
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual effects in single molecule tautomerization: hemiporphycene
Victoriya
Kim
 a,
Lukasz
Piatkowski
*b,
Maria
Pszona
b,
Regina
Jäger
a,
Jakub
Ostapko
b,
Jerzy
Sepioł
b,
Alfred J.
Meixner
*a and
Jacek
Waluk
*bc
a,
Lukasz
Piatkowski
*b,
Maria
Pszona
b,
Regina
Jäger
a,
Jakub
Ostapko
b,
Jerzy
Sepioł
b,
Alfred J.
Meixner
*a and
Jacek
Waluk
*bc
aInstitute of Physical and Theoretical Chemistry and LISA+, University of Tübingen, Auf der Morgenstelle 18, D-72076 Tübingen, Germany. E-mail: alfred.meixner@uni-tuebingen.de
bInstitute of Physical Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Kasprzaka 44/52, Poland. E-mail: lpiatkowski@ichf.edu.pl; jwaluk@ichf.edu.pl
cFaculty of Mathematics and Science, Cardinal Stefan Wyszyński University, Dewajtis 5, 01-815 Warsaw, Poland
First published on 10th October 2018
Abstract
Parent hemiporphycene, a recently obtained constitutional isomer of porphyrin, exists in room temperature solutions and polymer matrices in the form of two trans tautomers interconverting via double hydrogen transfer. Using confocal fluorescence microscopy, it was possible to monitor tautomerization in single hemiporphycene molecules embedded in a PMMA film by monitoring the spectral and temporal evolution of their fluorescence spectra. The emission spectra of the two tautomeric forms are similar to those obtained from ensemble studies. However, the analysis of temporal spectral evolution reveals effects not detected in the bulk. For some single molecules, a large decrease of tautomerization rate was observed. This is interpreted as an indication of multidimensional character of the tautomerization coordinate and coupling of the reaction with the polymer relaxation processes. In addition, fluorescence lifetimes obtained for single molecules are significantly shorter than those measured for the bulk. It is proposed that the shortening is caused by environment-induced distortion of the molecule, which enhances the S0 ← S1 internal conversion rate by lowering the barrier to excited state single hydrogen transfer. This effect seems to reflect the specific morphology of thin (30 nm) polymer samples, because it is not observed in ensemble studies carried out using thick (tens of micrometers or more) PMMA films.
Introduction
Structural (constitutional) isomers of porphyrins, such as porphycene1 or inverted2 (confused)3 porphyrin, are of interest to various chemical communities in the context of possible applications,4–6 as well as fundamental studies. Regarding the latter, porphycene and its derivatives emerged as very good models for intramolecular double hydrogen transfer.7 This process has been investigated not only in condensed phases,8,9 but also for molecules isolated in supersonic jets10,11 or helium nanodroplets,12 and finally, on a single molecule level, using three different techniques: (i) fluorescence microscopy;13–16 (ii) surface-enhanced Raman spectroscopy;17 and (iii) scanning probe microscopy.18–22 These studies have led to detailed characterization of the multidimensional reaction path, which includes specific effects such as tunneling or vibrational mode-specificity.23Recently, the synthesis of another parent porphyrin tautomer, hemiporphycene (HPc), has been reported.24 Due to low symmetry (Cs), the molecule can assume six different tautomeric forms. The two most stable ones, corresponding to trans tautomers (Scheme 1), have been observed in solution. Both experiment and DFT/TDDFT calculations indicate that they differ in energy by ca. 1 kcal mol−1 and even less in the first excited singlet state. Such a small energy difference suggests that interconversion between both forms can be observed in a single molecule. The observation of this process was the main goal of this work. Using confocal fluorescence microscopy, we were able to monitor tautomerization in single hemiporphycene molecules and to characterize the emission of each tautomer. It was also possible to measure the kinetics of fluorescence decay. Studies of single molecules revealed unusual effects, impossible to detect in routine studies based on ensemble averaging. Particularly impressive is the observation of “freezing” the tautomerization, i.e., decrease of the reaction rate by many orders of magnitude. The other effect is a significant decrease of fluorescence lifetime compared to the value obtained from ensemble studies. We propose to explain both effects using a model that considers coupling of the intramolecular potential energy surface with the dynamically changing properties of the surrounding matrix. Comparison of the present results with the data previously reported for porphycenes,14,25 molecules of higher symmetry, suggests an interesting possibility of accelerating tautomerization by matrix perturbation of a nonsymmetrical double minimum potential.
