
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nuclear spin singlet states as magnetic on/off probes in self-assembling systems
Salvatore
Mamone
ab and
Stefan
Glöggler
 *ab
*ab
aMax Planck Institute for Biophysical Chemistry, Am Faßberg 11, 37077 Göttingen, Germany. E-mail: stefan.gloeggler@mpibpc.mpg.de
bCenter for Biostructural Imaging of Neurodegeneration of UMG, Von-Siebold-Straße 3A, 37075 Göttingen, Germany
First published on 14th August 2018
Abstract
Self-assembling processes occur in a variety of compounds such as peptides, proteins and DNA. These processes have been linked to pathologies and have as well been exploited for designing responsive contrast agents for disease detection. Novel methods to investigate and detect self-assembly therefore hold promise to obtain more insights into disease progression or open pathways to the design of novel self-assembling materials. In this article we are introducing nuclear singlet states to probe self-assembly in the dipeptide isoleucine–phenylalanine (IF) as a thermoresponsive on/off switch for nuclear magnetic resonance (NMR). We have investigated the relaxation and singlet state properties of the β-protons of phenylalanine in the IF dipeptide in aqueous solutions. At IF concentrations of 2 wt% and above 308 K, a long lived nuclear singlet state, as compared to the longitudinal relaxation, was observed. At 308 K the dipeptide starts forming a gel and no singlet state is accessible at lower temperatures. Upon heating, the gel disassembles and an isotropic liquid forms making the singlet state accessible again. This demonstrates the thermoresponsive on–off character of the nuclear spin singlet state in the IF dipeptide.
1 Introduction
In a self-assembly process, various individual components arrange themselves into an ordered structure.1 Examples of materials that self-assemble include DNA, peptides, proteins, nanocrystals and nanoparticles.1–27 Triggered self-assembly of nanostructures or disassembly of these materials has been investigated in recent years for the design of magnetic resonance imaging (MRI) contrast agents.18–27 In particular, labelling self-assembling molecules with 19F nuclei has moved into the focus.23–27 Thereby, two concepts have been pursued.26,27 Firstly, hydrophilic molecules that have a reactive species are attached to a hydrophobic group containing fluorine and self-assemble into a nanostructure with the fluorine nuclei in the core. Interaction of the reactive group with e.g. an enzyme leads to disassembly of the nanostructure. Secondly, the reactive group in the described molecules is blocked but gets revealed and regains its reactivity upon a stimulus, subsequently leading to a self-assembled structure. Both processes can be observed via19F-NMR.26,27 If the molecule is in its disassembled state, short correlation times and isotropic molecular motion lead to a narrow observable peak in the 19F-NMR spectrum. Once self-assembled, the 19F-nuclei (in the core of the particle) are restricted in their motion and experience increased dipolar interactions and chemical shift anisotropy (CSA), leading to a broadening of the observable peak until it is not detectable anymore. In other words, the spin–spin relaxation time T2 becomes shorter in the self-assembled structure to the point that the NMR signal is not detectable anymore, hence resulting in an on/off switch depending on the assembly state. As an alternative that does not require labelling with 19F, we have explored on/off-switches for proton nuclear singlet states. Singlet states are nuclear spin states with effective spin 0 that can be formed between a pair of spin 1/2 nuclei which are dynamically isolated from the rest of the spin system.28–61 Interestingly, singlet states are immune to the direct dipole–dipole relaxation between the constituent spins in fast and isotropically tumbling molecules, which is often the main relaxation mechanism for the longitudinal and transverse magnetization modes. As such, singlet states are often characterized by singlet–triplet equilibration times Ts that exceed the spin–lattice relaxation times T1. Although the longest singlet lifetimes (over 1 hour),44 were found between spin pairs close to magnetic equivalence in strongly coupled systems, singlet states can be generated for systems in which the nuclei are far from magnetic equivalence and weakly coupled,28 for example in α-protons of glycine in small peptides and β-protons in amino acids and peptides.33,34,38 This observation has e.g. been explored in the development of a detection method for binding affinity, which has proven to be more sensitive than T1-based methods.38In this work we have explored the feasibility of utilizing nuclear singlet states as on/off switches and demonstrate that such a behaviour can be observed in β-protons of the dipeptide isoleucine–phenylalanine (IF), which is shown in Fig. 1 together with the respective NMR spectrum of the two protons of interest. For investigating the nuclear singlet state in H2O, we used a singlet filter sequence with water suppression. The sequence can be applied to weakly coupled spin systems (as it is the case here) and includes an extension of the previously published T00-filter.61 We show that the dipeptide acts as a thermoresponsive switch for singlet states, which can be populated (turned on) at a temperature of 308 K and are not accessible below this temperature (turned off). This is due to the fact that below the critical temperature a hydrogel62 is formed in which the correlation time is significantly increased. Our investigations on this molecule and its behaviour are presented in the following.
