
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ethanol, O, and CO adsorption on Pt nanoparticles: effects of nanoparticle size and graphene support†
L.
G. Verga
 ,
A. E.
Russell
and
C.-K.
Skylaris
*
,
A. E.
Russell
and
C.-K.
Skylaris
*
Department of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: c.skylaris@soton.ac.uk
First published on 1st October 2018
Abstract
Pt nanoparticles dispersed over carbonaceous supports are widely used as catalysts for different applications, making studies on the interplay between size and support effects indispensable for rational catalyst design. Here, we use DFT calculations to simulate the interaction between O, CO, and ethanol with free platinum cuboctahedral nanoparticles with up to 147 atoms and with the same Pt nanoparticles supported on a single layer of graphene with up to 720 carbon atoms. We compute adsorption energies for each adsorbate on different adsorption sites for supported and unsupported Pt nanoparticles. We show that as the Pt nanoparticle grows the adsorption energy decreases, and that the size effect is more important for O and CO adsorption than for ethanol. We observe that the generalized coordination number of each adsorption site controls the interaction strength for O and CO to a much larger extent than for ethanol. Electronic charge redistributions and density of states projected on the d band of the interacting Pt facets are used to obtain a better understanding of the differences between the electronic interactions for each adsorbate. For Pt nanoparticles supported on graphene, the support effects weaken the adsorption energies for all the adsorbates, but this effect rapidly decreases with larger nanoparticles, and it is only significant for our smallest nanoparticle Pt13. By demonstrating that the effects of nanoparticle size and support are different for ethanol as compared with O and CO, we conclude that it should be possible to modify different parameters in the catalyst design in order to tune the Pt nanoparticle to interact with specific adsorbates.
1 Introduction
The interest on clean, renewable and flexible sources of energy has driven many research fields during the last decade. In this drive, fuel cell technology is receiving substantial attention due to the high efficiency in the production of electricity, the low emission of pollutants, and the high capacity of acting as decentralized power plants, enabling autonomy and decreasing the energy losses associated with transmission of electricity.1 Fuel cells can be fed with several types of fuels, each one with its advantages and disadvantages. Nowadays, fuel cells fed with alcohols, such as methanol and ethanol, are extremely appealing, due to the easiness of handling, transporting and distributing such fuels.2,3The performance of fuel cells is greatly controlled by the efficiency of the catalysts for anodic and cathodic reactions.1 For ethanol fuel cells, the anodic reaction consists in the oxidation of ethanol molecules, which requires the complicated task of breaking C–H, C–O, O–H, and C–C bonds.3–6 Meanwhile, the cathodic catalyst performs the oxygen reduction reaction to form water.5,7 In addition to optimising these reactions, the chosen catalysts should also be resistant to poisoning by molecules like CO, in order to increase the lifetime and durability of the fuel cell.
The size, shape, distribution, and composition of metallic nanoparticles have been used to tune anodic and cathodic catalysts, as described in several recent review papers.5–10 In the literature, Pt, Rh, Ni, Co nanoparticles as well as several alloy structures are commonly studied as anodic catalysts for ethanol fuel cells.5,6 Particularly for Pt nanoparticles catalysts, a dependence of specific activity for ethanol oxidation on nanoparticle size was experimentally observed by Perez et al.,11 with an activity peak obtained for Pt nanoparticles of 2.6 nm.
The control of the shape of the nanoparticle is also significant to obtain the desired catalytic effect. For the ethanol oxidation reaction, the Pt surface orientation can control reaction pathways and rates. As an example, Colmati et al.12 showed experimentally that the main product obtained on Pt(111) surfaces after the ethanol oxidation is acetic acid, suggesting that C–C bond breaking is only possible on Pt(111) electrodes with the presence of defects. On the other hand, the C–C bond breaking is easier for Pt(100) and Pt(110) electrodes, with the first being easily poisoned by CO at low potentials.12 In addition, the theoretical work from Wang and Liu13 shows that Pt(100) surfaces are better than Pt(111) facets for a complete ethanol oxidation at low coverages and that acetic acid and acetaldehyde should be the main products for Pt(111) surfaces. Size and shape of Pt nanoparticles are also crucial parameters for the cathodic reaction ORR,7–10 with peaks of mass activity being found around 2.5 nm both theoretically and experimentally.14–17 The importance of the nanoparticle shape for ORR is used to explain the high activity for certain nanoparticles sizes and has been deeply studied for subnanoclusters,18–20 where just one Pt atom can modify the subnanoparticle structure and double the catalytic activity.20
In addition to increasing the catalytic efficiency, metallic nanoparticles can help to reduce the catalyst cost by decreasing the noble metal loading due to the increase in surface area to volume ratio. For fuel cell catalysts, the metallic nanoparticles can be dispersed over different types of supports, enabling another route for catalyst design.5–7,10,21 Metal oxides and carbides have been successfully used as supports for both anodic and cathodic reactions.22–25 However, carbonaceous materials are still widely used as catalyst supports on fuel cells applications. In particular, graphene seems an interesting option due to its large surface area, high electrical conductivity, and good thermal stability.21
Despite some advantages, the usage of graphene as a support for Pt nanoparticles also presents some challenges, such as the weak interaction between support and nanoparticle which could cause loss of catalytic surface area.26,27 Several theoretical studies have already sought to describe the Pt/graphene systems,26,28–41 showing a weak interaction between the nanoparticles and the graphene with a large contribution from van der Waals interactions and a small preference for Pt(111) facets in contact with the support. For small nanoparticles interacting with graphene, Pt–Pt bond lengths expansions (contractions) are observed for Pt facets close (distant) from the graphene, with an associated small charge redistribution and d-band centre changes.40
Given the complexity of treating fuel cell catalysts with computational simulations, theoretical studies are usually performed aiming for specific pieces of the problem, such as the interaction between adsorbed species and metallic surfaces. The adsorption of certain atoms and molecules on catalytic surfaces can provide information such as the changes induced in the molecules due to the interaction with the surface, adsorption sites preferences, and other characteristics of the interaction that can be useful to understand how the surface will react as a catalyst. Performing these studies with a computational approach enables control of catalyst variables which are usually unreachable on an experimental basis. As an example, quantum confinement and lattice strain changes due to size effects can be separated on theoretical studies by simply using a same Pt–Pt bond length for nanoparticles of different sizes, enabling us to quantify their effects on the adsorption energy of a given adsorbate.42,43 In this work, we assess O, CO, and ethanol as adsorbed species due to their importance for ethanol fuel cell catalysts.
The interaction of atomic oxygen with catalytic surfaces is widely studied in the literature and is commonly used as a predictive tool for reactivity in several chemical reactions. As an example, Nørskov et al.44 used the Sabatier Principle to correlate the activity of metallic surfaces for the oxygen reduction reaction to the adsorption energy of atomic oxygen and hydroxyl in the format of a volcano plot, providing valuable insights for designing catalysts for this reaction. Activation energies for the bond breaking reaction of C–OH on an ethanol molecule were also correlated with atomic oxygen adsorption energies in the work of Sutton and Vlachos,45 with the activation energies for C–C and β C–H bond breaking also being correlated with the O adsorption to a smaller extent.
Understanding the interaction of catalytic surfaces with ethanol molecules is also important to the design of new catalysts for ethanol fuel cells. First of all, the adsorption of ethanol is itself the first step in the ethanol oxidation reaction. Secondly, the ethanol adsorption can also be used as a descriptor for the first dehydrogenation step,46 which plays an important role on ethanol oxidation reaction. Moreover, analysing the position of the molecule on top of the catalyst surface and how the electronic density of the molecule changes with the interaction can help to explain dissociation steps of ethanol. In the literature, it is possible to find a few theoretical studies performed for ethanol molecules on metallic monolayers,47 slabs,48,49 small nanoparticles,50 and core–shell M@Pt nanoparticles.51 For Pt surfaces, all the computational results showed adsorption energies lower than 1.0 eV, with a clear adsorption site preference for on top sites, and a crucial role of van der Waals interactions, which is responsible for changing the ethanol configuration adsorbed on metallic nanoparticles, favouring configurations with the C–C bond parallel to Pt surfaces.48,52
The chemisorption of carbon monoxide molecules is also a subject of great interest for ethanol fuel cell catalysts. Carbon monoxide can poison the catalyst surface by blocking active sites and decreasing the catalyst activity.3,5–7 However, the theoretical description of CO adsorption on Pt(111) sites via DFT with LDA or GGA exchange–correlation functionals is widely known to be incompatible with experimental results. The “CO/Pt(111) puzzle” is characterized by DFT showing hollow sites as the preferred adsorption site, while experimental data show that CO is more stable on top sites.53 The interaction of CO with a Pt surface is characterized by the bond of the CO σ HOMO, a lone pair in the sp hybrid orbital from the C atom, with a d orbital from the metallic surface, and a back-bonding from the metal to an unoccupied π* orbital in the CO molecule.54 As the CO LUMO energy is lower than it should be when calculated with GGA DFT, this piece of the interaction starts to be stronger, and as this interaction is more important for hollow sites than for top sites, the error in the adsorption site preference occurs.55,56
Several researchers have tried to change the chosen XC functional and add dispersion interactions to solve this problem.57–64 Recently, the meta-GGA functional M06-L predicted the correct adsorption site for CO/Pt(111),61 however, when dispersion interactions were included, the hollow site started to be the preferred site again, indicating that the success of M06-L could be related to error cancellations effects.62 For Pt nanoparticles the error is still present for top and hollow sites in the centre of (111) facets, but less so near the edges, once finite size effects increase the adsorption strength, especially at on top and bridge adsorption sites on the edge of the nanoparticles.60,65
Here, we analyze via DFT simulations, how the adsorption of O, CO, and ethanol are affected by size effects for cuboctahedral Ptn nanoparticles with (n = 13–147). Electronic and structural changes induced by the interaction are investigated to explain the differences in the effects of nanoparticle size for each molecule. Moreover, by adsorbing O, CO, and ethanol on Pt nanoparticles on top of pristine graphene sheets, we assess how the interplay between size and support effects can change the adsorption of the same molecules. We finish with discussions about the observed phenomena, aiming to provide useful insights for rational catalyst design.