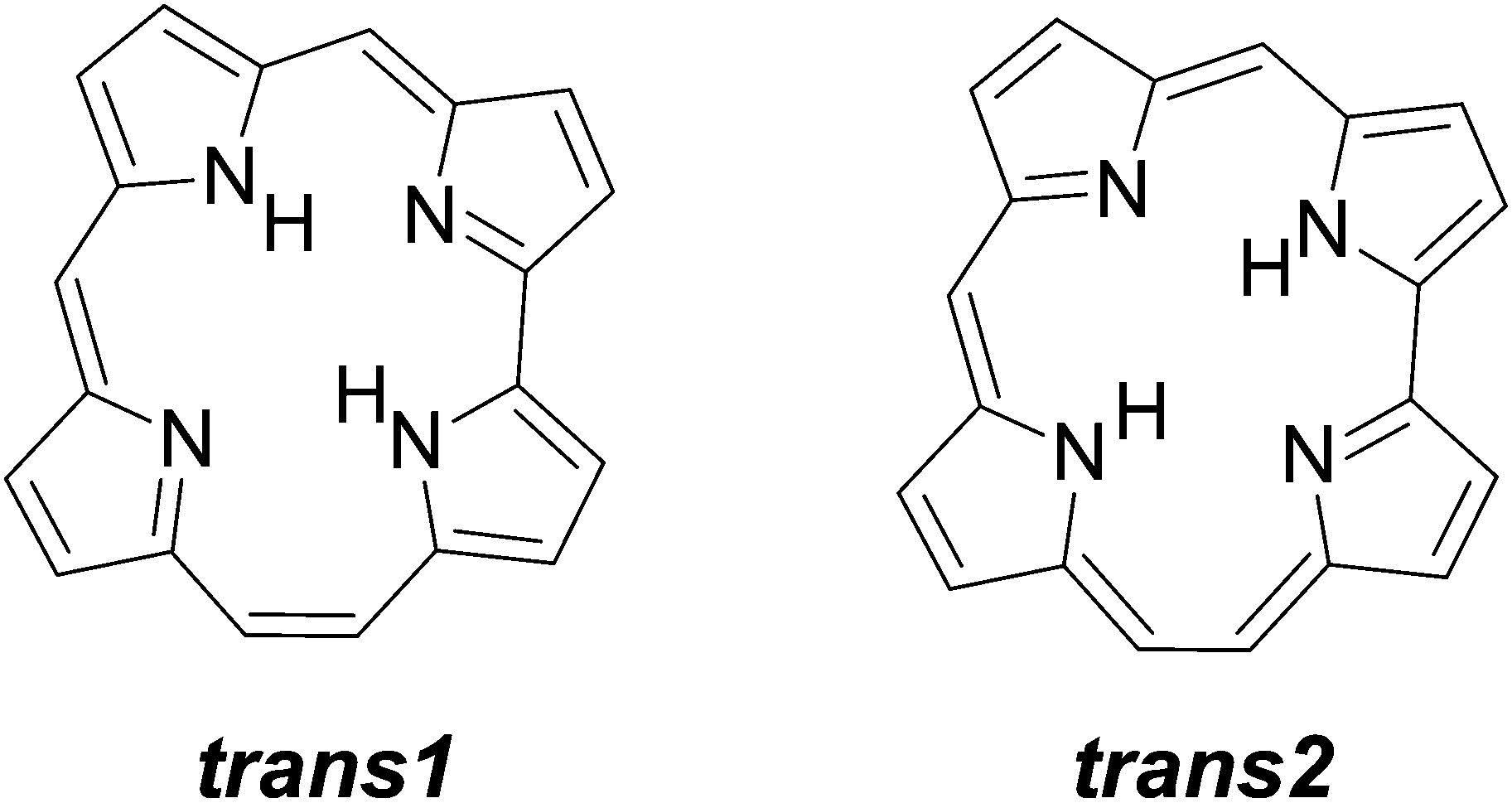 | ||
| Scheme 1 Two lowest energy tautomers of HPc. | ||
Experimental
The synthesis and purification of HPc have been described before.24 Samples for SMS studies were obtained by spin coating 25 μl of a cosolution of HPc in toluene containing 1% w/v of PMMA (Aldrich, medium molecular weight). Prior to spin coating, microscope cover slides were thoroughly cleaned in a mixture of chromic and sulfuric acid and rinsed with distilled water and methanol. Single molecule fluorescence spectra have been recorded using a SpectraPro spectrometer equipped with a nitrogen cooled SPEC-10 CCD-detector (Princeton Instruments) coupled to a home-built inverted confocal microscope which has already been described.25 Fluorescence lifetime traces were recorded using a time-correlated single-photon counting system (PicoQuant) with a photon counting module (PDM series, Micro Photon Devices). A pulsed supercontinuum laser (SuperK Extreme EXB-4, NKT Photonics) was used as an excitation source in combination with an acousto-optical tunable filter (AOTF, SuperK Select, NKT Photonics) and an optical filter to precisely select the excitation wavelength.PMMA samples containing HPc for ensemble studies were prepared by evaporating toluene solutions containing both the chromophore and polymer. Absorption of these samples was measured on a Shimadzu UV 3100 spectrophotometer. For the emission, an Avantes AvaSpec ULS RS-TEC spectrometer has been used. Fluorescence decay times were measured using a home-built setup, with a FIANIUM SC-400-6-PP-60 laser as the excitation source, a Digikröm CM112 monochromator, a Becker & Hickl HPM-100-40 hybrid detector, and an SPC 830 module for time-correlated single photon counting.
