
The reaction of alkyl hydropersulfides (RSSH, R = CH3 and tBu) with H2S in the gas phase and in aqueous solution†
Linxing
Zhang
 ab,
Xinhao
Zhang
a,
Yun-Dong
Wu
*ac,
Yaoming
Xie
b,
Jon M.
Fukuto
d and
Henry F.
Schaefer
*b
ab,
Xinhao
Zhang
a,
Yun-Dong
Wu
*ac,
Yaoming
Xie
b,
Jon M.
Fukuto
d and
Henry F.
Schaefer
*b
aLab of Computational Chemistry & Drug Design, State Key Laboratory of Chemical Oncogenomics, Peking University Shenzhen Graduate School, Shenzhen 518055, China. E-mail: wuyd@pkusz.edu.cn
bCenter for Computational Quantum Chemistry, University of Georgia, Athens, GA 30602, USA. E-mail: ccq@uga.edu
cCollege of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
dDepartment of Chemistry, Sonoma State University, Rohnert Park, CA 94928, USA
First published on 28th September 2018
Abstract
The RSSH + H2S → RSH + HSSH reaction has been suggested by numerous labs to be important in H2S-mediated biological processes. Seven different mechanisms for this reaction (R = CH3, as a model) have been studied using the DFT methods (M06-2X and ωB97X-D) with the Dunning aug-cc-pV(T+d)Z basis sets. The reaction of CH3SSH with gas phase H2S has a very high energy barrier (>45 kcal mol−1), consistent with the available experimental observations. A series of substitution reactions R1–S–S–H + −S–R2 (R1 = Me, tBu, Ad, R2 = H, S–Me, S–tBu, S–Ad) have been studied. The regioselectivity is largely affected by the steric bulkiness of R1, but is much less sensitive to R2. Thus, when R1 is Me, all −S–R2 favorably attack the internal S atom, leading to R1–S–S–R2. While for R1 = tBu, Ad, all −S–R2 significantly prefer to attack the external S atom to form −S–S–R2. These results are in good agreement with the experimental observations.
1. Introduction
In 1996 hydrogen sulfide (H2S) was suggested by Abe and Kimura as an endogenous neuromodulator in the brain.1 The endogenous metabolism and physiological functions of H2S make it the third gasotransmitter, in addition to the previously known gasotransmitters nitric oxide (NO) and carbon monoxide (CO).2 H2S has also been found to play an important role in cellular functions. It acts as a relaxant of smooth muscle and as a vasodilator.3 It is also active in the brain, altering hippocampal long-term potentiation, which is involved in the formation of memory.4Much of H2S signaling has been proposed to occur through modification of cysteine residues in proteins leading to the formation of hydropersulfides (–SSH groups),5 and this modification has been referred to as S-sulfhydration (or more appropriately, sulfuration or S-persulfidation). The S-sulfuration process can be a post-translational modification of specific proteins that regulate protein functions leading to either activation or inhibition of protein activity,6 and thus S-sulfuration can serve an important cellular regulatory role.7 It was found by mass spectrometry that, besides protein hydropersulfides, small molecule hydropersulfides such as cysteine hydropersulfide (CysSSH) and glutathione hydropersulfide (GSSH) are formed in mammalian cells and tissues.8 Some of the small molecule hydropersulfides are likely to be key intermediates in protein S-sulfuration. For example, polysulfide compounds, such as diallyl trisulfide,9 penicillamine-derived acyl disulfides,10 and dithioperoxyanhydride11 reacted with cysteine or glutathione in vivo.
However, since hydropersulfides are usually unstable species, especially in aqueous solution,12 only a limited number of small molecule hydropersulfides have been synthesized and characterized, although the first hydropersulfide was prepared as early as 1954.13 In recent years, many experimental studies of the reactivity of hydropersulfides have used hydropersulfides generated in situ rather than isolated persulfides.14 For example, Francoleon and coworkers studied protein hydropersulfides generated via the reaction of H2S with the papain-cysteine mixed disulfide (Papain–S–S–Cys) as the reactant.14a This reaction yielded inactive papain persulfide (Papain–SSH) via
| Papain–S–S–Cys + H2S → Papain–SSH (inactive) + Cys–SH | (1) |
With excess H2S, the inactive persulfide intermediate (Papain–SSH) was converted to the active thiol species (Papain–SH). This observation, the H2S-mediated generation of active thiol from the hydropersulfide, is consistent with the reaction of RSSH with H2S giving the presumed products thiol and hydrogen persulfide (H2S2)
| RSSH + H2S → RSH + H2S2 | (2) |
Recently, Bailey, Zakharov, and Pluth carried out experimental studies on a series of reactions of hydropersulfides with different reagents.15 Their experimental results showed that no reaction was observed when RSSH was treated with gas-phase H2S, while different products were found when RSSH with different R substituent groups reacted under the presence of various nucleophiles and bases in CD2Cl2 at room temperature. In essence, Trt–S–SH (trityl hydropersulfide) and Ad–S–SH (adamantyl hydropersulfide) lead to the formation of Trt–SH and Ad–SH, respectively, accompanied with the formation of S8. Bn–S–SH (benzyl hydropersulfide), on the other hand, reacted under various conditions to form polysulfides.
