
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAb initio molecular dynamics studies of formic acid dimer colliding with liquid water
Vesa
Hänninen
*a,
Garold
Murdachaew
 b,
Gilbert M.
Nathanson
c,
R. Benny
Gerber
de and
Lauri
Halonen
a
b,
Gilbert M.
Nathanson
c,
R. Benny
Gerber
de and
Lauri
Halonen
a
aDepartment of Chemistry, University of Helsinki, P. O. Box 55 (A.I. Virtasen aukio 1), FI-00014, Finland. E-mail: vesa.hanninen@helsinki.fi
bDepartment of Chemistry and Materials Science, Aalto University, P. O. Box 16100, FI-00076 Aalto, Finland
cDepartment of Chemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA
dInstitute of Chemistry and the Fritz Haber Research Center, The Hebrew University, 91904 Jerusalem, Israel
eDepartment of Chemistry, University of California, Irvine, California 92697, USA
First published on 30th August 2018
Abstract
Ab initio molecular dynamics simulations of formic acid (FA) dimer colliding with liquid water at 300 K have been performed using density functional theory. The two energetically lowest FA dimer isomers were collided with a water slab at thermal and high kinetic energies up to 68kBT. Our simulations agree with recent experimental observations of nearly a complete uptake of gas-phase FA dimer: the calculated average kinetic energy of the dimers immediately after collision is 5 ± 4% of the incoming kinetic energy, which compares well with the experimental value of 10%. Simulations support the experimental observation of no delayed desorption of FA dimers following initial adsorption. Our analysis shows that the FA dimer forms hydrogen bonds with surface water molecules, where the hydrogen bond order depends on the dimer structure, such that the most stable isomer possesses fewer FA–water hydrogen bonds than the higher energy isomer. Nevertheless, even the most stable isomer can attach to the surface through one hydrogen bond despite its reduced hydrophilicity. Our simulations further show that the probability of FA dimer dissociation is increased by high collision energies, the dimer undergoes isomerization from the higher energy to the lowest energy isomer, and concerted double-proton transfer occurs between the FA monomers. Interestingly, proton transfer appears to be driven by the release of energy arising from such isomerization, which stimulates those internal vibrational degrees of freedom that overcome the barrier of a proton transfer.
Introduction
Formic acid, HCOOH, is the simplest carboxylic acid and both highly acidic and soluble in water.1,2 In the gas phase, formic acid displays fascinating intramolecular dynamics through its ability to form one or two hydrogen bonds with itself. We ask in this simulation study how this dimerization alters the fate of a carboxylic acid when it collides with the surface of water with which it can also form hydrogen bonds. Upon collision, such fundamental pathways as adsorption, desorption, dissociation, entry, and proton transfer may all depend on the resiliency of these internal hydrogen bonds and its ability to make new hydrogen bonds with surface water molecules.Carboxylic acids, especially formic acid (FA), and their dimers have been studied experimentally and theoretically using various methods.3–16 Recently, the dynamics of interactions between FA and liquid water have been investigated both experimentally and computationally. In scattering experiments, FA and FA dimer were collided with a liquid water surface and each found to undergo nearly a complete uptake into salty water at 253 K.17 In order to yield detailed information about the molecular processes of FA at the air–water interface, ab initio molecular dynamics (AIMD) and metadynamics simulations were used to calculate collision trajectories.18 The simulations showed formation of hydrogen bonds between FA and water molecules which served to explain the disappearance of FA in the scattering experiments.17 Other fundamental dynamical events were also simulated, such as deprotonation of FA occurring during the 8–50 ps timeframe. On ice surfaces at the temperature of 250 K, earlier molecular dynamics simulations showed that FA and acetic acid form hydrogen bonds with two surface water molecules via the carbonyl group resulting in trapping of the acid molecule on the ice surface.19
The temperature dependence of the populations of six possible FA dimer isomeric forms has been studied computationally.20 It was estimated that the lowest energy isomer (structure I, see Fig. 1), in which the FA monomers are bound together with two strong internal O⋯H–O hydrogen bonds, is not the most populated species at high temperatures. Instead, at temperatures above 300 K, the isomer (structure II, see Fig. 1) bound by one strong O⋯H–O and one much weaker O⋯H–C hydrogen bond was nevertheless predicted to be more abundant due to entropic effects, even though its ground state energy was estimated to be about 18 kJ mol−1 higher than that of structure I. Experiments have not conclusively verified this prediction as it is difficult to identify spectral features of structure II due to its floppiness.11 However, in a Raman spectroscopy study,12 a weak vibrational band was assigned to structure II FA dimer, which supports its existence at room temperature.
