
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge-scale QM/MM free energy simulations of enzyme catalysis reveal the influence of charge transfer†
Heather J.
Kulik

Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: hjkulik@mit.edu; Tel: +1-617-253-4584
First published on 24th July 2018
Abstract
Hybrid quantum mechanical–molecular mechanical (QM/MM) simulations provide key insights into enzyme structure–function relationships. Numerous studies have demonstrated that large QM regions are needed to systematically converge ground state, zero temperature properties with electrostatic embedding QM/MM. However, it is not well known if ab initio QM/MM free energy simulations have this same dependence, in part due to the hundreds of thousands of energy evaluations required for free energy estimations that in turn limit QM region size. Here, we leverage recent advances in electronic structure efficiency and accuracy to carry out range-separated hybrid density functional theory free energy simulations in a representative methyltransferase. By studying 200 ps of ab initio QM/MM dynamics for each of five QM regions from minimal (64 atoms) to one-sixth of the protein (544 atoms), we identify critical differences between large and small QM region QM/MM in charge transfer between substrates and active site residues as well as in geometric structure and dynamics that coincide with differences in predicted free energy barriers. Distinct geometric and electronic structure features in the largest QM region indicate that important aspects of enzymatic rate enhancement in methyltransferases are identified with large-scale electronic structure.
1. Introduction
Mixed quantum mechanical (QM)/molecular mechanical (MM), i.e., QM/MM modeling1–9 remains the method of choice for simulating enzymes10 owing to its ability to balance accuracy in describing chemical rearrangements, polarization, and charge transfer at an enzyme active site with the low computational cost needed to enable sampling. Computational cost in QM/MM is typically minimized by employing small QM region sizes on the order of tens of atoms (i.e. ligands and a few direct residues),9,11,12 bringing to the fore the study and improvement of QM/MM boundary treatment7,13–22 and embedding (i.e., polarizable) approach.23–29 Nevertheless, QM region size minimization is limited by the inability to describe charge transfer between MM residues and the QM active site.30,31 At the same time, advances over the past decade30,32–39 in computational efficiency now enable fully ab initio, quantum chemical simulation of polypeptides32,40–46 as well as QM/MM treatments of enzymes with large (>100 atoms) QM regions. These advances have enabled systematic studies47–57 that have identified in static calculations a slow approach to asymptotic limits as QM regions are enlarged radially in QM/MM calculations. Such slow approach to asymptotic limit (at ca. 500–1000 atoms) has been observed for wide-ranging properties, including: NMR shieldings,47,48 proton transfer,58 solvation effects,49 barrier heights,50–52 forces,53 excitation energies,54,59,60 partial charges,55 bond critical points,61 and redox potentials.56 This strong sensitivity to QM region size has motivated ongoing method development for general50,62–65 or system-specific56 QM region determination, including through the response of QM active site residues to perturbation of surrounding environmental residues treated at the MM50,66 or QM52,67,68 level, to enable atom-economical, quantitative QM/MM.Although electronic structure calculations on over 1000 atoms have become increasingly routine, a potential of mean force requires hundreds of thousands of energy evaluations, meaning that large-QM (i.e., >100 atoms) QM/MM free energy calculations have overwhelmingly been carried out53,69–73 with semi-empirical methods that depend strongly on the quality of parameterization74 for adequate description of long-range charge transfer, hydrogen bonding, and electrostatic interactions. In order to assess (i) whether qualitative differences emerge from large-scale QM/MM and (ii) how sensitive QM/MM free energy simulations (FES) are to QM region size, we carried out the first large-scale, range-corrected, hybrid density functional theory (DFT) QM/MM free energy simulations with very large (>500 atom) QM regions. For this study, we select catechol O-methyltransferase (COMT), a representative member of a large class of S-adenosyl-L-methionine (SAM)-dependent75 methyltransferases (MTases) responsible for gene signaling,76 protein repair,77,78 and neurotransmitter regulation.75
The outline of the rest of this work is as follows. In Section 2, we review the computational details of the QM/MM free energy simulations. In Section 3, we present the reasoning for selected QM regions in QM/MM FES simulations of COMT and subsequent observations of energetic, structural, and electronic properties for differing QM regions. Finally, in Section 4, we present our conclusions.
2. Computational details
QM/MM dynamics with electrostatic embedding and hydrogen link atoms, i.e.:| EQM/MM = EQM + EQM/MM + EMM | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 852 atom starting structures were obtained from prior70 classical MD and semi-empirical QM/MM (SQM/MM) equilibration of COMT (PDB ID: 3BWM,88 protonation states given in ESI,† Table S1). In that prior work,70 the SQM region contained substrates, Mg2+ and its coordinating residues (D141, D169, N170, water), along with nearby E199 and K144 residues, whereas the remaining atoms were in the MM region. This −1 net charge, 147 atom SQM region is similar to the M2 region in the present work (see Section 3).
852 atom starting structures were obtained from prior70 classical MD and semi-empirical QM/MM (SQM/MM) equilibration of COMT (PDB ID: 3BWM,88 protonation states given in ESI,† Table S1). In that prior work,70 the SQM region contained substrates, Mg2+ and its coordinating residues (D141, D169, N170, water), along with nearby E199 and K144 residues, whereas the remaining atoms were in the MM region. This −1 net charge, 147 atom SQM region is similar to the M2 region in the present work (see Section 3).
The reaction coordinate in umbrella sampling was defined as an antisymmetric linear combination of distances (LCOD) between the S–C and C–O bonds, which break and form, respectively, during methyl transfer. Variable force constants ranging from 10 to 240 kcal mol−1 Å−2 were employed to minimize the number of windows required, which was 14 in total, while maximizing overlap over the LCOD range (details provided in ESI,† Table S2). The weighted-histogram analysis method (WHAM),89,90 as implemented in the Grossfield lab software package,91 was used to reconstruct one-dimensional free energy curves with 0.02 Å bin widths from umbrella sampling.92 Starting from the endpoint of SQM/MM dynamics (30 ps per window in ref. 70), a 2 ps equilibration at the QM/MM level was discarded before completing 15 ps production runs for the free energy calculations in five QM/MM regions ranging from 64 atoms to 544 atoms in size for a total of 1.05 ns production QM/MM dynamics (Fig. 1 and ESI,† Table S3). Mulliken partial charges, computed at each time step, were analyzed by summing over each residue to minimize errors in partial charge assignment intrinsic in the Mulliken approach. The methyl partial charge is partitioned between SAM and CAT according to progress along the reaction coordinate, as in previous work.52
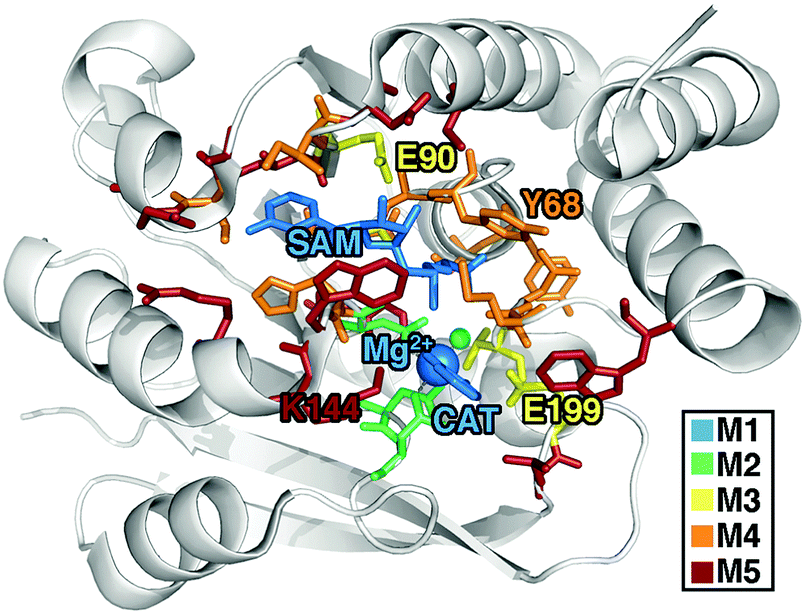 | ||
| Fig. 1 COMT structure with residues included in 5 QM region models shown as sticks, colored according to the smallest model in which they first appear: SAM, Mg2+, and CAT substrates-only model (64 atom, +2 net charge M1, blue), Mg2+ coordination sphere (109 atom, 0 net charge M2, green), critical residues (170 atom, −2 net charge M3, yellow), full systematic QM/MM region (325 atom, −3 net charge M4, orange), and a large QM model (544 atom, −1 net charge M5, red). | ||
3. Results and discussion
We investigate the effects of QM region size on dynamics and free energy evaluations in catechol O-methyltransferase (COMT), a representative member of a large class of methyltransferases (MTases).75 COMT is an S-adenosyl-L-methionine (SAM)- and Mg2+-dependent MTase75 that reacts with catecholamine substrates bound in a bidentate fashion to Mg2+ at the active site93 (Fig. 1). The rate-determining,94 direct SN2 methyl transfer95 from SAM96 to a Lys-deprotonated catecholate97,98 (CAT) has been the focus of kinetic studies93,99–101 that estimate a free energy barrier of 18.1100–19.2101 kcal mol−1 for the soluble, human form of the enzyme. COMT has also been the focus of numerous DFT52,102,103 and semi-empirical70–72,104,105 computational studies71,102–106 that have produced a wide range (i.e., 3–30 kcal mol−1) of free energy barriers that can be brought into agreement with experiment through various corrections (e.g., a spline that approximates differences between semi-empirical and MP2 energies107).We previously identified that static properties52 of COMT (e.g., barrier heights, and structures) obtained with range-separated hybrid DFT are slow to converge to asymptotic large-QM limits (ca. 300–500 atoms) through an evaluation of these properties with 10 increasing radial cutoffs for protein residue and water inclusion in the QM region. We then developed systematic methods to identify which residues were most essential in static calculations.52,67,68 Using these methods,52,67,68 we obtained 0 K QM/MM activation energies and reaction energies within 1 kcal mol−1 of the asymptotically converged result with around half the number of atoms in the radially converged region (around 200 vs. 500 atoms). The systematic selection methods focused on charge transfer between residues to reveal essential hydrogen bonding interactions and salt bridges as well as surprising nonpolar contributions to the enzyme active site.67 If interactions were captured across the QM/MM boundary, despite proximity (e.g., a lysine proximal to a glutamate near the active site), then those residues were not selected for the QM region. Now, we investigate if the regions that were shown to be asymptotically converged in 0 K QM/MM show similar or even reduced QM region size dependence than the prior 0 K studies.