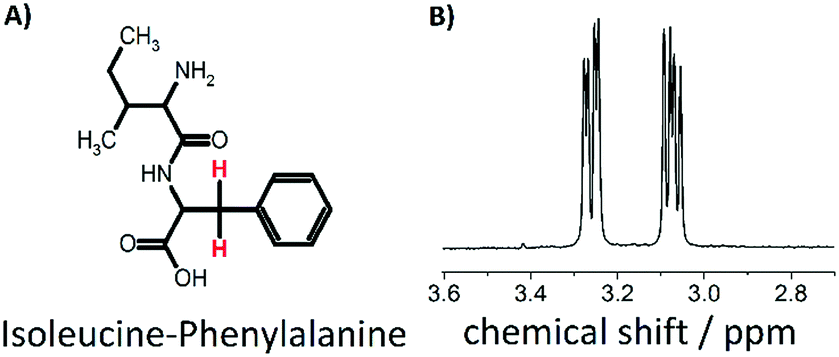 | ||
| Fig. 1 (A) IF dipeptide (B) 1H NMR spectrum of the two beta protons of F. | ||
2 Experimental section
The dipeptide isoleucine–phenylalanine was purchased from Bachem (product number 4001668.0001) and used without further purification. Two degassed NMR samples were prepared with either 0.5 wt% or 2 wt% dipeptide with respect to a mixture of 95% deionized H2O and 5% D2O. NMR experiments were performed on a AV600 HDIII Bruker system with variable temperature setup at 600 MHz proton frequency (corresponds to B0 = 14.1 T). To estimate the longitudinal relaxation times T1, an inversion recovery experiment was performed at different temperatures from 323 K to 283 K. TS was measured at the same temperatures with the recently introduced APSOC sequence including an extra PE-WATERGATE63 block for water suppression in the 1H NMR spectrum. Furthermore, we have added a singlet filter (T00) that can be utilized for weakly coupled spin systems. The whole sequence is shown in Fig. 2 with the respective timings. Compared to the T00 filter reported earlier,61 three 180 degree pulses have been added in order to remove the effect of offset evolution, and timings τr close to τ* = [J2 + Δν2]−0.5, which corresponds to a full rotation in the zero-quantum space, have been used. Here J and Δν represent the J-coupling and the chemical shift difference between the two nuclei measured in Hz, respectively. Further details of the T00 filter will be published elsewhere. Spin–spin relaxation times T2 were measured utilizing a CPMG sequence for both samples. CPMG echo times were 0.4 ms for the 0.5 wt% sample and for the 2 wt% sample above 308 K and 0.2 ms for the 2 wt% sample below 308 K.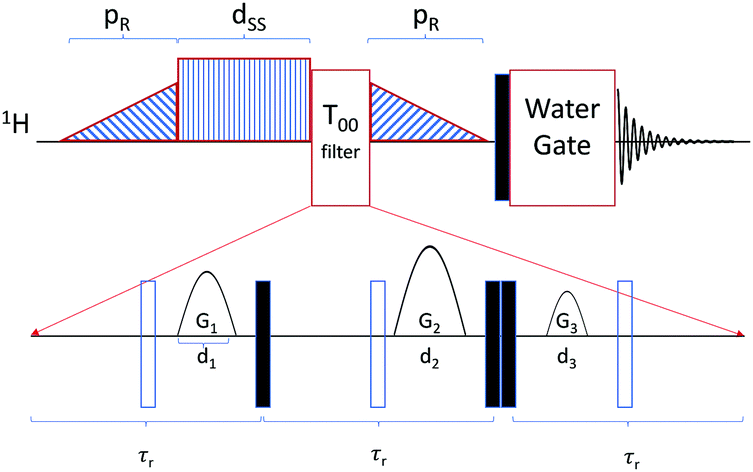 | ||
| Fig. 2 Schematic of the APSOC sequence used in this work. The filled and empty rectangles represent π/2 and π pulses, respectively. The sequence consists of a ramped RF passages with duration of pR = 200 ms and maximum RF field corresponding to a nutation frequency of 600 Hz which converts z-magnetization into the singlet state. The offset of the singlet excitation and reconversion ramps was ±23 Hz with respect to the resonances of the beta protons of the phenylalanine residue. The singlet sustaining block consists of a CW RF field with nutation frequency 1 kHz in order to quench chemical shifts induced oscillations in the singlet state which is turned on for the duration dSS. The T00 filter greatly reduces any signal orthogonal to the singlet state before it is converted back to z-magnetization by the ramped down RF passage. The acquisition block consists of a hard π/2 pulse, with nutation frequency of 25 kHz, followed by a PE-WATERGATE.63 In the T00 block, the duration τr of the spin echoes were 8 ms, the durations of the gradients d1 = d2 = d3 = 2 ms and the gradients were {G1, G2, G3} = {2, 27.5, 7} in G cm−1. | ||
