
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unimolecular decomposition kinetics of the stabilised Criegee intermediates CH2OO and CD2OO†
Daniel
Stone
 *a,
Kendrew
Au
b,
Samantha
Sime
a,
Diogo J.
Medeiros
a,
Mark
Blitz
a,
Paul W.
Seakins
a,
Zachary
Decker‡
b and
Leonid
Sheps
*b
*a,
Kendrew
Au
b,
Samantha
Sime
a,
Diogo J.
Medeiros
a,
Mark
Blitz
a,
Paul W.
Seakins
a,
Zachary
Decker‡
b and
Leonid
Sheps
*b
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: d.stone@leeds.ac.uk
bCombustion Research Facility, Sandia National Laboratories, Livermore, CA 94551, USA. E-mail: lsheps@sandia.gov
First published on 19th September 2018
Abstract
Decomposition kinetics of stabilised CH2OO and CD2OO Criegee intermediates have been investigated as a function of temperature (450–650 K) and pressure (2–350 Torr) using flash photolysis coupled with time-resolved cavity-enhanced broadband UV absorption spectroscopy. Decomposition of CD2OO was observed to be faster than CH2OO under equivalent conditions. Production of OH radicals following CH2OO decomposition was also monitored using flash photolysis with laser-induced fluorescence (LIF), with results indicating direct production of OH in the v = 0 and v = 1 states in low yields. Master equation calculations performed using the Master Equation Solver for Multi-Energy well Reactions (MESMER) enabled fitting of the barriers for the decomposition of CH2OO and CD2OO to the experimental data. Parameterisations of the decomposition rate coefficients, calculated by MESMER, are provided for use in atmospheric models and implications of the results are discussed. For CH2OO, the MESMER fits require an increase in the calculated barrier height from 78.2 kJ mol−1 to 81.8 kJ mol−1 using a temperature-dependent exponential down model for collisional energy transfer with 〈ΔE〉down = 32.6(T/298 K)1.7 cm−1 in He. The low- and high-pressure limit rate coefficients are k1,0 = 3.2 × 10−4(T/298)−5.81exp(−12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770/T) cm3 s−1 and k1,∞ = 1.4 × 1013(T/298)0.06exp(−10010/T) s−1, with median uncertainty of ∼12% over the range of experimental conditions used here. Extrapolation to atmospheric conditions yields k1(298 K, 760 Torr) = 1.1+1.5−1.1 × 10−3 s−1. For CD2OO, MESMER calculations result in 〈ΔE〉down = 39.6(T/298 K)1.3 cm−1 in He and a small decrease in the calculated barrier to decomposition from 81.0 kJ mol−1 to 80.1 kJ mol−1. The fitted rate coefficients for CD2OO are k2,0 = 5.2 × 10−5(T/298)−5.28exp(−11610/T) cm3 s−1 and k2,∞ = 1.2 × 1013(T/298)0.06exp(−9800/T) s−1, with overall error of ∼6% over the present range of temperature and pressure. The extrapolated k2(298 K, 760 Torr) = 5.5+9.2−5.5 × 10−3 s−1. The master equation calculations for CH2OO indicate decomposition yields of 63.7% for H2 + CO2, 36.0% for H2O + CO and 0.3% for OH + HCO with no significant dependence on temperature between 400 and 1200 K or pressure between 1 and 3000 Torr.
770/T) cm3 s−1 and k1,∞ = 1.4 × 1013(T/298)0.06exp(−10010/T) s−1, with median uncertainty of ∼12% over the range of experimental conditions used here. Extrapolation to atmospheric conditions yields k1(298 K, 760 Torr) = 1.1+1.5−1.1 × 10−3 s−1. For CD2OO, MESMER calculations result in 〈ΔE〉down = 39.6(T/298 K)1.3 cm−1 in He and a small decrease in the calculated barrier to decomposition from 81.0 kJ mol−1 to 80.1 kJ mol−1. The fitted rate coefficients for CD2OO are k2,0 = 5.2 × 10−5(T/298)−5.28exp(−11610/T) cm3 s−1 and k2,∞ = 1.2 × 1013(T/298)0.06exp(−9800/T) s−1, with overall error of ∼6% over the present range of temperature and pressure. The extrapolated k2(298 K, 760 Torr) = 5.5+9.2−5.5 × 10−3 s−1. The master equation calculations for CH2OO indicate decomposition yields of 63.7% for H2 + CO2, 36.0% for H2O + CO and 0.3% for OH + HCO with no significant dependence on temperature between 400 and 1200 K or pressure between 1 and 3000 Torr.
Introduction
Atmospheric oxidation initiated by ozone (O3) is a key removal mechanism for unsaturated hydrocarbons and volatile organic compounds (VOCs) emitted into the atmosphere, and proceeds via the addition of ozone to a carbon–carbon double bond in an ozonolysis reaction.1,2 Such reactions lead to the production of Criegee intermediates (R2COO), and are associated with high exothermicities (typically ∼250 kJ mol−1).1 Nascent Criegee intermediates produced in ozonolysis reactions thus contain an excess of internal energy, which can promote unimolecular decomposition, leading to production of key atmospheric species including OH, HO2 and CO, or can be quenched through collisional energy transfer to surrounding gas molecules, leading to the production of stabilised Criegee intermediates (SCIs) which can undergo further chemistry in the atmosphere.Recent developments have identified photolytic sources of stabilised Criegee intermediates, facilitating laboratory studies of SCI reaction kinetics.3,4 Subsequent experimental studies have largely focused on the bimolecular reactions of SCIs, often finding higher reactivity than previously expected, with relatively few studies placing an emphasis on SCI unimolecular decomposition reactions.5–8 Decomposition reactions of SCIs are potentially important in their own right; furthermore, analysis of SCI decomposition may provide insight to the decomposition of nascent excited CIs produced in atmospheric ozonolysis reactions. In addition, production of the CH2OO Criegee intermediate in the combustion of the biofuel dimethyl ether (DME, CH3OCH3) has been proposed, with unimolecular decomposition of CH2OO likely to be important under combustion conditions.9,10 Because CH2OO decomposition involves possible radical and closed-shell product pathways, the knowledge of its rate coefficient and product branching may explain why no evidence of Criegee intermediates has yet appeared in the combustion studies of DME or larger ethers.
Investigation of the potential energy surface (PES) for CH2OO decomposition (R1) using quantum chemical calculations at the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ level indicated the presence of two main reaction channels that produce formic acid and dioxirane, with barriers of 67.4 and 78.2 kJ mol−1, respectively, and with the formic acid channel proceeding via a roaming-like transition state.11 Both channels potentially lead to the final products H2 + CO2, H2O + CO, or HCO + OH.
| CH2OO → H2 + CO2 | (R1a) |
| → H2O + CO | (R1b) |
| → HCO + OH | (R1c) |
Theoretical studies of the decomposition of vibrationally excited nascent CH2OO produced in ethene ozonolysis, using high-accuracy calculations performed with HEAT-345(Q), have also revealed potential decomposition pathways involving dioxirane (with a barrier of 79.9 kJ mol−1 and ultimately leading to H2 and CO2) and formic acid (with final products H2 + CO2 or H2O + CO). An alternative pathway to that involving dioxirane was also reported, leading to production of OH and HCO, but with a calculated barrier of 133.1 kJ mol−1 and thus unlikely to compete with the dioxirane pathway.14,15
Experimental evidence for OH production from CH2OO also comes from studies in which the behaviour of OH radicals has been probed following the photolysis of CH2I2 in O2/Ar gas mixtures.16,17 These studies have successfully used OH measurements as proxies to determine the kinetics of CH2OO reactions, including with SO2. This finding implicates the decomposition of stabilised CH2OO as the OH radical source, although decomposition kinetics were not directly probed.16,17 Similarly, a study of SCIs generated by alkene ozonolysis has also identified the decomposition of stabilised CH2OO as a potential source of OH, but kinetic information was limited and a low OH yield was reported.18
Ozonolysis experiments performed in atmospheric simulation chambers have been used to infer the kinetics of stabilised CH2OO decomposition, but with large uncertainties, since first-order losses of CH2OO by physical processes such as wall reactions are difficult to distinguish from loss through unimolecular decomposition. A recent series of chamber experiments at atmospheric temperature and pressure reported an upper limit of 4.2 s−1 for the first-order loss of stabilised CH2OO, which includes a contribution from the decomposition reaction.19,20 This value was determined by measuring the ratio of the rate coefficient for first-order losses to that for the reaction of CH2OO with SO2, and the decomposition rate was indistinguishable from zero within the measurement uncertainties.19,20
Measurements of photolytically generated CH2OO using cavity ringdown spectroscopy (CRDS) have also been used to infer the unimolecular decomposition kinetics in N2 at 293 K in the pressure range 7 to 30 Torr, giving an upper limit to the decomposition rate coefficient of (11.6 ± 8.0) s−1. Similarly to the results obtained in the chamber experiments, the first-order removal of CH2OO in the CRDS measurements contains not only the contribution from the unimolecular decomposition but also contributions from physical processes such as diffusion and wall loss.21 A similar upper limit to the rate coefficient for decomposition of (9.4 ± 1.7) s−1 was reported from an investigation of the reactions of CH2OO with SO2 and water vapour at 293 K and atmospheric pressure.22
A further investigation of the decomposition of stabilised CH2OO was performed using a free-jet flow reactor, in which CH2OO was produced by ozonolysis of ethene in air and monitored by sampling from the flow reactor and titration to H2SO4 through reaction with SO2.23 The design of the free-jet flow reactor reduces wall losses, and the pressure used in the reactor inhibits losses through diffusion, thus minimising the contributions to CH2OO loss from physical processes. The study reported a rate coefficient for stabilised CH2OO decomposition of (0.19 ± 0.07) s−1 at 297 K and 1 bar, with quantum chemical calculations at the CCSD(T)/aug-cc-pVTZ level giving a calculated barrier height to decomposition of 78.9 kJ mol−1.23 A high pressure limiting rate coefficient of 0.25 s−1 was determined from master equation calculations at 297 K,23 similar to that obtained in an earlier theoretical study,24 with fall-off behaviour predicted at the pressure of the experiment and predictions for the rate coefficient at 1 bar between 0.037 s−1 and 0.12 s−1.23 Improved agreement with the experimental value was achieved by a reduction in the calculated barrier height to 76.8 kJ mol−1, resulting in an increase in the predicted high pressure limiting rate coefficient to 0.58 s−1 at 297 K and predictions for the rate coefficient at 1 bar and 297 K between 0.08 s−1 and 0.26 s−1.23
Thus, while there have been several attempts to determine the decomposition kinetics of stabilised CH2OO Criegee intermediates, the results have large uncertainties, and the most reliable measurements obtained using the free-jet flow reactor have only been reported at a single temperature and pressure. Full assessment of the impacts of stabilised CH2OO decomposition in the atmosphere, where it may be in competition with CH2OO + SO2 at low SO2 concentrations given the range of CH2OO decomposition rate coefficients reported in the literature (0.037–11.6 s−1), and in chamber studies of ozonolysis reactions, is therefore hindered by a lack of measurements of stabilised CH2OO kinetics over a wide range of temperatures and pressures. In this work we report a detailed study of the decomposition kinetics of stabilised CH2OO (k1) and CD2OO (k2) as a function of temperature and pressure.