In order to understand and provide insight into the reactions of RSSH with H2S or HS−, and R–S–S− in the present paper we perform theoretical studies to predict the mechanisms for these reactions under various conditions. Our theoretical results will be compared with available experiments, and may further shed light on the function of H2S as a signaling molecule in biochemical systems.
2. Computational details
Density functional theory (DFT) methods were employed, providing an approximate treatment of electron correlation effects. Two popular functionals adopted in the present study are M06-2X, which is a meta-GGA functional recommended by Zhao and Truhlar for the study of main-group thermochemistry and kinetics,16 and ωB97X-D, which including empirical atom-atom dispersion corrections reported by Chai and Head-Gordon.17 The M06-2X and ωB97X-D computations were performed with the Gaussian09 program package,18 using the ultrafine integration grid19 for geometry optimizations and vibrational frequency analyses.Dunning's correlation-consistent triple-zeta basis sets with augmented diffuse functions aug-cc-pVTZ were adopted for the C and H atoms.20 For the S atom, an additional set of d functions was added, denoted as aug-cc-pV(T+d)Z, which corrects some of the deficiencies of the standard correlation consistent basis sets for the second-row atoms Al–Cl.21 Solvation effects were taken into account via SMD, the Cramer–Truhlar solvation model based on the charge density.22 We would prefer to treat the solvent with explicit water molecules, but this is not feasible with our current resources. All structures shown here in figures were generated with the CYLview program.23
Since the potential energy surfaces predicted by M06-2X and ωB97X-D are in very good agreement with each other, only the M06-2X results are shown in the figures for clarity, while all DFT results are reported in Tables S1–S7 (in the ESI†) for comparison.
3. Results and discussion
3.1. Reaction between CH3SSH and H2S in the gas phase
Here we selected R = CH3 as a reasonable initial model for reaction (2) to perform the DFT study. First, a one-step four-membered ring transition state (TS1-1 in mechanism 1) was examined, in which the sulfur–hydrogen bond in H2S and the sulfur–sulfur bond in CH3SSH are about to break. Simultaneously, the sulfur–sulfur bond and the hydrogen–sulfur bond between the two molecules (H2S and CH3SSH) are being formed. In TS1-1, the distance of the H–S bond being formed is 1.34 Å, and the distance of the old H–S bond in H2S increases to 2.64 Å (Fig. 1), clearly indicating that a hydrogen atom of H2S is moving to CH3SSH. Similarly, the –SH group in CH3SSH is moving to H2S (Fig. 1). Intrinsic reaction coordinate (IRC) analysis shows that the transition state TS1-1 connects the two complexes (the IRC plot and representative structures along the reaction coordinate are shown in the ESI†). The reactant complex INT1-1 has an H⋯S hydrogen bond (2.81 Å) between one H atom in H2S and the internal S atom in CH3SSH. INT1-1 lies lower than the reactant (H2S and CH3SSH) by 3.2 kcal mol−1. The product complex INT1-2 has a hydrogen bond (2.64 Å) between one H atom in HSSH and the internal S atom in CH3SH. INT1-2 lies lower than the products (HSSH and CH3SH) by 4.6 kcal mol−1. Reaction (1) is endothermic by 3.8 kcal mol−1. However, for mechanism 1 the energy barrier (61.8 kcal mol−1 for electronic energy and 73.8 kcal mol−1 for the Gibbs free energy) is very high, suggesting that this mechanism is not feasible. | ||
| Fig. 1 Sketch of the potential energy surface of mechanism 1 for the CH3SSH + H2S reaction. The relative energies after ZVPE correction (ΔEZVPE) and the free energies at 298 K (ΔG298) are shown in parentheses. It is seen that entropy plays a significant role for INT1-1, TS1-1, and INT1-2. | ||
The second pathway in the gas phase (mechanism 2) is shown in Fig. 2. In this pathway, one H atom in H2S attacks the internal S atom in CH3SSH, and the remaining SH group approaches the external S atom in RSSH (Fig. 2). The IRC analysis shows that the transition state TS2-1 also connects two complexes. The reactant complex INT2-1, similar to INT1-1 in Fig. 1, has a hydrogen bond (H⋯S distance 2.80 Å) between one H atom in H2S and the internal S atom in CH3SSH. The other H atom in H2S points in the opposite direction. INT2-1 lies lower than the reactant (H2S and CH3SSH) by 2.8 kcal mol−1 (Fig. 2). The product complex INT2-2 does not have an obvious hydrogen bond, and INT2-2 lies lower than the products (HSSH plus CH3SH) by only 1.7 kcal mol−1. Again, the energy barrier for mechanism 2 is too high (67.3 kcal mol−1 for electronic energy and 77.8 kcal mol−1 for the Gibbs free energy), suggesting that this reaction in the gas phase will not take place at room temperature.
