
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A molecular dynamics model for glycosylphosphatidyl-inositol anchors: “flop down” or “lollipop”?†
Pallavi
Banerjee‡
,
Marko
Wehle‡§
,
Reinhard
Lipowsky
 and
Mark
Santer
*
and
Mark
Santer
*
Department of Theory and Biosystems, Max Planck Institute for Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: mark.santer@mpikg.mpg.de; Fax: +49 (0)331 567 9602; Tel: +49 (0)331 567 9610
First published on 9th November 2018
Abstract
We present a computational model of glycosylphosphatidyl-inositol (GPI) anchors for molecular dynamics studies. The model is based on state-of-the-art biomolecular force fields from the AMBER family, employing GLYCAM06 for carbohydrates and Lipid14 to represent fatty acid tails. We construct an adapted glycero-phosphatidyl-inositol unit to establish a seamless transition between the two domains of atom types. This link can readily be extended into a broad variety of GPI variants by applying either domain's building block scheme. As test cases, selected GPI fragments inserted into DMPC and POPC bilayer patches are considered. Our results suggest that the glycan part of the GPI anchor interacts strongly with the lipid head groups, partially embedding the carbohydrate moieties. This behaviour is supported by the conformational preferences of the GPI anchor, which in particular are conveyed by the strong interactions between the proximal amine and phosphate groups. In a similar way we can conclude that the extension of the anchor away from the lipid bilayer surface that could prevent the contact of the membrane with an attached protein (“lollipop picture”) is quite unfavorable. Indeed, when attaching green fluorescent protein to the GPI anchor, it is found to reside close to bilayer surface all the time, and the rather flexible phosphoethanolamine linker governs the extent to which the protein directly interacts not only with the head groups, but also with its own GPI core.
Introduction
Glycosylphosphatidylinositols (GPIs) are complex glycolipids present in eukaryotic cells, typically covalently bound to the C-terminus of proteins via a phospho-ethanolamine unit.1They primarily serve to anchor proteins to the outer leaflet of the cell membrane, where a Manα(1→2)-Man-α(1→4)-GlcN-α(1→6)-myo-inositol pseudopentasaccharide core bridges towards a lipid tail, which in turn is inserted into the plasma membrane. Fig. 1 shows a schematic of a minimal complete structure of a GPI-anchored protein. The sequence of four monosaccharides consisting of three mannoses and one glucosamine plus the trailing myo-inositol (blue) is referred to as the GPI core, the sequence without inositol is termed the GPI backbone. The core is a conserved part in almost all naturally occurring types of GPI-anchors, and it can be modified by various types of residues or side chains, the molecular weight of which may exceed that of the core itself.
 | ||
| Fig. 1 Chemical structure of a GPI anchored protein. The GPI anchor consists of three mannoses Man1–Man3, a glucosamine- (GlcN) and an myo-inositol (Ino) residue linked to a phospho-glycero-lipid (PGL). The highlighted inositol (Ino), phosphate (P) and glycerol (G) moieties indicate the bridge between force field domains, see text. | ||
Indeed, the vast number of possible side chain modifications including branched oligosaccharides that occur in nature suggests a rather broad functionality beyond the role of a mere anchoring device. GPIs are thought to be involved in localization of their proteins in membrane microdomains, commonly referred to as lipid rafts,2,3 and bring proteins into close proximity with other raft-associated species to enable their interactions, which underline and determine diverse processes such as signal transduction,4,5 cell adhesion protein trafficking and sorting,6,7 and antigen presentation.8 For all of these possible functions, a key aspect is how the GPI-anchored proteins distribute within the plasma membrane. In nerve cells, GPI-anchored Thy-1 and prion proteins have been suggested to form clusters in membrane domains with distinct lipid composition.9 In general, the usually saturated alkyl chains of the anchor should favor association with membrane domains rich in cholesterol or other saturated phospho- and glycolipids, glycosphingolipids and sphingomyelin,7 although there are indications that GPI-anchored proteins may also be distributed by non-equilibrium driving forces.10 Although intuitively appealing, pictures such as raft association or trafficking are still intensely debated,11,12 and in fact this conception has seriously been challenged by recent experiments of Schütz and coworkers.13
A major reason for the persisting controversies is that there is no clear molecular scale picture of how the complex of attached protein and the glycolipid anchor is embedded within and interacts with the local membrane environment. Experimental evidence for naturally occurring GPI-anchored proteins is scarce or ambiguous. From cryo-TEM studies with purified GPI-alkaline phosphatase (GPI-AP) inserted into liposomes14 it was concluded that the glycan part should fill the space between protein and lipid, reminiscent of the protective coat of Trypanosoma brucei.15 At ambient conditions in solution, a rather close proximity of AP to the lipid head group region has been inferred,16 suggesting a mutual interaction of glycan, lipid and protein.17–19 The seemingly opposing pictures are not necessarily a contradiction: in liposomes, the content of reconstituted protein or membrane composition and morphology are difficult to control. In addition, the glycan content of a GPI anchor is not exactly defined, for its composition may repetitively be remodeled during a protein's life cycle.20
Ideally a comprehensive characterization of GPI membrane embedding should proceed in a quasi-synthetic, bottom-up fashion starting from sufficiently simple and defined model systems. For instance, by creating a series of GPI-analogues attached to green fluorescent protein (GFP) and studying GPI–GFP diffusivities in supported lipid bilayers21 and cells in vivo,22 Bertozzi and coworkers conclude that high lateral mobility should be attributed to a rather stiff anchor preventing intermittent interactions with the membrane and increased protein–membrane contacts to more flexible anchor structures. In the former case GPI–GFP would roughly resemble a “lollipop”; in the latter, it can be thought to “flop down” onto the membrane, a picture introduced by Sharom and Lehto,16 see also Fig. 2. Exploring molecular scale details of GPI placement, however, even with elaborate NMR approaches inevitably requires computational methodology.23–25 With the present work we want to study the embedding of GPI anchors in lipid bilayers with atomistic, molecular dynamics (MD) simulations. To facilitate modular buildup and accurate representation of either molecular species, we shall construct a hybrid computational model by combining two state-of-the-art force fields from the AMBER family, GLYCAM0626 (glycans) and Lipid1427 (lipids). However, unlike simply “stitching together” a carbohydrate and a lipid via some ad hoc procedure, we shall specifically parametrise a molecular hub (highlighted in Fig. 1) that may readily be extended to various different GPI topologies simply by applying either force field's building block scheme. In this way, a coherent source of atomistic models is provided allowing to walk the “synthetic route” computationally. In order to keep the investigation at a manageable level in the spirit of a bottom-up approach, we shall restrict most of the exposition to anchor fragments up to and including Man3, compare Fig. 1. In the last section, we shall give a preliminary account of a fully fledged model of GPI-anchored green fluorescent protein (GFP) including the phosphoethanolamine (PE-) linker; the results largely support the major conclusions: the basic appearance of the embedded GPI-anchor can be understood in terms of rather simple molecular-mechanical arguments suggesting that a flop-down picture should dominate; in DMPC and POPC bilayers configurations with all glycan moieties stretching away from the solvent–headgroup interface are rare. Fig. 2 illustrates the “flop down” vs. the “lollipop” picture with specifically selected simulation snapshots of the GPI anchor model developed in this work. The flop down, however, can be realized in a number of ways, depending on the extent to which the glycan part intercalates with the head groups. Before analysing this behaviour in detail, we shall first discuss the computational model and how GPI conformations may be characterised before bilayer insertion. We will also comment on how the hybrid model may be validated and/or calibrated further especially along with our results on GPI–GFP, which gives some clues to experimental validation.
