
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolydopamine and eumelanin models in various oxidation states†
Chun-Teh
Chen
 and
Markus J.
Buehler
*
and
Markus J.
Buehler
*
Laboratory for Atomistic and Molecular Mechanics (LAMM), Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, Massachusetts 02139, USA. E-mail: mbuehler@MIT.EDU; Tel: +1 617 452 2750
First published on 9th October 2018
Abstract
We report a comprehensive ab initio structural investigation of more than 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 probable molecular structures of polydopamine (PDA) and eumelanin in various oxidation states. With the aid of a computational approach including a brute-force algorithmic generation of chemical isomers and density functional theory, all probable oxidized 5,6-dihydroxyindole (DHI) oligomers, ranging from dimers to tetramers, have been systematically generated and evaluated. We identify a set of the most stable molecular structures of PDA and eumelanin which represent the chemically diverse nature of these materials. Results show that more planar molecular structures have a tendency to be more stable. We also observe that, in some cases, forming cyclic molecular structures could reduce the energy of a DHI tetramer and make it more stable. This finding supports the hypothesis that cyclic molecules could exist in eumelanin-like materials. Additionally, the cyclic molecular models proposed in this work are energetically more favorable than the popular porphyrin-like models in this field.
000 probable molecular structures of polydopamine (PDA) and eumelanin in various oxidation states. With the aid of a computational approach including a brute-force algorithmic generation of chemical isomers and density functional theory, all probable oxidized 5,6-dihydroxyindole (DHI) oligomers, ranging from dimers to tetramers, have been systematically generated and evaluated. We identify a set of the most stable molecular structures of PDA and eumelanin which represent the chemically diverse nature of these materials. Results show that more planar molecular structures have a tendency to be more stable. We also observe that, in some cases, forming cyclic molecular structures could reduce the energy of a DHI tetramer and make it more stable. This finding supports the hypothesis that cyclic molecules could exist in eumelanin-like materials. Additionally, the cyclic molecular models proposed in this work are energetically more favorable than the popular porphyrin-like models in this field.
Introduction
Polydopamine (PDA) has become one of the most promising materials for surface functionalization since first being discovered by Lee et al. in 2007.1 PDA can be synthesized by the oxidative polymerization of dopamine under alkaline aqueous solutions without the need of any complicated equipment or harsh reaction conditions.1,2 Its extraordinarily robust adhesive ability comes from the coexistence of catechol and amine functional groups, the main components in mussel adhesive proteins.1 Due to its simplicity and versatility, PDA has been widely used for applications in the biomedical,3,4 energy,5,6 nanomaterials,7–9 and many other areas.10–12 Despite the growing interest in developing PDA-based materials, its molecular structures are still unclear to date, due to its high degree of disorder at all structural levels and insolubility in water and most organic solvents. For this reason, searching for its structure at the molecular scale is currently a fundamental challenge and an active research topic in this field, with many studies focusing on elucidating its molecular structures in many ways.13–18 Note that a very recent study showed that adding boric acid to dopamine solutions allows the termination of the autoxidation of dopamine into PDA. Thus, the structural characterization of PDA after a short reaction time becomes possible.19 Although there is currently no consensus on the molecular structures of PDA, it is widely accepted that dopamine-quinone and 5,6-dihydroxyindole (DHI) and uncyclized dopamine-like units are the essential building blocks.2,20,21Eumelanin, the most common type of melanin, shares various physiochemical properties with PDA due to their similarities in structures and functional groups. Eumelanin is found in almost every organism and provides many biological functions including colorization and photoprotection.22–25 The building blocks of eumelanin are 5,6-dihydroxyindole-2-carboxylic acid (DHICA) and DHI. Therefore, PDA is also referred to as a “eumelanin-like” material since DHI (and its oxidized forms) is the key building block of both PDA and eumelanin.26 However, as with PDA, eumelanin has a high degree of chemical and geometrical disorder and it is insoluble in most solvents.27,28 Additionally, there is no standard procedure of extraction and purification of eumelanin from its biological environment.29 Thus, the well-defined molecular structures of eumelanin are still unavailable to date.
The aim of this work is to propose probable molecular structures of PDA and eumelanin based on ab initio calculations for a better understanding of the structure–property–function relationship of these materials. However, this is a very difficult task as the number of possible molecular structures is enormous. To simplify the problem, here we only consider two oxidized forms of DHI as the building blocks in PDA and eumelanin. Thus, we are able to reduce the number of possible molecular structures to a reasonable number (around 43000 molecules). Note that a recent MALDI-MS study suggested that PDA does not contain the main structural components related to indole-based oligomers.30 For this reason, this work is primarily connected with eumelanin molecular structures and have an indirect relevance concerning PDA. This is a follow-up work to our previous study on PDA and eumelanin molecular structures,13 in which only the reduced forms of DHI oligomers were considered. Given the presence of high oxidation levels in PDA and eumelanin, considering oxidized forms of DHI oligomers in this work should provide more physical insight into the chemical nature of PDA and eumelanin. This work sheds light on the controversial molecular structures of eumelanin-like materials and opens new research directions in this field.
Methods
Generating DHI oligomers
Although the molecular structures of PDA and eumelanin are still ambiguous, more and more experimental evidence indicated that these materials are mixtures of low molecular weight oligomers.14,20,31 For this reason, three different sizes of oxidized DHI oligomers, namely dimers, trimers, and tetramers, are investigated, which should represent the majority molecular sizes of PDA and eumelanin. Based on our previous study, the covalent bonds connecting DHI monomers to generate DHI oligomers are most likely to form at the 2, 4, and 7 positions (Fig. S1, ESI†).13 Thus, there are 6 possible connections for a dimer, namely 2,2′, 2,4′, 2,7′, 4,4′, 4,7′, and 7,7′ connections. The reduced forms of those dimers were investigated in our previous study.13 In this work, we further consider oxidized forms of DHI oligomers. There are three oxidized forms of DHI, which are indolequinone (IQ), quinone-methide (MQ), and quinone-imine (NQ).13 To reduce the number of molecular structures that need investigation in this work, only IQ and MQ are considered, as NQ is less stable and has a negligible concentration compared to IQ and MQ.27,32 Therefore, there are 21 structurally unique dimers (constitutional isomers), as two types of monomers (i.e., IQ and MQ) are considered.To search for the most stable conformations of each structurally unique dimer, a total of 72 dimers are systematically generated. These dimers include the 21 structurally unique dimers, and also 51 redundant dimers with different conformations, for a quick conformation search. For trimers and tetramers, a total of 1728 trimers and 41472 tetramers are systematically generated, which include all the structurally unique trimers and tetramers, as well as redundant molecular structures with different conformations, for a quick conformation search. To generate oxidized DHI oligomers (i.e., dimers, trimers, and tetramers), their reduced forms are generated first by using a brute-force algorithmic generator. The details of the brute-force algorithmic generator are available in our previous study13 and also described briefly in the ESI.† In the first step, a total of 18 reduced dimers, 216 reduced trimers, and 2592 reduced tetramers are generated. To find the most stable configurations of those oligomers, their molecular structures are equilibrated using molecular dynamics (MD) simulations and then optimized using density functional theory (DFT) calculations.13 In the second step, oxidized DHI oligomers are systematically generated by modifying the molecular structures of their reduced forms (removing the corresponding hydrogen atoms).