For the largest-scale QM/MM free energy simulations in this work, we select a QM region of 544 atoms (28 protein residues) motivated by these prior52,67 convergence studies and employ range separated hybrid DFT in a polarized double-ζ basis set. Both basis and method are selected as a compromise for what is presently feasible to carry out the >400 k energy evaluations on the largest QM region selected (see Computational details and ESI,† Table S3). Our smaller QM regions are motivated by prior studies or our own systematic QM region construction results for convergence of static properties:67 (i) M1 (64 atoms, +1 net charge): CAT, SAM, and Mg2+ only, (ii) M2 (109 atoms, 0 net charge): added Mg2+ coordination sphere (D141, D169, and N170), (iii) M3 (170 atoms, −2 net charge): four additional key residues, V42, G66, E90, and E199, from static substrate-deletion analysis, (iv) M4 (325 atoms, −3 net charge): nine more residues from systematically-selected QM regions for static calculations,67 and (v) M5 (544 atoms, −1 net charge): a 28-residue region obtained from prior radial convergence studies52 (see Fig. 1 and ESI,† Table S3). The system sizes span an order of magnitude from 64 atoms with no link atoms for M1 to 544 atoms including 26 link atoms for M5 (see Computational details). Because each free energy simulation requires nearly half a million single point energy evaluations, we took inspiration from our prior studies to select QM regions ranging from minimal in size (M1 or M2) to systematically converged (M4 and M5) or nearly converged (M3) at the 0 K QM/MM limit observed for several 600–1000 atom QM regions.52,67 Thus, the central theme of this work is to identify if free energy simulations have similar or reduced sensitivity to QM region size as our prior 0 K QM/MM simulations.52,67 In the present work, the single water within 4 Å of the reacting atoms on SAM and catecholate is included in the QM region, motivated by (i) the fact that more distant (ca. 5–6 Å) water molecules were shown to have limited effect on 0 K barrier heights in prior work52 and (ii) that our fixed QM boundary approach would not be equipped to handle water diffusion across the boundary during the dynamics of the free energy simulations.
The 544 atom model (M5) predicts a ΔG‡ of 19.9 kcal mol−1 that is within 1–2 kcal mol−1 of best experimental estimates100,101 (Fig. 2). However, similar agreement with experimental ΔG‡ has been achieved with a number of levels of theory and QM region sizes in prior work. Using a combination of long-time classical and semi-empirical (i.e., PM6108) SQM/MM dynamics, we previously determined70 that semi-empirical barrier heights were sensitive to inclusion of the Mg2+ coordination sphere and residues adjacent to the substrates. The inclusion of both types of residues produced barrier heights (ca. 21–22 kcal mol−1) in nearly as good agreement with experiment as the present large-QM/MM simulations.109 Concurrent with our PM6 SQM/MM COMT study,70 a complementary SQM/MM study72 was published in which 15 PM6 SQM regions were compared to study methyl transfer between a partial model of SAM (i.e., S(CH3)3+) and catecholate in COMT. In both studies,70,72 the importance of the inclusion of Mg2+ and its coordination sphere in the SQM region were recognized. Therefore, in 13 of the models studied in ref. 72, Mg2+, sidechain-only models of the Mg2+ coordination sphere, and the sidechains of up to four additional protein residues were included in the SQM region. These 13 sub-100 atom SQM regions produced variations of the barrier height of approximately 5 kcal mol−1,72 with all models underestimating the experimental barrier height, as would be expected from the use of SQM/MM methods. Larger variations (∼20 kcal mol−1) were seen in the free energy of reaction.72 Both SQM/MM studies70,72 highlighted that reaction free energies were in fact even more sensitive to QM region size than the free energy barrier, consistent with expectations from Brønsted–Evans–Polanyi relations. However, they also seemed to indicate that relatively small SQM regions could be chosen to achieve reasonable agreement (i.e., within 5 kcal mol−1) with the experimental ΔG‡.70,72 Our present work differs from these prior studies because, despite these apparent successes, the reliability of SQM to predict essential energetic110 or density111 properties governed by an appropriate balance of non-covalent interactions, long-range charge transfer, and electrostatics remains an active area of research.74 Although corrections have been developed to address potential limitations, they are achieved by fitting to higher levels of theory on small gas phase test sets.112 The present work directly treats a full model of SAM and a much larger portion of the protein environment with range-separated hybrid DFT, which improves treatment of long-range charge transfer over both SQM methods and DFT with global hybrids or semi-local exchange–correlation functionals.40,52,113
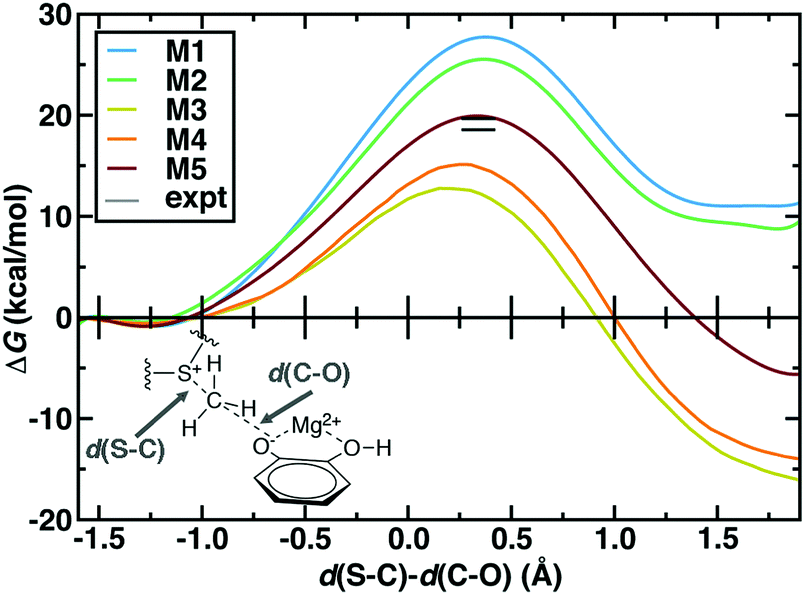 | ||
| Fig. 2 Potentials of mean force (kcal mol−1) for QM regions M1 through M5 plotted against the LCOD as defined in the main text and shown in bottom left inset. The experimental range100,101 is indicated by horizontal lines. | ||
First, rather than aiming for agreement of the theoretical and experimental free energy barriers, which might be arrived at without a complete description of the active site chemistry, we focus on electronic structure properties and dynamics at the active site of our large-scale QM/MM simulation. We will then return to the question of understanding the degree of holistic consistency of energetic, geometric, and electronic structure properties of the five QM regions we have studied. Based on gas phase models, it has been argued106 that the positively charged SAM and negatively charged CAT become neutral species at the transition state (TS), eliminating the electrostatic driving force for the reaction. The role of the greater enzyme environment in COMT has been debated,52,71,101,114 but the unexpectedly short non-bonded SAM–CAT distance observed experimentally88 and in QM/MM geometry optimizations52,114 appears electrostatically driven. Indeed, in our dynamics, we observe charge transfer mediated prolonged electrostatic attraction across the reaction coordinate defined by the difference in bond length of the cleaving S–C and forming C–O bonds (Fig. 3a). At the TS, the summed Mulliken partial charges on SAM (positive) and CAT (negative) fragments are roughly 2/3 of the magnitude they are in the Michaelis complex (d(S–C)–d(C–O), Δ ∼ −1 Å, see Fig. 3). Only when the product is fully formed do the SAM- and CAT-summed charges approach zero, even when taking into account dynamic fluctuations in partial charges on both substrates (shaded ranges in Fig. 3a). It is also noteworthy that the SAM substrate reaches a peak positive formal charge near the shortened non-bonded distances observed in crystal structures between the substrates (ca. Δ = −0.8 Å, d(S–C) = 1.82 Å d(C–O) = 2.62 Å) likely due to proximity and enhanced charge transfer to N41/V42 from the SAM carboxylate (ESI,† Fig. S2 and Tables S4–S6).
