
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pulse-shaped two-photon excitation of a fluorescent base analogue approaches single-molecule sensitivity†
Rachel S.
Fisher‡
a,
David
Nobis‡
 b,
Anders F.
Füchtbauer
c,
Mattias
Bood
d,
Morten
Grøtli
d,
L. Marcus
Wilhelmsson
c,
Anita C.
Jones
*a and
Steven W.
Magennis
*b
b,
Anders F.
Füchtbauer
c,
Mattias
Bood
d,
Morten
Grøtli
d,
L. Marcus
Wilhelmsson
c,
Anita C.
Jones
*a and
Steven W.
Magennis
*b
aEaStCHEM School of Chemistry, The University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: a.c.jones@ed.ac.uk
bWestCHEM School of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow, G12 8QQ, UK. E-mail: steven.magennis@glasgow.ac.uk
cDepartment of Chemistry and Chemical Engineering, Chemistry and Biochemistry, Chalmers University of Technology, SE-412 96 Gothenburg, Sweden
dDepartment of Chemistry and Molecular Biology, University of Gothenburg, SE-412 96 Gothenburg, Sweden
First published on 30th October 2018
Abstract
Fluorescent nucleobase analogues (FBAs) have many desirable features in comparison to extrinsic fluorescent labels, but they are yet to find application in ultrasensitive detection. Many of the disadvantages of FBAs arise from their short excitation wavelengths (often in the ultraviolet), making two-photon excitation a potentially attractive approach. Pentacyclic adenine (pA) is a recently developed FBA that has an exceptionally high two-photon brightness. We have studied the two-photon-excited fluorescence properties of pA and how they are affected by incorporation in DNA. We find that pA is more photostable under two-photon excitation than via resonant absorption. When incorporated in an oligonucleotide, pA has a high two-photon cross section and emission quantum yield, varying with sequence context, resulting in the highest reported brightness for such a probe. The use of a two-photon microscope with ultrafast excitation and pulse shaping has allowed the detection of pA-containing oligonucleotides in solution with a limit of detection of ∼5 molecules, demonstrating that practical single-molecule detection of FBAs is now within reach.
Introduction
Fluorescence spectroscopy and imaging are powerful techniques for studying the structure, dynamics and interactions of nucleic acids. As the naturally occurring nucleic acids are virtually non-emissive, they require a fluorescent label. The most common approach is to covalently attach external dyes by long linkers, but these are generally insensitive to local base interactions. An alternative is the use of a fluorescent analogue of one of the natural nucleobases, which can replace the nucleobase while maintaining hydrogen bonding and leaving the overall conformation of the duplex unaffected.1–3 A range of fluorescent nucleobase analogues (FBAs) for DNA and RNA have been developed,1–8 notably the archetypal base analogue 2-aminopurine (2-AP).9Although ensemble measurements of FBAs are very useful tools, single-molecule spectroscopy could provide unprecedented insight into the mechanism of processes such as DNA methylation at the individual base level,10 but this has proved challenging for nucleobase analogues for several reasons. Firstly, despite good emission quantum yields (ϕ), especially as monomeric units, the absorption coefficients of nucleobase analogues (ε) are much lower than for extrinsic dyes, limiting their brightness (εϕ). Secondly, nucleobase analogues absorb at short wavelength, often in the UV, and are prone to photobleaching. Thirdly, light penetration in biological media is poorer at short wavelength due to increased absorption and scattering. The optimised emission signal for single-molecule detection, given as count rate per molecule (CPM), for free 2-AP in water (unquenched and not incorporated in an oligonucleotide) with UV excitation was found to be ∼2 kHz,11 with a signal-to-background ratio (SBR) per molecule of 0.3. This CPM is very low in comparison with typical extrinsic organic labels (30–100 kHz). Similarly, the CPM of a guanine analogue, 3-MI, was reported as 4 kHz with a SBR of ∼5 following UV excitation.12 It has been reported that oligonucleotides labelled with 2-AP or pyrrolo-cytosine (Py-C) could be detected at the single-molecule level in nucleic acids via UV excitation on a surface.13 Although the authors did not report the CPM of 2-AP and Py-C, it is well known that the fluorescence of 2-AP is quenched between 10 and 100-fold due to base stacking interactions.9 In short, it seems unlikely that UV excitation of such bases will lead to a robust method of single-molecule detection of nucleic acids.
A way to overcome these problems could be to use multiphoton excitation, the simultaneous absorption of two or more longer wavelength photons.14 In addition to allowing deeper penetration into tissue in the near-IR, multiphoton excitation allows 3D localization of light absorption, without exciting out-of-focus chromophores; out-of-focus photobleaching is therefore also reduced. Furthermore, changes in the optical selection rules for multiphoton excitation can result in the more efficient population of the excited states, or completely new excited states being accessed.14 Other possible advantages of multiphoton excitation include reduced photobleaching in the excitation focus, a reduction of background fluorescence from optical components, greater spectral separation of excitation and emission bands, and the simultaneous excitation of multiple fluorophores.15
There have been few studies of multiphoton excitation of nucleobase analogs.16–20 The guanine analogue, 6-MI (6-methyl isoxanthopterin), is reported to have a two-photon (2P) cross section (σ2) of 2.5 GM units (Goeppert-Mayer, 1 GM = 10−50 cm4 s photon−1),16 while 6MAP (4-amino-8-(2-deoxy-β-D-ribofuranosyl)-6-methyl-7(8H)-pteridone), an adenine analog, has a cross section of 3.4 GM.17 The 2P (∼0.2 GM) and three-photon excitation of 2-AP was reported recently, along with the 2P excitation of the cytosine analogue tC in an oligonucleotide (1.5 GM).18 Although photobleaching was reduced for 2P excitation, the stability was not high enough to perform fluorescence correlation spectroscopy (FCS).18 In a very recent study, Mikhaylov et al. assessed the potential utility of several established isomorphic FBAs for multiphoton microscopy.19 As well as reporting 2P cross sections, they used a previously derived relationship between the 2P cross section and the speed of multiphoton imaging21 to estimate the maximum frame rate that would be achievable, in a generic scanning multiphoton microscope. The best-performing FBA in this context was found to be 6-MI, with an estimated frame rate of 200 frames per second.21
The potential for major improvements in 2P fluorescence brightness via systematic modifications was highlighted in a study of a series of structurally-related nucleosides.20 The 1P and 2P cross sections and spectra in that report also agree well with subsequent theoretical calculations,22 offering the prospect of the rational design of photophysical properties. Nevertheless, until recently, FBAs have either possessed a low two-photon cross section, a low quantum yield, an absorbance maximum outside the practical range of two-photon excitation, or a combination of all three.
A promising new development is the recent report that pentacyclic adenine, pA, a bright fluorescent adenine analogue (Chart 1a) can be excited via 2P excitation with a relatively high cross section of 6.6 GM.23 The 1P excitation maximum of pA is 391 nm with a quantum yield of 0.66 in water. This value is on a par with 2-AP and surpasses the brightness of many other FBAs. Unlike 2-AP (and most other fluorescent analogues), pA retains a relatively high quantum yield in oligonucleotides, in several sequence contexts. The cross section of pA in an oligonucleotide (GA, Chart 1b) was reported as 3 GM, which was encouragingly high. This was only the second example of the 2P detection of an FBA in DNA.18
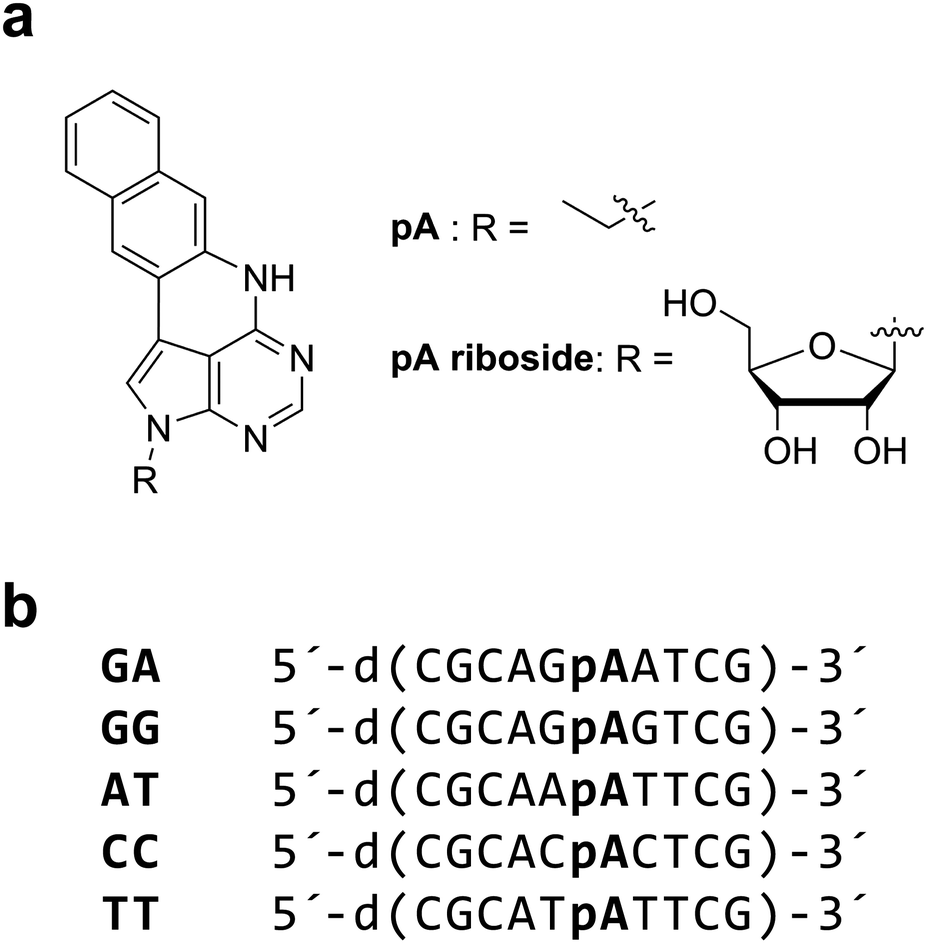 | ||
| Chart 1 (a) Structure of pentacyclic adenine (pA), as the free base (left) and the riboside (right). (b) pA-containing deoxyoligonucleotides (10mers). Sequences differ only by the neighbouring bases of pA, and are named accordingly. | ||
In this work we report a comparative analysis of the 1P and 2P photophysics of pA as the nucleobase, the riboside and incorporated in oligonucleotides (Chart 1). We demonstrate that pA has an enhanced photostability under 2P excitation. We then report the use of a 2P microscope with pulse-shaped, ultrafast laser excitation to establish the limits of detection for DNA containing pA. FCS allowed a determination of the CPM as 0.5 kHz per molecule. By optimizing the broadband laser pulse, as few as 5 molecules could be detected. We conclude with a discussion of some of the next developments that could result in the ultimate single-molecule level of sensitivity.
