. Ed., 2012, 51, 9572–9575 CrossRef PubMed.
- F. Schröder, M. Ojeda, N. Erdmann, J. Jacobs, R. Luque, T. Noël, L. V. Meervelt, J. Van der Eycken and E. V. Van der Eycken, Green Chem., 2015, 17, 3314–3318 RSC.
- F. Schröder, N. Erdmann, T. Noël, R. Luque and E. V. Van der Eycken, Adv. Synth. Catal., 2015, 357, 3141–3147 CrossRef.
- Z. L. Xia, C. Zheng, S. G. Wang and S. L. You, Angew. Chem., Int. Ed., 2018, 57, 2653–2656 CrossRef PubMed.
- S. Hajra, S. Roy and S. Maity, Org. Lett., 2017, 19, 1998–2001 CrossRef PubMed.
- B. M. Sharma, M. Yadav, R. G. Gonnade and P. Kumar, Eur. J. Org. Chem., 2017, 2603–2609 CrossRef.
- L. Cerisoli, M. Lombardo, C. Trombini and A. Quintavalla, Chem. – Eur. J., 2016, 22, 3865–3872 CrossRef PubMed.
- L. Zhu, Q. Chen, D. Shen, W. Zhang, C. Shen, X. Zeng and G. Zhong, Org. Lett., 2016, 18, 2387–2390 CrossRef PubMed.
- J. Xie, W. Xing, F. Sha and X. Wu, Eur. J. Org. Chem., 2016, 3983–3992 CrossRef.
- S. Hajra, S. Maity and S. Roy, Adv. Synth. Catal., 2016, 358, 2300–2306 CrossRef.
- M. Luo, R. Yuan, X. Liu, L. Yu and W. Wei, Chem. – Eur. J., 2016, 22, 9797–9803 CrossRef PubMed.
- S. Jia, Y. Lei, L. Song, A. Gopi, K. Reddy, D. Xing and W. Hu, Adv. Synth. Catal., 2017, 359, 58–63 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Leonid G.
Voskressensky
ab,
Leonid G.
Voskressensky