
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Why is the change of the Johari–Goldstein β-relaxation time by densification in ultrastable glass minor?†
K. L.
Ngai
*a,
Marian
Paluch
bc and
Cristian
Rodríguez-Tinoco
 *bc
*bc
aCNR-IPCF, Largo Bruno Pontecorvo 3, I-56127, Pisa, Italy. E-mail: kiangai@yahoo.com; kia.ngai@pi.ipcf.cnr.it
bSilesian Center for Education and Interdisciplinary Research, 75 Pulku Piechoty 1, 41-500 Chorzow, Poland. E-mail: crodrigueztinoco@gmail.com
cInstitute of Physics, University of Silesia, 75 Pulku Piechoty 1, 41-500 Chorzow, Poland
First published on 30th October 2018
Abstract
Ultrastable glasses (USG) formed by vapor deposition are considerably denser. The onset temperature of devitrification, Ton, is significantly higher than Ton or Tg of ordinary glass (OG) formed by cooling, which implies an increase of the structural α-relaxation time by many orders of magnitude in USG compared to that in OG at the same temperature. However, for a special type of secondary relaxation having properties strongly connected to those of the α-relaxation, called the Johari–Goldstein β-relaxation, its relaxation time in USG is about an order of magnitude slower than that in OG and it has nearly the same activation energy, Eβ. The much smaller change in τβ and practically no change in Eβ by densification in USG are in stark contrast to the behavior of the α-relaxation. This cannot be explained by asserting that the Johari–Goldstein (JG) β-relaxation is insensitive to densification in USG, since the JG β-relaxation strength is significantly reduced in USG to such a level that it would require several thousands of years of aging for an OG to reach the same state, and therefore the JG β-relaxation does respond to densification in USG like the α-relaxation. Here, we provide an explanation based on two general properties established from the studies of glasses and liquids at elevated pressures and applied to USG. The increase in density of the glasses formed under high pressure can be even larger than that in USG. One property is the approximate invariance of the ratio τα(Ton)/τβ(Ton) to density change at constant τα(Ton), and the other is the same ργ/T-dependence of τβ in USG and OG where ρ is the density and γ is a material constant. These two properties are derived using the Coupling Model, giving a theoretical explanation of the phenomena. The explanation is also relevant for a full understanding of the experimental result that approximately the same surface diffusion coefficient is found in USG and OG with and without physical aging, and ultrathin films of a molecular glass-former.
Introduction
Glasses produced by vapor deposition on a substrate at an optimal temperature below the bulk glass transition temperature (∼0.85Tg, where Tg is the glass transition temperature of the material) have density considerably higher than ordinary glasses even after aging for a realistically long time.1–7 These glasses are more stable than the ordinary glasses (OG), and hence are called ultrastable glasses (USG). Compared with OG, some dynamic and thermodynamic properties of USG are novel and challenging to explain.8–11 One such property and its explanation are the focus of the present paper.The stability of USG is measured by the onset temperature for the transformation into the supercooled liquid, Ton. Its value is significantly higher than Ton or Tg of the ordinary glass (OG) formed by cooling. These changes in Ton imply an increase of the structural α-relaxation time, τα, by many orders of magnitude in USG compared to that in OG at the same temperature. Often observed in OG and USG is not only the α-relaxation but also a secondary relaxation. Some, but not all, secondary relaxations have properties strongly connected or related to those of the α-relaxation, and hence they are fundamentally important.12 These secondary relaxations are called the Johari–Goldstein (JG) β-relaxations in order to distinguish them from the other trivial ones.12 In contrast to the α-relaxation time τα, the JG β-relaxation time, τβ, in USG of toluene,13 etoricoxib14 and telmisartan15 is about an order of magnitude longer than that in OG and has nearly the same activation energy Eβ. The data from these three cases are extracted from C. Rodríguez-Tinoco et al.15 and shown in Fig. 1. On the other hand, the secondary relaxations of maltose octa-acetate, carvedilol and celecoxib, also discussed in ref. 15, involve intramolecular degrees of freedom, bear no connection to the α-relaxation,15 do not belong to the class of JG β-relaxations, and are not considered in this paper.
 | ||
| Fig. 1 Adapted from ref. 15. Arrhenius plot of τβ in USG and OG of (A) toluene, (B) etoricoxib, and (C) telmisartan. Black and red closed symbols correspond to the USG and the OG obtained after transformation of the USG and subsequent cooling down, respectively. The red open points and the orange VFT fit to τα in (B and C) are from the literature (etoricoxib,14,16,17 telmisartan18,19). All the data of toluene are extracted from Yu et al.13 The black and red circles indicate the value of τβ at Ton. The horizontal blue lines in graphs a–c bring out the invariance of τβ at Ton. The sketch of each molecule is also depicted. | ||
On the other hand, the relaxation strength of the JG β-relaxation, Δεβ, is significantly reduced in USG in toluene to such a level that it would require several thousands of years of annealing for an ordinary glass to achieve the same state.13–15 The reduction of Δεβ indicates that the JG β-relaxation does respond to densification in the USG like the α-relaxation. Notwithstanding, the much smaller changes of τβ and Eβ by densification in USG are in stark contrast to the behavior of the α-relaxation time τα, and this remarkable experimental observation deserves an explanation. In this paper, we provide an explanation based on two general properties established from the studies of glasses and liquids at elevated pressures20–27 and applied it to USG. The increase in density of the glasses formed under high pressure can be even larger than that in USG, and hence the general properties should apply. One of the properties deduced and applied to USG is the approximate invariance of the ratio τα(Ton)/τβ(Ton) to density change at constant τα(Ton), and the other is the same ργ/T-dependence of τβ in USG and OG where ρ is the density and γ is a material constant. Furthermore, we show how these two general properties can be derived theoretically by applying the Coupling Model,28 and by this application we have completed our explanation of the observed minor changes of τβ and Eβ by densification in USG.