Results and discussion
Solutions of HPc exhibit dual fluorescence, with the maxima of the two emissions shifted by ca. 10 nm.24 The higher energy fluorescence was attributed to the lowest energy tautomer trans1, and the lower energy emission to the trans2 form. The relative intensities of the two emissions depend on the excitation wavelength, which shows that the excited state equilibrium between the two forms is not established. More detailed studies24 demonstrated that a small fraction (ca. 15% at 293 K) of excited trans2 is converted into trans1, but not vice versa.Essentially the same results are obtained for bulk HPc embedded in PMMA (Fig. 1). The spectra are somewhat broader than those in nonpolar solvents, so that the two absorption peaks corresponding to the origin of the S0–S1 transition in trans1 and trans2 (ca. 610 and 620 nm, respectively) are no longer resolved. However, the fluorescence dominated by either trans1 or trans2 can be observed if the excitation wavelength is set to preferentially excite one or the other tautomer, whose absorption spectra are also shifted.
 | ||
| Fig. 1 Room temperature ensemble absorption and emission spectra of HPc embedded in a PMMA film. Fluorescence was excited at 500 (red), 504 (green), and 532 (black) nm. | ||
Based on the results obtained for bulk measurements in PMMA, the 544 nm excitation wavelength was selected for single molecule experiments to ensure similar intensities of fluorescence from the two forms. The single molecule spectra obtained in this way exhibit a different behavior than those registered from bulk samples. In a few cases, dual emission from a single chromophore was observed, exhibiting peaks near 610 and 620 nm (Fig. 2a). However, most of the studied single molecules were characterized by broad emission, of which the maximum was shifting with time. A series of fluorescence spectra were recorded for the same molecule, with each spectrum collected for 10 s. An example of two consecutive spectra is shown in Fig. 2b and c. In each case, the largest intensity peak could be fitted with two Gaussians (P1 and P2) with different amplitudes; their ratio varied from 0.5 to 2.5, as shown in Fig. 3c.
 | ||
| Fig. 2 (a) Exemplary molecule exhibiting a clear splitting of the emission spectrum. (b and c) Exemplary emission spectra of one single molecule, recorded one after another with 10 s acquisition time. Red dashed lines represent fitted Gaussian profiles. Black dashed line is the total fitted emission spectrum comprising four Gaussian bands. | ||
 | ||
| Fig. 3 (a) Spectral position of fitted bands P1 (blue) and P2 (brown). (b) Full width half maximum of the fitted Gaussian profiles for the P1 (blue) and P2 (brown) bands. (c) Intensity ratio (P1/P2) between the fitted P1 and P2 bands. (d) Spectral position difference between the fitted P1 and P2 bands. | ||
Evolution of single molecule spectra with time is a well-documented effect and occurs at cryogenic as well as room temperature.26–28 It can occur by small random structural fluctuations in the surrounding host, leading to small energy level shifts of a chromophore and consequently to spectral diffusion characterized by Gaussian-like spectral broadening. This extrinsic behaviour is typical for single molecule spectra recorded at room temperature from chromophores embedded in an amorphous matrix such as a polymer, which lacks a unique energy minimum. Spectral switching between discrete spectral states, such as different conformers of the same chromophores that have different energies, is characterized by a bi- or multimodal distribution.29,30 This phenomenon leads to emission spectra with separated maxima specific for the different chromophores. When the switching rate is slower than the rate at which spectra are recorded, single-molecule spectra reveal spontaneous repeated spectral jumps between the spectra of the different conformers, whereas the spectrum of an ensemble would show a stable superposition of the spectra of the conformers.