| CH2OO → products | (R1) |
| CD2OO → products | (R2) |
Experimental
UV absorption
Time-resolved cavity-enhanced broadband UV absorption spectroscopy experiments were performed at the Sandia National Laboratories, Livermore, USA, using experimental apparatus described in detail in previous publications.26–28 Precursor gas mixtures containing CH2I2 (or CD2I2), entrained in He, O2, and He buffer gas were admitted into a quartz flow cell using calibrated mass flow controllers (MKS Instruments). Chemistry in the reaction cell was initiated by the photolysis of the di-iodo precursor at a wavelength of 266 nm, which was generated by the 4th harmonic of an Nd:YAG laser (Continuum Surelite III) with a typical fluence of ∼19 mJ cm−2, with O2 concentrations such that the production of CH2OO (or CD2OO) was rapid compared to the subsequent decay. Transient absorptions in the reaction cell were monitored using a Xe arc lamp (Newport Corp.), which was reflected between two concave high reflectivity mirrors (JDSU Inc.) forming an optical resonator cavity 1.6 m in length, operating over probe wavelengths between 300 and 450 nm simultaneously with total effective absorption path length of 40–56 m. Light exiting the optical cavity was directed into a time-resolved spectrometer, consisting of a rotating mirror, synchronized with the photolysis laser, that rapidly sweeps the probe beam vertically, followed by a ruled grating, which provides spectral dispersion in the horizontal plane. Spectral and temporal information contained in the probe beam are thus spatially mapped onto the horizontal and vertical position, respectively, at the focal plane of the spectrometer. The time evolution of the entire broadband cavity output is recorded by a TEC-cooled 1024 × 1024-element CCD camera for every laser shot, and averaged on-chip (300–600 shots in the present work), prior to transfer to a computer for data analysis.Transient absorption spectra were computed by Beer's Law from the difference between probe light intensities with (ION) and without (IOFF) the photolysis laser: OD(λ,t) = −ln(ION(λ,t)/IOFF(λ,t)). The mirror rotation was adjusted between 1 and 10 Hz (corresponding to total observation times between 13.5 and 1.35 ms, respectively) as needed to capture the kinetics under investigation. The experimental resolution of this spectrometer is ultimately determined by spatial focusing of the probe beam on the CCD sensor: ∼7 pixels (FWHM), corresponding to spectral resolution of ∼1.5 nm and temporal resolution of ∼9–90 μs, depending on the mirror rotation frequency. The total flow rate through the reaction cell was adjusted with changes in pressure and laser repetition rate to ensure a fresh sample of gas in the cell for each photolysis shot.
The pressure in the reaction cell was maintained by a roots pump and actively controlled by a butterfly valve throttling the exit of the cell. Temperatures in the reaction cell were controlled by a series of ceramic heaters (Watlow) surrounding the cell and monitored by K-type thermocouples situated along the length of the reaction cell. Experiments were performed in He (Matheson, 99.9999%) at pressures between 2 and 350 Torr and at temperatures in the range 450 to 650 K, with CH2I2 (Aldrich, 99%)/CD2I2 (Aldrich, 98%) concentrations in the range 8 × 1012 to 8 × 1013 cm−3 and O2 (Matheson, 99.9999%) concentrations varied between 1 × 1016 and 7 × 1018 cm−3. Gases and chemicals were used as supplied.
Concentrations of CH2OO were determined by fitting reference spectra for the CH2I2 precursor, CH2OO and IO (generated by secondary chemistry within the system) to the observed total absorbance between 300 and 440 nm for each time point throughout the reaction. Typical absorbance signals of 10−3–10−4 were measured in this work, which correspond to changes in concentration of 0.001–0.0001% (assuming 100% photodissociation on absorption of a photon), which is insignificant compared to the changes in concentration owing to reaction. Fig. 1 shows a typical concentration–time profile for CH2OO. Details regarding the fitting procedure are given in the ESI.†
 | ||
| Fig. 1 Normalised concentrations of CH2OO following photolysis of CH2I2/O2/He at a temperature of 525 K and pressure of 5 Torr (black points) and the result of a first-order kinetic fit to the data (red line), convoluted with a Gaussian instrument function, giving a decay rate coefficient k1,obs = (440 ± 10) s−1. The fit residuals are discussed in further detail in the ESI.† | ||
Laser-induced fluorescence
Laser-induced fluorescence (LIF) experiments were performed at the University of Leeds, UK, in a slow flow reactor which has been described in detail in previous work.29–31 Precursor gas mixtures (CH2I2/O2/N2) were prepared in a glass gas manifold and passed into a stainless steel six-way cross at known flow rates determined by calibrated mass flow controllers (MKS Instruments). Photolysis of CH2I2, leading to rapid production of CH2OO, was achieved at a wavelength of 355 nm using the 3rd harmonic of an Nd:YAG laser (Continuum Powerlite 8010). Experiments were typically performed at a repetition rate of 10 Hz, although the lasers were also operated at lower repetition rates to ensure that there were no interferences from photolysis products. The laser fluence was typically ∼20 mJ cm−2.The pressure in the reaction cell was monitored by a capacitance manometer, and was maintained by a rotary pump throttled by a needle valve on the exhaust line. Heating of the reaction cell was achieved by a series of cartridge heaters surrounding the cell, with temperatures monitored by K-type thermocouples situated close to the reaction zone.
OH radicals produced in the system were monitored by off-resonance laser-induced fluorescence following either A2Σ(v′ = 1) ← X2Π(v′′ = 0) excitation at a wavelength of 282 nm for detection of OH in the ground vibrational state, OH(v′′ = 0), or A2Σ(v′ = 1) ← X2Π(v′′ = 1) excitation at 288 nm for detection of OH in its first vibrationally excited state, OH(v′′ = 1). The 532 nm output of a Nd:YAG laser (Continuum Powerlite 8010) was used to pump a dye laser (Spectra Physics PDL-3) operating on either Rhodamine-6-G or pyromethene 597 dye, with the dye output frequency-doubled to generate light at 282 or 288 nm, respectively. For both excitation wavelengths, the off-resonant OH fluorescence at ∼308 nm was passed through an interference filter (Barr Associates, (308 ± 5) nm) and monitored by a channel photomultiplier (CPM, Perkin-Elmer C1943P) mounted perpendicular to the plane of photolysis and probe laser beams. The CPM signal was digitised and integrated on an oscilloscope (LeCroy LT262) prior to being passed to a computer for data analysis. The time delay between the photolysis and probe laser pulses was controlled by a digital delay generator (SRS DG535) and varied to enable monitoring of the OH profiles as a function of time following photolysis of the gas mixture. Kinetic traces typically consisted of 200 time points, with each time point averaged 5–10 times.
Experiments were performed in N2 (BOC, oxygen free, 99.99%) at pressures between 10 and 95 Torr and at temperatures in the range 480 to 570 K, with CH2I2 (Sigma-Aldrich, 99%) concentrations in the range 4 × 1012 to 2 × 1015 cm−3 and O2 (BOC, 99.999%) concentrations varied between 3.7 × 1016 and 5.8 × 1017 cm−3. Gases and chemicals were used as supplied.
