
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge transfer dynamics at the boron subphthalocyanine chloride/C60 interface: non-adiabatic dynamics study with Libra-X†
Kosuke
Sato
 a,
Ekadashi
Pradhan
b,
Ryoji
Asahi
a and
Alexey V.
Akimov
*b
a,
Ekadashi
Pradhan
b,
Ryoji
Asahi
a and
Alexey V.
Akimov
*b
aToyota Central Research and Development Laboratories, Inc., 41-1, Yokomichi, Nagakute, Aichi 480-1192, Japan
bDepartment of Chemistry, University at Buffalo, The State University of New York, Buffalo, New York 14260-3000, USA. E-mail: alexeyak@buffalo.edu
First published on 25th September 2018
Abstract
We report a study on the non-adiabatic molecular dynamics (NA-MD) of the charge transfer (CT) process in the boron subphtalocyanine chloride (SubPc)/fullerene (C60) interface using our newly implemented Libra-X software package, which is based on an interface of the Libra NA-MD library and the GAMESS electronic structure software. In particular, we address the following aspects of the simulation protocol: (a) the choice of the potential used to treat interatomic interactions and its effect on the structures of the complex and CT rates; (b) the choice of the electronic structure methodology used; and (c) the choice of the trajectory surface hopping (TSH) methodology used. From our analysis of the electronic structure, we suggest that the distortion of the SubPc conical structure affects orbital localization and that the “breathing” motion of SubPc drives the CT process in SubPc/C60. This study illustrates that the choice of the TSH methodology and electronic decoherence are crucial for the CT simulation. We extend our analysis of CT in SubPc/(C60)n models by increasing the number of C60 molecules up to n = 4. We find that the details of the interfacial SubPc/(C60)n geometry determine the CT rate. Finally, we find the computed CT timescale to be in the range of 2.2–5.0 ps, which is in agreement with the experimentally determined timescale in the order of magnitude of ∼10 ps. The developed open-source Libra-X package is freely available on the Internet at https://github.com/Quantum-Dynamics-Hub/Libra-X.
1. Introduction
Organic photovoltaic (OPV) materials are among the most studied systems owing to their potential for manufacturing efficient and inexpensive solar cells.1–14 Phthalocyanines constitute a class of organic chromophores that bear a close resemblance to the naturally occurring porphyrin molecules, which are employed in nature itself as the key components of photosynthetic complexes in plants and photosynthetic bacteria.15,16 Both porphyrins and phthalocyanines are remarkable for their high molar extinction coefficient,17,18 which is a pre-requisite for efficient solar energy harvesting. For these reasons, phthalocyanines and their derivatives have been attracting continuous interest as potential template chromophores that could be used in OPV materials.19–27Boron subphthalocyanine chloride (SubPc)/fullerene (C60) heterojunctions have been recently attracting a lot of interest because of the high open-circuit voltages and power conversion efficiencies of such systems.28–34 SubPc is composed of three nitrogen-fused diiminoisoindole rings that are connected around a boron atom and form a cone-shaped structure. SubPc has been widely employed as a donor material whose properties can be tuned by varying the atoms and anchoring molecules,35–41 while C60 and its derivatives have played the role of electron acceptors.42–45 Efficient charge transfer (CT) at the donor/acceptor interfaces is a pre-requisite for realizing OPV cells with a high power conversion efficiency. Thus, the CT process in the SubPc/C60 interface has been studied,46–48 as well as in other similar heterojunctions.14,22,49–53 Recently, Wilcox et al.47 reported that the CT process in a SubPc/C60 system occurred on the 10 ps timescale. The accompanying calculations for a tightly packed SubPc/C60 dimer based on the Fermi golden rule54 predicted duration of the CT process to be on the order of 500 fs, which is notably shorter than the experimental value. These results suggested that the experimentally observed longer timescale can be attributed to an alternative orientation of the SubPc molecule on the C60 surface. A following study carried out by Akimov48 focused on the CT dynamics in a larger model of the interface (nearly 700 atoms) using the non-adiabatic molecular dynamics (NA-MD) technique extended to fragment molecular orbitals (FMOs). The study reported timescales comparable to those observed in the previous experiment, but only when electronic decoherence effects were accounted for. Moreover, the study suggested that the longer timescales observed in the previous experiment can be attributed to the details of the interfacial structure that are present in the extended system but not necessarily in the minimal dimer model. The study relied on extended Hückel theory (EHT)55–57 for electronic structures, parameterized to reproduce the orbital alignment and energy gaps of the constituent fragments. Although the aforementioned FMO-NA-MD work revealed a number of important effects, it is known that the results of NA-MD calculations may be sensitive to the details of the methodologies and interactions employed.58–61 It is important to understand the possible dependencies of the results on the choice of methodologies used for treating nuclear dynamics, electronic structures, and CT dynamics.
In the present work, we aim to fill the noted knowledge gap and also extend our mechanism understanding of CT dynamics in SubPc/(C60)n systems. To address these goals, we have developed an open-source software for NA-MD simulations, dubbed Libra-X, which interfaces the Libra library62 for NA-MD simulations with a number of community electronic structure packages: in particular, with GAMESS.63,64 Our theoretical and computational methodology is presented and the program workflow is described.
Using the semiempirical methods available within the GAMESS electronic structure package interfaced with Libra within the Libra-X workflow, we undertake a comprehensive investigation of the role that the choice of computational methodologies may play in determining CT rates. In particular, we investigate the CT dynamics at the SubPc/C60 interface while avoiding the use of the fragmentation ansatz. In particular, we address the following aspects of the simulation protocol: (a) the choice of the potential used to treat interatomic interactions and its effects on the structures of the complex and the CT rates; (b) the choice of the electronic structure methodology used; and (c) the choice of the trajectory surface hopping (TSH) methodology used. Furthermore, via density functional theory (DFT), we assess the quality of the semiempirical molecular orbital theories used in the present NA-MD simulations.
We further extend our analysis of the CT process in SubPc/(C60)n by considering larger molecular models, with up to four (n = 4) C60 molecules. Such large-scale calculations are enabled by our newly developed software, Libra-X. By analyzing the correlation between the computed molecular/electronic structures and the computed electron transfer timescales, we illustrate how the details of the interfacial geometry can affect the CT rates in SubPc/(C60)n systems, confirming earlier suggestions of the need for an extended system for the accurate modeling of the CT process at SubPc/C60 interfaces.