2 Method and computational details
We performed our simulations with the ONETEP code,66 using the ensemble DFT method, implemented by Serrano and Skylaris,67 which allows simulations of metallic structures with thousands of atoms. We adopted the vdW functional rVV1068, 69 as our exchange–correlation functional and used the projector augmented wave (PAW) method70 to describe the electron–ion interaction.We set the psinc basis set71 kinetic energy cutoff to 550 eV for geometry optimizations and 850 eV for total energy and properties calculations. For each Pt atom we assigned 12 NGWFs, for C and O atoms we allocated 4 NGWFs and H atoms were calculated with only 1 NGWF. For C atoms in the graphene sheet we assigned 8 NGWFs. For all NGWFs, we used 9.0a0 radii. The NGWF conjugate gradient optimisation preconditioning parameter k072 used in our simulations was equal to 2.5a0−1.
We obtained the isolated and supported nanoparticles from a previous work,40 where we optimized the geometry of Pt nanoparticles and Pt monolayers of different sizes in contact with a pristine graphene sheet and compared the effect of the support over the properties of Pt structures. Here, for the supported nanoparticles, we only use cuboctahedral nanoparticles with the (111) facet interacting with the graphene sheet, as these were the most stable structures in our previous work.40 We performed our simulations in orthorhombic simulation boxes under periodic boundary conditions, with a minimum gap of 10 Å between the borders of the simulation box and the Pt atoms. Here, only the adsorbates geometries were relaxed, with a convergence threshold of 0.002 Eh/a0 on the atomic forces, while atoms from the isolated and supported nanoparticles remained fixed.
To perform a more detailed analysis of support and size effects for Pt nanoparticles interacting with O, CO and ethanol, we sampled different adsorption sites as illustrated in Fig. 1 for Pt147. Fig. 1(a) illustrates adsorption sites for a (111) facet, where 1 to 3 and 4 to 6, respectively represent the adsorbates on top of Pt atoms or on bridge sites located in the vertex, edge and centre of the nanoparticle facet, while 7 and 8 are HCP sites in the vertex and edge of the nanoparticle and 9 is an FCC adsorption site. Fig. 1(b) shows the chosen sites for a (100) facet, with a similar ordering as that presented for the (111) facet.
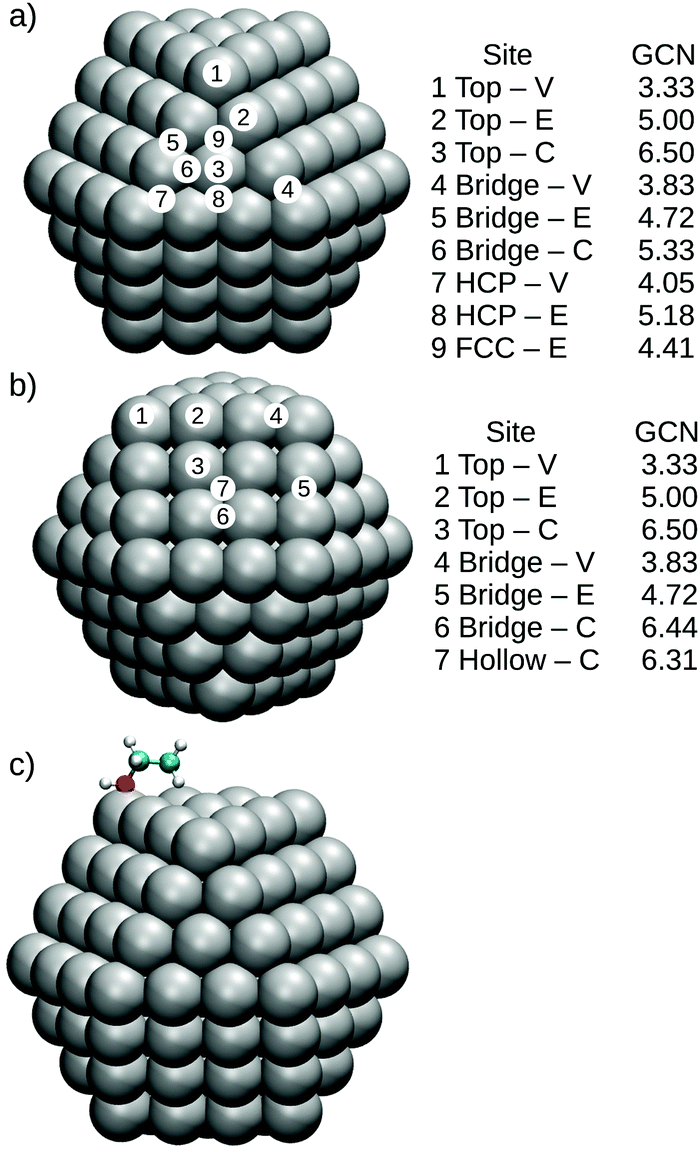 | ||
| Fig. 1 Adsorption sites for (a) Pt(111) facet of a Pt147, where 1 to 3 represent top adsorption sites located on the vertex, edge and centre of the nanoparticle facet, 4 to 6 are bridge sites also located on the vertex, edge and centre of the (111) facet, and 7 and 8 are HCP sites locate on the vertex and edge of the Pt(111) facet and 9 is an FCC site. (b) Shows a similar adsorption site distribution for the (100) facet of a Pt147, and (c) illustrates the initial configuration of an ethanol molecule on the top-V site of Pt147. For each adsorption site, we also present the generalized coordination number calculated as proposed by Calle-Vallejo.73 | ||
Meanwhile, Fig. 1(c) shows the initial configuration of an ethanol molecule on a top-vertex adsorption site. The ethanol molecules were initialised in the trans-ethanol configuration with the oxygen atom on top of the adsorption site and with the C–C bond parallel to the studied Pt facet. This configuration follows the results obtained in the literature for ethanol adsorption on Pt(111) slabs and extended Pt surfaces.47–49 Carbon monoxide molecules were placed with the C–O bond being perpendicular to the nanoparticle facet.
We used adsorption energies, EADS, to analyse the interaction between CO, O and ethanol with the catalyst model, i.e., supported or unsupported Pt nanoparticle, which is defined as follows:
| EADS = EAdsorbate/Catalyst − (ECatalyst + EAdsorbate) | (1) |
3 Results
3.1 Adsorption on isolated Pt nanoparticles
In this section, we present the adsorption energies for ethanol, atomic oxygen, and a CO molecule interacting with Pt nanoparticles of different sizes in several adsorption sites for (111) and (100) facets.Fig. 2 shows adsorption energies for each adsorbate in contact with a (111) facet for isolated Pt nanoparticles, with lower values of adsorption energies representing stronger interactions. For atomic oxygen, the observed adsorption site hierarchy is similar to that obtained in the literature with Pt nanoparticles.65,74–76 In comparison with the observations for extended surfaces in the literature,65,77,78 the most stable adsorption site changes from the FCC site on Pt(111) slabs to HCP and bridge sites near the vertices of the Pt nanoparticle facets. Moreover, the hierarchy obtained with our calculations for the Pt55 cluster is similar to that demonstrated by Han, Miranda, and Ceder74 with the adsorption energies ordered as follows: bridge-V < HCP-V < top-V < FCC-E < bridge-C < top-E. In addition to the adsorption site reordering, reducing the nanoparticle size strengthens the adsorption of oxygen on the nanoparticle surface, as previously demonstrated in the literature.42,79
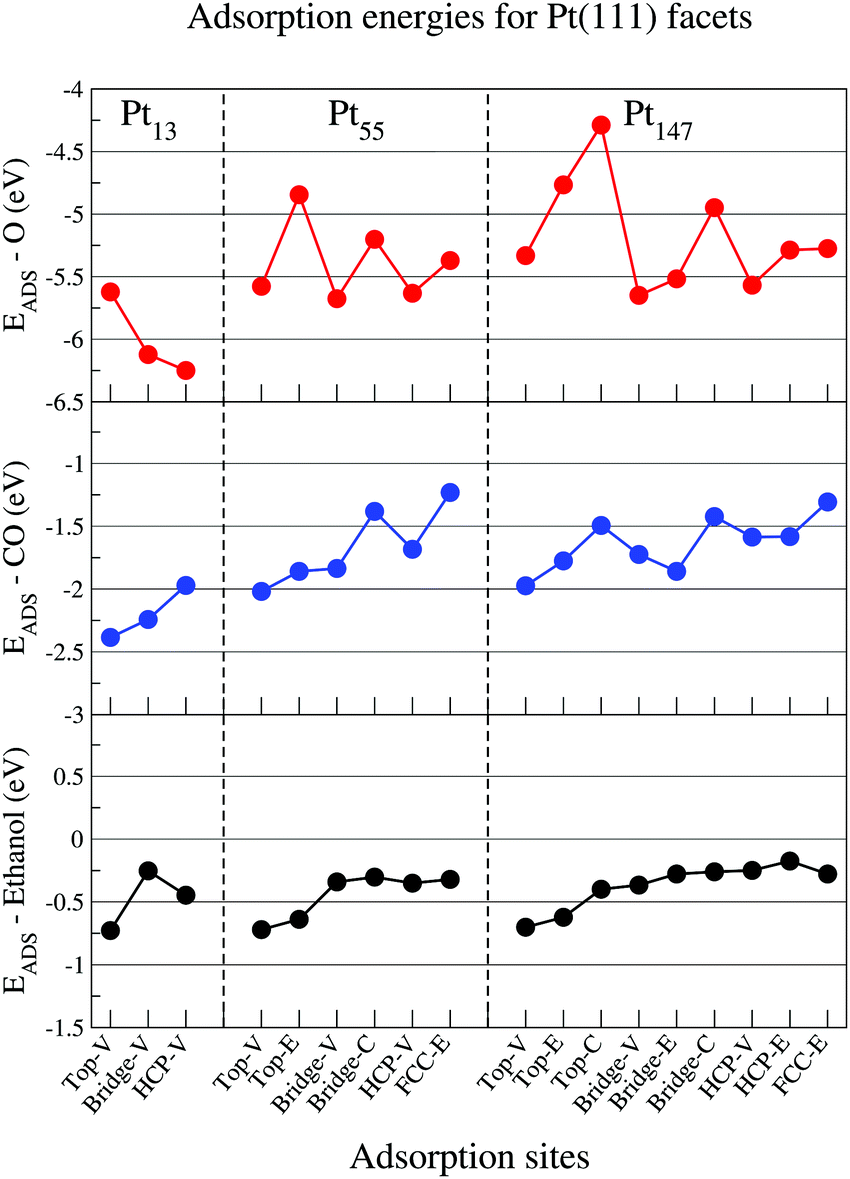 | ||