3 Results and discussion
3.1 Evaluation of the singlet filter
For the investigation of nuclear singlet states in a weakly coupled spin system (as it is the case in the IF peptide), we introduce a filter that is offset independent. The rational for the development is based on the idea that typically proton background signals occur in biological environments. Regarding the background signals, water is the most dominant, which is depicted in Fig. 3A. The concentration of water (≈55 M) often exceeds physiological metabolite concentrations by more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold.64 This may lead to masking of the desired signal to be observed. In order to increase the detectability from low-concentration component, water suppression techniques have been introduced for in vitro an in vivo experiments.63,65–67 To demonstrate the effect of a water suppression sequence on the investigated system we have performed a PE-WATERGATE experiment, that reduces the water signal by more than 1000-fold and the result is shown in Fig. 3B.63 From the spectrum it becomes evident that the water signal has significantly been suppressed but other 1H signals from the dipeptide are still observable. Under physiological conditions in e.g. cells or in vivo many more 1H signals may be present that may mask the signal of interest. In order to remove them we introduced a T00-filter for weakly coupled spin systems (see Fig. 2) that, in combination with WATERGATE, suppresses other proton signals than the desired one and also manages to suppress the water signal even further. The result is shown in Fig. 3C. This follows an idea presented in ref. 41 with the advantage that the T00-filter presented here is offset independent and may be combined with offset independent singlet NMR sequences such as the M2S–S2M sequence for imaging purposes.32
000-fold.64 This may lead to masking of the desired signal to be observed. In order to increase the detectability from low-concentration component, water suppression techniques have been introduced for in vitro an in vivo experiments.63,65–67 To demonstrate the effect of a water suppression sequence on the investigated system we have performed a PE-WATERGATE experiment, that reduces the water signal by more