Results
UV absorption
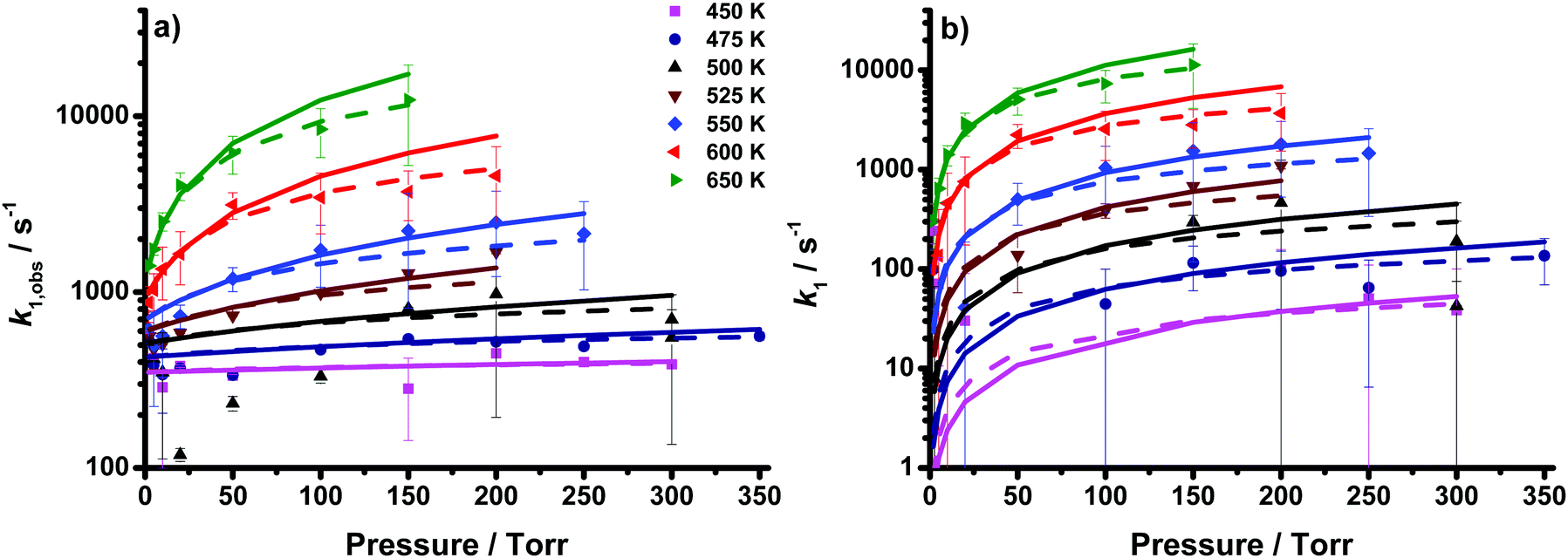 | ||
| Fig. 2 Rate coefficients describing (a) the observed decay of CH2OO (k1,obs) and (b) the decomposition of CH2OO (k1 = k1,obs − k1,bg) as a function of temperature and pressure (coloured points) determined from the UV experiments. Fits to the Troe equation (dashed lines) and MESMER simulations using optimised barrier heights and 〈ΔE〉down (solid lines) are also shown. Error bars are 1σ. | ||
| T/K | p/Torr | k 1,obs/s−1 | Fit to k1,obs/s−1 | k 1,Troe/s−1 | k 1,bg/s−1 | k 1,MESMER/s−1 |
|---|---|---|---|---|---|---|
| 450 | 2 | 590 ± 9 | 351 ± 60 | 1 | 350 | 0.5 ± 0.1 |
| 5 | 420 ± 7 | 352 ± 60 | 2 | 350 | 1.2 ± 0.2 | |
| 10 | 290 ± 190 | 354 ± 60 | 4 | 350 | 2.4 ± 0.4 | |
| 20 | 380 ± 6 | 357 ± 59 | 7 | 350 | 5 ± 1 | |
| 50 | 330 ± 6 | 365 ± 59 | 15 | 350 | 11 ± 2 | |
| 150 | 280 ± 140 | 381 ± 58 | 31 | 350 | 29 ± 5 | |
| 200 | 450 ± 10 | 386 ± 58 | 36 | 350 | 38 ± 7 | |
| 250 | 400 ± 11 | 391 ± 59 | 41 | 350 | 46 ± 9 | |
| 300 | 390 ± 18 | 395 ± 59 | 45 | 350 | 53 ± 10 | |
| ∞ | 2800 ± 2600 | |||||
| 475 | 2 | 530 ± 13 | 428 ± 58 | 2 | 426 | 2 ± 0 |
| 5 | 390 ± 11 | 431 ± 57 | 5 | 426 | 4 ± 1 | |
| 10 | 340 ± 130 | 436 ± 57 | 10 | 426 | 7 ± 1 | |
| 20 | 370 ± 9 | 445 ± 57 | 19 | 426 | 14 ± 2 | |
| 50 | 340 ± 8 | 466 ± 56 | 40 | 426 | 34 ± 5 | |
| 100 | 470 ± 10 | 491 ± 54 | 65 | 426 | 63 ± 9 | |
| 150 | 540 ± 10 | 510 ± 54 | 84 | 426 | 90 ± 14 | |
| 200 | 520 ± 10 | 524 ± 55 | 98 | 426 | 116 ± 19 | |
| 250 | 490 ± 20 | 537 ± 56 | 111 | 426 | 141 ± 23 | |
| 350 | 560 ± 30 | 558 ± 61 | 132 | 426 | 188 ± 33 | |
| ∞ | 9200 ± 8400 | |||||
| 500 | 3 | 520 ± 20 | 516 ± 55 | 8 | 508 | 6 ± 1 |
| 5 | 420 ± 10 | 521 ± 54 | 13 | 508 | 10 ± 1 | |
| 10 | 350 ± 240 | 534 ± 54 | 26 | 508 | 20 ± 2 | |
| 20 | 120 ± 10 | 555 ± 56 | 47 | 508 | 39 ± 4 | |
| 50 | 230 ± 20 | 608 ± 59 | 100 | 508 | 91 ± 10 | |
| 100 | 330 ± 30 | 669 ± 52 | 161 | 508 | 171 ± 21 | |
| 150 | 800 ± 20 | 714 ± 49 | 206 | 508 | 246 ± 32 | |
| 200 | 970 ± 780 | 750 ± 54 | 242 | 508 | 317 ± 44 | |
| 300 | 550 ± 410 | 807 ± 62 | 299 | 508 | 449 ± 66 | |
| 300 | 700 ± 100 | 807 ± 62 | 299 | 508 | 449 ± 66 | |
| ∞ |
27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000 ± 25000 000 ± 25000
|
|||||
| 525 | 3 | 560 ± 10 | 613 ± 56 | 16 | 597 | 13 ± 2 |
| 5 | 470 ± 110 | 628 ± 54 | 31 | 597 | 25 ± 3 | |
| 10 | 510 ± 10 | 655 ± 55 | 58 | 597 | 48 ± 4 | |
| 20 | 590 ± 10 | 705 ± 66 | 108 | 597 | 94 ± 8 | |
| 50 | 740 ± 10 | 823 ± 80 | 226 | 597 | 222 ± 21 | |
| 100 | 990 ± 10 | 961 ± 63 | 364 | 597 | 418 ± 44 | |
| 150 | 1280 ± 20 | 1062 ± 55 | 465 | 597 | 601 ± 67 | |
| 200 | 1690 ± 30 | 1143 ± 68 | 546 | 597 | 773 ± 90 | |
| ∞ | 70000 ± 64000 |
|||||
| 550 | 2 | 620 ± 20 | 717 ± 63 | 27 | 690 | 23 ± 3 |
| 5 | 500 ± 270 | 754 ± 60 | 64 | 690 | 55 ± 5 | |
| 10 | 560 ± 230 | 812 ± 68 | 122 | 690 | 107 ± 8 | |
| 20 | 730 ± 110 | 916 ± 95 | 226 | 690 | 209 ± 15 | |
| 50 | 1190 ± 190 | 1163 ± 128 | 473 | 690 | 493 ± 38 | |
| 100 | 1740 ± 670 | 1453 ± 98 | 763 | 690 | 929 ± 80 | |
| 150 | 2230 ± 1420 | 1664 ± 80 | 974 | 690 | 1336 ± 123 | |
| 200 | 2490 ± 1260 | 1832 ± 101 | 1142 | 690 | 1722 ± 167 | |
| 250 | 2150 ± 1120 | 1975 ± 131 | 1285 | 690 | 2092 ± 212 | |
| ∞ | 170000 ± 160000 |
|||||
| 600 | 2 | 870 ± 200 | 988 ± 97 | 97 | 891 | 87 ± 9 |
| 5 | 1030 ± 240 | 1125 ± 93 | 234 | 891 | 213 ± 15 | |
| 10 | 1350 ± 450 | 1336 ± 121 | 445 | 891 | 417 ± 25 | |
| 20 | 1650 ± 550 | 1714 ± 190 | 823 | 891 | 812 ± 45 | |
| 50 | 3130 ± 540 | 2613 ± 270 | 1722 | 891 | 1926 ± 108 | |
| 100 | 3450 ± 1300 | 3664 ± 210 | 2773 | 891 | 3641 ± 218 | |
| 150 | 3720 ± 1170 | 4427 ± 157 | 3536 | 891 | 5246 ± 333 | |
| 200 | 4580 ± 2140 | 5037 ± 178 | 4146 | 891 | 6774 ± 451 | |
| ∞ | 770000 ± 720000 |
|||||
| 650 | 2 | 1410 ± 10 | 1424 ± 150 | 318 | 1106 | 289 ± 29 |
| 5 | 1750 ± 130 | 1801 ± 123 | 695 | 1106 | 646 ± 50 | |
| 10 | 2530 ± 300 | 2429 ± 133 | 1323 | 1106 | 1268 ± 85 | |
| 20 | 4050 ± 730 | 3552 ± 213 | 2446 | 1106 | 2473 ± 151 | |
| 50 | 6190 ± 1500 | 6221 ± 322 | 5115 | 1106 | 5879 ± 335 | |
| 100 | 8440 ± 2620 | 9338 ± 288 | 8232 | 1106 | 11149 ± 615 |
|
| 150 | 12400 ± 7140 |
11599 ± 305 |
10493 |
1106 | 16103 ± 875 |
|
| ∞ | 2800000 ± 2600000 |
|||||
At temperatures below 500 K, there is little variation in the observed rate coefficients as a function of pressure, although there is an increase from 450 K to 475 K. At temperatures of 500 K and above, the rate coefficients increase with increasing temperature and pressure. The loss of CH2OO thus appears to contain contributions from two processes, a pressure- and temperature-dependent term, k(p,T), and a pressure-independent temperature-dependent term, k(T). Given the PES for CH2OO decomposition,11,12,14,23 we attribute the pressure-dependent term to CH2OO decomposition, and the pressure-independent term to other background losses of CH2OO, such that the observed rate coefficient, k1,obs, is given by the sum of k1(p,T) and k1,bg(T). A global fit using data at all temperatures and pressures was performed to determine k1(p,T) and k1,bg(T), with k1(p,T) described by the basic Troe equation32 as shown in eqn (1):
