
Designed transition metal catalysts for intracellular organic synthesis
Yugang
Bai†
,
Junfeng
Chen
and
Steven C.
Zimmerman
 *
*
Department of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois, USA. E-mail: sczimmer@illinois.edu
First published on 25th January 2018
Abstract
The development of synthetic, metal-based catalysts to perform intracellular bioorthogonal reactions represents a relatively new and important area of research that combines transition metal catalysis and chemical biology. The ability to perform reactions in cellulo, especially those transformations without a natural counterpart, offers a versatile tool for medicinal chemists and chemical biologists. With proper modification of the metal catalysts, it is even possible to direct a reaction to certain intracellular sites. This review highlights advances in this new area, from early work on intracellular functional group conversions to recent advances in intracellular synthesis of drugs, including cytotoxic agents. Both the fundamental and applied aspects of this approach to intracellular synthesis are reviewed.
 Yugang Bai | Yugang Bai obtained his BS in chemistry from Nanjing University, Nanjing, China in 2009, then he moved to the United States as a graduate student to study materials science and engineering at the University of Illinois at Urbana-Champaign. He switched back to chemistry and joined Steven C. Zimmerman's group in 2011, and obtained his PhD degree in chemistry in 2015. After two more years of postdoctoral training on the same campus, he began his independent academic career at Hunan University in Changsha, China in 2017, as a full time professor of chemistry at the Institute of Chemical Biology and Nanomedicine (ICBN). |
 Junfeng Chen | Junfeng Chen is currently a PhD student in the Department of Chemistry at the University of Illinois at Urbana-Champaign. He received his BS degree from Nanjing University in 2015. After graduation, he joined Dr. Zimmerman's group. His research interests include intracellular catalysis by single-chain organic nanoparticles and the polymer templated synthesis of metal nanoparticles. |
 Steven C. Zimmerman | Steven C. Zimmerman obtained his BS from the University of Wisconsin in 1979. After obtaining a PhD degree from Columbia University in the City of New York in 1984, he became an NSF-NATO Postdoctoral Fellow at the University of Cambridge in England. He began his academic career at the University of Illinois in 1985 where he rose through the ranks to become the Roger Adams Professor of Chemistry in 2004. He served for eight years as the Head of the Department of Chemistry and then three years as a Chancellor's Advisor on Diversity and Cultural Understanding. He returned to full time teaching and research in 2015. |
Introduction
In all life forms enzymes have evolved to perform the essential reactions needed for proper cell functioning and survival. For many of these enzymes, the folded polypeptide scaffold features one or more transition metals at their catalytic active sites. The combination of a metal and a polypeptide is a powerful one, allowing highly selective, on-demand synthetic transformations in a complex biological environment. However, defective or deficient enzymes cause disease and the range of reactions catalysed by naturally occurring enzymes is limited.Synthetic catalysts that can replace the function of missing natural enzymes and especially those that perform unnatural transformations would be a powerful addition to the medicinal chemistry and chemical biology tool kits. Transition metal catalysts have the potential to serve in this capacity, but there are many challenges in applying them in a biological context. First, the catalysts must be highly active with a high turnover number so that they can function at concentrations used in cellular assays that are often orders of magnitude lower than in typical synthetic organic reactions. Second, the metal-based catalysts must have high stability to be compatible with biomolecules that feature acidic, basic, coordinating, and redox active functional groups. Lastly, the catalysts and required additives must be nontoxic, water-soluble, active at physiological pH, and cell-permeable (Fig. 1).
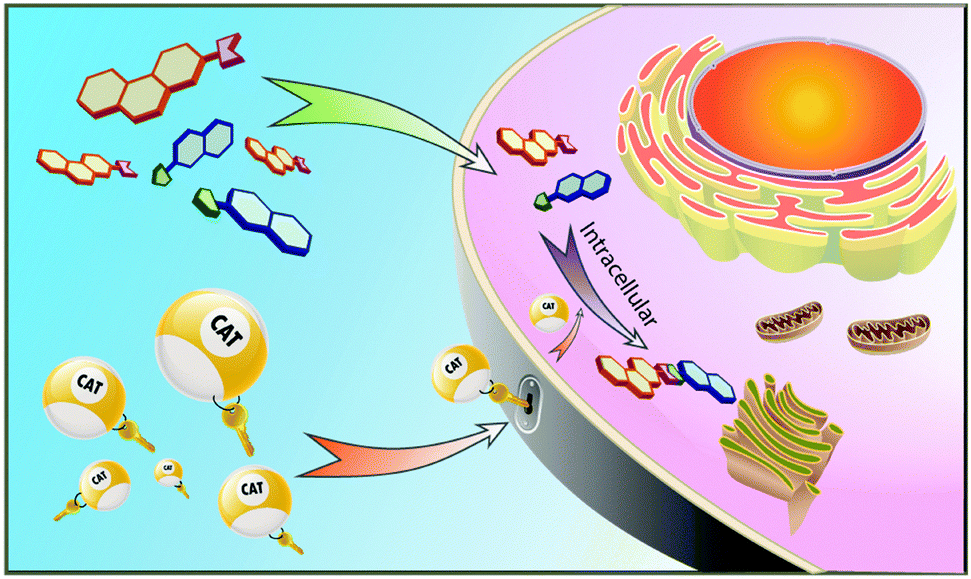 | ||
| Fig. 1 Cartoon illustration of the concept of targeted intracellular synthesis. | ||
Despite the challenges outlined above, the ability to perform intracellular catalysis, to do organic synthesis within cells, has such enormous potential that it is attracting increasing interest and is the subject of this review. The most obvious enabling power is the ability to use unnatural reactions to build a molecule on demand in a designated place within the cell. For example, an anti-cancer drug might be synthesized only in cancer cells if its precursor building blocks and the required catalyst selectively target cancer cells. Because a catalyst molecule functions repetitively generating multiple drug molecules, this approach is much more efficient than installing targeting moieties on bioactive small molecules. Furthermore, larger molecules are inherently less cell permeable, so that transporting their precursors into cells before assembling them can solve the uptake problem that has halted many drug development programs. A wide range of other applications to biology and medicine will likely be enabled by the successful development of intracellular organic synthesis.
Functional group interconversions
Overview of unmasking reactions
Arguably, the simplest intracellular transformation is a functional group deprotection, i.e., an unmasking reaction, in which a masked group is transformed into active functionality with the assistance of a metal catalyst. In such transformations, only one substrate is involved so that the reaction may be less affected by the low intracellular substrate concentration when compared to a bimolecular reaction. Thus, early efforts focused on functional group interconversions involving a single substrate and catalyst as either model reactions or sensing probes. Among these early examples, ruthenium-based catalysts were the most studied, although palladium and iron catalysts were also reported.Early work establishes proof of concept
Streu and Meggers introduced the concept of transition metal catalysis within a chemical biology/living cell setting in 2006, and in a compelling fashion summarized the future potential of the approach.1 Thus, they reported that the simple ruthenium complex, Cp*Ru(cod)Cl (1 in Fig. 2, Cp* = pentamethylcyclopentadienyl, COD = 1,5-cyclooctadiene), could unmask amino groups that are protected as allylcarbamates in air provided that thiols are present. In the presence of E. coli cell extracts and additional glutathione, the deprotection of allylcarbamate-protected rhodamine 110 occurred with a 6% yield, rising to 80% with the addition of thiophenol. Most significantly, live HeLa cells were incubated with the same, virtually non-fluorescent rhodamine derivative, washed with PBS buffer, and treated with 1 allowing the visualization of intracellular fluorescence using confocal microscopy. The results support the conclusion that the transition metal catalyst crosses the cell membrane and catalyses an abiotic reaction. The Meggers group used pyrene–ruthenium complex 2 (Fig. 2) to extend this strategy to a light triggered (≥330 nm) unmasking reaction.2 A 10 min irradiation afforded an overall 70-fold increase in HeLa fluorescence. | ||
| Fig. 2 Ruthenium catalysts (1) and (2) for, respectively, thermal and photoactivated cleavage of the allylcarbamate (alloc) group within live cells. | ||
Another major advance was reported by Bradley and coworkers in 2011,3 the first palladium-mediated intracellular reactions. Using a 500 nm, amino-functionalized polystryrene (PS) microsphere, the researchers added Pd(OAc)2 and then the bis acid chloride of racemic Fmoc-glutamic acid to trap the Pd2+ ions (Fig. 3). Pd2+ reduction with hydrazine afforded Pd0-loaded microspheres. Using the allylcarbamate protected rhodamine approach outlined in Fig. 2, the nanoparticle was reported to enter HeLa cells and catalyse the deprotection reaction, “lighting up” rhodamine fluorescence. The same group released detailed synthetic procedures for the microsphere synthesis and the intracellular catalysis in a follow-up report.4 In 2012 the Meggers group reported an iron-based catalyst that can reduce aromatic azido to the corresponding amino groups catalytically.5 The combination of [Fe(TPP)Cl] (TPP = 5,10,15,20-tetraphenylporphyrin) and a thiol efficiently transformed a non-fluorescent bisazido rhodamine derivative into fluorescent rhodamine 110 in HeLa cells (Fig. 4). However, it was discovered that the existing in vivo metabolic reduction of aromatic azides occurred in both nematodes and zebrafish, the catalyst providing no additional fluorescent enhancement.