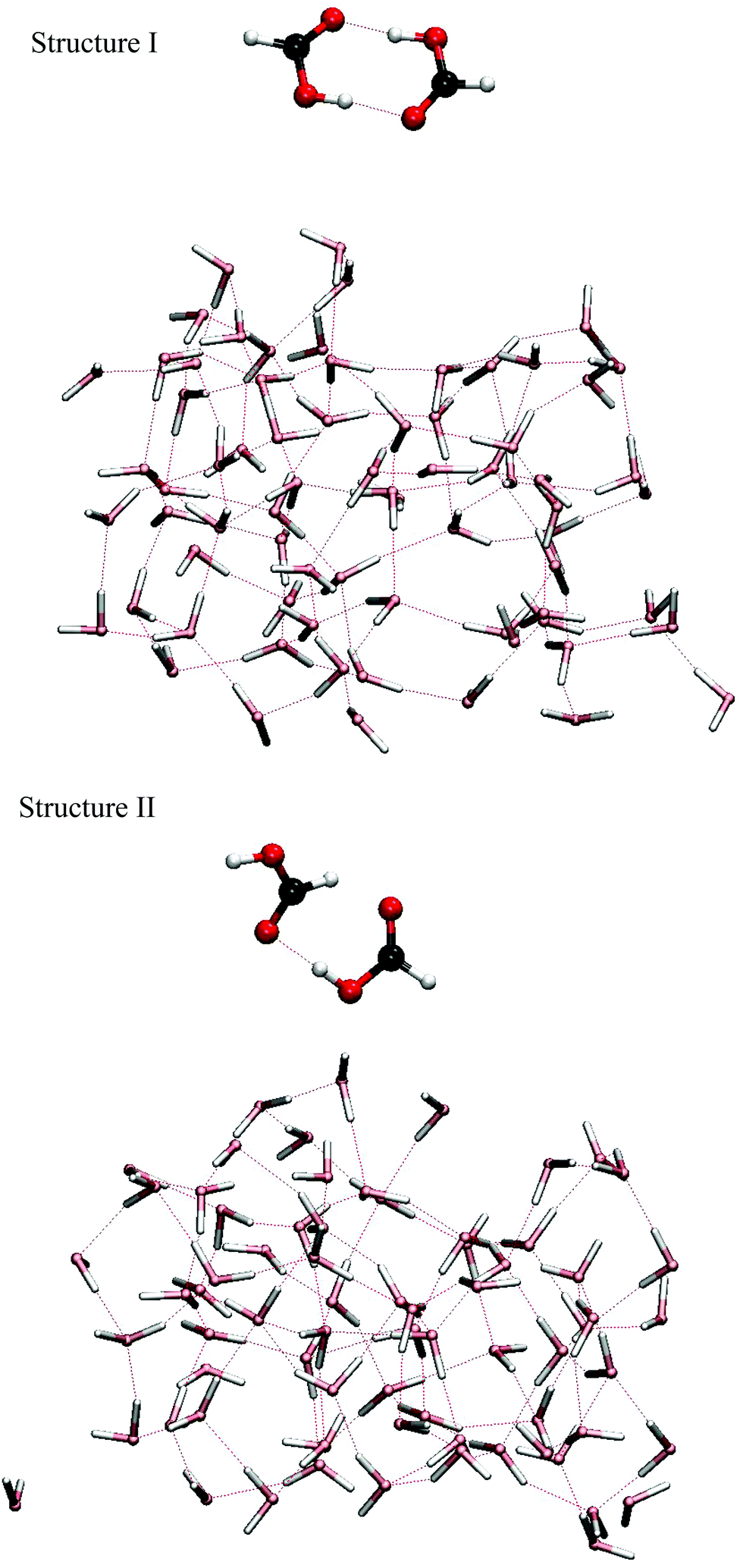 | ||
| Fig. 1 The two formic acid dimer structures and the liquid water slab. | ||
Proton transfer has been extensively studied in liquid water (see, e.g.ref. 21–23). Carboxylic acids and their dimers are also model systems in the studies of proton transfer.24,25 Whereas the monomer exchanges protons with solvent molecules such as water, cyclic dimers (e.g., structure I of FA dimer) facilitate intermonomer double proton transfer (DPT). Indeed, FA dimer is considered to be a prototype system of such internal DPT and, therefore, it has been a benchmark system for the study of DPT.26–29 The mechanism of DPT in FA dimer has been studied using path integral methods, where the proton motion around the DPT reaction path was determined to approach synchronous or concerted motion.28 A slight 8 fs time lag between the transferring proton motions was estimated using AIMD methods, where it was also noted that solvent effects can cause large differences between the two proton transfers.29 Solvent effects on DPT in FA dimer were studied using a hybrid quantum/classical approach in which the reactant species were treated with density functional theory (DFT) and the solvent with a classical method.30 The main results of this work showed that the presence of the solvent enhances DPT by a reduction of the DPT barrier. For the acetic acid dimer, it was found that DPT arises from the coupling between the O–H stretching vibrations and several low energy intermolecular vibrations.31 Thus, thermal fluctuations of the heavy atoms, such as O⋯O distances are an important ingredient in proton transfer processes, in both neat liquid water,21,23 and also in acids in water, including FA monomer in water, which some of us also noted in an earlier study.18
In this work, we employed AIMD to study dynamical processes, including scattering, adsorption, isomerization, dissociation, DPT, and energy transfer in collisions of FA dimers with a liquid water slab. Furthermore, our simulations are used to interpret and explain the experiments that show almost a full disappearance of FA dimer under similar conditions.