Experimental
Materials
The synthesis and characterization of the pA nucleobase and pA-modified oligodeoxynucleotides has been reported recently;23 the pA riboside24 was used instead of the nucleobase for photophysical measurements in aqueous solution, due to its higher solubility.For ensemble 1P and 2P spectroscopy, the free pA nucleobase was dissolved in ethanol (>99.9%, Fisher Scientific) and the pA riboside was dissolved in either ethanol or phosphate buffer (12.5 mM phosphate, 100 mM NaCl, pH 7.5 (Sigma-Aldrich)). pA-containing modified oligonucleotides were also dissolved in phosphate buffer and were annealed by combining each modified single strand with 10% excess of its complementary strand at RT followed by heating to 95 °C and leaving to cool overnight. Solution concentrations were ∼10−6 M with absorbance of ∼0.05 at the long-wavelength maximum.
For 2P microscopy, a pA-modified oligonucleotide was dissolved in buffer containing 20 mM Tris (Sigma-Aldrich), 150 mM NaCl (Fluka) at pH 7.4. Solutions were filtered through activated charcoal to remove fluorescent impurities. Ultrapure water was used for buffer preparation (Direct Q3, Merck Millipore). All solvents and buffers were checked for background fluorescence prior to use.
1P spectroscopy
Absorption spectra were recorded using a Cary 300 UV-visible spectrophotometer and fluorescence spectra were recorded using a Jobin Yvon Horiba FluoroMax-3 spectrofluorimeter running FluorEssence software v.3.5.Fluorescence lifetimes were determined using time-correlated single photon counting on an Edinburgh Instruments spectrometer equipped with TCC900 photon counting electronics. A mode-locked Ti:sapphire laser (Coherent Mira pumped by Coherent Verdi), producing pulses of duration ∼150 fs at a repetition rate of 76 MHz, was used as the excitation source. The pulse repetition rate was reduced to 4.75 MHz using a pulse picker (Coherent 9200) and the light was frequency doubled using a Coherent 5-050 harmonic generator. Fluorescence decay curves were recorded over 50 ns, 4096 channels and collected to a total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts in the peak channel. Decays were fitted by iterative reconvolution, assuming a multi-exponential function, given in eqn (1)
000 counts in the peak channel. Decays were fitted by iterative reconvolution, assuming a multi-exponential function, given in eqn (1)
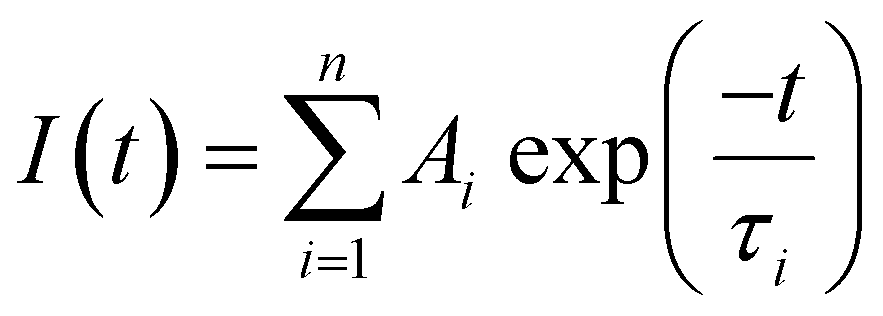 | (1) |
The average lifetime, 〈τ〉, of the emitting population is related to the average quantum yield, 〈ϕ〉 determined from steady-state intensity measurements, as given by eqn (2)
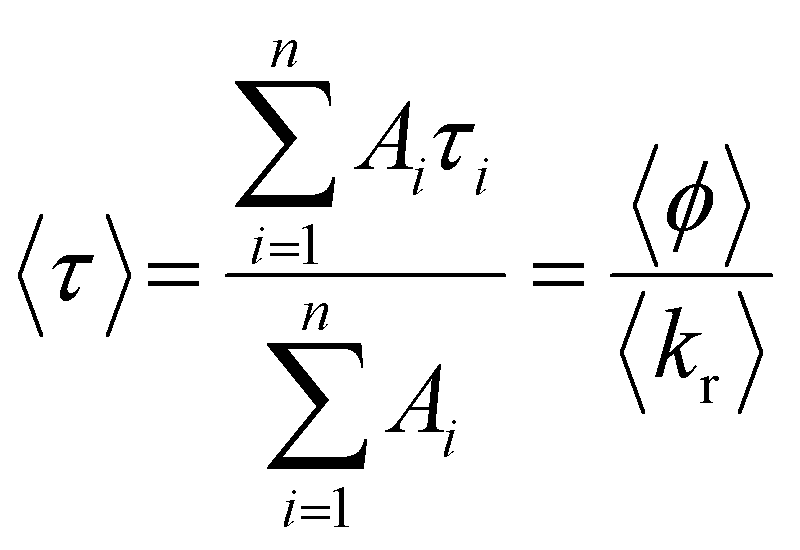 | (2) |
The fraction of the steady-state emission intensity due to each species i, SSi, is given by eqn (3).
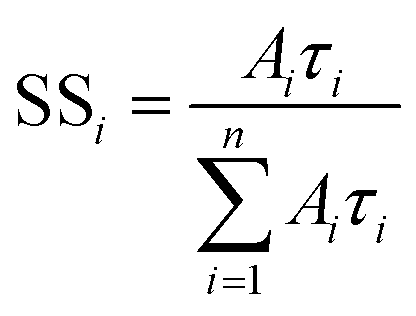 | (3) |
2P spectroscopy
See Fig. S1 (ESI†) for a schematic diagram of the experimental system. A mode-locked Ti:sapphire laser (see above) was used as the excitation source. A variable reflective neutral density filter was used to attenuate the excitation beam, which then passed through a dichroic mirror (Semrock Brightline FF735-Di02) and was focused by a 20× objective (Olympus) into the sample solution, which was contained in a 1 cm path-length cuvette. Fluorescence emission was collected by the same objective, reflected from the dichroic mirror, passed through a shortpass filter (Semrock Brightline FF01-720/SP-25) and detected by a fibre-coupled spectrometer (Ocean Optics USB2000+), with an acquisition time of 2 s. Measurements were made at between twenty and thirty different incident powers. Three spectra were collected at each power and an average taken. The incident power was measured using a Coherent FieldMaster power meter.2P cross sections were measured relative to a number of different standards (see ESI† for more details). The 2P cross sections have an estimated accuracy of 10% due to uncertainty in the cross sections of the standards and errors in the measurement of the spectral throughput, absorption spectra and emission spectra.
Fluorescence lifetime measurements using 2P excitation were recorded as described for 1P excitation, except that the pulse-picked fundamental output of the Ti:sapphire laser at 790 nm was used, and this was focused into the sample solution using a 10× microscope objective.
2P microscopy
The experimental system is shown in Fig. 1. The excitation source was a broadband Ti:Sapphire laser with a repetition rate of 80 MHz (Vitara UBB, Coherent). The laser spectrum was centered on 800 nm with a FWHM of 135 nm; the compressed pulses from the oscillator had a duration of 15 fs. The pulse shape at the sample was controlled via a pulse shaper (MIIPS-Box 640, Biophotonic Solutions Inc.) containing two liquid-crystal spatial light-modulators (SLM) in series (SLM-S640d, Jenoptik). After the pulse shaper the beam entered the inverted microscope (IX71, Olympus), where it was focussed onto the sample by a 60× water-immersion objective (UPlanSApo, Olympus). It was thus possible to independently shape amplitude and phase of the excitation pulse at the sample. The pulse compression and shaping utilised the method of multi-photon intra-pulse interference phase scan (MIIPS)25 whereby a reference phase function is applied to the dispersed beam. Through measurement of the second-harmonic generation (SHG) from a SHG crystal at the sample position (10 μm BBO, Biophotonic Solutions Inc.), phase distortions of the original beam can be measured and cancelled, yielding a transform-limited pulse or a desired pulse shape. The SHG spectrum was measured with a fibre-coupled spectrometer (USB4000, Ocean Optics). The optimal compensation phase and amplitude mask was then determined (MIIPS software version 2.0, Biophotonic Solutions Inc.).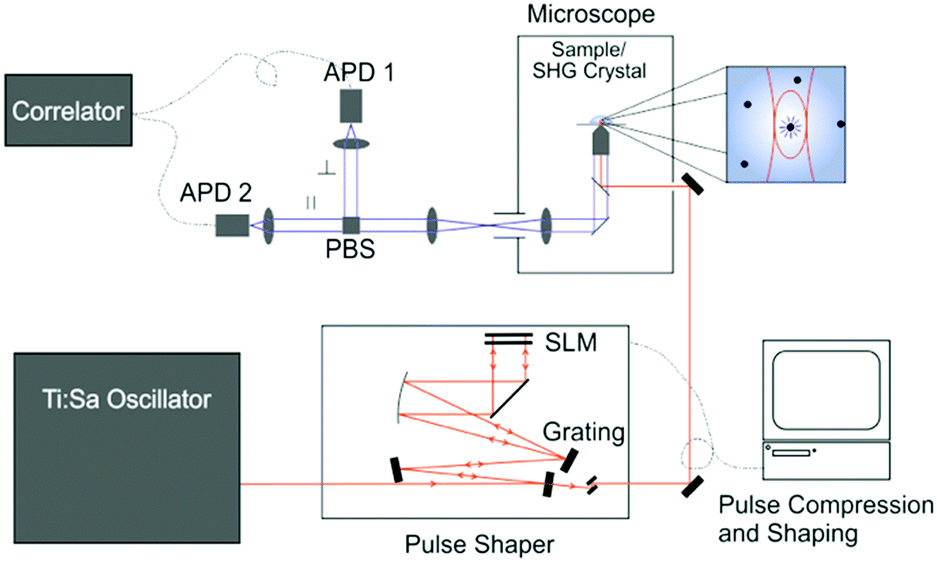 | ||
| Fig. 1 Schematic of the 2P microscopy setup. | ||
After pulse shaping, the SHG crystal was replaced by the sample. The solution samples were placed on a cover slip (Menzel Gläser, Thermo Scientific) and the solution temperature was controlled with an incubator (Live Cell Instrument, CU-501). All measurements were recorded at 22 ± 1 °C. Sample fluorescence was collected by the same objective and transmitted through a dichroic mirror (Chroma 675dcspxr), split by a polarising beamsplitter cube (PBS) and detected by two avalanche photodiodes (APD) (MPD PDM 50c and MPD $PD-050-CTB). The signal was subsequently detected by either a photon-counting card (SPC-132, Becker and Hickl GmbH) for multichannel scalar (MCS) analysis or a hardware correlator (ALV-7002, ALV GmbH) for FCS.