 | ||
| Fig. 3 Synthesis of Pd0-loaded polystyrene microspheres. Figure reproduced from ref. 3 with permission from Macmillan Publishers Ltd, copyright 2011. | ||
 | ||
| Fig. 4 Porphyrin-based iron catalyst for intracellular reduction of aromatic azide. The cleavage of allylcarbamate leads to the opening of the lactone structure in rhodamine 110, producing fluorescence. | ||
Usefulness of fluorogenic probes
From the discussion above, it is clear that these so-called “light-up” fluorescence probes provide an outstanding means of detecting and even quantifying intracellular transformations. As such, it is a natural extension to use these fluorogenic compounds as metal or metal ion detectors. Most aspects of this strategy have been reviewed recently6–9 so only a brief overview will be given here. Shown in Fig. 5 are masked dyes that reflect the range of fluorophore structures used for visualizing metal species inside cells. The earliest relevant report was from Ahn and coworkers in 2010.10 In this report, an O-propargylated fluorescein (4 in Fig. 5) was used to detect and visualize accumulated palladium species within zebrafish. In 2011, Du and Zhang et al. reported a 4-hydroxynaphthalimide-derived ratiometric fluorescent chemodosimeter (5 in Fig. 5) for imaging palladium (Pd0, Pd2+, and Pd4+) in living cells, with excellent selectivity for Pd over other metal ions.11 A closely related system with improved water solubility (6 in Fig. 5) was reported by Liu and coworkers in 2014 who further showed the system's ability to function as a ratiometric fluorescent and colorimetric probe.12 Fun and coworkers demonstrated the use of a cyano-adduct (7 in Fig. 5) in selective detection and visualization of Pd2+ in cells.13 The palladium assisted cyanide elimination results in the restoration of the full xanthene conjugation of the rhodamine 6G, with strong turn-on fluorescence.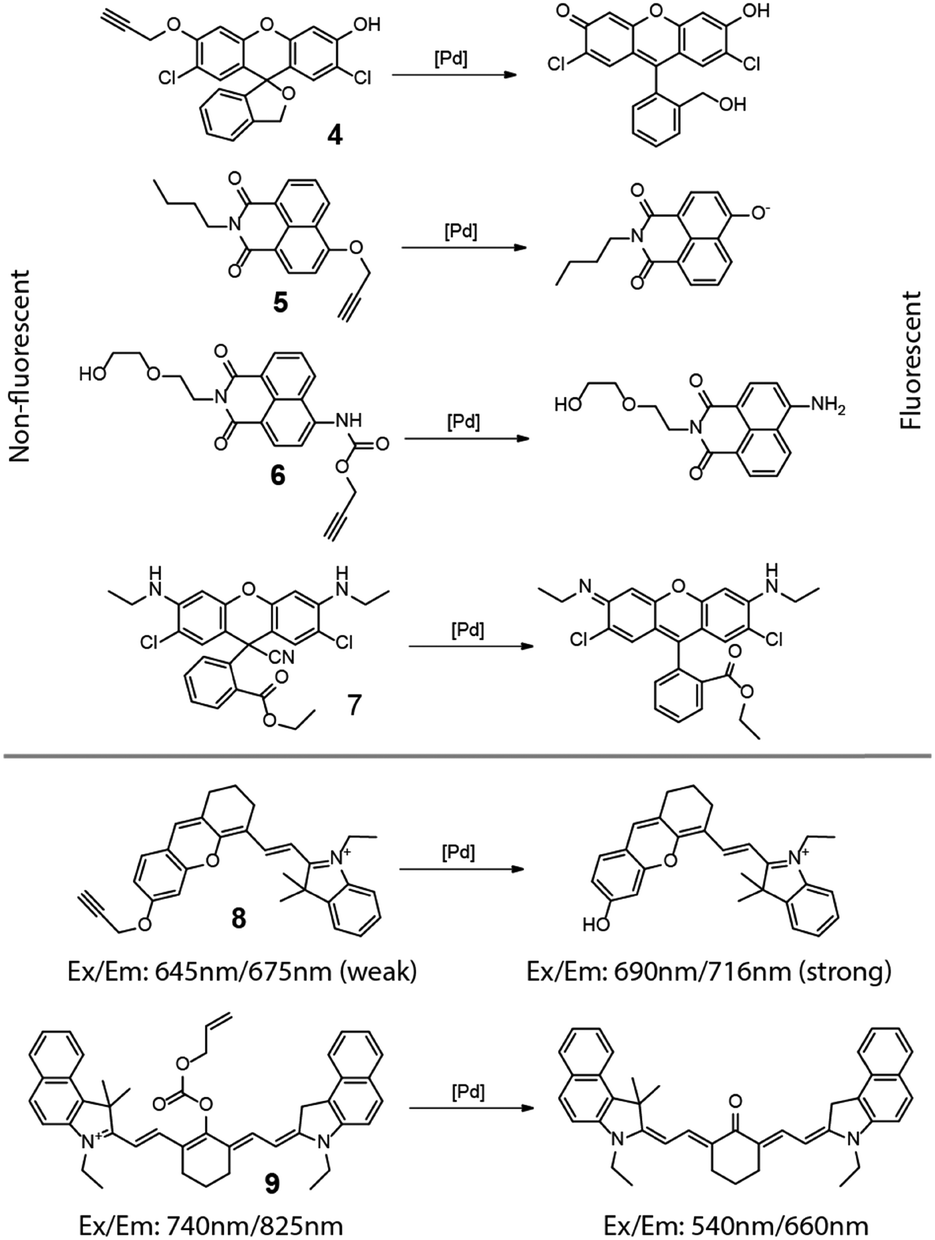 | ||
| Fig. 5 Various Pd probes and their working mechanisms. | ||
In addition to the fluorescence turn-on mechanism used by these probes, metal probes have also been designed to use a change in fluorescence wavelength caused by the metal-catalyzed unmasking reaction. Two cyanine-based intracellular probes 8 and 9 for palladium were reported by Lin in 2013,14 and Guo and Zhu in 2014,15 respectively, extending the detection wavelength into the near-infrared region. In these two examples, the removal of the “masking” groups can lead to a shift and enhancement in the fluorescence emission, providing a clear optical signal for the corresponding chemical reaction. It is worth noting that copper ions were also reported to cleave 1,1-dimethylpropargyl carbamates,16 but its intracellular application is yet to be explored.
