
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ab initio paramagnetic NMR shifts via point-dipole approximation in a large magnetic-anisotropy Co(II) complex†
Jiří
Mareš
 * and
Juha
Vaara
* and
Juha
Vaara
NMR Research Unit, University of Oulu, P.O. Box 3000, FI-90014 Oulu, Finland. E-mail: jiri.mares@oulu.fi
First published on 20th August 2018
Abstract
Transition metal complexes can possess a large magnetic susceptibility anisotropy, facilitating applications such as paramagnetic tags or shift agents in nuclear magnetic resonance (NMR) spectroscopy. Due to its g-shift and zero-field splitting (ZFS) we demonstrate on a Co(II) clathrochelate with an aliphatic 16-carbon chain, a modern approach for ab initio calculation of paramagnetic susceptibility. Due to its large anisotropy, large linear dimension but relatively low number of atoms, the chosen complex is especially well-suited for testing the long-range point-dipole approximation (PDA) for the pseudocontact shifts (PCSs) of paramagnetic NMR. A static structure of the complex is used to compare the limiting long-distance PDA with full first-principles quantum-mechanical calculation. A non-symmetric formula for the magnetic susceptibility tensor is necessary to be consistent with the latter. Comparison with experimental shifts is performed by conformational averaging over the chain dynamics using Monte Carlo simulation. We observe satisfactory accuracy from the rudimentary simulation and, more importantly, demonstrate the fast applicability of the ab initio PDA.
1 Introduction
Molecules with large magnetic anisotropy, often due to a paramagnetic metal center, find applications in chemistry, materials research, structural biology and medicine.1–5 Examples of effects that can be observed due to such centers are orientation in magnetic field6 and the unusually large range of NMR chemical shifts and rapid relaxation rate of the NMR signals.7,8 Among the compounds of 3d transition metals, some cobalt complexes feature the combination of large zero-field splitting (ZFS) and g-tensor parameters that are commonly expected to translate to a large magnetic anisotropy.9 Computational methods for the prediction and analysis of the magnetic properties of such paramagnetic systems have progressed during recent years. The NMR chemical shifts induced by a strongly anisotropic magnetic center have already for a long time been calculated by a long-distance PDA of the dipolar hyperfine coupling (HFC) interactions between the unpaired electrons and NMR nuclei.1 The PDA was recently implemented in a modern computational framework that allows ab initio pNMR shift calculation of large systems such as an entire metalloprotein,9 and which is based on the recently redressed version (ref. 10 and 11) of the classic Kurland–McGarvey theory.12To further investigate the performance of the method of ref. 9, we selected for the present work a large-anisotropy Co(II) complex (Fig. 1) studied by Novikov et al.,13 denoted in their paper as complex 2. Due to the large linear dimension of this S = ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) (quartet spin state) clathrochelate complex, it is well-suited for testing the methods for the long-distance effect of the paramagnetic center. For a system of this size, the required electron paramagnetic resonance (EPR) parameters can be calculated quite reliably using molecular quantum-chemical (QC) codes. pNMR modeling of 3d transition metal clusters has been by now well-probed10,11,14–16 and chemical shifts in such systems can be calculated by first-principles methods with a predictive accuracy. We retained only one of the two aliphatic 16-carbon chains of the complex. Since the presence of the chain does not significantly influence the EPR properties of the complex, as shown below, and the two chains are too far from each other to restrict their mutual conformational spaces, removing one of the chains from the model can be regarded as a safe approximation.
(quartet spin state) clathrochelate complex, it is well-suited for testing the methods for the long-distance effect of the paramagnetic center. For a system of this size, the required electron paramagnetic resonance (EPR) parameters can be calculated quite reliably using molecular quantum-chemical (QC) codes. pNMR modeling of 3d transition metal clusters has been by now well-probed10,11,14–16 and chemical shifts in such systems can be calculated by first-principles methods with a predictive accuracy. We retained only one of the two aliphatic 16-carbon chains of the complex. Since the presence of the chain does not significantly influence the EPR properties of the complex, as shown below, and the two chains are too far from each other to restrict their mutual conformational spaces, removing one of the chains from the model can be regarded as a safe approximation.
 | ||
| Fig. 1 Clathrochelate cage with the atomic pairs for which interatomic distances are listed in Table 1, indicated, as well as the complex including both the cage and one of the 16-carbon tails with numbering of the tail. Parallel and perpendicular directions of the cylindrically symmetric cage system are indicated as ‖ and ⊥, respectively. The cartesian X, Y, Z coordinate frame is also depicted. | ||
In the PDA, the HFC tensor appearing in the contemporary QC theory of pNMR shielding10–12 is approximated by its long-distance limit originating from the electron-spin magnetic moment, which is taken to be a point dipole. The magnetic moment of the NMR nucleus interacts with the field generated by the point dipole of the electron spin, giving rise to the approximate expression for the shielding tensor9
 | (1) |
| σPCS = Tr(σdip)/3; δPCS = −σPCS. | (2) |
2 Methods
For comparison with the PDA, we calculated quantum-chemically the full HFC tensors of the present Co(II) complex encompassing both the cage and the tail. In the used ORCA code,17 we employ nonrelativistic density-functional theory (DFT) with leading-order perturbational correction for the relativistic spin–orbit (SO) interaction.18 The importance of this correction for NMR shieldings in open-shell molecules has been recently pointed out, for example, by Marek and coworkers.19 The HFC tensors, together with g- and ZFS tensors, are incorporated in the expression for pNMR shielding tensors as detailed in ref. 11 and 20. A breakdown of the various terms appearing in the method of ref. 10–12 and featuring different contributions to the EPR parameters, is presented in Table S1 in the ESI.† To assess the spatial reach of the different mechanisms of hyperfine shielding originating from the paramagnetic center out to the ligand atom, the pNMR shielding tensors are broken down to scalar (rank-0) and dipolar (rank-2) terms.We use two different structures. First, we used a truncated Co(II) clathrochelate complex with both 16-carbon tails replaced by hydrogens. The cage structure was computationally optimised by Turbomole21 and Orca software using several settings of the DFT method to obtain a range of geometries (Table 1).