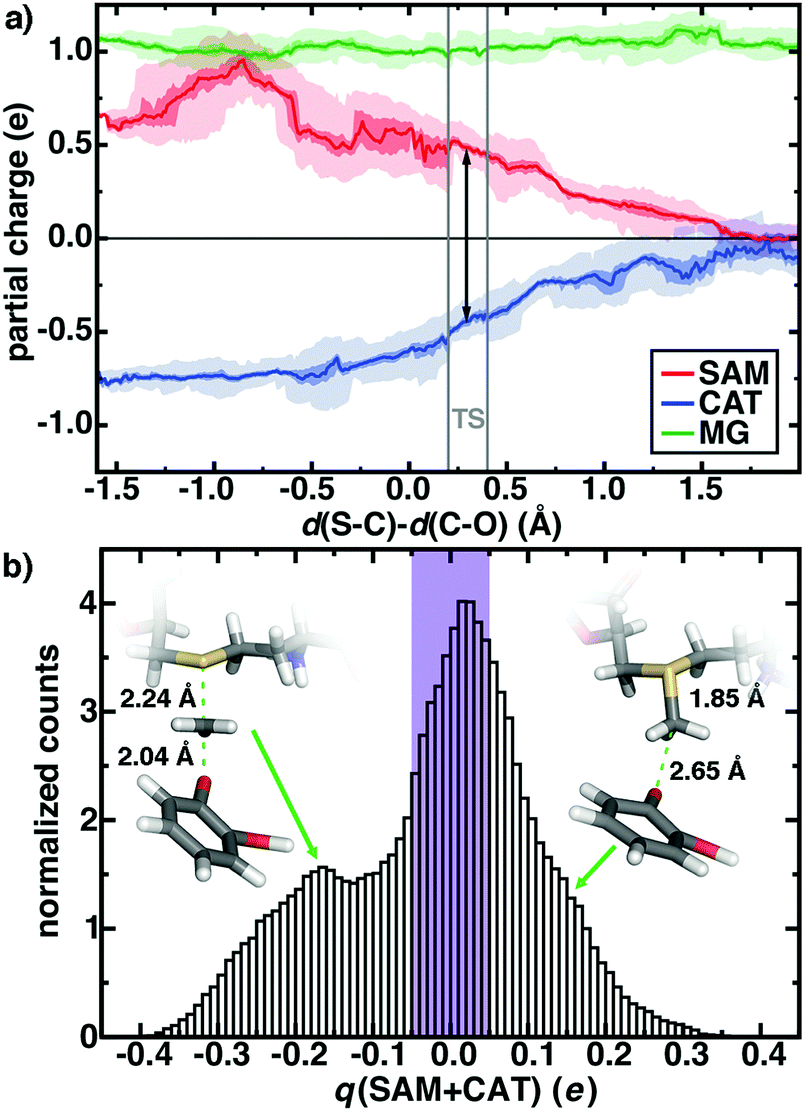 | ||
| Fig. 3 (a) Sum of charges on SAM (red), CAT (blue), and Mg2+ (green) during methyl transfer reaction in the M5 QM/MM model. Over 0.01 Å bins of simulation data, average charges are indicated by lines, the first to third interquartile range is indicated by the darker shaded region and the full range of charges are indicated by light shading. The methyl charge is partitioned between substrates as described in the main text, and the TS region is indicated as gray vertical bars with average charge separation shown as a black arrow. (b) Normalized histogram of partial charges summed over SAM and CAT (in e) across 200 ps of aggregated M5 free energy simulation. The purple bars represent ±0.05 e range and each bin is 0.01 e wide. | ||
The net charge on the combined positive SAM and negative CAT substrates represents the degree of charge transfer between substrates and the surrounding enzyme environment. If this quantity remains close to zero in the QM/MM free energy simulations, then we can expect that increasingly accurate embedding23–29 (i.e., polarizable) might be able to capture prolonged electrostatic attraction between residues. However, we observe a wide distribution of net charge from −0.4 to 0.4 on the substrates during our simulation, and only 1/3 of the simulation time is spent with the summed substrate charge within ±0.05 e (Fig. 3b). Decomposing the formal charge across the reaction coordinate reveals that a formal positive charge (charge transfer away from the substrates) mediates the shortened Michaelis complex structures observed earlier, whereas charge accumulates on the substrates leading up to the TS and in the product (Fig. 3b and ESI,† Fig. S3 and S4).
One might expect that the charge transfer between the substrates and the surrounding environment could be localized to only a few charged residues within hydrogen bonding distance of the substrates. We computed by-residue partial charges and compared the dependence of maximum fluctuations (i.e., the difference of maximum and minimum by-residue charges observed during the simulation) to the closest heavy atom distance between the residue and SAM/CAT/Mg2+ substrates averaged over the reactant, TS, and product geometries (Fig. 4 and ESI,† Table S4). We observe very large charge fluctuations (ca. 0.7 e) for some proximal and charged residues (e.g., E199) but moderate fluctuations for all residues in the QM region of this model (min. of 0.08 e) and surprisingly little closest-heavy-atom-distance dependence (ESI,† Fig. S2 and Tables S4–S6). To test if charge fluctuations are still localized between pairs of hydrogen bonding residues, we also summed the partial charges of the substrates and the 14 residues with closest heavy atom distances within 3.0 Å of the substrates. This portion of the M5 QM region exhibits just as large fluctuations in deviations from formal charge, with at least three distinct peak features in a wide −0.5 to +0.2 e deviation range and only 26% of all frames within 0.05 e of the expected formal charge (ESI,† Fig. S5).