Intracellular activation by unmasking
The usefulness of unmasking reactions is not limited to fluorescence activation and detection. By removing a masking/protecting group, the biological activity of a molecule can be significantly changed. As a result, a metal catalyst that is able to efficiently mediate unmasking reactions can be conveniently used to uncage a bioactive agent or modulate its activity within cells. For instance, using the catalyst [Cp*Ru(cod)Cl] (1 in Fig. 2), Vázquez, Mascarenas, and coworkers reported the catalytic unmasking of fluorogenic DNA-binding bisbenzamidine, DAPI, and ethidium units inside living cells (Fig. 6a).17 Protecting the amidine moieties and ethidium amino groups with the allylcarbamate group significantly attenuates their interaction with DNA as well as their fluorescence. These compounds were unmasked by ruthenium catalyst 1 (2.5–20 μM) and thiophenol (100 μM) within live chicken embryo fibroblast (CEF) cells with significant nuclear localization and fluorescent staining. | ||
| Fig. 6 (a) Alloc-masked DNA-binding bisbenzamidine, DAPI, and ethidium. (b) Intracellular activation of doxorubicin. (c) A method to evaluate metal-catalyzed reactions in real time in cells using a luciferase reporter system. Figure partially reproduced from ref. 20 with the permission from ACS Publications. | ||
Meggers and coworkers reported a new, highly active ruthenium complex with much higher activity and TON for the alloc protecting group removal (Fig. 6b).18 The catalyst used was based on an analogous catalyst initially reported by Kitramura for the allylation of nucleophiles.19 As illustrated in Fig. 6b, the catalyst was proposed to enter HeLa cells, remove the allylcarbamate group on the amino-glycoside unit of the anti-cancer drug doxorubicin, and induce cell death. Recently, Waymouth, Wender, and coworkers reported a general method to evaluate an intracellular allylcarbamate deprotection reaction in real time, using a luciferase reporter system (Fig. 6c).20 The authors used the same Kitamura catalyst as the Meggers group did but in a mouse mammary carcinoma cell line (4T1 cells), transfected with a plasmid that inserts the gene for click beetle luciferase. The pro-probe unmasking reaction to generate amino-luciferin and a subsequent bioluminescence optical readout was not observed when the cells were incubated with the catalyst and given a single wash, indicating that the ruthenium complex does not enter the cell or becomes extracellular rapidly and completely.
When the cells were incubated with the pro-probe and washed and then the catalyst added, intracellular bioluminescence was observed. However, the intensity decreased with the number of washes suggesting that the pro-probe was partitioning between intracellular and extracellular space, the unmasking likely occurring outside the cell. This result points to the great care that must be taken in conclusively demonstrating that catalysis occurs intracellularly. False positives can readily occur when reactants/reagents or reaction products are able to partition across the cell membrane, a key point returned to at the end of this review.
Rotello and coworkers reported21 a supramolecular assembly consisting of a gold nanoparticle (AuNP) coated with a layer of thiols (Fig. 7) containing three distinct parts: (1) an internal hydrophobic segment for encapsulating organometallic catalysts, (2) a tetra(ethylene glycol) unit to provide biocompatibility, and (3) a benzyl-ammonium end-group to complex cucurbituril[7] (CB[7]). The nanocatalyst performed the intracellular deprotection of bis-N,N′-allyloxycarbonyl rhodamine 110 with either Cp*Ru(cod)Cl or ((1,1′-bis(diphenylphosphino)ferrocene)palladium(II)dichloride) loaded in the hydrophobic layer. Importantly, the catalysis could be regulated by supramolecular interactions. Thus, addition of CB[7] blocks access to the catalyst molecules bound on the interior, effectively serving as “gatekeepers,” turning off the activity. Addition of adamantane derivatives that tightly complex CB[7] restores activity, mimicking the allosteric regulation of enzymes. The nanocatalyst was shown to unmask alloc-protected rhodamine and N-propargyl 5-fluorouracil in cells on demand.
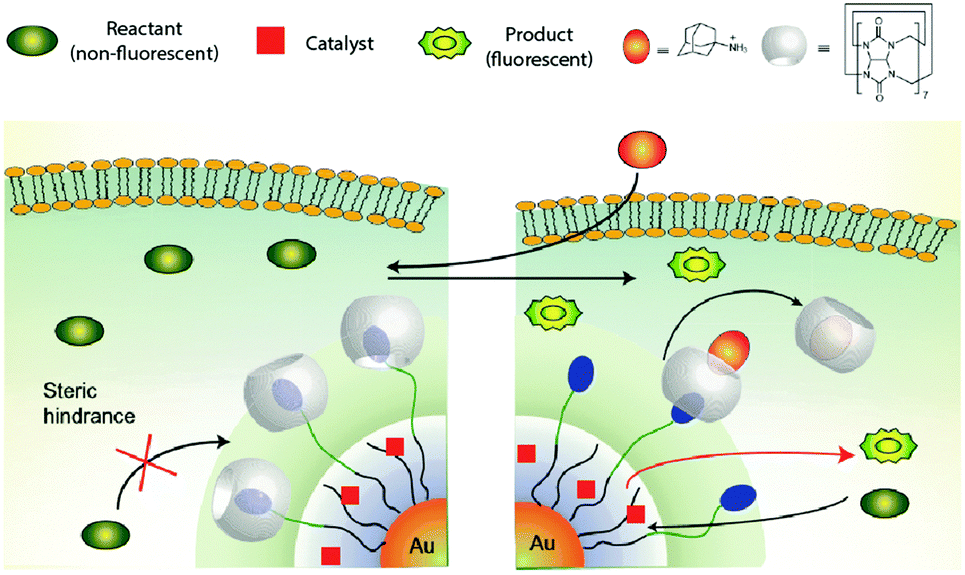 | ||
| Fig. 7 Working mechanism of the AuNP-based nanoreactor reported by Rotello and coworkers. Figure reproduced from ref. 21 with permission from Macmillan Publishers Ltd. | ||
The example above demonstrates temporal control over the production of a bioactive agent. Another powerful application of intracellular organic synthesis involves spatial control. Thus, by introducing targeting moieties on the catalyst, the activation of small molecules can be localized to a desired region within the cell. Mascarenas and coworkers reported the first example of mitochondria-targeted Ru catalysis using complexes 10 and 11 (Fig. 8).22 By linking a triphenylphosphonium moiety to the ruthenium complex, the catalyst preferentially accumulated in the mitochondria of HeLa cells, showing localized subcellular catalytic activity in releasing masked fluorophores. The pyrene unit in 11 allowed the cellular localization of the catalyst to be tracked. To test whether the synthesis of a bioactive agent on-site would produce a specific biological outcome, the investigators chose the mitochondrial membrane-uncoupler agent 2,4-dinitrophenol. Adding O-allyl 2,4-dinitrophenol to cells previously treated with 11 caused fast and strong depolarization whereas an analogue lacking the mitochondrial targeting did not.
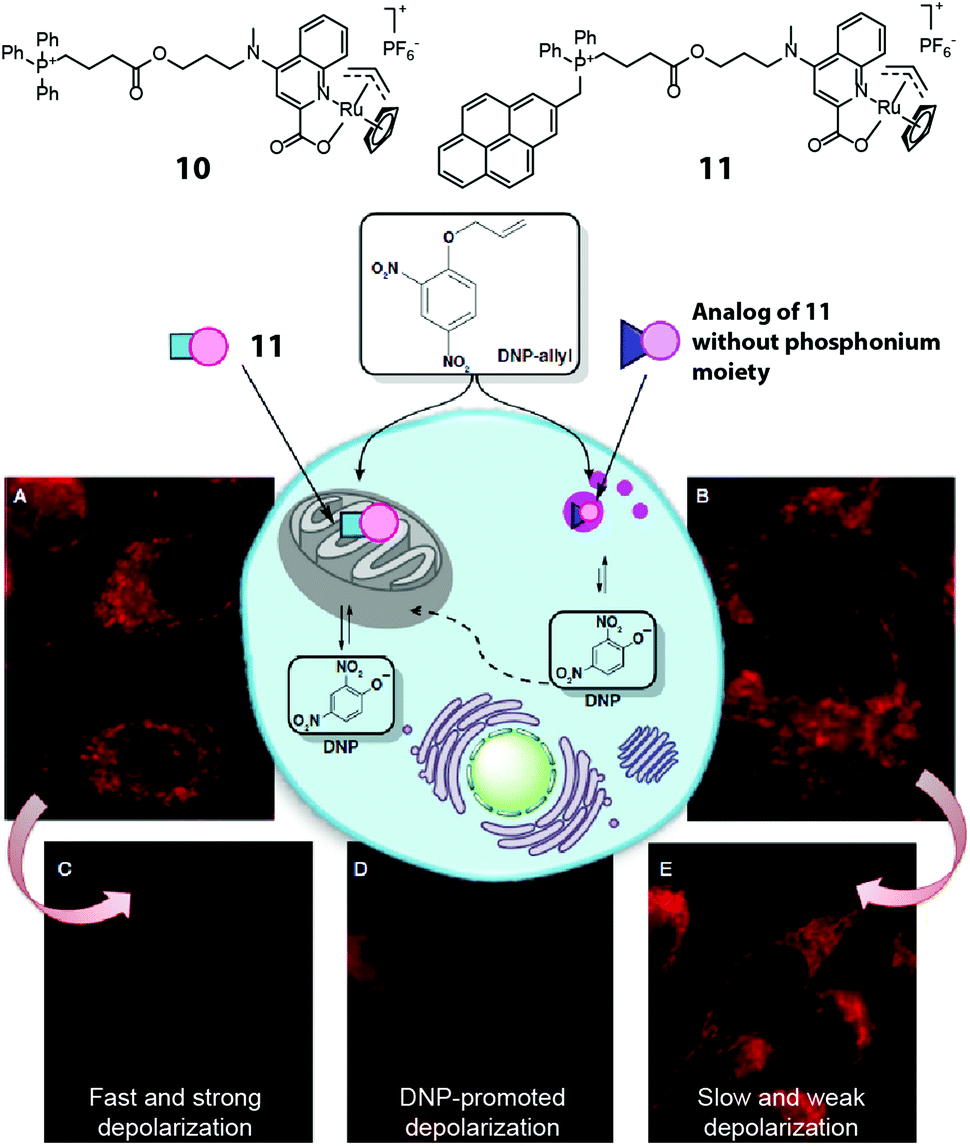 | ||
| Fig. 8 Two mitochondria-targeting ruthenium catalysts reported by Mascarenas et al. The phosphonium-bearing catalyst can accumulate in mitochondria and locally activate DNP-allyl in HeLa cells, leading to mitochondrial depolarization. Figure reproduced from ref. 22 with the permission from Macmillan Publishers Ltd. | ||
Chen and coworkers have pioneered the use of organometallic reagents to unmask proteins intracellularly. The approach involves genetically and site-specifically incorporating a ε-propargyloxycarbonyl protected Lys-residue (Proc-Lys) into a protein or an enzyme of interest in live cells.23 The efficiency of six different air-stable, common palladium reagents was tested for their ability to unmask a green fluorescent protein with Asn149 replaced by Proc-Lys GFP-N149-ProcLys in vitro. The common reagents Pd(dba)2 and allyl2Pd2Cl2 gave the best results with deprotection yields of 78% and 90%, respectively. The palladium reagents exhibited negligible cytotoxicity in several cell types at working concentrations. The intracellular efficiency was much lower, but nonetheless allowed several applications including unmasking a phosphothreonine lyase (OspF-K134-ProcLys) to elucidate its role as a virulence factor.