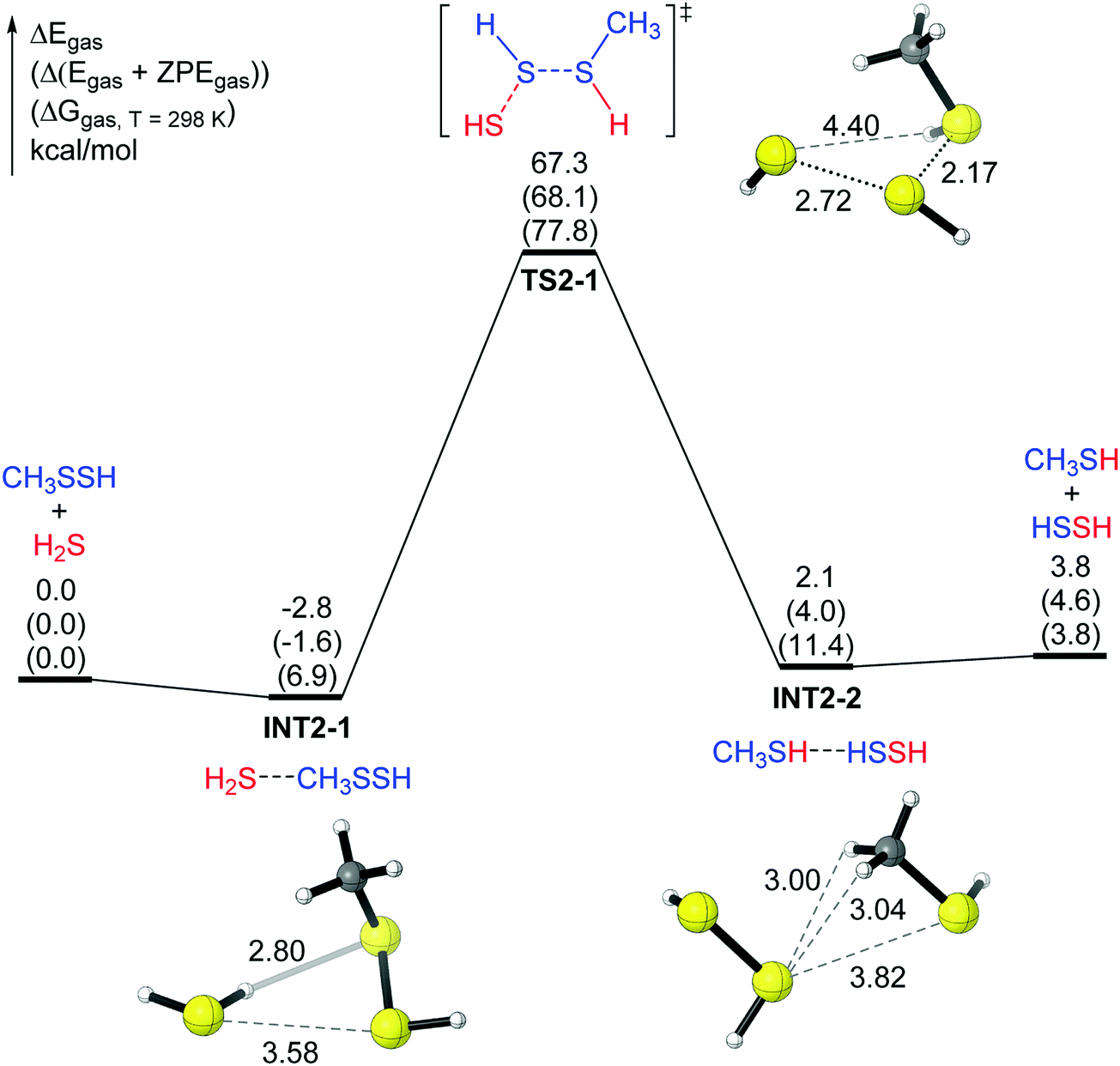 | ||
| Fig. 2 The potential energy surface of mechanism 2 for the CH3SSH + H2S reaction. The relative energies after ZVPE correction (ΔEZVPE) and the free energies at 298 K (ΔG298) are shown in parentheses. | ||
It was reported by mass spectrometry in 2000 by Gerbaux et al.24 that the thiosulfoxide species (R2SS, R = H, CH3, C2H5), which are tautomers of the disulfides (RSSR), are stable in the gas phase. The transition states for the tautomerizations between disulfides (RSSR) and thiosulfoxides (R2SS) have been considered in previous theoretical studies.24,25 The thiosulfoxides RS(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)H could be regarded as “singlet sulfur” sulfanes that are very electrophilic, and would be more reactive with H2S.6,12a Thus, a two-step mechanism (mechanism 3) was explored in the present study (Fig. 3). The first step is the tautomerization from the persulfide CH3SSH to its thiosulfoxide tautomer CH3S(S)H (INT3-1), which lies above CH3SSH by 21 kcal mol−1. The energy barrier for this tautomerization is predicted to be 44 kcal mol−1 (TS3-1). Following the tautomeric step is the nucleophilic attack by H2S. In that transition state (TS3-2), the sulfur–sulfur bond between H2S and CH3S(S)H is being formed (2.41 Å), while the sulfur–sulfur bond in CH3S(S)H is breaking (increasing to 2.53 Å). In the meantime, the proton in H2S is being transferred to the negatively charged (in terms of a Lewis structure) external sulfur of CH3S(S)H (Fig. 3). Although mechanism 3 has a lower overall energy barrier (46.0 kcal mol−1 for electronic energy and 54.8 kcal mol−1 for the Gibbs free energy) than mechanism 1 or mechanism 2, this barrier is still too high for reaction (2) to proceed.