In order to check whether the changes observed for HPc are caused by spectral diffusion or by tautomerization, we constructed the histograms showing the spectral positions and widths of the fitted bands, their intensity ratios, and the difference between the locations of the maxima. Even though we cannot completely exclude spectral diffusion, which, most likely also contributes to the observed effects, the analysis of these data provides strong arguments for tautomerization to be the dominant factor responsible for the observed behavior. The bimodal distribution of the maxima (Fig. 3a), as well as the difference between the positions of the P1 and P2 peaks (12–13 nm), is fully consistent with the results obtained for bulk solutions and PMMA matrices. A bimodal distribution should rather not be expected for sole spectral diffusion; it typically leads to Gaussian-like broadening of the emission. In the present case, it is necessary to use two Gaussian bands, which show that the spectra are quite asymmetric. Thus, the assignment of the observed changes to the presence of two tautomers is justified. However, a crucial difference exists with respect to the ensemble spectra recorded for solutions. Our unpublished data for hemiporphycenes indicate that in solution the reaction proceeds in about a microsecond. For such a case, the intensity ratio between the fitted P1 and P2 bands should exhibit a narrow distribution. In contrast, we observe a broad distribution of P1/P2, 0.5–2.5 (Fig. 3c), indicating that the residence time of the molecule in one or the other tautomeric form varies over long time periods.
Trivial causes of the unusual spectral behavior, e.g., a drastic difference of absorption coefficients at the wavelength of excitation or photoselection due to specific orientation of the chromophore, can be rejected, since the same molecule is able to reveal both trans1 and trans2 emissions on a longer timescale.
The tautomerization rate for hemiporphycene in solution is intermediate between those of porphycene, where the double hydrogen transfer occurs within a few picoseconds,31 and porphyrin, where it requires tens of microseconds.32 The present results show that, for a single chromophore, the large difference in the tautomerization rate in HPc and porphycene can disappear. The same effect of arresting the tautomerization as described here for HPc has been observed in the studies of fluorescence from single molecules of octaethylporphyrin,33 as well as for the parent porphycene and its two derivatives, 2,7,13,19-tetra-t-butylporphycene and 2,7,13,19-tetraphenylporphycene.14,25
In order to explain the similar behavior of two porphyrin isomers that strongly differ in their intrinsic potentials for tautomerization, we employ a model which takes into account the influence of the dynamics of the molecular environment on the reaction rate (Fig. 4). Thus, the potential energy surface includes both a (possibly multidimensional) tautomerization coordinate, which describes the hydrogen transfer path, and the reorganization coordinate, which includes the motion of the environment. The two coordinates are coupled, implying that the barrier for hydrogen transfer varies, depending on the arrangement of the molecular surroundings.
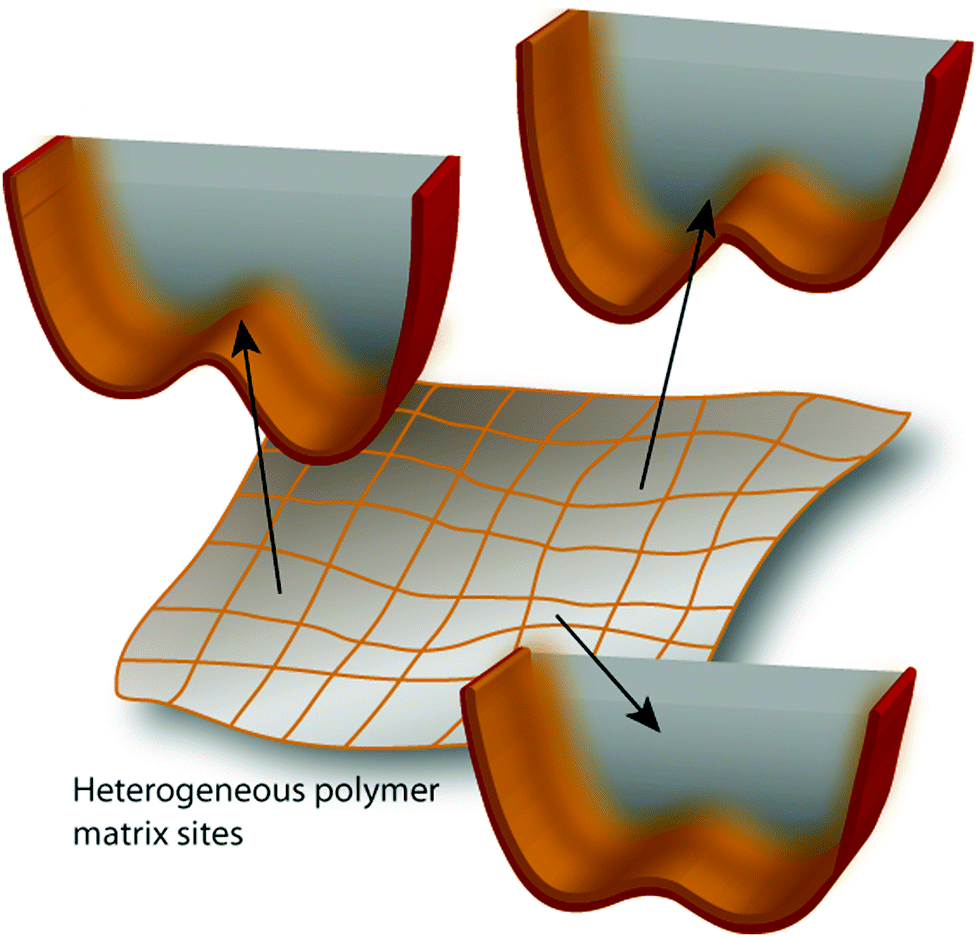 | ||