| Optimisation method | Co–Na (Å) | B–B (Å) | D (cm−1) | E/Db | g iso | Δgaxc |
|---|---|---|---|---|---|---|
a Optimised bond lengths, see illustration of the structure in Fig. 1.
b ZFS- and g-tensor calculations at the CASSCF/TZVP-DKH level.  and and  , where Dii are the eigenvalues of the ZFS-tensor. The ordering of the principal values is selected so that 0 ≤ E/D ≤ 1/3.22,23
c , where Dii are the eigenvalues of the ZFS-tensor. The ordering of the principal values is selected so that 0 ≤ E/D ≤ 1/3.22,23
c
 , where the gii are the g-tensor eigenvalues, see also eqn (S1)–(S3) (ESI) and the associated explanation.
d D3 dispersion correction.24
e With scalar relativistic effective core potential.25
f COSX approximation of Hartree–Fock exchange.26 , where the gii are the g-tensor eigenvalues, see also eqn (S1)–(S3) (ESI) and the associated explanation.
d D3 dispersion correction.24
e With scalar relativistic effective core potential.25
f COSX approximation of Hartree–Fock exchange.26
|
||||||
| PBE/def2-TZVP | 2.0552 | 6.146 | −85.19 | 0.0037 | 2.348 | 0.972 |
| PBE0/def2-SVP | 2.0594 | 6.117 | −104.50 | 0.0021 | 2.365 | 1.175 |
| PBE0/def2-TZVP | 2.0533 | 6.093 | −92.41 | 0.0032 | 2.355 | 1.047 |
| PBE0/def2-TZVP+D3d | 2.0517 | 6.097 | −92.03 | 0.0032 | 2.354 | 1.043 |
| PBE0/def2-TZVP/ECPe | 2.0610 | 6.094 | −96.06 | 0.0031 | 2.361 | 1.085 |
| PBE0/def2-TZVP/ECP+D3 | 2.0595 | 6.097 | −94.75 | 0.0033 | 2.359 | 1.072 |
| PBE0/def2-TZVP/ECP+COSXf | 2.0533 | 6.097 | −100.87 | 0.0026 | 2.361 | 1.137 |
The second structure used in this work consisted of Co(II) clathrochelate complex with one 16-carbon tail replaced by a hydrogen atom, retaining one tail. This was optimised by PBE27,28/def2-SVP29 combination of DFT functional and basis set, and served mainly in comparing methods for the HFC tensor calculation. If not stated otherwise, the geometry used in all calculations of this study was obtained at the DFT PBE027,28/def2-TZVP29 level with a quasirelativistic effective core potential25 on the Co centre and the COSX approximation26 for the Hartree–Fock exchange.
In the present work we use a combination of the multireference complete active space self-consistent field (CASSCF)30 method, augmented by dynamical correlation treatment by N-electron valence-state perturbation theory (NEVPT231–33), to calculate the EPR g- and ZFS tensors. In these computations, the scalar relativistic second-order Douglas–Kroll–Hess (DKH) Hamiltonian34,35 is employed, with SO effects treated by quasidegenerate perturbation theory.36,37 In state-averaged CASSCF wave function, the active space was selected to cover the d-orbitals of the metal center – meaning, for Co(II), 7 electrons in 5 orbitals. All the 10 quartet roots as well as 15 doublets possible within this active space were included. It has been shown9,15,38,39 that this methodology gives good results for the EPR parameters including the critical ZFS tensor, for systems with two or more unpaired electrons.