Geometry optimization
The ORCA quantum chemistry package33 is implemented to optimize the molecular structures of oxidized DHI oligomers and to calculate their molecular energies. To achieve high accuracy with acceptable computational cost, three steps of optimizations are applied. In the first step, the HF-3c method34 is adopted to optimize the molecular structures of oxidized DHI oligomers. The HF-3c method is based on a Hartree–Fock calculation with a small Gaussian atomic orbitals (AO) basis set, which provides reasonable results close to a large basis set DFT quality but requires only a tiny fraction of the computational cost. In the second step, the molecular structures optimized by using the HF-3c method in the first step are further optimized with the BLYP35,36 functional together with the def2-SVP37 basis set. Additionally, the DFT-D3 with Becke–Johnson damping (D3BJ)38 is implemented to consider dispersion energy. Since both PDA and eumelanin are synthesized in water, the SMD model,39 a continuum solvation model based on the quantum mechanical charge density of a solute molecule interacting with a continuum description of the solvent, is adopted to take solvent effects into account (using a dielectric constant of 80.4 for water). To reduce the so-called basis set superposition error (BSSE) when applying small basis sets,40 the recently developed geometrical counterpoise correction (gCP)41 is adopted to circumvent BSSE.The results of the 72 dimers and 1728 trimers using the HF-3c method and the BLYP/def2-SVP/SMD/gCP method are compared and shown in Fig. S2 (ESI†). The results show that the HF-3c method produces a similar rank as the BLYP/def2-SVP/SMD/gCP method produces, suggesting that the HF-3c method can be used as a filter to screen out molecular structures that are much less stable, thus reducing the number of molecular structures for more computationally expensive DFT calculations. In this work, all 72 dimers and 1728 trimers are optimized in the second step. However, to reduce computational cost, only the top 5184 tetramers (among a total of 41472 tetramers) identified by using the HF-3c method in the first step are optimized in the second step. In the third step, once the top 24 stable dimers, trimers, and tetramers are identified in the second step, those molecular structures optimized by using the BLYP/def2-SVP/SMD/gCP method are further optimized with the def2-QZVPP37 basis set. The large basis set (def2-QZVPP) adopted in the final step ensures that the energies of the most stable molecular structures proposed in this work have sufficient accuracy.
Results
Proposed molecular structures for dimers
The top 12 stable dimers identified in this work are shown in Fig. 1 after discarding higher-energy conformations for each structurally unique dimer. The most stable dimer is made of the 2,4′ position with two IQs, followed by the 2,7′ position with two IQs, the 2,2′ position with two IQs, the 4,4′ position with two IQs, the 2,7′ position with one IQ and one MQ, the 4,7′ position with two IQs, the 7,7′ position with two IQs, the 2,2′ position with one IQ and one MQ, the 2,4′ position with one IQ and one MQ, the 4,4′ position with one IQ and one MQ, the 7,7′ position with one IQ and one MQ, and the 4,7′ position with one IQ and one MQ. These results show that, with the same connection, the dimers consisting of two IQs are more stable than those consisting of one IQ and one MQ. These results are consistent with the fact that IQ is 9.79 kcal mol−1 more stable than MQ in our calculations. Note that none of the top 12 stable dimers consists of two MQs. Comparing the relative energies of the dimers consisting of two IQs, it is clear that 2,4′, 2,7′, and 2,2′ connections are energetically more favorable than 4,4′, 4,7′, and 7,7′ connections. The same statement can also apply to the dimers consisting of one IQ and one MQ. Consequently, 2,4′, 2,7′, and 2,2′ connections are referred to, for convenience, as the “favorite” connections in this work. In fact, the top 3 stable dimers (Fig. 1a–c), which are made of these three favorite connections, have been identified in experiments by oxidation of DHI.42,43 In contrast, 4,4′, 4,7′, and 7,7′ connections are referred to as the “unfavorable” connections in this work. This finding agrees with a recent NMR study, in which the authors showed that more than half of the C4/C7 positions remained protonated in polydopamine.18 Based on these results, we can conclude that the most important structural factors governing the stability of oxidized DHI dimers are the type of monomers (i.e., IQ and MQ) that a dimer consists of and the connection between the monomers.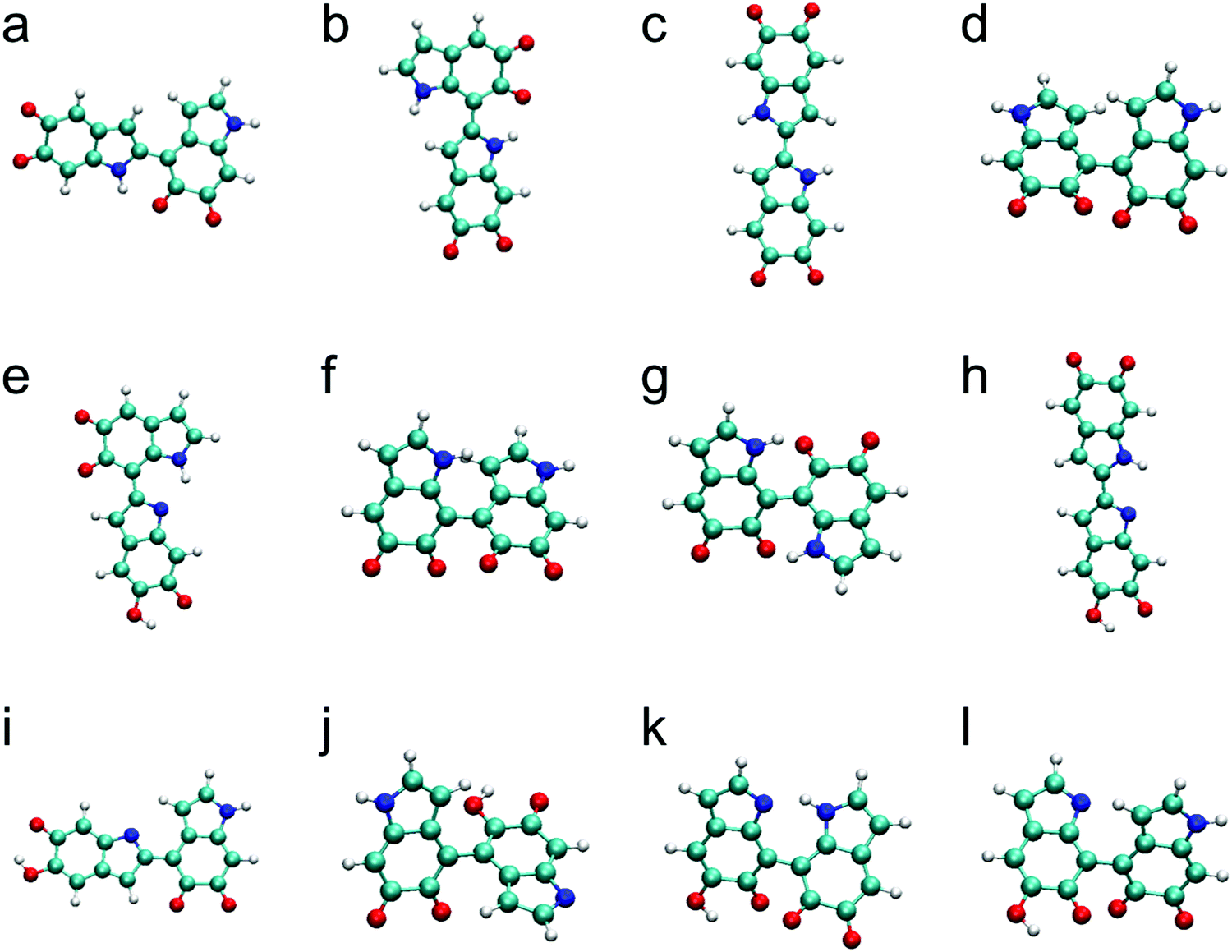 | ||
| Fig. 1 Molecular structures of top 12 stable dimers. The most stable dimer is made of (a) the 2,4′ position with two IQs, followed by (b) the 2,7′ position with two IQs (+1.48 kcal mol−1), (c) the 2,2′ position with two IQs (+1.61 kcal mol−1), (d) the 4,4′ position with two IQs (+6.22 kcal mol−1), (e) the 2,7′ position with one IQ and one MQ (+6.91 kcal mol−1), (f) the 4,7′ position with two IQs (+6.94 kcal mol−1), (g) the 7,7′ position with two IQs (+7.63 kcal mol−1), (h) the 2,2′ position with one IQ and one MQ (+8.38 kcal mol−1), (i) the 2,4′ position with one IQ and one MQ (+9.85 kcal mol−1), (j) the 4,4′ position with one IQ and one MQ (+16.75 kcal mol−1), (k) the 7,7′ position with one IQ and one MQ (+16.98 kcal mol−1), and (l) the 4,7′ position with one IQ and one MQ (+17.07 kcal mol−1). | ||
Proposed molecular structures for trimers
The top 12 stable trimers identified in this work are shown in Fig. 2 after discarding higher-energy conformations for each structurally unique trimer. The most stable trimer is made of the 2,4′ & 2,4′ position with three IQs, followed by the 2,4′ & 2,7′ position with three IQs, the 2,7′ & 2,4′ position with three IQs, the 2,2′ & 4,2′ position with three IQs, the 2,7′ & 2,7′ position with three IQs, the 2,4′ & 7,2′ position with three IQs, the 2,2′ & 7,2′ position with three IQs, the 4,2′ & 4,4′ position with three IQs, the 4,2′ & 4,7′ position with three IQs, the 2,4′ & 2,4′ position with two IQs and one MQ, the 2,4′ & 7,4′ position with three IQs, and the 4,2′ & 7,4′ position with three IQs. These results for oxidized DHI trimers share a similar feature with the results for oxidized DHI dimers. That is, with the same connection, the trimers consisting of more IQs are more stable than the trimers consisting of fewer IQs. In fact, among the top 12 stable trimers (Fig. 2), 11 of them consist of three IQs, and only one consists of two IQs and one MQ. Regarding the connectivity of monomers, while comparing the relative energies of the trimers consisting of three IQs, the trimers with more favorite connections (i.e., 2,4′, 2,7′, and 2,2′ connections) are energetically more favorable than the trimers with fewer favorite connections. Note that the top two stable trimers (Fig. 2a and b), which are made of 2,4′ & 2,4′ connection and 2,4′ & 2,7′ connection, have been identified in experiments by oxidation of DHI.42,43 Therefore, as with oxidized DHI dimers, the two structural factors can also be applied to explain the stability of oxidized DHI trimers. | ||
| Fig. 2 Molecular structures of top 12 stable trimers. The most stable trimer is made of (a) the 2,4′ & 2,4′ position with three IQs, followed by (b) the 2,4′ & 2,7′ position with three IQs (+2.69 kcal mol−1), (c) the 2,7′ & 2,4′ position with three IQs (+3.87 kcal mol−1), (d) the 2,2′ & 4,2′ position with three IQs (+5.40 kcal mol−1), (e) the 2,7′ & 2,7′ position with three IQs (+5.75 kcal mol−1), (f) the 2,4′ & 7,2′ position with three IQs (+6.68 kcal mol−1), (g) the 2,2′ & 7,2′ position with three IQs (+7.15 kcal mol−1), (h) the 4,2′ & 4,4′ position with three IQs (+9.14 kcal mol−1), (i) the 4,2′ & 4,7′ position with three IQs (+9.84 kcal mol−1), (j) the 2,4′ & 2,4′ position with two IQs and one MQ (+10.07 kcal mol−1), (k) the 2,4′ & 7,4′ position with three IQs (+10.11 kcal mol−1), and (l) the 4,2′ & 7,4′ position with three IQs (+10.12 kcal mol−1). | ||