| Fig. 2 Adsorption energies for oxygen (red), carbon monoxide (blue), and ethanol (black) interacting with the (111) facet of Pt13, Pt55, and Pt147 on different adsorption sites. The letters V, E, and C represent adsorption sites in the vertex, edge and centre of a nanoparticle facet. | ||
For the CO molecule, top and bridge sites near the edges and vertices of the nanoparticle facet are the favourable adsorption sites. A similar adsorption site preference was observed for icosahedral Pt55 nanoparticles,60 when the adsorption energies were computed with a vdW-DF functional and with the Grimme DFT+D380 semiempirical approach. The CO/Pt(111) problem, which is the failure of DFT to reproduce the experimental result of top or bridge sites being preferred over hollow sites for a CO molecule interacting with a Pt(111),53 is not as evident in our results as it is for Pt slabs. In nanoparticles, the adsorption energies are stronger near the edges and vertices, making the adsorption energies on top-V, top-E, bridge-V and bridge-E stronger than that obtained for HCP sites. However, for Pt147, the adsorption energy for the HCP sites is stronger than that obtained for top-C and bridge-C sites, showing the discrepancy commonly obtained when comparing DFT results with experimental data for extended surfaces.53 For CO molecules, we also observed that reducing the nanoparticle size strengthened the interaction between CO and the nanoparticle surface, which was also observed by Li et al.42
In contrast to O and CO adsorption, when ethanol interacts with Pt nanoparticles of different sizes, the adsorption site hierarchy and the strength of the adsorption remains almost constant. As an example, the adsorption energy of ethanol on a top-V site only changed from −0.73 eV for a Pt13 to −0.70 eV for a Pt147. In comparison, for oxygen atoms on HCP-V and CO on top-V sites, we see differences in adsorption energies of about 0.68 eV and 0.41 eV when comparing the adsorption on a Pt13 and a Pt147. For all the studied sizes, ethanol adsorbs preferentially at top-V and top-E adsorption sites. For Bridge, HCP, and FCC sites, the adsorption is unstable, with Pt–O distances ranging from 2.8 Å to 3.1 Å. The preference for top sites in the adsorption of ethanol was also observed for Pt slabs,48,49 extended Pt(111) monolayers,47 Pt1350 and core–shell M@Pt nanoparticles.51
Fig. 3 shows adsorption energies for adsorbates in contact with the (100) facet of isolated Pt nanoparticles. Again, our adsorption site hierarchy for atomic oxygen is comparable with previous studies for Pt nanoparticles,74,75 showing a clear preference for bridge sites over top and hollow sites, and stronger adsorptions for sites near the vertices and edges of the nanoparticle. The same size effect is observed, with stronger adsorptions obtained for smaller nanoparticles.
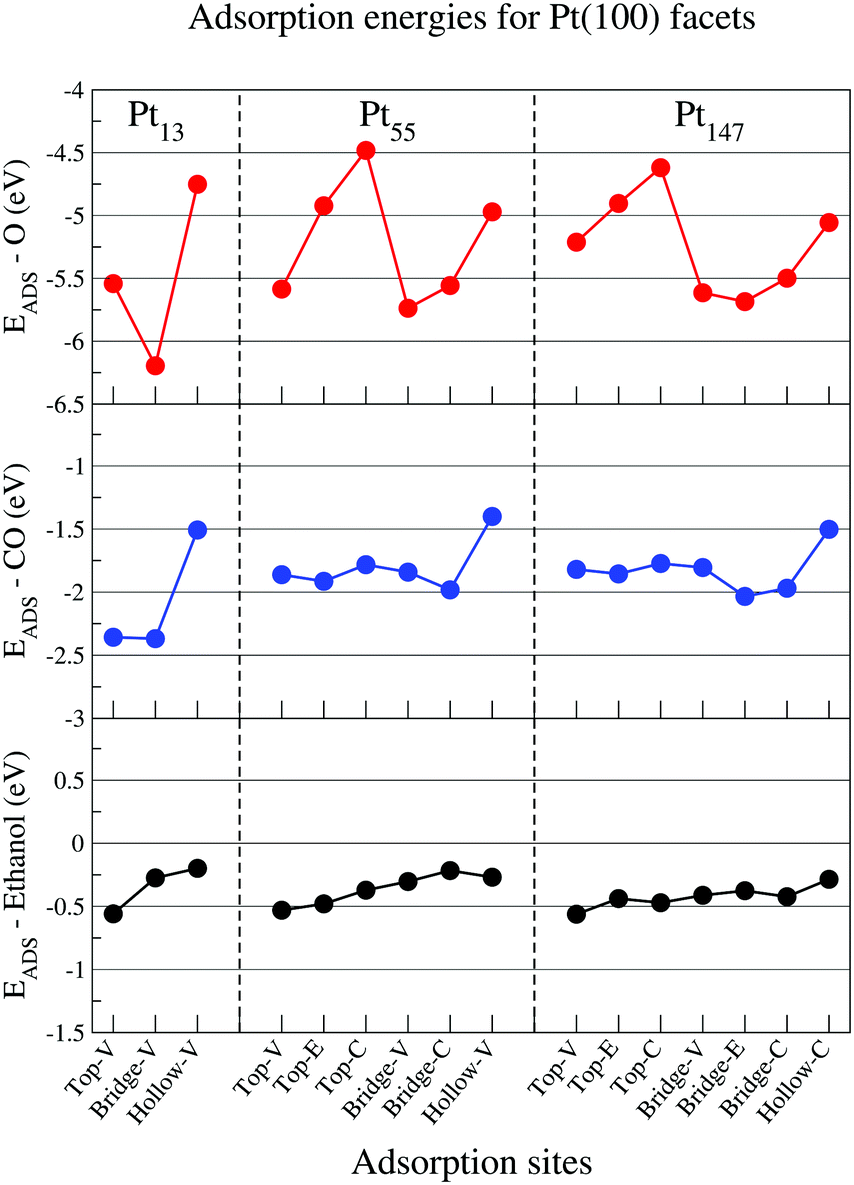 | ||
| Fig. 3 Adsorption energies for oxygen (red), carbon monoxide (blue), and ethanol (black) interacting with the (100) facet of Pt13, Pt55, and Pt147 on different adsorption sites. The letters V, E, and C represent adsorption sites in the vertex, edge and centre of a nanoparticle facet. | ||
For CO, there is a clear preference for bridge and top sites on (100) facets of Pt nanoparticles. The results for Pt55 and Pt147 are similar, showing weaker adsorptions as compared with Pt13. Moreover, most of the adsorption sites on a (100) facet show comparable adsorption energies with the most stable ones observed in Fig. 2. For ethanol molecules, the top adsorption sites still are preferred over the bridge and hollow sites. However, the adsorption energies obtained for top sites on Pt(100) facets are around −0.5 eV and considerably weaker than the −0.7 eV obtained for top sites on Pt(111). As observed for (111) facets, the size effects are weaker for ethanol than for atomic oxygen and CO.
Fajín et al.81 also observed almost constant values of adsorption energies for water molecules interacting with Pt nanoparticles of different sizes. In their work, the adsorption energy for a water molecule interacting with Ptn nanoparticles in the range 13 ≤ n ≤ 140 only changed from −0.58 eV to −0.47 eV, while for coadsorbed OH and H the adsorption energies varied from −0.90 eV to −0.41 eV.81 This effect was explained based on the changes in adsorption site preference of OH and H with nanoparticle size. Water molecules were adsorbed at on top sites for all the studied nanoparticles, while the most stable adsorption site for OH and H changed depending on the nanoparticle size. The authors concluded that the changes for adsorption energies of OH and H were not primarily related to size effects, but with the changes in the optimum adsorption site of OH and H for different nanoparticles.
In our case, we cannot use the explanation proposed by Fajín et al.81 to analyse the lack of size effects for ethanol adsorption energies. From our results, this effect is evident even when we compare a same adsorption site for all the adsorbed species interacting with different Pt nanoparticles. Therefore, it is necessary a more in-depth investigation of the structural and electronic changes induced by the interaction of the adsorbed species with Pt nanoparticles of different sizes to explain why size effects seem to be less important in the adsorption of ethanol.