Computational methods
Similarly to previous investigations of FA monomer,18 we have studied isomers I and II of FA dimer colliding with a liquid water slab consisting of 72 water molecules (see Fig. 1). These systems were simulated mainly with Born–Oppenheimer ab initio molecular dynamics (AIMD) employing the Kohn–Sham DFT framework. Additionally, molecular dynamics simulations using empirical potential energy surfaces were performed to provide more information of the systems. Three-dimensional periodic boundary conditions were adopted in all systems where the 13.4724 × 15.5566 × 40 Å3 sized rectangular supercell contained sufficient vacuum to separate the periodic replicas in the direction perpendicular to the water surface.32–34 All MD computations were performed using the CP2K software package.35 For AIMD, we employed the QUICKSTEP module with the BLYP-D2 exchange correlation functional including the Grimme dispersion correction.36 The utilized basis set was double-zeta valence polarization (DZVP), which together with the chosen DFT functional has been shown to be adequate in our earlier AIMD studies treating acid–water systems.18,37 The inner orbitals were represented by the Goedecker–Teter–Hutter pseudopotentials (GTH). The energy cutoff for the plane waves was 280 Ry (Ry = 2.1798 aJ) and the time step was 0.5 fs. The convergence threshold for the wave function was set to 3 × 10−7 Hartree (1 Hartree = 4.35974 aJ). Additional simulations with empirical potential energy surfaces were performed using the TIP4P/2005 model for water38 and the Jedlovszky et al. potential for FA dimer.39As a starting point, we first equilibrated the FA-free water system, the slab consisting of 72 water molecules. This choice mimics our earlier study.18 By placing FA dimer above the pre-equilibrated water slab (see Fig. 1), we performed equilibration in the canonical ensemble (NVT) at 300 K in durations ranging up to 5–10 ps with the assistance of a massive Nose–Hoover thermostat. In order to equilibrate the slab for the collision simulations, the dimer was placed approximately 5 Å (1 Å = 10−10 m) above the surface of the slab. The purpose of this step was to enable the approaching FA dimer to relax to its optimal structure and orientation relative to the liquid surface. This approach is justified because relaxation would naturally occur in respective experiments in which FA dimers are introduced above the surface and have time to adjust to interacting forces arising from the presence of the liquid water molecules. To enhance sampling, eleven different equilibrated geometries were produced with the method described above. From these, eight belonged to structure I and three to structure II. After this initial step, collision trajectories were calculated using the microcanonical ensemble (NVE) as FA dimer approached the water liquid slab at a 45 degree angle, which mimics the experimental conditions. Altogether 46 NVE trajectories with different starting conditions were calculated in which the simulation duration was typically between 7–14 ps. From these, 34 belonged to structure I and 12 to structure II. After a careful analysis of the trajectories, some of them were extended in length up to 30 ps. This procedure is elaborated in detail in the Results section. In order to vary the starting conditions, we selected different geometries from equilibrated NVT trajectories and chose a set of starting kinetic energies ranging from 0 to 68kBT for the formic acid dimer's center of mass motion, where T is the temperature of the slab and kB is the Boltzmann constant. Using these conditions, we performed two sets of trajectory calculations for the two different FA dimer conformers, structures I and II. Taking into account the 64 trajectories calculated with the empirical force field, the total number of calculated trajectories in this work is 121.