than 1000-fold and the result is shown in Fig. 3B.63 From the spectrum it becomes evident that the water signal has significantly been suppressed but other 1H signals from the dipeptide are still observable. Under physiological conditions in e.g. cells or in vivo many more 1H signals may be present that may mask the signal of interest. In order to remove them we introduced a T00-filter for weakly coupled spin systems (see Fig. 2) that, in combination with WATERGATE, suppresses other proton signals than the desired one and also manages to suppress the water signal even further. The result is shown in Fig. 3C. This follows an idea presented in ref. 41 with the advantage that the T00-filter presented here is offset independent and may be combined with offset independent singlet NMR sequences such as the M2S–S2M sequence for imaging purposes.32
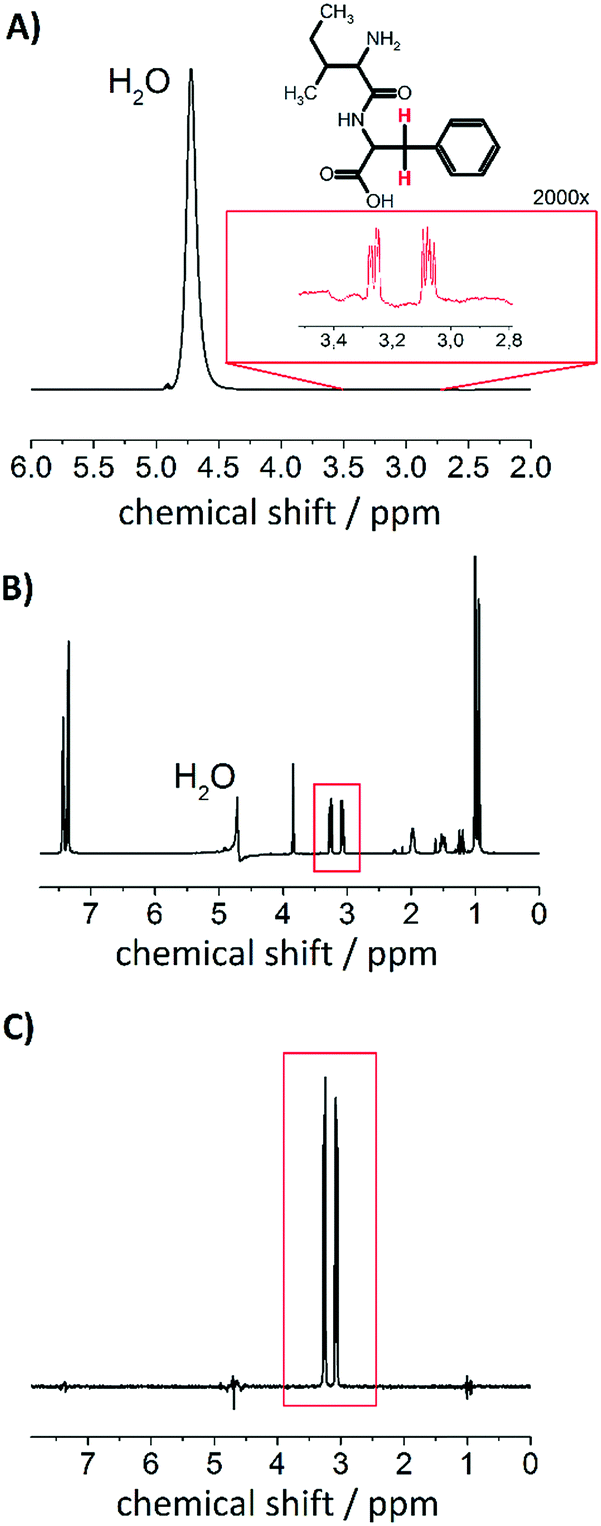 | ||
| Fig. 3 Effect of singlet filter. (A) Single scan 1H NMR spectrum of 0.5 wt% (16 mM) IF dipeptide dissolved in 95% H2O and 5% D2O (B0 = 14.1 T). The inset shows the proton signal of the two indicated protons with 2000-fold magnification. (B) 1H NMR WATERGATE spectrum of the same sample (8 scans). (C) 1H NMR after applying the singlet filter for weakly coupled spin systems with dss = 200 ms of singlet sustaining duration after the first APSOC ramp (32 scans). | ||
3.2 Investigation of the dipeptide isoleucine–phenylalanine (IF)
One of the IF properties includes the reversible gelling upon temperature changes62 above a critical concentration. If an IF sample with 2 wt% in water is investigated the peptide starts forming a gel at 303 K. This process can be monitored with NMR by observing a line broadening that occurs around this temperature.62Fig. 4(A) shows the spectra at 313 K at which the dipeptide is in its monomeric form with isotropic movement leading to narrow lines and after the gel has formed at 293 K in 4B with broadened lines. In case of a 0.5 wt% sample, no gelification occurs and the dipeptides keep up the isotropic movement.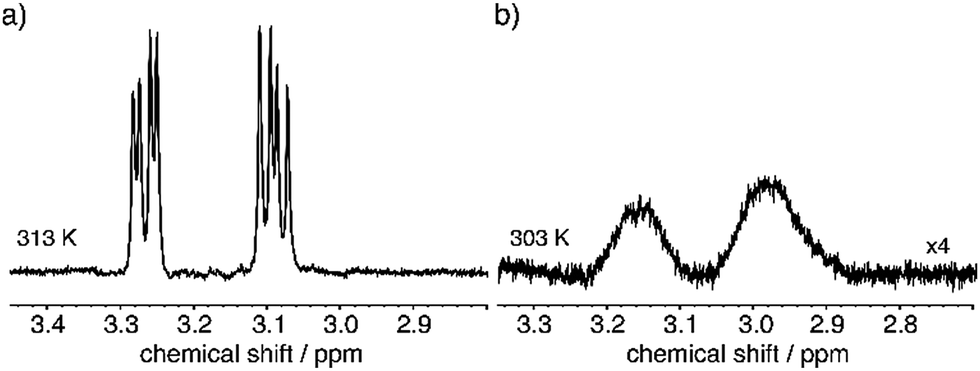 | ||
| Fig. 4 Comparison of the phenylalanine beta protons at (a) 313 K and (b) 303 K. Formation of the gel becomes evident by the observed line broadening. | ||