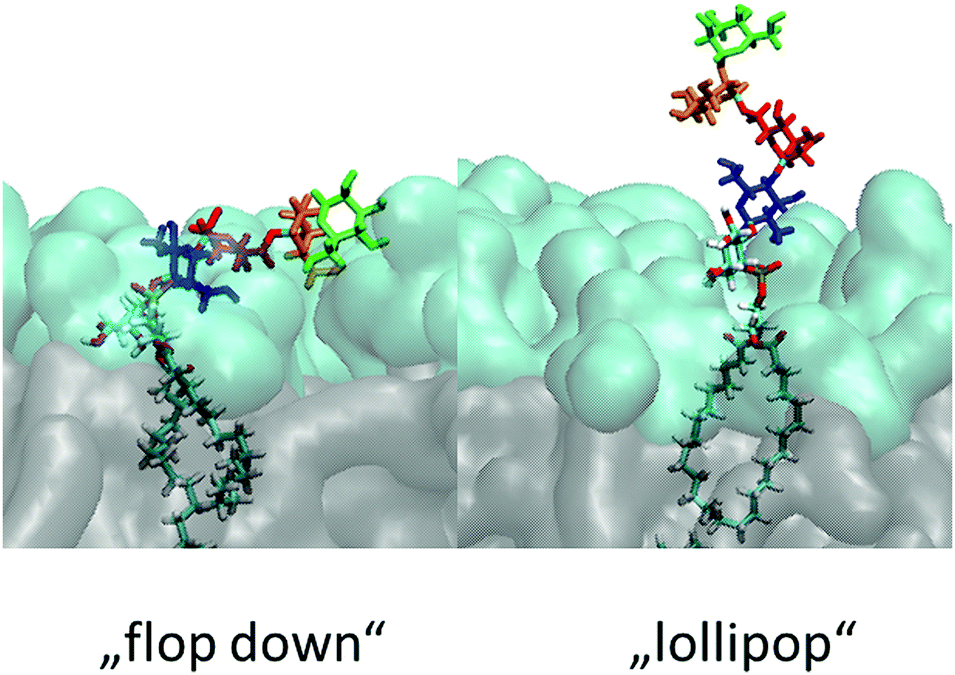 | ||
| Fig. 2 Illustration of the “lollipop” vs. the “flop down” picture as snapshots taken from the same MD simulation in a DMPC bilayer (upper leaflet). Color code (cmp. Fig. 1) green: Man3, orange: Man2, red: Man1, blue: GlcN, followed by phosphoinositol and a di-myristoyl-acylglycerol lipid tail. | ||
Computational approach and model validation
To our knowledge there are only very few computational studies involving GPI anchors or GPI anchored proteins. Homans et al. gave an early structural account of the GPI anchor covering the surface of Trypanosoma brucei,15 Zuegg and Gready28 investigated the flexibility of an isolated prion protein attached to a simplified model of a lipid monolayer. Lomás and coworkers have studied the conformational characteristics of the GPI core,23 and that of isolated GPI anchors inserted in small micelles,24 with short simulations in support of NMR data.In fact, providing a faithful model of a GPI anchored protein immersed in a cell membrane is a challenging endeavor, implying on the one hand that the characteristics of each type of biomolecule (protein, carbohydrate and lipid) must be represented with sufficient accuracy (with respect to stability, conformational preferences and phase behaviour, for instance).29 On the other, the mutual nonbonded interactions of the three species in close proximity must be calibrated, and the experimental evidence for such situations is scarce.30–33 A mere computational validation is rather difficult and subtle; in the scope of the present work we can only briefly indicate the state of the art of this subject (see, e.g., the discussion preceding the Conclusion section).
The former issue mainly requires specialization. In the Amber family of force fields, Lipid1427,34 has been developed to describe lipid bilayers in a stable and robust way; the focus of GLYCAM06 is devoted to modeling complex carbohydrates of almost arbitrary composition26,35 (compare the corresponding developments in the CHARMM force field36,37). The transferrability of GLYCAM06 (particularly important to the case of GPI anchors) required the adaption of bonded interactions and how they are encoded by a suitable (re-)definition of atom types. For creating a hybrid link that may be extended with both, GLYCAM06 and Lipid14 building blocks, we need to establish a smooth transition between the two domains of atom types that define torsion, angle bending and stretching potential terms. This is accomplished by selecting a suitable bridging fragment (highlighted in Fig. 1) and turning it into a molecule with appropriate methyl cappings, see Fig. 3(a). The transition between atom types occurs across the bond from the C1 carbon (type Cp, GLYCAM) to the phosphate oxygen (oT, Lipid14). χ0 denotes the corresponding torsion angle. Link (a) will be provided in two flavors: in addition to 6OMe-Ino-PGL(0) we define Ino-PGL(0) with bare inositol if the latter is the terminating head group moiety. This dual character is indicated in (b) with 6OMe-Ino-OMe and Ino-OMe, which play the key role for obtaining parameters of bonded interactions (e.g., χ0 and χ1) involving a mixed set of atom types (Cg, Cp pertaining to GLYCAM06, oT, pA and oP to Lipid14), and for defining partial charges as to comply with either domains building block principle. The path from (b) to (a) involves a number of somewhat lengthy and tedious steps and is outlined in detail in the ESI,† Section S1. To check for consistency, we compare the conformational preferences of χ0–χ2 obtained for Ino-POMe to either an all-GLYCAM06 or -Lipid14 parametrization presented in Section S2 (ESI†). These results prove to be rather valuable in order to categorize the plausible conformational preferences of the hybrid link, and then the full GPI anchor. We shall briefly summarize the essential findings:
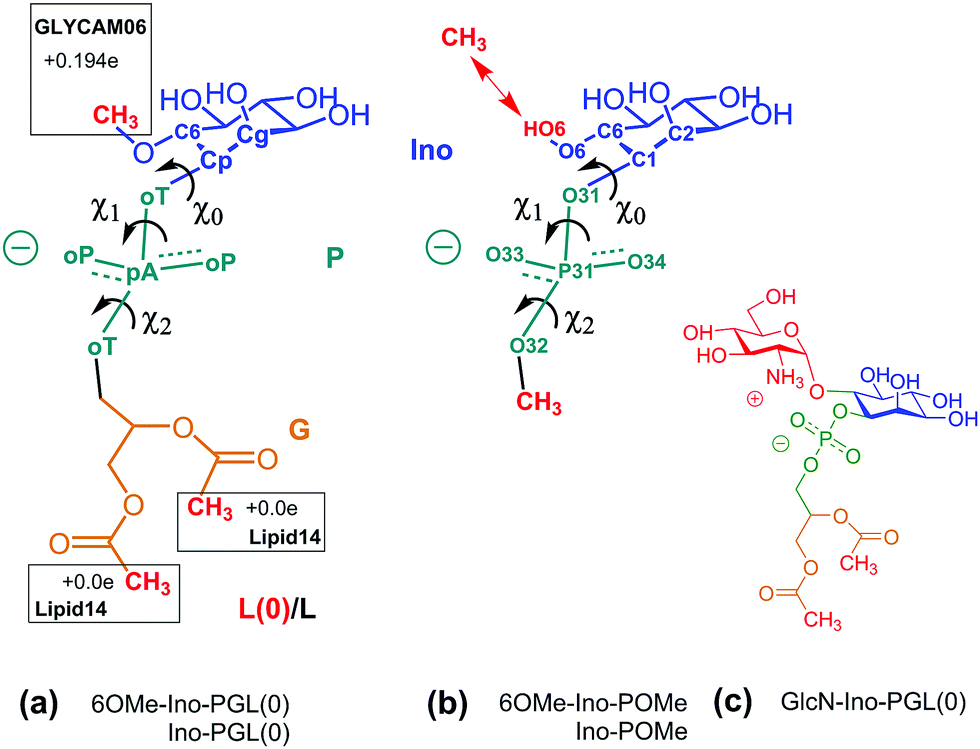 | ||
| Fig. 3 (a) 6OMe-Ino-PGL(0) and Ino-PGL(0) representing the desired hybrid link highlighted in Fig. 1, here with appropriate methyl cappings to be substituted with GLYCAM06 carbohydrate building blocks and various types of (Lipid14-) alkyl chains. L(0) indicates the bare link with methyl caps (the nomenclature with “L” will be used for a general alkyl tail extension). Net charges on caps are set as to comply with the respective building block principle. (b) Reduced fragment from (a), 6OMe-Ino-POMe and Ino-POMe. The schematic also illustrates the atom numbering used throughout this text. (c) GlcN-Ino-PGL(0), GlcN has been substituted for the methyl cap at the 6 position. | ||
(i) χ0–χ2 in the hybrid model of Ino-POMe are not affected by the type of partial charges assigned (ensemble averaged GLYCAM06 charges vs. AM1-BCC), see Fig. S3 (ESI†) for comparison. We use the AM1-BCC scheme as a convenient method to produce Lipid14-like partial charges, which rest on the standard two-stage RESP protocol in Amber.38 This observation is useful as it simplifies the compilation of reference compounds to compare effects of GLYCAM06 vs. Lipid14 parameters. In addition it facilitates further validation of fully fledged models for GPI anchors (see concluding discussion of the following section).
(ii) The behaviour of χ0 of the hybrid-, (obtained from MD in TIP3P solvent at ambient conditions), the all-GLYCAM06- and Lipid14 parametrisation is rather similar, major weights are given to intervals ∼[+60°,+90°] (within +synclinical or +sc orientation), and ∼[+150°,+180°] (within the antiperiplanar (ap) or trans-like domain of torsion angles). The distribution in χ0 exhibits two neighboring peaks, compare Fig. S4 and S5 in the ESI† (or S6, for direct comparison).
(iii) The distribution of χ1 in the hybrid model of Ino-POMe is very similar to that in the all-GLYCAM06 version exhibiting two peaks within −sc, +sc (close to ∼±90°), with slight preference for +sc (Fig. S4, ESI†). The all-Lipid14 version, by contrast, is different in that broad access is also given to torsions within ap. This can uniquely be attributed to the atom type assigned to the C1 carbon of the inositol ring: cA in Lipid14 (generic sp3 bonded carbon) and Cp in GLYCAM06 (sp3 carbon bonded to phosphate oxygen). The “cA–Cp exchange” does not impact χ0 see Fig. S6 and the corresponding discussion in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39
39
(iv) The torsions χ0–χ2 of the full link 6OMe-Ino-PGL(0) behave quite similarly to those of Ino-POMe; however, the substitution of glucosamine at the O6 oxygen (Fig. 3(c)) has a pronounced impact. The two peaks for χ0 merge and shift into ap; for χ1, major preference is now given +sc; for χ2, an asymmetry is established with a preference for +sc/−ac. This is solely due to the mutual intramolecular interaction of the proximal amine and phosphate groups, elucidated by a series of auxiliary simulations reported in Fig. S7 and the accompanying text in the ESI,† Section 2; see also the discussion related to Fig. S4–S6 (ESI†).