2. Computational methodology
2.1. Systems studied
As suggested in earlier studies,14,46,48,50–53 the details of the interfacial structure of SubPc/C60 systems may play an important role in determining the CT rates. In this work, we consider several such interfaces: SubPc/(C60)n with n = 1, 2, 3, and 4 (Fig. 1). The dimer model (n = 1), SubPc/C60, shown in Fig. 1a, has been previously used in the works of Wilcox47 and Akimov.48 The extended models (n = 2, 3, and 4), SubPc/(C60)n shown in Fig. 1b–d, are created by adding C60 to the dimer model. This addition makes the SubPc/C60 interfacial model more realistic. The computational techniques used in this work allow us to perform direct (without fragmentation) NA-MD simulations on such extended systems.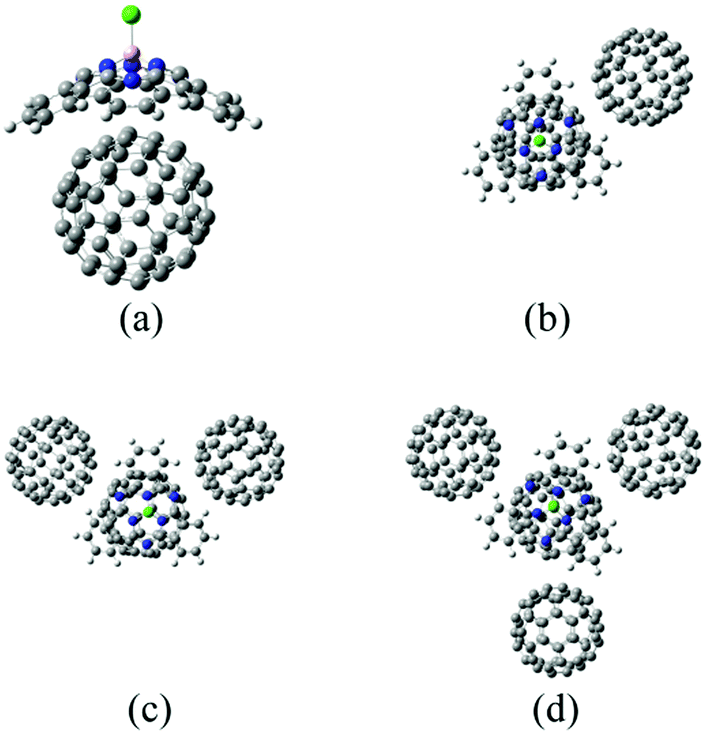 | ||
| Fig. 1 Molecular models for SubPc/(C60)n interfacial structures. (a) Dimer model (SubPc/C60). (b) SubPc/(C60)2. (c) SubPc/(C60)3. (d) SubPc/(C60)4. The balls in white, pink, gray, blue, and yellow-green depict hydrogen (H), boron (B), carbon (C), nitrogen (N), and chlorine (Cl) atoms, respectively. | ||
2.2. NA-MD methodology
The electronic state of the system is described by a time-dependent superposition of stationary states, ψi, parametrically dependent on the nuclear configurations, R(t):
 | (1) |
Here, r are the coordinates of electrons. The coefficient ci(t) represents the amplitudes of electronic state i in the superposition and acts as the electronic degree of freedom that determines the time evolution of the wavefunction of the system. The electronic degrees of freedom evolve according to the time-dependent Schrödinger equation (TD-SE):
 | (2) |
is the non-adiabatic (time-derivative) coupling (NAC) between states i and j.

Following earlier works,66–83 the stationary states, ψi, are chosen as 1-electron orbitals obtained from the corresponding electronic structure calculations. The use of the 1-electron orbitals is mainly motivated by the computational costs associated with the use of rigorous electronic states based on the superpositions of multi-electron Slater determinants.84 Such calculations are favorable for large systems and for long simulation timescales, such as those undertaken in the present work. At the same time, as our practice suggests, the transitions between rigorously-defined multi-electronic states are often dominated by the transitions between the frontier orbitals.85 Thus, the use of the 1-electron orbitals provides a practical way for obtaining qualitative and semi-quantitative insights into the charge and energy transfer dynamics in extended systems. Finally, one of the goals of the present work is to assess the potential differences in NA dynamics that may originate in the level of electronic structure treatment, nuclear dynamics, and intermolecular interactions used, as well as the NA-MD algorithm used. Focusing on these effects and preserving the 1-electron treatment of electronic states will allow us to relate the conclusions of this work to analogous NA-MD studies based on 1-electron approximations performed by other researchers.66–72,86 The accuracy of the approximation is assessed using TD-DFT in Section 3.3.
To study how the choice of electronic structure methodology affects the properties of 1-electron orbitals and electronic dynamics, we have considered two semiempirical approaches: (a) the Recife model 1 (RM1)87 and (b) extended Hückel theory (EHT).55–57 The Libra-X package developed in this work utilizes the GAMESS software63,64 as the electronic structure calculations driver. Thus, the range of semiempirical methods we can utilize at this point is limited by the methods available in the GAMESS code. Other methods such as MNDO,88 AM1,89 and PM390,91 are also available in GAMESS. We exclude the PM3 method due to the lack of the parameters for boron in the GAMESS implementation of PM3. In our preliminary tests, we found the AM1 method disagrees with ωB97XD92,93 results: the LUMO+1 and LUMO+2 of AM1 were localized on C60 and SubPc, respectively.
The RM1 method is based on the neglect of diatomic differential overlap (NDDO)94 approximation, and is defined by the reparametrization of the Austin model 1 (AM1) for some atomic species.89 NDDO-based methods, such as AM1 and RM1, have been used previously in a number of NA-MD studies.95–98 In this work we selected RM1 method in favour of MNDO for the sake of convenience of the dispersion energy adjustment. The MNDO may be another suitable methodology to simulate charge transfer if the dispersion energy is adjusted well to reproduce the SubPc/C60 binding energies of DFT.
EHT is a tight-binding Hamiltonian that allows for capturing the essential physics of many chemical systems, provides transparent routes for reparameterization, and is computationally efficient. Despite its approximate nature, it has been successfully utilized in numerous chemical applications.71,99–103 In the present calculations, it is available via the Libra package.62 For consistency with other studies, we have used the Calzaferri57 flavor of the EHT formula with its parameters taken from earlier work carried out by one of us.48 This parametrization reproduces the orbitals alignment and energy gaps of the SubPc and C60 fragments.
Eqn (2) is solved employing a unitary transformation that diagonalizes the vibronic Hamiltonian, as discussed elsewhere.48,104 Such a solution requires numerical values for Hvib at the mid-point of every nuclear integration timestep, Hvib(t + Δt/2). This quantity is evaluated by using the state energies averaged over two consecutive timesteps and the NACs computed at the mid-points:
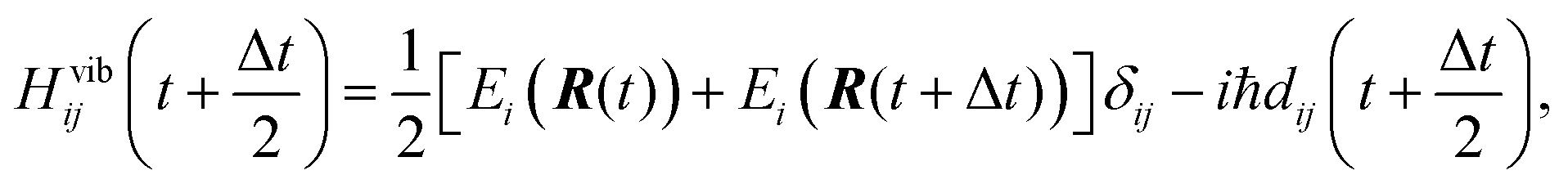
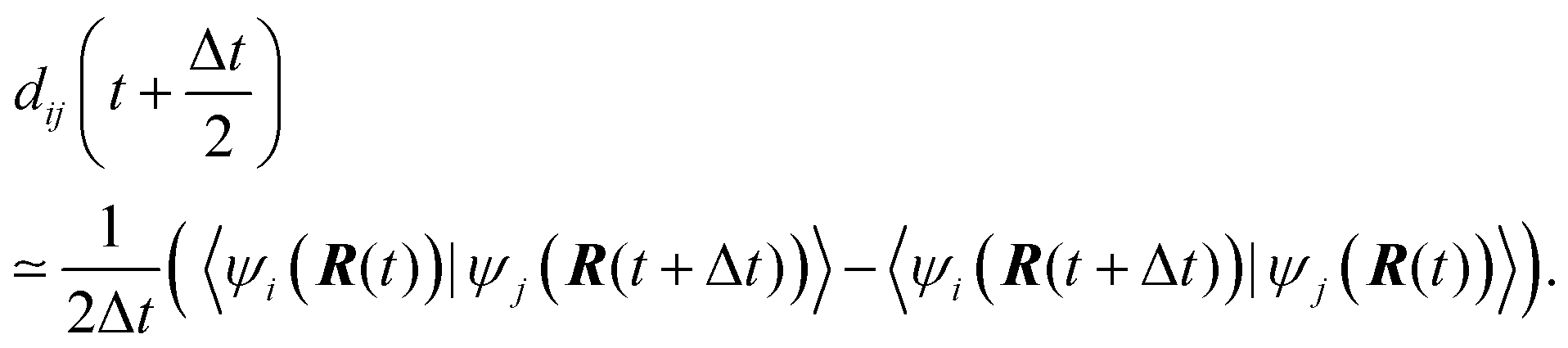 | (3) |
In the above notation, we have emphasized the parametric dependence of the vibronic Hamiltonian, energies, NACs, and wavefunctions on nuclear trajectories, R(t). This dependence constitutes the basis of the CPA. Apparently, any changes that affect the evolution of the nuclei and thus alter their trajectories R(t) also affect the electronic dynamics of the system. The nuclear trajectories can be affected notably by two factors: (a) whether or not the electron-nuclear back-reaction is included and (b) the details of the internuclear interaction potential employed.