We have investigated the longitudinal and transverse 1H relaxation properties as well as the singlet lifetime TS as a function of temperature in a H2O:D2O mixture (95%:5%) for the 0.5 wt% and 2 wt% dipeptide. The results are displayed in Fig. 5. For the 0.5 wt% sample, TS remains about three to four times longer than T1 over a temperature range from 313 K to 283 K. Considering that relaxation of protons in peptides is usually dominated by intramolecular dipolar relaxation, we used the 1H transverse relaxation times to estimate a correlation time τc(DD)68 according to the following formula previously used for the investigation in stretched hydrogels69 whereby ω0 represents the angular Larmor frequency:
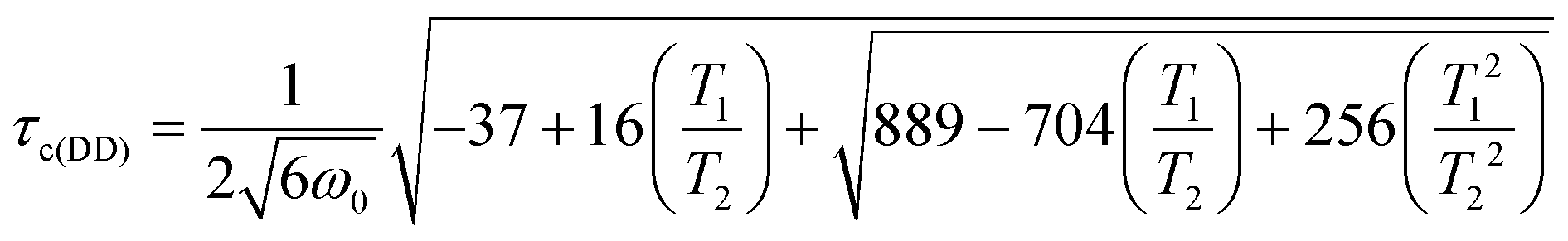 | (1) |
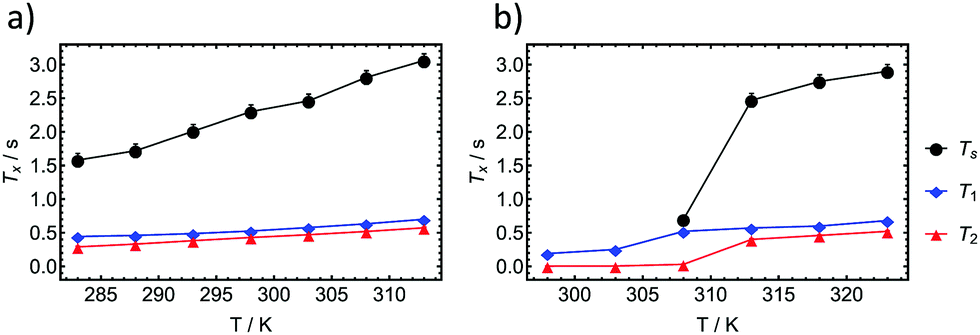 | ||
| Fig. 5 T s, T1 and T2 relaxation times of the beta protons in the F residue of the dipeptide IF, as a function of temperature for (a) 0.5 wt% and (b) 2 wt%. Below 308 K no singlet state was accessible in the 2 wt% sample. | ||
The equation was shown to be valid in regions where τc(DD) < 10 ns and is applicable if intramolecular dipolar interactions can be assumed the most dominant relaxation mechanism.69 At 313 K we estimate a correlation time of 92 ps and at 303 K of 94 ps for the 0.5 wt% sample in which the molecules undergoes isotropic motion at all times.
Upon investigation of the 2 wt% dipeptide at the same temperatures, it becomes evident that the relaxation times as well as the singlet lifetime behave similarly until the transition into a gel occurs. At 308 K we observed an intermediate state in which the singlet state can be still accessed is however almost on the order of T1 (T1 = 520 ms and TS = 700 ms). Below 303 K the singlet state is not accessible anymore. Investigations of the transverse relaxation time T2 show a drop from 400 ms at 313 K to 5.4 ms at 303 K. T1 is reduced from 570 ms to 250 ms. Using eqn (1) we estimate a τc(DD) of 132 ps at 313 K and 2.04 ns at 303 K. In the gel case, the molecular motion does not fall into the extreme narrowing limit anymore (ω0τc ≪ 1), for which most of the relaxation properties of the singlet states have been investigated so far. The utilized equation however was shown to be applicable for a gel in the past and supports the measured TS data.69 As TS is inverse proportional to the correlation time,70 we regard the increase in correlation time as the main reason for the reduction in the time for singlet–triplet equilibration. The ratio of the correlation times of the liquid state to the gel state amounts to 15.4. If TS is reduced by that factor in the gel we expect a drop from the measured TS (313 K) = 2470 ms to TS (303 K) = 160 ms. Since the sequence timings (two 200 ms APSOC ramps, sustaining period and filter) exceed three times the estimated TS at 303 K, relaxation effects may impede the effective observation of the singlet state.
