
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correction: Unravelling the impact of hydrocarbon structure on the fumarate addition mechanism – a gas-phase ab initio study
Vivek S.
Bharadwaj
 a,
Shubham
Vyas
ab,
Stephanie M.
Villano
a,
C. Mark
Maupin
a and
Anthony M.
Dean
*a
a,
Shubham
Vyas
ab,
Stephanie M.
Villano
a,
C. Mark
Maupin
a and
Anthony M.
Dean
*a
aChemical and Biological Engineering Department, Colorado School of Mines, 1500 Illinois Street, Golden, CO 80401, USA. E-mail: amdean@mines.edu; Fax: +1-303-273-3730; Tel: +1-303-273-3643
bDepartment of Chemistry and Geochemistry, Colorado School of Mines, 1500 Illinois Street, Golden, CO 80401, USA
First published on 22nd March 2018
Abstract
Correction for ‘Unravelling the impact of hydrocarbon structure on the fumarate addition mechanism – a gas-phase ab initio study’ by Vivek S. Bharadwaj et al., Phys. Chem. Chem. Phys., 2015, 17, 4054–4066.
In the case of toluene, our calculations indicate a small energy difference between the transition states of the R and S stereoisomers for the fumarate addition step. This arises due to the separate optimizations of the transition states and inherent uncertainties in the calculation (cf. Fig. 2 of the manuscript). This energy difference is translated into the reactant and product complexes due to the independent IRC calculations for each isomer. We expect that the energy difference between the two transition states for the stereoisomers in an unperturbed environment should be zero. However, this does not impact the results or the conclusions of the study, as evidenced by a re-evaluation of the kinetic model (Fig. 1) with this energy difference set to zero. The figure below compares the kinetic modelling results with the (original) small energy difference (left) and zero energy difference (right) in the transition states for the R and S stereoisomers of toluene.
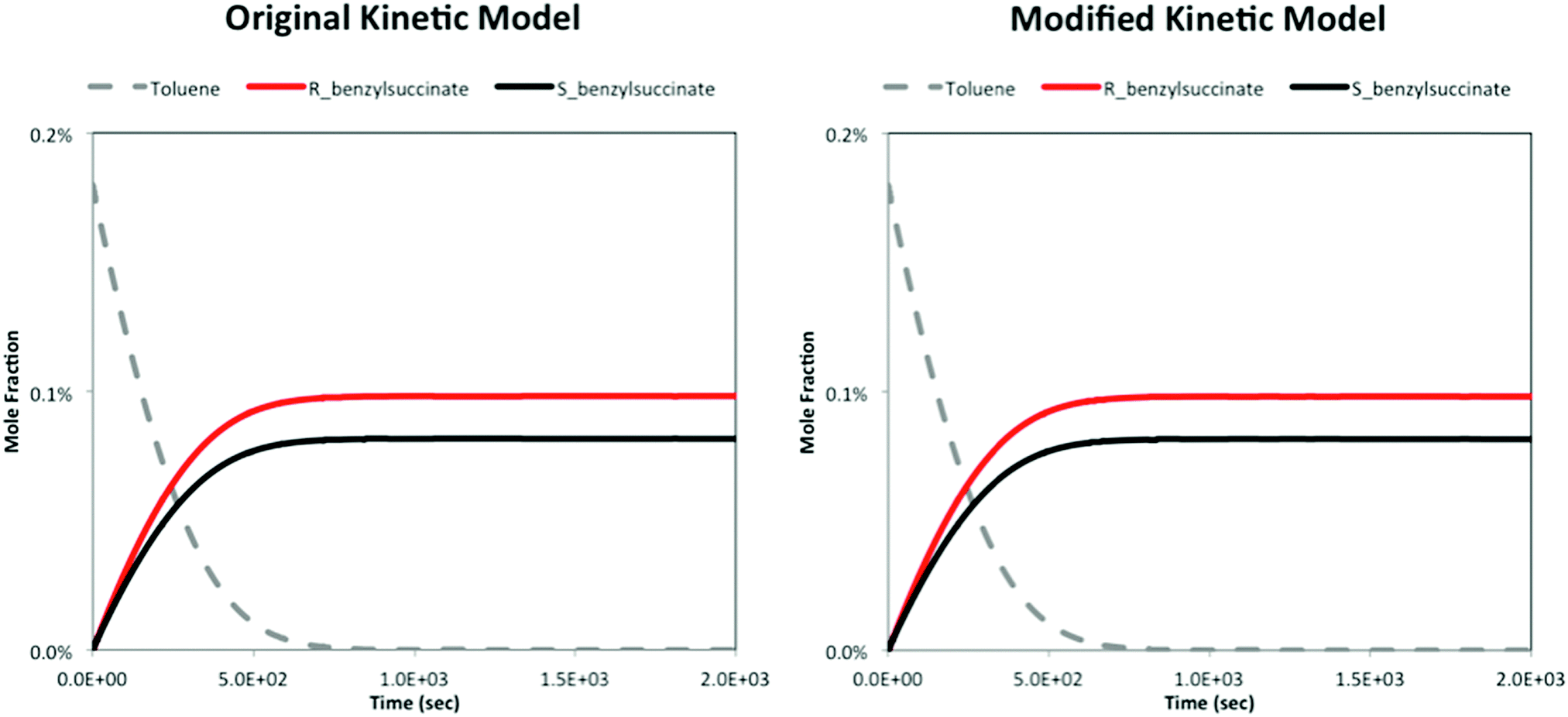 | ||
| Fig. 1 Comparison of product distributions between the R and S isomers from the kinetic model with k-tst values of 2.36 × 10−3 s−1 (left) and 4.82 × 10−4 s−1 (right) for the fumarate addition step. The other rate constant values are the same as those reported in Table 1 of the article. | ||
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
| This journal is © the Owner Societies 2018 |