 | (1) |
Fits to eqn (1) were performed with k1,bg(T) either constrained to Arrhenius behaviour or unconstrained, i.e. allowed to float independently at each temperature. The fits with k1,bg(T) constrained to Arrhenius behaviour gave a lower χ2 value, although the parameterisations from each fit were in agreement within the fit uncertainties. The fit with k1,bg(T) constrained to Arrhenius behaviour gives k1,0 = (1.3 ± 4.1) × 10−8exp(−(9130 ± 2080)/T) cm3 s−1 and k1,∞ = (2.4 ± 9.8) × 1010exp(−(8460 ± 2660)/T) s−1, with Fc fixed to a value of 0.6,33,34 and kbg = (1.5 ± 1.1) × 104exp(−(1680 ± 400)/T) s−1. While the uncertainties in the individual Arrhenius parameters describing k1,0(T), k1,∞(T) and k1,bg are large, inspection of the covariance matrix (given in the ESI†) indicates that the fit parameters are highly correlated. A complete uncertainty analysis (described in the ESI†), incorporating the correlations between the fit parameters shows that the overall uncertainty in the fit ranges from 3% at the highest temperatures and pressures to 17% at the lowest temperatures and pressures, with a median of 10%. The full results are given in Table 1. While this parameterisation can be used to provide a value for k1 at 298 K and 760 Torr, the extrapolation is subject to significant uncertainties since the experiments do not cover a sufficiently broad range of pressures in the fall-off regime. Instead, the parameterisation is performed primarily to determine the contributions to the total loss from decomposition and background losses, with the pressure and temperature dependence of the decomposition best described by the Master Equation treatment discussed below.
The pressure-independent background losses of CH2OO, k1,bg(T), demonstrate the presence of removal processes other than CH2OO decomposition, including wall losses and secondary chemical loss of CH2OO. Given the magnitude and temperature dependence of the pressure-independent contribution to the loss, chemical reactions of CH2OO are likely to be the dominant factor. Results from mixed-order fits to the observed decays indicated little sensitivity to second-order processes, and thus a negligible contribution from CH2OO self-reaction. At 450 K, the data suggest a contribution from reaction between CH2OO and CH2I2 (see ESI†), with a bimolecular rate coefficient of (8.2 ± 1.7) × 10−12 cm3 s−1. The reaction has also recently been observed by Liu et al.,17 with a rate coefficient of (5.2 ± 2.6) × 10−14 cm3 s−1 at 298 K. At temperatures above 450 K, concentrations of CH2I2 were not varied over a sufficient range to fully assess the role of CH2OO + CH2I2; however, the results overall are consistent with the pressure-independent loss term at all temperatures coming largely from the pseudo-first-order loss of CH2OO through reaction with CH2I2.
 | ||
| Fig. 3 Normalised absorption spectra for CH2OO (blue) and CD2OO (red) determined in this work at 295 K and 10 Torr. Details regarding the characterisation of the spectra are given in the ESI.† | ||
Decays for CD2OO were fit to first-order loss kinetics, convolved with a Gaussian instrument function, and the observed rate coefficients are shown in Fig. 4 and Table 2. Similarly to CH2OO, the rate coefficients describing the decays of CD2OO exhibit pressure dependence at temperatures of 500 K and above, but not at 450 K, indicating contributions from both the pressure-dependent CD2OO decomposition (k2) and pressure-independent secondary background losses (k2,bg). The observed rate coefficients for the CD2OO decays were thus fit to an analogous expression to eqn (1) given for CH2OO. We propose that the background loss of CD2OO is, at least in part, a result of reaction with CD2I2. However, constraining k2,bg(T) to Arrhenius behaviour gave poor fits to data at 450 K, where the background loss dominates the observed decay, potentially owing to fewer data points compared to CH2OO. The fits with k2,bg unconstrained to Arrhenius behaviour give k2,0 = (1.5 ± 4.0) × 10−11exp(−(4640 ± 1800)/T) cm3 s−1 and k2,∞ = (6.4 ± 86.7) × 1015exp(−(14750 ± 7800)/T) s−1 with Fc fixed at a value of 0.6. Values for k2,bg are summarised in Table 2 and can be approximated by the expression k2,bg = (2.4 ± 6.9) × 104exp(−(2080 ± 1570)/T) s−1. Similarly to the parameterisation for CH2OO decomposition, the uncertainties in the individual fit parameters for k2,obs are deceptively large, owing to correlations between the fit parameters. Again, the fits are performed largely to determine k2,bg, rather than to extrapolate k2 to 298 K and 760 Torr. Consideration of these correlations between the fit parameters in the uncertainty analysis, as described in the ESI† for CH2OO, indicates a median total uncertainty of 21% in the fits to k2,obs. The total uncertainties in the fits to CD2OO decays are larger than for CH2OO, and the fits display greater variability, since there are fewer data points for CD2OO compared to CH2OO, particularly at the lower pressures. Fit results and uncertainties for CD2OO are given in Table 2.