Proposed molecular structures for tetramers
The top 12 stable tetramers identified in this work are shown in Fig. 3 after discarding higher-energy conformations for each structurally unique tetramer. The most stable tetramer is made of the 2,4′ & 2,4′ & 2,7′ position with three IQs and one MQ, followed by the 2,4′ & 2,4′ & 2,4′ position with four IQs, the 2,4′ & 2,4′ & 2,7′ position with four IQs, the 2,7′ & 2,4′ & 2,4′ position with four IQs, the 2,4′ & 2,7′ & 2,4′ position with four IQs, the 2,7′ & 2,4′ & 2,7′ position with three IQs and one MQ, the 2,7′ & 2,4′ & 2,4′ position with three IQs and one MQ, the 2,4′ & 2,7′ & 2,7′ position with four IQs, the 2,2′ & 4,2′ & 4,2′ position with four IQs, the 2,7′ & 2,4′ & 2,7′ position with four IQs, the 2,4′ & 2,4′ & 2,4′ position with three IQs and one MQ, and the 2,4′ & 2,4′ & 7,2′ position with four IQs. Based on the results of oxidized DHI dimers and trimers, one might expect the most stable tetramer to consist of four IQs. However, the most stable tetramer identified here consists of three IQs and one MQ. This is an interesting finding as IQ is more stable than MQ. To explain this result, we have to take a close look at its molecular structure. In the optimized molecular structure of the most stable tetramer (Fig. 3a), we notice that there is a new covalent bond formed between the first and fourth monomers, making the molecular structure cyclic (see Fig. S3, ESI† for more details). In fact, the sixth-most stable tetramer, consisting of three IQs and one MQ (Fig. 3f), also forms a cyclic molecular structure. We suspect that, in some cases, forming this type of cyclic molecular structures could reduce the energy of a tetramer and make it more stable. Regarding the connectivity of monomers, the top 12 stable tetramers are all made of favorite connections (i.e., 2,4′, 2,7′, and 2,2′ connections). Although it is surprising to find that the most stable tetramer does not consist of four IQs, the two structural factors, which are proposed based on the results of oxidized DHI dimers and trimers, can still be applied to explain the stability of oxidized DHI tetramers.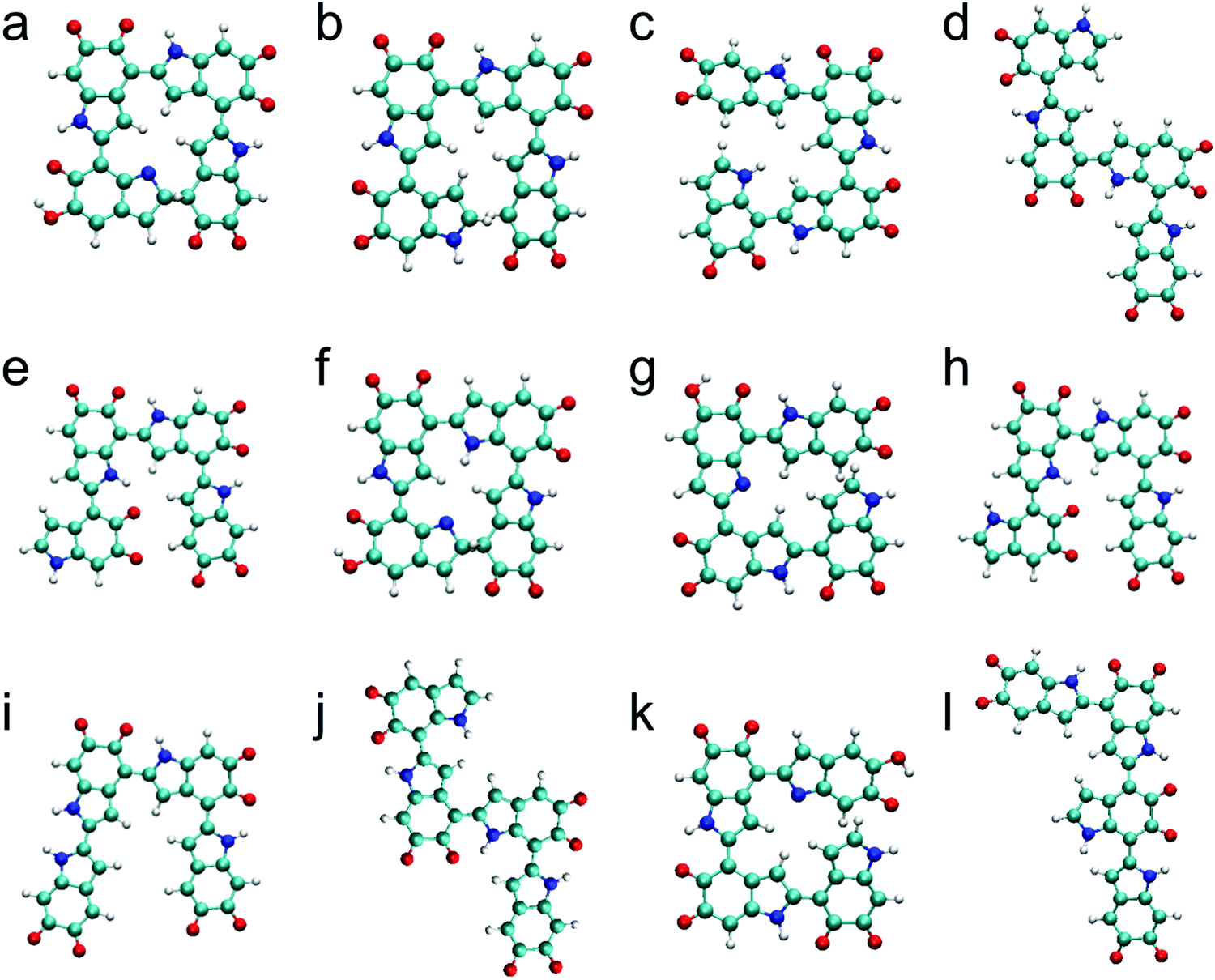 | ||
| Fig. 3 Molecular structures of top 12 stable tetramers. The most stable tetramer is made of (a) the 2,4′ & 2,4′ & 2,7′ position with three IQs and one MQ, followed by (b) the 2,4′ & 2,4′ & 2,4′ position with four IQs (+0.57 kcal mol−1), (c) the 2,4′ & 2,4′ & 2,7′ position with four IQs (+5.33 kcal mol−1), (d) the 2,7′ & 2,4′ & 2,4′ position with four IQs (+7.58 kcal mol−1), (e) the 2,4′ & 2,7′ & 2,4′ position with four IQs (+7.61 kcal mol−1), (f) the 2,7′ & 2,4′ & 2,7′ position with three IQs and one MQ (+7.69 kcal mol−1), (g) the 2,7′ & 2,4′ & 2,4′ position with three IQs and one MQ (+9.09 kcal mol−1), (h) the 2,4′ & 2,7′ & 2,7′ position with four IQs (+9.74 kcal mol−1), (i) the 2,2′ & 4,2′ & 4,2′ position with four IQs (+9.78 kcal mol−1), (j) the 2,7′ & 2,4′ & 2,7′ position with four IQs (+10.48 kcal mol−1), (k) the 2,4′ & 2,4′ & 2,4′ position with three IQs and one MQ (+10.53 kcal mol−1), and (l) the 2,4′ & 2,4′ & 7,2′ position with four IQs (+11.45 kcal mol−1). | ||
Statistical characteristics of data for trimers and tetramers