In this work, we chose to use the neglect of back-reaction approximation (NBRA) of Duncan et al.,106 according to which nuclear dynamics is not affected by the changes of electronic states. This approximation has been widely used in NA dynamics studies in many extended systems,71,74,79,107–114 including SubPc/(C60)n.48 The utility of this approach is justified by the relative rigidity of our molecular system as well as by its extent.77,79,80,108 The NBRA is often a good approximation for relatively rigid structures, where excited states gradients may only slightly change the magnitudes of internal degrees of freedom, but not lead to qualitative new geometries via photoinduced nuclear reorganization. The slight changes of internal coordinates could be comparable to the fluctuations induced by a thermostat that accounts for the environmental effects. The earlier work of Wilcox et al. also suggests that the ground and excited state geometries of SubPc/C60 could be assumed to be identical. Based on the above data, we don’t expect NBRA would notable affect the computed electronic dynamics, although careful studies of excited state geometries of such complexes may be useful in the future.
One of the goals of the present work is to explore the extent to which electronic dynamics are sensitive to the details of intermolecular interactions within the NBRA. We systematically investigate this effect by considering the molecular dynamics of SubPc/(C60)n systems described using various intermolecular potentials. Namely, we explore three options: (a) the universal force field (UFF);115 (b) the RM1;87 and (c) the RM1 interactions combined with the van der Waals (vdW) terms originating from the UFF. For the last model, we consider two further options: (a) the default UFF parameters for vdW interactions and (b) the scaled UFF parameters with the scaling defined on the basis of our DFT calculations. The UFF has been used to investigate several molecules, including C60 and other systems.116–124 Although the UFF is often considered a generic but low-accuracy force field,125,126 it is useful for our purposes, because it can help to highlight the qualitative changes in electronic dynamics that are induced by changes in the internuclear interaction potentials.
According to the TSH algorithms, N stochastic processes are executed. Each process represents a single realization of electronic state hopping in the manifold of adiabatic basis states. At every time step, the active state i may change to any other state j, with probability given by:
 | (4a) |
| gi→j(t) = |cj|2. | (4b) |
Here, eqn (4a) and (4b) describe the FSSH and MSSH surface hopping probabilities, respectively.
The TSH-based population of state i at time t is defined as a fraction of the processes (trajectories) in state i,

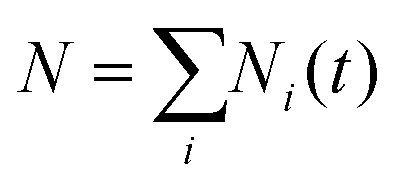 at any time. The execution of the stochastic processes described above depends only on the sequences of ci(t) and dij(R(t)) parameterized by the nuclear trajectories. In our calculations, several nuclear trajectories, {R(t)}, are considered to account for thermal (ensemble) averaging. For each nuclear trajectory, we execute N = 1000 stochastic realizations of TSH to ensure an adequate sampling of electronic transitions.
at any time. The execution of the stochastic processes described above depends only on the sequences of ci(t) and dij(R(t)) parameterized by the nuclear trajectories. In our calculations, several nuclear trajectories, {R(t)}, are considered to account for thermal (ensemble) averaging. For each nuclear trajectory, we execute N = 1000 stochastic realizations of TSH to ensure an adequate sampling of electronic transitions.
To account for the detailed balance in the electron-nuclear system, not all the attempted (proposed) electronic transitions are accepted. The original prescription65 considers the initial and final electronic energies, Ein and Ef, as well as the initial nuclear kinetic energy Kin. For the proposed hop to be accepted, the final nuclear kinetic energy Kf should be non-negative: Kf = Kin + Ein − Ef ≥ 0. If this condition is satisfied, the proposed hop is accepted and the velocities are rescaled to preserve the total energy.96 Otherwise, the proposed hop is rejected and the directions of the velocities are reverted.
In the Libra-X implementation, we enable the following three opportunities to satisfy the energy conservation and detailed balance criteria. (a) Rescaling velocities along the direction of the derivative coupling vectors, as prescribed by the original TSH algorithm. This is the most rigorous approach,65,96 but can be executed only when the derivative coupling vectors are available. (b) Rescaling velocities uniformly. This option may be necessary when the derivative couplings are not readily available. (c) Not rescaling velocities, but rescaling the hopping probabilities by the Boltzmann factor, fB = min{1,exp[−(Ef − Ein)/kBT]}, where T is temperature and kB is the Boltzmann constant. This option is typically used within the NBRA framework. As it follows from the definition of the probability rescaling factor, only the probabilities of the uphill energy transitions are affected. Note that option “c” does not conserve the total energy and therefore effectively accounts for the dissipation of the excitation energy to the environment. A comparison of the three options suggests that options “a” and “b” have the largest computational complexity, because each realization of the stochastic surface hopping process is tightly associated with a separate nuclear trajectory {R(t)}, whereas option “c” allows for running multiple stochastic realizations of the surface hopping process for each single nuclear trajectory. Options “a” and “b” are largely distinct because of the type of the forces that are used to drive the nuclear dynamics: option “a” implies the use of the electronic-state-specific forces, whose selection depends on the execution of the surface hopping algorithm, whereas option “b” implies the use of forces for a fixed electronic state (the ground electronic state by default).
Eqn (2) propagates the electronic degrees of freedom under the assumption of the classical nuclei. However, the quantum nature of nuclei leads to divergence in the history of their evolution, causing the electronic states to decohere. This effect is not accounted for in the TSH prescriptions described above, making them overcoherent.128–131 To account for decoherence effects, the instantaneous decoherence at attempted hops (ID-A) technique of Nelson and Tretiak132 is used. According to the ID-A, the amplitudes of the basis adiabatic states, ci(t), are reset to 0 or 1 at every proposed transition (whether successful or not) to represent the wavefunction collapse onto one of the pure states. If the transition is successful, the wavefunction collapses onto the state where the system hops to. If the proposed transition is not accepted (unsuccessful transitions), the wavefunction collapses onto the currently active state – the system is associated with the state from which the attempted hop occurs. More details of the algorithm description can be found elsewhere.48,132
2.3. NA-MD protocols
In this work, we use three NA-MD simulation protocols defined by combinations of the methods used to treat nuclear dynamics and electronic structure: (a) RM1+D/RM1, (b) UFF/RM1, and (c) UFF/EHT. Here, the first label corresponds to the nuclear dynamics methodology, whereas the second label denotes the electronic structure method used to compute the adiabatic orbitals. The label RM1+D corresponds to the RM1 internuclear interactions corrected by adding the dispersion interactions of the vdW type. Technically, these interactions are added as the vdW potential of the UFF (as implemented in the Libra package), but with the parameters derived from the DFT calculations as described below. The introduced correction is important because the RM1, as well as other NDDO-based methods,62 lacks the dispersion interactions. The introduced correction is found using the DFT calculations with the ωB97XD functional,92,93 which is known to account well for dispersion interactions.133 To obtain structures and energies in good agreement with those found via the ωB97XD calculations, the vdW carbon interaction parameters are rescaled with respect to the UFF values as follows: εC–C(this work) = 2εC–C(UFF), σC–C(this work) = 0.9σC–C(UFF).The UFF/EHT approach has already been used to investigate the CT process in SubPc/(C60)n systems,48 although within the fragmentation ansatz. Note that the re-scaling of the carbon–carbon vdW interaction parameters used in that work was as follows: εC–C(UFF/EHT) = 3εC–C(UFF). In the present work, the approach does not involve any fragmentation and relies on the use of the adiabatic molecular orbitals of entire systems. The UFF/RM1 approach employs the same carbon–carbon vdW interaction parameters as those in UFF/EHT.