Results
The MD calculations described above were used to investigate the fate of FA dimers as they collided with the water slab. We focused on gas-surface energy transfer as the initial collision energy was varied. We were also interested in hydrogen bond formation and adsorption and evaporation, and bond dissociation and proton transfer. Each of these topics is discuss below.Energy transfer in high energy collisions
Eight trajectories of structure I FA dimers were calculated in high-energy collisions with the water slab at 300 K. A kinetic FA dimer center of mass energy of 68kBT (170 kJ mol−1) was selected to mimic recent experiments.17 We estimated the escape probability of the directly scattering FA dimer by using two points of views. First, we qualitatively counted the number of trajectories that qualify as scattering events. For this, we adopted a criterion that the dimer must immediately rebound to a position above the starting point and does not readsorb. Two of the trajectories had sufficient escape velocity, which at the defined position were on average about half of the starting velocities, and therefore met the criterion of scattering. Second, to quantify the energy transfer of the collisions, we also monitored the change of the average kinetic energy of the FA dimers using all eight high energy collisional trajectories (see Fig. 2). We calculated the magnitude of the collisional kinetic energy transfer of FA dimers immediately following contact with the water surface. Our calculations showed that only 5 ± 4% of the FA dimer initial kinetic energy was retained after a collision. The standard deviation of ±4% indicates that all trajectories consistently show that the dimer experiences over 90% transfer of kinetic energy to potential energy resulting in various dynamical processes which will be discussed in the following sections. These two analyses can be compared to the impulsive energy transfer of FA dimers of structure I directly after a collision, i.e., the kinetic energy of directly scattering FA dimers relative to the initial kinetic energy of the incoming FA dimer molecular beam. The experimental impulsive energy transfer was measured to be 10%,17 which is in good agreement with both the qualitative outlook of our simulations and with our quantitative computational result. We also note that in the trajectories, in the case of less added kinetic energies and the ones having structure II dimer, the direct scattering from the surface was absent because there is not enough kinetic energy retained for the dimer to escape from the surface. | ||
| Fig. 2 Kinetic energies of structure I FA dimers in high energy trajectories 1–8. | ||
We also performed MD simulations with empirical force fields, referred to here as classical MD simulations. Added kinetic energies for FA dimer ranged from 1 to 32kBT in these simulations. These calculations were thought to provide more sampling of the systems and to increase the length of the simulations. However, they turned out to be not directly as useful as first thought as they showed almost full direct scattering of structure I dimers, which does not agree with experiments. We discuss implications of this outcome shortly here. The near elastic scattering occurs because the empirical force field we used models the monomers as rigid entities, and, thus, do not allow kinetic energy redistribution to internal vibrational degrees of freedom. Even though the internal hydrogen bonds in the dimer can absorb energy, the rigidity of FA monomers makes the collisions unrealistically over-elastic. This overestimates the remaining translational energies which allows FA dimers to escape from the surface directly after collisions. The typical initial internal kinetic energy of the dimer is around 30 kJ mol−1 in our AIMD calculations and can fluctuate up to around 80 kJ mol−1 in high energy collisions. We performed some preliminary calculations with empirical force fields that do not treat molecules as rigid entities but this work is unfinished.
The adsorption of FA dimer on water surface
As noted above, our calculations indicate that over 90% of FA dimers with high kinetic energies of 68kBT remain adsorbed over the simulation time on the liquid water slab after the collision. Our simulations also show no scattering and complete adsorption when FA dimers are collided thermally or with 32kBT or lower initial kinetic energies. Analogously, the previous study,18 where the collisions of FA monomer with water were simulated, confirmed the experimental observation of no scattering of FA into the vapor. This result was expected because the FA molecule is hydrophilic due its ability to form strong hydrogen bonds with water making it soluble to water. This property of FA monomer alone cannot explain the adsorption of FA dimers on the water surface because the FA dimer is less hydrophilic than the monomer in its lowest energy form structure I due to occupation of the H-bonding sites by intermonomer hydrogen bonds. However, if the dimer's internal hydrogen bonds broke due to a collision, it would be able to form strong hydrogen bonds that would increase its hydrophilic character. Our calculations showed three trajectories resulting in dissociation of FA dimer internal hydrogen bonds. One dimer break-up event occurred in trajectory with starting structure I. In this calculation, the dimer possessed high starting kinetic energy of 68kBT. The dissociation occurred immediately after the collision between the dimer with the water surface. Two of these FA dimer internal hydrogen bond break-up events belonged to the trajectories starting from FA dimer structure II. The findings are in accordance with the experimental results. Thus, the experimentally observed adsorption of FA dimers must have another explanation.Here, we adopt a model in which hydrogen bonding describes the main attraction between FA and water molecules. Formic acid can form four different types of hydrogen bonds as both hydroxyl and carbonyl groups can act as a proton acceptor and donor (see Fig. 3). Because both hydroxyl and carbonyl group oxygens have two lone pair electrons, they can form two acceptor H-bonds, making the total number of H-bonds sites equal to six. The strongest H-bonds are the hydroxyl donor (O–H⋯), hydroxyl acceptor (⋯O–H), and carbonyl acceptor (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯) H-bonds. The strength of the carbonyl proton donor H-bond (⋯H–C) is only around 10% of the strongest hydrogen bonds for the pure liquid formic acid.40 In dimer structure I, the hydroxyl donor vacancies and other carbonyl acceptor vacancies are absent, which leaves the dimer with three possible different hydrogen bonds with water. Here, the total number of available liquid water–FA dimer H-bonding sites is just six.