Chen and coworkers further showed that the same simple palladium reagents could effect the intracellular uncaging of tyrosine residues protected with allene (Alle) groups (Fig. 9).24 First, GFP-Y40-AlleY was expressed in HEK293T cells and deprotected in 51% yield by allyl2Pd2Cl2 after a 2 h incubation. Two additional examples demonstrated the power of this approach as a tool for studying signal transduction in living HEK293T cells. One involved the chemical rescue of a Tyr phosphorylation site on an oncogenic variant of Src kinase that plays an important role in cancer. The other protein, anthrax lethal factor (LF), is a protease that operates on MEK3 in infected cells. The caged protein LF-Y728-AlleY was found to be inactive, but incubation with 20 μM Pd(dba)2 and allyl2Pd2Cl2 restored the activity.
 | ||
| Fig. 9 Cartoon illustration of in cellulo activation of proteins by “decaging” of the masked amino acid residues. Figure reproduced from ref. 24 with the permission from ACS Publications. | ||
These advances in protein caging use carbamate and allene moieties to mask lysine and tyrosine residues, respectively. Both groups are removed with the same simple palladium reagents. Future efforts will doubtlessly focus on expanding the intracellular toolkit so that two different proteins might be uncaged using orthogonal unmasking reactions25 that provide spatial and temporal control for even more complex multi-protein gain-of-function studies. Likewise, it can be expected that methods for masking of additional amino acid residues intracellularly will be developed in the future.
Intracellular bond forming reactions
Overview
The intracellular unmasking reactions described above involve bond cleavage transformations. Far fewer examples have been reported where metal-based catalysts effect bond forming reactions within cells. Coupling reactions in particular are quite challenging because of the available concentrations of catalysts and substrates are often low, necessitated by toxicity and uptake issues. Thus, not surprisingly, the early examples of bond forming reactions featured intracellular cyclization reactions.Fluorescent probes based on cyclization reactions
Yoon and coworkers reported in 200926 a highly selective fluorescent probe for intracellular Au3+ based on cyclization of the nonfluorescent N-propargylamide derivative of tetraethyl-rhodamine (12) to give the highly fluorescent oxazole-carbaldehyde product (Fig. 10). The human keratinocyte cell line HaCaT was treated with 20 μM 12, washed with PBS, and incubated with 10 μM AuCl3 for 1 h at which time significant intracellular fluorescence was observed. The Tae group reported independently an N-propargyloxyamide derivative of rhodamine 6G that works in a similar manner to detect Au3+ ions.27 Coumarin and BODIPY based probes for Au3+ were later reported by Kim and Song, respectively, in 2010 and 2011,28,29 utilizing similar chemistry (Fig. 10). In Kim's report, a coumarin derivative was synthesized in cellulo by Au3+ mediated ring-closing of aryl phenylpropiolate (13). Song and coworkers showed that the Au3+-mediated ring-closing reaction of 14 removes the aniline amino group, thereby efficiently turning off the photoinduced electron transfer (PET) quenching process, restoring the fluorescence of the BODIPY core.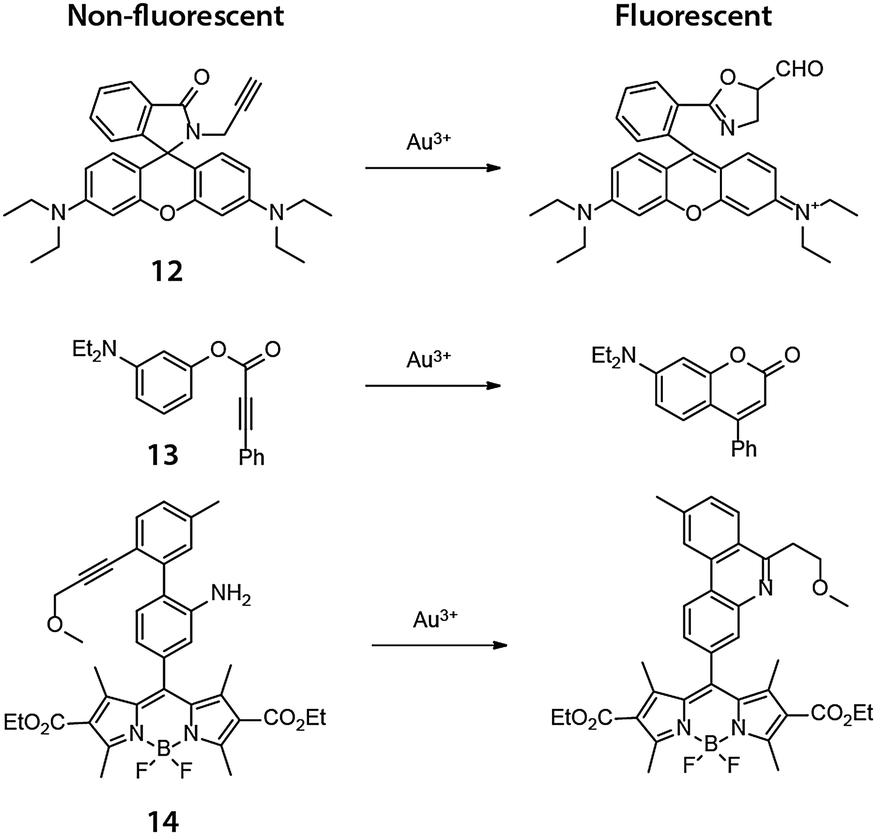 | ||
| Fig. 10 Chemical structures of the fluorogenic, intramolecular reactions mediated by Au(III) in cellulo. | ||
Intermolecular bond formation – metal catalysed coupling reactions
A metal-mediated intermolecular reaction typically involves two substrates and a catalyst, thus the reaction rate can be highly sensitive to concentration. Further requirements include the need for each component to be compatible with the cellular environment and for each to be co-located at the same time or in the correct reaction sequence. For these reasons, in cellulo intermolecular reactions have typically required catalysts of much higher activity compared to the ones used in intramolecular reactions, and such catalysts were discovered later in this field.An early example of an in cellulo CO-coupling that is stoichiometric in metal was reported by Chang and coworkers in 2012.30 In this work, palladium was pre-loaded onto a borondipyrromethene difluoride (BODIPY) fluorophore to form a complex (15). The palladium quenched the BODIPY fluorescence but carbonylation in the presence of carbon monoxide turned on fluorescence (Fig. 11). In HEK293T cells incubated with 1 μM 15 and 50 μM [Ru(CO)3Cl(glycinate)] as a CO source, an approximately 2.5-fold increase in fluorescence was observed. By introducing the metal complex to the fluorescent probe, what would ordinarily be a three-component coupling reaction (two substrates and one metal complex) required only two species (15 and CO).