The observed behaviour in the gels is completely reversible i.e. the detectable singlet-states can be switched “on” or “off” by controlling the temperature. The accessibility of the singlet state after its back conversion from the gel holds therefore promise to develop stimuli responsive contrast agents with an on/off switch. This mechanism is not based on line broadening effects but utilizes the storage of magnetization in proton singlet states in combination with an efficient filter to suppress background signals.
4 Conclusions
In conclusion, we have introduced an extension of the T00 filter that can be utilized for weakly coupled spin systems. In combination with water suppression techniques, proton signals other than the desired ones are effectively removed in the context of singlet NMR. Additionally, we have demonstrated the feasibility of a stimuli-responsive on/off switch for nuclear singlet states based on a thermo-responsive gel formed from the isoleucine–phenylalanine dipeptide. Overall, our investigations demonstrate the possibility of switchable contrast agents that utilize nuclear singlet states of protons without the need of isotopic labelling.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge generous funding from the Max-Planck-Society and Prof. Christian Griesinger for access to his equipment and facilities. Open Access funding provided by the Max Planck Society.Notes and references
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220 CrossRef PubMed.
- L. Sun, C. Zheng and T. J. Webster, Int. J. Nanomed., 2017, 12, 73 CrossRef PubMed.
- T. Fan, X. Yu, B. Shen and L. Sun, J. Nanomater., 2017 Search PubMed , 4562474.
- A. M. Nyström, J. W. Bartels, W. Du and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1023 CrossRef PubMed.
- M. Grzelczak, J. Verman, E. M. Furst and L. M. Liz-Marzan, ACS Nano, 2010, 4, 3591 CrossRef PubMed.
- J. J. McManus, P. Charbonneau, E. Zaccarelli and N. Asherie, Curr. Opin. Colloid Interface Sci., 2016, 22, 73 CrossRef.
- Y. Bai, Q. Luo and J. Liu, Chem. Soc. Rev., 2016, 45, 2756 RSC.
- R. Faas, A. Pohle, K. Moß, M. Henkel and R. Hausmann, Biotechnol. Rep., 2017, 16, 1 CrossRef PubMed.
- H. Li, J. D. Cater and T. H. LaBean, Mater. Today, 2009, 12, 24 CrossRef.
- G. Whitsides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef PubMed.
- L. L. Ong, N. Hanikel, O. K. Yaghi, C. Grun, M. T. Strauss, P. Bron, J. Lai-Kee-Him, F. Schueder, B. Wang, P. Wang, J. Y. Kishi, C. Myhrvold, A. Zhu, R. Jungmann, G. Bellot, Y. Ke and P. Yin, Nature, 2017, 552, 72 CrossRef PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297 CrossRef PubMed.
- E. Winfree, F. Liu, L. A. Wenzler and N. C. Seeman, Nature, 1998, 394, 539 CrossRef PubMed.
- Y. Taniguchi, M. A. Bin Sazali, Y. Kobayashi, N. Arai, T. Kawai and T. Nakshima, ACS Nano, 2017, 11, 9312 CrossRef PubMed.
- D. Vanmaekelbergh, Nano Today, 2011, 6, 419 CrossRef.
- Y.-C. Lin, C.-Y. Chen, H.-L. Chem, T. Hashimoto, S.-A. Chen and Y.-C. Li, J. Chem. Phys., 2015, 142, 214905 CrossRef PubMed.
- N. Li, A. Z. Panagiotopoulos and A. Nikoubashman, Langmuir, 2017, 33, 6021 CrossRef PubMed.
- C. Frangville, Y. Li, C. Billotey, D. R. Talham, J. Taleb, P. Roux, J.-D. Marty and C. Mingotaud, Nano Lett., 2016, 16, 4069 CrossRef PubMed.
- G. Liang, J. Ronald, Y. Chen, D. Ye, P. Pandit, M. L. Ma, B. Rutt and J. Rao, Angew. Chem., Int. Ed., 2011, 50, 6283 CrossRef PubMed.
- Z. Zheng, H. Sun, C. Hu, G. Li, X. Liu, P. Chen, Y. Cui, J. Liu, J. Wang and G. Liang, Anal. Chem., 2016, 88, 3363 CrossRef PubMed.
- S. Bo, C. Song, Y. Li, W. Yu, S. Chen, X. Zhou, Z. Yang, X. Zheng and Z.-X. Jiang, J. Org. Chem., 2015, 80, 6360 CrossRef PubMed.
- A. T. Preslar, T. Tantakitti, K. Park, S. Zhang, S. I. Stupp and T. J. Meade, ACS Nano, 2016, 10, 7376 CrossRef PubMed.