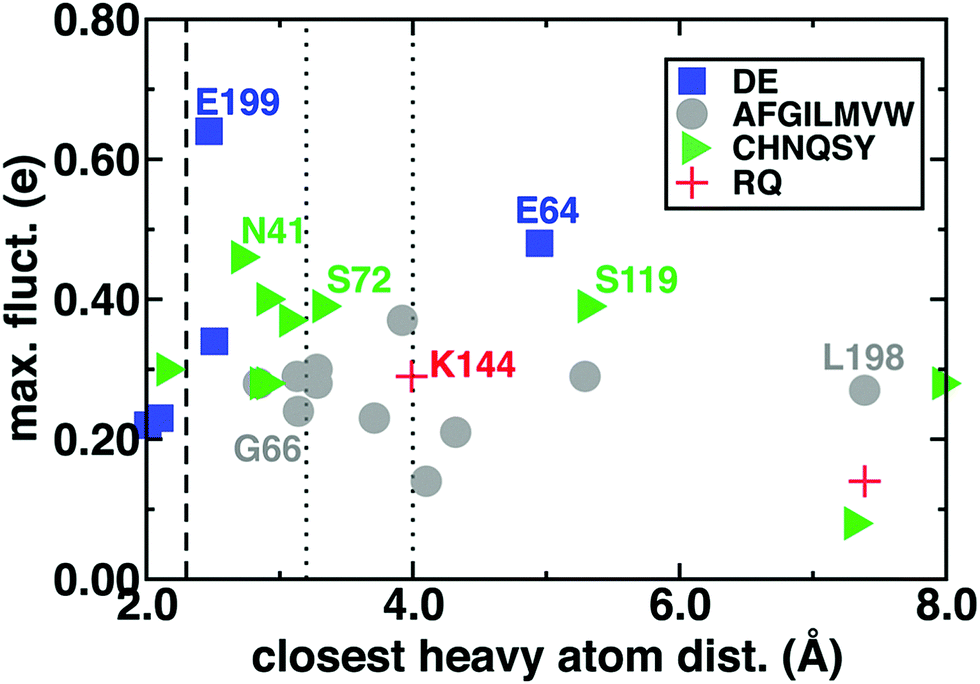 | ||
| Fig. 4 Range of by-residue summed partial charges including link atoms (max. fluct., e) sampled for each residue grouped by negatively charged (blue squares), nonpolar (gray circles), polar (green triangles), and positively charged (red crosses), with residue types indicated in legend plotted against the average closest heavy atom distance between the residue and SAM, CAT, or Mg2+. Vertical lines indicate approximate covalent bond (dashed), moderate hydrogen bond (dotted), and weak hydrogen bond (dotted) distance cutoffs. | ||
As an example of weak distance dependence, D141 and D169, which form the Mg2+ coordination sphere, exhibit lower fluctuations than nonpolar L198, which is 7 Å from the closest substrate heavy atoms (Fig. 4). Perhaps surprisingly, similar magnitude fluctuations are observed for both polar and nonpolar residues, and a comparison of charge standard deviations preserves trends (ESI,† Fig. S6–S8). Large fluctuations in residues beyond typical hydrogen bonding distances to the substrates (i.e., ∼3.25 Å, see Fig. 4) can be rationalized by proximity to substrate-neighboring residues that have large fluctuations. For example, the relatively high fluctuations for substrate-distantce L198 are a consequence of its being adjacent to E199, and the somewhat distant E64 hydrogen bonds with S72, which in turn hydrogen bonds with SAM (see ESI†). On the other hand, D141 and D169 have among the lowest fluctuations for charged residues, despite significant deviations from formal charges (ESI,† Fig. S9). This highlights that fluctuations arise predominantly in mobile residues that vary in active site proximity along the reaction coordinate rather than protein residues that remain covalently coordinated to the substrates (ESI,† Table S4).
The delicate interplay of charge transfer between the substrate and the enzyme environment can be observed through the structure of the reacting substrates and Mg2+, also highlighting the crucial importance of Mg2+ in COMT catalysis.75 The four Mg2+–O bonds with the protein, i.e., D141, D169, N170, and a water molecule are relatively unchanged across the reaction coordinate but differ in length (Fig. 5). Although D169 and D141 Mg2+–O− bonds are shorter than the neutral N170 bond (2.02–2.10 Å vs. 2.17 Å) as expected, the Mg2+–water bond is substantially shorter (2.02 Å) than observed for hexa-aqua complexes115 (Fig. 5 and ESI,† Text S1). Across the reaction coordinate, charge transfer mediated prolonged electrostatic attraction is evident in the Mg2+–O bond of the catecholate oxygen that carries out nucleophilic attack on the SAM methyl group. Initially, this bond is the shortest (ca. 2.00 Å) of all Mg2+–O bonds and remains in the range of the D141 and D169 Mg2+–carboxylate bond lengths until well past the TS (ca. Δ = 0.7 Å, d(S–C) = 2.59 Å d(C–O) = 1.89 Å). Thus, Mg2+ plays the critical role not only of positioning catechol for nucleophilic attack but in maintaining the strong nucleophilic character of the catecholate oxygen. This observation is consistent with prior work that revealed70 the importance of a QM description of Mg2+ to capture this interaction to reproduce experimental structural observations, which was frequently neglected in prior SQM/MM studies.107
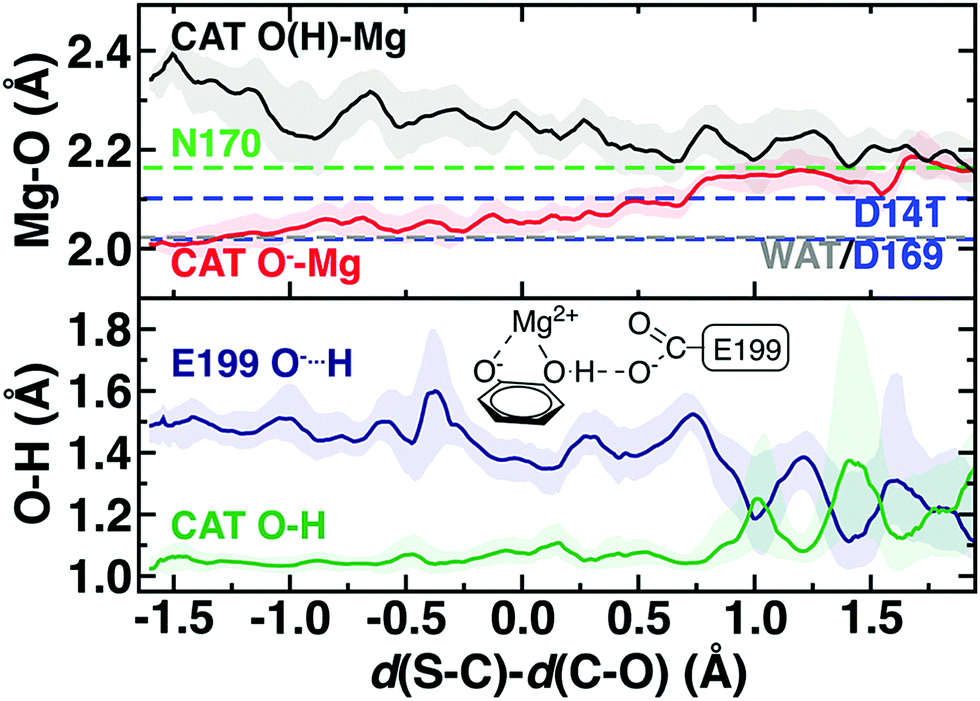 | ||
| Fig. 5 Mg2+ coordination sphere (top) and catecholate (bottom) structural properties in the M5 QM/MM model across the reaction coordinate. At top, the median, averaged distance is reported as dashed lines for N170 (green), D141 and D169 (blue), and water (WAT, gray) as a reference for the CAT O(H) (dark gray) and CAT O− (red) distances to Mg2+ across the reaction coordinate. At bottom, catechol hydrogen atom distance to neighboring E199 (dark blue) and to the catechol oxygen atom (dark green) are plotted along the reaction coordinate. The remaining quantities are the median values obtained at 0.01 Å increments and averaged over a 0.1 Å window with the range of variations shown as a shaded region. A schematic of E199, Mg2+, and CAT is shown in inset at bottom. | ||
In comparison, the catechol oxygen distance to Mg2+ is the longest in the octahedral coordination sphere, becoming comparable in length to the methylated oxygen–Mg2+ distance only in the product (Fig. 5). The long catechol–Mg2+ bond length is likely due to competition with a strong hydrogen bond that forms with the E199 carboxylate, leading to a shared hydrogen atom between the two species in the product state (both O–H ca. 1.2 Å) typically indicative of a low-barrier hydrogen bond116 that would be unlikely to be suitably described across a QM/MM boundary (Fig. 5). Thus, it would appear that charge transfer effects only observable with large QM regions can influence both energetics and structure/dynamics across the reaction coordinate.
The remaining question is to what extent these large-scale QM/MM observations of geometric and electronic structure trends are preserved in smaller models in spite of moderate discrepancies in energetics (Fig. 2). In order to evaluate QM region dependence in dynamics, we now compare our four increasingly large QM regions (M1–M4) to our largest QM/MM simulation (M5). Wide ranges of both ΔG‡ and ΔGr as well as TS character are observed across M1–M5 (Fig. 2 and 6 and ESI,† Table S7). Consistent with prior SQM/MM70,72 and trends in our static results,52 both M1 and M2 substantially overestimate the ΔG‡, predict an uphill ΔGr, and underestimate the charge separation at the transition state with respect to M5 (Fig. 6 and ESI,† Table S7). The 170 atom M3 region incorporates four proximal residues beyond those in the Mg2+ coordination sphere, overstabilizing the transition state and product (i.e., ΔG‡(M3) < ΔG‡ (M5) or expt.), despite underestimating the separation of the substrate charges in the transition state and predicting substrate neutralization in advance of the products (Fig. 6 and ESI,† Fig. S10). Analysis of partial charges reveals that the absence of N41/V42 interactions with the SAM carboxylate likely accounts for the more neutral overall SAM charge but an absence of charge transfer with a QM K144 adjacent to the catecholate overestimates the nucleophilic character of the attacking catecholate oxygen.