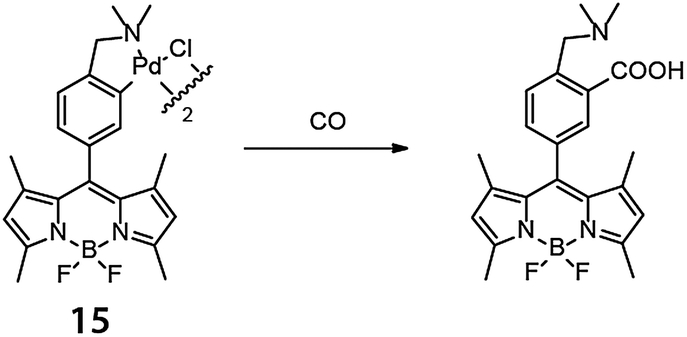 | ||
| Fig. 11 BODIPY-based palladium complex for in cellulo reaction with carbon monoxide reported by Chang and coworkers (ref. 30). | ||
The first reported example of a catalytic, intracellular, palladium-mediated coupling involving two substrates was reported by Bradley and coworkers in 2011.3,4 Thus, HeLa cells were incubated for 24 h with the Pd0 microspheres described in Fig. 3 at a ratio of ca. 105 microspheres per cell. After washing, the cells were incubated with 20 μM of a nonfluorescent fluorescein triflate derivative and an arylboronate for 48 h (Fig. 12). The Suzuki–Miyaura cross-coupling product undergoes opening of the lactone ring, turning on fluorescence, whereas the triphenylphosphonium group targets mitochondria allowing its visualization.31
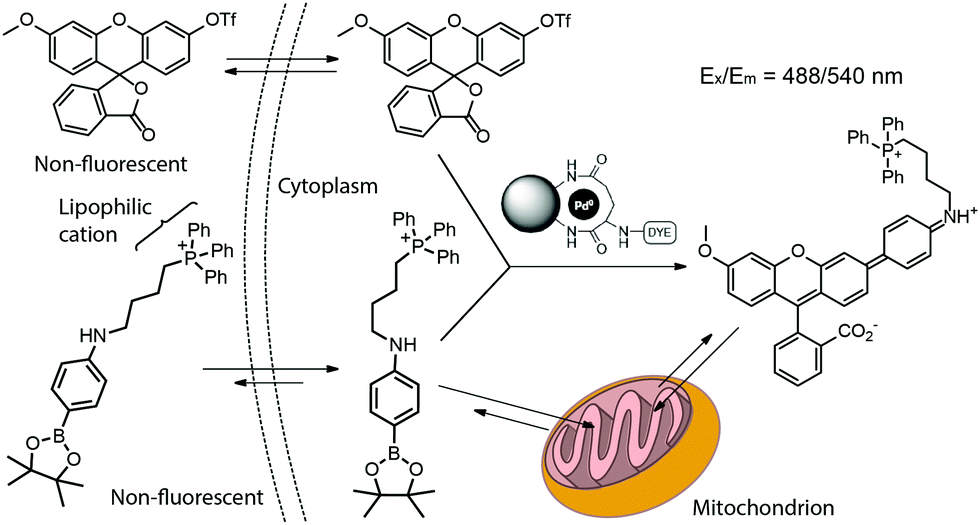 | ||
| Fig. 12 Pd0-Catalyzed intracellular cross-coupling of fluorescein triflate and triphenylphosphonium-bearing arylboronate. The coupling activates the fluorescence and targeting ability toward mitochondria. Figure adapted from ref. 3 with permission from Macmillan Publishers Ltd. | ||
In the same year, Lin and coworkers reported the copper-free Sonogashira cross-coupling reaction of a homopropargyl-glycine (HPG)-encoded ubiquitin protein with various aryl iodides in E. coli (Fig. 13).32 The copper-free coupling reaction was effected by a robust aminopyrimidine–Pd(II) complex 16, analogous to the dihydroxypyrimidine–palladium catalysts pioneered by Davis and coworkers for aqueous and cell-surface Suzuki–Miyaura cross-coupling reactions.33,34 Thus, the addition of the 1 mM aminopyrimidine–Pd(II) complex, 100 μM fluorescein iodide and 5 mM sodium ascorbate to HPG Ub-overexpressing M15A cells gave the coupled product after 4 h. In 2013, Chen and coworkers reported a ligand-free intracellular Sonogashira coupling in Gram-negative bacteria (Fig. 13).35 Thus, GFP with an alkyne-bearing lysine expressed in E. coli underwent an efficient intracellular Sonogashira coupling reaction with an iodophenyl rhodamine in 1 h by simply using 200 μM Pd(NO3)2 or Na2PdCl4 in the medium. Not only did the coupling occur more effectively without the aminopyrimidine-based ligands, the ligands actually reduced the amount of Pd taken up by the cells.
 | ||
| Fig. 13 (a) Functionalization of HPG-Ub protein using aryl iodide substrates or iodophenyl-functionalized PEG5k, mediated by Pd(OAc)2 and catalyst 16. (b) Fluorescence labelling of HPG-Ub by fluorescein iodide in E. coli mediated by 16. Left: Fluorescence of the bacteria pallets upon 365 nm excitation. Middle/right: In-gel fluorescence and Coomassie blue staining of SDS-PAGE analysis of the proteins from the pallets, respectively. Figure reproduced from ref. 32 with the permission of ACS Publications. | ||
As can be seen from the above examples, the reactions that form C–C bonds are currently limited to Pd-catalyzed Suzuki–Miyaura and Sonogashira reactions. It can be expected that other cross-coupling reactions will ultimately be developed in the intracellular environment, particularly as new catalysts are developed with improved biocompatibility and activity. There are other strategies for intracellular conjugation that do not involve C–C bond formation. In particular, Cu(I)-assisted alkyne–azide cycloaddition (CuAAC), also known as click chemistry, is arguably the most important extracellular conjugation method because of its high substrate selectivity, bioorthogonality, rapid reaction rate and high turnover number. Although the use of CuAAC in protein functionalization36 and cell surface modification37,38 is well established, highly efficient and biocompatible intracellular CuAAC reactions were reported only recently because of copper toxicity.31,39 In 2012, Ting and coworkers reported a fast, cell-compatible strategy that used copper-chelating azides for biomolecular labelling inside cells.40 By introducing a pyridinyl group in proximity to the azide functionality, the reactivity is greatly increased as a result of Cu(I) coordination by the pyridyl unit (Fig. 14). The reaction rate could be further accelerated by the addition of known CuAAC ligands such as tris(hydroxypropyltriazolylmethyl)amine (THPTA). These improvements significantly reduced the amount of Cu needed for the CuAAC conjugation while retaining a sufficient reaction rate such that the ligation of small molecules to proteins inside cells could be effected with ≤100 μM of Cu(I).
 | ||
| Fig. 14 Top: Reaction scheme for chelation-assisted CuAAC by using picolyl azide. Middle and bottom: Labelling of newly synthesized, alkyne-bearing RNAs and proteins using chelation-assisted CuAAC. Figure reproduced from ref. 40 with the permission of Wiley-VCH. | ||
Chen and coworkers developed a new protein-based pH indicator for measuring intracellular acidities as low as pH = 2.41 The indicator was prepared using CuAAC chemistry to conjugate a fluorophore, 4-N,N-dimethylamino-1,8-naphthalimide (4-DMN) to HdeA, an acid-stress chaperon carrying a site-specifically introduced azide handle. Using the Cu(I) ligand 2-[4-[[bis[(1-tert-butyltriazol-4-yl)methyl]amino]methyl]triazol-1-yl] acetic acid (BTTAA) developed by Wu and coworkers,38 the CuAAC reaction with 100 μM Cu(I), 500 μM alkyne, and 3 mM NaAsc was found to occur between the HdeA azide and the 4-DMN alkyne in the periplasmic space of living E. coli cells. HdeA changes conformation upon pH change from 7 to 2 altering the fluorescence of the conjugated solvatochromic 4-DMN (Fig. 15). The same group expanded the strategy to the measurement of compartment-specific pH values inside E. coli in 2014.42 In this work, HdeA–dye conjugates were produced in both the periplasm and cytoplasm of E. coli, allowing the resulting pH indicator to determine the pH gradient across the cytoplasmic membrane.