| Fig. 4 A model that allows rationalizing slowing down of the tautomerization rate. The shape of the double minimum potential for hydrogen transfer depends on the (fluctuating) nanoenvironment. The figure has been inspired by the work of Limbach and coworkers.45 | ||
A similar model, based on the Marcus theory approach to proton transfer, has been proposed for enzymes.34 The term “work function” was used to describe the energy required to attain the configuration most favorable for the reaction. In the present case, such a configuration corresponds to a specific arrangement of the polymer chains around HPc. Since the polymer relaxation can be slow, this explains the drastic reduction of the tautomerization rate. In other words, in condensed phases, a scenario is possible where the reaction rate is controlled not by the intrinsic properties of the reactant, but by the timescale of reorganization of the environment. One should note that, for the same molecule, this effect can disappear if the timescale of matrix reorganization becomes shorter than that given by the tautomerization coordinate. This is usually the case for non-viscous solvents.
The other spectacular difference between the results obtained for ensembles and single HPc molecules was provided by the values of fluorescence lifetimes. In the bulk, the decay time at room temperature amounts to 8–9 ns, with the lifetime of excited trans1 being slightly shorter.24 In a more viscous solvent (dodecane), the lifetime increases to about 12 ns. As could have been expected from this trend, for bulk PMMA samples the measured lifetime is even longer – 13 ns.
Surprisingly, the lifetimes observed for single molecules are significantly shorter, ranging from 2 to 6 ns. In principle, one could postulate that the increase in the S1 radiative constant is responsible for lifetime shortening; this leads to a larger brightness of the short-lived species, which naturally biases the statistics towards stronger emitters. However, such a scenario seems very unlikely. Using the value of the radiative constants (∼2 × 107 s−1) obtained from solution studies, combined with the fluorescence lifetime obtained for bulk studies, one can estimate that the radiative constant would have to increase by an order of magnitude to account for the observed lifetime shortening. It would be extremely difficult to find reasons for such a large increase. Therefore, we postulate that the shorter fluorescence decay is caused by an increase in the rate of nonradiative depopulation of the lowest excited singlet state. We cannot definitely state which of the two possible pathways, intersystem crossing (ISC) to T1 or internal conversion (IC) to S0, are more strongly affected. Based on the knowledge of photophysics of HPc and porphycene, enhanced IC looks more probable. In previous studies, we postulated, for both porphycene35 and hemiporphycene,24 that the channel responsible for efficient S0 ← S1 internal conversion may involve a single hydrogen transfer coupled with distortion from planarity. It may be that the polymer environment affects the tautomerization potential in a way that favors such distortion. In fact, the same model as proposed above to account for fluctuations in the tautomerization rate can be used to explain the enhancement of IC. Now, however, the intramolecular coordinate has a more complex character, encompassing both single hydrogen shift and geometry distortion.