- M. Buzhor, L. Avram, L. Frish, Y. Cohen and R. J. Amir, J. Mater. Chem. B, 2016, 4, 3072 RSC.
- X. Huang, G. Huang, S. Zhang, K. Sagiyama, O. Togao, X. Ma, Y. Wang, Y. Li, T. C. Soesbe, B. D. Sumer, M. Takahashi, A. D. Sherry and J. Gao, Angew. Chem., Int. Ed., 2013, 52, 8074 CrossRef PubMed.
- K. Matsuo, R. Kamada, K. Mizusawa, H. Imai, Y. Takayama, M. Narazaki, T. Matsuda, Y. Takaoka and I. Hamachi, Chem. – Eur. J., 2013, 19, 12875 CrossRef PubMed.
- Y. Takaoka, T. Sakamoto, S. Tsukiji, M. Narazaki, T. Matsuda, H. Tochio, M. Shirakawa and I. Hamachi, Nat. Chem., 2009, 1, 557 CrossRef PubMed.
- Y. Yuan, S. Ge, H. Sun, X. Dong, H. Zhao, L. An, J. Zhang, J. Wang, B. Hu and G. Liang, ACS Nano, 2015, 9, 5117 CrossRef PubMed.
- M. Carravetta and M. H. Levitt, J. Am. Chem. Soc., 2004, 126, 6228–6229 CrossRef PubMed.
- M. Carravetta, O. G. Johannessen and M. H. Levitt, Phys. Rev. Lett., 2004, 92, 153003 CrossRef PubMed.
- M. Carravetta and M. H. Levitt, J. Chem. Phys., 2005, 122, 214505 CrossRef PubMed.
- G. Pileio and M. H. Levitt, J. Chem. Phys., 2009, 130, 214501 CrossRef PubMed.
- G. Pileio, M. Carravetta and M. H. Levitt, Proc. Natl. Acad. Sci. U. S. A., 2011, 107, 17135–17139 CrossRef PubMed.
- M. C. D. Tayler, S. Marie, A. Ganesan and M. H. Levitt, J. Am. Chem. Soc., 2010, 132, 8225 CrossRef PubMed.
- A. N. Pravdivtsev, A. V. Yurkovskaya, H. Zimmermann, H.-M. Vieth and K. L. Ivanov, Phys. Chem. Chem. Phys., 2014, 16, 7584 RSC.
- R. Sarkar, P. R. Vasos and G. Bodenhausen, J. Am. Chem. Soc., 2007, 129, 328–334 CrossRef PubMed.
- P. Ahuja, R. Sarkar, P. R. Vasos and G. Bodenhausen, J. Chem. Phys., 2007, 127, 134112 CrossRef PubMed.
- R. Buratto, D. Mammoli, E. Chiarparin, G. Williams and G. Bodenhausen, Angew. Chem., Int. Ed., 2014, 53, 11376–11380 CrossRef PubMed.
- N. Salvi, R. Buratto, A. Bornet, S. Ulzega, I. R. Rebollo, A. Angelini, C. Heinis and G. Bodenhausen, J. Am. Chem. Soc., 2012, 134, 11076–11079 CrossRef PubMed.
- R. Buratto, A. Bornet, J. Milani, D. Mammoli, B. Vuichoud, N. Salvi, M. Singh, A. Laguerre, S. Passemard, S. Gerber-Lemaire, S. Jannin and G. Bodenhausen, ChemMedChem, 2014, 9, 2509–2515 CrossRef PubMed.
- R. Buratto, D. Mammoli, E. Canet and G. Bodenhausen, J. Med. Chem., 2016, 59, 1960–1966 CrossRef PubMed.
- A. S. Kiryutin, A. N. Pravdivtsev, A. V. Yurkovskaya, H.-M. Vieth and K. L. Ivanov, J. Phys. Chem. B, 2016, 120, 11978–11986 CrossRef PubMed.
- G. Pileio, M. Carravetta, E. Hughes and M. H. Levitt, J. Am. Chem. Soc., 2008, 130, 12582–12583 CrossRef PubMed.
- M. C. D. Tayler and M. H. Levitt, Phys. Chem. Chem. Phys., 2011, 13, 5556–5560 RSC.
- G. Pileio, M. Concistre, M. Carravetta and M. H. Levitt, J. Magn. Reson., 2006, 182, 353–357 CrossRef PubMed.
- T. Theis, Y. Feng, T. Wu and W. S. Warren, J. Chem. Phys., 2014, 140, 014201 CrossRef PubMed.