O⋯) H-bonds. The strength of the carbonyl proton donor H-bond (⋯H–C) is only around 10% of the strongest hydrogen bonds for the pure liquid formic acid.40 In dimer structure I, the hydroxyl donor vacancies and other carbonyl acceptor vacancies are absent, which leaves the dimer with three possible different hydrogen bonds with water. Here, the total number of available liquid water–FA dimer H-bonding sites is just six.
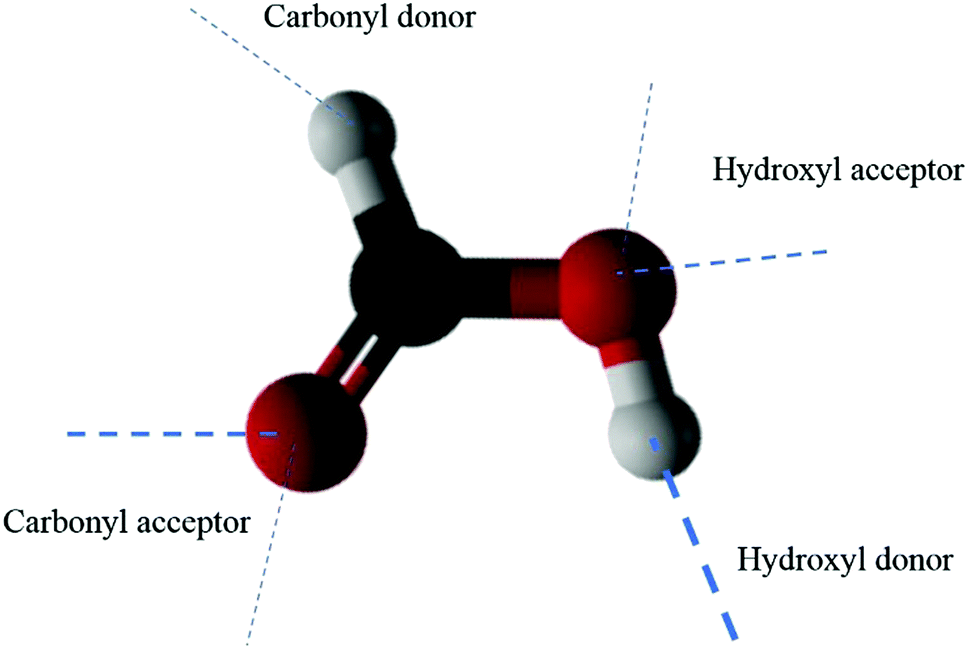 | ||
| Fig. 3 The six hydrogen bonds that a formic acid molecule can form. The strongest H-bonds are marked with thick lines. | ||
We have calculated the occupancies of three types (defined above) of hydrogen bonds from 27 trajectories for FA dimer structure I. The H-bond occupancy is defined as an average number of hydrogen bonds. These occupancies reflect the strength of interaction between FA dimer and the surface water molecules, as their strong interaction most likely leads to H-bonding. The higher the occupancy of the given H-bond is, the more it contributes to the attraction between FA dimer and the water surface. The H-bond occupancies extracted from trajectory sets are given in Table 1. The occupancies stay reasonably consistent through the different starting conditions and numbers of trajectories in given sets. Our results show that the carbonyl acceptor H-bonds are approximately one order of magnitude more occupied than the carbonyl donor H-bonds, which gives a similar trend when compared to the importance and relative strengths of these types of H-bonds previously estimated in pure liquid formic acid.39,40 Our calculated H-bond occupancies indicate that in FA dimer structure I acceptor H-bonds give the majority of the attractive interaction between FA dimer and the water surface and, thus, are mainly responsible of the attachment of the dimers to the water surface in collisions.