To compare the energy differences between the 1728 trimers, the relative energies (compared to the most stable trimer) and ranks of the trimers are shown in Fig. 4a. In the figure, blue represents the trimers consisting of three IQs, red represents the trimers consisting of two IQs and one MQ, yellow represents the trimers consisting of one IQ and two MQs, and purple represents the trimers consisting of three MQs. The results show a clear trend that the trimers consisting of more IQs are more stable than the trimers consisting of fewer IQs. The trend also indicates that, regarding the molecular energy, the type of monomers that a trimer consists of is more significant than the connection between the monomers. Additionally, the projection products against the relative energies of the trimers are shown in Fig. 4b. The projection products quantify the planarity of DHI oligomers, ranging from zero to one, being the later a completely planar molecular structure (see the ESI† for more details). The dashed lines separate the results into three regions: the first, with trimers having two favorable connections; the second, with trimers having one favorable connection and one unfavorable connection; and the third, with trimers having two unfavorable connections (see Fig. S4a, ESI† for additional information). For the trimers consisting of the same type of monomers (shown in the same color), those having more favorable connections are more stable than the others. Moreover, those made of more favorable connections have a higher projection product, meaning that they have a more planar molecular structure.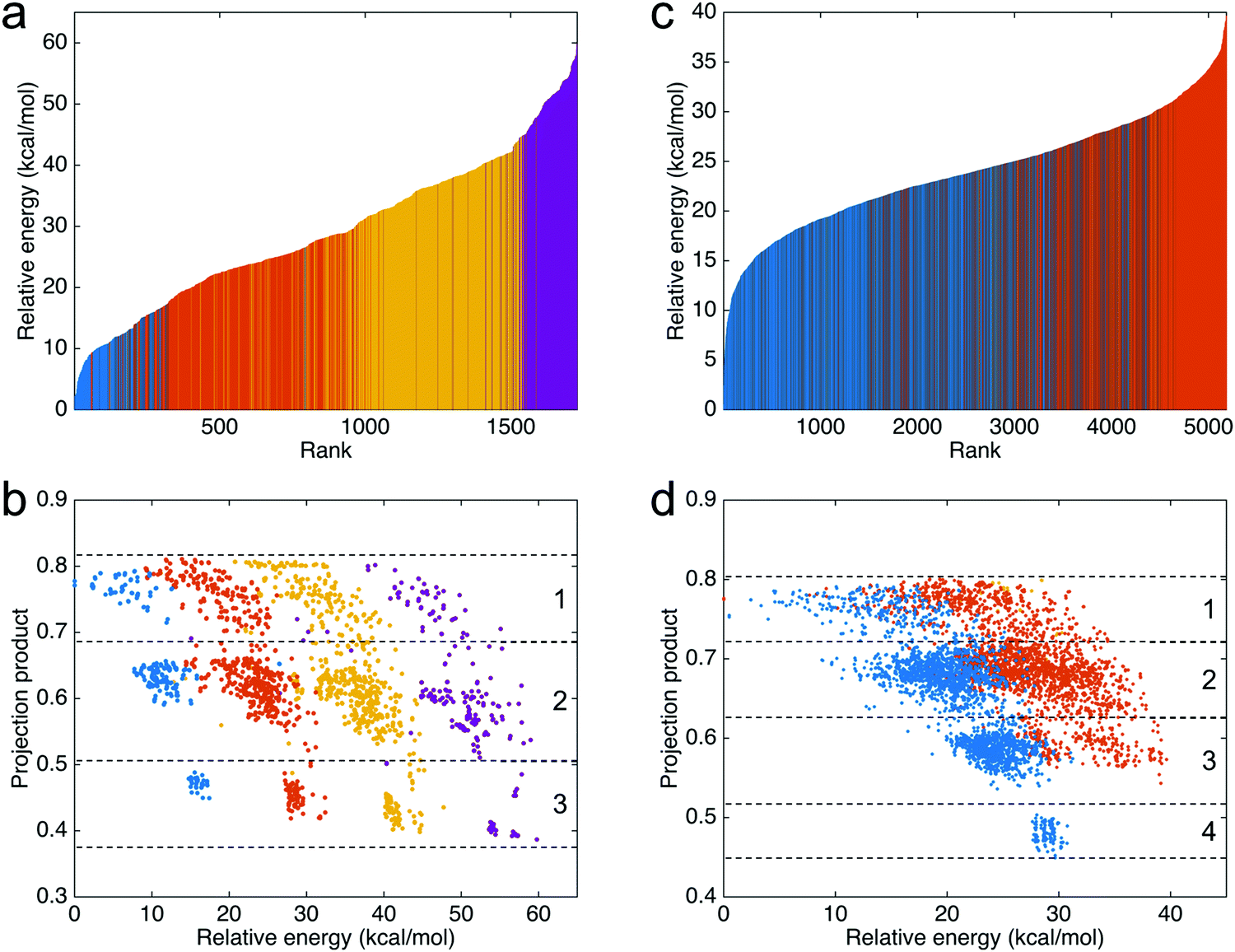 | ||
| Fig. 4 Statistical characteristics of data for trimers and tetramers. (a) The relative energies and ranks of the 1728 trimers, depending on their relative energies, from the most to least stable one. (b) The projection products against the relative energies of the 1728 trimers. The dashed lines separate the results into three regions: the first, with trimers having two favorable connections; the second, with trimers having one favorable connection and one unfavorable connection; and the third, with trimers having two unfavorable connections. (c) The relative energies and ranks of the top 5,184 tetramers, depending on their relative energies, from the most to least stable one. (d) The projection products against the relative energies of the top 5184 tetramers. The dashed lines separate the results into four regions: the first, with tetramers having three favorable connections; the second, with tetramers having two favorable connection and one unfavorable connection; the third, with tetramers having one favorable connection and two unfavorable connection; and the fourth, with tetramers having three unfavorable connections. For (a) and (b), blue represents the trimers consisting of three IQs, red represents the trimers consisting of two IQs and one MQ, yellow represents the trimers consisting of one IQ and two MQs, and purple represents the trimers consisting of three MQs. For (c) and (d), blue represents the tetramers consisting of four IQs and red represents the tetramers consisting of three IQs and one MQ. | ||
For the tetramers, the relative energies (compared to the most stable tetramer) and ranks of the top 5184 tetramers are shown in Fig. 4c. In the figure, blue represents the tetramers consisting of four IQs and red represents the tetramers consisting of three IQs and one MQ. As the figure only shows the top 5184 tetramers, the tetramers consisting of more than one MQ are not shown in the figure since those tetramers have a relatively higher energy (less stable). The results show that the tetramers consisting of four IQs (shown in blue) are more stable than those consisting of three IQs and one MQ (shown in red). Additionally, the projection products against the relative energies of the tetramers are shown in Fig. 4d. The dashed lines separate the results into four regions: the first, with tetramers having three favorable connections; the second, with tetramers having two favorable connection and one unfavorable connection; the third, with tetramers having one favorable connection and two unfavorable connection; and the fourth, with tetramers having three unfavorable connections (see Fig. S4b (ESI†) for additional information). For the tetramers consisting of the same type of monomers (shown in the same color), the results clearly show that more planar molecular structures have a tendency to be more stable, consistent with the results of the timers.