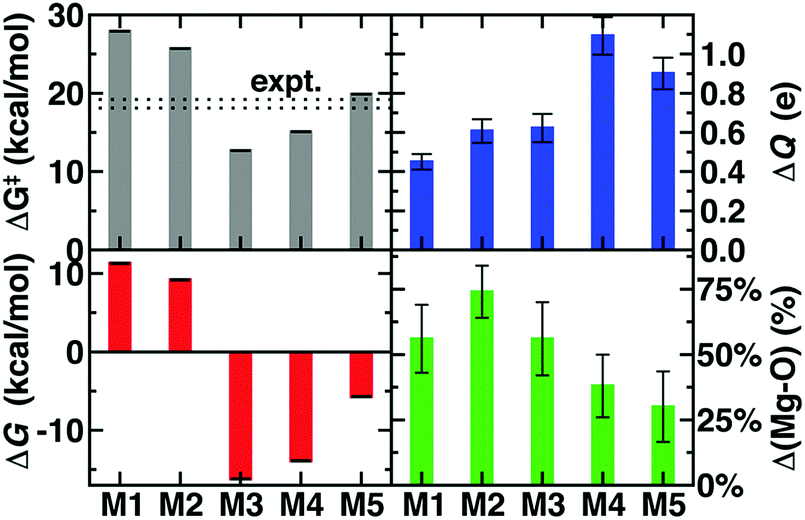 | ||
| Fig. 6 Comparison of ΔG‡ (in kcal mol−1), ΔGr (in kcal mol−1), charge separation of SAM and CAT at the TS (ΔQ in e), and anionic catecholate–Mg2+ bond length progress in the TS with respect to difference between the reactant and product (in %) for five QM regions. Error bars for top and bottom left hand side panels come from bootstrapping and they correspond to standard deviations in quantities on the top and bottom right. | ||
Finally, M4 was selected systematically52,67,68 on the basis of charge/Fukui shift analysis on 20 0 K snapshots of COMT and confirmed to have a 0 K barrier height within 1 kcal mol−1 of 0 K converged (i.e., M5) values.67 Agreement of QM/MM dynamics, energetics, and charge density between M4 and M5 is slightly worse than the equivalent regions in prior study67 but by far in the best agreement of any two regions (Fig. 6). Like M3, M4 overstabilizes the TS and products with respect to M5 but does so by overestimating the charge separation at the TS, suggesting enhanced electrostatic attraction as the driving force for M4 barrier underestimation (Fig. 6). The differences in characteristics between M4 and M5 can likely be attributed to the absence of K144 from the M4 QM region, which was distant from the substrate in the selected 0 K snapshots but proximal throughout the 300 K dynamics in this work (ESI,† Table S4). The inclusion of K144 in the M5 QM region has the additional effect of producing a shorter Mg2+–O− CAT bond in the TS for M5 than in the other 4 QM regions (Fig. 6 and ESI,† Table S8). Overall, the static-QM-selected regions M3 and M4 bear the best qualitative agreement with the large-scale M5 region. Overall, we find greater disagreement (i.e., 3–5 kcal mol−1 free energy differences vs. 0–1 kcal mol−1 0 K electronic energy differences) among QM regions that had been previously systematically converged for 0 K properties,67 emphasizing that deviations are likely due to enhanced sensitivity during dynamics. This observation over QM regions that differ by as much as 375 atoms in size is consistent with prior observations made of 5 kcal mol−1 free energy barrier fluctuations for varying SQM regions of much more similar (ca. 100 atoms) size.72 Remaining differences highlight that quantitative agreement through systematic QM region selection will require sampling117 a wider range of possible configurations relevant during dynamics. Furthermore, as computing power continues to increase, even larger QM regions and longer timescales in QM/MM dynamics should be employed to identify the limits of convergence beyond this study.
4. Conclusions
In conclusion, we have leveraged accelerated electronic structure approaches to carry out a large-scale study of over 1 ns of ab initio, range-separated hybrid DFT QM/MM dynamics with QM regions from 64 to 544 atoms in size. These large-scale QM/MM studies revealed QM-region-dependent differences in electronic and geometric structure. In addition to accurate prediction of the free energy barrier for methyl transfer, charge separation and neutralization only in the product structure are observed with large QM/MM free energy simulations. Differences in geometry (e.g., relative Mg2+–catecholate oxygen bond lengths) and >0.4 e fluctuations of summed charge on residues 5 Å away from the substrates (i.e., E64 or S119) indicate the extent to which substrate–active site charge transfer can play a key role in enzyme mechanism. This long-range substrate–active site charge transfer is consistent with notions that the enzyme creates an electrostatic driving force for the reaction by stabilizing the transition state but highlights that in some cases large QM regions may be needed to fully observe this effect. These studies also reveal that differences in electronic environments give rise to differences in dynamics and structure in a manner that will likely enhance not decrease QM region dependence for free energy simulations and dynamics. Such observations should motivate the continued improvement of QM/MM through (i) improved embedding schemes and boundary treatment, including to allow charge transfer, (ii) incorporation of large, beyond-DFT QM regions (e.g., through reduced-scaling correlated wavefunction theory), and (iii) development of efficient sampling techniques and free energy decomposition. We note, however, that the large number of hydrogen bonds between charged residues and substrates in the active site of COMT and homologous MTases likely increase QM region dependence over a number of cases where small QM-region QM/MM studies may still encapsulate essential physics.Although the best agreement with the largest QM region was obtained from systematically constructed QM/MM regions (325 atoms vs. 544 atoms in size) obtained from analysis of two dozen 0 K snapshots, variations between the M4 and M5 QM regions in energetics and properties were still apparent. These differences were observed to be due to fluctuations in the orientations of residues during dynamics absent from the 0 K geometry optimizations and barrier evaluations in previous work that had shown smoother convergence with QM region size. As accelerated electronic structure calculations and computing power become increasingly available, it is anticipated that these calculations could be repeated with alternative QM regions of even larger size to confirm stricter property convergence (e.g., free energies within 1 kcal mol−1). Overall, this work also reinforces the importance of systematic QM region construction including dynamic benchmarking of substrate electronic properties to identify minimal sufficient QM regions in future QM/MM studies. Methods that directly incorporate fluctuations of electronic properties during dynamics to construct QM regions are currently being developed in our group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by an MIT Research Support Committee NEC Corporation grant. Support for this research was provided by a core center grant P30-ES002109 from the National Institute of Environmental Health Sciences, National Institutes of Health. H. J. K. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund, which also supported this work. This work was carried out in part using computational resources from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. This work used the XStream computational resource, supported by the National Science Foundation Major Research Instrumentation program (ACI-1429830). The authors thank Adam H. Steeves for providing a critical reading of the manuscript.References
- M. J. Field, P. A. Bash and M. Karplus, A Combined Quantum-Mechanical and Molecular Mechanical Potential for Molecular-Dynamics Simulations, J. Comput. Chem., 1990, 11, 700–733 CrossRef.
- D. Bakowies and W. Thiel, Hybrid Models for Combined Quantum Mechanical and Molecular Mechanical Approaches, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef.
- T. Z. Mordasini and W. Thiel, Combined Quantum Mechanical and Molecular Mechanical Approaches, Chimia, 1998, 52, 288–291 Search PubMed.
- G. Monard and K. M. Merz, Combined Quantum Mechanical/Molecular Mechanical Methodologies Applied to Biomolecular Systems, Acc. Chem. Res., 1999, 32, 904–911 CrossRef.
- J. Gao and D. G. Truhlar, Quantum Mechanical Methods for Enzyme Kinetics, Annu. Rev. Phys. Chem., 2002, 53, 467–505 CrossRef PubMed.
- E. Rosta, M. Klahn and A. Warshel, Towards Accurate Ab Initio QM/MM Calculations of Free-Energy Profiles of Enzymatic Reactions, J. Phys. Chem. B, 2006, 110, 2934–2941 CrossRef PubMed.
- H. Lin and D. Truhlar, QM/MM: What Have We Learned, Where Are We, and Where Do We Go from Here?, Theor. Chem. Acc., 2007, 117, 185–199 Search PubMed.
- A. Warshel and M. Levitt, Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme, J. Mol. Biol., 1976, 103, 227–249 CrossRef PubMed.
- H. M. Senn and W. Thiel, QM/MM Methods for Biomolecular Systems, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef PubMed.