Results and discussion
1P photophysics
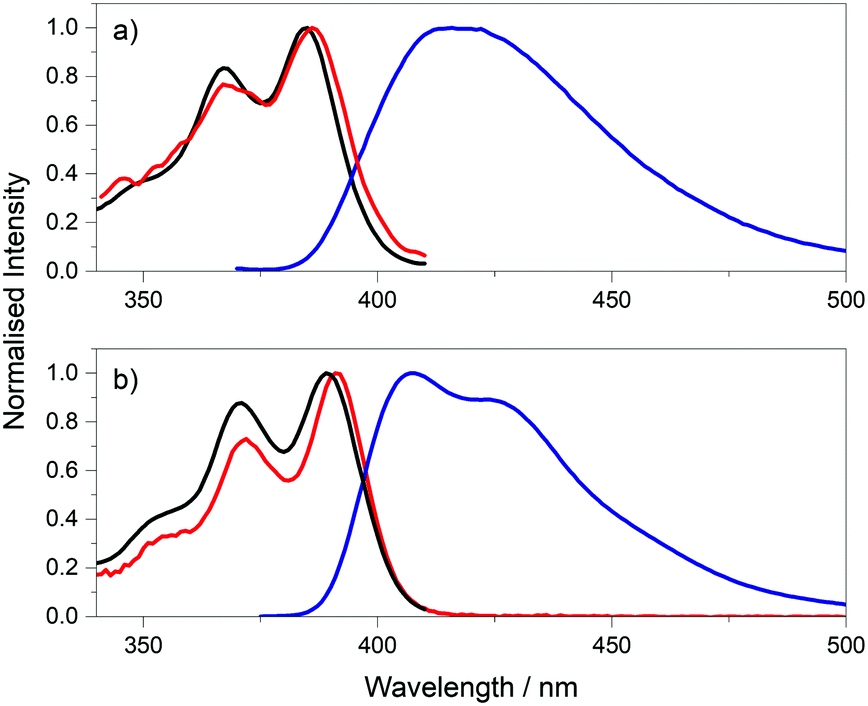 | ||
| Fig. 2 Absorption (red), excitation spectra (black), at emission wavelength of 420 nm, and the emission spectra (blue), at excitation wavelength of 360 nm, of pA riboside in (a) phosphate buffer and (b) ethanol. | ||
Further evidence of more than one emitting species comes from fluorescence lifetime measurements. In both phosphate buffer and ethanol, the pA riboside exhibits a bi-exponential fluorescence decay (Table S1, ESI†) when excited at 390 nm. The average lifetimes in phosphate buffer and ethanol are very similar at 3.9 ns and 3.8 ns, respectively. As discussed above, the quantum yield is the same in both solvents, with a value of 0.66.23 The consistency between the average lifetimes and quantum yields implies that the radiative lifetimes of the two emitting species are very similar, and hence the A-factors are indicative of the fractional populations. The dominant component has a lifetime of 5.97 ns in buffer and 4.82 ns in ethanol and accounts for the majority of the emitting population (65% and 79%, respectively). A short lifetime of 0.19 ns in buffer and 0.16 ns in ethanol accounts for the remaining population.
From the decay parameters we find that, in both solvents, the short-lifetime component contributes less than 2% of the steady-state emission intensity, i.e. the steady-state spectrum is due almost entirely to the longer-lifetime species (Table S1, ESI†). This is consistent with the observed discrepancy between the excitation and absorption spectra (Fig. 2). Comparison of the absorption and excitation spectra suggest that the short-lifetime (low-quantum-yield) species has an absorption spectrum that is slightly red-shifted compared with that of the long-lifetime (high-quantum-yield) species. The fractional populations (A-factors) show negligible dependence on emission wavelength over the measurement range of 430 to 470 nm, (Table S2, ESI†) indicating that the two species have very similar emission spectra over this range, but they may differ at shorter wavelengths.
These observations point to the existence of two ground-state species with quantum yields that differ by more than an order of magnitude. Consideration of the structure of pA (Chart 1a) suggests that the only likely cause of this is N–H tautomerism, as illustrated in Fig. S3 (ESI†). Tautomerism is a well-known phenomenon in the natural nucleobases and has also been identified in fluorescent adenine analogues like 2-aminopurine26 and the tC-family.27 There is also a precedent for a large difference in excited-state lifetimes between tautomers. For adenine in aqueous solution, the lifetime of the 7H tautomer is about 40 times longer than that of the more abundant (78%) 9H tautomer, and it is thus the minor tautomer that is responsible for the (very weak) fluorescence of adenine.28
The excitation and emission spectra of the pA-containing oligonucleotides, both single- and double-stranded, are shown in Fig. 3. The excitation spectra of the single strands, in the wavelength range 310 to 410 nm, where pA is excited exclusively, closely resemble the corresponding absorption spectra (Fig. S5, ESI†) (at wavelengths below 310 nm there is large discrepancy between excitation and absorption spectra as a result of absorption by the natural bases). The emission spectrum of pA in the single strands more closely resembles that of the free riboside in ethanol than that of the riboside in buffer (see Fig. S6, ESI†), consistent with a less polar, base-stacked environment.
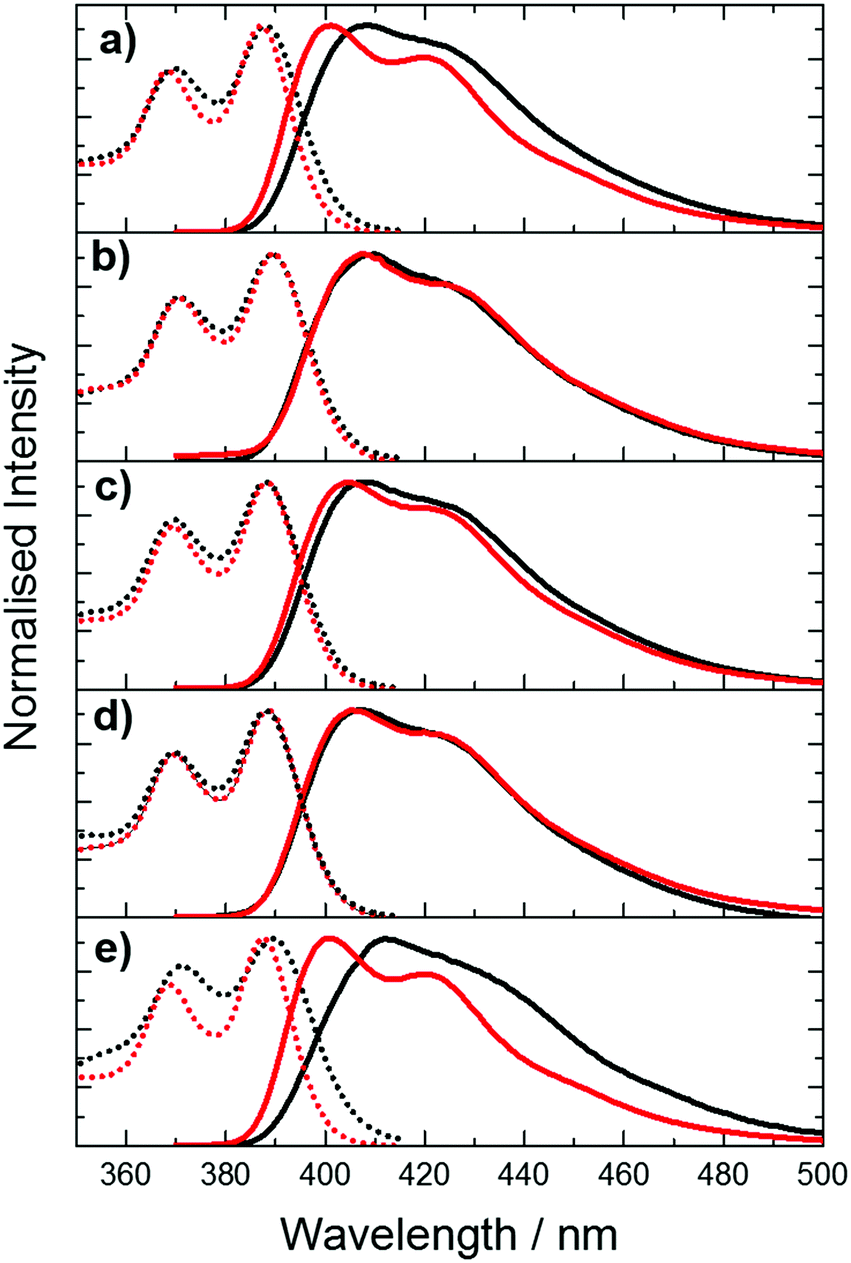 | ||
| Fig. 3 Normalised excitation (dotted lines) and emission spectra (solid lines) of pA in single- (black) and double-stranded (red) oligonucleotides with different combinations of nearest neighbours. (a) AT (b) CC (c) GA (d) GG (e) TT. For emission spectra, the excitation wavelength was 360 nm. For excitation spectra, the emission wavelength was 420 nm. | ||
Comparison of the spectra of single and double strands reveals a sequence-dependent effect. For CC, GA and GG, duplex formation has little effect on either excitation or emission spectra. However, for AT and TT, duplex formation results in a noticeable blue-shift in both the emission and the excitation spectrum, and a sharpening of the bands in both spectra.