3.2 Structural and electronic changes for adsorbates interacting with isolated Pt nanoparticles
So far we have investigated the adsorption site preference and the adsorption energies of O, CO and ethanol interacting with Pt nanoparticles of different sizes. Here, we study in more detail these effects by analysing structural and electronic changes happening in the nanoparticle and adsorbate after the interaction. We focus our attention on selected adsorption sites on (111) facets. As the size effects for the (100) facets are similar to those obtained for (111) facets, we expect that the conclusions drawn from our analysis will be transferable to the size effects trends for the (100) facets.Metallic nanoparticles present a large variety of adsorption sites when compared to extended surfaces. Moreover, as shown in Fig. 2 and 3 and in the literature,60,65,73,74,82,83 adsorption energies can dramatically vary when an adsorbate interacts with a different adsorption site. Recently, Calle-Vallejo73 proposed the generalized coordination number descriptor, which can encode the changes in adsorption energies caused by placing adsorbates on different adsorption sites. Coordination numbers are commonly used in chemistry to describe the ability of an atom to form a bond, where low coordinated sites are usually more capable of forming new bonds. The generalized coordination number accounts for changes in the coordination not only for the adsorption site, but also for its first neighbours which are weighted by their own coordination numbers as follows:
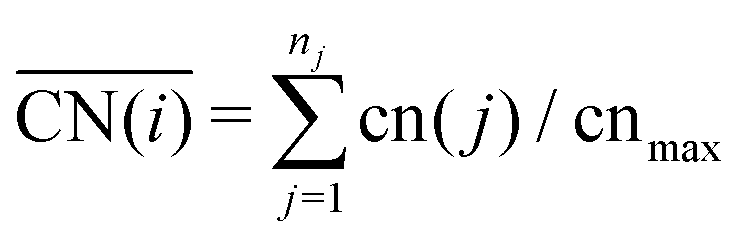 | (2) |
Fig. 4 shows the correlation between adsorption energies obtained for top sites placed on the (111) facet of different Pt nanoparticles and the calculated generalized coordination numbers. We are restraining our correlation to top sites on (111) facets to illustrate better how the nanoparticle size affects the coordination for the same type of adsorption site. Moreover, ethanol adsorption is more stable on top sites, while dispersion interactions dominate the adsorption on bridge and hollow sites, as evidenced by the large Pt–O distances, ranging from 2.8 Å to 3.1 Å and by the adsorption energy values obtained in our calculations without dispersion interactions on Fig. 5.
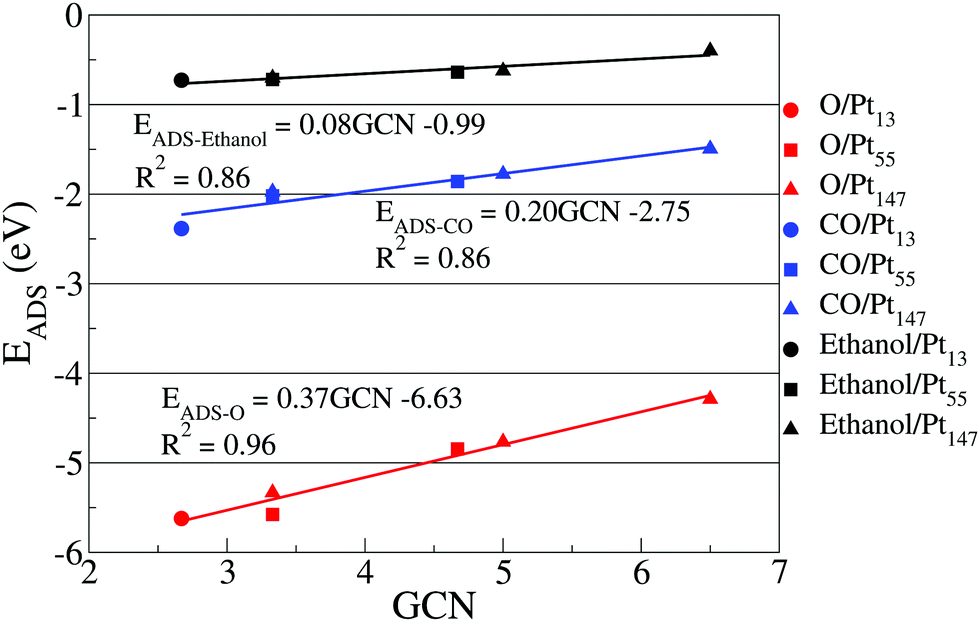 | ||
| Fig. 4 Adsorption energies for oxygen (red), carbon monoxide (blue), and ethanol (black) interacting with different top sites on (111) facets versus the generalized coordination number relative to each adsorption site. Circles, squares, and triangles represent Pt13, Pt55, and Pt147 nanoparticles. | ||
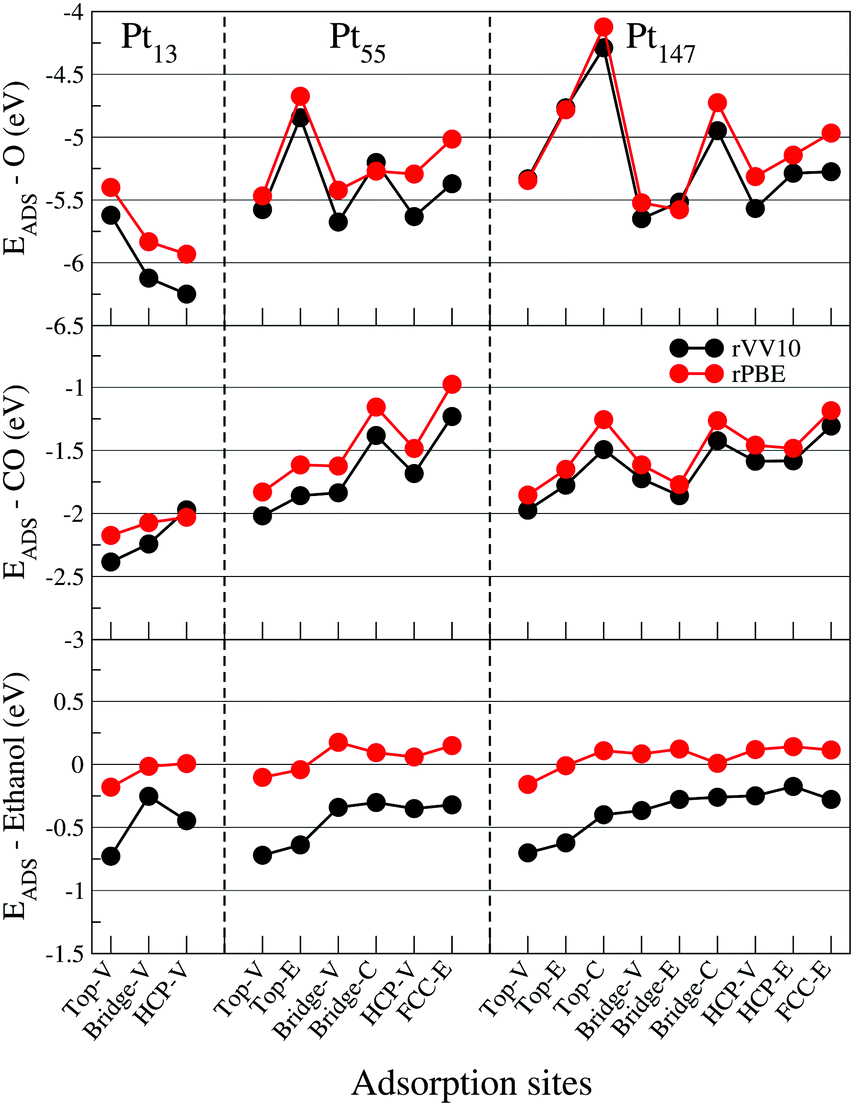 | ||
| Fig. 5 Adsorption energies for oxygen, carbon monoxide, and ethanol interacting with the (111) facet of Pt13, Pt55, and Pt147 on different adsorption sites. Red (black) circles are adsorption energies obtained with the rPBE (rVV10) functional. The letters V, E, and C represent adsorption sites in the vertex, edge and centre of a nanoparticle facet. | ||
The correlation between adsorption energies and GCN illustrates how size effects can alter the interactions of the nanoparticle and adsorbates by modifying the environment around similar adsorption sites. It shows that the ethanol adsorption is less affected by changes in the generalized coordination number as compared with O and CO adsorptions. The equations obtained through the linear correlation between adsorption energies and generalized coordination numbers also show a larger slope for the oxygen adsorption than for CO, which is different from the trends observed in a recent work by Jørgensen et al.,84 where the slopes for O and CO were respectively 0.22 eV and 0.25 eV.
The differences in the slopes can be associated to the different methodology for choosing the adsorption sites to obtain the linear correlations. In our work, we are using the same types of adsorption sites to obtain the correlations for each adsorbate. As discussed before, the motivation for that is showing the differences caused by size effects on a single type of adsorption site and not the differences between adsorption sites. Meanwhile, Jørgensen et al.84 obtained the correlation using the most stable adsorption sites for each adsorbate on four different slab models. For CO, the authors have used only top sites, while for O they have used bridge sites on Pt(100), Pt(110), and Pt(211) and an FCC site for Pt(111). As we are only using top sites in Fig. 4, only the slope for CO is comparable with Jørgensen et al.84
Similar plots containing the data from hollow and bridge sites can be found in the ESI,† together with a discussion about the changes in the slopes and correlation coefficients for each case. As an example, in the ESI,† we show that the slopes for ethanol adsorption on hollow and bridge sites are close to zero, helping to strengthen the argument that dispersion interactions dominate the ethanol adsorption in these sites.
Another effect of changing the nanoparticle size and shape are the changes in the ratio of different adsorption sites. Thus, finding relations between a purely geometrical descriptor, such as the generalized coordination number, and the adsorption energies for different adsorbates can greatly contribute for faster predictions about the efficiency of a given nanoparticle size and shape as a catalyst for a chemical reaction.82,83
The correlation between the Pt nanoparticle size effects and the level of coordination of an adsorption site is also compatible with the findings from Li et al..42 By studying size effects on Pt and Au nanoparticles ranging from 13 to 1415 atoms, the authors observed that the spacing in the d-band projected density of states near the Fermi level, caused by quantum size effects, only played a role for Au13 clusters. For Pt clusters, the discreteness in the d-band DOS was much smaller and the size effects observed for Pt were also explained in terms of the coordination numbers of adsorption sites and existence of nearest neighbours in edges and vertices of the nanoparticle facet.