We show that the, for relativistic purposes recontracted variant40 of the valence triple-ζ + polarization (TZVP) basis set41 (denoted here as TZVP-DKH), is for current calculations of the g- and ZFS tensors equally good as the much larger def2-QZVPP-DKH basis40 (Table 2). The related def2-TZVP-DKH basis has been found to represent a good compromise between the computational cost and accuracy in previous pNMR computations.11,15,20
| Structure | Structure optimisation | Method | Basis (g, ZFS calc.) | D | E/Da | g iso | Δgaxa |
|---|---|---|---|---|---|---|---|
| a See footnotes b, c of Table 1. b This structure, optimised at PBE0(def2-SVP) level, has been used only for this comparison. c Structure obtained by removing the 16-carbon chain and terminating with a hydrogen atom. d g- and ZFS tensors obtained using this structure and combination of method and basis set were used for the production results presented for example in Tables 3, 4 and Table S2 (ESI). | |||||||
| Cage + tailb | PBE0/def2-SVP | CASSCF | TZVP-DKH | −101.35 | 0.00206 | 2.360 | 1.138 |
| Cage onlyc | PBE0/def2-SVP | CASSCF | TZVP-DKH | −101.19 | 0.00208 | 2.362 | 1.141 |
| Cage only | PBE0/def2-TZVP, ECP | CASSCF | TZVP-DKH | −100.87 | 0.00262 | 2.361 | 1.137 |
| Cage only | PBE0/def2-TZVP, ECP | CASSCF | def2-QZVPP-DKH | −101.22 | 0.00271 | 2.362 | 1.132 |
| Cage onlyd | PBE0/def2-TZVP, ECP | NEVPT2 | TZVP-DKH | −85.50 | 0.00281 | 2.325 | 0.979 |
In accordance with our previous experience,9,20 there is a significant impact on the g- and ZFS tensors of dynamical electron correlation accounted for via the NEVPT2 method, as compared to the CASSCF results (Table 2). Both the D – parameter of the ZFS tensor and the anisotropy of the g-tensor decrease upon adopting the NEVPT2 correction. For comparison with experimental data, it is important to use the NEVPT2 results if possible.15,20 For production results, the g- and ZFS tensors were calculated using ORCA by the CASSCF/NEPVT2 methods and the TZVP-DKH basis. These numbers, obtained from the cage-only model unless otherwise stated, were subsequently used for the PDA calculations including the 16-carbon chain.
In conformational averaging calculations, the energy difference of 3.9 kJ mol−1 between the trans and gauche conformers of the carbon chain (with the gauche conformer higher in energy) for each bond was adopted from the literature.13,42 The bond lengths were kept at the values obtained from the present QC (PBE/def2-SVP) optimisation, approximately 1.53 Å for the C–C bonds. The temperature was set to 300 K both for the MC simulation as well as for all the pNMR calculations of this study. It was assumed that the conformations of the bonds are independent of the conformations of all the other bonds. However, conformations involving overlap of the atomic radii were excluded. Pseudocontact shifts were calculated using PDA, eqn (2) for every atom and for each cycle of the Monte Carlo simulation, with the g- and ZFS-tensors of the rigid cage-only model calculated only once.
3 Results and discussion
3.1 Structure
It has been seen before9,15,38 that the geometry of the paramagnetic center can dramatically influence the resulting ZFS and g-tensors. Table 1 shows the effect of the detailed structure as optimised using the PBE and PBE0 functionals, with particularly the PBE0 functional being not only first-principles-based but also performing well for structures of transition metal complexes.9,43 Various other choices are elaborated in Table 1. It can be concluded that, in this case, applying the dispersion correction44 or using a relativistic pseudopotential25 at the cobalt centre does not change the geometry or the EPR parameters significantly. For the structure optimisation, the resulting effects are smaller than those arising from the choice of the DFT functional.3.2 Comparison of full theory and the point-dipole approximation
Full QC HFC tensors were calculated at the PBE0/def2-TZVP level with the shielding results shown in Table 3 and Fig. 2. | ||
| Fig. 2 Calculated shielding constant (13C, 1H, 11B) contributions at T = 300 K as a function of the distance of the nucleus from the paramagnetic centre, according to Table 3. The different panels (top, middle, down) represent the same data in increasing magnification of the vertical axis. | ||
In the vicinity of the paramagnetic center, the electron spin density distribution (Fig. 3) translates into, among others, the Fermi contact part of the hyperfine coupling,45,46 which dominates the pNMR shielding in this region. This necessitates a full QC approach involving the evaluation of HFC at the NMR nuclei. At larger distances, where the spin density vanishes, the contact contribution to the HFC decays to zero. This leaves the dipolar contribution, manifested in the isotropic pNMR chemical shift as PCS,47 as the dominant mechanism. Since in the long-distance limit, the dipolar interaction takes a form familiar from classical magnetostatics and, hence, is independent on the bonding structure, the pseudocontact shifts can equally well be calculated for the 1-dimensional C16 chain of this study and, for example, for a 3D protein structure.
 | ||
| Fig. 3 Optimised structure of a Co(II) clathrochelate cage compound with the tail. Left panel: The isosurfaces of the computed pseudocontact shift are shown. The red surface depicts +5 ppm and blue −5 ppm pseudocontact shifts, calculated using the present point-dipole approximation and the NEVPT2/TZVP-DKH data on the cage-only structure. Right panel: Spin density isosurfaces. The red transparent/opaque surface represents the isovalue of 1 × 10−6/1 × 10−5, the blue surfaces correspond to negative values of the same magnitudes. | ||
For a doublet system, comparison of the PDA- and QC-calculated shifts has been recently discussed in ref. 48. In ref. 9, a formulation of PDA consistent with the full QC treatment of arbitrary spin state of ref. 10–12 and 20 was elaborated. According to that, the magnetic susceptibility tensor is written as
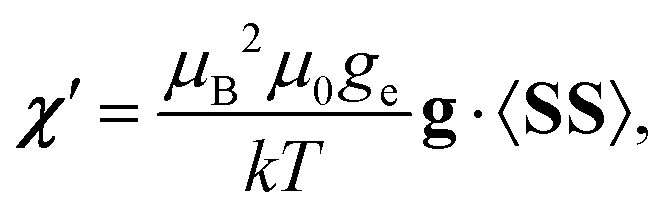 | (3) |
Using the breakdown of the pNMR shielding into different mechanisms, presented in Table S1 of the ESI,† the PDA includes the sum of hyperfine term 2 – a combination of the dipolar hyperfine coupling and the free electron g-value ge, term 7 due to the isotropic g-shift and term 9 due to the anisotropy of the g-tensor. All of these contributions to the full pNMR shielding expression involve the dipolar hyperfine interaction.