The explanation is also relevant in achieving a better understanding than before29 of another remarkable experimental finding, which is about the same surface diffusion coefficient, DS, in USG and OG with and without physical aging, and ultrathin films of the molecular glass, N,N′-bis(3-methylphenyl)-N,N′-diphenylbenzidine (TPD) by Fakhraai and coworkers.30–32
1. Two general properties applied to USG
As mentioned in the Introduction, to explain the properties of the JG β-relaxation in USG compared with the OG we shall apply two general dynamic and thermodynamic properties of the α-relaxation and JG β-relaxation established before from studies at elevated pressures in molecular glass-formers.20–27 The application is justified by the fact that density is increased at elevated pressure, and the increase can be comparable to or even larger than that in USG. Through the application, we are able to explain why the Johari–Goldstein (JG) β-relaxation time, τβ, in USG of toluene,13 etoricoxib14 and telmisartan15 is about an order of magnitude longer than that in OG, and has nearly the same activation energy Eβ. Later we shall show how the two general properties are derived from theoretical considerations. Thus, a theoretical explanation of the properties of the JG β-relaxation in USG is obtained.A. General property leading to the same τβ(Ton) for USG and OG
By elevating pressure P to hundreds of MPa or more, one can increase the glass transition temperature from Tg at ambient pressure, P0 = 0.1 MPa, of many molecular glass-formers by several tens of degrees to TgP and the density by a few per cent. These changes are comparable to or larger than those achieved by producing USG. Found in general in many molecular glass-formers20–28 is the approximate invariance of τβ(T,P) to variations of P and T provided τα(T,P) are kept constant. Examples of experimental evidence for this property in OG are shown in Fig. S1 and S2 in the ESI.† Since τα(TgP,P) are approximately the same at TgP for all P by definition of glass transition temperature, the property specified at TgP states that τβ(TgP,P) is invariant to variations of P. In turn, this can be restated as approximate invariance of τβ(TgP,ρgP) to variations of ρgP in OG, where ρgP is the density at temperature equal to TgP and pressure P determined from the equation of state connecting P, T, and specific volume V or density ρ of the glass-former. Pressure P is just a parameter used to vary TgP and ρgP to bring out the property of the invariance of τβ(TgP,ρgP). It can be replaced by density ρ in some cases where ρ is changed directly by some operation instead of applied pressure. Producing USG by vapor deposition is such an operation whereby the density is increased compared to OG. The equivalence of effects due to change in density either by the route of USG or by application of pressure on OG can be appreciated from the fact that Ton of the USG of indomethacin is larger than that of the OG at ambient pressure, but Ton of the two glasses increases with pressure and becomes nearly the same upon reaching 300 MPa.33 Thus, the approximate invariance of τβ(TgP,ρgP) to variations of ρgP translates to the approximate invariance of τβ(Ton) with the change from OG to USG. It is expressed explicitly by| τβ,USG(Ton,USG) ≈ τβ,OG(Ton,OG), | (1) |
B. General property leading to similar Arrhenius T-dependence of τβ,USG and τβ,OG
The structural α-relaxation time, τα, of non-associated small molecular and polymeric glass-formers obeys thermodynamic scaling, i.e. τα is a function of the product variable, ργ/T, where γ is a material dependent parameter.34–36 This property in many glass-formers was deduced from measurements of τα at ambient and elevated pressures analyzed in conjunction with P–V–T data. The property of the invariance of the ratio τα(T,P)/τβ(T,P) to variations of T and P while keeping τα(T,P) constant20–27 is key to arriving at the results in Section (A). It also has the immediate consequence that τβ is also a function of ργ/T, if τα is a function of ργ/T, although the two functions τβ = fβ(ργ/T) and τα = Fα(ργ/T) are different. This is an example of the dynamic and thermodynamics properties of the JG β-relaxation similar to those of the α-relaxation,20–27 which follows from the approximate relation,23,27 , where tc (∼1 ps) is a constant and βKWW is the stretch exponent of the Kohlrausch–Williams–Watts correlation function for the α process. From this approximate relation and the fact that the JG β-relaxation occurs before the α-relaxation in time (i.e., its precursor), it follows that the ργ/T-dependence of τα = Fα(ργ/T) originates from τβ = fβ(ργ/T)27 as required by causality. Another piece of evidence for ργ/T-dependence originating from τβ = fβ(ργ/T) can be drawn from the remarkably small value of rs at which the steepness of the repulsive part of the intermolecular potential U(r) determines the scaling exponent γ.27 The short distance rs was found by molecular dynamics simulations of several Lennard-Jones liquids37 and in cis 1,4-polybutadiene as shown in Fig. S3 in the ESI,†
, where tc (∼1 ps) is a constant and βKWW is the stretch exponent of the Kohlrausch–Williams–Watts correlation function for the α process. From this approximate relation and the fact that the JG β-relaxation occurs before the α-relaxation in time (i.e., its precursor), it follows that the ργ/T-dependence of τα = Fα(ργ/T) originates from τβ = fβ(ργ/T)27 as required by causality. Another piece of evidence for ργ/T-dependence originating from τβ = fβ(ργ/T) can be drawn from the remarkably small value of rs at which the steepness of the repulsive part of the intermolecular potential U(r) determines the scaling exponent γ.27 The short distance rs was found by molecular dynamics simulations of several Lennard-Jones liquids37 and in cis 1,4-polybutadiene as shown in Fig. S3 in the ESI,†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 and reviewed in ref. 27.
38 and reviewed in ref. 27.