Time-resolved fluorescence measurements, exciting at 390 nm, reveal the presence of multiple emitting species in both single and double strands; in most cases three lifetime components are observed. The decay parameters together with the fractional contribution of each decay component to the steady-state emission intensity, are summarised graphically in Fig. 4. The multi-exponential decays reflect the heterogeneity of inter-base interactions in the oligonucleotides; pA exists in a variety of base-stacked environments and hence experiences a variety of quenching rates. The decay parameters are influenced by sequence context and duplex formation. A detailed interpretation of these results is not relevant to the two-photon study that is the main focus of this paper and will be addressed in a future publication. However, it is pertinent to consider the contributions of the different lifetime components to the steady-state emission intensities. For three of the single strands, GA, GG and AT, >70% of the steady-state intensity is due to the decay component with the longest lifetime (τ3 ∼ 7 ns), which also constitutes >70% of the emitting population. In contrast, each of the three decay components makes a substantial contribution to the emission intensity for TT and CC, and, in the latter case, a component with a much shorter lifetime (0.8 ns) is dominant. In spite of this, the emission spectra of TT and CC do not differ significantly from those of the other single strands. This suggests that all of the emitting species have similar emission spectra. This is supported by the observation that, in all cases, the fraction populations (A-factors) are essentially independent of emission wavelength between 410 and 450 nm.
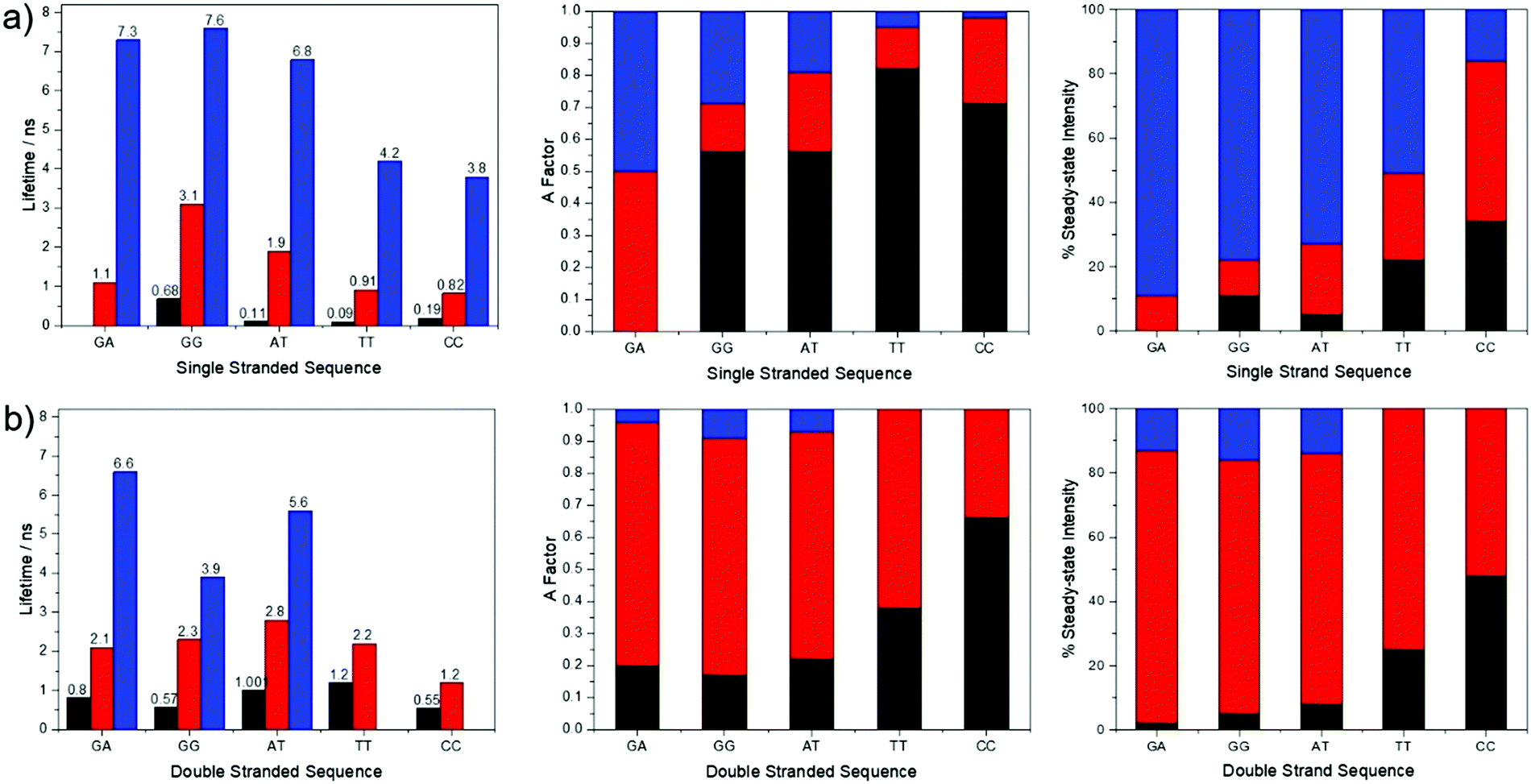 | ||
| Fig. 4 Graphical representation of the fluorescence decay parameters (lifetimes and A-factors) and the fractional contribution of each decay component to the steady-state emission intensity, for pA in (a) single-stranded and (b) double-stranded oligonucleotides, with different combinations of nearest neighbours. Excitation was at 390 nm and emission was detected at 430 nm. | ||
For four of the double strands, GA, GG, AT and TT, one decay component (τ2 ∼ 2 ns) contributes >70% of the steady-state intensity. For CC, two components (τ1 = 0.5 ns and τ2 = 1.2 ns) contribute almost equally to the emission intensity; nevertheless, the emission spectrum of CC is very similar to those of GA and GG. As in the case of the single strands, the A-factors for all of the double strands are wavelength-independent over the emission range 410–450 nm. It is evident that in both single and double strands the emission spectra show little evidence of the heterogeneity of the emitting population. This is partly because the steady-state intensity tends to be dominated by one species, but also because the different emitting species appear to have similar spectra.
2P photophysics
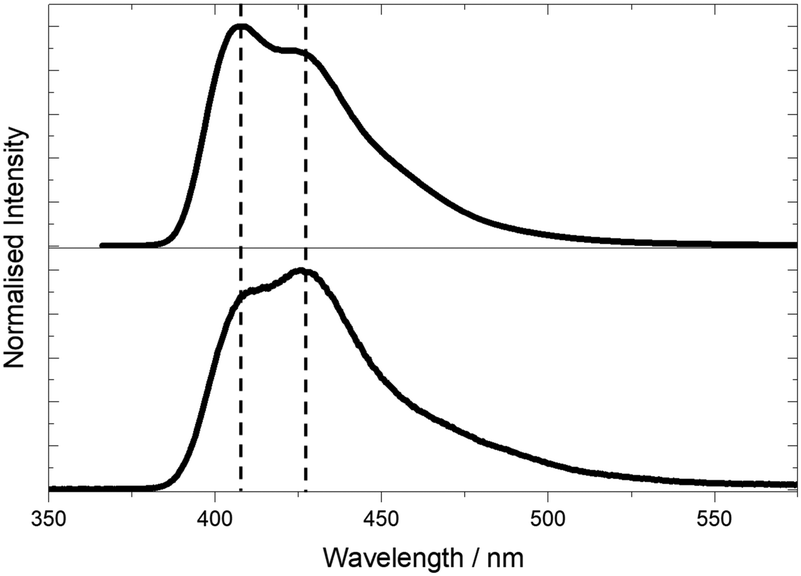 | ||
| Fig. 5 Emission spectra of pA nucleobase in ethanol following 1P excitation at 360 nm (top) and 2P excitation at 780 nm (bottom). | ||
Spectral differences are also apparent in the 1P and 2P excitation spectra (Fig. 6). The 2P excitation spectrum was recorded both at a single emission wavelength (corresponding to that used for the 1P spectrum) and for the integrated emission intensity over the entire spectrum. As can be seen, these spectra are identical, ruling out any effects that might arise from the selective detection of different emitting species.