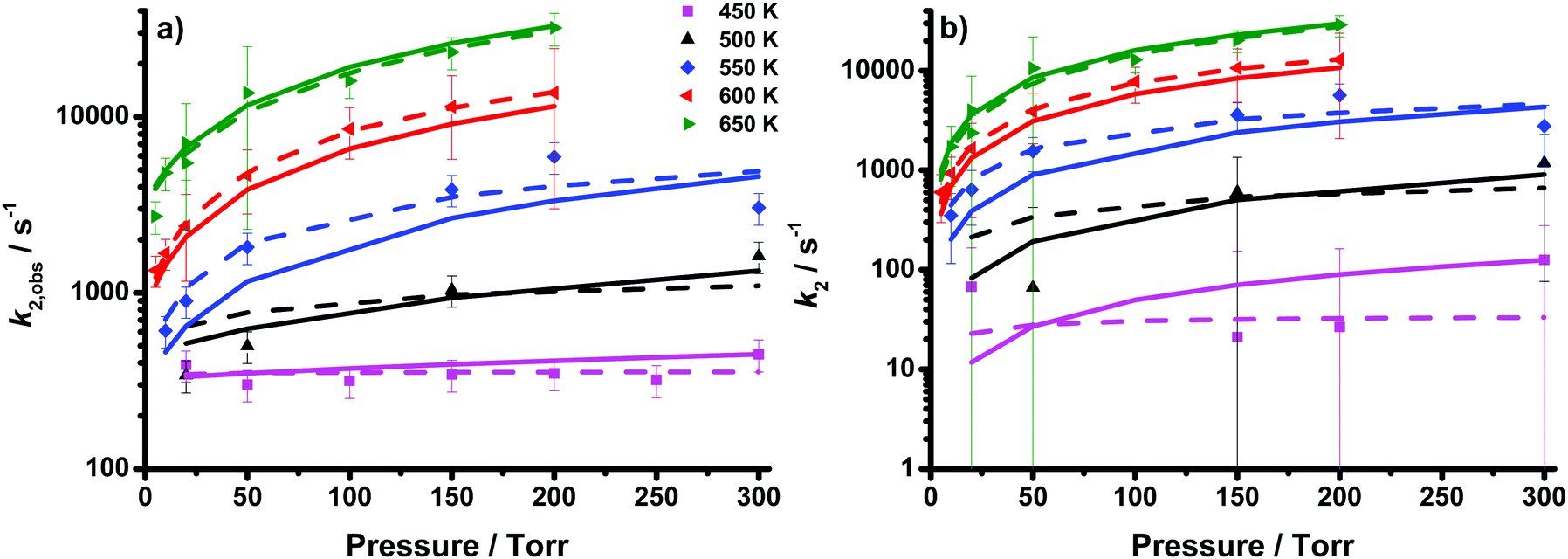 | ||
| Fig. 4 Rate coefficients describing (a) the observed decay of CD2OO (k2,obs) and (b) the decomposition of CD2OO (k2 = k2,obs − k2,bg) as a function of temperature and pressure (coloured points) determined from the UV experiments. Fits to the Troe equation (dashed lines) and MESMER simulations using optimised barrier heights and 〈ΔE〉down (solid lines) are also shown. Error bars are 1σ. | ||
| T/K | p/Torr | k 2,obs/s−1 | Fit to k2,obs/s−1 | k 2,Troe/s−1 | k 2,bg/s−1 | k 2,MESMER/s−1 |
|---|---|---|---|---|---|---|
| 450 | 20 | 390 ± 10 | 345 ± 60 | 23 | 322 | 12 ± 1 |
| 50 | 300 ± 10 | 350 ± 88 | 28 | 322 | 27 ± 3 | |
| 100 | 320 ± 10 | 353 ± 105 | 31 | 322 | 50 ± 5 | |
| 150 | 340 ± 10 | 344 ± 112 | 22 | 322 | 70 ± 7 | |
| 200 | 350 ± 10 | 354 ± 116 | 32 | 322 | 90 ± 9 | |
| 250 | 320 ± 20 | 355 ± 119 | 33 | 322 | 108 ± 12 | |
| 300 | 450 ± 20 | 355 ± 121 | 33 | 322 | 125 ± 14 | |
| ∞ | 4100 ± 3200 | |||||
| 500 | 20 | 340 ± 20 | 645 ± 186 | 213 | 432 | 83 ± 5 |
| 50 | 500 ± 20 | 772 ± 343 | 340 | 432 | 192 ± 13 | |
| 150 | 1040 ± 20 | 975 ± 715 | 543 | 432 | 505 ± 38 | |
| 300 | 1610 ± 60 | 1096 ± 1056 | 664 | 432 | 905 ± 76 | |
| ∞ | 36000 ± 25000 |
|||||
| 550 | 10 | 610 ± 20 | 707 ± 200 | 448 | 259 | 202 ± 10 |
| 20 | 900 ± 30 | 1075 ± 308 | 816 | 259 | 388 ± 17 | |
| 50 | 1810 ± 60 | 1913 ± 457 | 1654 | 259 | 902 ± 41 | |
| 150 | 3860 ± 90 | 3510 ± 987 | 3251 | 259 | 2398 ± 128 | |
| 200 | 5920 ± 210 | 4043 ± 1217 | 3784 | 259 | 3073 ± 172 | |
| 300 | 3050 ± 150 | 4901 ± 1593 | 4642 | 259 | 4329 ± 262 | |
| ∞ | 220000 ± 140000 |
|||||
| 600 | 5 | 1340 ± 10 | 1223 ± 139 | 481 | 742 | 365 ± 16 |
| 10 | 1680 ± 20 | 1665 ± 261 | 923 | 742 | 693 ± 25 | |
| 20 | 2390 ± 1130 | 2528 ± 473 | 1786 | 742 | 1333 ± 40 | |
| 50 | 4650 ± 1590 | 4895 ± 918 | 4153 | 742 | 3121 ± 94 | |
| 100 | 8520 ± 2170 | 8312 ± 1314 | 7570 | 742 | 5844 ± 194 | |
| 150 | 11420 ± 5200 |
11247 ± 1576 |
10505 |
742 | 8370 ± 298 | |
| 200 | 13690 ± 5160 |
13818 ± 1914 |
13076 |
742 | 10756 ± 406 |
|
| ∞ | 990000 ± 550000 |
|||||
| 650 | 5 | 2720 ± 170 | 3892 ± 114 | 811 | 3081 | 970 ± 38 |
| 10 | 4800 ± 320 | 4685 ± 213 | 1604 | 3081 | 1882 ± 59 | |
| 20 | 7080 ± 4600 | 6240 ± 380 | 3159 | 3081 | 3634 ± 93 | |
| 20 | 5460 ± 110 | 6240 ± 380 | 3159 | 3081 | 3634 ± 93 | |
| 50 | 13640 ± 5510 |
10733 ± 720 |
7652 | 3081 | 8550 ± 201 | |
| 100 | 15990 ± 690 |
17816 ± 996 |
14735 |
3081 | 16093 ± 382 |
|
| 150 | 23300 ± 1210 |
24504 ± 1318 |
21423 |
3081 | 23126 ± 569 |
|
| 200 | 31990 ± 2024 |
30864 ± 2052 |
27783 |
3081 | 29798 ± 762 |
|
| ∞ | 3500000 ± 1800000 |
|||||
Laser-induced fluorescence
 | ||
| Fig. 5 Normalised OH(v = 0) LIF signal following photolysis of CH2I2/O2/N2 at T = 570 K and p = 20 Torr (black points) with kinetic fit (eqn (2), red line), giving k1 = (1120 ± 30) s−1. The inset shows the first 4000 μs following photolysis in greater detail. | ||
The rate of the OH(v = 0) decay was dependent on the concentration of CH2I2, indicating removal of OH(v = 0) through the expected reaction of OH with CH2I2. However, the observed loss of OH was not well described by a single exponential decay. Instead, it was better described by a biexponential function, indicating a slower growth of OH(v = 0) in the system on a timescale similar to the loss through reaction with CH2I2. The slow growth of OH(v = 0) in the system was attributed to production through the decomposition of CH2OO. The production and loss of OH in the system was thus assigned to the mechanism in reactions (R1) and (R3)–(R7):
| CH2I2 + hν → CH2I* + I | (R3) |
| CH2I* + O2 → CH2OO | (R4) |
| CH2I* + O2 → OH(v = 0,n) + HCO + I | (R5) |
| OH(v = 1) + M → OH(v = 0) + M | (R6) |
| CH2OO + M → OH(v = 0,n) + other products | (R1) |
| OH(v = 0) + CH2I2 → products | (R7) |
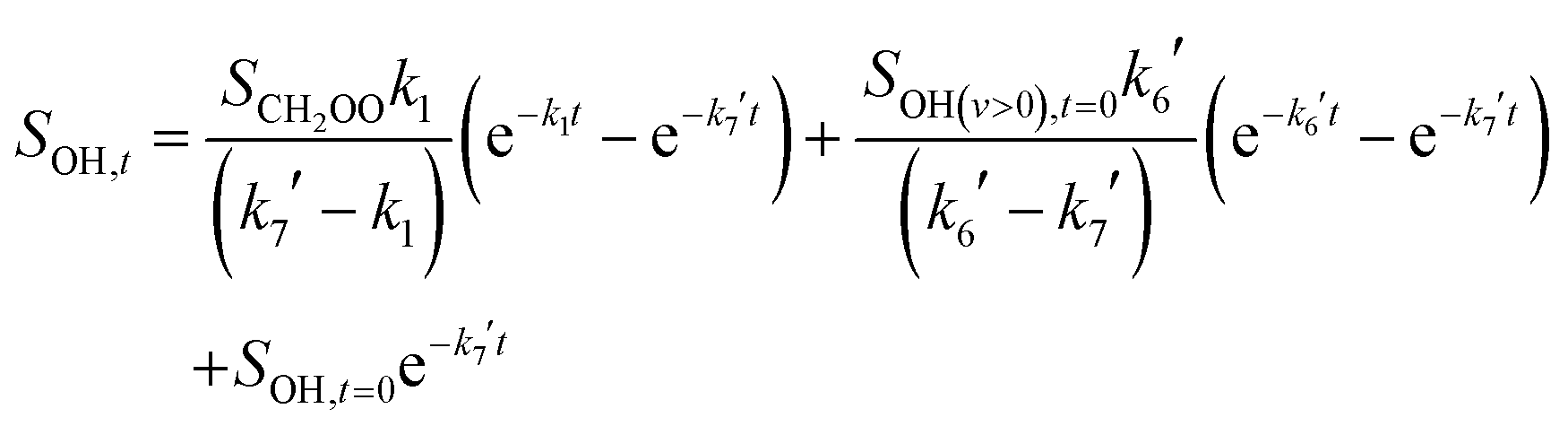 | (2) |
While good fits to the observed OH(v = 0) signals could be achieved, as shown in Fig. 5, the fits displayed poor sensitivity to individual rate coefficients. The complexity of the mechanism controlling OH(v = 0) in the system thus led to difficulties in obtaining reliable CH2OO decomposition kinetics, although the temperatures and pressures over which the slow growth of OH(v = 0) in the system was apparent are consistent with those where UV absorption experiments observed CH2OO decomposition.