Energy comparison of cyclic tetramers
Based on the top 12 stable tetramers identified in this work, it can be seen that forming cyclic tetramers is probable. In fact, the first- and sixth-most stable tetramers (Fig. 3a and f) have a cyclic molecular structure, as noted previously. In addition to these two tetramers, it is also possible that the second-, third-, seventh-, and eleventh-most stable tetramers (Fig. 3b, c, g, and k) can form a new covalent bond between the first and fourth monomers at the 2,4′ or 2,7′ position to make a cyclic molecular structure. Although this bond forming does not happen to these tetramers in our DFT calculations, it could occur in ambient conditions as we perform static DFT calculations (temperature effects are not considered), and kinetic energy plays an important role in overcoming energy barriers when forming new covalent bonds. Therefore, we manually create a cyclic molecular structure for each of the six tetramers (Fig. 3a–c, f, g, and k) by removing the corresponding hydrogen atoms in the first and fourth monomers. Their cyclic forms are shown in Fig. 5a. Note that these cyclic tetramers have different molecular structures compared to those porphyrin-like tetramers proposed by Kaxiras et al. in 2006 as shown in Fig. S5 (ESI†).27 In those porphyrin-like tetramers, the connections are all 2,7′ connections with the four nitrogen atoms in the center. However, the connections in the cyclic tetramers proposed in this work are a mixture of 2,4′ and 2,7′ connections. Fig. 5b shows that these cyclic tetramers have a higher formation energy compared to those porphyrin-like tetramers, suggesting that these cyclic tetramers (Fig. 5a) are energetically more favorable than those porphyrin-like tetramers (Fig. S5, ESI†). Note that those porphyrin-like tetramers showed positive formation energies (exothermic reaction, energy released) in the work of Kaxiras et al. in the gas phase.27 However, our results show that those porphyrin-like tetramers actually have negative formation energies (endothermic reaction, energy absorbed) when taking solvent effects into consideration (see the Methods section for more details). Interestingly, even in the gas phase (Fig. S6, ESI†), the cyclic tetramers proposed in this work still have a higher formation energy compared to those porphyrin-like tetramers. These results indicate that our cyclic tetramers are indeed more stable than those porphyrin-like tetramers.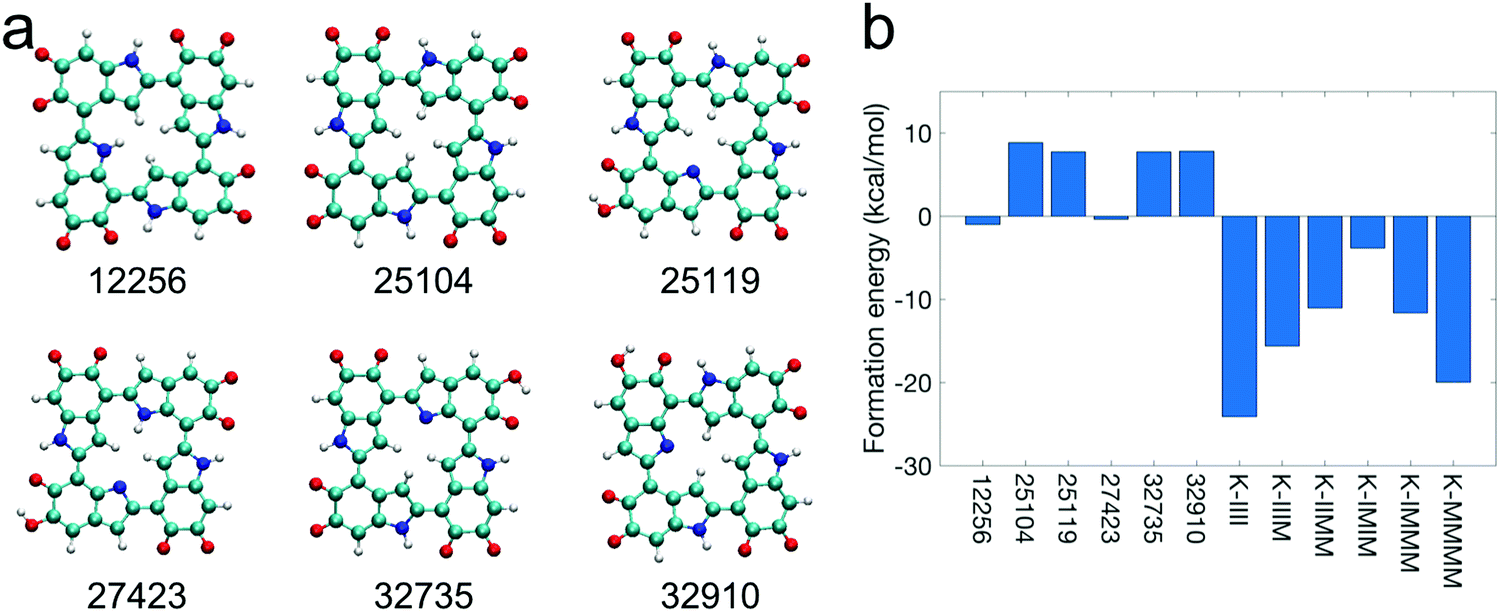 | ||
| Fig. 5 Molecular structures and formation energies of cyclic tetramers. (a) The molecular structures of the cyclic tetramers proposed in this work. The numbers represent the molecular IDs of the tetramers in our calculations. (b) The comparison of formation energies of the cyclic tetramers shown in (a) and those porphyrin-like tetramers proposed in the literature,27 suggesting that the cyclic tetramers shown in (a) are energetically more favorable than those porphyrin-like tetramers. The formation energy of a tetramer is defined as the difference between the ground state energy of the four isolated monomers and the summation of the ground state energy of the tetramer and the ground state energy of four isolated hydrogen molecules. | ||
Polymerization path for forming cyclic tetramers
Based on the results discussed above, it can be seen that IQs and MQs tend to form planar oligomers and that it is probable that a portion of tetramers are cyclic. To understand how cyclic tetramers could be formed, a probable polymerization path is proposed and shown in Fig. 6. In the first step, most monomers start to form dimers and the most stable dimer (i.e., two IQs connecting at the 2,4′ position), as shown at the left of Fig. 6, could be the majority type of dimers. In the second step, dimers start to interact with each other and form tetramers. For example, two of the most stable dimers can form a non-cyclic tetramer at the 2,4′ position, as shown in the center of Fig. 6. This non-cyclic tetramer is 9 kcal mol−1 energetically more favorable than the two isolated dimers. Note that this polymerization process involves the loss of two hydrogen atoms. A hydrogen molecule is used as the reservoir for a pair of the removed hydrogen atoms when comparing the energies of the systems with different numbers of hydrogen atoms. The 9 kcal mol−1 reported here is the energy difference between the ground state energy of the two isolated dimers and the summation of the ground state energy of the non-cyclic tetramer and the ground state energy of a hydrogen molecule. As most monomers are probably be consumed in the first step to form dimers, there might not be many monomers available for dimers to form trimers in the second step. Thus, it is probable that the concentration of trimers is lower than that of tetramers in this step. In the third step, the non-cyclic tetramers could form cyclic tetramers through the 2,4′ position between the first and fourth monomers, as shown at the right of Fig. 6. This cyclic tetramer is 4 kcal mol−1 energetically more favorable than the non-cyclic tetramer. The 4 kcal mol−1 reported here is the energy difference between the ground state energy of the non-cyclic tetramer and the summation of the ground state energy of the cyclic tetramer and the ground state energy of a hydrogen molecule.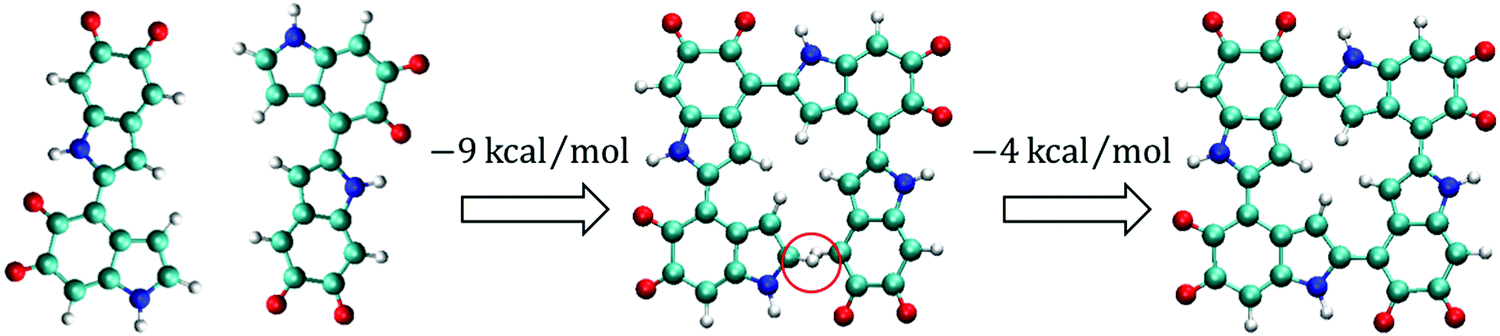 | ||