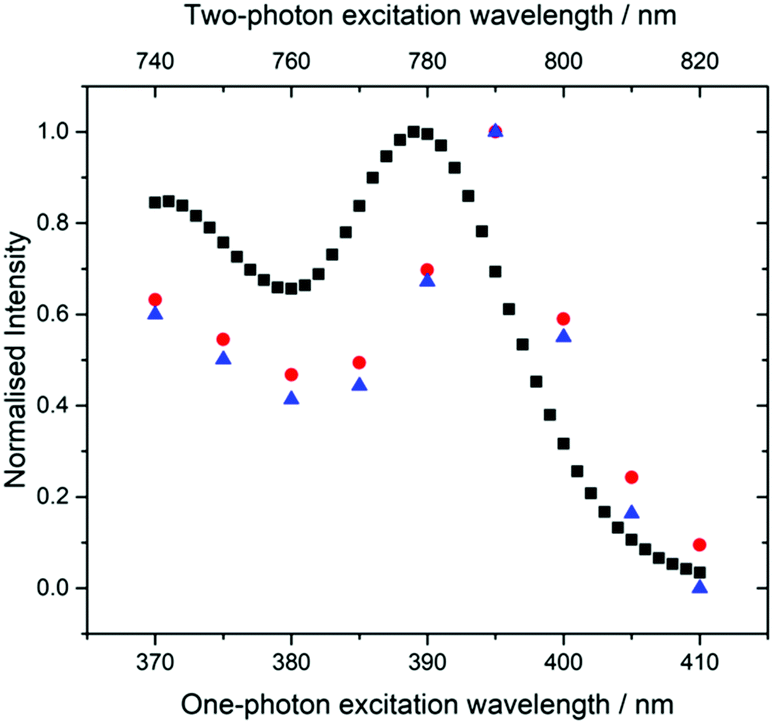 | ||
| Fig. 6 Normalised fluorescence excitation spectra of the pA nucleobase in ethanol for 1P excitation (black squares, bottom axis) and 2P excitation (red circles and blue triangles, top axis). For the 1P spectrum, emission intensity was recorded at 420 nm (bandpass 3 nm). For the 2P spectra, emission intensity was recorded at 420 nm (blue triangles) and integrated over the whole spectrum (red circles). | ||
The 2P excitation maximum is red-shifted by around 5 nm compared to the one-photon maximum (the 2P peak is at 790 nm, whereas the 1P peak is at 390 nm). This is not a shift of the entire spectrum, but a change in the spectral profile, since, in the 2P spectrum, the second maximum (370/740 nm) is not red-shifted and is also more intense relative to the short-wavelength band. It is notable that the 2P excitation spectrum more closely resembles the 1P absorption spectrum than the 1P excitation spectrum (Fig. 2).
These observations are consistent with the presence of two tautomers whose relative 2P cross sections differ from their relative one-photon absorption coefficients. This is confirmed by the results of time-resolved fluorescence measurements under 2P excitation (Table S3, ESI†) which show the same two lifetime components as those seen for 1P excitation, but with significantly different A-factors. Under 2P excitation, the short-lifetime (τ1) species constitutes 80% of the emitting population, compared with only 21% under 1P excitation. Consequently, the contribution of the τ1 tautomer to the steady-state emission intensity is increased from 1% under 1P excitation to 12% (at 435 nm) under 2P excitation, resulting in the observed change in the profile of the emission spectrum. A greater contribution to the emission from the τ1 species also accounts for the discrepancy between 2P and 1P excitation spectra, and the resemblance of the 2P spectrum to the 1P absorption spectrum. The emission spectral profile is essentially independent of excitation wavelength (Fig. S8, ESI†), but there are small differences in the relative intensities of the two maxima. The spectrum excited at 810 nm, on the red edge of the excitation spectrum, shows the greatest intensity of the longer wavelength band and is likely to be most characteristic of the τ1 tautomer.
Unlike the 1P absorption coefficient, which is determined by the transition dipole moment between ground and excited states, the 2P cross section, σ2, depends on both the transition dipole moment and the change in permanent dipole moment on excitation (in a two-level model for a dipolar chromophore), as expressed in eqn (4)
σ2 ∝ |![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) 10|2|Δ10|2 10|2|Δ10|2 | (4) |
10 is the transition dipole moment between the ground state (S0) and the excited state (S1), and Δ10 is the difference between the permanent dipole moments of S1 and S0.29 The difference in relative 2P cross sections of the two tautomers, compared with their relative 1P cross sections can, therefore, be attributed to a greater change in permanent dipole moment on excitation for the τ1 tautomer.
We end this section with a caveat regarding the calculation of the 2P cross section from the experimentally measured parameter, the 2P brightness, σ2ϕ, which is the product of the quantum yield and the 2P cross section. Calculation of the cross section assumes that the quantum yield is independent of the route to excitation. Where there are multiple species in a sample, the steady-state quantum yield is an average over the various absorbing populations. Should these populations have different 2P cross sections relative to 1P absorption coefficients, then the average quantum yield will change accordingly. This applies in the case of pA. The 2P brightness of 5.3 GM (at 780 nm) is the true value of the average 2P brightness of the two tautomers and, in the context of fluorescence detection, this is the parameter of interest. However, it is now evident that the previously quoted (average) 2P cross section, calculated using the average 1P quantum yield,23 was considerably underestimated. On the basis of the average lifetimes under 1P and 2P excitation, 3.8 ns and 1.2 ns, respectively (Tables S1 and S3, ESI†), we now obtain a revised value of 21 GM for the (average) 2P cross section of pA. This is almost three times greater than the highest value reported previously for a FBA, 7.6 GM for 5-(thiophen-2-yl)-2′-deoxyuridine.20
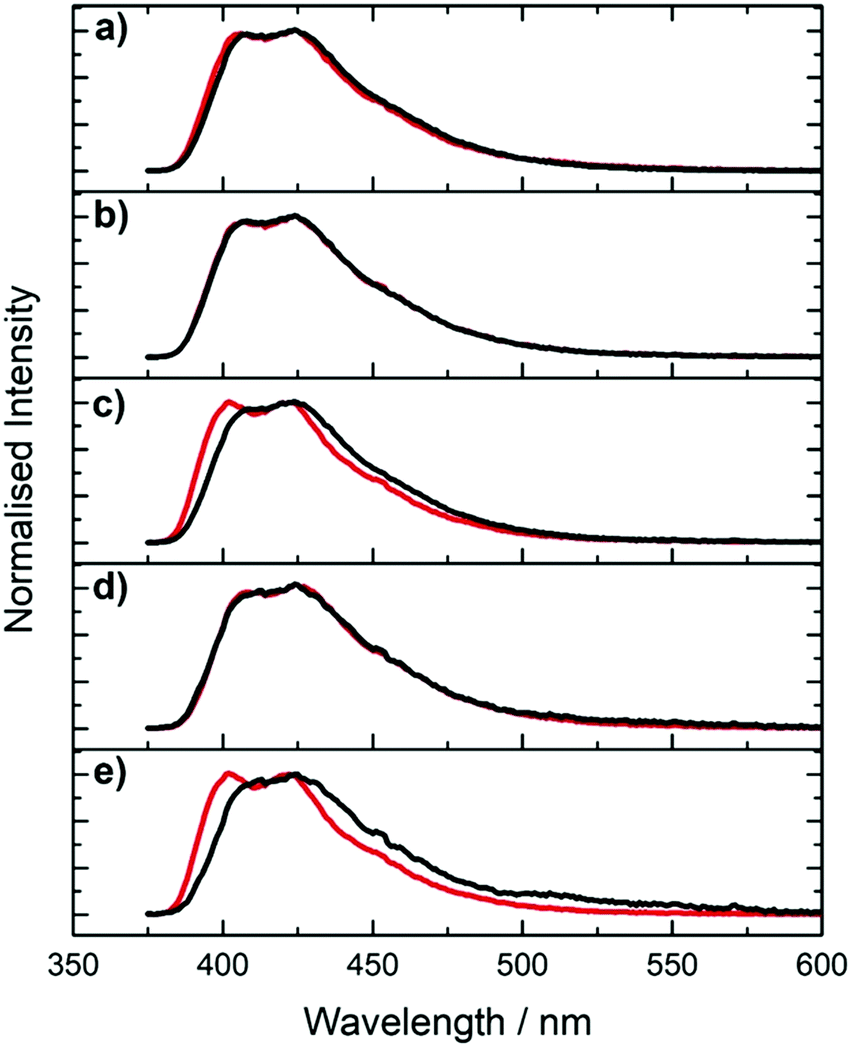 | ||
| Fig. 7 2P-excited emission spectra of pA in single- (red) and double-stranded DNA (black) with different combinations of nearest neighbours. (a) AT (b) CC (c) GA (d) GG (e) TT. Excitation was at 780 nm. | ||
The measured 2P brightness values together with the derived cross sections of the five pA-modified oligonucleotides (ssDNA and dsDNA) are presented in Table 1 (an extended version (Table S4, ESI†) also gives the previously reported values for the 1P quantum yield, molar absorption coefficient (ε390) and 1P brightness (ε390ϕ)).23 In deriving the cross section values, we again need to be mindful of the presence of multiple species. The brightness values are sequence-dependent, particularly for the single strands. As shown in Table S4 and Fig. S10 (ESI†), the 2P brightness correlates very closely with the sequence-dependent 1P (average) quantum yield reported previously,23 across the entire set of samples. This implies that there is little discrepancy between the average quantum yields under 2P and 1P excitation. There could be a systematic difference, but this seems unlikely, given the variation in the composition of the emitting populations across the range of samples that is evident in the decay parameters.
The cross sections are similar for pA in ssDNA and dsDNA and do not show any significant dependence on sequence context (in view of the possible uncertainty in quantum yield values under 2P excitation, we consider the values of 2P cross sections for the various oligonucleotides to be within error of each other). However, they are substantially lower, by about a factor of 2, than the value for the pA nucleobase, 6.6 ± 0.5 GM (at 780 nm), that we reported previously.23 In comparison, the 1P absorption coefficient of pA decreases by only about 20% on incorporation in oligonucleotides.23 A decrease in 2P cross section on incorporation in an oligonucleotide has been reported for an intercalating dye,30 but, to our knowledge, this phenomenon has not been reported previously for a FBA. This effect appears to be analogous to the influence of molecular environment on 2P absorption in fluorescent proteins, that has been investigated in detail by Rebane and coworkers.31 As shown in their studies, this can be explained by the influence of the local electrostatic field on the difference in permanent electric dipole moment between ground and excited states, Δ10 and hence on the 2P cross section (see eqn (4)). Application of an external electric field results in an additional induced component of the dipole moment, as a result of the polarizability of the π-electron system of the chromophore. If the polarizabilities of the ground and excited states are different, the change of induced dipole moment will contribute to the total change of dipole moment. For pA in oligonucleotides, it appears that the local electrostatic field acts to decrease Δ10. If this field were due predominantly to the charged phosphate groups, this could account for the lack of dependence on sequence context or duplex formation.