- J. Gao, S. Ma, D. T. Major, K. Nam, J. Pu and D. G. Truhlar, Mechanisms and Free Energies of Enzymatic Reactions, Chem. Rev., 2006, 106, 3188–3209 CrossRef PubMed.
- P. Vidossich, G. Florin, M. Alfonso-Prieto, E. Derat, S. Shaik and C. Rovira, On the Role of Water in Peroxidase Catalysis: A Theoretical Investigation of Hrp Compound I Formation, J. Phys. Chem. B, 2010, 114, 5161–5169 CrossRef PubMed.
- P. Carloni, U. Rothlisberger and M. Parrinello, The Role and Perspective of Ab Initio Molecular Dynamics in the Study of Biological Systems, Acc. Chem. Res., 2002, 35, 455–464 CrossRef PubMed.
- K. P. Eurenius, D. C. Chatfield, B. R. Brooks and M. Hodoscek, Enzyme Mechanisms with Hybrid Quantum and Molecular Mechanical Potentials. I. Theoretical Considerations, Int. J. Quantum Chem., 1996, 60, 1189–1200 CrossRef.
- H. M. Senn and W. Thiel, QM/MM Studies of Enzymes, Curr. Opin. Chem. Biol., 2007, 11, 182–187 CrossRef PubMed.
- A. Monari, J.-L. Rivail and X. Assfeld, Advances in the Local Self-Consistent Field Method for Mixed Quantum Mechanics/Molecular Mechanics Calculations, Acc. Chem. Res., 2012, 46, 596–603 CrossRef PubMed.
- Y. Wang and J. Gao, Projected Hybrid Orbitals: A General QM/MM Method, J. Phys. Chem. B, 2015, 119, 1213–1224 CrossRef PubMed.
- P. Slavicek and T. J. Martinez, Multicentered Valence Electron Effective Potentials: A Solution to the Link Atom Problem for Ground and Excited Electronic States, J. Chem. Phys., 2006, 124, 084107 CrossRef PubMed.
- R. B. Murphy, D. M. Philipp, R. A. Friesner and A. Mixed, Quantum Mechanics/Molecular Mechanics (QM/MM) Method for Large Scale Modeling of Chemistry in Protein Environments, J. Comput. Chem., 2000, 21, 1442–1457 CrossRef.
- Y. Zhang, T.-S. Lee, W. Yang and A. Pseudobond, Approach to Combining Quantum Mechanical and Molecular Mechanical Methods, J. Chem. Phys., 1999, 110, 46–54 CrossRef.
- G. A. DiLabio, M. M. Hurley and P. A. Christiansen, Simple One-Electron Quantum Capping Potentials for Use in Hybrid QM/MM Studies of Biological Molecules, J. Chem. Phys., 2002, 116, 9578–9584 CrossRef.
- O. A. von Lilienfeld, I. Tavernelli, U. Rothlisberger and D. Sebastiani, Variational Optimization of Effective Atom-Centered Potentials for Molecular Properties, J. Chem. Phys., 2005, 122, 14113 CrossRef PubMed.
- B. Wang and D. G. Truhlar, Combined Quantum Mechanical and Molecular Mechanical Methods for Calculating Potential Energy Surfaces: Tuned and Balanced Redistributed Charge Algorithm, J. Chem. Theory Comput., 2010, 6, 359–369 CrossRef PubMed.
- N. M. Thellamurege and H. Hirao, Effect of Protein Environment within Cytochrome P450cam Evaluated Using a Polarizable-Embedding QM/MM Method, J. Phys. Chem. B, 2014, 118, 2084–2092 Search PubMed.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht and R. A. DiStasio Jr., Current Status of the AMOEBA Polarizable Force Field, J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef PubMed.
- T. A. Halgren and W. Damm, Polarizable Force Fields, Curr. Opin. Struct. Biol., 2001, 11, 236–242 CrossRef PubMed.
- L. J. Nåbo, J. M. H. Olsen, T. J. Martínez and J. Kongsted, The Quality of the Embedding Potential Is Decisive for Minimal Quantum Region Size in Embedding Calculations: The Case of the Green Fluorescent Protein, J. Chem. Theory Comput., 2017, 13, 6230–6236 CrossRef PubMed.
- A. Ganguly, E. Boulanger and W. Thiel, Importance of Mm Polarization in QM/MM Studies of Enzymatic Reactions: Assessment of the QM/MM Drude Oscillator Model, J. Chem. Theory Comput., 2017, 13, 2954–2961 CrossRef PubMed.
- Q. Li, B. Mennucci, M. A. Robb, L. Blancafort and C. Curutchet, Polarizable QM/MM Multiconfiguration Self-Consistent Field Approach with State-Specific Corrections: Environment Effects on Cytosine Absorption Spectrum, J. Chem. Theory Comput., 2015, 11, 1674–1682 CrossRef PubMed.
- D. Loco, L. Lagardère, S. Caprasecca, F. Lipparini, B. Mennucci and J.-P. Piquemal, Hybrid QM/MM Molecular Dynamics with AMOEBA Polarizable Embedding, J. Chem. Theory Comput., 2017, 13, 4025–4033 CrossRef PubMed.
- I. S. Ufimtsev, N. Luehr and T. J. Martínez, Charge Transfer and Polarization in Solvated Proteins from Ab Initio Molecular Dynamics, J. Phys. Chem. Lett., 2011, 2, 1789–1793 CrossRef.
- G. Nadig, L. C. Van Zant, S. L. Dixon and K. M. Merz, Charge-Transfer Interactions in Macromolecular Systems: A New View of the Protein/Water Interface, J. Am. Chem. Soc., 1998, 120, 5593–5594 CrossRef.
- H. J. Kulik, N. Luehr, I. S. Ufimtsev and T. J. Martinez, Ab Initio Quantum Chemistry for Protein Structure, J. Phys. Chem. B, 2012, 116, 12501–12509 CrossRef PubMed.
- I. S. Ufimtsev and T. J. Martínez, Quantum Chemistry on Graphical Processing Units. 1. Strategies for Two-Electron Integral Evaluation, J. Chem. Theory Comput., 2008, 4, 222–231 CrossRef PubMed.
- I. S. Ufimtsev and T. J. Martínez, Quantum Chemistry on Graphical Processing Units. 2. Direct Self-Consistent-Field Implementation, J. Chem. Theory Comput., 2009, 5, 1004–1015 CrossRef PubMed.
- I. S. Ufimtsev and T. J. Martínez, Quantum Chemistry on Graphical Processing Units. 3. Analytical Energy Gradients, Geometry Optimization, and First Principles Molecular Dynamics, J. Chem. Theory Comput., 2009, 5, 2619–2628 CrossRef PubMed.
- C. M. Isborn, N. Luehr, I. S. Ufimtsev and T. J. Martinez, Excited-State Electronic Structure with Configuration Interaction Singles and Tamm-Dancoff Time-Dependent Density Functional Theory on Graphical Processing Units, J. Chem. Theory Comput., 2011, 7, 1814–1823 CrossRef PubMed.
- C. Ochsenfeld, J. Kussmann and D. S. Lambrecht, Linear-Scaling Methods in Quantum Chemistry, Rev. Comput. Chem., 2007, 23, 1 Search PubMed.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Auxiliary Basis Sets for Main Row Atoms and Transition Metals and Their Use to Approximate Coulomb Potentials, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Auxiliary Basis Sets to Approximate Coulomb Potentials, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef.
- E. Rudberg, E. H. Rubensson and P. Salek, Kohn-Sham Density Functional Theory Electronic Structure Calculations with Linearly Scaling Computational Time and Memory Usage, J. Chem. Theory Comput., 2011, 7, 340–350 CrossRef PubMed.
- M. Challacombe and E. Schwegler, Linear Scaling Computation of the Fock Matrix, J. Chem. Phys., 1997, 106, 5526–5536 CrossRef.
- C.-K. Skylaris, P. D. Haynes, A. A. Mostofi and M. C. Payne, Introducing ONETEP: Linear Scaling Density Functional Simulations on Parallel Computers, J. Chem. Phys., 2005, 122, 084119 CrossRef PubMed.
- D. R. Bowler and T. Miyazaki, O(N) Methods in Electronic Structure Calculations, Rep. Prog. Phys., 2012, 75, 036503 CrossRef PubMed.
- J. VandeVondele, U. Borštnik and J. Hutter, Linear Scaling Self-Consistent Field Calculations with Millions of Atoms in the Condensed Phase, J. Chem. Theory Comput., 2012, 8, 3565–3573 CrossRef PubMed.