The result, τβ = fβ(ργ/T), is the general property we now use to deduce why the Arrhenius temperature dependence of τβ in USG is not much different from that in OG. Although the ργ/T-dependences of τα = Fα(ργ/T) and τβ = fβ(ργ/T) are mostly considered in the liquid state, it is applicable also in the glassy state. An example demonstrating this is the study of diglycidyl ether of bisphenol-A (DGEBA) with Mw = 380 g mol−1.23 In the glassy state, the temperature dependence of fβ(ργ/T) becomes effectively Arrhenius. The difference between the activation energy Eβ,USG in USG and Eβ,OG in OG comes from the factor ργ and the difference between the density ρUSG of USG and ρOG of OG. Hence the ratio Eβ,USG/Eβ,OG can be estimated from the value of (ρUSG/ρOG)γ. Ultrastable glasses of IMC have a density up to 1.4% more than that of the liquid-cooled OG,4 and a similar value is obtained for USG of TPD.39 The ratio Eβ,USG/Eβ,OG is equal to (1.014)γ. Typical values of γ fall within the range of 2 to 7, and toluene has γ = 7. The largest value of Eβ,USG/Eβ,OG = (1.014)γ is 1.1 for γ = 7. In other words, Eβ,USG is larger than Eβ,OG by merely 10%, and therefore the small increase of activation energy in USG compared to OG is a consequence of the general property of the ργ/T-dependence of τβ.
There are other experimental results that prove the fact that the ργ/T-dependence of τα and τβ is the same in both USG and OG because the repulsive part of the intermolecular potential is unchanged. Wide-angle X-ray scattering (WAXS) experiments were performed on USG and OG of indomethacin (IMC)3 and etoricoxib.14 In both cases, the intensity as a function of the scattering wave vector q exhibits no difference at q values higher than that of the peak, indicating that the relation of g(r) with the repulsive part of the intermolecular potential is the same for USG and OG. Moreover, molecular dynamics simulations of USG40,41 show that the changes of its g(r) from that of OG occur at larger values of r, which are irrelevant for the determination of γ from the slope of the repulsive part of the intermolecular potential. Therefore, from these two facts we conclude the same repulsive part of the potential, and hence the same ργ/T-dependence of τβ for USG and OG. In a recent report,7 the authors used the calorimetric transformation time of glasses with different thermodynamic stability to infer the structural relaxation times τα of the glassy systems. Interestingly, they found that for all the glasses, USG and OG, the relaxation data have the same ργ/T-dependence, with a unique γ exponent (see Fig. S4 in ESI†). This is another indication that γ is practically the same in USG and OG.
C. Derivation of the two general properties to complete the explanation
The two general properties of the JG β-relaxation time τβ and its relation to τα have been applied to compare USG with OG in Sections (A) and (B) with two results: (i) the value of τβ,USG(T) at T = Ton,USG is the same as τβ,OG(T) at T = Ton,OG, i.e., eqn (1), and (ii) the activation energy Eβ,USG of the Arrhenius temperature dependence of τβ,USG(T) for T < Ton,USG is only slightly increased from Eβ,OG of τβ,OG(T) for T < Ton,OG. The two results combined explain why the difference between τβ,USG(T) and τβ,OG(T) over the entire common temperature range below Ton,OG is minor as observed experimentally in toluene,13 etoricoxib,14 and telmisartan.15 However, the explanation is not complete until we have given the theoretical basis of the two general properties. This is our next task and we use the Coupling Model (CM).The CM starts from the primitive relaxation which is independent and local. The time honored CM equation,27,28
| τα(T,P) = [t−n(T,P)cτ0(T,P)]1/[1−n(T,P)] | (2) |
 | (3) |
The independent and local nature of the primitive relaxation suggests that it is similar to the JG β-relaxation. The two are not identical because the JG β-relaxation is composed of a distribution of processes42,43 and the primitive relaxation is only the leading part. Notwithstanding, approximate correspondence of the two relaxation times τ0(T,P) and τβ(T,P) is expected and predicted.27,28,44 Written as
| τβ(T,P) ≈ τ0(T,P), | (4) |
The CM is based on classical chaos engendered by the anharmonic intermolecular potential.28,45 In the model, it is the intermolecular potential in conjunction with the primitive relaxation time τ0(T,P) which exclusively determines φK(t). Upon varying P and T while keeping τ0(T,P) the same, the intermolecular potential is also unchanged,37,38,46 therefore φK(t) is the same, or both τα(T,P) and n(T,P) are the same. Therefore, the exact co-invariance to changes of P and T of the three quantities, τ0(T,P), τα(T,P) and n(T,P), is immediately the consequence of the CM. Combining this property with τβ(T,P) ≈ τ0(T,P), we have derived the approximate co-invariance of τβ(T,P), τα(T,P) and n(T,P). From this result, the general property used in Section (A) (i.e., approximate invariance of τβ(T,P) to variations of P and T at constant τα(T,P)) is now derived from the CM. The reader may recall the application of this general property to USG and OG in showing that τβ,USG(Ton,USG) is approximately equal to τβ,OG(Ton,OG).14,15
Derived also from the CM is the invariance of n(T,P) or the α-relaxation frequency dispersion to variations of P and T at constant τα(T,P), which has not been utilized in this paper in the relation between USG and OG. Nevertheless, it is worthwhile to point out that it is a remarkable prediction. Equally remarkable is that this property has been verified in so many glass-formers, molecular or polymeric.47,48 The importance of the frequency dispersion is brought out by the property.
In Section (B) we use another property and give a reason to show that the Arrhenius activation energies Eβ,USG of USG and Eβ,OG of OG are not much different. The property is that τβ is a function f(ργ/T) of ργ/T with the same γ as the function F(ργ/T) of τα. The reason is that γ is determined by the repulsive part of the intermolecular potential which is the same in USG and OG, and hence the same f(ργ/T) for USG and OG. We have demonstrated in Section (B) that the property, τβ a function of ργ/T, is a consequence of the approximate invariance of the ratio τα(T,P)/τβ(T,P) to variations of T and P while keeping τα(T,P) constant. The latter is just part of the approximate co-invariance of τβ(T,P), τα(T,P) and n(T,P) derived above from the CM, and hence the property is justified as well.