It is worth noting that the rate of S0 ← S1 IC in some porphycenes was found to be very strongly viscosity-dependent;36 the results obtained for HPc in ensemble studies also suggest such a behavior. The present observation of different fluorescence lifetimes: “normal” for bulk, thick samples, and much shorter for single molecules embedded in thin films, suggests different environments in the two cases. In the bulk, the majority of molecules are located inside a rigid polymer, experiencing a large viscosity. On the other hand, single HPc molecules behave as if immersed in a medium of lower viscosity. This could suggest the location at the polymer–air interface. It should be recalled, however, that significant differences in morphology have been suggested for thin and thick polymer films.37–42 It may thus happen that a molecule embedded in a polymer changes the photophysics depending on the exact location within the matrix.
Summary
Hemiporphycene is the second porphyrin isomer for which the possibility of monitoring the intramolecular double hydrogen transfer on a single molecule level using confocal fluorescence microscopy has now been demonstrated. In porphycenes, which were the subject of previous studies,13,14,16,25 the energy profile is described by a symmetrical double minimum and the reaction proceeds between two chemically identical trans forms. Therefore, special techniques that exploit polarized light had to be used to detect the reaction.43,44 Single molecule studies of porphycenes embedded in PMMA revealed “localization” of the two internal hydrogens on a timescale of seconds or even longer.14,25 This effect can be explained by a Marcus-type model which assumes that double hydrogen transfer occurs with the intrinsic rate, once the molecule, embedded in a polymer, has attained a symmetric double minimum configuration along the reaction coordinate. Fluctuations of the environment (described by a generalized “reorganization” coordinate) may lead to localization of the hydrogens, i.e., to a slower tautomerization rate. The dynamics of reorganization is determined by the matrix properties, which explains a huge difference between solutions and polymers. It should be noted that our present results on single molecules nicely illustrate the postulate put forward by Limbach, on the basis of NMR experiments, that the equilibrium and rate constants of hydrogen transfer in strong intramolecular hydrogen bonds embedded in solids can be environment-dependent.45–47For hemiporphycene, because of lower molecular symmetry and the inequivalence of trans1 and trans2, it is possible to monitor tautomerization on the basis of the spectral profile of fluorescence. Interestingly, the experimental results lead to similar conclusions for both types of porphyrin isomers. In each case, molecules exhibiting “unusual” behavior, i.e., temporal decrease of the tautomerization rate, were found. It should be stressed that such an observation is only possible for the regime of single molecule studies; in the bulk, the “exotic” behavior would have been averaged.
The same model as applied earlier to porphycenes25 can now be used for HPc. However, a significant difference has to be pointed out. Since the intrinsic tautomerization coordinate is non-symmetrical in HPc, fluctuations of the polymer matrix may not necessarily slow down the reaction. The opposite effect of increasing the rate is also possible, if the fluctuations make the potential more symmetrical. Such an effect cannot be excluded, although, using our methodology, we cannot directly confirm it because of insufficient time resolution. However, a hint is provided by the observation of a shorter fluorescence decay, i.e., an increase of the S1 decay rate. We postulated, for both porphycene and hemiporphycene, that the channel responsible for efficient S0 ← S1 internal conversion may involve a single hydrogen transfer coupled with distortion from planarity. It may thus be that the polymer environment affects the tautomerization potential in a way that favors either the hydrogen transfer or geometrical distortion, or both.
The implications of the current work are twofold. First, the observed fluctuations of the tautomerization rate in single molecules not only indicate a complex, multidimensional character of the double hydrogen transfer path, but also show an important (dominant, in specific cases) role of the environment. Second, our results show that tautomerization may be used as a tool for monitoring the polymer relaxation dynamics. Both of these aspects clearly suggest the directions of future studies: investigation of tautomerization in a particular chromophore in the single molecule regime using different polymer matrices. Such studies may contribute to a better understanding of both the mechanisms of tautomerization and polymer relaxation dynamics. Hopefully, they may also help to explain in more detail the effect of the large decrease of fluorescence decay time observed for hemiporphycene. This phenomenon seems to be molecule-specific: the ongoing measurements of fluorescence decay times carried out for substituted porphycenes did not reveal significant differences between single molecule and ensemble regimes.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Polish National Science Centre (NCN) grants 2017/26/M/ST4/00072, 2011/03/D/ST4/05258 and 2016/22/A/ST4/00029. The project has received funding from the National Science Centre, Poland, grant 2015/19/P/ST4/03635, POLONEZ 1 and from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 665778.References
- E. Vogel, M. Köcher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed., 1986, 25, 257–259 CrossRef.