We have also tested the effects of Pt–Pt bond length strain due to the size effects, calculating O, CO and ethanol adsorption on cuboctahedral nanoparticles created with Pt–Pt bond lengths similar to those obtained for experimental Pt bulk, with a lattice parameter of 3.92 Å.85,86 For all nanoparticle sizes and adsorbates, the effect of Pt nanoparticle deformation caused shifts in the adsorption energies of around 0.1 eV, showing that the contribution of this effect is small. These results are shown in more details in the ESI.†
Up to now, we have investigated the size effects for the adsorption of O, CO and ethanol on Pt nanoparticles, and encoded these results in terms of geometrical descriptors. It is clear, that the interactions of O and CO with Pt nanoparticles are more affected by size effects than the interaction between ethanol and Pt. This effect is also evident in the slope of the correlation between adsorption energies and generalized coordination numbers, showing that, despite some similarities, the bonding mechanism for ethanol on Pt surfaces differs from the one for O and CO.
To assess the importance of dispersion interactions for each adsorbate, we calculated the adsorption energies with the rPBE87 functional and compared the results with our calculations with the vdW functional rVV10.68,69Fig. 5 shows similar adsorption energy hierarchies obtained with rPBE and rVV10 for all adsorbates. Moreover, the dispersion interactions are much more important for ethanol adsorption than for O and CO, being one of the main contributions for stabilizing the ethanol molecule on Pt surfaces. This importance of the dispersion interactions for the ethanol adsorption was also observed by Tereschuck and Da Silva,48 where the ratio between the adsorption energies obtained with and without dispersion interaction was approximately 3.25 for ethanol molecules.
To obtain more information about how interactions between adsorbates and Pt surfaces occur, we show in Fig. 6 electronic density differences and Mulliken charge differences for each adsorbate interacting with Pt nanoparticles. For this analysis, we selected energetically favoured sites for each adsorbate, namely HCP-V adsorption sites for O and top-V for CO and ethanol.
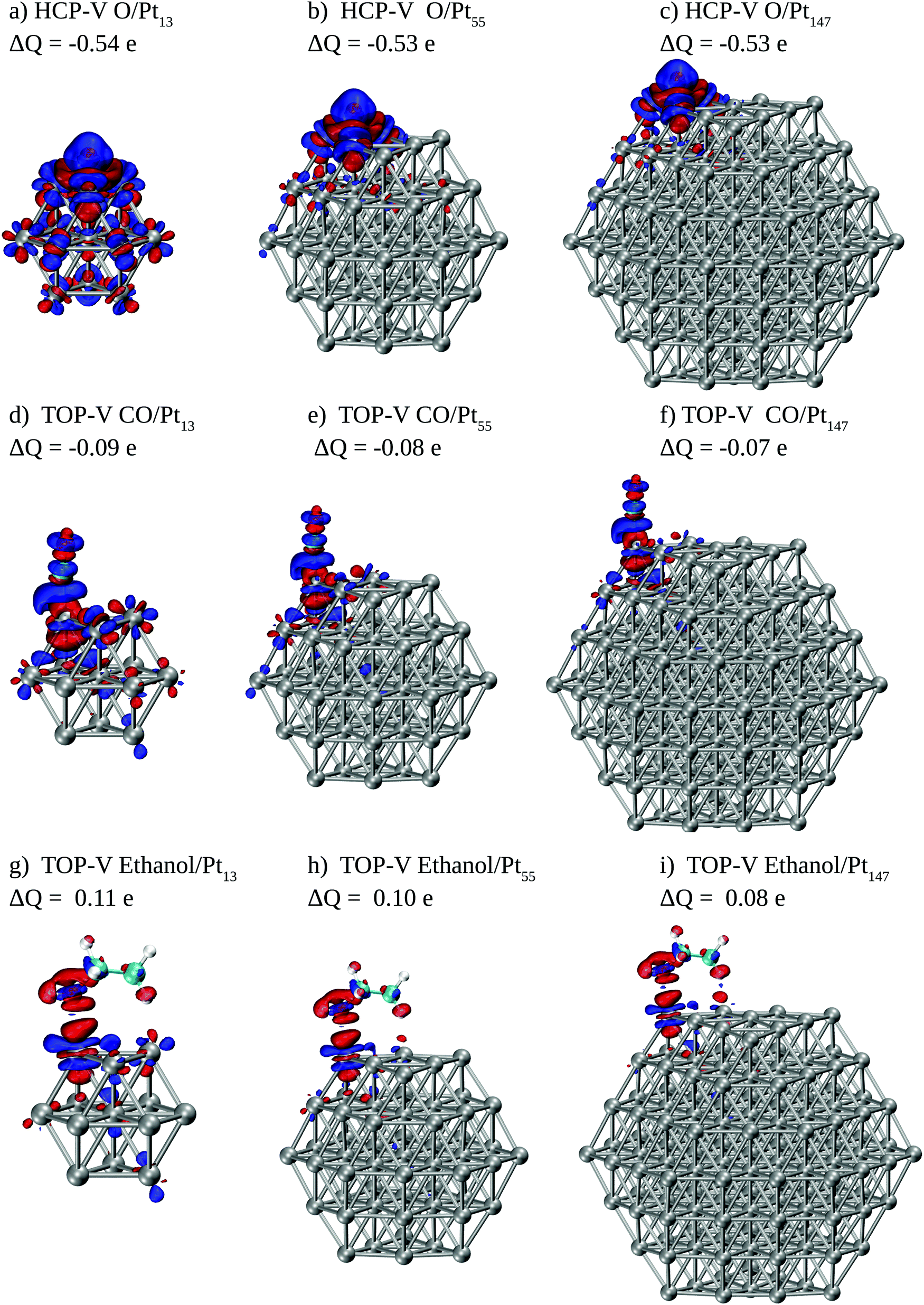 | ||
| Fig. 6 Electronic density differences plots for O, CO and ethanol interacting with Pt13, Pt55, and Pt147 nanoparticles (isosurface at 0.02 e Å−3), where blue (red) represents electron accumulation (depletion). The numbers associated with each plot are overall Mulliken charge differences calculated for each adsorbate, where a negative number represents adsorbate receiving electrons. We show the results for O interacting in the HCP-V adsorption site and CO and ethanol adsorbed on top-V sites. | ||
For O and CO we see significant electronic rearrangements, with Mulliken charges indicating that electrons flow from the metallic surface to the adsorbates. The overall electronic density rearrangement does not change with the Pt nanoparticle size. For the atomic oxygen adsorption, we observe that the electronic density change is more local for larger nanoparticles. Moreover, the electronic density rearrangement caused by the adsorption is slightly smaller for ethanol than that observed for CO, and considerably smaller as compared to the changes caused by the interaction with O. Regarding Mulliken charges, only two atoms in the ethanol molecule show considerable differences, the O atom in contact with the Pt surface and the H atom in the β C–H bond near the Pt surface. While the O atom loses electrons after the interaction, the H atom receives it, with an overall electron donation from ethanol to the Pt surface.
According to the d-band model,44,88,89 the interaction of an adsorbate and a metallic surface can be separated in two terms. The first is the interaction of the adsorbate states with the s–p metallic states, which should be similar for different metallic surfaces due to the delocalisation character of the metallic s–p states. The second term is the interaction of the metallic d-states and the valence states from the adsorbate. Differently from the s–p states, the d-band is localised and considered to be the main reason for differences in the adsorption energies when comparing different metallic surfaces. According to this model, the centre of the d-band can also be used as a descriptor for the adsorption energies.
Fig. 7 shows the density of states projected on the d-band of surface Pt atoms in the interacting region with the adsorbates. We show the density of states before and after the interaction with each adsorbate, using the adsorption sites presented on Fig. 6 for Pt13 and Pt147. The analysis of the changes caused in the d-band after interaction with an adsorbate can provide qualitative information about how the adsorption happens, helping to explain the lack of size effects observed in the ethanol adsorption.