| Carbonyl acceptor (CO⋯) |
Carbonyl donor (⋯H–C) | Hydoxyl acceptor (⋯O–H) | Hydoxyl donor (O–H⋯) | Total | |
|---|---|---|---|---|---|
| Structure I | 0.29 | 0.03 | 0.13 | 0.00 | 0.45 |
Desorption of FA dimer after trapping on the surface of water
The desorption after trapping of FA dimer is a μs timescale phenomenon. These timespans are beyond the reach of AIMD simulations, which are limited to at most 100 ps. However, we can study the desorption after trapping by sampling multiple trajectories, calculated in parallel, and by varying the initial conditions in a way described in Computational methods section. In this approach, we varied FA dimer's initial kinetic energies to mimic local thermal fluctuations which would naturally occur as more time passes. These hot spots on the surface provide the kinetic energy for FA dimer to escape from the surface. The different conditions of our system have been discussed in the Computational details section. Interestingly, none of the simulated trajectories showed clear evaporation from the surface even though several “close calls” were seen in which the dimer hovered over the surface seemingly without any hydrogen bonding to the liquid surface. In Fig. 4, it is shown that the total number of hydrogen bonds of four representative trajectories of structure I maintains low numbers, i.e., from 0 to 3, throughout the simulations. Fig. 4 also shows snapshots of these representative trajectories. Excluding the scattering events observed in some high energy collision trajectories, all simulations show that despite the hydrophobic character of structure I, FA dimer sticks to the surface as even the low H-bond order yields sufficient attractive interaction between FA dimer and water molecules. In addition to the NVE simulations of 7–14 ps durations, we performed a longer NVT simulation of 35 ps duration, which also showed that the dimer species stay on the top of the surface. This is an important result in three aspects. Firstly, it supports the experimental observation of disappearance of the formic acid dimers when they collide with water liquid.17 In these experiments, the desorption after trapping was monitored in around a 100 μs timeframe. Almost full disappearance of structure I FA dimer was observed; on average only 0.9 ± 0.4% of the FA dimer species were found to desorb after collision. Secondly, according to Fig. 4, it can be seen that FA dimer in its most abundant conformer in low temperature molecular beam experiments (structure I) forms on average fewer than one hydrogen bond with the surface water molecules. Thirdly, this result contradicts the implications of an earlier experiment, where due to the assumed hydrophobicity of FA dimers these species would have to surmount a barrier to adsorption that requires concerted FA–FA hydrogen bond breaking and FA–water hydrogen bond making.41 Our simulations indicate that this concerted rearrangement is unnecessary for longtime trapping and that there is no barrier to adsorption. | ||
| Fig. 4 The number of H-bonds with water molecules when starting with the formic acid dimer in structure I and snapshots in trajectories I1–I4. The added kinetic energies for the dimer are 1kBT, 1kBT, 4kBT, and 36kBT for the trajectories I1, I2, I3, and I4, respectively. The symbols FA1 and FA2 refer to two formic acid monomers in the dimer. | ||
FA dimer proton transfer dynamics
We have also investigated how collisions of FA dimer with the water slab induce internal dynamics of FA dimer, including proton transfer, dimer breakup, and isomerization. Because structure I FA dimer possesses two strong internal hydrogen bonds the trajectories with 32kBT or less added kinetic energy showed no breaking of these internal hydrogen bonds as the system was not energetic enough for H-bond dissociation. The interaction energy between FA monomers in the dimer was estimated to be 82.4 kJ mol−1, which is around 33kBT, using the CCSD(T)/aug-cc-pVTZ level of theory.42 However, two trajectories with added kinetic energy of 68kBT showed partial and even full dissociation of the intermonomer hydrogen bonds. This is an important result because, as noted earlier, this kinetic energy corresponds to the average experimental kinetic energy of the dimers in the molecular beam experiments. Thus, our simulations suggest that a significant portion of FA dimers in the molecular beam will break the hydrogen bond ring structure upon collisions, making them a target for hydrogen bonding with water and, therefore, significantly raising the attractive interaction between FA dimer and the water surface. However, the experiments also show that the uptake of FA dimer is nearly complete even at low collisional energies,17 which indicates that hydrogen bond breaking caused by high collision energies is not essential for uptake.FA's structure II possesses weaker internal hydrogen bonds than the energetically lower-lying structure I. Here, our simulations show two interesting processes: isomerization from structure II to structure I followed by concerted proton transfer and FA dimer dissociation. These processes occur independently of the approaching dimer's kinetic energy as we observe two of these events in both low and mid (1kBT and 32kBT) kinetic energies. In the isomerization process, the weak CO⋯H hydrogen bond breaks and the lower energy structure I forms. Due to this isomerization, FA dimer becomes relatively hydrophobic, as is shown by low number of hydrogen bonds in the first and second rows of the top graph of Fig. 5. Here, the hydrogen bonding dynamics shows similarities with the trajectories illustrated in Fig. 4. Isomerization from the higher energy isomer to the lower energy isomer adds internal energy to the system. This extra energy stimulates intermonomer concerted proton transfer, as shown in the first two snapshots of Fig. 5.