S)H could be regarded as “singlet sulfur” sulfanes that are very electrophilic, and would be more reactive with H2S.6,12a Thus, a two-step mechanism (mechanism 3) was explored in the present study (Fig. 3). The first step is the tautomerization from the persulfide CH3SSH to its thiosulfoxide tautomer CH3S(S)H (INT3-1), which lies above CH3SSH by 21 kcal mol−1. The energy barrier for this tautomerization is predicted to be 44 kcal mol−1 (TS3-1). Following the tautomeric step is the nucleophilic attack by H2S. In that transition state (TS3-2), the sulfur–sulfur bond between H2S and CH3S(S)H is being formed (2.41 Å), while the sulfur–sulfur bond in CH3S(S)H is breaking (increasing to 2.53 Å). In the meantime, the proton in H2S is being transferred to the negatively charged (in terms of a Lewis structure) external sulfur of CH3S(S)H (Fig. 3). Although mechanism 3 has a lower overall energy barrier (46.0 kcal mol−1 for electronic energy and 54.8 kcal mol−1 for the Gibbs free energy) than mechanism 1 or mechanism 2, this barrier is still too high for reaction (2) to proceed.
 | ||
| Fig. 3 A two-step potential surface (mechanism 3) for the CH3SSH + H2S reaction. The relative energies after ZVPE correction (ΔEZVPE) and the free energies at 298 K (ΔG298) are shown in parentheses. | ||
3.2. Reaction between CH3SSH and H2S in solvent
The high energy barriers of the above mechanisms indicate that the CH3SSH + H2S reaction in the gas phase is unfavorable. This is consistent with the experimental facts reported by Pluth and co-workers.15 They studied the reactivity of a series of persulfides in the presence of gas phase H2S, and found that no reaction happened BnSSH, TrtSSH or AdSSH.It is known that proton transfer steps are often facilitated by solvation in water, and the solvent effect may be very important for reaction (2). However, for reaction (2) in aqueous solution, either hydropersulfide or H2S may ionize depending on their acidity. To compare the deprotonation abilities of CH3SSH and H2S, at first we studied the following reaction with the SMD solvation model22
| CH3SSH + HS− → CH3SS− + H2S | (3) |
This reaction energy ΔE for (3) is −0.4 kcal mol−1, and the corresponding ΔG298 is −1.5 kcal mol−1, revealing that CH3SSH is more acidic than H2S in water. This is consistent with previous studies.12a,26
In light of the differences in acidities between a hydropersulfide and H2S, the reactants in the aqueous solution model are CH3SS− and H2S. The main feature of the potential energy surface for the reaction of CH3SS− + H2S → CH3SH + HSS− is illustrated in Fig. 4 and 5. The reaction begins with a barrierless formation of a reactant complex INT4-1 (CH3SS−⋯HSH), which is predicted to lie below the reactants (CH3SS− + H2S) by 5.4 kcal mol−1. Subsequently the reactant complex experiences a small energy barrier (TS4-1, 3.2 kcal mol−1) for proton transfer to form a second complex CH3SSH⋯SH− (INT4-2), leading to the activation of the nucleophile. The HS− moiety in INT4-2 then undergoes a nucleophilic attack on the external S atom of CH3SSH, over an energy barrier of 14.0 kcal mol−1 (TS4-2), to form the intermediate CH3S−⋯S(H)SH (INT4-3). A very quick proton transfer from the HSSH moiety to the CH3S− part follows, forming the product complex CH3SH⋯SSH− (INT4-4). The release of the final products (CH3SH + HSS−) from the complex INT4-4 requires 5.1 kcal mol−1 energy. The overall reaction (3) is endothermic by 2.5 kcal mol−1, and the overall barrier with respect to the lowest-lying structure is only 15.0 kcal mol−1 (20.6 kcal mol−1 for the Gibbs free energy), which makes this reaction feasible in aqueous solution.
 | ||
| Fig. 4 The potential energy surface (mechanism 4) for the CH3SS− + H2S → CH3SH + HSS− reaction in aqueous solution. The relative energies after ZVPE correction (ΔEZVPE) and the free energies at 298 K (ΔG298) are shown in parentheses. | ||
 | ||
| Fig. 5 The geometries for the stationary points in Fig. 4. | ||
Inspired by proposed mechanisms in which thiols react with disulfides via α carbon nucleophilic attack,9a,27 an alternative mechanism for the reaction between CH3SS− and H2S was taken into account (mechanism 5, Fig. 6). After the formation of INT4-2, which follows that in mechanism 4, the nucleophilic attack of HS− can take place at the α-C atom of CH3SSH as an SN2 reaction to give the final products (CH3SH + HSS−). The energy barrier with respect to the lowest-lying structure for mechanism 5 is 35.2 kcal mol−1 (TS5-2) (39.4 kcal mol−1 for the Gibbs free energy), which is much higher than that for mechanism 4. Related results were reported in a 2017 theoretical study,27 which showed that nucleophilic attacks on the α-C atoms of disulfides (RSSR) and trisulfides (RSSSR) involve higher barriers than that on the S atoms.