D. Accounting for the minor difference in τβ(T) between USG and OG
Having derived in (C) the two properties in (A and B), we are ready to put them together to account for the minor change of τβ(T) in USG compared to OG observed experimentally. The change is given by the ratio, τβ,USG(T)/τβ,OG(T), which we only need to calculate at T = Ton,OG because the activation energies Eβ,USG and Eβ,OG of the Arrhenius T-dependences of τβ,USG(T) and τβ,OG(T) in the glassy states are about the same. Since the ratio is the main interest, it is denoted by![[R with combining circumflex]](https://www.rsc.org/images/entities/i_char_0052_0302.gif) and written explicitly as
and written explicitly as| ≡ τβ,USG(Ton,OG)/τβ,OG(Ton,OG). | (5) |
≈ [τβ,USG(Ton,OG)/τβ,USG(Ton,USG)], which can be calculated from the change of the factor, exp(Eβ,USG/RT), of the temperature dependence of τβ,USG(T). The final result is | (6) |
) = 0.7. It is slightly less than a decade, and in approximate agreement with the experimental data of Yu et al.13
For etoricoxib, with Eβ = 51 kJ mol−1, Ton,OG = 327 K, Ton,USG = 360 K, and Λ = 18.76,14 we have log10() = 0.75, which compares well with the experimental value of ∼0.84 at T = 293 K. The small value of Λ for etoricoxib is because the secondary relaxation is not the usual JG β-relaxation.14,17 Notwithstanding, this secondary relaxation has some of the properties of the JG β-relaxation such as a significant dependence of its relaxation time on pressure. This is the reason why it is used for comparison between USG and OG since the usual JG β-relaxation of etoricoxib is not resolved.
The difference between Ton,USG of USG and Ton,OG of OG is 18 degrees for TPD.39 Taking Ton,OG = 330 K and Ton,USG = 348 K, and assuming Eβ/RTon,OG ≡ Λ = 24, the predicted log10() = 0.54. This small increase of τβ,USG(T) in USG of TPD with reference to τβ,OG(T) of OG is consistent with the CM explanation29 of the invariant surface diffusion observed in the two glasses by Fakhraai and coworkers.31,32
2. Discussion
The main objective of this paper is to explain why the differences in magnitude and temperature dependence of τβ between USG and OG are minor compared with the huge change of τα. Our goal is accomplished by the theoretical treatment given in Section 1. The explanation is beneficial to complete a previous rationalization of another remarkable experimental finding,29 which is approximately the same surface diffusion coefficient DS in USG, OG, and nanometer thin films of the same glass-former,30–32 while their structural α-relaxation times differ by many orders of magnitude. The rationalization is based on the relation DS(T) ≈ d2/4τ(T), where d is the size of the molecule, used previously to account for the enhancement of the surface diffusion29 (further support for the use of this expression is shown in the ESI,† Fig. S5), and the experimental evidence showing that τβ(T) is approximately the same in USG, aged OG, OG, and ultrathin films. The development in the present paper justifies on theoretical grounds the rationalization given before.Glass with density higher than USG can be realized by applying pressure P much higher than ambient pressure P0 = 0.1 MPa. If the minor changes in the magnitude and activation energy of τβ in USG compared to OG is general, it should be observed by elevating pressure from P0 to P. The answer is positive from the results of studies at elevated pressures.20–28 An example is the isobaric data of DGEBA (diglycidyl ether of bisphenol-A, Mw = 380 g mol−1, also known as EPON 828). Shown in Fig. 2 are the α-relaxation times τα and JG β-relaxation times τβ at ambient pressure P0 of 0.1 MPa and at P = 400 MPa. Defining glass transition temperatures by τα = 100 s, we have Tg = 252.8 K at P0 and TgP = 306.7 K at P = 400 MPa. The increase of 54 K is significantly larger than that expected for USG. The value of τβ(Tg,P0) is about the same as τβ(TgP,P). The activation energy is Eβ = 52.2 kJ mol−1 at ambient pressure, and it is about the same as the value at P = 400 MPa. Thus, the relation of τβ at high pressure to ambient pressure is the same as that of USG to OG. The ratio τβ(T,P)/τβ(T,P0) ≈ 100 is larger, and this is consistent with the density increase by elevating pressure, although the value of γ of DGEBA is 3.5, which is a factor of 2 smaller than that of toluene.