 | ||
| Fig. 5 The number of H-bonds with water molecules when starting with the formic acid dimer in structure II and snapshots in trajectories II1–II3. The added kinetic energies for the dimer are 1kBT, 36kBT, and 36kBT for the trajectories II1, II2, and II3, respectively. The symbols FA1 and FA2 refer to two formic acid monomers in the dimer. | ||
The dissociation of FA dimer shows diverse hydrogen bonding dynamics. Snapshots of structure II dimer break-ups are pictured in Fig. 5 (the last snapshot) and Fig. 6. The correlation between the dimer break-up and hydrogen bonding is also seen in Fig. 6 where, in trajectory II4, the label b points to the dissociation event which coincides with the rise of the number of water⋯FA hydrogen bonds by one. This new hydrogen bond is located in the carbonyl group that already possessed an H-bond with water before the breakup event. Shortly after dissociation, the other FA monomer continues to dissolve in water mediated by hydrogen bonding. This H-bond is located in the hydroxyl group losing the internal dimer hydrogen bond as is shown in the first and third snapshots of Fig. 6. Later after the dimer dissociation, trajectory II4 shows a near protonation event caused by solvation with water. The total number of H-bonds rises after 6 ps, which results in trajectory point d as a near deprotonation of the other FA monomer. This is shown in a shared proton with the FA monomer and water in the last snapshot in Fig. 6.
 | ||
| Fig. 6 The number of H-bonds with water molecules when starting with the formic acid dimer in structure II and snapshots in trajectory II4. The added kinetic energy for the dimer is 1kBT for the trajectory II4. The active monomer is the one which has hydrogen bonds with water before a dimer breakup. | ||
An interesting observation from our simulations is that FA dimer proton transfer dynamics was absent in the thermal or in the low to mid kinetic energy collision trajectories (up to 32kBT) where the dimer was initially in structure I. This is because the FA dimer did not gain enough vibrational energy from a collisional impact to overcome the barrier of the proton transfer.26,28 Typically, we observed several near transfers of protons, of which some examples are depicted in snapshots and pointed out by arrows in Fig. 4. These events do not seem to correlate with hydrogen bonding with the water molecules. Moreover, we did not observe dissolution of FA dimer or dissociated monomer in these short simulations.
Conclusions
Several ab initio and empirical force field molecular dynamics simulations have been performed with durations of 7 to 35 ps to investigate scattering, adsorption, desorption, proton transfer, and dissociation of FA dimer in its collisions with liquid water. This study confirms the latest experimental observation of negligible direct scattering and escape of FA dimers after collisions. The computed trajectories show that scattering is rare and that no desorption occurs after trapping on the surface. Thus, we conclude that all calculations started from the two energetically lowest lying FA conformers at thermal energy, as well as at all kinetic energies up to 68kBT show that FA dimer–liquid water hydrogen bonding leads to trapping on the surface of liquid water. The hydrophobic, lowest energy FA dimer conformer (structure I) formed on average less than one hydrogen bond with the surface water molecules, built from either the FA hydroxyl or carbonyl group acting as an acceptor, or the carbonyl H atom acting as a weak donor. Even, this low hydrogen bond order is enough to keep the dimer attached to the outermost water molecules. With so few hydrogen bonds between the dimer and water molecules, it is intriguing to postulate that this longtime trapping should occur with other weakly hydrogen bonding species. In trajectories involving high added kinetic energies for structure I and the less stable structure II, there are more possibilities due to the larger energy available. In particular, the weaker internally H-bonded structure II FA dimer is likely to break up due to solvation by water molecules. The simulations starting with structure II FA dimer displayed (i) a concerted double proton transfer arising from energy redistribution to internal vibrational motions from a conformer change, (ii) dissociation of the dimer due to solvation with water molecules via hydrogen bonding, and (iii) a high bond order of hydrogen bonds with water molecules, resulting in the opening up of the dimer structure and formation of cyclic structures with water. These structures are known to be precursors of proton transfers between formic acid and water.18 The AIMD simulations performed here provide new insights into the elementary steps leading up to this proton transfer, and therefore ultimately shed light on the acidification of water even when these protons are tied up by hydrogen bonds within the gas-phase acid.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Finland's Center for Scientific Computing (CSC) for providing computational resources. R. B. G. and G. M. N. gratefully acknowledge support of their research by the US National Science Foundation through CAICE, the Center for Aerosol Impacts on the Chemistry of the Environment (CHE-1305427). Our CAICE research contributed to the analysis of findings in the present study. L. H. thanks the Academy of Finland (grant number 294752) and the Finnish Cultural Foundation for financial support.Notes and references
- CRC Handbook of Chemistry and Physics, ed. J. Rumble, CRC Press, Boca Raton, 98th edn, 2017 Search PubMed.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. de Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, Atmos. Chem. Phys., 2015, 15, 6283–6304 CrossRef.
- K. Mackeprang, Z. H. Xu, Z. Maroun, M. Meuwly and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2016, 18, 24654–24662 RSC.
- I. Yokoyama, Y. Miwa and K. Machida, J. Am. Chem. Soc., 1991, 113, 6458–6464 CrossRef.
- Y. Qi, R. Zhu and D. Zhang, J. Mol. Model., 2014, 20, 2264 CrossRef PubMed.
- C. H. Turner, J. Phys. Chem. B, 2004, 108, 11716–11721 CrossRef.