It can be seen from Table 3 as well as Fig. 2 and 4 that the PDA is for the dipolar part of the hyperfine interaction very well valid already for the C1 carbon of the 16-carbon chain, where the PCS still reaches a high value of 74 ppm (Table 3 and Fig. 3). In fact, already for the boron atom of the cage, at 3.1 Å from the metal center, the PDA (−266 ppm) differs from the PCS obtained by the QC calculation (−237 ppm) only by 12%. In a singly-bonded, aliphatic hydrocarbon chain such as in the present model complex, the spin density does not extend over a large distance from the paramagnetic centre. The disappearance of the spin density far away from the paramagnetic center is discussed further for example in ref. 9 and 49. According to Table 3, at the C3 position of the 16-carbon chain, the contact term still amounts to 2.32 ppm, which represents 12% of the total shielding constant. The contact term becomes negligible at around C4 with 0.3 ppm contribution remaining in the total σ (Fig. 2). It is worth noting that the shielding terms 2 and 9 are of a very similar magnitude (Fig. S1 in the ESI†). The nonrelativistic dipolar term 2, which would have a vanishing contribution in the S = ½ case, is represented here (due to the effect of the 〈SS〉 dyadic) as equally important as the traditional pseudocontact term 9 (arising due to the g-tensor anisotropy).
| Atom | Distance to Co (Å) | Full QC | Contact QCb | Contact %c | PCS QCd | PCS %e | PDAf | Error of PDA%g |
|---|---|---|---|---|---|---|---|---|
| a Full quantum-chemical pNMR shielding constant according to the method of ref. 10–12. The g- and ZFS-tensors were calculated at the NEVPT2/TZVP-DKH level for the cage-only model optimised at the PBE0/def2-TZVP level (the last line of Table 2). The molecular cage structure with the C16 chain was geometry-optimised at the PBE/def2-SVP level. The QC calculations of the HFC tensors were carried out at the PBE0/def2-TZVP level using the Orca programme. For PDA, the dipolar HFC tensors (column “PDA”) were calculated using the same structure. b Sum of the pNMR shielding terms from the full calculation, which include the rank-0 contact hyperfine coupling. Contact QC = σcon + σcon,2 + σcon,3 + σc,aniso. See Table S1 (ESI) for the details of the breakdown of the pNMR shielding tensors to the various physical contributions and the paragraph “Anisotropy parameters” in the ESI for clarification of the tensorial ranks of the HFC tensor. c Relative contribution of the contact terms (in %) to the total pNMR shielding. d Sum of the pNMR shielding terms from the full quantum-chemical calculation, involving the second-rank hyperfine coupling contribution, PCS QC = σdip + σdip,2 + σdip,3 + σpc. e Relative contribution of the dipolar terms (%) to the total pNMR shielding. f Sum of the pNMR shielding terms involving the second-rank (dipolar) hyperfine coupling contribution calculated using the point-dipole approximation: PDA = σdip + σdip,3 + σpc. The same g- and ZFS-tensors were used as for other calculations (see footnote a). g Relative error calculated as (“PDA” – “Full QC”)/“Full QC”. | ||||||||
| Cage N | 2.05 | −29690.50 | −29706.52 | 100.1 | 16.02 | −0.1 | −86.81 | −99.7 |
| Cage C | 2.87 | −796.44 | −836.05 | 105.0 | 39.60 | −5.0 | 132.99 | −116.7 |
| Cage B | 3.09 | −142.38 | 94.87 | −66.6 | −237.25 | 166.6 | −266.60 | 87.2 |
| C1 | 4.70 | −3.47 | 70.25 | −2024.5 | −73.72 | 2124.5 | −76.13 | 2093.7 |
| C2 | 5.49 | −51.98 | −9.61 | 18.5 | −42.37 | 81.5 | −42.57 | −18.1 |