| Fig. 6 Proposed polymerization path for forming cyclic tetramers. In the first step, the most stable dimers (i.e., the dimers made of the 2,4′ position with two IQs) are formed. In the second step, two of the most stable dimers form a non-cyclic tetramer through the 2,4′ position. This non-cyclic tetramer is 9 kcal mol−1 energetically more favorable than the two isolated dimers. The red circle indicates that the first and fourth monomers are not connected. In the third step, the non-cyclic tetramer forms a cyclic tetramer through the 2,4′ position between the first and fourth monomers. This cyclic tetramer is 4 kcal mol−1 energetically more favorable than the non-cyclic tetramer shown in the center. Note that a hydrogen molecule is used as the reservoir for a pair of the removed hydrogen atoms when comparing the energies of the systems with different numbers of hydrogen atoms. | ||
Discussion
Although our DFT calculations indicate that forming cyclic tetramers is probable as it is an energetically favorable process, we do not propose these cyclic tetramers to be the majority type of tetramers in PDA and eumelanin. In reality, the probability of two dimers made of 2,4′ or 2,7′ connection happen to interact with each other and form two new covalent bonds at the specific positions to become a cyclic tetramer could be very low.30 Consequently, the presence of these cyclic tetramers in eumelanin-like materials is still skeptical and should be examined in experiments. It is worth mentioning that the results presented in this work are based on some assumptions. One of them is the assumption that PDA and eumelanin consist of only oxidized DHI oligomers. In fact, PDA and eumelanin are not the same materials and they also consist of other building blocks besides DHI. For example, it is commonly assumed that PDA consists of uncyclized dopamine-like units and eumelanin consists of DHICA. Another assumption is that the molecular structures with a lower energy are more likely to exist. Their activation energy in the polymerization process is not considered because the intermediates during the polymerization process are still unclear. Nevertheless, we believe that this work brings forth the tools necessary to elucidate the molecular structures of eumelanin-like materials. With more computational recourses, other building blocks of PDA and eumelanin can also be considered in the future. Therefore, the molecular structures of eumelanin-like materials will be clearer, and their structure–property–function relationship will eventually be fully understood.Conclusion
In this work, more than 43000 oxidized DHI oligomers have been systematically generated and evaluated. Among these oligomers, a set of the most stable molecular structures ranging from dimers to tetramers in various oxidation states have been identified. The results show that more planar molecular structures have a tendency to be more stable. This finding is consistent with our previous study on reduced DHI oligomers, meaning that no matter if DHI oligomers are in the reduced or oxidized forms, more planar molecular structures are energetically more favorable. We also find that, in some cases, forming cyclic molecular structures could reduce the energy of a tetramer and make it more stable. Based on this finding, we propose a set of cyclic tetramers that are more stable than those porphyrin-like tetramers proposed in the literature.27 In summary, this work brings insight into the molecular structures of PDA and eumelanin, showing that cyclic molecular structures are likely to exist, and providing a set of molecular models for more accurate modeling of eumelanin-like materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by ONR (N000141612333) and AFOSR (FATE MURI FA9550-15-1-0514). This research used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575.References
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. d'Ischia, A. Napolitano, V. Ball, C. T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef PubMed.