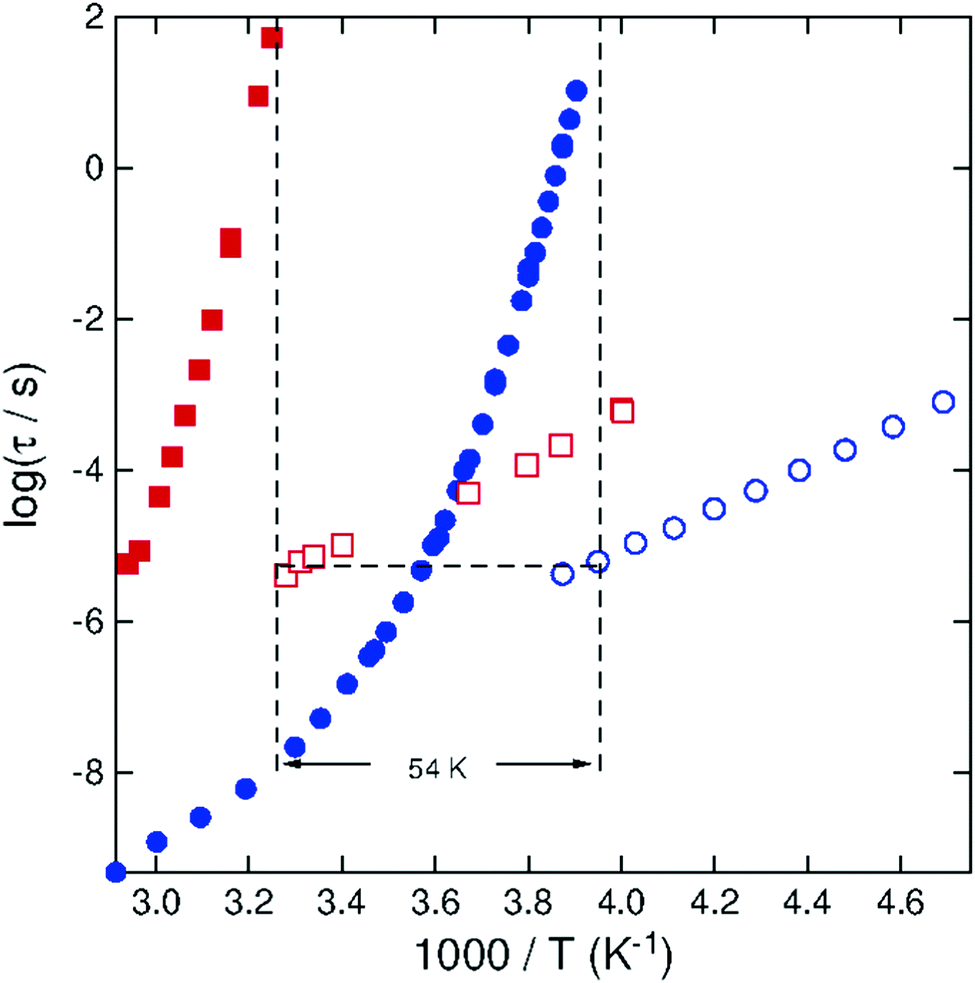 | ||
| Fig. 2 Logarithm of characteristic time of dielectric loss maximum of DGEBA (diglycidyl ether of bisphenol-A, Mw = 380 g mol−1, also known as EPON 828) for the α-relaxation and JG β-relaxation under isobaric conditions versus reciprocal temperature. Red symbols, P = 400 MPa; blue symbols, P0 = 0.1 MPa. The difference in Tg is 54 K, which is matched by the separation of the JG β-relaxation time as indicated by the dashed line. | ||
In this paper we have exclusively considered toluene, etoricoxib, and telmisartan with regard to the change of their secondary relaxations upon densification in USG compared to OG. The properties of the secondary relaxation in these three glass-formers are connected to or correlated with that of the α-relaxation, as discussed before in ref. 15. They belong to the class of JG β-relaxations if the relaxation time τβ(P,T) satisfies the criteria set forth in ref. 12 and 44, including the approximate relation (4), i.e. τβ(P,T) ≈ τ0(P,T). This criterion together with the CM eqn (2) led to the approximate relation,
| τα(P,T) ≈ [t−ncτβ(P,T)]1/(1−n). | (7) |
The secondary relaxation with properties uncorrelated with those of the α-relaxation and that does not satisfy the criteria is unimportant and is referred to as a non-JG relaxation. This is the case for the well-resolved secondary δ-relaxation of celecoxib, and γ-relaxations of D-maltose octa-acetate and carvedilol, discussed also in ref. 15. Their τγ(T,ρ) becomes faster while τα(T,ρ) becomes slower on densification of the glass by vapour deposition. Thus, the change of τγ(T,ρ) upon densification is opposite to that of τα(T), and hence the γ-relaxations in D-maltose octa-acetate and carvedilol are the non-JG relaxations. For celecoxib, it was shown before49 that it is the fast δ-relaxation, and its relaxation time τδ is much shorter than the calculated primitive relaxation time τ0 ≈ τβ. The JG β-relaxation is present but it has low dielectric strength and is detected as a broad shoulder in the glassy state. Its relaxation time τβ is in agreement with τ0. It was not considered in ref. 15. For D-maltose octa-acetate and carvedilol, the non-JG nature of the resolved secondary relaxation is further justified by the fact that the observed relaxation time τγ(T) is much shorter than τ0(T) calculated by eqn (2). The JG β-relaxation of D-maltose octa-acetate and carvedilol cannot be resolved due to its proximity to the α-relaxation. Nevertheless, this is irrelevant to this work because we are considering the resolved JG β-relaxations of toluene, etoricoxib, and telmisartan in the change by densification in USG.
Some of the results in Sections (B) and (C) are based on the approximate invariance of τβ(T,P) to variations of T and P while keeping τα(T,P) constant, which follows from relation (7). Only approximate invariance of τβ(P,T) is predicted because the JG β-relaxation is composed of a distribution of processes involving increasing number of molecules with increasing time,42,43 and the value of τβ(P,T) determined from experimental data for different combinations of P and T may not be associated with the same process in the distribution. Fortunately, in all previous experiments involving non-polymeric glass-formers in ref. 15 and 20–28, the JG β-relaxation was resolved in the dielectric spectra as a pronounced loss peak, and the peak frequency fβ(P,T) provides a characteristic time τβ(P,T) of the distribution. No fit is needed to determine fβ(P,T) and test the approximate invariance. Nearly exact invariance of fβ(P,T) or τβ(P,T) was found in all cases published in ref. 15 and 20–28 as well as in DGEBA (diglycidyl ether of bisphenol-A, Mw = 380 g mol−1) shown in Fig. 2, and thus overwhelmingly verifying the approximate invariance of τβ(P,T) for molecular glass-formers.