Observation (iv) is very useful as it allows us to single out a few particular GPI conformations that can be expected to occur with high probability. Fig. 4 illustrates the corresponding geometries, which have been obtained in a “molecular mechanics”-like fashion as follows: from an equilibrated DMPC bilayer a representative phospholipid tail was selected with both alkyl chains pointing along the z-direction, and the O32–P31 bond tilted by 45° with respect to the bilayer normal. This tail is common to structures (a)–(d). Structure (a) was then obtained by adding inositol and GlcN with (χ0,χ1,χ2) set to (175°,90°,90°) according to observation (iv). The (Ψ,Φ) glycosidic angles from GlcN to inositol exhibit only one narrow minimum around (70°,−150°). The distance distribution between amine and phosphate group (distance between nitrogen and phosphorous atom) has a maximum at 3.5 Å and a secondary one at 5.0 Å. The remaining three mannoses were added with glycosidic angles set according to their global free energy minima.41 Selecting the lowest energy values for all torsions furnishes the anchor with a hook-like appearance shown in (a) or (b), where the latter is obtained simply by setting the Ω-glycosidic angle in the Manα(1→6)Man linkage (captured by the orange ellipse in Fig. 4(a and b)) to the gt configuration, which should be equally or a little less populated than gg. The transition gg→gt twists the hydroxy-methyl group of Man3 approximately from −z to +z-direction.
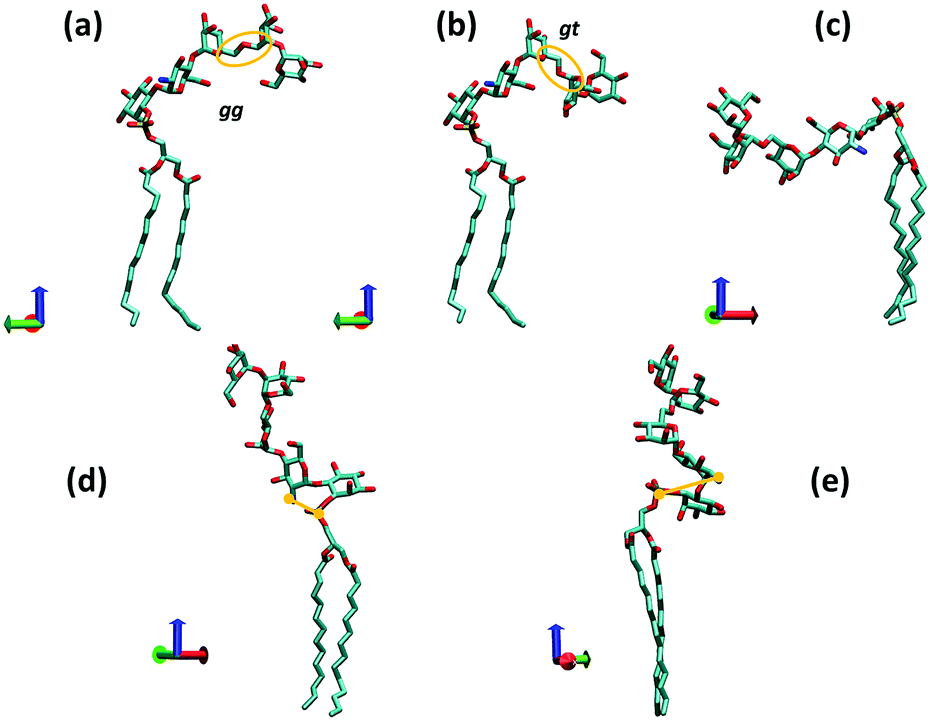 | ||
| Fig. 4 Various conformations of a complete GPI anchor derived from the torsional preferences of the hybrid fragment 6OMe-Ino-PGL(0) and those of the backbone. The red, green and blue coordinate axes indicate positive x, y and z directions, respectively (visualization with VMD40) (z indicating the direction of the bilayer normal). The bond P31–O31 is oriented roughly at 45° w.r.t. the z-axis. In (a) and (c), the Ω glycosidic angle of the 1→6 linkage (orange ellipse) is set to the gauche–gauche (gg), in (b) and (e) to gauche–trans (gt) and in (d) to the trans–gauche (tg) orientation. In (d) and (e), the distance between phosphorous atom P31 and the nitrogen of GlcN is indicated. | ||
(c) is derived from (a) by setting (χ0,χ1,χ2) to (175°,−90°,−90°) that is, choosing for both, χ1 and χ2 the values that are less likely (compare Fig. S7, ESI†). In (d) we have stretched out the GPI core as much as possible (Ω in Manα(1→6)Man linkage set to the unfavorable trans gauche and Manα(1→2)Man tweaked to a high energy conformation) and setting (χ0,χ1,χ2) to (90°,90°,90°) that is, unfavorable for χ0. In this conformation the amine and phosphate group are rather close (nitrogen–phosphorous distance about 3.1 Å) such that steric conflicts can be expected. Twisting the two charged groups away from each other as shown in (e) requires adjustment of the GlcN-Ino glycosidic angles, elevating the internal molecular energy as well. From these considerations one may predict that a lollipop-like conformation (d and e) should be rare or rather overwhelmed by variations of the hook-like appearance (a and b). In the following section we will compare the corresponding behavior with molecular dynamics and lipid bilayer systems.
GPI-anchors embedded in lipid bilayer patches
Simulation setup
For MD simulations in small DMPC- and POPC bilayer patches we consider the GPI-anchor fragments displayed in Fig. 5. (a) has been modeled from the link Ino-PGL(0), (b) and (c) from 6OMe-Ino-PGL(0). As simulation engines Amber1442 as well as GROMACS v. 4.6.4 were employed43,44 the latter in the majority of cases for running long (microsecond) bilayer simulations. All simulations pertaining to GROMACS were run using the CPU as well as the GPU version. To translate Amber/GLYCAM- to GROMACS molecule topologies and input files we utilized a script originally devised by Sorin and Pande45 and adapted by us41 to cope with the specificities of GLYCAM06. All simulations were carried out under standard conditions at 303 K with the TIP3P water model with pressure maintained at 1 bar using semi-isotropic Berendsen rescaling46 in an orthorhombic simulation box. Removal of center of mass motion was applied to the bilayer system as a whole. The species (a–c) were inserted through a number of separate steps. First a quadratic bilayer patch of 2 × 64 (8 by 8) phospholipid molecules was prepared with surface area of 100 Å2 per lipid. At this stage one lipid per leaflet was deleted and replaced with a corresponding GPI molecule, keeping the symmetry of the simulation setup. The latter was pulled out a few Å in z-direction as to avoid any initial steric clashes of the glycan part with the PC head groups. In addition, the glycan portion was twisted away from the bilayer surface as much as possible by manually adjusting the torsion angles χ0–χ2 and all subsequent glycosidic linkages, obtaining a structure close to that shown in Fig. 4(e). In this way some excess internal energy is provided at the start of the simulation procedure. The whole system is then solvated with TIP3P waters taken from pre-equilibrated solvent boxes. Waters placed within the region of alkyl chains were subsequently removed. This fact and the different sizes of (a) to (c) lead to slight variations in the final system size with average extensions of 6.5 nm, 6.5 nm and 13 nm in x, y and z-direction, respectively, ∼66500 atoms in total, and ∼17500 water molecules. In the case of Ino-PGL (a) one Na+ counterion was added to the system. We used a plain cutoff of 1 nm for vdW interactions and the same value was used as the real space cutoff in the particle mesh ewald (PME) electrostatics, which was used througout.
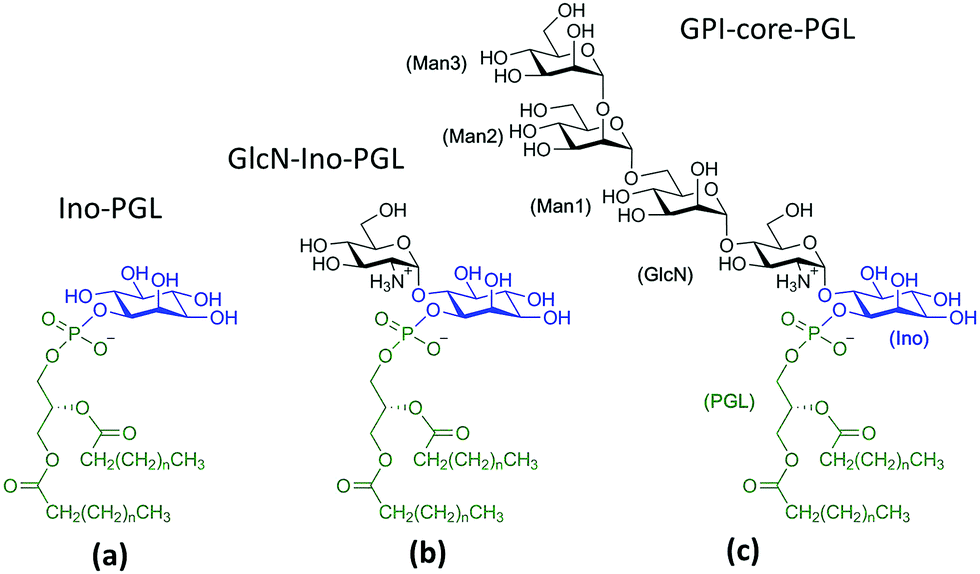 | ||
| Fig. 5 GPI fragments studied within DMPC and POPC lipid bilayers, created by replacing the methyl cappings of the respective links with saturated myristoyl alkyl chains (corresponding to n = 11 in the schematic) taken from the Lipid14 database. Parameters for GlcN have been taken from Singh et al.61 All other glycan building blocks have either been assembled from the GLYCAM06 force field modification file or taken from the GLYCAM web database (http://www.glycam.org). | ||
Initially, all lipids were subject to soft harmonic restraints with respect to their center-of-mass motion to allow the bilayer to soak with solvent during an initial period of 100 ps at 100 K during which bad contacts were resolved, using a Langevin-type stochastic integrator47 with a collision time of 1 ps. Subsequently, the temperature was ramped up to 300 K during another 100 ps, again removing water molecules that penetrated into the domain of alkyl chains. Then the system was allowed to relax with pressure control on (pressure relaxation time of 2 ps) at 1 bar. The area per lipid quickly (well within 10 ns) relaxed to its equilibrium value, and remained there for the total simulation time in each case. Reference simulations with temperature controlled by a Nosé–Hoover chain thermostat48 resulted in the same behavior. Snapshots were written out at an interval of 0.25 ns resulting in a total of 4000 frames per μs, of which we omit the first 50 ns (equilibration period) leaving 3801 for data analysis.