 | ||
| Fig. 15 A two-step labelling strategy by CuAAC for site-specific attachment of a dye onto HdeA protein under living conditions. The protein–dye conjugates change fluorescence intensity with pH changes. Figure reproduced from ref. 41 with the permission of Wiley-VCH. | ||
In addition to protein labelling, intracellular click reactions can also be used to probe the localization of DNA. The Bierbach group demonstrated that an alkyne-bearing Alexa Fluor 488 dye could be conjugated to an azide-bearing platinum–acridine complex bound to DNA.43 The intracellular click reaction allowed mapping the subcellular localization of this platinum-containing pharmacophore in fixed cancer cells (Fig. 16). Because the fluorescence of the acridine moiety is significantly quenched when bound to DNA, this method provided an indirect yet efficient way to visualize the platinum-based anticancer agent. In 2014, Taran and coworkers reported the ligation and visualization of an alkyne-functionalized paclitaxel using TAMRA linked to copper-chelating azides in HuH-7 human liver carcinoma cells.44 Conjugation of the TAMRA to the paclitaxel indicated the cellular localization of the drug following cell fixation. The paclitaxel substrate concentration used was quite low (62.5 nM), whereas the copper–azide complex was 50 μM (pretreated with 50 equiv. of NaAsc). The excellent kinetics of the reaction enabled the efficient click reaction using such a low drug concentration with a 2 h incubation. Similar imaging methods may be extended to the visualization of a wide range of biomolecules within live cells especially if even faster click chemistry becomes available.
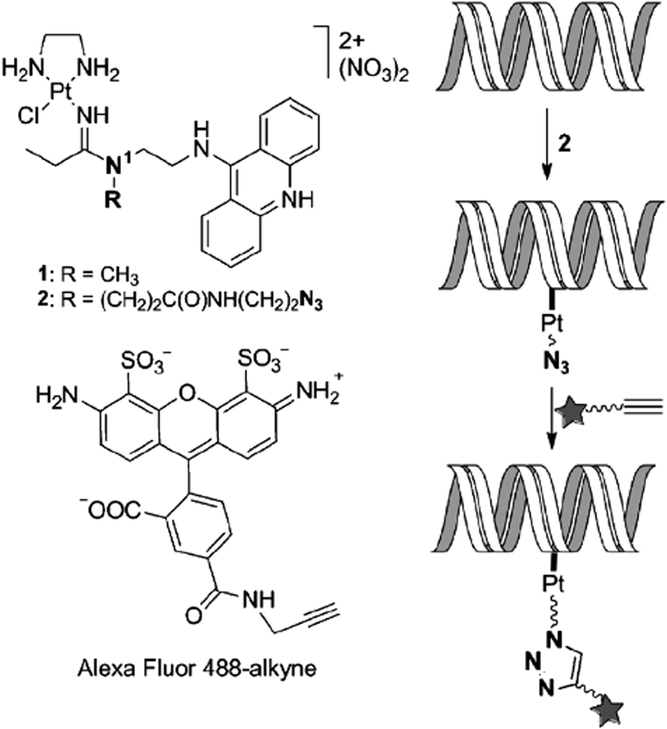 | ||
| Fig. 16 Illustration of the bioorthogonal fluorescent labelling of DNA-targeted Pt–acridine agent using alkyne-bearing dye and intracellular click chemistry. Figure reproduced from ref. 43 with the permission of Wiley VCH. | ||
Another recent example of an intracellular CuAAC reaction was reported by Cai and coworkers. It used a 4-{[bis-(1-tert-butyl-1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}O-[1,2,3]tri-azol-1-yl-acetic acid (BTTAA) analog functionalized by oligoPEG and TAT moieties (Fig. 17).45 This PEGylated catalyst could be readily taken into cells and was found to efficiently catalyse the in cellulo ligation of an azido-functionalized biotin. By using a conjugation-hydrolysis strategy, the authors were able to apply an LC-MS technique to quantify the yield of the intracellular CuAAC reaction, an important accomplishment. Indeed, it would significantly advance this area of research if more yield determinations were reported. In addition to reporting the first intracellular CuACC reaction without a copper ligating azide, the authors showed the ligation to occur even with serum present, significantly broadening the potential application of the approach.
 | ||
| Fig. 17 Top: Structures of BTTAA-based ligands. Bottom: Illustration of the incorporation of HPG to proteins, CuAAC reaction in live cells, and hydrolysis of the biotinylated proteins to HPG-PEG7-amine for LC-ESI-MS/MS quantification for the study of intracellular click yields. Figure reproduced from ref. 45 with the permission from The Royal Society of Chemistry. | ||
In cellulo assembly of therapeutic agents
The intracellular chemical synthesis of therapeutic agents is an important new direction for intracellular metallocatalysis. Preparing bioactive agents intracellularly offers a number of potential benefits, including bypassing uptake issues that plague large molecules and potentially increasing cell and tissue selectivity (Fig. 18).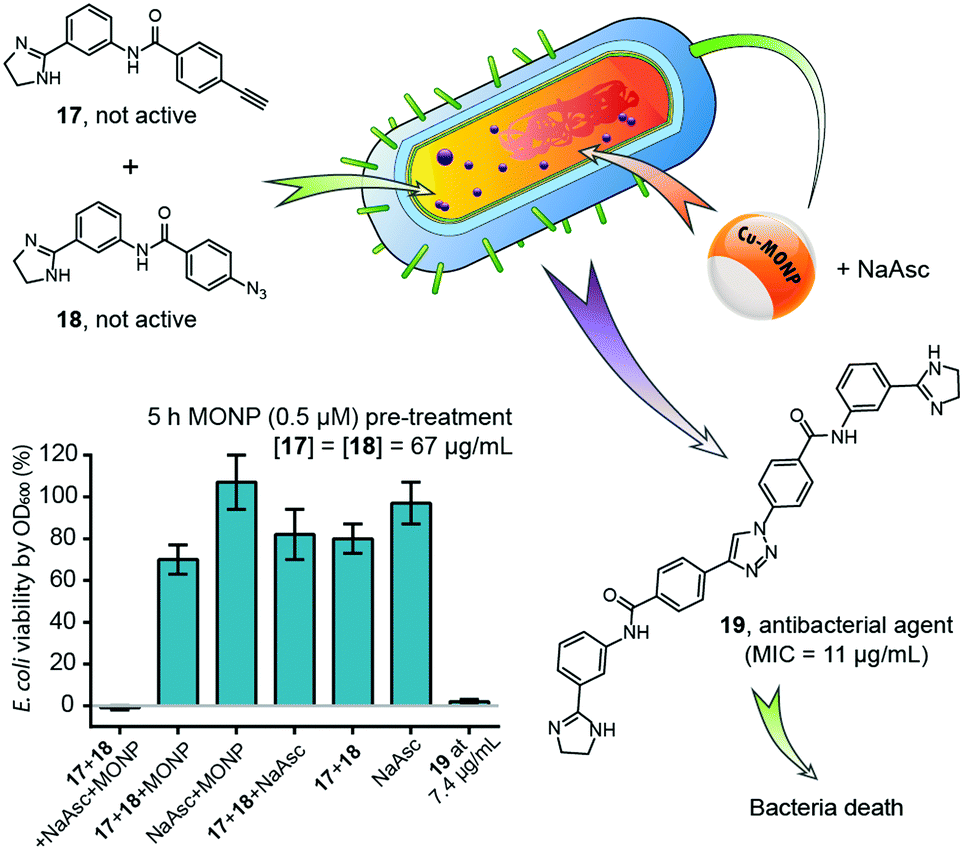 | ||
| Fig. 18 Intracellular synthesis of an antimicrobial bisamidine compound inside E. coli mediated by Cu-MONP, resulting in death of bacteria. Figure partially reproduced from ref. 46 with the permission of ACS Publications. NaAsc = sodium L-ascorbate. | ||
In 2016, Zimmerman and coworkers reported a single-chain copper-containing metal-organic nanoparticle (Cu-MONP) that efficiently catalysed intracellular click reactions in bacteria and mammalian cells.46 The Cu-MONPs were prepared by the intramolecular chain collapse of an aspartate-bearing polymer mediated by Cu2+ ions. In vitro experiments showed high catalytic activity at room temperature for a wide range of azide and alkyne substrates at ppm levels of copper. NCI-H460 and MDA-MB-231 human cancer cells treated with a 500 nM solution of Cu-MONP in PBS took up the nanoparticles and effected an intracellular click reaction between 3-azidocoumarin and 4-methoxyphenylacetylene with light-up fluorescence detection. A bisamidine-based antimicrobial compound was successfully synthesized inside E. coli from 2-phenylimidazoline azide and alkyne precursors of low toxicity, and killed the bacteria upon intracellular synthesis. This work was the first reported example of the intracellular synthesis of a bioactive compound through intermolecular coupling.