On the other hand, the dielectric spectra of the polymers, polyisoprene,43 polymethylmethacrylate50 (PMMA), and 1,4 polybutadiene,51 are not as ideal or straightforward as in the case of molecular glass-formers to test invariance of τβ(P,T). The α-relaxation of PMMA has low dielectric strength and its loss peak is not well resolved for many P and T combinations. Consequently, an assumption of the frequency dispersion of the α-relaxation had to be made,50 and used to fit the loss spectra with further assumption that the α-relaxation and the JG β-relaxation represented by a Cole–Cole function are additive. It is remarkable but questionable that practically the same value of τα(P,T) was obtained from the fits for different P and T despite the lack of well defined α-loss peaks. Nevertheless, the deviation of the deduced values of τβ(P,T) from exact invariance is only ± half a decade, while the τα(P,T) values change over 7 orders of magnitude. The results therefore are consistent with the approximate invariance of τβ(P,T) predicted from relation (7).
In the case of 1,4 polybutadiene51 the data at ambient pressure of 0.1 MPa show a long plateau or a broad shoulder, and fβ(P,T) was deduced from the fit by assuming the Cole–Cole function representing the JG β-relaxation and the data can be represented by the sum of the Cole–Cole function and the α-loss represented by the Fourier transform of the Kohlrausch function with its intensity adjusted in the global fit. This is common practice, but no one can be sure whether the JG β-relaxation is a Cole–Cole function and the assumption of additivity is correct or not, and thus the value of τβ(P,T) deduced for 0.1 MPa is debatable. The same fits were made to data taken at elevated pressures, where the JG β-relaxation shows up as broad loss peaks. Despite the loss peaks at elevated pressures being very broad, the loss peak frequencies fβ(P,T) are approximately the same. Thus, except for the questionable value of τβ(P,T) at ambient pressure deduced by fitting data without a β-loss peak, the peak frequencies of data at elevated pressures are approximately the same and consistent with the predicted approximate invariance of τβ(P,T). Only studies of other polymers having both well resolved α and β loss peaks in the future, as in the molecular glass-formers, can critically test the approximate invariance of τβ(P,T) for polymers. The present paper deals exclusively with molecular glass-formers, toluene, etoricoxib, and telmisartan. Since all data of molecular glass-formers in ref. 15 and 20–28, as well as those of DGEBA shown in Fig. 2, show consistency with the approximate invariance of τβ(P,T), the unsettled issue of polymers are not relevant for the present paper.
3. Conclusions
The onset temperature Ton of ultrastable glass (USG) is significantly higher than that of ordinary glass (OG) due to the increase in density and this suggests an increase in the α-relaxation time τα by many orders of magnitude. The JG β-relaxation also responds to the higher density by a large reduction in relaxation strength, which would take thousands of years of aging for OG to achieve. However, experimental studies of USG and OG show the increase of the relaxation time τβ(T) of USG compared to OG is within one decade over the entire common temperature range. Thus, the activation energy of τβ(T) is practically the same in the two glasses. The insensitivity of these dynamic properties of the JG β-relaxation to the density increase in USG is remarkable and challenging to explain. We have successfully explained these properties by the Coupling Model (CM) after considering that the intermolecular potential in USG remains the same as in OG. High pressure can facilitate the formation of glass with higher density than at ambient pressure, and we demonstrate that the change in properties of τβ is similar to that in USG compared to OG. This proves that the effects are general and the explanation is given. The explanation also applies to another remarkable finding of approximately the same surface diffusion coefficient DS in USG, OG, and nanometer thin films of the same glass former.29–32Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. R.-T. and M. P. acknowledge the support from the National Science Centre through the Polonez scheme (Grant No. DEC-2015/19/P/ST3/03540/2). This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 665778.References
- S. F. Swallen, K. L. Kearns, M. K. Mapes, Y. S. Kim, R. J. McMahon, M. D. Ediger, T. Wu, L. Yu and S. Satija, Organic Glasses with Exceptional Thermodynamic and Kinetic Stability, Science, 2007, 315, 353–356 CrossRef CAS PubMed.
- E. Leon-Gutierrez, A. Sepúlveda, G. Garcia, M. T. Clavaguera-Mora, J. Rodríguez-Viejo, T. Matsuo, L. H. Allen, T. L. Yu and S. Satija, Stability of thin film glasses of toluene and ethylbenzene formed by vapor deposition: an in situ nanocalorimetric study, Phys. Chem. Chem. Phys., 2010, 12, 14693–14698 RSC.
- K. J. Dawson, K. L. Kearns, L. Yu, W. Steffen and M. D. Ediger, Physical vapor deposition as a route to hidden amorphous states, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15165–15170 CrossRef CAS PubMed.
- S. S. Dalal, Z. Fakhraai and M. D. Ediger, High-throughput ellipsometric characterization of vapor-deposited indomethacin glasses, J. Phys. Chem. B, 2013, 117, 15415–15425 CrossRef CAS PubMed.
- C. Rodriguez-Tinoco, M. Gonzalez-Silveira, J. Rafols-Ribe, A. F. Lopeandia and J. Rodriguez-Viejo, Transformation kinetics of vapor-deposited thin film organic glasses: the role of stability and molecular packing anisotropy, Phys. Chem. Chem. Phys., 2015, 17, 31195–31201 RSC.
- E. A. A. Pogna, C. Rodríguez-Tinoco, G. Cerullo, C. Ferrante, J. Rodríguez-Viejo and T. Scopigno, Probing equilibrium glass flow up to exapoise viscosities, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2331–2336 CrossRef PubMed.
- C. Rodríguez-Tinoco, J. Ràfols-Ribé, M. González-Silveira and J. Rodríguez-Viejo, Relaxation dynamics of glasses along a wide stability and temperature range, Sci. Rep., 2016, 6, 35607 CrossRef PubMed.
- T. Perez-Castaneda, C. Rodriguez-Tinoco, J. Rodriguez-Viejo and M. A. Ramos, Suppression of tunneling two-level systems in ultrastable glasses of indomethacin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11275–11280 CrossRef CAS PubMed.