- L. Turi, J. Phys. Chem., 1996, 100, 11285–11291 CrossRef.
- J. Granatier, M. Pitonak and P. Hobza, J. Chem. Theory Comput., 2012, 8, 2282–2292 CrossRef PubMed.
- L. E. Fernández, A. C. Gomez Marigliano and E. L. Varetti, Vib. Spectrosc., 2005, 37, 179–187 CrossRef.
- V. V. Matylitsky, C. Riehn, M. F. Gelin and B. Brutschy, J. Chem. Phys., 2003, 119, 10553–10562 CrossRef.
- R. M. Balabin, J. Phys. Chem. A, 2009, 113, 4910–4918 CrossRef PubMed.
- J. M. Briggs, T. B. Nguyen and W. L. Jorgensen, J. Phys. Chem., 1991, 95, 3315–3322 CrossRef.
- R. W. Gora, S. J. Grabowski and J. Leszczynski, J. Phys. Chem. A, 2005, 109, 6397–6405 CrossRef PubMed.
- C. Colominas, J. Teixido, J. Cemeli, F. J. Luque and M. Orozco, J. Phys. Chem. B, 1998, 102, 2269–2276 CrossRef.
- C. M. Nordstrom, A. J. McGrath and A. J. Thakkar, Can. J. Chem., 2010, 88, 736–743 CrossRef.
- T. B. Sobyra, M. P. Melvin and G. M. Nathanson, J. Phys. Chem. C, 2017, 121, 20911–20924 CrossRef.
- G. Murdachaew, G. M. Nathanson, R. B. Gerber and L. Halonen, Phys. Chem. Chem. Phys., 2016, 18, 29756–29770 RSC.
- M. Compoint, C. Toubin, S. Picaud, P. N. M. Hoang and C. Girardet, Chem. Phys. Lett., 2002, 365, 1–7 CrossRef.
- J. Chocholoušová, J. Vacek and P. Hobza, Phys. Chem. Chem. Phys., 2002, 4, 2119–2122 RSC.
- D. Marx, ChemPhysChem, 2007, 8, 209–210 CrossRef.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef.
- D. Marx, M. E. Tuckermann, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef.
- R. A. Bachorz, M. Haranczyk, I. Dabkowska, J. Rak and M. Gutowski, J. Chem. Phys., 2005, 122, 204304 CrossRef PubMed.
- K. Marushkevich, L. Khriachtchev, J. Lundell and M. Räsänen, J. Am. Chem. Soc., 2006, 128, 12060–12061 CrossRef PubMed.
- N. Shida, P. F. Barbara and J. Almlöf, J. Chem. Phys., 1991, 94, 3633–3643 CrossRef.
- Y. Kim, J. Am. Chem. Soc., 1996, 118, 1522–1528 CrossRef.
- S. Miura, M. E. Tuckerman and M. L. Klein, J. Chem. Phys., 1998, 109, 5290–5299 CrossRef.
- H. Ushiyama and K. Takatsuka, J. Chem. Phys., 2001, 115, 5903–5912 CrossRef.
- J. Kohanoff, S. Koval, D. A. Estrin, D. Laria and Y. Abashkin, J. Chem. Phys., 2000, 112, 9498–9508 CrossRef.
- P. Durlak, S. Berski and Z. Latajka, J. Mol. Model., 2011, 17, 2995–3004 CrossRef PubMed.
- A. D. Hammerich and V. Buch, J. Phys. Chem. A, 2012, 116, 5637–5652 CrossRef PubMed.
- A. D. Hammerich, B. J. Finlayson-Pitts and R. B. Gerber, J. Phys. Chem. Lett., 2012, 3, 3405–3410 CrossRef PubMed.
- A. D. Hammerich, B. J. Finlayson-Pitts and R. B. Gerber, Phys. Chem. Chem. Phys., 2015, 17, 19360–19370 RSC.
- The CP2K developers group, http://www.cp2k.org, 2000–2014.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef PubMed.
- L. Partanen, G. Murdachaew, R. B. Gerber and L. Halonen, Phys. Chem. Chem. Phys., 2016, 18, 13432–13442 RSC.
- J. L. F. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef PubMed.
- P. Jedlovszky and L. Turi, J. Phys. Chem. B, 1997, 101, 5429–5436 CrossRef.
- R. Chelli, R. Righini and S. Califano, J. Phys. Chem. B, 2005, 109, 17006–17013 CrossRef PubMed.
- M. Faubel and Th. Kisters, Nature, 1989, 339, 527–529 CrossRef.
- P. Krishnakumar and D. K. Maity, Comput. Theor. Chem., 2017, 1099, 185–194 CrossRef.
| This journal is © the Owner Societies 2018 |