Very recently, Bradley and coworkers demonstrated the concept of targeted, intracellular drug synthesis by using a fluorescent, PdNP-loaded microsphere with targeting moieties for brain cancer cells (cRGDfE-PdNP, Fig. 19a).47 Flow cytometry studies showed the targeting effect of the peptide, which enhanced U87-MG cell uptake of the catalyst. The catalyst maintained its activity in the cells and could mediate both in cellulo coupling and decaging, generating two cytotoxic agents simultaneously in the targeted cells and leading to enhanced cell death (Fig. 19b and c). This impressive example shows the power of the intracellular synthetic strategy for cancer therapy.
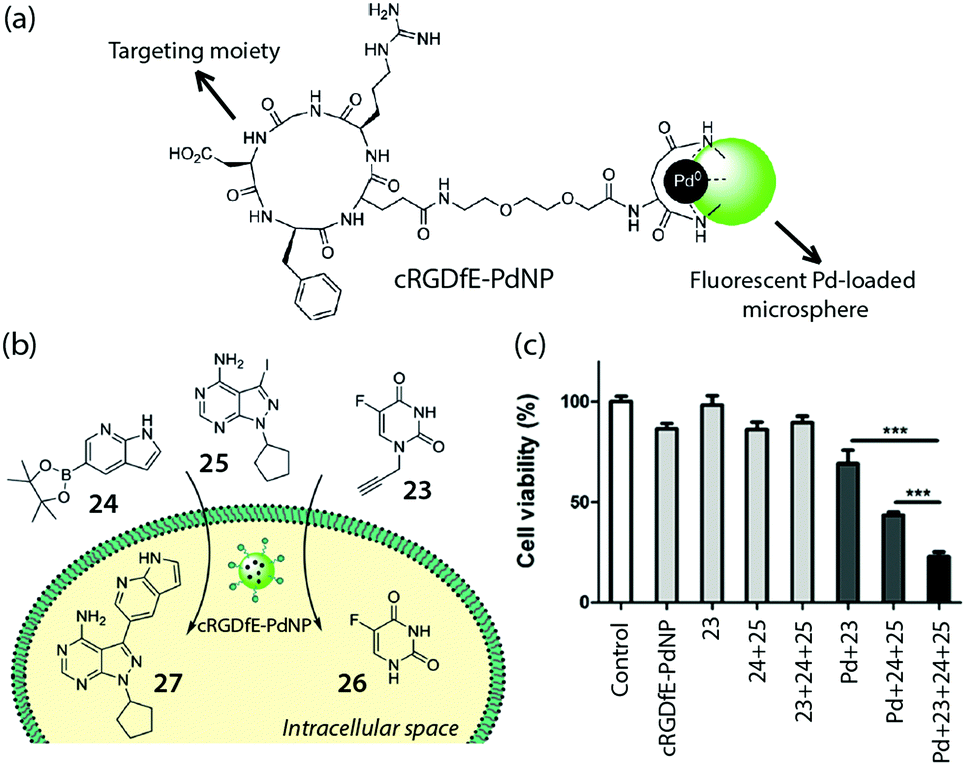 | ||
| Fig. 19 (a) Structure of the fluorescent Pd catalyst with cancer targeting capability, cRGDfE-PdNP, reported by Bradley et al. (b) Conceptual illustration of intracellular decaging and synthesis of drugs mediated by cRGDfE-PdNP. (c) Cell viability changes after treating U87-MG cells with compound 23–25 and cRGDfE-PdNP. Figure reproduced from ref. 48 with the permission from Wiley VCH. | ||
The vista
Bioorthogonal metal-based catalysis within cells is indeed an emerging field of chemistry. This review has highlighted a number of pioneering, early efforts that establish the promise for this new field. It is clear that progress in using intracellular chemistry to probe the chemical biology of the cell has advanced much more rapidly than its application to drug synthesis. In part, this is likely a result of the more stringent requirements for therapeutic applications both with regard to catalyst toxicity and its “drug-like” properties.As exciting as progress is in this area, there are numerous challenges facing the field, both technical and in the scope. The technical challenges are many and only a few can be highlighted here. One major difficulty is in firmly establishing a reaction as occurring within the confines of the cell. One of the advantages of protein labelling is the high likelihood that the protein remains within the cell as it reacts with the metal catalyst and the label. In the case of intracellular small molecule synthesis, one must always consider the possibility that the reaction is occurring outside the cell and the product subsequently migrates in, thus mimicking a true intracellular process. This concern is magnified when fixed cells are imaged because of the ability of molecules to migrate during or after the fixation process. The careful study by Weymouth, Wender, and coworkers demonstrates20 how even with live cells extracellular reaction products may readily migrate into the cell mimicking an intracellular transformation. It is quite possible that some of the intracellular reactions presented herein actually took place in the extracellular space.
Another challenge is in quantifying and comparing the efficiency of different intracellular metal-catalysed reactions. Often evidence for the reaction is simply a light-up cellular response, but a 2-fold increase in fluorescence when >100 μM substrate and catalyst are used is arguably less impressive than a 10-fold increase at 100 nM reagent concentration. Calculating the equivalent of a reaction yield based on a reasonable choice for limiting reagent would help standardize experiments and provide a measure of “quality control.” At the same time, many of the transition metal “catalysts” reported herein have not been firmly demonstrated to show turnover intracellularly.
A final technical challenge is related to cellular delivery. Small, hydrophobic metal catalysts tend to pass the cell membrane more readily, but are likely to be more susceptible to decomplexation and both deleterious ligation and redox side reactions. In this regard, the nano- and microparticle platforms described above have the advantage that they can help protect the metal catalyst. But they are typically more reluctant to enter the cell and those surface groups that accelerate uptake often lead to an increase in cytotoxicity.
Regarding the scope of the field, the intracellular metal-catalysed chemistry described herein is largely limited to copper, ruthenium and palladium. In large part this results from the limited number of highly efficient, biocompatible metal catalysts available. The development of intracellular metal catalysis is very difficult because of the low substrate and catalyst concentrations achievable in cells, the inherent cytotoxicity of transition metals, and the competitive environment of the cell where a number of small molecules and biomacromolecules can effectively poison the catalyst. Compared to natural enzymes, nearly all of the artificial metal catalysts reported to date still require a relatively high concentration to compensate for their lower activity and turnover number.
Recent advances in organometallic chemistry have featured “hybrid” catalysts that derive from natural proteins but with artificially introduced metal centers. By exploiting protein engineering, directed evolution and metal screening, the scope of metalloenzyme-catalyzed reactions is no longer limited to the inherent reactivity of metal centers in native metalloenzymes. Instead, artificially introduced transition metals within protein pockets can catalyze abiological reactions not seen in the natural biological systems, such as olefin metathesis.
In 2016, Hartwig and coworkers successfully installed various noble metals in haem proteins, and tested these artificially metalloproteins for their catalytic activities for C–H insertion and cyclopropanation reactions.48 Ir-containing proteins showed the best efficiency in both cases, and the directed evolution strategy was further applied to introduce enantioselectivity in the catalysis. In the same year, Ward and coworkers reported the use of directed evolution to obtain an efficient artificial “metathase” in E. coli periplasm.49 The Ru-metalloenzyme was prepared by introducing a biotinylated Hoveyda–Grubbs catalyst into a periplasm-localizing streptavidin. After demonstrating the activity of the resulting Ru-metalloenzyme in olefin metathesis, the authors used directed evolution to further promote the catalytic activity, characterized conveniently by the coumarin-based fluorescence assay.
