
First-principles study of rocksalt early transition-metal carbides as potential catalysts for Li–O2 batteries
 a
Hao
Huang
*a
a
Hao
Huang
*a
Abstract
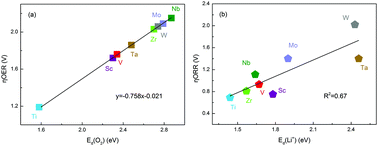
A series of early transition-metal carbides (TMCs) in the NaCl structure have been constructed to compare the catalytic activity in Li–O2 batteries by first-principles calculations. The reasonable interfacial models of LixO2 (x = 4, 2, and 1) molecules adsorbed on early TMCs surfaces were used to simulate oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) processes. Taking overpotentials as a merit parameter of catalytic activity, more relationships between material properties relative to the adsorption/desorption behavior of active molecules and catalytic activity are constructed for early TMCs. The equilibrium and charging potentials used to calculate the OER overpotentials of early TMCs are inversely proportional to the adsorption energies of (Li2O)2 and LiO2, respectively. The ORR overpotentials are inversely proportional to the adsorption energies of (Li2O)2 and LiO2 for early TMCs, but the relationship between OER overpotentials and the adsorption energies of reactive intermediates is unclear. Additionally, the overpotentials of early TMCs for ORR and OER are proportional to the desorption energies of Li+ and O2, respectively. In general, both the adsorption energy of (Li2O)2/LiO2 and desorption energy of Li+/O2 are effective characterization parameters of catalytic activity. By providing the comprehensive valuable parameters on electrochemical performance to compare the catalytic activity of early TMCs and establishing more correlations between material properties relative to the adsorption/desorption behavior of active molecules with their catalytic activity, our investigation is helpful for knowing more about the catalytic process and beneficial to screen and design novel highly active catalysts for Li–O2 batteries.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