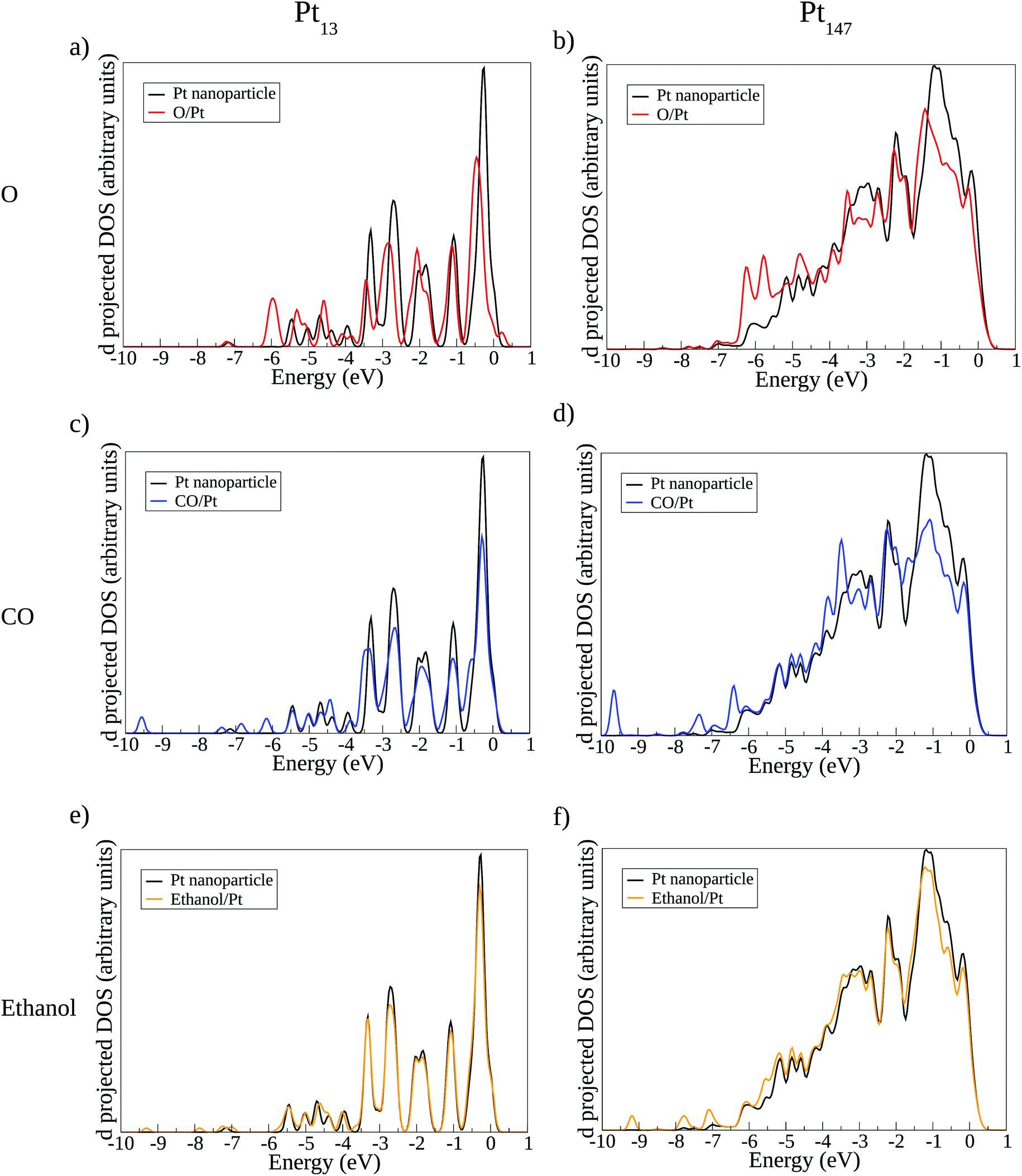 | ||
| Fig. 7 Local density of states projected on the d-band of surface Pt atoms in the interacting region of the (111) facet. (a, c and e) Show the density of states for Pt13 interacting with O, CO, and ethanol. (b, d and f) Show O, CO, and ethanol interacting with Pt147. | ||
Fig. 7(a–d) shows new peaks in the density of states in the Pt facet after the interaction with O and CO, indicating the appearence of overlapping orbitals at the adsorbate/Pt interface. Atomic oxygen induces extra peaks around −6 eV, showing the overlap of p oxygen states with d states from the Pt surface. For the CO adsorption, extra peaks are observed around −10 eV and between −7 eV and −6 eV, arising from the overlap between the Pt surface d band and CO orbitals. Other small peaks due to the interaction with the adsorbates are observed for lower values of energy. Such orbital overlaps were already observed and discussed in more details in previous theoretical studies, such as the work developed by Lynch and Hu.77
For ethanol adsorption, the changes in the d-band density of states are much smaller as compared with the changes caused by O and CO adsorption. The main differences in the density of states are the intensity of the peaks due to the electronic repopulation and the appearance of small peaks that are not changing the overall d-band DOS. The results from density of states show that the interaction between the d-band of the Pt surface with adsorbates states is much more relevant for O and CO adsorption than for ethanol. As changes in the d-band are consequences of altering the nanoparticle size and the coordination of the adsorption site, we should expect that the adsorbates with stronger interactions with the d-band would suffer more these effects, which helps to explain the small slopes found for ethanol adsorption as compared with O and CO in Fig. 4.
Thus, as a large amount of the ethanol adsorption energy arises from dispersion interactions, which are not largely affected by the Pt nanoparticle size, and as we see a small interaction between ethanol and d states from the Pt surface, it is understandable that nanoparticle size effects are less important for ethanol adsorption. Additionally, a recent work from Kakekhani et al.90 demonstrated that the covalent contributions can be much less important for the adsorption energies of closed-shell adsorbates such as water, alcohols, ammonia, on metallic surfaces than for unsaturated intermediates such as O, OH, CO. The authors show that the correlation slope between adsorption energies of water and O is small, helping to explain the small slope in the correlation between ethanol adsorption energies and descriptors such as the generalized coordination number or d-band centres, which can successfully describe the O adsorption.
Analysis of geometric changes in the adsorbate can also provide valuable information about the interaction and its effects. Table 1 shows Pt–O, Pt–C, β C–H, and C–O distances for adsorbates interacting with the nanoparticles. For atomic oxygen and carbon monoxide, we observe a small increase in the distances between the metallic surface and the interacting atom from the adsorbate as we increase the nanoparticle size. These changes in bond lengths agree with the weakening in adsorption energies previously observed. For adsorbed ethanol no substantial difference is observed in the Pt–O distances, which also agrees with the lack of size effects observed in the energetic profile.
| System | Pt–O (Å) |
|---|---|
| HCP-V O/Pt13(111) | 2.08 |
| HCP-V O/Pt147(111) | 2.10 |
| System | Pt–C (Å) | C–O (Å) |
|---|---|---|
| CO isolated | — | 1.14 |
| Top-V CO/Pt13(111) | 1.85 | 1.16 |
| Top-V CO/Pt147(111) | 1.89 | 1.16 |
| System | Pt–O (Å) | C–O (Å) | β C–H (Å) |
|---|---|---|---|
| Ethanol isolated | — | 1.45 | 1.10 |
| Top-V ethanol/Pt13(111) | 2.25 | 1.49 | 1.11 |
| Top-V ethanol/Pt147(111) | 2.24 | 1.50 | 1.12 |
For CO and ethanol adsorbed at Pt nanoparticles we also observe changes in the C–O bond lengths, increasing from 1.45 Å (1.14 Å) for isolated ethanol (CO) to up to 1.50 Å (1.16 Å). For the ethanol adsorption, most of the other internal bond lengths remain almost constant, with the β C–H bond, increasing up to 0.02 Å due to the interaction of the hydrogen atom with the nanoparticle surface as evidenced in Fig. 6, and with the C–C bond decreasing 0.01 Å. However, the changes for β C–H and C–C bonds are minimal when compared with the change in the C–O bond. We also found small increases in CCO and HOC angles as compared with the isolated ethanol molecule. A table with detailed structural data can be found in the ESI.†
3.3 O, CO, and ethanol interacting with Pt nanoparticles supported on graphene
After studying size effects on the adsorption of O, CO, and ethanol on Pt nanoparticles, we simulated the same Pt nanoparticles in contact with a pristine graphene sheet, aiming to describe how the interplay between support and nanoparticle size affects the adsorption properties of the Pt nanoparticles. Here, we only show the systems where the Pt nanoparticle interacts with the graphene support via the (111) facet, as this interaction is stronger than the one obtained when Pt(100) facet is in contact with the support.79 In the ESI,† we show some cases for O adsorption on nanoparticles supported on graphene via the (100) facet.Adsorption energies were calculated only for facets far from the support, aiming to assess effects of the support in the nanoparticle properties and avoid direct interactions between support and adsorbate. We reduced the number of calculated adsorption sites for Pt147 nanoparticles due to the high computational demand of such simulations and due to the reduced support effect with increasing system size previously observed.40,41
Fig. 8 illustrates the effect of the support by showing adsorption energies for different adsorption sites on a Pt(111) facet. In general, the presence of support weakens the interaction between adsorbates and the Pt surface. No meaningful change in the adsorption site preference is observed due to the support effects, which can also be seen through the correlation between adsorption energies and GCN for supported nanoparticles present in the ESI.† In all the simulated cases, the change in adsorption energy due to the pristine graphene support was smaller than 0.3 eV. This change is smaller than the differences observed for isolated Pt nanoparticles when comparing different adsorption sites, and smaller than the changes due to size effects for CO and O, but larger than the differences observed due to size effects for ethanol molecules. Similar trends were obtained for adsorbates interacting with the Pt(100) facet of supported nanoparticles, which can be found in the ESI.†
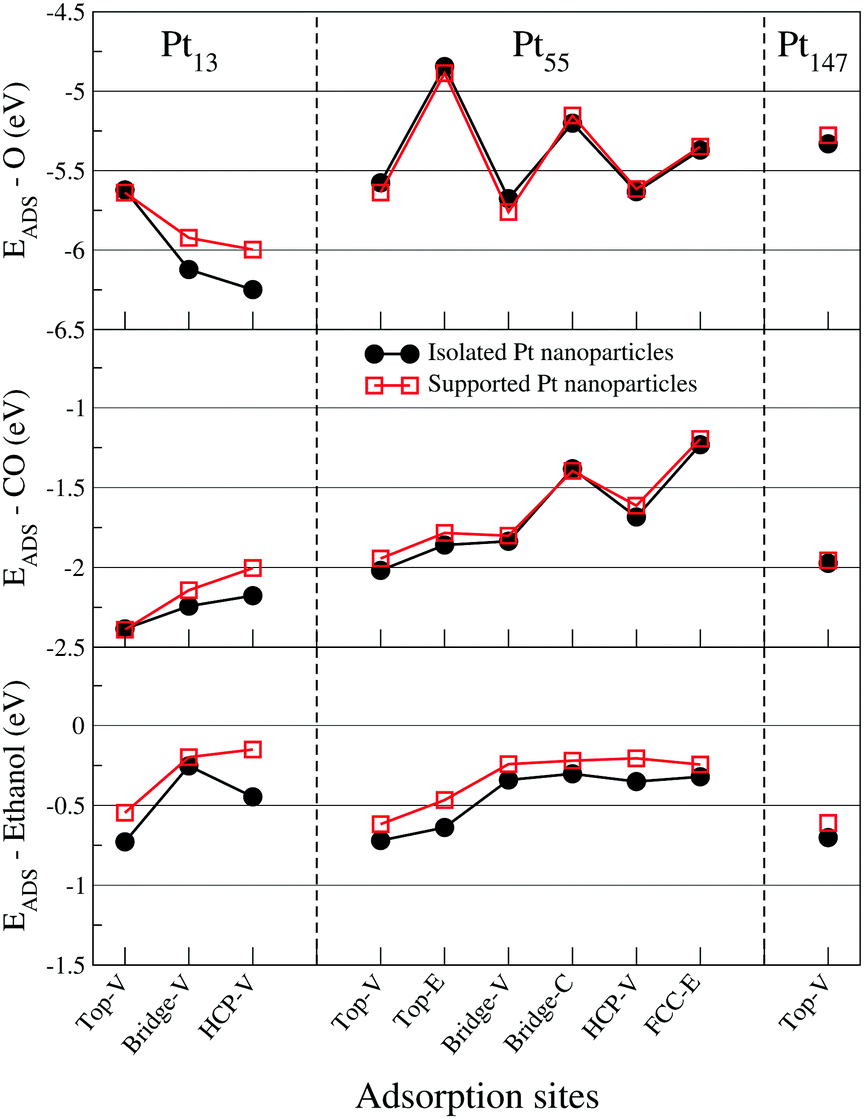 | ||
| Fig. 8 Adsorption energies for oxygen, carbon monoxide, and ethanol interacting with the (111) facet of Pt13, Pt55, and Pt147 on different adsorption sites. Black dots are adsorption energies obtained with isolated Pt nanoparticles, where red squares are the adsorption energies for Pt nanoparticles supported on graphene. The letters V, E, and C represent adsorption sites in the vertex, edge and centre of a nanoparticle facet. | ||
Similar effects were also observed by Lim and Wilcox32 for an O2 molecule, and by Fampiou and Ramasubramaniam31 for CO molecules interacting with Pt13/graphene systems. In both cases, the authors also included defects on the graphene support that enhanced the interaction between Pt and graphene and, consequently, the effects of the support in the interaction with the adsorbates. To analyse how the support effect alters the interaction between adsorbates and Pt nanoparticles, we show on Table 2 the main changes that the interaction with graphene induced on the studied nanoparticle facet. For the farthest Pt facet from the support, the interaction with graphene causes Pt–Pt bond length contractions, downshifts in the d-band centre and makes the Pt atoms more anionic.40
| ΔPt–Pt (Å) | ΔQ (e per atom) | ΔEd (eV) | |
|---|---|---|---|
| Pt13 | −0.02 | −0.047 | −0.08 |
| Pt55 | −0.01 | −0.033 | −0.01 |
| Pt147 | 0.00 | −0.015 | −0.01 |
The bond lengths contractions and d-band centre downshifts are associated with weaker adsorption energies and can be used to explain the support effect for O and CO. For ethanol, our previous results demonstrated that the Pt–Pt bond lengths and d-band centre changes induced by size effects are not able to change the ethanol adsorption energies to this extent. Recently, Seminovski et al.91,92 and Kakekhani et al.90 demonstrated that the ethanol adsorption is stronger on cationic adsorption sites and that in some cases the electrostatic effects can be dominant.90 Thus, a possible explanation for the support effect for ethanol adsorption can be the change in the atomic charges for the surface Pt atoms.