- M. E. Lynge, R. van der Westen, A. Postma and B. Stadler, Nanoscale, 2011, 3, 4916–4928 RSC.
- S. K. Madhurakkat Perikamana, J. Lee, Y. B. Lee, Y. M. Shin, E. J. Lee, A. G. Mikos and H. Shin, Biomacromolecules, 2015, 16, 2541–2555 CrossRef CAS PubMed.
- M. H. Ryou, D. J. Lee, J. N. Lee, Y. M. Lee, J. K. Park and J. W. Choi, Adv. Energy Mater., 2012, 2, 645–650 CrossRef CAS.
- W. Zhou, X. Xiao, M. Cai and L. Yang, Nano Lett., 2014, 14, 5250–5256 CrossRef CAS PubMed.
- C.-T. Chen, F. J. Martin-Martinez, S. Ling, Z. Qin and M. J. Buehler, Nano Futures, 2017, 1, 011003 CrossRef.
- J. Mu, C. Hou, H. Wang, Y. Li, Q. Zhang and M. Zhu, Sci. Adv., 2015, 1 Search PubMed.
- W. Xu, Z. Qin, C.-T. Chen, H. R. Kwag, Q. Ma, A. Sarkar, M. J. Buehler and D. H. Gracias, Sci. Adv., 2017, 3, e1701084 CrossRef PubMed.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- T. G. Barclay, H. M. Hegab, S. R. Clarke and M. Ginic-Markovic, Adv. Mater. Interfaces, 2017, 4, 1601192 CrossRef.
- C.-T. Chen, F. J. Martin-Martinez, G. S. Jung and M. J. Buehler, Chem. Sci., 2017, 8, 1631–1641 RSC.
- J. R. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- S. C. Lin, C. T. Chen, I. Bdikin, V. Ball, J. Gracio and M. J. Buehler, Soft Matter, 2014, 10, 457–464 RSC.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- M. Cîrcu and C. Filip, Polym. Chem., 2018, 9, 3379–3387 RSC.
- A. Schneider, J. Hemmerle, M. Allais, J. Didierjean, M. Michel, M. d’Ischia and V. Ball, ACS Appl. Mater. Interfaces, 2017, 10, 7574–7580 CrossRef PubMed.
- Y. Ding, L.-T. Weng, M. Yang, Z. Yang, X. Lu, N. Huang and Y. Leng, Langmuir, 2014, 30, 12258–12269 CrossRef CAS PubMed.
- R. Mrówczyński, R. Markiewicz and J. Liebscher, Polym. Int., 2016, 65, 1288–1299 CrossRef.
- C. T. Chen, V. Ball, J. J. D. Gracio, M. K. Singh, V. Toniazzo, D. Ruch and M. J. Buehler, ACS Nano, 2013, 7, 1524–1532 CrossRef CAS PubMed.
- C. T. Chen, C. Chuang, J. S. Cao, V. Ball, D. Ruch and M. J. Buehler, Nat. Commun., 2014, 5, 3859 CrossRef CAS PubMed.
- M. Brenner and V. J. Hearing, Photochem. Photobiol., 2008, 84, 539–549 CrossRef CAS PubMed.
- P. Ghosh and D. Ghosh, J. Phys. Chem. B, 2017, 121, 5988–5994 CrossRef CAS PubMed.
- C.-T. Chen, Eumelanin and polydopamine: self-assembly, structure, and properties, Massachusetts Institute of Technology, 2016 Search PubMed.
- E. Kaxiras, A. Tsolakidis, G. Zonios and S. Meng, Phys. Rev. Lett., 2006, 97, 218102 CrossRef PubMed.
- S. Meng and E. Kaxiras, Biophys. J., 2008, 94, 2095–2105 CrossRef CAS PubMed.
- L. Huang, M. Liu, H. Huang, Y. Wen, X. Zhang and Y. Wei, Biomacromolecules, 2018 Search PubMed.
- M. L. Alfieri, R. Micillo, L. Panzella, O. Crescenzi, S. L. Oscurato, P. Maddalena, A. Napolitano, V. Ball and M. d’Ischia, ACS Appl. Mater. Interfaces, 2017, 10, 7670–7680 CrossRef PubMed.
- P. Díaz, Y. Gimeno, P. Carro, S. González, P. L. Schilardi, G. Benítez, R. C. Salvarezza and A. H. Creus, Langmuir, 2005, 21, 5924–5930 CrossRef PubMed.
- B. Powell, T. Baruah, N. Bernstein, K. Brake, R. H. McKenzie, P. Meredith and M. Pederson, J. Chem. Phys., 2004, 120, 8608–8615 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- R. Sure and S. Grimme, J. Comput. Chem., 2013, 34, 1672–1685 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- H. Kruse, L. Goerigk and S. Grimme, J. Org. Chem., 2012, 77, 10824–10834 CrossRef CAS PubMed.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed.
- L. Panzella, A. Pezzella, A. Napolitano and M. d'Ischia, Org. Lett., 2007, 9, 1411–1414 CrossRef CAS PubMed.
- M. d’Ischia, A. Napolitano, A. Pezzella, E. J. Land, C. A. Ramsden and P. A. Riley, in Advances in Heterocyclic Chemistry, ed. R. K. Alan, Academic Press, 2005, vol. 89, pp. 1–63 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp05037f |
| This journal is © the Owner Societies 2018 |