| C3 | 6.99 | −19.52 | 2.32 | −11.9 | −21.85 | 111.9 | −21.53 | 10.3 |
| C4 | 7.96 | −12.37 | 0.33 | −2.7 | −12.70 | 102.7 | −12.48 | 0.9 |
| C5 | 9.42 | −8.25 | −0.08 | 1.0 | −8.17 | 99.0 | −8.04 | −2.5 |
| C6 | 10.48 | −5.28 | −0.15 | 2.8 | −5.13 | 97.2 | −5.06 | −4.2 |
| C7 | 11.90 | −3.88 | −0.11 | 2.9 | −3.76 | 97.1 | −3.70 | −4.5 |
| C8 | 13.02 | −2.61 | −0.08 | 3.1 | −2.53 | 96.9 | −2.49 | −4.5 |
| C9 | 14.42 | −2.06 | −0.06 | 3.1 | −2.00 | 96.9 | −1.97 | −4.5 |
| C10 | 15.56 | −1.46 | −0.05 | 3.5 | −1.41 | 96.5 | −1.40 | −4.2 |
| C11 | 16.95 | −1.20 | −0.02 | 1.4 | −1.18 | 98.6 | −1.16 | −3.1 |
| C12 | 18.12 | −0.88 | −0.02 | 1.9 | −0.86 | 98.1 | −0.86 | −2.6 |
| C13 | 19.49 | −0.74 | 0.00 | 0.0 | −0.74 | 100.0 | −0.74 | −1.1 |
| C14 | 20.68 | −0.58 | −0.02 | 2.9 | −0.57 | 97.1 | −0.56 | −3.1 |
| C15 | 22.04 | −0.49 | 0.00 | −0.7 | −0.50 | 100.7 | −0.50 | 0.7 |
| C16 | 23.23 | −0.39 | 0.00 | −0.9 | −0.40 | 100.9 | −0.39 | −1.0 |
| H1 | 5.16 | −53.44 | 1.38 | −2.6 | −54.81 | 102.6 | −54.00 | 1.1 |
| H2 | 5.39 | −42.49 | −4.03 | 9.5 | −38.46 | 90.5 | −37.78 | −11.1 |
| H3 | 7.32 | −21.07 | −1.48 | 7.0 | −19.59 | 93.0 | −19.17 | −9.0 |
| H4 | 7.89 | −11.65 | −0.40 | 3.4 | −11.25 | 96.6 | −11.10 | −4.7 |
| H5 | 9.67 | −8.26 | −0.31 | 3.7 | −7.95 | 96.3 | −7.80 | −5.6 |
| H6 | 10.42 | −4.77 | −0.17 | 3.5 | −4.61 | 96.5 | −4.55 | −4.7 |
| H7 | 12.10 | −3.94 | −0.15 | 3.7 | −3.79 | 96.3 | −3.72 | −5.4 |
| H8 | 12.97 | −2.39 | −0.09 | 3.6 | −2.30 | 96.4 | −2.27 | −4.9 |
| H9 | 14.58 | −2.13 | −0.08 | 4.0 | −2.05 | 96.0 | −2.01 | −5.6 |
| H10 | 15.53 | −1.35 | −0.05 | 3.6 | −1.30 | 96.4 | −1.29 | −5.0 |
| H11 | 17.09 | −1.27 | −0.05 | 4.1 | −1.21 | 95.9 | −1.19 | −5.7 |
| H12 | 18.09 | −0.81 | −0.01 | 0.7 | −0.80 | 99.3 | −0.80 | −1.1 |
| H13 | 19.61 | −0.77 | −0.01 | 0.8 | −0.76 | 99.2 | −0.76 | −1.2 |
| H14 | 20.65 | −0.54 | 0.00 | 0.8 | −0.53 | 99.2 | −0.53 | −1.5 |
| H15 | 22.14 | −0.52 | −0.01 | 1.3 | −0.52 | 98.7 | −0.51 | −2.1 |
| H16 | 23.57 | −0.37 | 0.00 | 1.2 | −0.37 | 98.8 | −0.36 | −2.0 |
 | ||
| Fig. 4 pNMR shielding of the clathrochelate cage + tail nuclei shown as a correlation between hyperfine shielding calculated using the full QC hyperfine couplings and using only the dipolar part of hyperfine coupling calculated using the point-dipole approximation (PDA). All common NMR nuclei (1H, 11B, 13C), are included in the plot. The shielding constant of nitrogen nuclei directly bonded to Co remain outside of the scale of the plot, however. For the H1 nucleus, the Fermi contact and the relativistic spin–orbit correction terms (see columns 1 and 3 of Table S2, ESI†) largely cancel each other. | ||
3.3 Susceptibility
Using eqn (3) and NEVPT2 calculation with the selected computational parameters listed in Table 2 (hydrogen-terminated cage unit obtained at the PBE0/def2-TZVP level, NEVPT2 calculation with DKH Hamiltonian and TZVP-DKH basis), the susceptibility volume tensor reads | (4) |
 | (5) |
with the axiality parameter Δχax = 27.3 × 10−32 m3. For comparison, Δχax = 10 × 10−32 m3 is the experimental value obtained by Novikov et al.13 using superconducting quantum interference device (SQUID) magnetometry. The parameters of ZFS and g-tensors obtained by fitting the temperature-dependent SQUID data by these authors are D = −64 cm−1 and Δgax = 0.2. For comparison, our best results from the cage-only model are −85.5 cm−1 and 0.98, respectively. The experimental EPR parameters of ref. 13 give slightly different results when plugged into the present PDA, resulting in is Δχax′ = 7.1 × 10−32 m3.