The RM1+D/RM1 approach is used to examine the both effects of the simulation protocol and the interfacial modeling. On the other hand, UFF/RM1 and UFF/EHT are employed only to examine the effects of the simulation protocol. As we illustrate later, these methods lack consistency with the DFT results, and thus are not considered as the “production” methods. However, they are useful as atomically resolved model potentials and help us to make important inferences about the mechanisms of the CT process as well as about the methodology-dependence of the NA-MD results. The NA-MD calculation of the UFF/EHT type is performed using a script in Libra.62
2.4. Implementation in Libra-X
The NA-MD calculation schemes outlined above are implemented in an open-source Libra-X software package freely available on the Internet at https://github.com/Quantum-Dynamics-Hub/Libra-X. The program is written in the Python language.134 Python scripts organize the computational workflow by preparing and updating the input files of external electronic structure packages, reading their output, and performing the necessary transformations. The scripts also take care of various stages of computations, such as annealing, thermalization, and NA-MD production run. The NA-MD functionality as well as chemical structures preparation and manipulations are available in Libra-X via the Libra library.62 The latter is a library written mostly in the C/C++ language, but which exposes a lot of its functionalities to Python with the help of Boost.Python wrappers.135,136The computational workflow of Libra-X is illustrated schematically in Fig. 2. The calculations are initialized by the run_X.py script, which serves as an interactive input file. This file allows the user to define all simulation parameters in an intuitive way using the dictionary argument with self-explanatory sets of recognized keywords. Some of the input parameters are, in fact, objects of specifically designed data types. Such objects are used, for instance, to define the basis of excitations further used in the calculations.
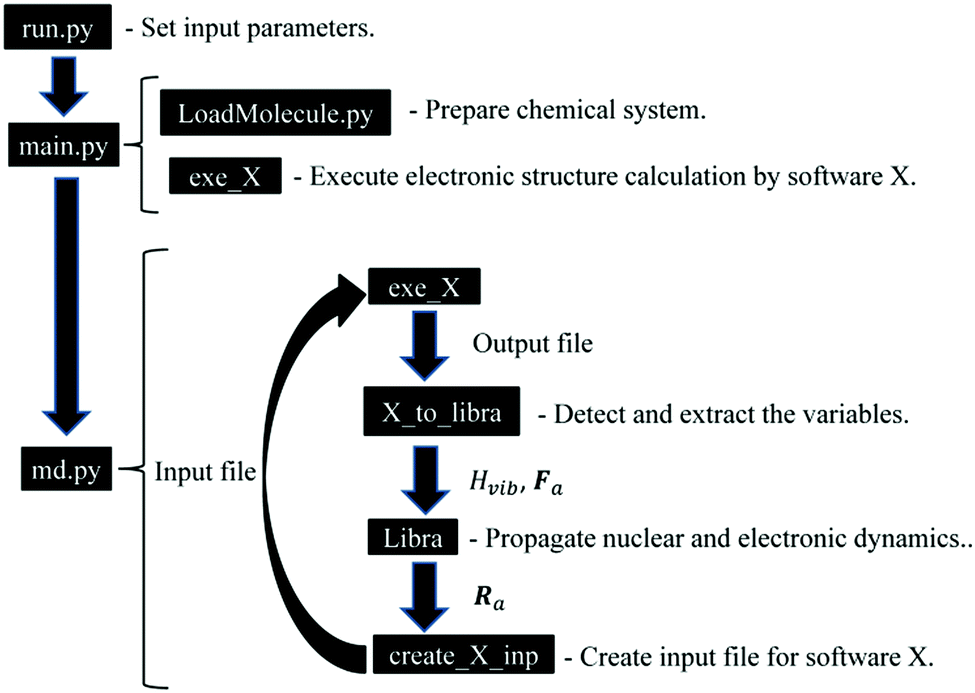 | ||
| Fig. 2 Computational workflow in the Libra-X software. | ||
The input script calls the main.py module, which creates a chemical system (System) object and loads the actual molecule information into it using a user-defined format based on the regular expressions and patterns defined in the LoadMolecule.py module of the Libra program. The script comes with a number of pre-defined patterns for common chemical structure formats, such as “xyz” or “pdb”, and their customized variations. The loaded copy of the molecular system is replicated to create several instances, each of which will evolve independently. The initial geometry of each instance can be perturbed randomly to sample the initial distribution of nuclear configurations from a Wigner distribution. In addition, the nuclear momenta are sampled from the Maxwell–Boltzmann distribution for a given temperature. Thus, the generated trajectories allow one to perform thermal averaging over the initial distributions. Finally, each trajectory may evolve according to different histories of TSH or decoherence processes and these histories are needed to determine the thermal (over initial conditions) and statistical (over realizations of stochastic processes) averages of the properties of interest.
The main.py module also calls the initial execution of the external electronic structure package via the exe_X function. To date, we have implemented interfaces with X = GAMESS (gms),63,64 X = Quantum Espresso (qe),137 and X = Gaussian09 (g09).138 This module extracts the output information and sets up the volumetric information about the system of interest, such as the number of 1-electron orbitals, atoms, and electrons. Together with minimal input from the user, it constructs the active space and the basis of time-independent configurations that will be used in the NA-MD simulations. The script allocates all necessary memory and prepares the output files. Finally, the module performs a minimal sanity check to ensure the consistency of various input options.
Once the indicated preparatory steps are finished, the module calls the run_MD function of the md.py module. This function performs the essential part of the NA-MD simulations via a workflow between the native components of the Libra library that take care of the TSH and TD-SE calculations and external packages (collectively denoted by X) that take care of the stationary electronic structure calculations. The workflow is outlined in Fig. 2.
First, the electronic structure calculations are performed by software X via the exe_X function. The output of these calculations contains the information of the molecular orbitals, φμ, the orbital energies, εμ, the total energies, Etot, and the forces, Fa. Here, μ and a are the indices of the eigenstates and atoms, respectively. The extraction of these variables from the output file is conducted using the X_to_libra function, with X = gamess, qe, or g09. The functions are customized for each type of external software in the corresponding X_to_libra.py modules (X = gamess, qe, g09). The functions detect and extract the variables essential for the dynamics calculations. Namely, εμ and Etot are used to define the energies of many-electron states (Slater determinants), Ei, while φμ is used to define NAC dij, using eqn (3), with the stationary states ψj constructed as Slater determinants of 1-electron orbitals included in the active space, φμ.84 The total energies, Ei, and couplings, dij, are used to construct the vibronic Hamiltonian, Hvibij. The forces, Fa, and Hvibij are used by the intrinsic Libra functions for evolving the nuclear and electronic dynamics and to perform the surface hops for each trajectory. The evolved nuclear coordinates, Ra, are written to the input files for the external programs via the functions of the create_input_X.py modules. Once the input files are updated, the electronic structure calculations are re-submitted via the exe_X function.