In Fig. 6 we show the time-dependent area per lipid for systems (a–c) in DMPC and POPC bilayers, respectively. The panel for DMPC indicates the evolution of GPI-core-PGL from the initial erected- (snapshot taken at 0.3 ns) to a flop-down conformation where the GPI backbone appears to point roughly parallel to the bilayer plane.
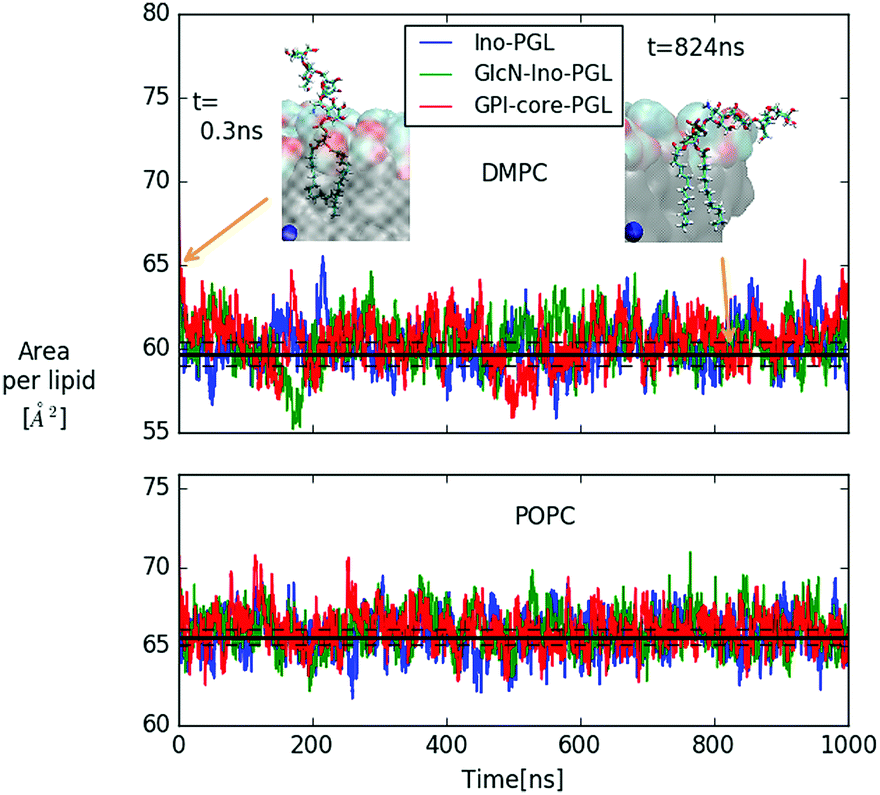 | ||
| Fig. 6 Area per lipid of DMPC and POPC bilayer systems with embedded GPI-anchor fragments displayed in Fig. 5; color coding for POPC is the same as in the upper panel. The black lines denote average and error quoted by Dickson et al.27 for the case of pure DMPC (59.7 ± 0.7 Å2) and POPC (65.6 ± 0.5 Å2) bilayers, respectively. For DMPC, the snapshots of GPI-core-PGL of the upper leaflet are displayed for the time points indicated by arrows. Blue spheres mark the bilayer center. To improve visibility, in the snapshot at 824 ns half of the bilayer has been removed. | ||
General appearance of GPI conformations within bilayers
In the previous section it was argued that for GPI-core-PGL hook-like conformations arise through the special preferences of torsion angles χ0–χ2, induced by intramolecular interactions between amine and phosphate group. We can essentially make the same observation as described in (iv) with GPI fragments (a–c) in Fig. 5, but now embedded within the bilayers, see Fig. 7. As soon as GlcN is included, the bimodal distribution for χ0 merges and shifts into +ap, almost complete emphasis for χ1 on +sc, and an asymmetry of χ2 with emphasis on +ac/+ap configurations, the orientation of the O31–P31 bond shows its maximum around 45° (see insets). Indeed, in the simulation runs we observe (with the exception of the “submarine”, see below) a pronounced occurrence of hook-like conformations for GPI-core-PGL in each leaflet, that is, the GPI core mostly tends to “flop down” onto the bilayer surface. Fig. 8 displays how similar conformations to the ones shown in Fig. 4 are assumed within the lipid environment.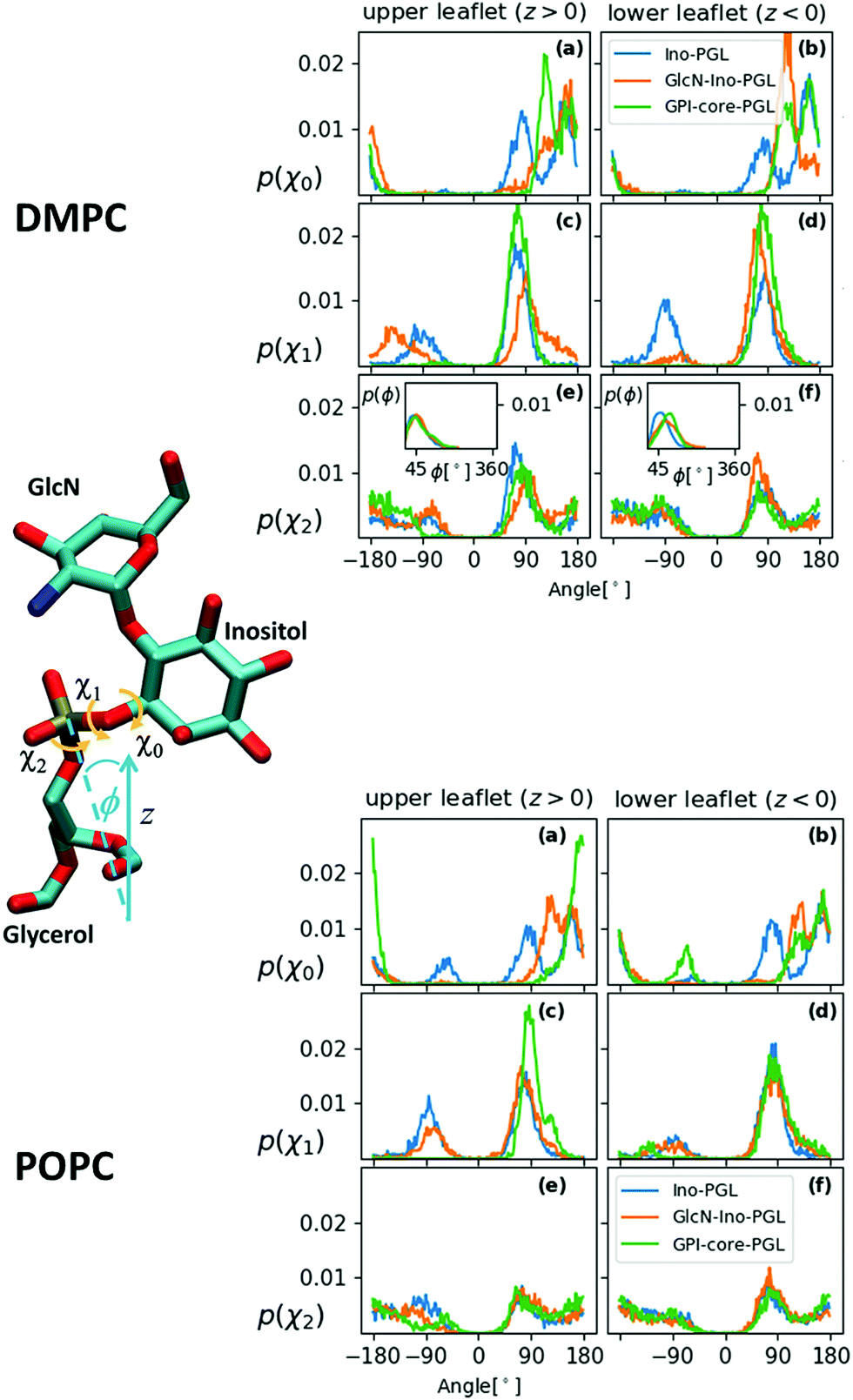 | ||
| Fig. 7 Distribution of the torsion angles χ0, χ1 and χ2 for all three GPI anchor derivatives embedded in the DMPC- (top panel) and POPC- (bottom panel) bilayer system. The bin width of all histograms in is 0.1°, each acquired from data of 3801 simulation snapshots. The insets in (e) and (f) in the DMPC case show the distribution of the inclination angle ϕ of the bond defining χ2 with the z-axis (bilayer normal), see schematic to the left. | ||
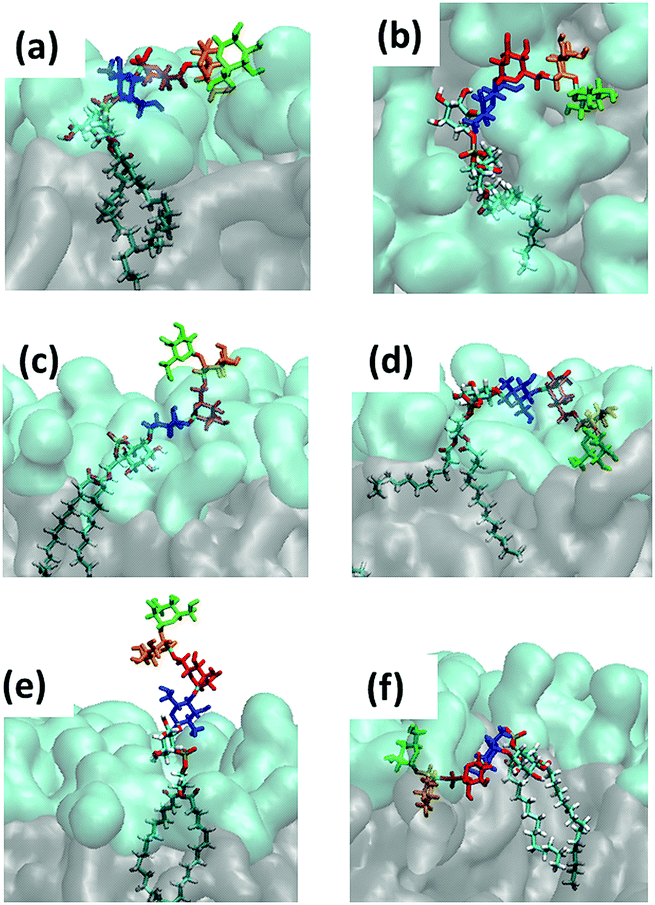 | ||
| Fig. 8 Snapshots of the characteristic GPI configurations characterized in the text, the time points at which they were acquired are indicated in Fig. 9. Surface representation: lipid layer excluding GPI-core-PGL, with PC head groups in cyan and glycerol and fatty acid chains in grey. (a) “swimmer”, viewed in vertical cross section through the bilayer, (b) same, but top view; (c) “submarine”; (d) “diver”; (e) phoenix; (f) “mole”. Color coding of carbohydrate moieties: Man3/green, Man2/orange, Man1/red, GlcN/blue. The “flop down” and “lollipop” in Fig. 2 are reproduced by (a) and (e), respectively. | ||
(a) reproduces the snapshot shown in Fig. 2, the conformation belongs to a class that we shall coin “swimmer”, because all carbohydrate moieties are well exposed to the solvent, situated at the interface between PC head groups and aqueous subphase, see also (b) for the corresponding top view. The swimmer can be realized by a number of variations of the conformations (a)–(c) in Fig. 4, in combination with the flexible glycerol moiety. However, there are also embeddings with stronger contact to the surrounding lipids, see for instance (d) where Man3 and Man2 insert into the head group region (“diver”). (f) shows an extreme situation (with a conformation close to Fig. 4(c)) where the GPI core is completely buried within the head groups, contacting the hydrophobic core of the bilayer with minimum exposure to water. This “mole” actually persists over the full simulation time of the lower leaflet of the POPC bilayer, and appears to be stabilized or kinetically trapped. A similar situation is observed with extended conformations. Fig. 8(c) shows the “submarine”: the stretched out GPI core persists for more than 500 ns, but only Man3 is well exposed to the solvent subphase. The submarine eventually turns into the “diver” at the end of the simulation run. The “phoenix” (e), which is close to the conformation (d) in Fig. 4, lasts as expected only for a few nanoseconds.