1P vs. 2P photostability
In addition to possessing a high brightness, it is essential for many applications that FBAs have high photostability. The fluorescence intensity of pA during 1P and 2P excitation was measured as a function of irradiation time, with laser power adjusted to give the same initial, absolute emission intensity for both excitation regimes. The experimental arrangement is shown in Fig. S1 (ESI†). As shown in Fig. 8, over the 35 minute timescale investigated, the 2P emission intensity remained constant, whereas the 1P signal decreased to about a third of its initial value. The 1P photobleaching decay is non-exponential, suggesting a time-dependent rate constant that may be due to the depletion of oxygen concentration in the irradiated volume during the measurement, since the solution was not stirred. The dependence of photobleaching rate on the excitation regime suggests that the photochemical process does not occur from the initially excited state, but as a result of further 1P absorption by the excited singlet state (during the initial excitation pulse) and/or a longer-lived triplet state (during subsequent excitation pulses).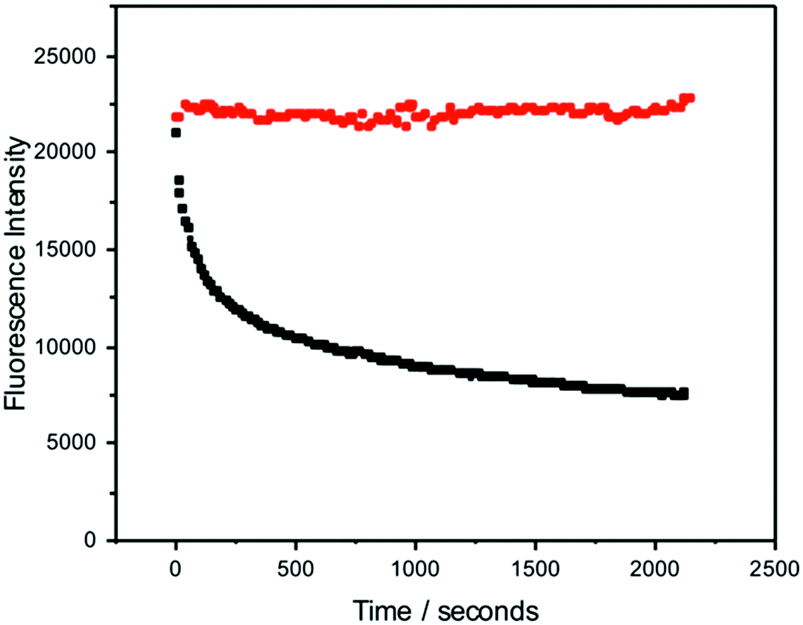 | ||
| Fig. 8 Comparison of the dependence of emission intensity of the free pA nucleobase on irradiation time for 2P excitation at 780 nm (red symbols) and 1P excitation at 390 nm (black symbols). Excitation intensities were adjusted to give the same initial, absolute emission intensity in each case. 2P excitation used the fundamental output of the Ti:sapphire laser, repetition rate 76 MHz, average power 930 mW. 1P excitation used the second-harmonic, pulse-picked output of the same laser, repetition rate 4.75 MHz, average power 690 μW. | ||
The high photostability under 2P excitation encouraged us to attempt ultrasensitive detection, as described in the next section.
Pulse-shaped 2P microscopy of pA-containing DNA
The GA single-strand oligonucleotide was identified as having the highest 2P brightness (Table 1) and was used for subsequent measurements. For microscopy, we used a broadband Ti:sapphire laser as the excitation source (Fig. 1). In order to generate a transform-limited pulse, or other user-defined pulse shapes, at the sample it is necessary to measure and control dispersion in the optical setup. In particular, the higher-order dispersion terms become significant as the bandwidth increases (and pulse duration decreases).32The need for dispersion control can be understood by considering 2P excitation of a two-level system with energy difference ħω0
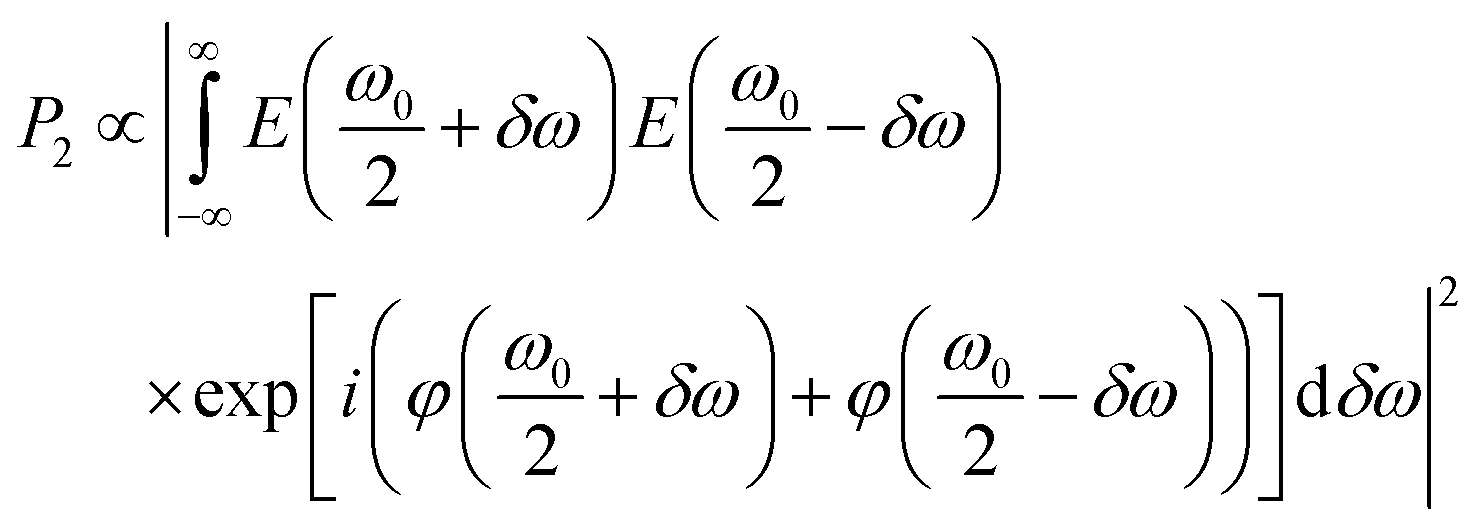 | (5) |
The full spectrum of the laser extends from ∼650–950 nm (Fig. 9). For each pulse shape, the signal and background were recorded as a function of laser power. Intensity was proportional to the square of the laser power until a threshold was reached, the value of which depended on the particular laser spectrum and phase (Fig. 10). Firstly, we investigated the effect of phase shaping on the signal. Using the full laser spectrum, we found that the best contrast was achieved using transform-limited pulses with SHG signal centred on 410 nm and pulse width of 8 fs (Fig. 9).