To date, the interface with the qe package allows for computing the excited state forces in molecular and solid-state systems via the delta-SCF approach. The details of such a method, as well as its benchmarks, will be reported in a separate work. Although both gms and g09 provide an access to the excited state energies, Ei, and state-specific forces computed directly at various levels of theory, such computations are often limited to molecular systems and are too computationally demanding for extended systems. Thus, the focus of the present implementation is on a simplistic description of energies, couplings, and forces, with the potential of extending this approach to more sophisticated treatments.
In the present approach, we adopt the 1-electron treatment of excited state energies, but we use a multi-electron formulation to compute the NACs. Namely, the energies of excited states are approximated based on the orbital energies and the total energy of the ground state:
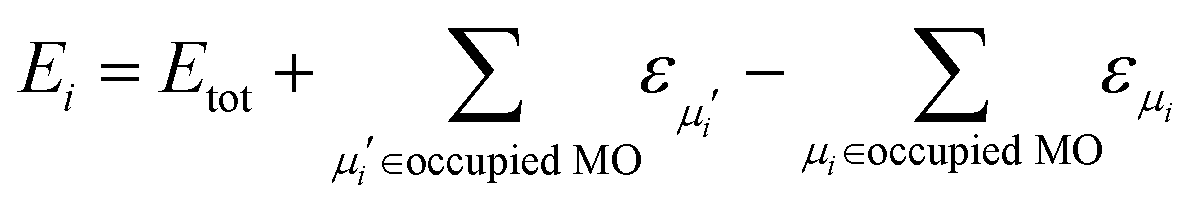 | (5) |
Here, the second and third terms express the summation over the energies of the orbitals, εμi, occupied in the ground (μi) and excited (μi′) Slater determinants. The computations of the NACs between the N-electron Slater determinants, ψi = |ϕi1ϕi2…ϕiN|, involve the overlaps of such determinants at different times, which are computed as:139
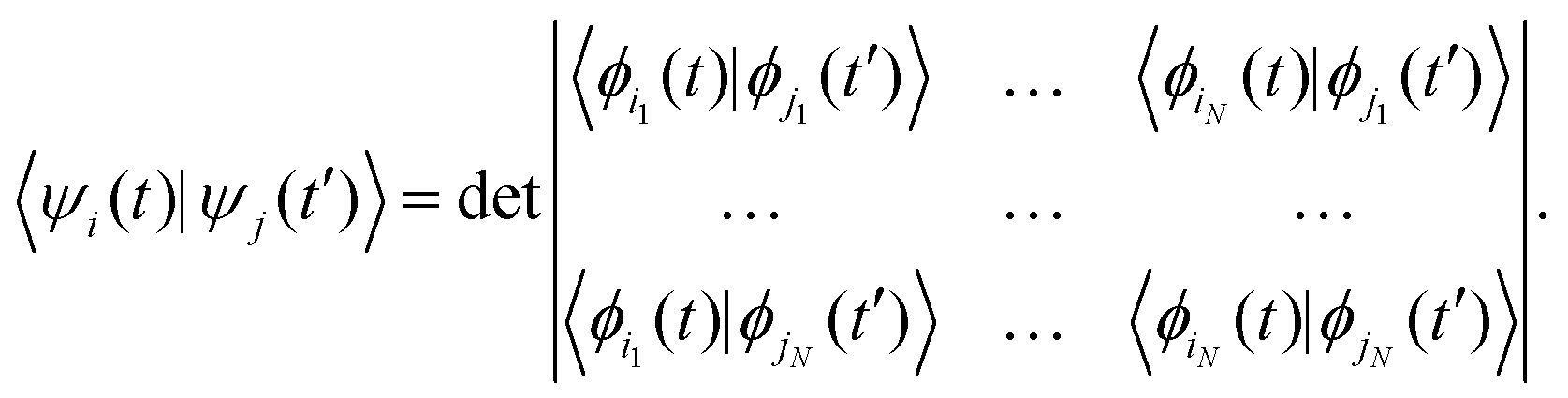 | (6) |
Assuming that the 1-electron functions, {φμ}, are expressed as the linear combination of atomic orbital basis {χα}:
 | (7) |
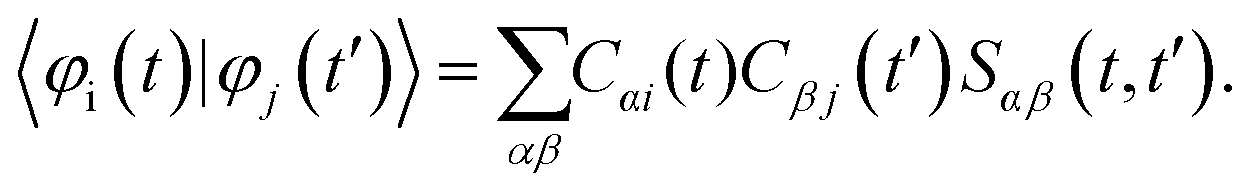 | (8) |
Here, Sαβ(t,t′) = 〈χα(t)|χβ(t′)〉. According to the NDDO-approximation (which is the focus of the present work), the matrix of the atomic orbital overlaps is assumed to be the identity matrix. Hence, the elements Sαβ(t,t′) also stay equal to δαβ. Thus, the computation of the 1-electron overlaps is simplified to:
 | (9) |
These quantities form the matrix in eqn (6) and are used to compute the time overlaps of the Slater determinants, 〈ψi(t)|ψj(t′)〉, which are in turn used to compute the NACs between the multi-electron states. Note that this formulation is distinct from the one formerly used in another NBRA-based code, PYXAID.69,104
2.5. Computational details
The initially relaxed structures are annealed by running 20 cycles of 500 MD steps with a 0.5 fs time step under a microcanonical (NVE) ensemble. In the end of each cycle, the atomic momenta are reset to zero to remove residual kinetic energy from the system. The annealing procedure ensures that the systems are not metastable as when they are accidentally trapped in local minima during zero-temperature relaxation. This helps avoiding instabilities during the MD simulations. The annealed systems are further thermalized to 300 K by running 5000 MD steps with a 0.5 fs time step (for a total of 2.5 ps) under a canonical (NVT) ensemble. The temperature is maintained by a Nose–Hoover chain thermostat,141–145 which ensures that the proper thermal fluctuations are sampled. We use the Nose–Hoover chain thermostat of length 5, and the characteristic time of 24 fs.
The resulting structures as well as the momenta sampled by the end of the NVT (thermalization) simulations are used to carry out the NVE MD followed by the NBRA-based NA-MD calculations. Five-picosecond MD trajectories are computed using an integration time step of 0.5 fs for each system. Thermal averaging is performed using eight starting geometries (distinct nuclear trajectories) in RM1+D/RM1 and UFF/RM1 simulations and sixteen in UFF/EHT simulations. The MD trajectories are used to propagate the electronic dynamics and perform the surface hopping simulations. For each nuclear trajectory, 1000 realizations of the stochastic surface hopping procedure are computed.
The index of the MO to which the electron is excited is determined based on the orbital localization properties at the thermalized molecular structure. This index of the orbital localized dominantly on SubPc may vary depending on the methodology used. For the SubPc/C60 dimer, such initial indices vary in the range of 4 to 8, where index 0 corresponds to the HOMO. Analogously, in the SubPc/(C60)2 system, the orbitals localized on SubPc have indices 7 and 12; in the SubPc/(C60)3 system they have index 10; and in the SubPc/(C60)4 system they have indices 13 and 14. Because the localization properties of an orbital with a given index may vary depending on geometry, we compute the NA-MD starting with all of the noted states as initially populated. For each initial excitation, eight or sixteen distinct nuclear trajectories are computed. The CT is evaluated by averaging the population/energy dynamics computed with each of these initial excitation levels.