- G. E. Scuseria, Linear Scaling Density Functional Calculations with Gaussian Orbitals, J. Phys. Chem. A, 1999, 103, 4782–4790 CrossRef.
- M. Guidon, J. Hutter and J. VandeVondele, Robust Periodic Hartree-Fock Exchange for Large-Scale Simulations Using Gaussian Basis Sets, J. Chem. Theory Comput., 2009, 5, 3010–3021 CrossRef PubMed.
- D. Flaig, M. Beer and C. Ochsenfeld, Convergence of Electronic Structure with the Size of the QM Region: Example of QM/MM NMR Shieldings, J. Chem. Theory Comput., 2012, 8, 2260–2271 CrossRef PubMed.
- J. D. Hartman, T. J. Neubauer, B. G. Caulkins, L. J. Mueller and G. J. Beran, Converging Nuclear Magnetic Shielding Calculations with Respect to Basis and System Size in Protein Systems, J. Biomol. NMR, 2015, 62, 327–340 Search PubMed.
- S. J. Fox, C. Pittock, T. Fox, C. S. Tautermann, N. Malcolm and C. K. Skylaris, Electrostatic Embedding in Large-Scale First Principles Quantum Mechanical Calculations on Biomolecules, J. Chem. Phys., 2011, 135, 224107 CrossRef PubMed.
- R. Z. Liao and W. Thiel, Convergence in the QM-Only and QM/MM Modeling of Enzymatic Reactions: A Case Study for Acetylene Hydratase, J. Comput. Chem., 2013, 34, 2389–2397 Search PubMed.
- K. Sadeghian, D. Flaig, I. D. Blank, S. Schneider, R. Strasser, D. Stathis, M. Winnacker, T. Carell and C. Ochsenfeld, Ribose-Protonated DNA Base Excision Repair: A Combined Theoretical and Experimental Study, Angew. Chem., Int. Ed., 2014, 53, 10044–10048 CrossRef PubMed.
- H. J. Kulik, J. Zhang, J. P. Klinman and T. J. Martinez, How Large Should the QM Region Be in QM/MM Calculations? The Case of Catechol O-Methyltransferase, J. Phys. Chem. B, 2016, 120, 11381–11394 CrossRef PubMed.
- I. Solt, P. Kulhanek, I. Simon, S. Winfield, M. C. Payne, G. Csanyi and M. Fuxreiter, Evaluating Boundary Dependent Errors in QM/MM Simulations, J. Phys. Chem. B, 2009, 113, 5728–5735 CrossRef PubMed.
- C. M. Isborn, A. W. Goetz, M. A. Clark, R. C. Walker and T. J. Martinez, Electronic Absorption Spectra from Mm and Ab Initio QM/MM Molecular Dynamics: Environmental Effects on the Absorption Spectrum of Photoactive Yellow Protein, J. Chem. Theory Comput., 2012, 8, 5092–5106 CrossRef PubMed.
- D. E. Vanpoucke, J. Oláh, F. De Proft, V. Van Speybroeck and G. Roos, Convergence of Atomic Charges with the Size of the Enzymatic Environment, J. Chem. Inf. Model., 2015, 55, 564–571 CrossRef PubMed.
- T. V. Harris and R. K. Szilagyi, Protein Environmental Effects on Iron-Sulfur Clusters: A Set of Rules for Constructing Computational Models for Inner and Outer Coordination Spheres, J. Comput. Chem., 2016, 37, 1681–1696 CrossRef PubMed.
- L. Hu, P. r. Söderhjelm and U. Ryde, On the Convergence of QM/MM Energies, J. Chem. Theory Comput., 2011, 7, 761–777 CrossRef PubMed.
- S. Roßbach and C. Ochsenfeld, Influence of Coupling and Embedding Schemes on QM Size Convergence in QM/MM Approaches for the Example of a Proton Transfer in DNA, J. Chem. Theory Comput., 2017, 13, 1102–1107 CrossRef PubMed.
- M. R. Provorse, T. Peev, C. Xiong and C. M. Isborn, Convergence of Excitation Energies in Mixed Quantum and Classical Solvent: Comparison of Continuum and Point Charge Models, J. Phys. Chem. B, 2016, 120, 12148–12159 CrossRef PubMed.
- J. M. Milanese, M. R. Provorse, E. Alameda and C. M. Isborn, Convergence of Computed Aqueous Absorption Spectra with Explicit Quantum Mechanical Solvent, J. Chem. Theory Comput., 2017, 13, 2159–2171 CrossRef PubMed.
- A. Morgenstern, M. Jaszai, M. E. Eberhart and A. N. Alexandrova, Quantified Electrostatic Preorganization in Enzymes Using the Geometry of the Electron Charge Density, Chem. Sci., 2017, 8, 5010–5018 RSC.
- R.-Z. Liao and W. Thiel, Comparison of QM-only and QM/MM Models for the Mechanism of Tungsten-Dependent Acetylene Hydratase, J. Chem. Theory Comput., 2012, 8, 3793–3803 CrossRef PubMed.
- P. A. Bash, M. J. Field, R. C. Davenport, G. A. Petsko, D. Ringe and M. Karplus, Computer Simulation and Analysis of the Reaction Pathway of Triosephosphate Isomerase, Biochemistry, 1991, 30, 5826–5832 CrossRef PubMed.
- L. Hu, J. Eliasson, J. Heimdal and U. Ryde, Do Quantum Mechanical Energies Calculated for Small Models of Protein-Active Sites Converge?, J. Phys. Chem. A, 2009, 113, 11793–11800 CrossRef PubMed.
- L. Hu, P. Söderhjelm and U. Ryde, Accurate Reaction Energies in Proteins Obtained by Combining QM/MM and Large QM Calculations, J. Chem. Theory Comput., 2012, 9, 640–649 CrossRef PubMed.
- S. Sumner, P. Söderhjelm and U. Ryde, Effect of Geometry Optimizations on QM-Cluster and QM/MM Studies of Reaction Energies in Proteins, J. Chem. Theory Comput., 2013, 9, 4205–4214 CrossRef PubMed.
- M. Karelina and H. J. Kulik, Systematic Quantum Mechanical Region Determination in QM/MM Simulation, J. Chem. Theory Comput., 2017, 13, 563–576 CrossRef PubMed.
- H. W. Qi, M. Karelina and H. J. Kulik, Quantifying Electronic Effects in QM and QM/MM Biomolecular Modeling with the Fukui Function, Acta Phys.-Chim. Sin., 2018, 34, 81–91 Search PubMed.
- K. Meier, W. Thiel and W. F. van Gunsteren, On the Effect of a Variation of the Force Field, Spatial Boundary Condition and Size of the QM Region in QM/MM MD Simulations, J. Comput. Chem., 2012, 33, 363–378 CrossRef PubMed.
- N. Patra, E. I. Ioannidis and H. J. Kulik, Computational Investigation of the Interplay of Substrate Positioning and Reactivity in Catechol O-Methyltransferase, PLoS One, 2016, 11, e0161868 CrossRef PubMed.
- J. Lameira, R. P. Bora, Z. T. Chu and A. Warshel, Methyltransferases Do Not Work by Compression, Cratic, or Desolvation Effects, but by Electrostatic Preorganization, Proteins: Struct., Funct., Bioinf., 2015, 83, 318–330 CrossRef PubMed.
- G. Jindal and A. Warshel, Exploring the Dependence of QM/MM Calculations of Enzyme Catalysis on the Size of the QM Region, J. Phys. Chem. B, 2016, 120, 9913–9921 CrossRef PubMed.
- M. A. Olsson and U. Ryde, Comparison of QM/MM Methods to Obtain Ligand-Binding Free Energies, J. Chem. Theory Comput., 2017, 13, 2245–2253 CrossRef PubMed.
- J. Řezáč and P. Hobza, Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods, J. Chem. Theory Comput., 2011, 8, 141–151 CrossRef PubMed.
- J. Axelrod and R. Tomchick, Enzymatic O-Methylation of Epinephrine and Other Catechols, J. Biol. Chem., 1958, 233, 702–705 Search PubMed.
- A. Razin and A. D. Riggs, DNA Methylation and Gene Function, Science, 1980, 210, 604–610 CrossRef PubMed.
- M. M. Skinner, J. M. Puvathingal, R. L. Walter and A. M. Friedman, Crystal Structure of Protein Isoaspartyl Methyltransferase: A Catalyst for Protein Repair, Structure, 2000, 8, 1189–1201 CrossRef PubMed.