These (and in particular the last) examples show that it is not enough to distinguish between different conformations, but also between different levels of hydration. To fully capture the dynamics of the embedding the following observation is useful: the vertical positioning of all GPI species seems to be quite independent of conformation or carbohydrate content, see Fig. S8 and the corresponding discussion in the ESI,† Section S3. That is, in z-direction, the GPI phosphate resides at the level defined by all other phosphates in the lipid layers. We may thus picture the GPI core to move and reorient with respect to this reference level and to produce some characteristic time-dependence of the hydration pattern, complementary to conformational dynamics. This is analysed in the following.
Aspects of conformational dynamics
In addition to tracking the hydration level of all carbohydrate moieties we also consider the hydrogen bridges formed with the PC heads in each frame, see Fig. 9(a) and (b), respectively, where the time points of the snapshots in Fig. 8 have been annotated. The swimmer is characterized by an overall even exposure of all monosaccharide entities to the solvent, or very few h-bonds towards the PC heads. This situation is seen to prevail for the upper leaflets of the DMPC and POPC bilayer. The “phoenix” Fig. 8(e) observed in the upper DMPC leaflet (reproduced from Fig. 2) lasts for a few ns only; its hydration level is similar to that of the swimmer, yet its placement is unfavorable not only because of the molecular conformation, but also because the alkyl chains have to enter the hydrophilic region to some extent.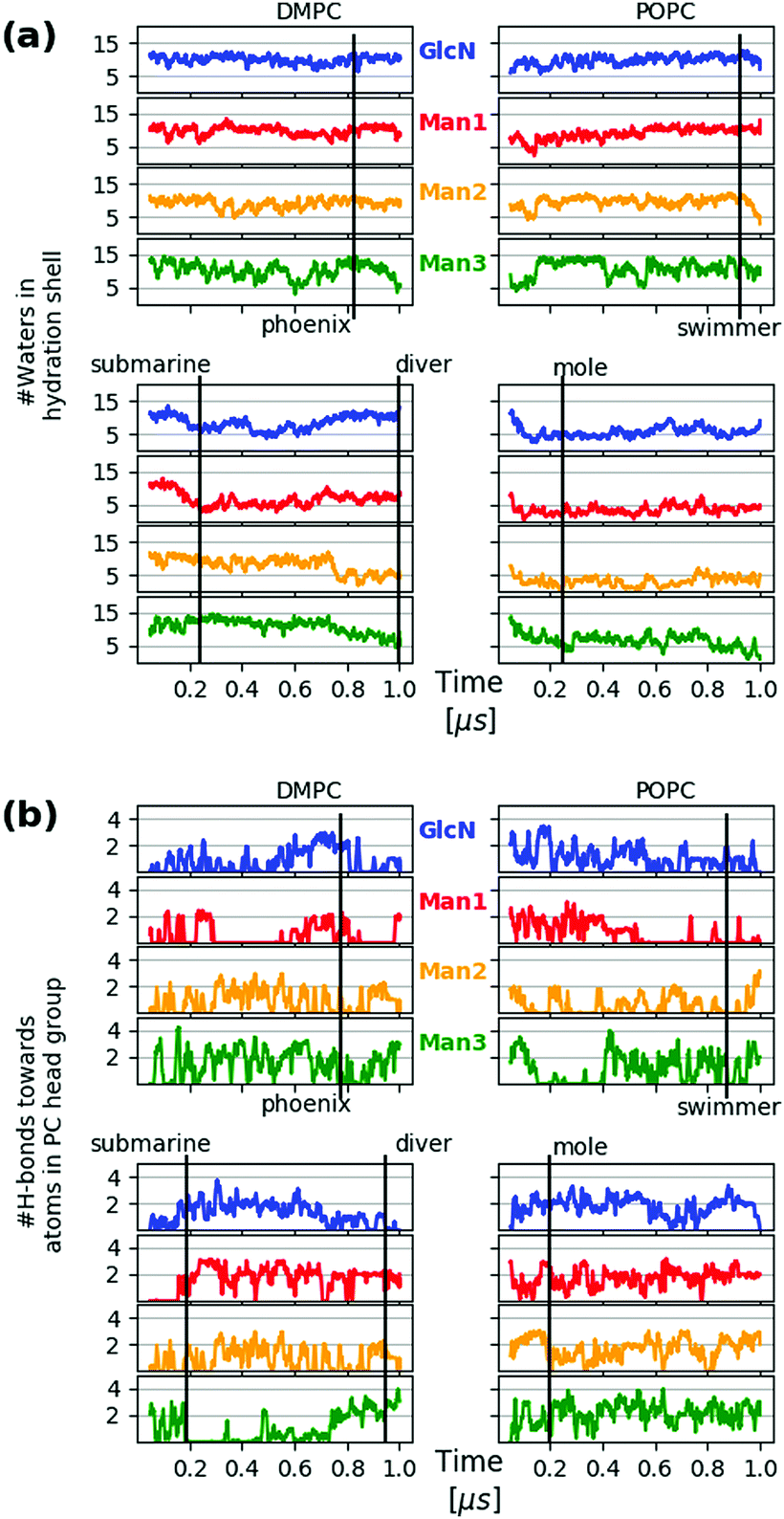 | ||
| Fig. 9 Hydration level of GPI core moieties Man3, Man2, Man1 and GlcN, color coding as in Fig. 8. The upper two panels of each block refer to the upper leaflet (z > 0) of the respective bilayer system. The hydration level for a specific frame at time t is defined as the number of water molecules within a shell of 3 Å thickness around the selected residue. The distance criterion is applied to water oxygen only. (b) Number of hydrogen bonds of each monosaccharide towards the PC head group atoms per frame. Both quantities in (a) and (b) have been subjected to a running average with a 20-frame window in order to remove excessive noise. | ||
The transition from submarine to diver is clearly visible in the lower left panels of (a) and (b), respectively: hydration is traded for h-bonds towards PC heads. The mole exhibits an even stronger interaction with the surrounding lipids and in both cases we expect that the conformational freedom of the GPI backbone itself is affected. This can be verified with the evolution of the end-to-end distance d(C4,C6) and the tilt angle θz of the core with respect to a vector defined by the C4 ring carbon of Man3, and C6 of inositol, see Fig. 10. For the submarine the tilt towards the z-axis persists for more than 0.6 μs (see inset) corresponding to the course of the hydration level in Fig. 9(a), until it gradually relaxes towards a lateral orientation. The mole shows little variation in tilt angle. Both configurations are additionally restrained with respect to end-to-end distance, which spans an interval of only 10–15 Å (see lower panel in Fig. 10) whereas the “swimmers” (green and blue graphs) can explore values of d(C4,C6) as low as 6 Å.