The OH(v = 0) signal attributed to production from CH2OO indicates a low yield of OH from stabilised CH2OO decomposition. Previous experiments indicate that photolysis of CH2I2 at a wavelength of 248 nm leads to near-instant production of HCHO, via generation of excited CH2I* or CH2OO*, followed by subsequent growth of HCHO produced via chemistry of CH2OO.40,41 These experiments indicated eventual 100% yield of HCHO from CH2OO (through reactions including CH2OO + CH2OO, CH2OO + I and CH2OO + SO2), with the near-instant yield of HCHO representing approximately 5–10% of the total HCHO, or CH2OO, yield. Assuming that the near-instant OH signal observed in this work is produced via a similar mechanism to the near-instant HCHO signal observed previously (i.e. via generation of excited CH2I* or CH2OO*), and with similar yields to the near-instant HCHO signal, we can estimate that the near-instant yield of OH also represents only 5–10% of the total CH2OO in the system. The OH signals observed in this work were typically dominated by the near-instant signal, comprising both the instant OH(v = 0) signal and the relaxation of OH(v > 0), with the OH(v = 0) produced via CH2OO decomposition being only a fraction of the total OH signal. Thus, the yields of OH(v = 0) from CH2OO decomposition are low. For example, for the data shown in Fig. 5, the fits to eqn (2) indicate that SCH2OO is ∼(46 ± 5)% of the total OH(v = 0) signal (i.e. SCH2OO + SOH(v=1),t=0 + SOH,t=0). If we estimate that the near-instant OH signal (SOH,t=0 and SOH(v=1),t=0 combined) represents only 5–10% of the total CH2OO produced in the system, the yield of OH(v = 0) from CH2OO decomposition is ∼4–8%. However, similarly to the kinetic analysis, a fully quantitative analysis of the OH yields in the system is not possible owing to the complexity of the mechanism and poor sensitivity of the fits to individual processes, and the data allow only a qualitative analysis of the OH yields from stabilised CH2OO decomposition.
| SOH(v = 1),t = SOH(v = 1),t=0e−k6′t + Sge−kgt | (3) |
 | ||
| Fig. 6 Normalised OH(v = 1) LIF signal following photolysis of CH2I2/O2/N2 at T = 570 K and p = 20 Torr (black points) with kinetic fit (eqn (3), red line), giving k1 = (1130 ± 30) s−1. The inset shows the first 500 μs in greater detail. The fit to the data was limited to t > 100 μs after photolysis to ensure complete collisional relaxation of OH(v > 1) states. | ||
The kinetics of the fast component of the OH(v = 1) decay were consistent with collisional relaxation to OH(v = 0), principally by O2,42 and are provided in the ESI.† The kinetics of the slow component to the decay displayed a dependence on temperature and total pressure similar to that observed for CH2OO decomposition in the UV experiments, as shown in Fig. 7. Thus, we propose that the apparent biexponential decay of OH(v = 1) results from a combination of OH(v = 1) relaxation to OH(v = 0) and direct production of OH(v = 1) from decomposition of CH2OO. Thus, Sg in eqn (3) represents the amplitude of the OH(v = 1) signal arising from decomposition of CH2OO and kg is equivalent to k1, the rate coefficient for CH2OO decomposition. The results for k1 determined from the OH(v = 1) experiments are summarised in Table 3 and compare well to those obtained in the UV experiments in which CH2OO was monitored directly.
 | ||
| Fig. 7 Rate coefficients describing the decay of CH2OO (k1) as a function of temperature and pressure (coloured points) determined from the OH(v = 1) LIF experiments (open data points) and MESMER simulations using the optimised parameters from the fits to the OH data (dotted lines). Data from UV experiments monitoring CH2OO at nearby temperatures are also shown (filled data points) alongside the corresponding Troe fits to the UV data (dashed lines) and MESMER simulations (solid lines). Error bars are 1σ. | ||
| T/K | p/Torr | k 1/s−1 |
|---|---|---|
| 480 | 20 | 500 ± 60 |
| 50 | 420 ± 200 | |
| 75 | 640 ± 440 | |
| 95 | 1310 ± 320 | |
| 530 | 10 | 510 ± 110 |
| 20 | 730 ± 30 | |
| 50 | 770 ± 80 | |
| 75 | 830 ± 70 | |
| 95 | 930 ± 70 | |
| 570 | 10 | 770 ± 110 |
| 20 | 1130 ± 30 | |
| 30 | 1060 ± 80 | |
| 40 | 1300 ± 150 | |
| 50 | 1600 ± 60 | |
| 75 | 2000 ± 100 | |
| 95 | 2190 ± 140 | |
The yields of OH(v = 1) from decomposition of CH2OO are thus low, since there is little perturbation to the OH(v = 0) signal that we attribute to OH(v = 1) relaxation. For the data shown in Fig. 6, the fitted yield of OH(v = 1) from CH2OO decomposition is approximately 30% of the total OH(v = 1) signal. Examination of all fits for v = 0 and v = 1 OH signals leads us to conclude that if the v = 0 signal is ∼4–8% of the total CH2OO then v ≥ 1 is on the order of 1%.
Master equation analysis
Master equation calculations for the decomposition of CH2OO were performed using the Master Equation Solver for Multi-Energy well Reactions (MESMER), which has been described in detail in previous work.25,43,44 The energies of each species, including reactants, transition states and products, are divided into a number of levels, known as grains, which contain a defined number of states. These grains are assigned populations, average energies, and, where appropriate, average values of microcanonical rate coefficients, forming the basis for the master equation analysis. Changes in the population distribution among the grains occur through collisional energy transfer via interactions with a thermal bath gas or via transfer from one species to another via reactions governed by the microcanonical rate coefficients in the system.The equation of motion of the grain population probabilities is represented by:
| dp/dt = Mp | (4) |
| 〈ΔE〉down,T = 〈ΔE〉down,298K(T/298)n | (5) |
For the master equation calculations presented in this work, geometries, frequencies and rotational constants for CH2OO, transition states to decomposition and the decomposition products were provided by the calculations of Nguyen et al.11 at the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ level of theory, with the roaming channel leading to formic acid excluded, as suggested by recent improved calculations.12 If the roaming channel were active, a strong kinetic isotope effect might be expected between CH2OO and CD2OO, which is not supported by the experimental data or the calculations reported in this work. Geometries, frequencies and rotational constants for CD2OO, the transition state to decomposition and the initial intermediate leading to decomposition products were calculated using the Gaussian 09 suite of programs45 at the M06-2X/aug-cc-pVTZ46–51 level of theory. The barrier to decomposition was improved via single point energy computations (SPE) of the stationary structures using coupled cluster calculations with single, double and perturbative triple excitations (CCSD(T)).52 The SPEs were extrapolated to the complete basis set limit (CBS) with the use of correlation-consistent basis sets (aug-cc-pVXT, X = D, T, Q)47–51 and the extrapolation scheme presented by Peterson et al.48 Barrier heights and stationary point energies were corrected for zero point energies (ZPEs), and although the deuterated reactant does have a lower ZPE compared to the non-deuterated reactant, the same effect is observed in the respective transition states, such that the barrier height is similar between the deuterated and non-deuterated systems. The barrier calculated at the CCSD(T)/CBS//M06-2//aug-cc-pVTZ level of theory (81.04 kJ mol−1) is in agreement with the barrier of 78.24 kJ mol−1 obtained by Nguyen et al.11 for CH2OO.
Pressure dependent rate coefficients for CH2OO and CD2OO were calculated by MESMER using an inverse Laplace transformation to determine microcanonical rate coefficients (k(E)), with molecular densities of states calculated by a rigid rotor-harmonic oscillator approximation.43 A grain size of 100 cm−1 was used in the calculations described here. The molecular constants and further details regarding the calculations are given in the ESI.†
The master equation calculations were optimised by varying the parameters 〈ΔE〉down,298K and n in eqn (5), as well as the barrier height to decomposition. A fit to the rate coefficients for CH2OO and CD2OO determined from the UV experiments, was performed using a Levenburg–Marquardt algorithm to minimise the merit function χ2, as defined by eqn (6):
 | (6) |
Since the decomposition of CH2OO is thought to proceed via a single barrier,11,12,14,23 optimisation of 〈ΔE〉down,298K, n and the barrier to decomposition can be achieved through consideration of the simplified potential energy surface shown in blue in Fig. 8, consisting of only CH2OO, the first transition state (TS2), and the cyclic intermediate (dioxirane). An analogous PES was used for CD2OO, in which a further simplification was made such that it considers only the energies of CD2OO and the initial transition state which ultimately leads to product formation.
 | ||
| Fig. 8 Potential energy surface for CH2OO decomposition, showing the optimised barrier to decomposition (blue) and the calculated barrier (red).11 The simplified potential energy surface used to optimise the MESMER simulations is shown in blue (i.e. comprising CH2OO, TS2 and the intermediate cyc-H2COO (dioxirane)), with the full PES used to estimate product yields shown in black. Names of transition states and intermediates are analogous to those reported by Nguyen et al.11 | ||
Fig. 8 shows the results of the optimisation of the barrier to decomposition of CH2OO, with the comparison between the experimentally observed rate coefficients for CH2OO decomposition and the output from the MESMER optimisation given in Fig. 2 and Table 1. The MESMER fits to the data yield 〈ΔE〉down = (32.6 ± 13.7)(T/298 K)(1.7±0.4) cm−1, and require an increase of 3.6 kJ mol−1 in the calculated barrier height from 78.2 kJ mol−1 to 81.8 kJ mol−1, giving k1 = 1.1+1.5−1.1 × 10−3 s−1 in He at T = 298 K and p = 760 Torr. For the experimental conditions surveyed in this work, the value for 〈ΔE〉down ranges from 65 cm−1 at T = 450 K to 121 cm−1 at T = 650 K. The optimised parameters are given in Table 4. Although the increase in the barrier height is greater than the estimated uncertainty of ∼2 kJ mol−1 in calculations of this nature,7 the optimised barrier in MESMER is also subject to uncertainties of several kJ mol−1, and the calculations may be influenced by multireference effects which could result in additional uncertainty. Optimisation of 〈ΔE〉down in N2 was also performed using the data obtained from measurements of OH (shown in Table 3) with the barrier to decomposition constrained to the value of 81.8 kJ mol−1 as indicated by the UV experiments. Fig. 7 shows the results of the optimisation, which gave 〈ΔE〉down = (125.4 ± 32.2)(T/298 K)(0.5±0.4) cm−1 and k1 = 0.01 s−1 at 298 K and 760 Torr in N2. Uncertainties in the value of k1 in N2 at 298 K and 760 Torr determined from the OH experiments, determined by propagation of errors in the MESMER fits, are on the order of ∼200%. However, as shown in Fig. 7 the optimisation tends to overpredict the observed rate coefficients, and the results may be subject to larger uncertainties than indicated by the statistical error propagation owing to the complexity of the mechanism controlling the production and loss of OH in the system.