- C. Rodríguez-Tinoco, M. Gonzalez-Silveira, J. Ràfols-Ribé, G. Garcia and J. Rodríguez-Viejo, Highly stable glasses of celecoxib: Influence on thermo-kinetic properties, microstructure and response towards crystal growth, J. Non-Cryst. Solids, 2015, 407, 256–261 CrossRef.
- J. Ràfols-Ribé, P.-A. Will, C. Hänisch, M. Gonzalez-Silveira, S. Lenk, J. Rodríguez-Viejo and S. Reineke, High-performance organic light-emitting diodes comprising ultrastable glass layers, Sci. Adv., 2018, 4, eaar8332 CrossRef PubMed.
- Y. Qiu, L. W. Antony, J. J. de Pablo and M. D. Ediger, Photostability Can Be Significantly Modulated by Molecular Packing in Glasses, J. Am. Chem. Soc., 2016, 138, 11282–11289 CrossRef CAS PubMed.
- K. L. Ngai and M. Paluch, Classification of secondary relaxation in glass-formers based on dynamic properties, J. Chem. Phys., 2004, 120, 857–873 CrossRef CAS PubMed.
- H. B. Yu, M. Tylinski, A. Guiseppi-Elie, M. D. Ediger and R. Richert, Suppression of β Relaxation in Vapor-Deposited Ultrastable Glasses, Phys. Rev. Lett., 2015, 115, 185501 CrossRef CAS PubMed.
- C. Rodríguez-Tinoco, M. Rams-Baron, K. L. Ngai, K. Jurkiewicz, J. Rodríguez-Viejo and M. Paluch, Secondary relaxation in ultrastable etoricoxib: evidence of correlation with structural relaxation, Phys. Chem. Chem. Phys., 2018, 20, 3939–3945 RSC.
- C. Rodríguez-Tinoco, K. L. Ngai, M. Rams-Baron, J. Rodríguez-Viejo and M. Paluch, Distinguishing different classes of secondary relaxations from vapour deposited ultrastable glasses, Phys. Chem. Chem. Phys., 2018, 20, 21925–21933 RSC.
- M. Rams-Baron, Z. Wojnarowska, K. Grzybowska, M. Dulski, J. Knapik, K. Jurkiewicz, W. Smolka, W. Sawicki, A. Ratuszna and M. Paluch, Toward a Better Understanding of the Physical Stability of Amorphous Anti-Inflammatory Agents: The Roles of Molecular Mobility and Molecular Interaction Patterns, Mol. Pharmaceutics, 2015, 12, 3628–3638 CrossRef CAS PubMed.
- M. Rams-Baron, Z. Wojnarowska, J. Knapik-Kowalczuk, K. Jurkiewicz, A. Burian, M. Wojtyniak, J. Pionteck, M. Jaworska, C. Rodríguez-Tinoco and M. Paluch, The dielectric signature of glass density, Appl. Phys. Lett., 2017, 111, 121902 CrossRef.
- K. Adrjanowicz, M. Paluch and K. L. Ngai, Determining the structural relaxation times deep in the glassy state of the pharmaceutical Telmisartan, J. Phys.: Condens. Matter, 2010, 22, 125902 CrossRef CAS PubMed.
- K. Adrjanowicz, Z. Wojnarowska, P. Wlodarczyk, K. Kaminski, M. Paluch and J. Mazgalski, Molecular mobility in liquid and glassy states of Telmisartan (TEL) studied by Broadband Dielectric Spectroscopy, Eur. J. Pharm. Sci., 2009, 38, 395–404 CrossRef CAS PubMed.
- M. Mierzwa, S. Pawlus, M. Paluch, E. Kaminska and K. L. Ngai, Correlation between primary and secondary Johari–Goldstein relaxations in supercooled liquids: Invariance to changes in thermodynamic conditions, J. Chem. Phys., 2008, 128, 44512 CrossRef CAS PubMed.
- K. Kessairi, S. Capaccioli, D. Prevosto, M. Lucchesi, S. Sharifi and P. A. Rolla, Interdependence of primary and Johari-Goldstein secondary relaxations in glass-forming systems, J. Phys. Chem. B, 2008, 112, 4470–4473 CrossRef CAS PubMed.
- M. Shahin Thayyil, K. L. Ngai, D. Prevosto and S. Capaccioli, Revealing the rich dynamics of glass-forming systems by modification of composition and change of thermodynamic conditions, J. Non-Cryst. Solids, 2015, 407, 98–105 CrossRef CAS.
- K. L. Ngai, J. Habasaki, D. Prevosto, S. Capaccioli and M. Paluch, Thermodynamic scaling of α-relaxation time and viscosity stems from the Johari-Goldstein β-relaxation or the primitive relaxation of the coupling model, J. Chem. Phys., 2012, 137, 034511 CrossRef CAS PubMed.
- S. Capaccioli, M. Paluch, D. Prevosto, L.-M. Wang and K. L. Ngai, Many-Body Nature of Relaxation Processes in Glass-Forming Systems, J. Phys. Chem. Lett., 2012, 3, 735–743 CrossRef CAS PubMed.
- D. Prevosto, S. Capaccioli, M. Lucchesi, P. A. Rolla and K. L. Ngai, Does the entropy and volume dependence of the structural α-relaxation originate from the Johari–Goldstein β-relaxation?, J. Non-Cryst. Solids, 2009, 355, 705–711 CrossRef CAS.
- S. Valenti, S. Capaccioli and K. L. Ngai, Contrasting two different interpretations of the dynamics in binary glass forming mixtures, J. Chem. Phys., 2018, 148, 054504 CrossRef CAS PubMed.
- K. L. Ngai and M. Paluch, Corroborative evidences of TVγ -scaling of the α-relaxation originating from the primitive relaxation/JG β relaxation, J. Non-Cryst. Solids, 2017, 478, 1–11 CrossRef CAS.