 | ||
| Fig. 6 The potential energy surface (mechanism 5) for the CH3SS− + H2S → CH3SH + HSS− reaction in aqueous solution. The relative energies after ZVPE correction (ΔEZVPE) and the free energies at 298 K (ΔG298) are shown in parentheses. | ||
3.3. Further explorations: adding RSS− to the Brew
The product HSS−, predicted by the mechanisms 4 and 5, was not observed in the 2015 experiments of Bailey and Pluth.15b This may be because HSS− is not an isolable species, but able to further react to give H2S and elemental sulfur.12b However, the experiments show that different products were observed when different RSSH molecules were treated with [NBu4+][HS−],15b giving a hint that some competitive mechanisms may exist. Therefore, additional possible mechanisms involving different nucleophiles should be explored. Since the reactant RSS− has been reported to be a nucleophile,12b,28 it could react with hydropersulfides. The target of the nucleophilic attack of RSS− could be either of the two S atoms in RSSH, leading to two possible mechanisms. The target of the α-C atom in RSSH is less plausible and will not be considered, as it may be similar to the case of mechanism 5, compared with mechanism 4.Starting from the complex INT4-2 (CH3SSH⋯SH− in mechanisms 4 and 5), a more thermodynamically favorable complex CH3SSH⋯−SSCH3 (INT6-1) is found to combine with CH3SS− and release HS− (Fig. 7). Then the CH3SS− moiety in INT6-1 may attack the CH3SSH moiety at either the internal S atom or the external S atom. In the former case (mechanism 6, black line in Fig. 7), it goes over a transition state (TS6-1) with a barrier of 10.1 kcal mol−1 to produce CH3SSSCH3⋯HS− (INT6-2), releasing an energy of 0.3 (5.9–5.6) kcal mol−1. In the latter case (mechanism 7, red line in Fig. 7), it goes over a transition state (TS7-1) with a slightly higher energy barrier (10.9 kcal mol−1) but to produce CH3SH⋯SSSCH3− (INT7-1), which is lower than INT6-2 by 3.8 (9.7–5.9) kcal mol−1. Fig. 7 shows that mechanism 6 (black line) is a kinetically favored mechanism, while mechanism 7 (red line) is a thermodynamically favored mechanism. A similar case was earlier considered for the nucleophilic reaction of CN− and RSSH,29 in which the nucleophilic attack by CN− onto the internal sulfur atom of RSSH is kinetically favored, while attack on the external sulfur atom is thermodynamically favored.
 | ||
| Fig. 7 Sketch of the PESs for mechanisms 6 and 7 for the reaction of CH3SS− and H2S when the intermediate CH3SSH is attacked by the additional nucleophile CH3SS−. The aqueous solution is taken into count. The relative energies after ZVPE correction (ΔEZVPE) and the free energies at 298 K (ΔG298) are shown in parentheses. Note that the left section of this sketch is the same as that shown in mechanisms 4 and 5. In order to keep the PESs continuous before and after the ion exchange, additional energy of CH3SS− is added before the ion exchange, and additional energy of HS− is added after the exchange. | ||
As described above, the products of the reactions between RSSH and [NBu4+][HS−] depend on the R groups. For example, when BnSSH (benzyl persulfide) reacts with [NBu4+][HS−] the products were reported to be H2S and polysulfides (mainly BnSSSBn),15b and this corresponds to mechanism 6 (black line in Fig. 7). When TrtSSH reacts with [NBu4+][HS−], the products were reported to be TrtSH, S8, and H2S.15b Since H2S and elemental sulfur could be obtained from the further reaction of RSS−,12b this reaction may correspond to mechanism 7 (red line in Fig. 7). For the same RSSH + HS− reaction, why did the reactants with different R groups, such as BnSSH and TrtSSH, go through different mechanisms? Is it because of steric hindrance as a function of the group size? For those with less sterically-hindered groups, such as BnSSH, it may be less difficult to attack the internal sulfur atom (via mechanism 6); while for those with the more sterically-hindered groups, such as TrtSSH, it may be more likely to attack the external sulfur atom (via mechanism 7).