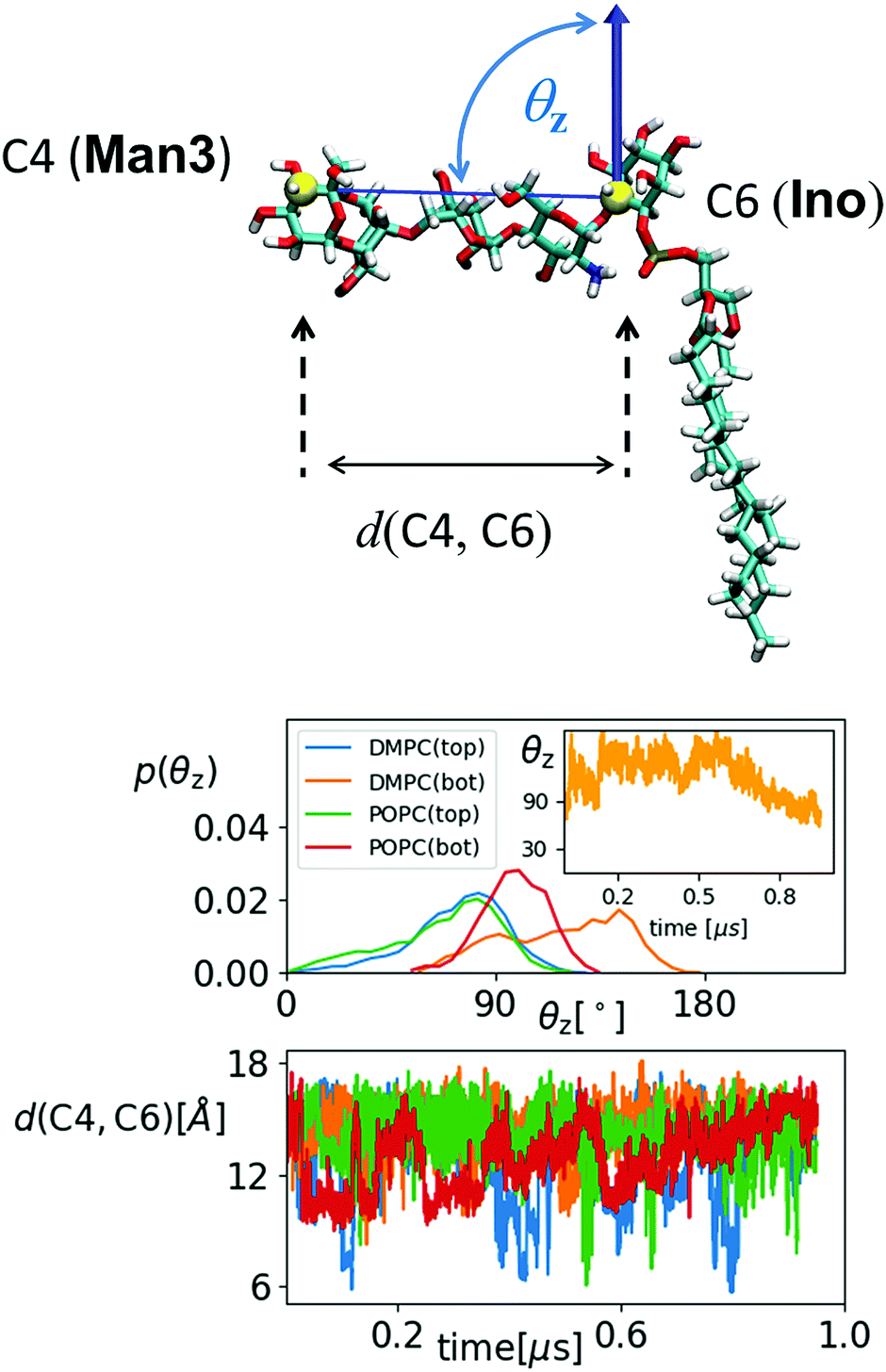 | ||
| Fig. 10 Top: Schematic for defining the end-to-end distance d(C4,C6) and the tilt angle θz of the glycan GPI core with respect to the bilayer normal. The distribution of θz is shown below, separately for upper (top) and lower (bot) leaflet. The inset shows the trajectory for DMPC (bot). The lower panel shows the time dependence of d(C4,C6) for each case using the same color code. | ||
How to validate the force field for embedded GPIs?
In the previous assessment it became clear that a flop-down appearance of the GPI core prevails. In terms of steric/geometric accessibility by a protein the swimmer as a specific example naturally appears as a more plausible placement than the mole. But to determine the relative stability between a buried and a more flexible placement certainly requires advanced sampling strategies49 beyond the scope of the present account. To judge whether the “mole” could eventually be ruled out requires yet additional work. As has already been pointed out in the computational approach, the mutual interaction of aminoacids/protein, carbohydrates and lipids is extraordinarily difficult to characterise experimentally and thus poorly explored computationally. Consequently, one cannot expect every aspect to be calibrated with highest accuracy. The general problem of adequately tuning nonbonded interactions in crowded environments has been identified as one of the major challenges of current force field development.50–52Variation of force field parameters within admissible limits is a one viable strategy to test the sensitivity (and thereby the plausibility) of a certain phenomenology.53,54 For instance, in discussing how the conformational preferences of the hybrid link Ino-POMe emerge, extensive use has been made of the fact that use of either GLYCAM06-type or AM1-BCC charges led to indistinguishable results, see e.g. Fig. S3 in the ESI.† That is, without seriously affecting aspects of molecular mechanics, the hybrid link to bridge GLYCAM06 and Lipid14 domains of atom types may also be viewed as a vehicle to test nonbonded interactions systematically for instance, by altering the charge scheme on the glycan moieties, exploring different vdW combination rules between carbohydrates and lipids,55etc.
Another possibility is to immediately go on with the synthetic route. In what follows, we shall explore what we can learn from suitably extending GPI-core-PGL and provide a first account of a fully fledged model of GPI-anchored green fluorescent protein (GFP). GFP is not a natural candidate as it lacks the typical motifs of GPI anchored proteins around their C-termini,56 but it is rather useful for several reasons. It is known to interact very little with other lipid components, and is frequently used as a marker for in vivo studies of cell membranes such as in the investigation of GPI analogues in supported lipid bilayers21,22 or different re-assembled GPI–GFP into live cells.57 Thus, GPI–GFP is a convenient starting point to study how the protein would act back on the GPI anchor and how the latter would influence orientation and proximity of the protein with respect to a membrane.
GPI-anchored green fluorescent protein
To construct a GPI anchored GFP we proceeded through the following steps. Coordinates for GFP were taken from the X-ray structure with PDB code 1EMA, and rendered a complete molecular topology using the tLeaP facility from the Amber suite. The phosphoethanolamine (PE) linker was parametrized with the GLYCAM06 force field defining a transition region between linker and GFP's C-terminus (Threonine) similar to the way the hybrid building block between GLYCAM06 and Lipid14 was derived. This is fully outlined in the ESI,† Section 4. The creation of the simulation setup followed along the same lines as described for the free GPI-anchor, except that now the bilayer patches are 16 by 16 phospholipids in size with only one phospholipid per bilayer (upper leaflet) replaced by a GPI-anchored GFP. The total number of atoms was 309634 and 311789 for the setup with DMPC and POPC lipids, respectively, and additional 7 Na+ ions were introduced to compensate GFP's charged residues. All other simulation conditions and parameters are as in the case of free GPIs. Initially, the GPI–GFP points away from the bilayer surface with its long axis parallel to the bilayer normal (z-direction). The long axis is relatively well defined by a vector running through the center of masses of residues 76 (glutamine) and 135 (histidine), see Fig. 11. Since a twisted configuration of GPI-core-PGL was chosen as initial configuration (compare Fig. 6), GFP and phosphoethanolamine linker initially had no contact to the bilayer surfaces. In the course of 1 μs long simulations for each bilayer system, the GPI core within 200–300 ns flops down onto the bilayer assuming a hook-like conformation reminiscent of the “diver”, with the GFP barrel resting on the head group region with the long axis pointing along the bilayer surface, compare Fig. 11(a); the “flop down”, remarkably, also allows for a completely upright orientation conveyed by the PE linker (b). Panel (c) shows the evolution of GFP orientation for DMPC and POPC. Both simulations indicate that the parallel orientation (the “sleeping GFP”) should dominate, but the re-orientational dynamics is quite slow, occurring roughly over 200 ns. We therefore restarted the simulation of GPI–GFP embedded in POPC from 6 different time points along the trajectory indicated by the dashed lines in (c), with new initial velocities (simulation time 200 ns each). The corresponding trajectories are shown in (d). Four of these show a tilt angle of smaller than 60° initially. The trajectory in orange starts from the same initial configuration as the one in panel (c), but now relaxing somewhat faster to parallel orientation. After 200 ns, only in one run the protein still stands upright, in all other cases the sleeping GFP appears to evolve. In this way, our current model makes a rather clear prediction about the orientation of the GFP, and this could be verified experimentally by employing anisotropic fluorescence spectroscopy58 because the GFP determines the orientation of the chromophore inside.