- P. J. Chmielewski, L. Latos-Grażyński, K. Rachlewicz and T. Glowiak, Angew. Chem., Int. Ed., 1994, 33, 779–781 CrossRef.
- H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767–768 CrossRef CAS.
- M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Drain, Coord. Chem. Rev., 2010, 254, 2297–2310 CrossRef CAS PubMed.
- J. H. Chou, M. E. Kosal, H. S. Nalwa, N. A. Rakow and K. S. Suslick, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 6, pp. 43–131 Search PubMed.
- E. Alves, M. A. F. Faustino, M. G. P. M. S. Neves, A. Cunha, H. Nadais and A. Almeida, J. Photochem. Photobiol., C, 2015, 22, 34–57 CrossRef CAS.
- J. Waluk, Chem. Rev., 2017, 117, 2447–2480 CrossRef CAS PubMed.
- P. Ciąćka, P. Fita, A. Listkowski, M. Kijak, S. Nonell, D. Kuzuhara, H. Yamada, C. Radzewicz and J. Waluk, J. Phys. Chem. B, 2015, 119, 2292–2301 CrossRef PubMed.
- P. Ciąćka, P. Fita, A. Listkowski, C. Radzewicz and J. Waluk, J. Phys. Chem. Lett., 2016, 7, 283–288 CrossRef PubMed.
- E. T. Mengesha, J. Sepioł, P. Borowicz and J. Waluk, J. Chem. Phys., 2013, 138, 174201 CrossRef PubMed.
- E. T. Mengesha, A. Zehnacker-Rentien, J. Sepioł, M. Kijak and J. Waluk, J. Phys. Chem. B, 2015, 119, 2193–2203 CrossRef CAS PubMed.
- A. Vdovin, J. Waluk, B. Dick and A. Slenczka, ChemPhysChem, 2009, 10, 761–765 CrossRef CAS PubMed.
- H. Piwoński, A. Hartschuh, N. Urbańska, M. Pietraszkiewicz, J. Sepioł, A. Meixner and J. Waluk, J. Phys. Chem. C, 2009, 113, 11514–11519 CrossRef.
- H. Piwoński, A. Sokołowski, M. Kijak, S. Nonell and J. Waluk, J. Phys. Chem. Lett., 2013, 4, 3967–3971 CrossRef.
- H. Piwoński, A. Sokołowski and J. Waluk, J. Phys. Chem. Lett., 2015, 6, 2477–2482 CrossRef PubMed.
- H. Piwoński, C. Stupperich, A. Hartschuh, J. Sepioł, A. Meixner and J. Waluk, J. Am. Chem. Soc., 2005, 127, 5302–5303 CrossRef PubMed.
- S. Gawinkowski, M. Pszona, A. Gorski, J. Niedziółka-Jönsson, I. Kamińska, W. Nogala and J. Waluk, Nanoscale, 2016, 8, 3337–3349 RSC.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- J. N. Ladenthin, L. Grill, S. Gawinkowski, S. Liu, J. Waluk and T. Kumagai, ACS Nano, 2015, 9, 7287–7295 CrossRef CAS PubMed.
- H. Böckmann, S. Liu, J. Mielke, S. Gawinkowski, J. Waluk, L. Grill, M. Wolf and T. Kumagai, Nano Lett., 2016, 16, 1034–1041 CrossRef PubMed.
- J. Ladenthin, T. Frederiksen, M. Persson, J. Sharp, S. Gawinkowski, J. Waluk and T. Kumagai, Nat. Chem., 2016, 8, 935–940 CrossRef CAS PubMed.
- M. Koch, M. Pagan, M. Persson, S. Gawinkowski, J. Waluk and T. Kumagai, J. Am. Chem. Soc., 2017, 139, 12681–12687 CrossRef CAS PubMed.
- P. Fita, L. Grill, A. Listkowski, H. Piwoński, S. Gawinkowski, M. Pszona, J. Sepioł, E. T. Mengesha, T. Kumagai and J. Waluk, Phys. Chem. Chem. Phys., 2017, 19, 4921–4937 RSC.
- J. Ostapko, K. Nawara, M. Kijak, J. Buczyńska, B. Leśniewska, M. Pietrzak, G. Orzanowska and J. Waluk, Chem. – Eur. J., 2016, 22, 17311–17320 CrossRef CAS PubMed.