These two efforts suggest a new way to generate biocompatible transition metal catalysts with diverse metals without the need for synthesizing complicated ligands and shells. Natural proteins have provided a limitless library of macromolecular skeletons within which metal catalytic centers can safely reside. These skeletons are not only innately biocompatible, but can also help prevent the poisoning from intracellular inhibitors, as well as provide the regioselectivity and enantioselectivity desirable in modern catalysis. The directed evolution process further adds promise to this strategy, as being a straightforward and convenient way to increase the catalyst performance.
To date, the scope of intracellular catalysis has been limited to single step reactions. Given the power of orthogonal chemistry, more complicated, multi-step assembly processes can be readily envisioned.25 Indeed, an advantage of the synthetic polymeric scaffold approach is the ability to increase the catalyst load and to create materials with multiple metallocatalysts. Of course, variations in the scaffold surface can potentially provide cell permeability, selectivity and targeting capability.
All in all, as more and more intracellular catalysts are developed and reported, it can be expected that this on-demand, in cellulo synthesis will become a powerful strategy not just for exploring the chemical biology of the cell, but ultimately for creating highly personalized drug nano-factories that are both responsive and able to target the relevant disease.
Note added in proof
Sadler and coworkers very recently reported an osmium-based catalyst that performs asymmetric transfer hydrogenation reactions in cancer cells. See: Nat. Chem., 2018, DOI: 10.1038/NCHEM.2918.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (NSF CHE 1709718).Notes and references
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 118, 5773–5776 CrossRef.
- P. K. Sasmal, S. Carregal-Romero, W. J. Parak and E. Meggers, Organometallics, 2012, 31, 5968–5970 CrossRef CAS.
- R. M. Yusop, A. Unciti-Broceta, E. M. Johansson, R. M. Sanchez-Martin and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- A. Unciti-Broceta, E. M. Johansson, R. M. Yusop, R. M. Sanchez-Martin and M. Bradley, Nat. Protoc., 2012, 7, 1207–1218 CrossRef CAS PubMed.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliott, R. W. Köster, W. J. Parak and E. Meggers, ChemBioChem, 2012, 13, 1116–1120 CrossRef CAS PubMed.
- M. P. Tracey, D. Pham and K. Koide, Chem. Soc. Rev., 2015, 44, 4769–4791 RSC.
- Y. L. Pak, K. M. K. Swamy and J. Yoon, Sensors, 2015, 15, 24374–24396 CrossRef PubMed.
- A. T. Aron, K. M. Ramos-Torres, J. A. J. Cotruvo and C. J. Chang, Acc. Chem. Res., 2015, 48, 2434–2442 CrossRef CAS PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- M. Santra, S.-K. Ko, I. Shin and K. H. Ahn, Chem. Commun., 2010, 46, 3964–3966 RSC.
- B. Zhu, C. Gao, Y. Zhao, C. Liu, Y. Li, Q. Wei, Z. Ma, B. Du and X. Zhang, Chem. Commun., 2011, 47, 8656–8658 RSC.
- W. Liu, J. Jiang, C. Chen, X. Tang, J. Shi, P. Zhang, K. Zhang, Z. Li, W. Dou, L. Yang and W. Liu, Inorg. Chem., 2014, 53, 12590–12594 CrossRef CAS PubMed.
- S. Goswami, A. Manna, A. K. Maity, S. Paul, A. K. Das, M. K. Das, P. Saha, C. K. Quah and H.-K. Fun, Dalton Trans., 2013, 42, 12844–12848 RSC.
- H. Chen, W. Lin and L. Yuan, Org. Biomol. Chem., 2013, 11, 1938–1941 CAS.
- X. Wang, Z. Guo, S. Zhu, H. Tian and W. Zhu, Chem. Commun., 2014, 50, 13525–13528 RSC.
- A. A. Kislukhin, V. P. Hong, K. E. Breitenkamp and M. G. Finn, Bioconjugate Chem., 2013, 24, 684–689 CrossRef CAS PubMed.
- M. I. Sanchez, C. Penas, M. E. Vazquez and J. L. Mascarenas, Chem. Sci., 2014, 5, 1901–1907 RSC.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- H. Saburi, S. Tanaka and M. Kitamura, Angew. Chem., Int. Ed., 2005, 44, 1730–1732 CrossRef CAS PubMed.
- H.-T. Hsu, B. M. Trantow, R. M. Waymouth and P. A. Wender, Bioconjugate Chem., 2016, 27, 376–382 CrossRef CAS PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- M. Tomas-Gamasa, M. Martinez-Calvo, J. R. Couceiro and J. L. Mascarenas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- J. Wang, S. Zheng, Y. Liu, Z. Zhang, Z. Lin, J. Li, G. Zhang, X. Wang, J. Li and P. R. Chen, J. Am. Chem. Soc., 2016, 138, 15118–15121 CrossRef CAS PubMed.
- C.-H. Wong and S. C. Zimmerman, Chem. Commun., 2013, 49, 1679–1695 RSC.
- M. J. Jou, X. Chen, K. M. K. Swamy, H. N. Kim, H.-J. Kim, S.-G. Lee and J. Yoon, Chem. Commun., 2009, 7218–7220 CAS.
- Y.-K. Yang, S. Lee and J. Tae, Org. Lett., 2009, 11, 5610–5613 CrossRef CAS PubMed.
- J. H. Do, H. N. Kim, J. Yoon, J. S. Kim and H.-J. Kim, Org. Lett., 2010, 12, 932–934 CrossRef CAS PubMed.
- J.-B. Wang, Q.-Q. Wu, Y.-Z. Min, Y.-Z. Liu and Q.-H. Song, Chem. Commun., 2012, 48, 744–746 RSC.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed.
- R. A. J. Smith, C. M. Porteous, A. M. Gane and M. P. Murphy, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5407–5412 CrossRef CAS PubMed.
- N. Li, R. K. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316–15319 CrossRef CAS PubMed.
- C. D. Spicer, T. Triemer and T. B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800–803 CrossRef CAS PubMed.
- J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 CrossRef CAS PubMed.
- J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. R. Chen, J. Am. Chem. Soc., 2013, 135, 7330–7338 CrossRef CAS PubMed.
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS PubMed.
- D. Soriano del Amo, W. Wang, H. Jiang, C. Besanceney, A. C. Yan, M. Levy, Y. Liu, F. L. Marlow and P. Wu, J. Am. Chem. Soc., 2010, 132, 16893–16899 CrossRef CAS PubMed.
- C. Besanceney-Webler, H. Jiang, T. Zheng, L. Feng, D. Soriano del Amo, W. Wang, L. M. Klivansky, F. L. Marlow, Y. Liu and P. Wu, Angew. Chem., Int. Ed., 2011, 50, 8051–8056 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., 2009, 121, 10063–10067 CrossRef.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Angew. Chem., Int. Ed., 2012, 51, 5852–5856 CrossRef CAS PubMed.
- M. Yang, Y. Song, M. Zhang, S. Lin, Z. Hao, Y. Liang, D. Zhang and P. R. Chen, Angew. Chem., Int. Ed., 2012, 51, 7674–7679 CrossRef CAS PubMed.
- M. Yang, A. S. Jalloh, W. Wei, J. Zhao, P. Wu and P. R. Chen, Nat. Commun., 2014, 5, 4981 CrossRef CAS PubMed.
- S. Ding, X. Qiao, J. Suryadi, G. S. Marrs, G. L. Kucera and U. Bierbach, Angew. Chem., Int. Ed., 2013, 52, 3350–3354 CrossRef CAS PubMed.
- V. Bevilacqua, M. King, M. Chaumontet, M. Nothisen, S. Gabillet, D. Buisson, C. Puente, A. Wagner and F. Taran, Angew. Chem., 2014, 126, 5982–5986 CrossRef.
- S. Li, L. Wang, F. Yu, Z. Zhu, D. Shobaki, H. Chen, M. Wang, J. Wang, G. Qin, U. J. Erasquin, L. Ren, Y. Wang and C. Cai, Chem. Sci., 2017, 8, 2107–2114 RSC.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Angew. Chem., Int. Ed., 2017, 48, 6974–6976 Search PubMed.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef CAS PubMed.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed.
Footnote |
| † Current address: Institute of Chemical Biology and Nanomedicine, Hunan University, Changsha, Hunan 410082, China. |
| This journal is © The Royal Society of Chemistry 2018 |