To verify this conjecture, we carried out a parallel study on another model persulfide with a larger R group, namely, tert-butyl persulfide (tBuSSH), for comparison with the above study of CH3SSH. The potential surfaces for the nucleophilic attack of tBuSS− onto tBuSSH are shown in Fig. 8. Indeed, the energy barriers for the two mechanisms are reversed in comparison with those in Fig. 7. For the smaller methyl group, the nucleophilic attack at the internal sulfur atom of RSSH has the lower energy barrier (10.1 kcal mol−1vs. 10.9 kcal mol−1). For the larger tert-butyl group, the nucleophilic attack at the external sulfur atom of RSSH has a lower energy barrier (11.7 kcal mol−1vs. 17.4 kcal mol−1). This comparison shows that the regio-selectivity depends on the R group size in RSS−, and this is consistent with the experimental results for BnSSH and TrtSSH.
 | ||
| Fig. 8 The PESs for the reaction of tBuSS− and H2S in aqueous solvent. The relative energies after ZVPE correction (ΔEZVPE) and the free energies at 298 K (ΔG298) are shown in parentheses. In order to keep the PESs continuous before and after the ion exchange, additional energy of tBuSS− is added before the ion exchange, and additional energy of HS− is added after the exchange. | ||
To have a more complete understanding of the reactivities and selectivities of various reactions, we carried out a systematic study as shown in Scheme 1. The computed activation free energies are given in Table 1. The results may be summarized as follows:
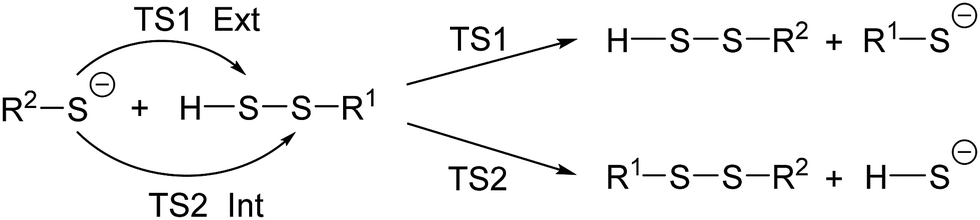 | ||
| Scheme 1 Two possible pathways of the reaction between R2–S− and H–S–S–R1. | ||
| ΔEwater (ΔGwater) | R2 = H | R2 = S–Me | R2 = S–tBu | R2 = S–Ad | ||||
|---|---|---|---|---|---|---|---|---|
| TS1 | TS2 | TS1 | TS2 | TS1 | TS2 | TS1 | TS2 | |
| R1 = Me | 16.1 (16.5) | 13.8 (13.9) | 11.0 (11.5) | 8.8 (9.2) | 10.4 (12.7) | 9.5 (11.6) | 11.8 (11.6) | 11.1 (11.0) |
| R1 = tBu | 17.1 (18.2) | 21.1 (21.8) | 12.7 (14.6) | 17.0 (18.7) | 11.8 (12.5) | 16.2 (16.6) | 13.0 (13.2) | 17.9 (18.4) |
| R1 = Ad | 17.1 (17.6) | 21.0 (22.3) | 13.8 (12.9) | 18.6 (18.4) | 12.9 (13.8) | 18.1 (18.0) | 10.2 (12.1) | 16.7 (16.3) |
1. −S–S–R groups are intrinsically more reactive than −SH, despite of the steric effect of R.
2. When R1 is a methyl group (representing a primary alkyl), −S–R2 all prefer to attack the internal S atom. This is because steric effects are not significant, but the formation of −SH is more favorable than the formation of −S–R1.
3. When R1 is tBu or Ad, all −S–R2 favorably attack the external S atom. In this case, the steric interaction between R1 and the incoming nucleophile is significant (Scheme 2).
 | ||
| Scheme 2 The steric interaction between R1 and the incoming nucleophile. | ||
4. The barrier for the attack of nucleophile on the external S atom is not sensitive to the size of the nucleophile −S–R2.
4. Concluding remarks
We have studied the reaction of RSSH and H2S, which reaction is suggested to be important in the S-sulfuration process of the gasotransmitter H2S. Two different DFT methods, M06-2X and ωB97X-D gave similar results, which made us more confident about our results. Our theoretical results predict:1. The energy barrier for the gas phase reaction CH3SSH + H2S → CH3SH + HSSH (mechanisms 1–3) is very high, and in the gas phase this reaction would be unlikely.
2. Although the reaction CH3SS− + H2S → CH3SH + HSS− (mechanisms 4–5) in aqueous solvent has a lower energy barrier, the product HSS− is not a favorable species, and other more favorable mechanisms should be explored.
3. CH3SS− is a reasonable nucleophile to attack either of the S atoms of CH3SSH (mechanisms 6–7). Mechanism 6 has a lower energy barrier than mechanism 4, and the products are consistent with the experimental observations.
4. The size of the R group in RSSH will affect the reaction mechanisms. Smaller R groups with less steric hindrance are apt to attack the internal S atom of RSSH (mechanism 6), while larger R groups are likely to attack at the external S atom (mechanism 7).
5. Our research supports the mechanism for sterically hindered R–S–S–H proposed by Bayley et al.15b For sterically unhindered R–S–S–H, the theory predict the mechanism shown in Scheme 3.