- L. Piatkowski, C. Schanbacher, F. Wackenhut, A. Jamrozik, A. J. Meixner and J. Waluk, J. Phys. Chem. Lett., 2018, 9, 1211–1215 CrossRef CAS PubMed.
- W. P. Ambrose and W. E. Moerner, Nature, 1991, 349, 225–227 CrossRef CAS.
- A. Kiraz, M. Ehrl, C. Bräuchle and A. Zumbusch, J. Chem. Phys., 2003, 118, 10821–10824 CrossRef CAS.
- F. Stracke, C. Blum, S. Becker, K. Müllen and A. J. Meixner, Chem. Phys. Chem., 2005, 6, 1242–1246 CrossRef CAS PubMed.
- F. Stracke, C. Blum, S. Becker, K. Müllen and A. J. Meixner, Chem. Phys. Lett., 2000, 325, 196–202 CrossRef CAS.
- F. Stracke, C. Blum, S. Becker, K. Müllen and A. J. Meixner, Chem. Phys., 2004, 300, 153–164 CrossRef CAS.
- P. Fita, N. Urbańska, C. Radzewicz and J. Waluk, Chem. – Eur. J., 2009, 15, 4851–4856 CrossRef CAS PubMed.
- B. Wehrle, H. H. Limbach, M. Köcher, O. Ermer and E. Vogel, Angew. Chem., Int. Ed. Engl., 1987, 26, 934–936 CrossRef.
- A. M. Chizhik, R. Jäger, A. I. Chizhik, S. Bär, H. G. Mack, M. Sackrow, C. Stanciu, M. Hanack and A. J. Meixner, Phys. Chem. Chem. Phys., 2011, 13, 1722–1733 RSC.
- D. N. Silverman, Biochim. Biophys. Acta, 2000, 1458, 88–103 CrossRef CAS.
- A. Sobolewski, M. Gil, J. Dobkowski and J. Waluk, J. Phys. Chem. A, 2009, 113, 7714–7716 CrossRef CAS PubMed.
- M. Gil, J. Dobkowski, G. Wiosna-Sałyga, N. Urbańska, P. Fita, C. Radzewicz, M. Pietraszkiewicz, P. Borowicz, D. Marks, M. Glasbeek and J. Waluk, J. Am. Chem. Soc., 2010, 132, 13472–13485 CrossRef CAS PubMed.
- R. D. Priestley, C. J. Ellison, L. J. Broadbelt and J. M. Torkelson, Science, 2005, 309, 456–459 CrossRef CAS PubMed.
- S. Habuchi, T. Oba and M. Vacha, Phys. Chem. Chem. Phys., 2011, 13, 7001–7007 RSC.
- T. Oba and M. Vacha, ACS Macro Lett., 2012, 1, 784–788 CrossRef CAS.
- H. Kobayashi, S. Onda, S. Furumaki, S. Habuchi and M. Vacha, Chem. Phys. Lett., 2012, 528, 1–6 CrossRef CAS.
- D. Wöll, E. Braeken, A. Deres, F. C. De Schryver, H. Uji-i and J. Hofkens, Chem. Soc. Rev., 2009, 38, 313–328 RSC.
- B. M. I. Flier, M. C. Baier, J. Huber, K. Müllen, S. Mecking, A. Zumbusch and D. Wöll, J. Am. Chem. Soc., 2011, 134, 480–488 CrossRef PubMed.
- T. Züchner, A. V. Failla and A. J. Meixner, Angew. Chem., Int. Ed. Engl., 2011, 50, 5274–5293 CrossRef PubMed.
- A. I. Chizhik, A. Chizhik, A. Huss, R. Jäger and A. J. Meixner, J. Phys. Chem. Lett., 2011, 2, 2152–2157 CrossRef CAS.
- B. Wehrle, H. Zimmermann and H. H. Limbach, J. Am. Chem. Soc., 1988, 110, 7014–7024 CrossRef CAS.
- B. Wehrle, H. H. Limbach and H. Zimmermann, Ber. Bunsenges. Phys. Chem., 1987, 91, 941–950 CrossRef CAS.
- H. H. Limbach, H. Rumpel, L. Meschede, B. Wehrle, M. Schlabach and G. Scherer, J. Mol. Struct., 1988, 177, 143–155 CrossRef.
| This journal is © the Owner Societies 2018 |