Fig. 5 illustrates the dependence of Δχax′ on the D parameter of the ZFS-tensor, as well as on giso and Δgax, according to eqn (3). The plot assumes cylindrical symmetry for both the g- and ZFS tensors for this quartet-state molecule. Furthermore, it assumes that the unique axes of these tensors coincide. These assumptions are valid for an idealised geometry of the studied cage complex. The same dependence of Δχax′ on the different magnetic parameters is shown in separate graphs in Fig. 6, where only one variable is changed at a time while the others are kept at the experimental or computational results. Δχax′ depends linearly on Δgax and giso, as well as (in the presented range of parameters) slightly non-linearly on the D parameter of the ZFS-tensor. This shows that there is no simple dependence of Δχax′ on any single magnetic parameter. Instead, the resulting Δχax′ depends principally on the combination of the tensors 〈SS〉‖g‖ − 〈SS〉⊥g⊥, where the component along the cylindrical symmetry axis is denoted with ‖ and the perpendicular component with ⊥.
 | ||
| Fig. 5 Calculated susceptibility anisotropy Δχax′ in the point-dipole approximation for pNMR shielding in a Co(II) clathrochelate system. Dependence on the D-parameter of zero-field splitting, isotropic g-factor, giso, and g-tensor anisotropy, Δgax. The reddish surface is calculated with the value Δgax = 0.98 as obtained presently for the cage complex using the NEPVT2 calculation. The blue surface is obtained with Δgax = 0.2 used by Novikov et al.13 The highlighted blue point corresponds to values of D = −65 cm−1 and giso = 2.27 in ref. 13, whereas the red point corresponds to the presently calculated values of D = −85.5 cm−1 and giso = 2.32. The highlighted red and blue lines correspond to vanishing Δχax′. | ||
 | ||
| Fig. 6 Two-dimensional slices of the plot of Fig. 5. The panels show the dependence of Δχax′ on the D-parameter of the ZFS tensor and the parameters giso and Δgax of the g-tensor. | ||
Even though our Δgax is larger than that used by Novikov et al. by a factor of five, the corresponding, presently calculated Δχax′ is only roughly twice as large as the one obtained using their parameters. It is also worth noting that the vanishing of magnitude of Δχax′ (corresponding to nodes of the PCS surface) occurs when the condition 〈SS〉‖g‖ = 〈SS〉⊥g⊥ holds. The interplay of the value of D and temperature in forming the 〈SS〉 matrix has been elaborated in ref. 11.
3.4 Conformational averaging
The PDA is crucial in modelling biological or materials systems of real applications interest, such as the pseudocontact shifts of distant ligands in a protein, as demonstrated elsewhere.9 In the case of the present Co(II) clathrochelate complex, we demonstrate the utility of the PDA in a dynamical averaging over the conformationally flexible chain part of the molecule. An advanced analytical and numerical method to calculate pseudocontact shifts caused by a mobile paramagnetic center has been proposed, together with the solution of the inverse problem – finding the position of the paramagnetic center from the known pseudocontact shifts.51 For our problem it was natural to adopt a simple model and sample the conformational space of the hexadecyl chain by the Metropolis Monte Carlo method52 over the trans/gauche conformers at each of the C–C bonds.Averages over the conformers are calculated from the PCSs based on the PDA and listed in Table 4. The same averaging is also performed using the experimental g- and ZFS tensors. Since the simulation has been started from the conformation representing the global minimum of the potential energy, there was no need for any equilibration run in the MC simulation. The achieved error, estimated by the method described in ref. 53, remains similar in size along the length of the chain. This means that the relative error with respect to the diminishing shielding constant increases with the distance from the paramagnetic center (Table 4). The total length of the MC simulation was 4.5 × 106 cycles as compared to altogether 315 ≈ 14.3 × 106 possible conformers of the 16-carbon chain.
| Atom | PCS-calc.a | PCS-calc. with experimental Δχaxb | PCS-expc |
|---|---|---|---|
| a Fully theoretical results using the susceptibility tensor χ′ of eqn (4). Note that the results are given as contributions to chemical shifts (opposite sign as compared to shieldings) for consistency with the experimental results. All values are given in ppm. b Results obtained from the MC simulation using the experimental Δχax = 10 × 10−32 m3.13 c Table S3 of ref. 13. For the calculated results, the error margins were calculated using the “data halving” method described by Flyvbjerg and Petersen.53 | |||
| H1 | 53.9207 ± 0.0003 | 36.2131 ± 0.0003 | 34.78 |
| H2 | 35.573 ± 0.004 | 24.025 ± 0.004 | 25.76 |
| H3 | 23.262 ± 0.007 | 15.717 ± 0.006 | 14.21 |
| H4 | 13.120 ± 0.004 | 8.862 ± 0.004 | 9.11 |
| H5 | 9.159 ± 0.005 | 6.196 ± 0.005 | 5.90 |
| H6 | 6.152 ± 0.004 | 4.164 ± 0.003 | 3.99 |
| H7 | 4.189 ± 0.004 | 2.839 ± 0.003 | 2.70 |
| H8 | 2.815 ± 0.004 | 1.910 ± 0.004 | 1.84 |
| H9 | 1.952 ± 0.004 | 1.328 ± 0.004 | 1.26 |