The TSH trajectories are used to compute the SH populations pSHi(t) and the expectation values of electronic energy 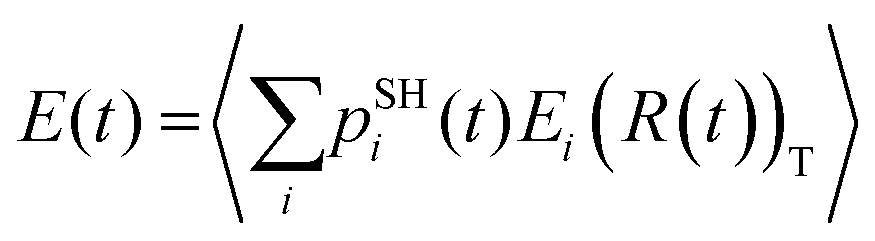 , where 〈
, where 〈![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 〉T denotes thermal averaging (over distinct nuclear geometries). The timescale of the charge transfer, τ, is evaluated by fitting pSHi(t) and E(t) functions with a single exponential function: pSHi(t) = exp(−t/τ) or exp(−t2/τ2); E(t) = E0exp(−t/τ) or exp(−t2/τ2), where E0 is the initial energy level. The fitting function is determined depending on the methodologies used.
〉T denotes thermal averaging (over distinct nuclear geometries). The timescale of the charge transfer, τ, is evaluated by fitting pSHi(t) and E(t) functions with a single exponential function: pSHi(t) = exp(−t/τ) or exp(−t2/τ2); E(t) = E0exp(−t/τ) or exp(−t2/τ2), where E0 is the initial energy level. The fitting function is determined depending on the methodologies used.
To assess the 1-electron approximation employed in our study, we conduct TD-DFT calculation with the range-separated hybrid functionals such as ωB97XD, LC-ωPBE146–148 and CAM-B3LYP.149 In the TD-DFT calculation, environmental effects are also examined. This effect is treated by the polarizable continuum model (PCM)150 with a dielectric constant of 4.2,47 which is close to the constants of SubPc (3.9)44 and C60 (4.0–4.5).33,151–153 All the TD-DFT calculations are performed at the structure optimized using ωB97XD functional.
3. Results and discussion
3.1. Assessment of intermolecular interaction potentials
The optimized molecular structures of SubPc/C60 obtained with different methodologies are shown in Fig. 3. We summarize the Cl–B bond lengths, the B–N bond lengths, the Cl–B–N bond angles, the intermolecular distances, d, and the binding energies, EB, in Table 1. The B–N lengths and Cl–B–N angles are obtained by averaging the three lengths and angles, respectively. The intermolecular distance, d, is defined as the minimal distance between the boron atom of SubPc and the closest carbon atom of C60. The binding energy, EB, is defined as the difference in the energies of the optimized dimer structure and the isolated SubPc and C60 molecules optimized individually, EB = E(SubPc) + E(C60) − E(SubPc/C60).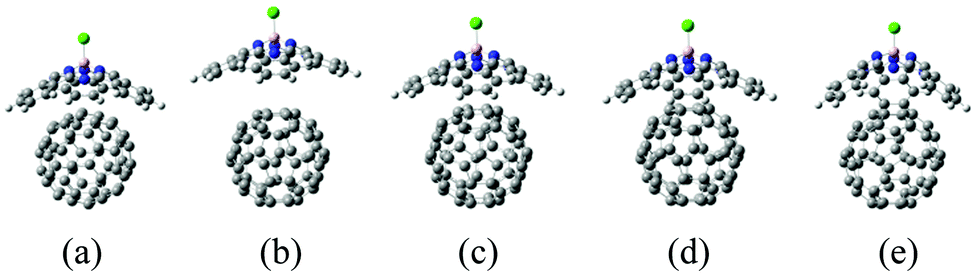 | ||
| Fig. 3 Molecular structures of SubPc/C60 dimer models computed with different interaction potentials: (a) DFT/ωB97XD; (b) RM1; (c) RM1 plus vdW dispersion terms with default UFF parameters, RM1+D (default); (d) same as (c), but with scaled vdW parameters; and (e) bare UFF dispersion with the parameters suggested in ref. 48. | ||
The Cl–B and B–N lengths are 1.9 Å and 1.5–1.6 Å in all methods, respectively; they are comparative to experimental values (1.8 and 1.5).21 The Cl–B–N angle of DFT is 114°, which is in good agreement with experimental value.21 Those of RM1-related methods (RM1 and RM1+D) are slightly underestimated from 114° by 3°. The angle of UFF is underestimated by 9°, which is larger than the error of RM1-related methods.
As expected, the dimer optimized with the ωB97XD functional is predicted to be well bound, with a binding energy of 1.3 eV and an intermolecular distance of 3.9 Å (Fig. 3a). The resulting energy compares well with that of the reported DFT results (1.2 eV) obtained at the ωB97XD level (Table 1).21 Additionally, the resulting distance compares well with that from the experiment (3.7–3.8 Å), based on the X-ray crystal structure analysis of the SubPc-derivative/C60 interface (Table 1).21 The dimer optimized using solely RM1 is predicted to be very weakly bound with EB = 0.014 eV, which is lower than the thermal energy. Thus, the bare RM1 method is not suitable for modeling the MD of the system, because the system would readily dissociate into two molecular components at room temperature. The weak binding at the RM1 level of theory leads to a notably larger intermolecular separation of d = 5.7 Å (Fig. 3b).
The failure of the RM1 method is a consequence of the general deficiency of NDDO-based methods in regard of a description of dispersion interactions. Such a deficiency has been highlighted earlier62 for the Intermediate Neglect of Differential Overlap (INDO) method,154 where the lack of repulsive dispersion interactions erroneously favored the internal cyclization of the azo-benzene molecule. The addition of a repulsive Lennard-Jones potential was required to stabilize the non-cyclized structure. Here, we follow the same strategy and correct the RM1 potential with the UFF vdW term, which accounts empirically for the dispersion interactions. We consider two sets of the vdW parameters for C atoms, as described in Section 2.3.
The RM1+D method with the original (default) UFF parameters shows a notable improvement for describing interactions in the dimer in comparison to bare RM1: d decreases to 4.5 Å (Fig. 3c) and EB increases up to 0.98 eV, both approaching the order of magnitude of the corresponding DFT-derived properties. Because both UFF and RM1 are parameterized interactions, we have considered further tuning of the UFF interactions by scaling the vdW strength and radii parameters of C atoms, resulting in the RM1+D (scaled) approach. This approach decreases the intermolecular distance down to 4.2 Å (Fig. 3d) and increases the binding energy up to 1.5 eV; these values are within 0.3 Å and 0.2 eV of the their respective DFT values. All the RM1, RM1+D (default), and RM1+D (scaled) methods preserve the structure of the SubPc unit such that it remains relatively flat, “open”. In the following discussions, we will imply the RM1+D (scaled) variant when referring to the RM1+D method, unless otherwise noted.
The dimer optimization with UFF (with the scaling of vdW parameters as explained in Section 2.3) leads to a relatively distant placement of the SubPc and C60 molecules, with d = 4.9 Å, which is longer than the DFT value by 1.0 Å (Fig. 3e). The increased intermolecular separation does not lead to a smaller binding energy but a larger one than the DFT-derived value by 0.6 eV. This effect originates from a notable bending of the conical structure of SubPc, leading to its more “closed” configuration. Such configuration maximizes the number of surface contacts between SubPc and C60 yet keeping the B atom of SubPc distant from C60. This bending of SubPc may be attributed to the underestimated out-of-plane interactions present in UFF in comparison to the quantum mechanical treatment of the π-conjugation effects possible at the RM1 level of theory.