| Barrier to decomposition/kJ mol−1 | 〈ΔE〉down/cm−1 |
k
(T=298K,p=760Torr)/10−3 s−1 |
|
|---|---|---|---|
| CH2OO | 81.8 ± 6.2 | (32.6 ± 13.7)(T/298 K)(1.7±0.4) | 1.1+1.5−1.1 |
| CD2OO | 80.1 ± 3.0 | (39.6 ± 7.8)(T/298 K)(1.3±0.2) | 5.5+9.2−5.5 |
The results of Berndt et al.23 required a decrease in the calculated barrier height from 78.9 kJ mol−1 to 76.8 kJ mol−1 to improve the agreement between the master equation calculation and the measured rate coefficient for decomposition of (0.19 ± 0.07) s−1 at 298 K and 760 Torr in N2 using the free-jet flow reactor. The higher barrier height determined in this work from the UV observations of CH2OO in He lead to a lower value of k1 = 1.1 × 10−3 s−1 at 298 K and 760 Torr compared to the work of Berndt et al.23 The difference in the barrier heights is significant, but it is worth noting that the barrier height determined by Berndt et al. was fitted to a single measurement of k1, while that determined in this work fitted over a range of temperatures and pressures, providing greater constraint in the fit to eqn (6). The experiments reported in this work also use direct detection of CH2OO, while the experiments of Berndt et al. rely on titration of CH2OO to H2SO4, with subsequent ionisation and detection of H2SO4.
Simulations in MESMER using the full PES by Nguyen et al.,11 shown in Fig. 8, with the optimised values for 〈ΔE〉down and TS2 energy, determined from the UV experiments, were performed at p = 1–3040 Torr and T = 400–1200 K to investigate the product distribution. There was little variation in the product distribution over the pressure and temperature ranges investigated, with yields of 63.7% for H2 + CO2 and 36.0% for H2O + CO, on average. The yields of OH + HCO is predicted to be 0.3%, on average, and is lower than the estimates based on the OH measurements reported in this work and those indicated by the use of OH as a proxy to CH2OO in experiments by Liu et al.16 and Li et al.17
The optimised TS2 energy and 〈ΔE〉down were also used in MESMER simulations to calculate k1 at temperatures between 200 K and 850 K and pressures up to 10 atm. The calculated rate coefficients were subsequently parameterised using the Troe expression for broad falloff curves53 (eqn (7)–(9)) for use in kinetic models:
 | (7) |
 | (8) |
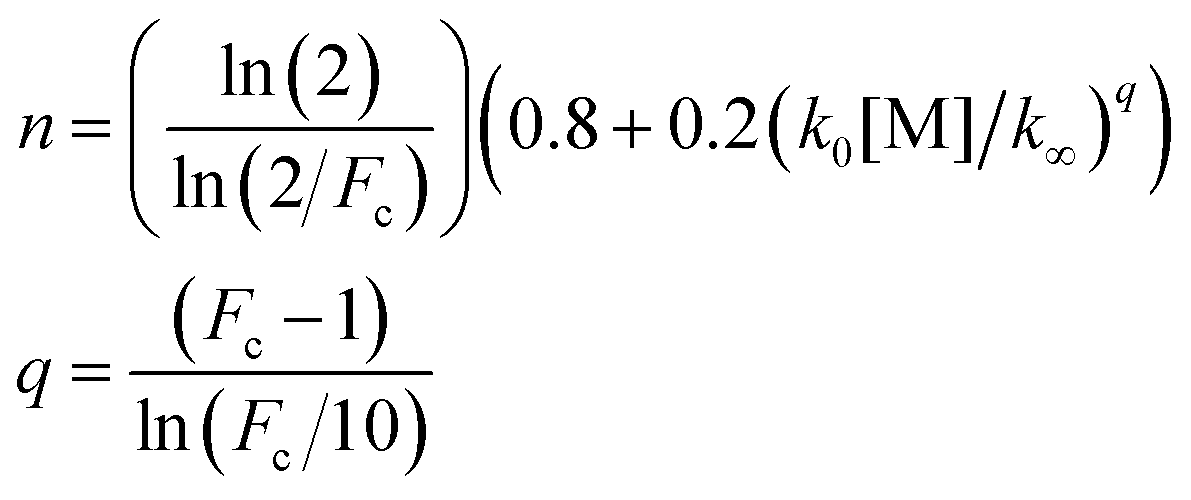 | (9) |
770/T) cm3 s−1, k1,∞ = 1.4 × 1013(T/298)0.06exp(−10010/T) s−1 and Fc = 0.447.
Analogous results for CD2OO give 〈ΔE〉down = (39.6 ± 7.8)(T/298 K)(1.3±0.2) cm−1 and a barrier to decomposition of 80.1 kJ mol−1, a decrease of 0.9 kJ mol−1 from the calculated barrier of 81.0 kJ mol−1, with 〈ΔE〉down thus ranging from 67 cm−1 at 450 K to 109 cm−1 at 650 K. The fits give a value of k2 = 5.5+9.2−5.5 × 10−3 s−1 in He at T = 298 K and p = 760 Torr. The comparison between the experimentally observed rate coefficients and the MESMER output is given in Fig. 4 and Table 2, with the optimised parameters summarised in Table 4. Fits to eqn (7)–(9), using the optimised parameters for CD2OO in MESMER to calculate k2 at temperatures between 200 K and 850 K and pressures up to 10 atm, give k2,0 = 5.2 × 10−5(T/298)−5.28exp(−11610/T) cm3 s−1, k2,∞ = 1.2 × 1013(T/298)0.06exp(−9800/T) s−1 and Fc = 0.427.
The optimised PES thus indicates that there is no significant change in the barrier height to decomposition upon deuteration of CH2OO. However, comparison of Fig. 2 and 4 shows that CD2OO decomposes faster than CH2OO under equivalent conditions, which is also confirmed by the MESMER calculations. Given the similar electronic barriers to decomposition for CH2OO and CD2OO, such differences likely result from an increased density of states in CD2OO near the transition state, which promotes the high pressure limit at lower pressures. A similar effect has been observed in a recent study of deuterated Criegee intermediate kinetics, in the reactions of (CH3)3COO and (CD3)3COO with SO2,54 and was attributed to the potential impact of increased collisional stabilisation of the deuterated association complex between (CD3)3COO and SO2 compared to (CH3)3COO and SO2 owing to the increased density of vibrational states in the deuterated system.
Conclusions
The decomposition kinetics of CH2OO Criegee intermediate have been investigated at temperatures between 450 K and 650 K and pressures in the range 2–350 Torr of He using flash photolysis of CH2I2 in O2 and a combination of time-resolved cavity enhanced broadband UV absorption spectroscopy, for direct monitoring of CH2OO, and laser-induced fluorescence, for monitoring of OH decomposition products. Kinetics of CD2OO decomposition were also investigated using flash photolysis of CD2I2 with time-resolved cavity enhanced broadband UV absorption spectroscopy.The decomposition of CH2OO is expected to be slow under ambient conditions, and thus is not a significant sink for CH2OO in the atmosphere or in chamber experiments of ozonolysis reactions, despite reports in previous work. Master equation fits using MESMER give k1 = 1.1+1.5−1.1 × 10−3 s−1 at 298 K and 760 Torr in He, using an exponential down model to describe the collisional energy transfer, where 〈ΔE〉down = (32.6 ± 13.7)(T/298 K)(1.7±0.4) cm−1, and requiring an increase in the calculated barrier height to decomposition from 78.2 kJ mol−1 to 81.8 kJ mol−1. Product yields, determined from MESMER simulations using the increased barrier height to decomposition, are predicted to be 63.7% for H2 + CO2, 36.0% for H2O + CO and 0.3% for OH + HCO. For CD2OO, the master equation fits give k2 = 5.5+9.2−5.5 × 10−3 s−1 at 298 K and 760 Torr in He, and give values of 〈ΔE〉down = (39.6 ± 7.8)(T/298 K)(1.3±0.2) cm−1 and a barrier height of 80.1 kJ mol−1 compared to the calculated value of 81.0 kJ mol−1. We observed no kinetic isotope effect between the decomposition kinetics of CH2OO and CD2OO.
Results from this work provide a detailed description of CH2OO decomposition kinetics that can be applied to the analysis of the decomposition and stabilisation of nascent CH2OO Criegee intermediates produced in ozonolysis reactions, and to assess the contributions of wall losses to larger SCI species produced by ozonolysis in chamber experiments, which decompose more rapidly under ambient conditions owing to the existence of alternative decomposition pathways. Measurements reporting combined kinetics of CH2OO decomposition and wall loss are likely dominated by wall losses,19–22 and can therefore provide an estimate for SCI wall loss rates that could be applied to other SCI species, enabling separation of the wall loss rate and decomposition rate in chamber experiments.