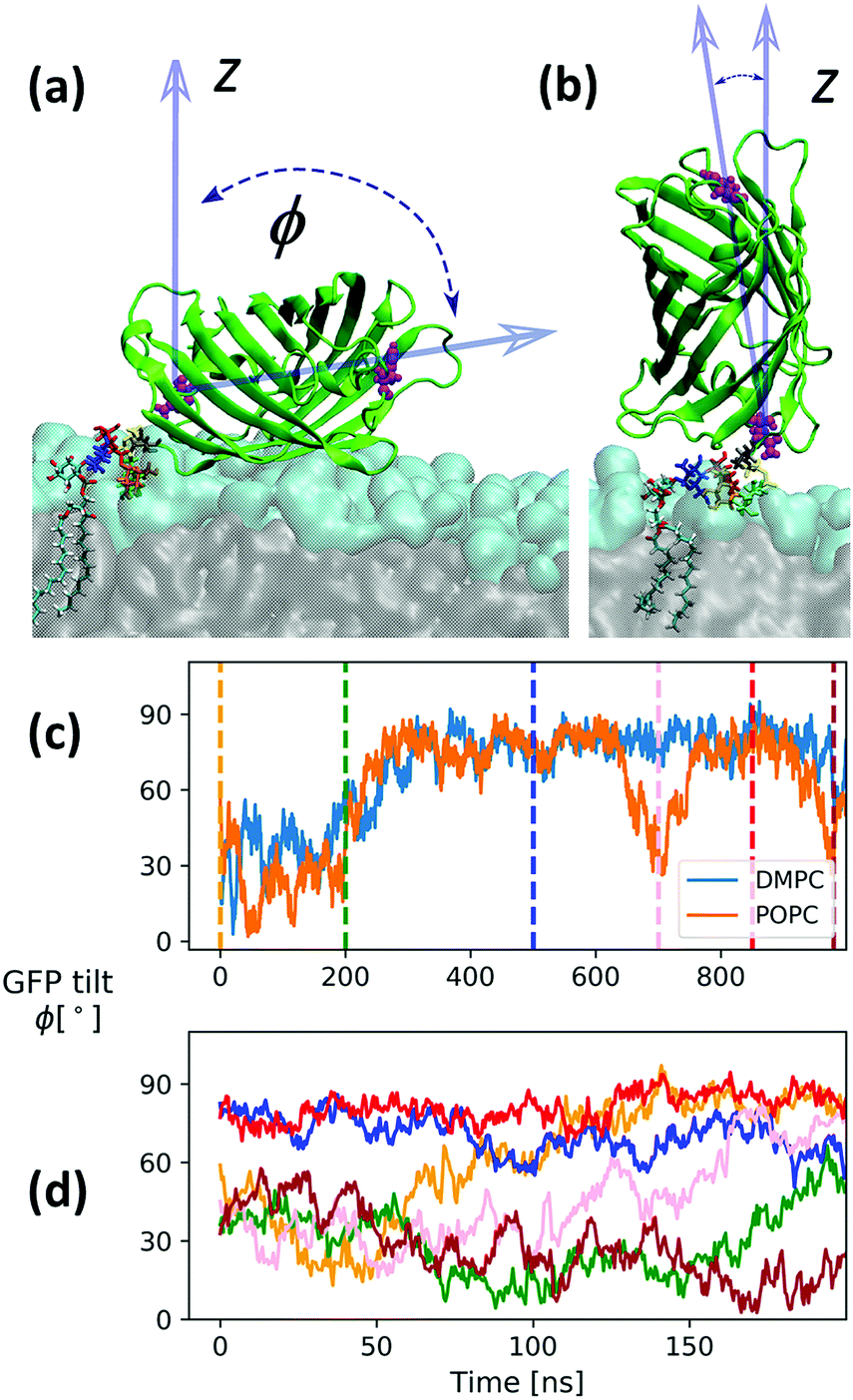 | ||
| Fig. 11 Snapshots from a simulation with a full GPI–GFP inserted into a POPC bilayer as described in the text where the GFP “sleeps” on the head groups with its long axis pointing laterally (a) or stands upright (b), respectively. Panel (c) shows the corresponding evolution of long axis orientation for the GPI–GFP in POPC and DMPC, respectively. Panel (d) shows the dynamics of the long axis for trajectories restarted from the time points indicated in (c) (same color code). The times are 0 ns (initial, tweaked GPI configuration), 200 ns, 500 ns, 700 ns (snapshot (b)), 850 ns and 980 ns. | ||
We note that we verified that interaction of GFP with lipid–water interface is weak. When brought into initial contact with the head group region (sleeping or upright orientation) the GFP detaches from the bilayer surface within 100–200 ns driven by diffusion and bilayer undulations.
By analysing all trajectories of Fig. 11(d), we can make further observations:
(i) Fig. S9(a) in the ESI† shows that taken together, the orientation distribution of the end-to-end vector from C6 (inositol) to C4 (Man3) is in accord with those of the swimmer-like configurations seen to dominate in the POPC and DMPC top leaflet of the free GPI anchor GPI-core-PGL, compare Fig. 10; the attachment to the protein appears to stabilizes the end-to-end distance d(C4,C6) at ∼1.4 nm, see Fig. S9(b) (ESI†). However, unlike the swimmer of GPI-core-PGL where the Ω glycosidic angle of the αMan1→6Manα linkage between Man2 and Man1 exhibits mostly the gg rotamer, in GPI–GFP it is exclusively gt, compare also Fig. 4. This actually causes Man1 to stick out of the headgroups (compare red-colored Man1 in Fig. 11(a)) more than Man2 or Man3.
(ii) The GPI core does not notably influence GFP's orientation; it is by contrast the PE linker and its flexibility that allows the GFP to explore a range of different orientations. It may even “twist back” to interact with in principle all glycan moieties. In Fig. S10 (ESI†) we characterise these interactions by displaying the dynamic formation of hydrogen bonds. The fact that an attached protein may quite extensively interact with its own GPI glycan could be a very important hint for possible NMR experiments where, for instance, a GPI-anchored protein is inserted into a well defined and suitable environment such as a small micelle24 or a phospholipid nanodisc.59
Conclusions
We can state that the hybrid GPI anchor models developed in this work turn out to be quite useful. They exhibit a broad variety of conformational modes by which the GPI glycan can interact with the headgroup–solvent interface facilitated by the dominating hook-like appearance of GPI-core-PGL, which causes the GPI core glycan to flop down onto the lipid bilayer but nevertheless retains a high degree of internal flexibility. This may be contrasted to other glycolipid species such as the ganglioside GM3, which is inherently stiffer and rich in β-type glycosidic linkages, making the glycan headgroup point away from the bilayer surface.25 In contrast we can conclude that a “lollipop” picture of a GPI anchor (represented, e.g., by the “phoenix”) is rather implausible. The preference of GPI-core-PGL for hook-like conformations can be expected to be a robust and generic phenomenon, as it is in particular conveyed by the proximity and mutual intramolecular interaction of amine- and phosphate groups, a mechanism retained after attaching GFP. The proximity to the bilayer surface and the flexibility of orientation conveyed by the phospho-ethanolamine linker permit the GFP to interact quite extensively with its own GPI-glycan. Naturally the question arises about the consequences of adding various side chains to Man1 such as in the case of, e.g., Toxoplasma gondii.60 Whether a specific side chain would impose a certain preference of orientation or stabilize a favorable protein fold are then indeed aspects that could be studied numerically and experimentally giving valuable insights into the general purpose of GPI anchoring.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the International Max Planck Research School (IMPRS) for Multiscale Biosystems for support. Open Access funding provided by the Max Planck Society.References
- M. G. Paulick and C. R. Bertozzi, Biochemistry, 2008, 47, 6991–7000 CrossRef CAS PubMed.
- K. Simons and D. Toomre, Nat. Rev. Mol. Cell Biol., 2000, 1, 31–39 CrossRef CAS PubMed.
- E. Klotzsch and G. J. Schütz, Philos. Trans. R. Soc., B, 2013, 368, 20120033 CrossRef PubMed.
- M. G. Low and A. R. Saltiel, Science, 1988, 239, 268–275 CrossRef CAS PubMed.
- S. D. Tachado, S. D. Gerold, R. Schwarz, S. Novakovich, M. McConvill and L. Schofield, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4022–4027 CrossRef CAS.