3.2. Assessment of electronic structure methods
The electronic structure methods used are compared to each other based on the molecular orbitals (MOs) and orbital energies they produce. The isosurfaces for several frontier orbitals and their energies are summarized in Fig. 4 and Table 2, respectively. As Fig. 4 suggests, all four combinations of optimization and electronic structure methods considered predict the HOMO (orbital index 0) localization on the SubPc fragment, whereas the triply degenerate LUMO (orbitals 1, 2, and 3) is always localized on the C60 fragment. The degeneracies may be slightly lifted, depending on the particular method used, but remain clearly defined. The HOMO–LUMO gap varies from 3.94 eV to 5.05 eV for all protocols other than UFF/EHT (Table 3). These values are notably overestimated with respect to the 1.9 eV gap between isolated SubPc and C60 orbitals, as reported by Wilcox et al.47 The UFF/EHT method predicts a 0.7 eV HOMO–LUMO gap, which is somewhat closer to the abovementioned value. It should be noted that in an earlier work,48 the HOMO–LUMO gap was assessed based on the orbital energies of the two individual fragments. The estimated value was on the same order of magnitude as those presently obtained. For our purposes, the exact value of the HOMO–LUMO gap is unimportant, because the electronic transition of interest does not involve the HOMO. | ||
| Fig. 4 Frontier orbitals of the SubPc/C60 dimer, computed with the methods considered in this work. The orbital index 0 corresponds to the HOMO. The assignment of the orbitals depends on the method used, but the HOMO–1 (orbitals –5, –4, –3, –2, and –1), HOMO (orbital 0), and LUMO (orbitals 1, 2, and 3) can be identified with all methods. | ||
| DFT/DFT | RM1+D/RM1 | RM1/RM1 | DFT/RM1 | UFF/RM1 | UFF/EHT | |
|---|---|---|---|---|---|---|
| 8, L+2 | −0.33 | −1.94 | −1.91 | −1.95 | −2.20 | −10.6 |
| 7, L+2 | −0.36 | −1.94 | −1.91 | −1.96 | −2.23 | −10.7 |
| 6, L+2 | −0.36 | −1.95 | −1.91 | −1.99 | −2.58 | −10.7 |
| 5, L+1 | −1.17 | −2.05 | −2.02 | −2.00 | −2.62 | −10.8 |
| 4, L+1 | −1.17 | −2.06 | −2.02 | −2.01 | −2.68 | −10.8 |
| 3, L | −1.72 | −2.62 | −2.58 | −2.67 | −3.21 | −12.0 |
| 2, L | −1.74 | −2.62 | −2.58 | −2.68 | −3.27 | −12.0 |
| 1, L | −1.75 | −2.62 | −2.58 | −2.69 | −3.30 | −12.1 |
| 0, H | −6.80 | −7.46 | −7.44 | −7.60 | −7.24 | −12.8 |
| −1, H−1 | −7.74 | −9.36 | −9.40 | −9.35 | −8.81 | −12.9 |
| −2, H−1 | −7.75 | −9.37 | −9.40 | −9.35 | −8.85 | −13.2 |
| −3, H−1 | −7.76 | −9.37 | −9.40 | −9.36 | −8.88 | −13.3 |
| −4, H−1 | −7.77 | −9.38 | −9.40 | −9.36 | −8.91 | −13.6 |
| −5, H−1 | −7.78 | −9.48 | −9.40 | −9.36 | −8.92 | −13.6 |
| DFT/DFT | RM1+D/RM1 | RM1/RM1 | DFT/RM1 | UFF/RM1 | UFF/EHT | |
|---|---|---|---|---|---|---|
| H–H−1 | 0.94 | 1.90 | 2.04 | 1.74 | 1.57 | N/A |
| H–L | 5.06 | 4.84 | 4.86 | 4.92 | 3.94 | 0.7 |
| L+1–L | 0.55 | 0.55 | 0.56 | 0.67 | 0.53 | 1.0 |
| L+2–L+1 | 0.80 | 0.1 | 0.11 | N/A | N/A | N/A |
The lower energy orbitals (indices from −1 to −5) computed with all the approaches are fivefold degenerate. However, UFF/EHT predicts their localization on the SubPc fragment, whereas the other approaches predict the orbital localization on the C60 fragment. The HOMO–HOMO−1 gap varies from 0.9 eV to 2.0 eV for all methods except UFF/EHT (Table 3). The values are generally overestimated with respect to the experimental value (0.6 eV) reported in Wilcox's work.47 On the other hand, UFF/EHT predicts almost a zero gap between HOMO–HOMO−1. Similar to the HOMO–LUMO gap, the HOMO–HOMO−1 gap is also not of interest in our CT study.
The higher energy orbitals computed with the DFT/DFT, RM1+D/RM1, and RM1/RM1 (the orbitals are shown in Fig. S1 of ESI†) methods form two groups: a doubly degenerate LUMO+1 orbital (indices 4 and 5) and a triply degenerate LUMO+2 orbital (indices 6, 7, and 8). These groups of orbitals are clearly localized on the SubPc and C60 molecules, respectively, when the DFT/DFT, RM1+D/RM1, and RM1/RM1 methods are used. Thus, the charge transfer from the photoexcited SubPc to C60 corresponds to the LUMO+1 → LUMO transition, as previously suggested by Wilcox et al.47 The LUMO+1–LUMO orbitals gaps computed using the three abovementioned protocols are approximately 0.55 eV (Table 3), which is somewhat higher than the value (0.1 eV) reported by Wilcox et al. The agreement of the LUMO+1–LUMO gaps computed at the DFT and RM1 levels indicates that the RM1 method may be a reasonably accurate alternative to the computationally demanding DFT method for studying electronic dynamics in SubPc/(C60)n systems.
The character of the LUMO+1 and LUMO+2 changes in the UFF/RM1 and DFT/RM1 (shown in Fig. S2 of ESI†) methods with respect to the RM1+D/RM1 or DFT/DFT methods. We observe that higher index orbitals (indices 7 and 8 in Fig. 4) are localized on SubPc, whereas the lower index states (indices 4, 5, and 6 in Fig. 4) correspond to electrons localized on the C60 moiety. A similar situation is observed when the UFF/EHT method is used. In the three methods, the SubPc electron localization corresponds to what can be classified as LUMO+2, whereas LUMO+1 orbitals are localized on C60. In addition, one can observe a mixing and mutual delocalization of the intermediate orbitals (e.g. orbital index 6 in the UFF/EHT method). The analysis of the character of the orbitals shown in Fig. 4 and Fig. S1, S2 (ESI†) suggests that it is dictated not only by the molecular structure of the system, but also the electronic structure method used. In our study, both the DFT and RM1 methods predict a similar character of the LUMO+1 and LUMO+2 orbitals as long as the geometries of the systems are similar (e.g. compare DFT/DFT and DFT/RM1 results in Tables 2 and 3, Fig. 4a and b). The HOMO–LUMO+2 orbitals obtained with RM1 and EHT are similar to each other (e.g.Fig. 4c and b). At the same time, both the UFF/RM1 and UFF/EHT methods predict the characters of LUMO+1 and LUMO+2 orbitals to be opposite to such orbitals obtained at the DFT-optimized structures (e.g. compare Fig. 4a and c). This effect can be clearly attributed to the UFF-optimized geometries, which show a notable conical distortion of SubPc structure in comparison to the DFT-optimized ones. The orbitals at relatively “open” SubPc structures computed by DFT and RM1 are similar. These orbitals are different from those at the relatively “closed” structures computed by RM1 and EHT. This makes us suggest that the out-of-plane bending degree of the SubPc “plane” that determines the degree of the molecular conical-like distortion may be the main reaction mode (we call this a “breathing” mode) that drives the CT process as discussed later in detail.