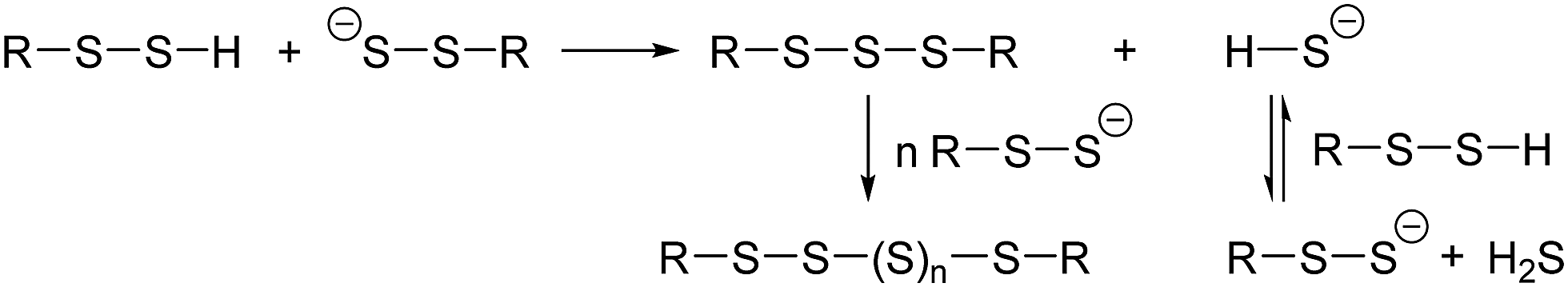 | ||
| Scheme 3 Mechanism for the reaction of unhindered R–S–S–H. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Shenzhen STIC (Grant JCYJ20170412150507046). The research at the Center for Computational Quantum Chemistry was supported by the U.S. National Science Foundation, Grant CHE-1661604.References
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066 CrossRef CAS.
- (a) O. Kabil and R. Banerjee, J. Biol. Chem., 2010, 285, 21903–21907 CrossRef CAS; (b) R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS; (c) R. U. I. Wang, FASEB J., 2002, 16, 1792–1798 CrossRef CAS PubMed; (d) R. Wang, Antioxid. Redox Signaling, 2003, 5, 493–501 CrossRef CAS PubMed.
- (a) L. Li, P. Rose and P. K. Moore, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 169–187 CrossRef CAS PubMed; (b) D. J. Lefer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17907 CrossRef CAS; (c) G. Yang, L. Wu, B. Jiang, W. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587 CrossRef CAS.
- H. Kimura, Mol. Neurobiol., 2002, 26, 13–19 CrossRef CAS.
- B. D. Paul and S. H. Snyder, Nat. Rev. Mol. Cell Biol., 2012, 13, 499 CrossRef CAS.
- C.-M. Park, L. Weerasinghe, J. J. Day, J. M. Fukuto and M. Xian, Mol. BioSyst., 2015, 11, 1775–1785 RSC.
- (a) N. Krishnan, C. Fu, D. J. Pappin and N. K. Tonks, Sci. Signaling, 2011, 4, ra86 CrossRef; (b) A. K. Mustafa, G. Sikka, S. K. Gazi, J. Steppan, S. M. Jung, A. K. Bhunia, V. M. Barodka, F. K. Gazi, R. K. Barrow, R. Wang, L. M. Amzel, D. E. Berkowitz and S. H. Snyder, Circ. Res., 2011, 109, 1259 CrossRef CAS; (c) Y. Liu, R. Yang, X. Liu, Y. Zhou, C. Qu, T. Kikuiri, S. Wang, E. Zandi, J. Du, S. Ambudkar Indu and S. Shi, Cell Stem Cell, 2014, 15, 66–78 CrossRef CAS; (d) A. K. Mustafa, M. M. Gadalla, N. Sen, S. Kim, W. Mu, S. K. Gazi, R. K. Barrow, G. Yang, R. Wang and S. H. Snyder, Sci. Signaling, 2009, 2, ra72 Search PubMed; (e) N. Sen, B. D. Paul, M. M. Gadalla, A. K. Mustafa, T. Sen, R. Xu, S. Kim and S. H. Snyder, Mol. Cell, 2012, 45, 13–24 CrossRef CAS PubMed; (f) G. Yang, K. Zhao, Y. Ju, S. Mani, Q. Cao, S. Puukila, N. Khaper, L. Wu and R. Wang, Antioxid. Redox Signaling, 2012, 18, 1906–1919 CrossRef.
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7606–7611 CrossRef CAS.
- (a) G. A. Benavides, G. L. Squadrito, R. W. Mills, H. D. Patel, T. S. Isbell, R. P. Patel, V. M. Darley-Usmar, J. E. Doeller and D. W. Kraus, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17977–17982 CrossRef CAS; (b) D. Liang, H. Wu, M. W. Wong and D. Huang, Org. Lett., 2015, 17, 4196–4199 CrossRef CAS.
- Y. Zhao, S. Bhushan, C. Yang, H. Otsuka, J. D. Stein, A. Pacheco, B. Peng, N. O. Devarie-Baez, H. C. Aguilar, D. J. Lefer and M. Xian, ACS Chem. Biol., 2013, 8, 1283–1290 CrossRef CAS.