| H10 | 1.259 ± 0.004 | 0.849 ± 0.004 | 0.85 |
| H11 | 0.889 ± 0.004 | 0.602 ± 0.004 | 0.58 |
| H12 | 0.571 ± 0.004 | 0.391 ± 0.004 | 0.38 |
| H13 | 0.390 ± 0.003 | 0.267 ± 0.003 | 0.25 |
| H14 | 0.232 ± 0.004 | 0.164 ± 0.004 | 0.13 |
| H15 | 0.126 ± 0.005 | 0.091 ± 0.004 | 0.07 |
| H16 | 0.049 ± 0.005 | 0.038 ± 0.004 | 0.03 |
| C1 | 76.1258 ± — | 51.1885 ± — | — |
| C2 | 42.7531 ± 0.0003 | 28.8329 ± 0.0003 | 34.51 |
| C3 | 25.331 ± 0.004 | 17.101 ± 0.004 | 17.59 |
| C4 | 14.834 ± 0.003 | 10.021 ± 0.003 | 9.84 |
| C5 | 9.806 ± 0.003 | 6.630 ± 0.004 | 6.44 |
| C6 | 6.505 ± 0.003 | 4.403 ± 0.003 | 4.33 |
| C7 | 4.433 ± 0.003 | 3.004 ± 0.003 | 2.92 |
| C8 | 2.995 ± 0.003 | 2.031 ± 0.003 | 2.01 |
| C9 | 2.038 ± 0.004 | 1.385 ± 0.003 | 1.38 |
| C10 | 1.351 ± 0.003 | 0.918 ± 0.003 | 0.96 |
| C11 | 0.925 ± 0.003 | 0.628 ± 0.003 | 0.66 |
| C12 | 0.612 ± 0.003 | 0.418 ± 0.003 | 0.41 |
| C13 | 0.408 ± 0.003 | 0.281 ± 0.003 | 0.33 |
| C14 | 0.253 ± 0.003 | 0.177 ± 0.003 | 0.23 |
| C15 | 0.141 ± 0.003 | 0.101 ± 0.003 | 0.16 |
| C16 | 0.068 ± 0.004 | 0.051 ± 0.003 | 0.12 |
The results are depicted in Fig. 7 and listed in Table 4. The simulated PCS results with experimental Δχax agree very well with the experimental PCSs starting at around the position C3/H3 of the 16-carbon chain. This finding is in accordance with the static structure calculation reported in Table 3, where the error of PDA compared to the full QC pNMR shielding constant decreases to around 10% at this position in the chain. The simulation results obtained with the QC-computed χ′ tensor systematically overshoot the experimental PCS values, in complete accordance with the overestimation of the computed Δχax as compared to the experimental value. The fact that the calculations of the ZFS tensor using state-average CASSCF with the minimal active space, even when augmented with the NEVPT2 method, tend to overestimate the D parameter, has been discussed previously.9,54,55 However, the fact that the experimental trend is reproduced by the PDA confirms that for this application, conformational averaging by the Monte Carlo simulation over the discrete trans/gauche conformers represents the conformational space of the real system well enough – regardless of its simplicity and a lack of incorporation of any solvent effects.
 | ||
| Fig. 7 Pseudocontact shifts of the 16-carbon chain calculated in a Monte Carlo simulation using theoretical and experimental axiality parameter of the susceptibility tensor, Δχax′ and Δχax, respectively. Experimental PCSs13 are plotted for reference. See Table 4 for numerical values. | ||
4 Conclusion
We have investigated the point-dipole approximation (PDA) for the pseudocontact shifts of paramagnetic NMR in a Co(II) clathrochelate complex with a flexible 16-carbon chain. The PDA method was recently rendered consistent9 with the modern formulation10,11 of the full quantum-chemical pNMR shielding theory.12 We have used ab initio CASSCF/NEVPT2 methods to compute the ZFS and g-tensors necessary for the calculation of, first, the susceptibility tensor using the non-symmetric formula of ref. 9 and, secondly, the resulting pseudocontact shifts. Using the example complex, we have illustrated the interdependence on the ZFS and g-tensor parameters of the resulting Δχax and, consequently, the pseudocontact shift. The results indicate that Δχax′ depends principally on the difference between the products 〈SS〉‖g‖ and 〈SS〉⊥g⊥. We have further confirmed the usefulness of the combination of the PDA and a simple trans/gauche conformational space averaging to simulate the dynamics of the aliphatic 16-carbon chain. The PDA makes it possible to compute a very large number of pseudocontact shifts for simulation snapshots, based on a precalculated susceptibility tensor of the metal coordination center, and the mere coordinate data of the flexible part of the model. There is great promise of the method in the prediction and analysis of pNMR shifts in complex materials and biological systems, including those with conformationally flexible parts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank to Finnish Cultural Foundation (JM), Academy of Finland (grant no. 296292) and University of Oulu (Kvantum Institute) for financial support, as well as CSC-IT Center for Science (Espoo, Finland) and Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533) for computational resources.References
- I. Bertini, C. Luchinat and G. Parigi, Solution NMR of Paramagnetic Molecules: Applications to Metallobiomolecules and Models, Elsevier Science, 1st edn, 2001, p. 376 Search PubMed.
- G. M. Clore and J. Iwahara, Chem. Rev., 2009, 109, 4108 CrossRef PubMed.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901 CrossRef.
- O. V. Yazyev and L. Helm, Eur. J. Inorg. Chem., 2008, 201 CrossRef.
- D. V. Hingorani, A. S. Bernstein and M. D. Pagel, Contrast Media Mol. Imaging, 2015, 10, 245 CrossRef PubMed.
- I. Bertini, V. Calderone, L. Cerofolini, M. Fragai, C. F. G. C. Geraldes, P. Hermann, C. Luchinat, G. Parigi and J. M. C. Teixeira, FEBS Lett., 2012, 586, 557 CrossRef PubMed.
- J. Kowalewski, D. Krug and G. Parigi, Adv. Inorg. Chem., 2005, 57, 41 CrossRef.