- J. E. Visick, H. Cai and S. Clarke, The L-Isoaspartyl Protein Repair Methyltransferase Enhances Survival of Aging Escherichia Coli Subjected to Secondary Environmental Stresses, J. Bacteriol., 1998, 180, 2623–2629 Search PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, An Overview of the Amber Biomolecular Simulation Package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 Search PubMed.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham, III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, Amber 2015, University of California, San Francisco, 2015 Search PubMed.
- Petachem, http://www.petachem.com, (accessed January 1, 2018) Search PubMed.
- M. A. Rohrdanz, K. M. Martins, J. M. Herbert and A. Long-Range-Corrected Density, Functional That Performs Well for Both Ground-State Properties and Time-Dependent Density Functional Theory Excitation Energies, Including Charge-Transfer Excited States, J. Chem. Phys., 2009, 130, 054112 CrossRef PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-Type Basis for Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1971, 54, 724 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich, H. Krieg and A. Consistent, and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters, Proteins: Struct., Funct., Bioinf., 2006, 65, 712–725 CrossRef PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, Ff14sb: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99sb, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef.
- K. Rutherford, I. Le Trong, R. E. Stenkamp and W. W. Parson, Crystal Structures of Human 108v and 108m Catechol O-Methyltransferase, J. Mol. Biol., 2008, 380, 120–130 CrossRef PubMed.
- M. Souaille and B. Roux, Extension to the Weighted Histogram Analysis Method: Combining Umbrella Sampling with Free Energy Calculations, Comput. Phys. Commun., 2001, 135, 40–57 CrossRef.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef.
- A. Grossfield, “Wham: The Weighted Histogram Analysis Method”, Version 2.0.9, http://membrane.urmc.rochester.edu/content/wham, (accessed January 1, 2018) Search PubMed.
- G. M. Torrie and J. P. Valleau, Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- T. Lotta, J. Vidgren, C. Tilgmann, I. Ulmanen, K. Melen, I. Julkunen and J. Taskinen, Kinetics of Human Soluble and Membrane-Bound Catechol O-Methyltransferase: A Revised Mechanism and Description of the Thermolabile Variant of the Enzyme, Biochemistry, 1995, 34, 4202–4210 CrossRef PubMed.
- M. F. Hegazi, R. T. Borchardt and R. L. Schowen, Alpha.-Deuterium and Carbon-13 Isotope Effects for Methyl Transfer Catalyzed by Catechol O-Methyltransferase. Sn2-Like Transition State, J. Am. Chem. Soc., 1979, 101, 4359–4365 CrossRef.
- R. W. Woodard, M. D. Tsai, H. G. Floss, P. A. Crooks and J. K. Coward, Stereochemical Course of the Transmethylation Catalyzed by Catechol O-Methyltransferase, J. Biol. Chem., 1980, 255, 9124–9127 Search PubMed.
- J. Axelrod, J. Methylation Reactions in the Formation and Metabolism of Catecholamines and Other Biogenic Amines, Pharmacol. Rev., 1966, 18, 95–113 Search PubMed.
- J. K. Coward, E. P. Slisz and F. Y. H. Wu, Kinetic Studies on Catechol O-Methyltransferase, Product Inhibition and the Nature of the Catechol Binding Site, Biochemistry, 1973, 12, 2291–2297 CrossRef PubMed.
- J. Vidgren, L. A. Svensson and A. Liljas, Crystal Structure of Catechol O-Methyltransferase, Nature, 1994, 368, 354–358 CrossRef PubMed.
- E. Schultz and E. Nissinen, Inhibition of Rat Liver and Duodenum Soluble Catechol-O-Methyltransferase by a Tight-Binding Inhibitor or-462, Biochem. Pharmacol., 1989, 38, 3953–3956 CrossRef PubMed.
- P. Lautala, I. Ulmanen and J. Taskinen, Molecular Mechanisms Controlling the Rate and Specificity of Catechol O-Methylation by Human Soluble Catechol O-Methyltransferase, Mol. Pharmacol., 2001, 59, 393–402 CrossRef PubMed.
- J. Zhang and J. P. Klinman, Enzymatic Methyl Transfer: Role of an Active Site Residue in Generating Active Site Compaction That Correlates with Catalytic Efficiency, J. Am. Chem. Soc., 2011, 133, 17134–17137 CrossRef PubMed.
- T. H. Rod and U. Ryde, Quantum Mechanical Free Energy Barrier for an Enzymatic Reaction, Phys. Rev. Lett., 2005, 94, 138302 CrossRef PubMed.
- T. H. Rod and U. Ryde, Accurate QM/MM Free Energy Calculations of Enzyme Reactions: Methylation by Catechol O-Methyltransferase, J. Chem. Theory Comput., 2005, 1, 1240–1251 CrossRef PubMed.
- M. Roca, V. Moliner, I. Tuñón and J. T. Hynes, Coupling between Protein and Reaction Dynamics in Enzymatic Processes: Application of Grote−Hynes Theory to Catechol O-Methyltransferase, J. Am. Chem. Soc., 2006, 128, 6186–6193 CrossRef PubMed.
- G. D. Ruggiero, I. H. Williams, M. Roca, V. Moliner and I. Tuñón, QM/MM Determination of Kinetic Isotope Effects for COMT-Catalyzed Methyl Transfer Does Not Support Compression Hypothesis, J. Am. Chem. Soc., 2004, 126, 8634–8635 CrossRef PubMed.
- B. Kuhn and P. A. Kollman, QM–Fe and Molecular Dynamics Calculations on Catechol O-Methyltransferase: Free Energy of Activation in the Enzyme and in Aqueous Solution and Regioselectivity of the Enzyme-Catalyzed Reaction, J. Am. Chem. Soc., 2000, 122, 2586–2596 CrossRef.
- M. Roca, V. Moliner, J. J. Ruiz-Pernía, E. Silla and I. Tuñón, Activation Free Energy of Catechol O-Methyltransferase. Corrections to the Potential of Mean Force, J. Phys. Chem. A, 2006, 110, 503–509 CrossRef PubMed.
- J. J. P. Stewart, Optimization of Parameters for Semiempirical Methods V: Modification of NDDO Approximations and Application to 70 Elements, J. Mol. Model., 2007, 13, 1173–1213 CrossRef PubMed.
- N. Kanaan, J. J. Ruiz Pernia and I. H. Williams, QM/MM Simulations for Methyl Transfer in Solution and Catalysed by COMT: Ensemble-Averaging of Kinetic Isotope Effects, Chem. Commun., 2008, 6114–6116 RSC.
- M. Marianski, A. Supady, T. Ingram, M. Schneider and C. Baldauf, Assessing the Accuracy of across-the-Scale Methods for Predicting Carbohydrate Conformational Energies for the Examples of Glucose and A-Maltose, J. Chem. Theory Comput., 2016, 12, 6157–6168 CrossRef PubMed.
- P. Zhang, P. Bao and J. Gao, Dipole Preserving and Polarization Consistent Charges, J. Comput. Chem., 2011, 32, 2127–2139 CrossRef PubMed.
- P. Jurečka, J. Šponer, J. Černý and P. Hobza, Benchmark Database of Accurate (MP2 and CCSD (T) Complete Basis Set Limit) Interaction Energies of Small Model Complexes, DNA Base Pairs, and Amino Acid Pairs, Phys. Chem. Chem. Phys., 2006, 8, 1985–1993 RSC.
- C. M. Isborn, B. D. Mar, B. F. Curchod, I. Tavernelli and T. J. Martinez, The Charge Transfer Problem in Density Functional Theory Calculations of Aqueously Solvated Molecules, J. Phys. Chem. B, 2013, 117, 12189–12201 CrossRef PubMed.
- J. Zhang, H. J. Kulik, T. J. Martinez and J. P. Klinman, Mediation of Donor–Acceptor Distance in an Enzymatic Methyl Transfer Reaction, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7954–7959 CrossRef PubMed.
- D. Rutkowska-Zbik, M. Witko and L. Fiedor, Ligation of Water to Magnesium Chelates of Biological Importance, J. Mol. Model., 2013, 19, 4661–4667 CrossRef PubMed.
- W. W. Cleland, P. A. Frey and J. A. Gerlt, The Low Barrier Hydrogen Bond in Enzymatic Catalysis, J. Biol. Chem., 1998, 273, 25529–25532 CrossRef PubMed.
- U. Ryde, How Many Conformations Need to Be Sampled to Obtain Converged QM/MM Energies? The Curse of Exponential Averaging, J. Chem. Theory Comput., 2017, 13, 5745–5752 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03871f |
| This journal is © the Owner Societies 2018 |