As previously observed,40,41 the interaction between Pt nanoparticles and the graphene support decreases with the Pt nanoparticle size. Moreover, as the Pt nanoparticle grows, more Pt layers are added between the interface Pt/graphene and the Pt facets far from the graphene. These effects decrease the graphene support effect on the adsorption properties of the Pt nanoparticle as we increase the Pt system size, being as small as 0.1 eV for Pt55 and Pt147. A similar decrease of support effect within the system size was also observed by Ramos-Sánchez and Balbuena93 for smaller Pt nanoparticles interacting with graphite supports. Furthermore, the small effect of the support for Pt facets far from the interface was recently observed even for metal oxide supports,94 which have much stronger interactions with Pt nanoparticle than pristine graphene. However, even with a weak interacting support, such as pristine graphene, we have managed to show that the support effects can be used to control the adsorption energies of ethanol, which is not strongly dependent on the nanoparticle size and shape. We also demonstrated that the support effect has a size dependency, showing that nanoparticle size and support need to be simultaneously controlled to design catalysts for specific reactions.
4 Conclusions
We performed DFT simulations to study how support and size can affect the adsorption strength of O, CO and ethanol on Pt surfaces. We computed Pt cuboctahedral nanoparticles free and supported on graphene, where we have simulated nanoparticles with up to 147 atoms interacting with graphene with up to 720 carbon atoms. We sampled adsorption sites in (111) and (100) Pt facets for all nanoparticles, obtaining adsorption sites hierarchies similar to the ones observed in the literature for all the adsorbates.The energetically favourable adsorption sites for atomic O were bridge and HCP sites near the edges of the nanoparticle facet. For the CO adsorption, bridge and top sites near the vertex of the nanoparticle presented stronger adsorption energies, while for ethanol top sites near the edge and vertex of the nanoparticle are the most stable adsorption sites. For O and CO, as the Pt nanoparticle size increases the adsorption energies weaken. The same effect was not observed when ethanol interacted with Pt nanoparticles. We encoded the effects of size and the multiplicity of adsorption sites via a geometrical descriptor, showing a linear correlation between the adsorption energies and the generalized coordination number, which controlled the adsorption strength of O and CO to a greater extent as compared with ethanol.
We analysed electronic density difference plots, charge differences obtained via Mulliken populations and densities of states projected on the d-band of interacting Pt atoms. The electronic redistributions due to the interaction with the adsorbate are much more local for ethanol than for O and CO. In the density of states plots, we observed the appearance of extra peaks related to overlapping states between the Pt surface and adsorbate for O and CO, while the ethanol interaction only induced small changes in the d-band peaks intensities. These characteristics of the ethanol interaction and our observations regarding the importance of dispersion interaction in the adsorption energies of ethanol on Pt surfaces helped to explain the smaller size effect observed for ethanol on Pt nanoparticles as compared with O and CO.
Finally, we observed weakenings in the adsorption energies of O, CO and ethanol due to support effects. We show that the weakening happens due to the Pt lattice deformation, d-band centre downshifts and electronic redistribution associated with interaction between Pt nanoparticle and support.40 The support effect is also size dependent, being only significant for Pt13 nanoparticles. The fact that the ethanol adsorption energies remained stable for different nanoparticle sizes, but were changed due to support effects show possible ways of tuning the adsorption energies for specific adsorbates. As atomic oxygen adsorption is widely used as a descriptor for several chemical reactions; CO is a key contaminant for Pt catalysts; and ethanol adsorption is the first step in the ethanol oxidation reaction and can be used as a descriptor for ethanol dehydrogenation, we expect that our results will be useful for rational nanocatalyst design, providing insights about the importance of controlling Pt nanoparticle size and support concurrently in order to develop better catalysts.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
L. G. V. acknowledges the support of Brazilian Government's Science Without Borders Programme (CNPQ: 206419/2014-7). We are grateful for computational support from the UK national high performance computing service, ARCHER, for which access was obtained via the UKCP consortium and funded by EPSRC grant ref EP/P022561/1. We are also grateful to the UK Materials and Molecular Modelling Hub for computational resources (Thomas supercomputer), which is partially funded by EPSRC (EP/P020194/1). We are also grateful for access to the University of Southampton supercomputers Iridis4 and Irirdis5.References
- O. Z. Sharaf and M. F. Orhan, Renewable Sustainable Energy Rev., 2014, 32, 810–853 CrossRef CAS.
- S. Song and P. Tsiakaras, Appl. Catal., B, 2006, 63, 187–193 CrossRef CAS.
- M. Kamarudin, S. Kamarudin, M. Masdar and W. Daud, Int. J. Hydrogen Energy, 2013, 38, 9438–9453 CrossRef CAS.
- D. Zanchet, J. B. O. Santos, S. Damyanova, J. M. R. Gallo and J. M. C. Bueno, ACS Catal., 2015, 5, 3841–3863 CrossRef CAS.
- M. Akhairi and S. Kamarudin, Int. J. Hydrogen Energy, 2016, 41, 4214–4228 CrossRef CAS.
- D. Li, X. Li and J. Gong, Chem. Rev., 2016, 116, 11529–11653 CrossRef CAS PubMed.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- M. T. M. Koper, Nanoscale, 2011, 3, 2054–2073 RSC.
- F. Baletto and R. Ferrando, Rev. Mod. Phys., 2005, 77, 371–423 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2016, 181, 298–313 CrossRef CAS.
- J. Perez, V. A. Paganin and E. Antolini, J. Electroanal. Chem., 2011, 654, 108–115 CrossRef CAS.
- F. Colmati, G. Tremiliosi-Filho, E. R. Gonzalez, A. Berna, E. Herrero and J. M. Feliu, Faraday Discuss., 2009, 140, 379–397 RSC.
- H.-F. Wang and Z.-P. Liu, J. Am. Chem. Soc., 2008, 130, 10996–11004 CrossRef CAS PubMed.
- G.-F. Wei and Z.-P. Liu, Phys. Chem. Chem. Phys., 2013, 15, 18555–18561 RSC.
- V. Viswanathan and F. Y.-F. Wang, Nanoscale, 2012, 4, 5110–5117 RSC.
- V. Tripković, I. Cerri, T. Bligaard and J. Rossmeisl, Catal. Lett., 2014, 144, 380–388 CrossRef.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS PubMed.
- S. Proch, M. Wirth, H. S. White and S. L. Anderson, J. Am. Chem. Soc., 2013, 135, 3073–3086 CrossRef CAS PubMed.
- R. Siburian, T. Kondo and J. Nakamura, J. Phys. Chem. C, 2013, 117, 3635–3645 CrossRef CAS.
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089–13095 CrossRef CAS PubMed.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Coord. Chem. Rev., 2016, 312, 99–148 CrossRef CAS.
- X. Xia, G. Jones, M. Sarwar, Q. Tang, I. Harkness and D. Thompsett, J. Mater. Chem. A, 2015, 3, 24504–24511 RSC.
- X. Xia, J. L. R. Yates, G. Jones, M. Sarwar, I. Harkness and D. Thompsett, J. Mater. Chem. A, 2016, 4, 15181–15188 RSC.
- J. L. R. Yates, G. H. Spikes and G. Jones, Phys. Chem. Chem. Phys., 2015, 17, 4250–4258 RSC.
- K. Miecznikowski and P. J. Kulesza, J. Power Sources, 2011, 196, 2595–2601 CrossRef CAS.