- J. Kowalewski and L. Mäler, Nuclear Spin Relaxation in Liquids: Theory, Experiments, and Applications, Taylor & Francis, 1st edn, 2006, p. 440 Search PubMed.
- L. Benda, J. Mareš, E. Ravera, G. Parigi, C. Luchinat, M. Kaupp and J. Vaara, Angew. Chem., Int. Ed., 2016, 55, 14713 CrossRef PubMed.
- A. Soncini and W. Van den Heuvel, J. Chem. Phys., 2013, 138, 021103 CrossRef PubMed.
- J. Vaara, S. A. Rouf and J. Mareš, J. Chem. Theory Comput., 2015, 11, 4840 CrossRef PubMed.
- R. J. Kurland and B. R. McGarvey, J. Magn. Reson., 1970, 2, 286 Search PubMed.
- V. V. Novikov, A. A. Pavlov, A. S. Belov, A. V. Vologzhanina, A. Savitsky and Y. Z. Voloshin, J. Phys. Chem. Lett., 2014, 5, 3799 CrossRef PubMed.
- B. Martin and J. Autschbach, J. Chem. Phys., 2015, 142, 054108 CrossRef PubMed.
- S. A. Rouf, J. Mareš and J. Vaara, J. Chem. Theory Comput., 2017, 13, 3731 CrossRef PubMed.
- R. Di Remigio, M. Repisky, S. Komorovsky, P. Hrobarik, L. Frediani and K. Ruud, Mol. Phys., 2017, 115, 214 CrossRef.
- F. Neese, ORCA, An ab initio, density functional and semiempirical program package, Version 3.0.1, 2012.
- A. V. Arbuznikov, J. Vaara and M. Kaupp, J. Chem. Phys., 2004, 120, 2127 CrossRef PubMed.
- J. Novotný, D. Přichystal, M. Sojka, S. Komorovsky, M. Nečas and R. Marek, Inorg. Chem., 2018, 57, 641 CrossRef PubMed.
- S. A. Rouf, J. Mareš and J. Vaara, J. Chem. Theory Comput., 2015, 11, 1683 CrossRef PubMed.
- TURBOMOLE V7.0.2 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, TURBOMOLE GmbH: 1989–2007, since 2007. Available from http://www.turbomole.com, accessed January 30, 2018.
- W. E. Blumberg, in Magnetic Resonance in Biological Systems, ed. A. Ehrenberg and B. Malmström, Pergamon Press, Oxford, 1st edn, 1967, p. 110 Search PubMed.
- F. Neese, in Calculation of NMR and EPR Parameters: Theory and Applications, ed. M. Kaupp, M. Bühl and V. G. Malkin, Wiley-VCH, 1st edn, 2004, p. 541 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157 CrossRef.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138 CrossRef.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297 CrossRef.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252 CrossRef.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89 Search PubMed.
- B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3742 CrossRef.
- D. Ganyushin and F. Neese, J. Chem. Phys., 2006, 125, 024103 CrossRef PubMed.
- D. Ganyushin and F. Neese, J. Chem. Phys., 2013, 138, 104113 CrossRef PubMed.
- A. Kubica, J. Kowalewski, D. Kruk and M. Odelius, J. Chem. Phys., 2013, 138, 064304 CrossRef PubMed.
- S. Khan, A. Kubica-Misztal, D. Kruk, J. Kowalewski and M. Odelius, J. Chem. Phys., 2015, 142, 034304 CrossRef PubMed.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908 CrossRef PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef.
- S. Tsuzuki, T. Uchimaru and K. Tanabe, Chem. Phys. Lett., 1995, 246, 9 CrossRef.
- M. Bühl, C. Reimann, D. A. Pantazis, T. Bredow and F. Neese, J. Chem. Theory Comput., 2008, 4, 1449 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef PubMed.
- J. Harriman, Theoretical Foundations of Electron Spin Resonance, Academic Press Inc, 1978 Search PubMed.
- M. Kaupp and F. H. Köhler, Coord. Chem. Rev., 2009, 253, 2376 CrossRef.
- H. M. McConnell and D. B. Chesnut, J. Chem. Phys., 1958, 28, 107 CrossRef.
- J. Chyba, M. Novák, P. Munzarová, J. Novotný and R. Marek, Inorg. Chem., 2018, 57, 8735 CrossRef PubMed.
- P. Hrobárik, R. Reviakine, A. V. Arbuznikov, O. L. Malkina, V. G. Malkin, F. H. Köhler and M. Kaupp, J. Chem. Phys., 2007, 126, 024107 CrossRef PubMed.
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Mag. Res. Sp., 2018 DOI:10.1016/j.pnmrs.2018.05.001.
- E. A. Suturina and I. Kuprov, Phys. Chem. Chem. Phys., 2016, 18, 26412 RSC.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087 CrossRef.
- H. Flyvbjerg and H. G. Petersen, J. Chem. Phys., 1989, 91, 461 CrossRef.
- M. Sundararajan, D. Ganyushin, S. Ye and F. Neese, Dalton Trans., 2009, 6021 RSC.
- D. Maganas, S. Sottini, P. Kyritsis, E. J. J. Groenen and F. Neese, Inorg. Chem., 2011, 50, 8741 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular structures, tables and plots with numerical results, additional theory definitions. See DOI: 10.1039/c8cp04123g |
| This journal is © the Owner Societies 2018 |