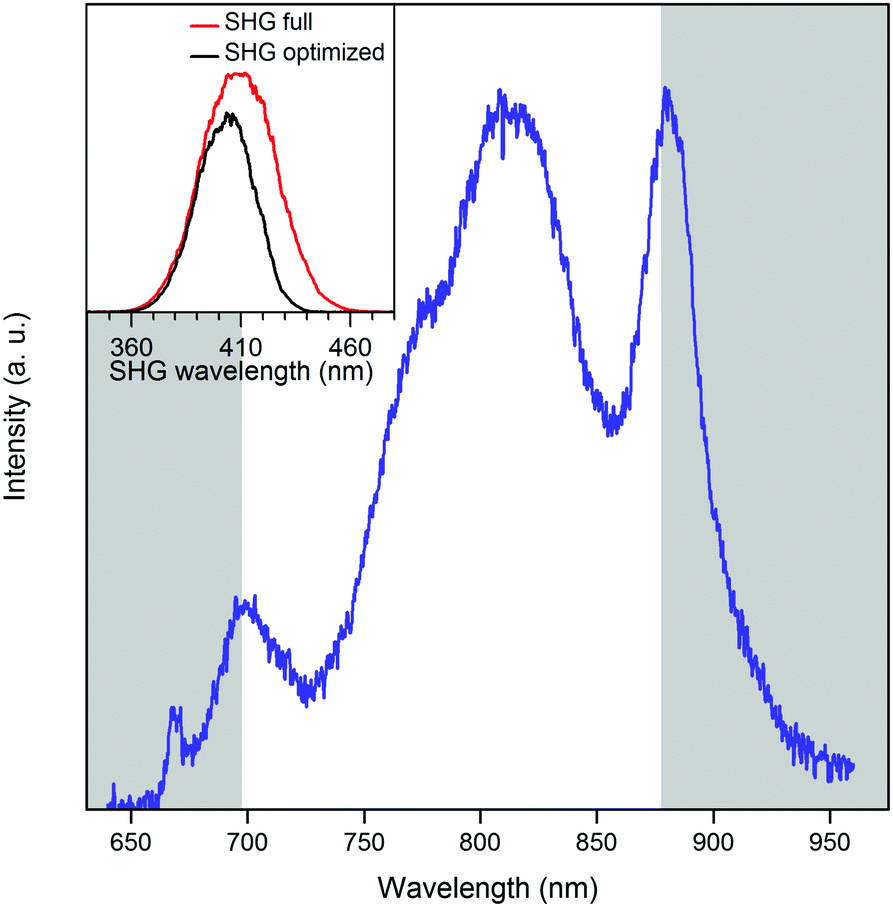 | ||
| Fig. 9 The full spectrum of the laser source (blue line). Grey areas indicate the spectral components, blocked for the optimal pulse shape. The inset graph shows the second harmonic generation (SHG) spectrum for the full laser spectrum (red) and the SHG spectrum from the masked laser spectrum (black). | ||
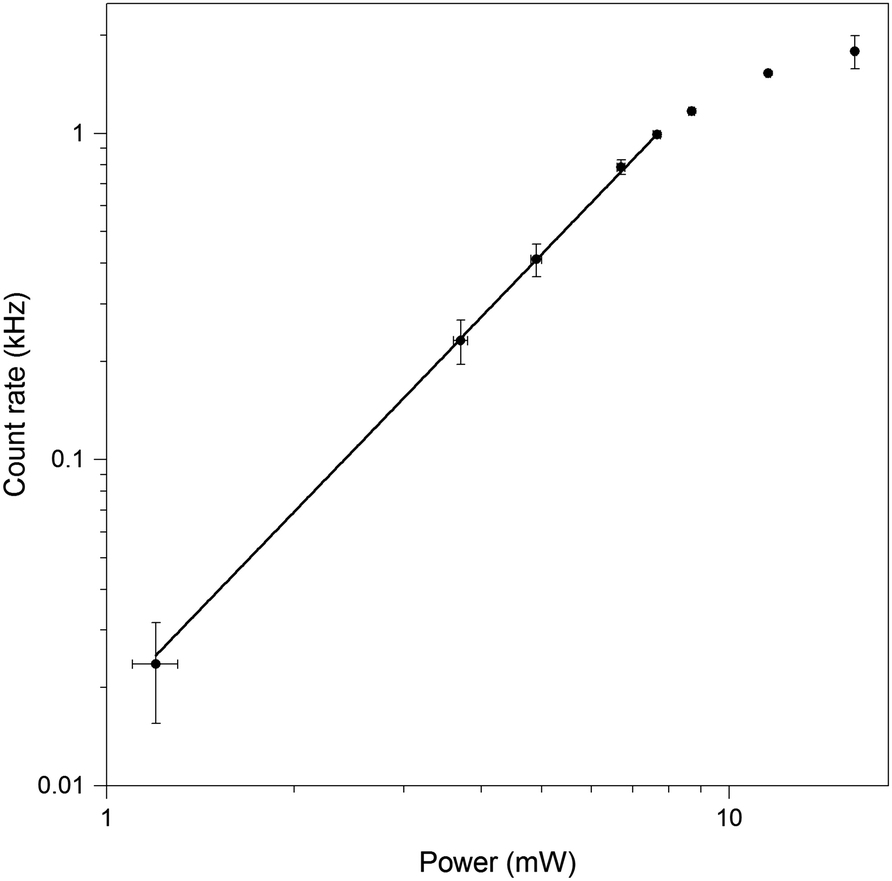 | ||
| Fig. 10 Logarithmic power dependence of the fluorescence intensity of GA oligonucleotide, with no spectral masking. Dependence is linear, with a slope of 1.98 ± 0.04, up to a power of 7.5 mW (1.7 TW cm−2). Loss of linearity occurs at higher powers. Error bars represent the standard error of the mean (N = 4). | ||
Further improvements were achieved by adjusting the centre and the width of the spectrum, removing regions of the spectrum that lead predominantly to background signal rather than two-photon absorption by pA. The best SBR ratio was obtained with an amplitude mask centred at 787 nm and a width of 182 nm (Fig. 9). This increased the SBR from 7.1 to 11.7, with respect to the full spectrum. This is consistent with the long-wavelength maximum identified in the two-photon excitation spectra (Fig. 6) and with the SHG spectrum (Fig. 9), which shows that the amplitude shaping shifts the maximum towards the pA excitation maximum. Compared to a compressed pulse with no additional shaping, the SBR ratio increases by almost 65% when a shaped pulse is used. The pA count rate decreases slightly when using the shaped pulse. Therefore, we attribute the increase in the SBR to background suppression, i.e. more selective excitation of pA.
The SBR was highest for a laser power of 9.7 mW with phase and amplitude shaping (Fig. 9). Having optimized the signal, we used FCS to probe the freely-diffusing pA-modified oligonucleotide and assess the 2P brightness. The setup was first optimised by measuring the 2P-FCS of the dye rhodamine 110 (Fig. S11, ESI†). A representative FCS curve recorded for pA in the GA oligonucleotide is shown in Fig. 11. Background measurements of the buffer were made under identical conditions to ensure that the contribution of fluorescence background was negligible (Fig. 11, inset). There was no evidence of photobleaching during the confocal transit (stable signal for 30 minutes) at 11 mW (Fig. S12, ESI†). Furthermore, the number of molecules measured in the focus scales linearly with sample concentration (Fig. S13, ESI†). The emission is resolved according to the polarization with respect to the laser alignment. The collection efficiency for the two detection channels are matched, so it is apparent that there is a higher signal in the parallel channel (Fig. S12, ESI†), which is due to the restricted mobility of the pA in the 10mer ssDNA and 2P photoselection.43
 | ||
| Fig. 11 Fluorescence correlation spectroscopy for GA oligonucleotide at 9.8 mW with the optimum pulse shape. The correlation curve is shown in blue. The fit (red curve) gave an average of 6.7 molecules in the excitation volume with a diffusion time of 81 μs. The inset shows the correlation curve for pure buffer, recorded at the same conditions. IB = 0.725 kHz; S = 4.889 kHz. The temperature of the solution was 22 ± 1 °C, the sample concentration was 54 nM and the acquisition time was 30 minutes. | ||
The correlation curve G(t) was fitted (Fig. 11) to a model of diffusion through a 3D Gaussian volume (eqn (6)), taking the background contribution IB and the strength of the signal S into account44
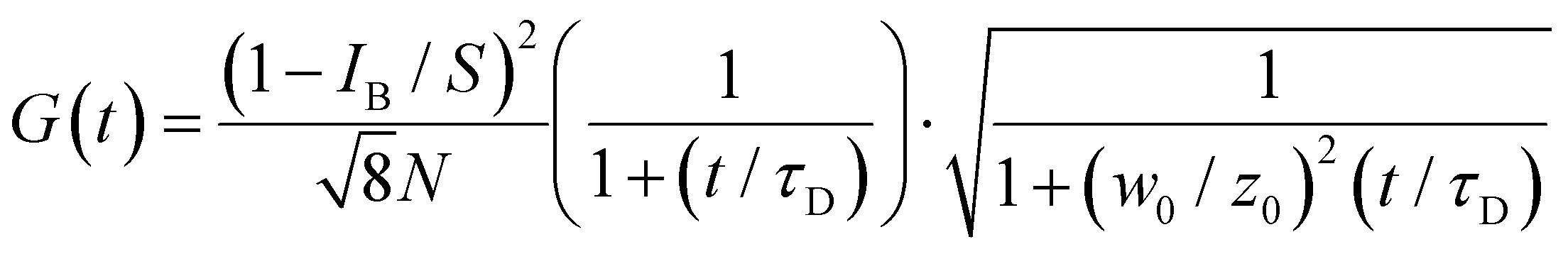 | (6) |
The FCS model (eqn (6)) assumes a homogeneous sample. To examine the possibility of sub-populations of pA states with different brightness, we diluted the solution to the ∼10 pM level and collected MCS traces (Fig. S14, ESI†). The MCS traces for sample (Fig. S14a, ESI†) and buffer (Fig. S14b, ESI†), recorded for 30 minutes, are similar with slightly more high-intensity bursts apparent for the sample. The burst frequency histogram (Fig. S13c, ESI†) from these MCS traces confirms that most of the bursts are of a similar size and distribution to the buffer, as expected for a mean CPM of ∼0.50 kHz, with no evidence of any highly-emissive populations (e.g. due to aggregate formation).
We attempted FCS of the GA duplex, but the signal was too weak to achieve a reliable correlation curve. Nevertheless, since we know that the 2P brightness of the dsDNA is ca. 3 time lower than for the ssDNA (Table S4, ESI†), this is further proof that we are detecting pA in ssDNA, rather than the free base or some other impurity.
Conclusions
The 1P and 2P fluorescence properties of the FBA pentacyclic adenine, pA, have been characterised for the nucleobase, nucleoside and in a series of oligonucleotides. Time-resolved fluorescence measurements indicate that the nucleoside in aqueous solution exists as two emitting species with quantum yields that differ by more than an order of magnitude, and it is likely that these are tautomers. The two tautomers differ in their relative 2P and 1P cross sections, giving rise to different spectral profiles, both excitation and emission, for 2P and 1P excitation regimes. Consequently, the average (steady-state) fluorescence quantum yield is significantly lower for 2P excitation than for 1P excitation.In oligonucleotides, pA exhibits multi-exponential fluorescence decays indicative of different inter-base interactions arising from the conformational heterogeneity of DNA oligomers. There is little evidence of the heterogeneity of the emitting population in the emission spectra, partly because the steady-state intensity tends to be dominated by one species, but also because the different emitting species have similar spectra.
Although the 2P cross section of pA in oligonucleotides is lower than that of the nucleobase, the 2P brightness of pA is the highest reported for a FBA in an oligonucleotide. The 2P brightness depends on sequence context as a consequence of the sensitivity of the quantum yield to interaction with different neighbouring bases. The 2P cross section values are similar for single and double strands and do not show any significant dependence on sequence context.
The decrease in 2P cross section on incorporation in DNA can be attributed to the influence of the local electrostatic field on the change in dipole moment on excitation. This is expected to be a general effect for FBAs and highlights the importance of measuring 2P cross sections in oligonucleotides when characterising new FBAs. Although this effect results in a decrease in cross section for pA, it could, in principle, result in an increase for other FBAs, depending on the projection of the local electric field vector on Δ10.
The high 2P brightness of pA, its photostability under 2P excitation, compared with resonant excitation, and the favourable excitation and emission wavelengths, led us to examine its potential for ultrasensitive analysis. Using a 2P microscope with shaped broadband laser pulses, we were able to detect as few as five pA-modified oligonucleotide molecules free in solution.