- K. L. Ngai, Relaxation and Diffusion in Complex Systems, Springer New York, New York, NY, 2011 Search PubMed.
- K. L. Ngai, M. Paluch and C. Rodríguez-Tinoco, Why is surface diffusion the same in ultrastable, ordinary, aged, and ultrathin molecular glasses?, Phys. Chem. Chem. Phys., 2017, 19, 29905–29912 RSC.
- Y. Zhang, R. Potter, W. Zhang and Z. Fakhraai, Using tobacco mosaic virus to probe enhanced surface diffusion of molecular glasses, Soft Matter, 2016, 12, 9115–9120 RSC.
- Y. Zhang and Z. Fakhraai, Invariant Fast Diffusion on the Surfaces of Ultrastable and Aged Molecular Glasses, Phys. Rev. Lett., 2017, 118, 066101 CrossRef PubMed.
- Y. Zhang and Z. Fakhraai, Decoupling of surface diffusion and relaxation dynamics of molecular glasses, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4915–4919 CrossRef CAS PubMed.
- C. Rodríguez-Tinoco, M. González-Silveira, M. Barrio, P. Lloveras and J. L. Tamarit, J.-L. Garden and J. Rodríguez-Viejo, Ultrastable glasses portray similar behaviour to ordinary glasses at high pressure, Sci. Rep., 2016, 6, 34296 CrossRef PubMed.
- R. Casalini and C. M. Roland, Thermodynamical scaling of the glass transition dynamics, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 69, 062501 CrossRef CAS PubMed.
- G. Tarjus, D. Kivelson, S. Mossa and C. Alba-Simionesco, Disentangling density and temperature effects in the viscous slowing down of glassforming liquids, J. Chem. Phys., 2004, 120, 6135–6141 CrossRef CAS PubMed.
- C. M. Roland, S. Hensel-Bielowka, M. Paluch and R. Casalini, Supercooled dynamics of glass-forming liquids and polymers under hydrostatic pressure, Rep. Prog. Phys., 2005, 68, 1405–1478 CrossRef CAS.
- D. Coslovich and C. M. Roland, Thermodynamic Scaling of Diffusion in Supercooled Lennard-Jones Liquids, J. Phys. Chem. B, 2008, 112, 1329–1332 CrossRef CAS PubMed.
- G. Tsolou, V. A. Harmandaris and V. G. Mavrantzas, Atomistic molecular dynamics simulation of the temperature and pressure dependences of local and terminal relaxations in cis-1,4-polybutadiene, J. Chem. Phys., 2006, 124, 084906 CrossRef PubMed.
- D. M. Walters, R. Richert and M. D. Ediger, Thermal stability of vapor-deposited stable glasses of an organic semiconductor, J. Chem. Phys., 2015, 142, 134504 CrossRef PubMed.
- P.-H. Lin, I. Lyubimov, L. Yu, M. D. Ediger and J. J. de Pablo, Molecular modeling of vapor-deposited polymer glasses, J. Chem. Phys., 2014, 140, 204504 CrossRef PubMed.
- S. Singh and J. J. de Pablo, A molecular view of vapor deposited glasses, J. Chem. Phys., 2011, 134, 194903 CrossRef PubMed.
- G. D. Smith and D. Bedrov, Relationship between the α- and β-relaxation processes in amorphous polymers: Insight from atomistic molecular dynamics simulations of 1,4-polybutadiene melts and blends, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 627–643 CrossRef CAS.
- S. Kołodziej, S. Pawlus, K. L. Ngai and M. Paluch, Verifying the Approximate Coinvariance of the α and Johari–Goldstein β Relaxation Times to Variations of Pressure and Temperature in Polyisoprene, Macromolecules, 2018, 51, 4435–4443 CrossRef.
- K. L. Ngai, Relation between some secondary relaxations and the α relaxations in glass-forming materials according to the coupling model, J. Chem. Phys., 1998, 109, 6982–6994 CrossRef CAS.
- K. L. Ngai and K. Y. Tsang, Similarity of relaxation in supercooled liquids and interacting arrays of oscillators, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 60, 4511–4517 CrossRef CAS.
- D. Bedrov and G. D. Smith, Secondary Johari–Goldstein relaxation in linear polymer melts represented by a simple bead-necklace model, J. Non-Cryst. Solids, 2011, 357, 258–263 CrossRef CAS.
- K. L. Ngai, R. Casalini, S. Capaccioli, M. Paluch and C. M. Roland, Do theories of the glass transition, in which the structural relaxation time does not define the dispersion of the structural relaxation, need revision?, J. Phys. Chem. B, 2005, 109, 17356–17360 CrossRef CAS PubMed.
- K. L. Ngai, R. Casalini, S. Capaccioli, M. Paluch and C. M. Roland, Fractals, Diffusion, and Relaxation in Disordered Complex Systems, John Wiley & Sons, Inc., 2006, pp. 497–593 Search PubMed.
- K. Grzybowska, M. Paluch, A. Grzybowski, Z. Wojnarowska, L. Hawelek, K. Kolodziejczyk and K. L. Ngai, Molecular Dynamics and Physical Stability of Amorphous Anti-Inflammatory Drug: Celecoxib, J. Phys. Chem. B, 2010, 114, 12792–12801 CrossRef CAS PubMed.
- R. Casalini and C. M. Roland, Density Scaling of the Structural and Johari–Goldstein Secondary Relaxations in Poly(methyl methacrylate), Macromolecules, 2013, 46, 6364–6368 CrossRef CAS.
- T. C. Ransom, D. Fragiadakis and C. M. Roland, The α and Johari–Goldstein Relaxations in 1,4-Polybutadiene: Breakdown of Isochronal Superpositioning, Macromolecules, 2018, 51, 4694–4698 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S4. See DOI: 10.1039/c8cp05107k |
| This journal is © the Owner Societies 2018 |