The low yield of OH radicals observed indicates that decomposition of CH2OO cannot be responsible for any potential OH interferences in field instruments measuring ambient OH concentrations using the LIF-FAGE (laser-induced fluorescence with fluorescence assay by gas expansion) technique, as has been postulated in the literature.18 Under combustion conditions, decomposition of CH2OO will be rapid, with a fraction of decomposition leading to production of OH and HCO radicals, and thus contributing to chain-branching processes. The role of CH2OO in combustion, however, has yet to be fully established.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank T.-N. Nguyen, R. Putikam and M. C. Lin for providing details of their calculations on the potential energy surface for CH2OO decomposition. DS would like to thank the Natural Environment Research Council (NERC) for the award of an Independent Research Fellowship (grant reference NE/L010798/1) and New Investigator grant (grant reference NE/P012876/1) for funding. This material is based upon work supported by the Division of Chemical Sciences, Geosciences and Biosciences, Office of Basic Energy Sciences (BES), U.S. Department of Energy (USDOE). Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the USDOE's National Nuclear Security Administration under contract DE-NA0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the USDOE or the United States Government. LS was supported by the Division of Chemical Sciences, Geosciences and Biosciences, BES/USDOE, through the Argonne-Sandia Consortium on High Pressure Combustion Chemistry. ZD was supported by the DOE Office of Science Workforce Development Program for Teachers and Scientists Summer Undergraduate Laboratory Internship. DJM would like to thank the Brazilian National Council for Scientific and Technological Development (CNPq, grant reference 206527/2014-4).References
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699 RSC.
- T. B. Nguyen, G. S. Tyndall, J. D. Crounse, A. P. Teng, K. H. Bates, R. H. Schwantes, M. M. Coggon, L. Zhang, P. Feiner, D. O. Milller, K. M. Skog, J. C. Rivera-Rios, M. Dorris, K. F. Olson, A. Koss, R. J. Wild, S. S. Brown, A. H. Goldstein, J. A. de Gouw, W. H. Brune, F. N. Keutsch, J. H. Seinfeldcj and P. O. Wennberg, Phys. Chem. Chem. Phys., 2016, 18, 10241 RSC.
- D. L. Osborn and C. A. Taatjes, Int. Rev. Phys. Chem., 2015, 34, 309 Search PubMed.
- C. A. Taatjes, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2014, 16, 1704 RSC.
- M. C. Smith, W. Chao, K. Takahashi, K. A. Boering, J. J.-M. Lin and J. Jr-Min, J. Phys. Chem. A, 2016, 120, 4789–4798 CrossRef PubMed.
- N. M. Kidwell, H. Li, X. Wang, J. M. Bowman and M. I. Lester, Nat. Chem., 2016, 8, 509 CrossRef PubMed.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, J. Chem. Phys., 2016, 144 Search PubMed.
- Y. Fang, F. Liu, S. J. Klippenstein and M. I. Lester, J. Chem. Phys., 2016, 145, 044312 CrossRef PubMed.
- A. Andersen and E. A. Carter, J. Phys. Chem. A, 2003, 107, 9463 CrossRef.
- A. Andersen and E. A. Carter, Mol. Phys., 2008, 106, 367 CrossRef.
- T.-N. Nguyen, R. Putikam and M. C. Lin, J. Chem. Phys., 2015, 142 Search PubMed.
- L. B. Harding and S. J. Klippenstein, J. Chem. Phys., 2015, 143 Search PubMed.
- C. F. Goldsmith, L. B. Harding, Y. Georgievskii, J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2015, 119, 7766 CrossRef PubMed.
- T. L. Nguyen, H. Lee, D. A. Matthews, M. C. McCarthy and J. F. Stanton, J. Phys. Chem. A, 2015, 119, 5524 CrossRef PubMed.
- M. Pfeifle, Y.-T. Ma, A. W. Jasper, L. B. Harding, W. L. Hase and S. J. Klippenstein, J. Chem. Phys., 2018, 148, 174306 CrossRef PubMed.
- Y. Liu, K. D. Bayes and S. P. Sander, J. Phys. Chem. A, 2014, 118, 741 CrossRef PubMed.
- Y. Q. Liu, F. H. Liu, S. Y. Liu, D. X. Dai, W. R. Dong and X. M. Yang, Phys. Chem. Chem. Phys., 2017, 19, 20786 RSC.
- A. Novelli, L. Vereecken, J. Lelieveld and H. Harder, Phys. Chem. Chem. Phys., 2014, 16, 19941 RSC.
- M. J. Newland, A. R. Rickard, M. S. Alam, L. Vereecken, A. Munoz, M. Rodenas and W. J. Bloss, Phys. Chem. Chem. Phys., 2015, 17, 4076 RSC.
- M. J. Newland, A. R. Rickard, L. Vereecken, A. Munoz, M. Rodenas and W. J. Bloss, Atmos. Chem. Phys., 2015, 15, 9521 CrossRef.
- R. Chhantyal-Pun, A. Davey, D. E. Shallcross, C. J. Percival and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2015, 17, 3617 RSC.
- T. Berndt, J. Voigtlander, F. Stratmann, H. Junninen, R. L. Mauldin, M. Sipila, M. Kulmala and H. Herrmann, Phys. Chem. Chem. Phys., 2014, 16, 19130 RSC.
- T. Berndt, R. Kaethner, J. Voigtlander, F. Stratmann, M. Pfeifle, P. Reichle, M. Sipila, M. Kulmala and M. Olzmann, Phys. Chem. Chem. Phys., 2015, 17, 19862 RSC.
- M. Olzmann, E. Kraka, D. Cremer, R. Gutbrod and S. Andersson, J. Phys. Chem. A, 1997, 101, 9421 CrossRef.
- D. R. Glowacki, C.-H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545 CrossRef PubMed.
- L. Sheps, J. Phys. Chem. Lett., 2013, 4, 4201 CrossRef PubMed.
- L. Sheps, A. M. Scully and K. Au, Phys. Chem. Chem. Phys., 2014, 16, 26701 RSC.
- Z. C. J. Decker, K. Au, L. Vereecken and L. Sheps, Phys. Chem. Chem. Phys., 2017, 19, 8541 RSC.
- D. R. Glowacki, J. Lockhart, M. A. Blitz, S. J. Klippenstein, M. J. Pilling, S. H. Robertson and P. W. Seakins, Science, 2012, 337, 1066 CrossRef PubMed.
- S. A. Carr, M. T. B. Romero, M. Blitz, M. J. Pilling, D. E. Heard and P. W. Seakins, Chem. Phys. Lett., 2007, 108 CrossRef.
- M. T. Baeza-Romero, D. R. Glowacki, M. A. Blitz, D. E. Heard, M. J. Pilling, A. R. Rickard and P. W. Seakins, Phys. Chem. Chem. Phys., 2007, 9, 4114 RSC.
- J. Troe, J. Phys. Chem., 1979, 83, 114 CrossRef.
- J. B. Burkholder, S. P. Sander, J. Abbatt, J. B. Barker, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, D. M. Wilmouth and P. H. Wine, JPL Publication 15-10 2015, Jet Propulsion Laboratory, http://jpldataeval.jpl.nasa.gov.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2004, 4, 1461 CrossRef.
- A. W. L. Ting and J. J. M. Lin, J. Chin. Chem. Soc., 2017, 64, 360 CrossRef.
- W.-L. Ting, Y.-H. Chen, W. Chao, M. C. Smith and J. J.-M. Lin, Phys. Chem. Chem. Phys., 2014, 16, 10438 RSC.
- E. S. Foreman, K. M. Kapnas, Y. Jou, J. Kalinowski, D. Feng, R. B. Gerber and C. Murray, Phys. Chem. Chem. Phys., 2015, 17, 32539 RSC.
- J. H. Lehman, H. W. Li and M. I. Lester, Chem. Phys. Lett., 2013, 590, 16 CrossRef.
- J. H. Lehman, H. W. Li, J. M. Beames and M. I. Lester, J. Chem. Phys., 2013, 139 Search PubMed.
- D. Stone, M. Blitz, L. Daubney, T. Ingham and P. Seakins, Phys. Chem. Chem. Phys., 2013, 15, 19119 RSC.
- D. Stone, M. Blitz, L. Daubney, N. U. M. Howes and P. Seakins, Phys. Chem. Chem. Phys., 2014, 16, 1139 RSC.
- D. C. McCabe, I. W. M. Smith, B. Rajakumar and A. R. Ravishankara, Chem. Phys. Lett., 2006, 421, 111 CrossRef.
- R. J. Shannon, A. S. Tomlin, S. H. Robertson, M. A. Blitz, M. J. Pilling and P. W. Seakins, J. Phys. Chem. A, 2015, 119, 7430 CrossRef PubMed.
- K. W. McKee, M. A. Blitz, P. A. Cleary, D. R. Glowacki, M. J. Pilling, P. W. Seakins and L. M. Wang, J. Phys. Chem. A, 2007, 111, 4043 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- A. K. Wilson, T. van Mourik and T. H. Dunning, J. Mol. Struct., 1996, 388, 339 CrossRef.
- K. A. Peterson, D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 7410 CrossRef.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Headgordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef.
- J. Troe and V. G. Ushakov, Z. Phys. Chem., 2014, 228, 1 Search PubMed.
- R. Chhantyal-Pun, O. Welz, J. D. Savee, A. J. Eskola, E. P. F. Lee, L. Blacker, H. R. Hill, M. Ashcroft, M. A. H. Khan, G. C. Lloyd-Jones, L. Evans, B. Rotavera, H. F. Huang, D. L. Osborn, D. K. W. Mok, J. M. Dyke, D. E. Shallcross, C. J. Percival, A. J. Orr-Ewing and C. A. Taatjes, J. Phys. Chem. A, 2017, 121, 4 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp05332d |
| ‡ Present address: University of Colorado, Boulder, CO 80302, USA. |
| This journal is © the Owner Societies 2018 |