- S. Mayor and H. Riezmann, Mol. Cell. Biol., 2004, 5, 110 CAS.
- D. A. Brown and J. K. Rose, Cell, 1992, 6, 533–544 CrossRef.
- Y. Maeda and T. Kinoshita, Prog. Lipid Res., 2011, 50, 411–424 CrossRef CAS PubMed.
- B. Brügger, C. Graham, I. Leibrecht, E. Mombelli, A. Jen, F. Wieland and R. Morris, J. Biol. Chem., 2004, 279, 7530–7536 CrossRef PubMed.
- D. Goswami, K. Gowrishankar, S. Bilgrami, S. Ghosh, R. Raghupathy, R. Chadda, R. Vishwakarma, M. Rao and S. Mayor, Cell, 2008, 135, 1085–1097 CrossRef CAS PubMed.
- S. Schuck and K. Simons, J. Cell Biol., 2006, 172, 963–965 CrossRef CAS PubMed.
- C. Eggeling, C. Ringemann, R. Medda, G. Schwarzmann, K. Sandhoff, S. Polyakova, V. N. Belov, B. Hein, C. von Middendorf, A. Schönle and S. W. Hell, Nature, 2009, 457, 1159–1163 CrossRef CAS PubMed.
- E. Sevcsik, M. Brameshuber, M. Fölser, J. Weghuber, A. Honigmann and G. J. Schütz, Nat. Commun., 2015, 6, 6969 CrossRef CAS PubMed.
- F. Ronzon, S. Morandat, B. Roux and M. Bortolato, J. Membr. Biol., 2004, 197, 167–177 CrossRef PubMed.
- S. W. Homans, C. J. Edge, M. A. J. Ferguson, R. A. Dwek and T. W. Rademacher, Biochemistry, 1989, 28, 2881–2887 CrossRef CAS PubMed.
- M. T. Lehto and F. J. Sharom, Biochemistry, 2002, 41, 8368–8376 CrossRef CAS PubMed.
- E. Barboni, B. P. Rivero, A. J. T. George, S. R. Martin, D. V. Renouf, D. F. Hounsell, P. C. Barber and R. J. Morris, J. Cell Sci., 1995, 108, 487–497 CAS.
- P. Bütikofer, T. Malherbe, M. Boschung and I. Roditi, FASEB J., 2001, 15, 545–548 CrossRef PubMed.
- F. J. Sharom, I. Lorimer and M. P. Lamb, Can. J. Biochem. Cell Biol., 1985, 63, 1049–1057 CrossRef CAS PubMed.
- M. Fujita and T. Kinoshita, Biochim. Biophys. Acta, 2012, 1821, 1050–1058 CrossRef CAS PubMed.
- M. G. Paulick, M. B. Forstner, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2007, 129, 11543–11550 CrossRef CAS PubMed.
- M. G. Paulick, M. B. Forstner, J. T. Groves and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20332–20337 CrossRef CAS PubMed.
- F. Chevalier, J. Lopez-Prados, S. Perez, M. Martín-Lomas and P. M. Nieto, Eur. J. Org. Chem., 2006, 3489–3498 Search PubMed.
- F. Chevalier, J. Lopez-Prados, P. Groves, S. Perez, M. Martín-Lomas and P. M. Nieto, Glycobiology, 2006, 16, 969–980 CrossRef CAS PubMed.
- M. L. DeMarco and R. J. Woods, Glycobiology, 2008, 18, 426 CrossRef CAS PubMed.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. González-Outeiriño, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS PubMed.
- C. J. Dickson, B. D. Madej, A. A. Skjevik, R. M. Betz, K. Teigen, I. R. Gould and R. C. Walker, J. Chem. Theory Comput., 2014, 10, 865–879 CrossRef CAS PubMed.
- J. Zuegg and J. E. Gready, Glycobiology, 2000, 10, 959–974 CrossRef CAS PubMed.
- This is the reason why we did not stay with GLYCAM06 to describe lipids as well (presented in Tessier 2008): under ambient conditions, the GLYCAM lipid was awkward to work with. Bilayers frequently entered gel-like phases, even after careful preparation.
- C. Stefaniu, I. Vilotijevic, M. Santer, D. Varon-Silva, G. Brezesinski and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 12874–12878 CrossRef CAS PubMed.
- C. Stefaniu, I. Vilotijevic, M. Santer, G. Brezesinski, P. H. Santer and D. Varón-Silva, Langmuir, 2014, 30, 5185–5192 CrossRef CAS PubMed.
- E. Schneck, T. Schubert, O. V. Konovalov, B. E. Q. T. Gutsmann, K. Brandenburg, R. G. Oliviera, D. A. Pink and M. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9147–9151 CrossRef CAS PubMed.
- K. N. Kirschner, R. D. Lins, A. Maass and T. A. Soares, J. Chem. Theory Comput., 2012, 8, 4719–4731 CrossRef CAS PubMed.
- A. A. Skjevik, B. D. Madej, R. C. Walker and K. Teigen, J. Phys. Chem. B, 2012, 116, 11124–11136 CrossRef CAS PubMed.
- R. J. Woods, R. A. Dwek, C. J. Edge and B. Fraser-Reid, J. Phys. Chem., 1995, 99, 3832–3846 CrossRef CAS.
- O. Guvench, E. Hatcher, R. M. Venable, R. W. Pastor and A. D. MacKerell Jr., J. Chem. Theory Comput., 2009, 5, 2353–2370 CrossRef CAS PubMed.
- R. W. Pastor and J. A. D. MacKerell, J. Phys. Chem. Lett., 2011, 2, 1526–1532 CrossRef CAS PubMed.
- A. Jakalian, B. L. Bush, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2000, 21, 132–146 CrossRef CAS.
- The corresponding carbon (bonded to the phosphate O32) of the glycerol moiety is left as cA because the division between force fields is defined as one between atom types; the specialization to a type such as Cp would be part of a general revision of the lipid force field.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. Wehle, I. Vilotijevic, R. Lipowsky, P. H. Seeberger, D. Varón-Silva and M. Santer, J. Am. Chem. Soc., 2012, 134, 18964–18972 CrossRef CAS PubMed.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, I. T. E. Cheatham,. T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. J. J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo,. B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui,. R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. W. J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER14, University of California, San Francisco, 2014 Search PubMed.
- D. V. der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef PubMed.
- B. Hess, C. Kutzner, D. V. der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- E. J. Sorin and V. S. Pande, Biophys. J., 2005, 88, 2472–2493 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- W. F. van Gunsteren and H. J. C. Berendsen, Mol. Simul., 1988, 1, 173–185 CrossRef.
- G. J. Martyna, M. L. Klein and M. J. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- L. Monticelli, E. J. Sorin, D. P. Tieleman and V. S. P. G. Colombo, J. Comput. Chem., 2008, 29, 1740–1752 CrossRef CAS PubMed.
- D. Petrov and B. Zagrovic, PLoS Comput. Biol., 2014, 10, e1003638 CrossRef PubMed.
- S. Rauscher, V. Gapsys, M. J. Gajda, M. Zweckstetter, B. L. de Groot and H. Grubmüller, J. Chem. Theory Comput., 2015, 11, 5513–5524 CrossRef CAS PubMed.
- D. J. Cole, J. Z. V. J. Tirado-Rives, M. C. Payne and W. L. Jorgensen, J. Chem. Theory Comput., 2016, 12, 2312–2323 CrossRef CAS PubMed.
- S. Abel, F.-Y. Dupradeau, E. P. Raman, A. D. MacKerell Jr. and M. Marchi, J. Phys. Chem. B, 2011, 115, 487–499 CrossRef CAS PubMed.
- J. A. Hadden, A. D. French and R. J. Woods, Cellulose, 2014, 21, 879–884 CrossRef CAS PubMed.
- T. A. Halgren, J. Am. Chem. Soc., 1992, 114, 7827–7843 CrossRef CAS.
- M. A. J. Ferguson, T. Kinoshita and G. W. Hart, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbour, New York, 2nd edn, 2009, ch. 11, pp. 143–161 Search PubMed.
- D. F. Legler, M.-A. Doucey, P. Schneider, L. Chapatte, F. C. Bender and C. Bron, FASEB J., 2005, 19 Search PubMed.
- J. V. Rocheleau, M. Edidin and D. W. Piston, Biophys. J., 2003, 84, 4078–4086 CrossRef CAS PubMed.
- I. G. Denisov, Y. V. Grinkova, A. A. Lazarides and S. G. Sligar, J. Am. Chem. Soc., 2004, 126, 3477–3487 CrossRef CAS PubMed.
- S. Götze, N. Azzouz, Y. H. Groß, A. Reinhardt, C. A. P. H. Seeberger and D. V. Silva, Angew. Chem., Int. Ed., 2014, 53, 13701–13705 CrossRef PubMed.
- A. Singh, M. B. Tessier, K. Pederson, A. P. Wang, A. P. Venot, G.-J. Boons, J. H. Prestegaard and R. J. Woods, Can. J. Chem., 2016, 94, 927–935 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp04059a |
| ‡ These authors contributed equally to this work. |
| § Present address: ITK Engineering GmbH, Karl-Marx-Allee 90A, D-10243 Berlin, Germany. E-mail: marko.wehle@itk-engineering.de |
| This journal is © the Owner Societies 2018 |