- G. Stevanato, J. T. Hill-Cousins, P. Håkansson, S. S. Roy, L. J. Brown, R. C. D. Brown, G. Pileio and M. H. Levitt, Angew. Chem., Int. Ed., 2015, 54, 3740–3743 CrossRef PubMed.
- S. J. Elliott, L. J. Brown, J.-N. Dumez and M. H. Levitt, Phys. Chem. Chem. Phys., 2016, 18, 17965–17972 RSC.
- Y. Feng, R. M. Davis and W. S. Warren, Nat. Phys., 2012, 8, 831–837 Search PubMed.
- A. K. Grant and E. Vinogradov, J. Magn. Reson., 2008, 194, 46–57 CrossRef PubMed.
- W. S. Warren, E. Jensita, R. T. Branca and X. Chen, Science, 2009, 323, 1711–1714 CrossRef PubMed.
- G. Pileio, S. Bowen, C. Laustsen, M. C. D. Tayler, J. T. Hill-Cousins, L. J. Brown, R. C. D. Brown, J. H. Ardenkjaer-Larsen and M. H. Levitt, J. Am. Chem. Soc., 2013, 135, 5084–5088 CrossRef PubMed.
- Y. Feng, T. Theis, X. Liang, Q. Wang and W. S. Warren, J. Am. Chem. Soc., 2013, 135, 9632–9635 CrossRef PubMed.
- T. Theis, G. X. Ortiz Jr., A. W. J. Logan, K. E. Claytor, Y. Feng, W. P. Huhn, V. Blum, S. J. Malcolmson, E. Y. Chekmenev, Q. Wang and W. S. Warren, Sci. Adv., 2016, 2, e1501438 Search PubMed.
- Y. Zhang, X. Duan, P. C. Soon, V. Sychrovsky, J. W. Canary and A. Jerschow, Chem. Phys. Chem., 2016, 17, 2967–2971 CrossRef PubMed.
- Y. Zhang, P. C. Soon, A. Jerschow and J. W. Canary, Angew. Chem., Int. Ed., 2014, 53, 3396–3399 CrossRef PubMed.
- S. Cavadini, J. Dittmer, S. Antonijevi and G. Bodenhausen, J. Am. Chem. Soc., 2005, 127, 15744–15748 CrossRef PubMed.
- P. Ahuja, R. Sarkar, P. R. Vasos and G. Bodenhausen, J. Am. Chem. Soc., 2009, 131, 7498–7499 CrossRef PubMed.
- S. J. DeVience, R. Walsworth and M. S. Rosen, NMR Biomed., 2013, 26, 1204–1212 CrossRef PubMed.
- M. C. D. Tayler and M. H. Levitt, Phys. Chem. Chem. Phys., 2011, 13, 9128–9130 RSC.
- S. Glöggler, S. J. Elliott, G. Stevanato, R. C. D. Brown and M. H. Levitt, RSC Adv., 2017, 7, 34574 RSC.
- M. C. D. Tayler and M. H. Levitt, J. Am. Chem. Soc., 2013, 135, 2120 CrossRef PubMed.
- N. S. de Groot, T. Parella, F. X. Aviles, J. Vendrell and S. Ventura, Biophys. J., 2007, 92, 1732 CrossRef PubMed.
- R. W. Adams, C. M. Holroyd, J. A. Aguilar, M. Nilsson and G. A. Morris, Chem. Commun., 2013, 49, 358 RSC.
- V. Govindaraju, K. Young and A. A. Maudsley, NMR Biomed., 2000, 13, 129 CrossRef PubMed.
- G. Zheng and W. S. Price, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 56, 267 CrossRef PubMed.
- A. Haase, J. Frahm, W. Hanicke and D. Matthaei, Phys. Med. Biol., 1985, 30, 341 CrossRef PubMed.
- M. Mescher, H. Merkle, J. Kirsch, M. Garwood and R. Gruetter, NMR Biomed., 1998, 11, 266 CrossRef PubMed.
- W. R. Carper and C. E. Keller, J. Phys. Chem., 1997, 101, 3246 CrossRef.
- K. Nahashima, D. Krishna Rao, G. Pagès, S. S. Velan and P. W. Kuchel, J. Biomol. NMR, 2014, 59, 31 CrossRef PubMed.
- G. Pileio, J. T. Hill-Cousins, S. Mitchell, I. Kuprov, L. J. Brown, R. C. D. Brown and M. H. Levitt, J. Am. Chem. Soc., 2012, 134, 17494 CrossRef PubMed.
| This journal is © the Owner Societies 2018 |