A number of theoretical studies of the 1P and 2P photophysics of fluorescent nucleobases have appeared recently.22,45–48 However, the influence of the local electric field on the 2P cross section in DNA has not been addressed. We hope that the results of the present work will stimulate theoretical studies of this effect. As design principles emerge, structural modifications will be implemented to systematically improve the 2P properties of FBAs. Other gains in signal are likely to come from the use of strategies to increase the photostability. For example, antifade reagents which can reduce bleaching at high powers and increase the brightness are commonly used for other organic dyes.49 Through such developments, we believe that the goal of routine single-molecule detection of FBA-modified nucleic acids will soon be realised.
Although the 2P brightness of pA falls short of that required for single-molecule spectroscopy, it is an exceptionally promising fluorescent probe for multiphoton microscopy. In combining high detection sensitivity, high photostability, optimal wavelength for Ti:Sapphire laser-excitation, and an estimated achievable imaging rate of ∼300 frames per second (under the generic conditions defined in ref. 21), pA comprehensively outperforms other FBAs. We anticipate that the application of pA in 2P microscopy will greatly enhance the capability for imaging nucleic acids with minimal structural perturbation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by EPSRC (studentships for RSF and DN) and the University of Edinburgh (RSF). This work was also supported by the Swedish Foundation for Strategic Research (SSF, grants no. ID14-0036 (MG), IS14-0041 (LMW) and IRC15-0065 (LMW)) and the Swedish Research Council (VR, grant No. 2017-03707 (LMW)). We would like to thank Dr Dmitry Pestov (Biophotonic Solutions Inc.) for technical support. Research data supporting this publication can be found at http://dx.doi.org/10.7488/ds/2457.Notes and references
- L. M. Wilhelmsson, Q. Rev. Biophys., 2010, 43, 159–183 CrossRef CAS PubMed.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS PubMed.
- W. Xu, K. M. Chan and E. T. Kool, Nat. Chem., 2017, 9, 1043–1055 CrossRef CAS PubMed.
- D. D. Burns, K. L. Teppang, R. W. Lee, M. E. Lokensgard and B. W. Purse, J. Am. Chem. Soc., 2017, 139, 1372–1375 CrossRef CAS PubMed.
- D. Shin, R. W. Sinkeldam and Y. Tor, J. Am. Chem. Soc., 2011, 133, 14912–14915 CrossRef CAS PubMed.
- A. R. Rovira, A. Fin and Y. Tor, J. Am. Chem. Soc., 2015, 137, 14602–14605 CrossRef CAS PubMed.
- M. S. Wranne, A. F. Füchtbauer, B. Dumat, M. Bood, A. H. El-Sagheer, T. Brown, H. Gradén, M. Grøtli and L. M. Wilhelmsson, J. Am. Chem. Soc., 2017, 139, 9271–9280 CrossRef CAS PubMed.
- J. H. Han, S. Yamamoto, S. Park and H. Sugiyama, Chem. – Eur. J., 2017, 23, 7607–7613 CrossRef CAS PubMed.
- A. C. Jones and R. K. Neely, Q. Rev. Biophys., 2015, 48, 244–279 CrossRef CAS PubMed.
- R. K. Neely, D. Daujotyte, S. Grazulis, S. W. Magennis, D. T. F. Dryden, S. Klimašauskas and A. C. Jones, Nucleic Acids Res., 2005, 33, 6953–6960 CrossRef CAS PubMed.
- S. Wennmalm, H. Blom, L. Wallerman and R. Rigler, Biol. Chem., 2001, 382, 393–397 CAS.
- J. E. Sanabia, L. S. Goldner, P. A. Lacaze and M. E. Hawkins, J. Phys. Chem. B, 2004, 108, 15293–15300 CrossRef CAS.
- E. A. Aleman, C. de Silva, E. M. Patrick, K. Musier-Forsyth and D. Rueda, J. Phys. Chem. Lett., 2014, 5, 777–781 CrossRef CAS PubMed.
- G. S. He, L. S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1368–1376 CrossRef PubMed.
- E. Katilius and N. W. Woodbury, J. Biomed. Opt., 2006, 11, 044004 CrossRef PubMed.
- R. J. Stanley, Z. J. Hou, A. P. Yang and M. E. Hawkins, J. Phys. Chem. B, 2005, 109, 3690–3695 CrossRef CAS PubMed.
- R. S. K. Lane and S. W. Magennis, RSC Adv., 2012, 2, 11397–11403 RSC.
- A. Mikhaylov, S. de Reguardati, J. Pahapill, P. R. Callis, B. Kohler and A. Rebane, Biomed. Opt. Express, 2018, 9, 447–452 CrossRef PubMed.
- R. S. K. Lane, R. Jones, R. W. Sinkeldam, Y. Tor and S. W. Magennis, ChemPhysChem, 2014, 15, 867–871 CrossRef CAS PubMed.
- C. R. Stoltzfus and A. Rebane, Biomed. Opt. Express, 2016, 7, 1768–1782 CrossRef PubMed.
- P. K. Samanta and S. K. Pati, Phys. Chem. Chem. Phys., 2015, 17, 10053–10058 RSC.
- M. Bood, A. F. Füchtbauer, M. S. Wranne, J. J. Ro, S. Sarangamath, A. H. El-Sagheer, D. L. M. Rupert, R. S. Fisher, S. W. Magennis, A. C. Jones, F. Höök, T. Brown, B. H. Kim, A. Dahlén, L. M. Wilhelmsson and M. Grøtli, Chem. Sci., 2018, 9, 3494–3502 RSC.
- M. Bood, E. Weis, A. F. Füchtbauer, A. Dahlén, L. M. Wilhelmsson and M. Grøtli, manuscript in preparation.
- V. V. Lozovoy, I. Pastirk and M. Dantus, Opt. Lett., 2004, 29, 775–777 CrossRef PubMed.
- R. K. Neely, S. W. Magennis, D. T. F. Dryden and A. C. Jones, J. Phys. Chem. B, 2004, 108, 17606–17610 CrossRef CAS.
- G. Stengel, B. W. Purse, L. M. Wilhelmsson, M. Urban and R. D. Kuchta, Biochemistry, 2009, 48, 7547–7555 CrossRef CAS PubMed.
- B. Cohen, P. M. Hare and B. Kohler, J. Am. Chem. Soc., 2003, 125, 13594–13601 CrossRef CAS PubMed.
- A. Rebane, G. Wicks, M. Drobizhev, T. Cooper, A. Trummal and M. Uudsemaa, Angew. Chem., Int. Ed., 2015, 54, 7582–7586 CrossRef CAS PubMed.
- P. H. Doan, D. R. G. Pitter, A. Kocher, J. N. Wilson and T. Goodson, III, J. Am. Chem. Soc., 2015, 137, 9198–9201 CrossRef CAS PubMed.
- M. Drobizhev, N. S. Makarov, S. E. Tillo, T. E. Hughes and A. Rebane, Nat. Methods, 2011, 8, 393–399 CrossRef CAS PubMed.
- A. M. Larson and A. T. Yeh, Opt. Lett., 2006, 31, 1681–1683 CrossRef PubMed.
- D. Meshulach and Y. Silberberg, Nature, 1998, 396, 239–242 CrossRef CAS.
- Y. Silberberg, Annu. Rev. Phys. Chem., 2009, 60, 277–292 CrossRef CAS PubMed.
- V. V. Lozovoy and M. Dantus, ChemPhysChem, 2005, 6, 1970–2000 CrossRef CAS PubMed.
- I. Pastirk, J. M. Dela Cruz, K. A. Walowicz, V. V. Lozovoy and M. Dantus, Opt. Express, 2003, 11, 1695–1701 CrossRef PubMed.
- P. Xi, Y. Andegeko, D. Pestov, V. V. Lovozoy and M. Dantus, J. Biomed. Opt., 2009, 14, 7 CrossRef PubMed.
- P. Xi, Y. Andegeko, L. R. Weisel, V. V. Lozovoy and M. Dantus, Opt. Commun., 2008, 281, 1841–1849 CrossRef CAS.
- R. S. K. Lane, A. N. Macpherson and S. W. Magennis, Opt. Express, 2012, 20, 25948–25959 CrossRef CAS PubMed.
- J. M. Dela Cruz, I. Pastirk, M. Comstock, V. V. Lozovoy and M. Dantus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16996–17001 CrossRef CAS PubMed.
- J. P. Ogilvie, D. Debarre, X. Solinas, J. L. Martin, E. Beaurepaire and M. Joffre, Opt. Express, 2006, 14, 759–766 CrossRef PubMed.
- D. C. Flynn, A. R. Bhagwat, M. H. Brenner, M. F. Nunez, B. E. Mork, D. Cai, J. A. Swanson and J. P. Ogilvie, Opt. Express, 2015, 23, 3353–3372 CrossRef PubMed.
- J. R. Lakowicz, I. Gryczynski, Z. Gryczynski, E. Danielsen and M. J. Wirth, J. Phys. Chem., 1992, 96, 3000–3006 CrossRef CAS.
- L. Brand, C. Eggeling, C. Zander, K. H. Drexhage and C. A. M. Seidel, J. Phys. Chem. A, 1997, 101, 4313–4321 CrossRef CAS.
- M. Gedik and A. Brown, J. Photochem. Photobiol., A, 2013, 259, 25–32 CrossRef CAS.
- Y. J. Lee, Y. H. Jang, Y. Kim and S. Hwang, Bull. Korean Chem. Soc., 2012, 33, 4255–4257 CrossRef CAS.
- T. Pesnot, L. M. Tedaldi, P. G. Jambrina, E. Rosta and G. K. Wagner, Org. Biomol. Chem., 2013, 11, 6357–6371 RSC.
- P. K. Samanta, A. K. Manna and S. K. Pati, J. Phys. Chem. B, 2012, 116, 7618–7626 CrossRef CAS PubMed.
- T. Cordes, A. Maiser, C. Steinhauer, L. Schermelleh and P. Tinnefeld, Phys. Chem. Chem. Phys., 2011, 13, 6699–6709 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp05496g |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2018 |