- B. H. Morrow and A. Striolo, Nanotechnology, 2008, 19, 195711 CrossRef PubMed.
- X. Liu, Y. Han, J. W. Evans, A. K. Engstfeld, R. J. Behm, M. C. Tringides, M. Hupalo, H.-Q. Lin, L. Huang, K.-M. Ho, D. Appy, P. A. Thiel and C.-Z. Wang, Prog. Surf. Sci., 2015, 90, 397–443 CrossRef CAS.
- P. Błoński and J. Hafner, J. Chem. Phys., 2011, 134, 154705 CrossRef PubMed.
- T. Daio, A. Staykov, L. Guo, J. Liu, M. Tanaka, M. S. Lyth and K. Sasaki, Sci. Rep., 2015, 5, 13126 CrossRef CAS PubMed.
- I. Fampiou and A. Ramasubramaniam, J. Phys. Chem. C, 2012, 116, 6543–6555 CrossRef CAS.
- I. Fampiou and A. Ramasubramaniam, J. Phys. Chem. C, 2013, 117, 19927–19933 CrossRef CAS.
- D.-H. Lim and J. Wilcox, J. Phys. Chem. C, 2011, 115, 22742–22747 CrossRef CAS.
- A. Maiti and A. Ricca, Chem. Phys. Lett., 2004, 395, 7–11 CrossRef CAS.
- Y. Okamoto, Chem. Phys. Lett., 2006, 420, 382–386 CrossRef CAS.
- W. B. Schneider, U. Benedikt and A. A. Auer, ChemPhysChem, 2013, 14, 2984–2989 CrossRef CAS PubMed.
- G. Ramos-Sanchez and P. B. Balbuena, Phys. Chem. Chem. Phys., 2013, 15, 11950–11959 RSC.
- C. F. Sanz-Navarro, P.-O. Åstrand, D. Chen, M. Rønning, A. C. T. van Duin, T. Jacob and W. A. Goddard, J. Phys. Chem. A, 2008, 112, 1392–1402 CrossRef CAS PubMed.
- K. Okazaki-Maeda, Y. Morikawa, S. Tanaka and M. Kohyama, Surf. Sci., 2010, 604, 144–154 CrossRef CAS.
- Q. Qi, H. Liu, W. Feng, H. Tian, H. Xu and X. Huang, Comput. Mater. Sci., 2015, 96(part A), 268–276 CrossRef CAS.
- L. G. Verga, J. Aarons, M. Sarwar, D. Thompsett, A. E. Russell and C.-K. Skylaris, Phys. Chem. Chem. Phys., 2016, 18, 32713–32722 RSC.
- H. Shi, S. M. Auerbach and A. Ramasubramaniam, J. Phys. Chem. C, 2016, 120, 11899–11909 CrossRef CAS.
- L. Li, A. H. Larsen, N. A. Romero, V. A. Morozov, C. Glinsvad, F. Abild-Pedersen, J. Greeley, K. W. Jacobsen and J. K. Nørskov, J. Phys. Lett., 2013, 4, 222–226 CAS.
- F. Calle-Vallejo, P. Sautet and D. Loffreda, J. Phys. Lett., 2014, 5, 3120–3124 CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. E. Sutton and D. G. Vlachos, Ind. Eng. Chem. Res., 2015, 54, 4213–4225 CrossRef CAS.
- H. Li and G. Henkelman, J. Phys. Chem. C, 2017, 121, 27504–27510 CrossRef CAS.
- A. O. Pereira and C. R. Miranda, Appl. Surf. Sci., 2014, 288, 564–571 CrossRef CAS.
- P. Tereshchuk and J. L. F. Da Silva, J. Phys. Chem. C, 2012, 116, 24695–24705 CrossRef CAS.
- R. L. H. Freire, A. Kiejna and J. L. F. Da Silva, Phys. Chem. Chem. Phys., 2016, 18, 29526–29536 RSC.
- L. Zibordi-Besse, P. Tereshchuk, A. S. Chaves and J. L. F. Da Silva, J. Phys. Chem. A, 2016, 120, 4231–4240 CrossRef CAS PubMed.
- V. A. Rigo, C. R. Miranda and F. Baletto, 2018, arXiv:1804.07631.
- W. Liu, A. Tkatchenko and M. Scheffler, Acc. Chem. Res., 2014, 47, 3369–3377 CrossRef CAS PubMed.
- P. J. Feibelman, B. Hammer, J. K. Nørskov, F. Wagner, M. Scheffler, R. Stumpf, R. Watwe and J. Dumesic, J. Phys. Chem. B, 2001, 105, 4018–4025 CrossRef CAS.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS.
- G. Kresse, A. Gil and P. Sautet, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 073401 CrossRef.
- A. Gil, A. Clotet, J. M. Ricart, G. Kresse, M. García-Hernández, N. Rösch and P. Sautet, Surf. Sci., 2003, 530, 71–87 CrossRef CAS.
- I. Grinberg, Y. Yourdshahyan and A. M. Rappe, J. Chem. Phys., 2002, 117, 2264–2270 CrossRef CAS.
- A. Stroppa, K. Termentzidis, J. Paier, G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 195440 CrossRef.
- A. Stroppa and G. Kresse, New J. Phys., 2008, 10, 063020 CrossRef.
- J. B. A. Davis, F. Baletto and R. L. Johnston, J. Phys. Chem. A, 2015, 119, 9703–9709 CrossRef CAS PubMed.
- S. Luo, Y. Zhao and D. G. Truhlar, J. Phys. Lett., 2012, 3, 2975–2979 CAS.
- P. Janthon, F. Viñes, J. Sirijaraensre, J. Limtrakul and F. Illas, J. Phys. Chem. C, 2017, 121, 3970–3977 CrossRef CAS.
- Y.-W. Huang and S.-L. Lee, Chem. Phys. Lett., 2010, 492, 98–102 CrossRef CAS.
- T. M. Soini, A. Genest and N. Rösch, J. Phys. Chem. A, 2015, 119, 4051–4056 CrossRef CAS PubMed.
- G. Peng and M. Mavrikakis, Nano Lett., 2015, 15, 629–634 CrossRef CAS PubMed.
- C.-K. Skylaris, P. D. Haynes, A. A. Mostofi and M. C. Payne, J. Chem. Phys., 2005, 122, 084119 CrossRef PubMed.
- A. Ruiz-Serrano and C.-K. Skylaris, J. Chem. Phys., 2013, 139, 054107 CrossRef PubMed.
- O. A. Vydrov and T. Van Voorhis, J. Chem. Phys., 2010, 133, 244103 CrossRef PubMed.
- R. Sabatini, T. Gorni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 041108 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- C.-K. Skylaris, A. A. Mostofi, P. D. Haynes, C. J. Pickard and M. C. Payne, Comput. Phys. Commun., 2001, 140, 315–322 CrossRef CAS.
- A. A. Mostofi, P. D. Haynes, C.-K. Skylaris and M. C. Payne, J. Chem. Phys., 2003, 119, 8842 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, P. Sautet and D. Loffreda, Angew. Chem., Int. Ed., 2014, 53, 8316–8319 CrossRef CAS PubMed.
- B. C. Han, C. R. Miranda and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075410 CrossRef.
- L. Wang, A. Roudgar and M. Eikerling, J. Phys. Chem. C, 2009, 113, 17989–17996 CrossRef CAS.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and R. L. Johnston, Phys. Chem. Chem. Phys., 2014, 16, 26539–26545 RSC.
- M. Lynch and P. Hu, Surf. Sci., 2000, 458, 1–14 CrossRef CAS.
- Z. Gu and P. B. Balbuena, J. Phys. Chem. C, 2007, 111, 9877–9883 CrossRef CAS.
- L. G. Verga, J. Aarons, M. Sarwar, D. Thompsett, A. E. Russell and C.-K. Skylaris, Faraday Discuss., 2018, 208, 497–522 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. L. C. Fajín, A. Bruix, M. N. D. S. Cordeiro, J. R. B. Gomes and F. Illas, J. Chem. Phys., 2012, 137, 034701 CrossRef PubMed.
- J. Aarons, L. Jones, A. Varambhia, K. E. MacArthur, D. Ozkaya, M. Sarwar, C.-K. Skylaris and P. D. Nellist, Nano Lett., 2017, 17, 4003–4012 CrossRef CAS PubMed.
- T. Ellaby, J. Aarons, A. Varambhia, L. Jones, P. Nellist, D. Ozkaya, M. Sarwar, D. Thompsett and C.-K. Skylaris, J. Phys.: Condens. Matter, 2018, 30, 155301 CrossRef PubMed.
- M. Jørgensen and H. Grönbeck, ACS Catal., 2017, 7, 5054–5061 CrossRef.
- J. W. Arblaster, Platinum Met. Rev., 1997, 41, 12–21 CAS.
- J. W. Arblaster, Platinum Met. Rev., 2006, 50, 118–119 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- B. Hammer and J. Nørskov, Impact of Surface Science on Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- A. Kakekhani, L. T. Roling, A. Kulkarni, A. A. Latimer, H. Abroshan, J. Schumann, H. AlJama, S. Siahrostami, S. Ismail-Beigi, F. Abild-Pedersen and J. K. Nørskov, Inorg. Chem., 2018, 57, 7222–7238 CrossRef CAS PubMed.
- Y. Seminovski, R. C. Amaral, P. Tereshchuk and J. L. D. Silva, Surf. Sci., 2018, 667, 84–91 CrossRef CAS.
- Y. Seminovski, P. Tereshchuk, A. Kiejna and J. L. F. Da Silva, J. Chem. Phys., 2016, 145, 124709 CrossRef PubMed.
- G. Ramos-Sánchez and P. Balbuena, J. Electroanal. Chem., 2014, 716, 23–30 CrossRef.
- A. Moradabadi, S. Ahmadi and P. Kaghazchi, Nanoscale, 2017, 9, 4478–4485 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp04798g |
| This journal is © the Owner Societies 2018 |