- T. Roger, F. Raynaud, F. Bouillaud, C. Ransy, S. Simonet, C. Crespo, M. P. Bourguignon, N. Villeneuve, J. P. Vilaine, I. Artaud and E. Galardon, ChemBioChem, 2013, 14, 2268–2271 CrossRef CAS.
- (a) K. Ono, T. Akaike, T. Sawa, Y. Kumagai, D. A. Wink, D. J. Tantillo, A. J. Hobbs, P. Nagy, M. Xian, J. Lin and J. M. Fukuto, Free Radical Biol. Med., 2014, 77, 82–94 CrossRef CAS PubMed; (b) S. Kawamura, T. Kitao, T. Nakabayashi, T. Horii and J. Tsurugi, J. Org. Chem., 1968, 33, 1179–1181 CrossRef CAS.
- (a) H. Böhme and G. Zinner, Justus Liebigs Ann. Chem., 1954, 585, 142–149 CrossRef; (b) T. Nakabayashi and J. Tsurugi, J. Org. Chem., 1963, 28, 811–813 CrossRef CAS; (c) J. Tsurugi, T. Nakabayashi and T. Ishihara, J. Org. Chem., 1965, 30, 2707–2710 CrossRef CAS; (d) S. Kawamura, Y. Otsuji, T. Nakabayashi, T. Kitao and J. Tsurugi, J. Org. Chem., 1965, 30, 2711–2714 CrossRef CAS; (e) S. Kawamura, T. Nakabayashi, T. Kitao and J. Tsurugi, J. Org. Chem., 1966, 31, 1985–1987 CrossRef CAS; (f) T. Nakabayashi, S. Kawamura, T. Kitao and J. Tsurugi, J. Org. Chem., 1966, 31, 861–864 CrossRef CAS; (g) N. E. Heimer and L. Field, J. Org. Chem., 1984, 49, 1446–1449 CrossRef CAS; (h) N. E. Heimer, L. Field and R. A. Neal, J. Org. Chem., 1981, 46, 1374–1377 CrossRef CAS; (i) N. E. Heimer, L. Field and J. A. Waites, J. Org. Chem., 1985, 50, 4164–4166 CrossRef CAS; (j) J. Tsurugi, S. Kawamura and T. Horii, J. Org. Chem., 1971, 36, 3677–3680 CrossRef CAS.
- (a) N. E. Francoleon, S. J. Carrington and J. M. Fukuto, Arch. Biochem. Biophys., 2011, 516, 146–153 CrossRef CAS; (b) R. Millikin, C. L. Bianco, C. White, S. S. Saund, S. Henriquez, V. Sosa, T. Akaike, Y. Kumagai, S. Soeda, J. P. Toscano, J. Lin and J. M. Fukuto, Free Radical Biol. Med., 2016, 97, 136–147 CrossRef CAS PubMed; (c) A. Vasas, É. Dóka, I. Fábián and P. Nagy, Nitric oxide, 2015, 46, 93–101 CrossRef CAS PubMed.
- (a) T. S. Bailey, L. N. Zakharov and M. D. Pluth, J. Am. Chem. Soc., 2014, 136, 10573–10576 CrossRef CAS; (b) T. S. Bailey and M. D. Pluth, Free Radical Biol. Med., 2015, 89, 662–667 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- S. E. Wheeler and K. N. Houk, J. Chem. Theory Comput., 2010, 6, 395–404 CrossRef CAS.
- (a) T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS; (b) R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- T. H. Dunning, K. A. Peterson and A. K. Wilson, J. Chem. Phys., 2001, 114, 9244–9253 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- P. Gerbaux, J.-Y. Salpin, G. Bouchoux and R. Flammang, Int. J. Mass Spectrom., 2000, 195-196, 239–249 CrossRef.
- (a) R. Steudel, Y. Drozdova, K. Miaskiewicz, R. H. Hertwig and W. Koch, J. Am. Chem. Soc., 1997, 119, 1990–1996 CrossRef CAS; (b) C. J. Marsden and B. J. Smith, J. Phys. Chem., 1988, 92, 347–353 CrossRef CAS.
- E. Cuevasanta, M. Lange, J. Bonanata, E. L. Coitiño, G. Ferrer-Sueta, M. R. Filipovic and B. Alvarez, J. Biol. Chem., 2015, 290, 26866–26880 CrossRef CAS.
- Y.-R. Cai and C.-H. Hu, J. Phys. Chem. B, 2017, 121, 6359–6366 CrossRef CAS.
- J. Tsurugi, Y. Abe, T. Nakabayashi, S. Kawamura, T. Kitao and M. Niwa, J. Org. Chem., 1970, 35, 3263–3266 CrossRef CAS.
- S. S. Saund, V. Sosa, S. Henriquez, Q. N. N. Nguyen, C. L. Bianco, S. Soeda, R. Millikin, C. White, H. Le, K. Ono, D. J. Tantillo, Y. Kumagai, T. Akaike, J. Lin and J. M. Fukuto, Arch. Biochem. Biophys., 2015, 588, 15–24 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp05503c |
| This journal is © the Owner Societies 2019 |