For the purposes of this study, the UFF/RM1 and UFF/EHT models are regarded as atomically resolved model potentials that helped us to reveal information about the possible nuclear modes that drive the CT process. However, the deviation of the electronic structure of the SubPc/C60 system from the reference DFT calculations makes us conclude that the UFF/RM1 and UFF/EHT methods should not be regarded as “production” methods to study CT dynamics in SubPc/(C60)n systems. In subsequent studies, the results obtained with these methods should be regarded as the qualitative exploration and not as the predictive description of the systems of interest. For the qualitative purpose, the RM1+D/RM1 method will be suitable as it reproduces the DFT results mostly.
Finally, it is worth commenting on the combination of the UFF and EHT methods used earlier in studies of CT dynamics in SubPc/(C60)n.48 This earlier work considered CT process in the basis of quasi-diabatic fragment states. Such states have clear localization properties, as follows straight from their definition. This helped to avoid the artificial mixing or re-ordering of orbitals, even when the erroneously bent SubPc molecular structure was used. This effect is analogous to the fortuitous cancellation of DFT delocalization errors within the fragmentation ansatz, reported by the Pavanello group.155
3.3. Assessment of the environmental effects and 1-electron approximation
As described in Section 2.2. A, we employ 1-electron approximation to simulate CT dynamics. To assess its accuracy, we compute the transition energy spectra using TD-DFT with several functionals (Fig. 5). The structure optimized with the ωB97XD functional is utilized in all calculations to delineate the effects of geometry change. First, we observe that not all density functionals used together with TD-DFT can predict the state ordering consistent with the experimental reality – that is when the CT state is lower in energy than the SubPc-localized excitation. Among the tested functionals (ωB97XD, LC-ωPBE, CAM-B3LYP), only CAM-B3LYP predicts such ordering of states (Fig. 5, panels a–c). Earlier, Wilcox et al.47 suggested that the Baer–Neuhauser–Livshits (BNL) functional156,157 was suitable for modeling CT states.158–161 Unfortunately, this functional is not presently available in the Gaussian package we utilized. The molecular orbitals computed with the LC-ωPBE and CAM-B3LYP functionals are shown in Fig. S3 and S4 of ESI,† respectively. The corresponding orbital energies are summarized in Tables S1 and S2 of ESI.†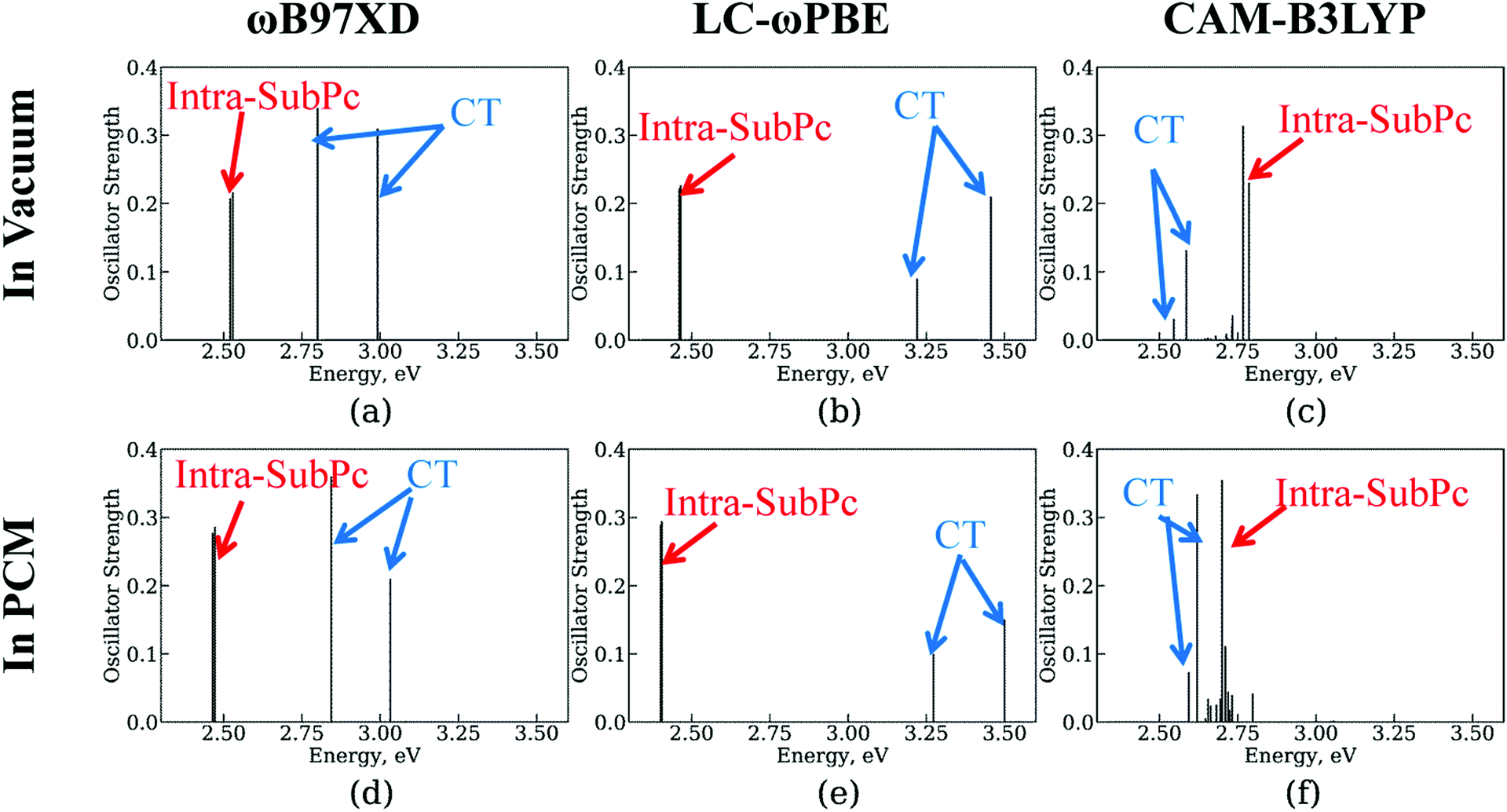 | ||
| Fig. 5 Transition energy spectra at TD-DFT level with several functionals and environments at the structure optimized by DFT with ωB97XD functional. (a), (b), and (c) are in vacuum, whereas (d), (e), and (f) are in PCM. The functionals are ωB97XD in (a) and (d); LC-ωPBE in (b) and (e); CAM-B3LYP in (c) and (f). For convenience, the strengths of charge transfer (CT) excitation spectra increase 100, 10, and 5 times larger in (a)–(d), (e), and (f), respectively. | ||
Second, we test the effect of polarization due to the solid state environment using polarized continuum model (PCM) approach. Following the work of Wilcox et al.,47 we utilize the dielectric constant of 4.2 which is close to that of C60 (4.0–4.5)33,151–153 and SubPc (3.9)44 molecules. We find that in comparisons to the vacuum, the solid state environment slightly stabilizes the inta-SubPc excitations and destabilizes the CT state. However, the changes of the state energies are less than 0.1 eV, which is expected since the dielectric constant of 4.2 is relatively close to the vacuum level (1.0) in comparison to the dielectric constants of polar environments such as water. Thus, the environmental effects can be considered of little importance to the CT dynamics and can be neglected in the NA-MD simulations.
Third, in order to assess the accuracy of the one-electron picture used in the NA-MD simulations, we (a) compare the energy offsets between TD-DFT many-electron states and 1-electron orbital energies (Table 4); (b) analyze the amplitudes of the 1-electron transitions in the computed many-electron states.
| Transition\method | TD-DFT/CAM-B3LYP | 1-Electron | ||
|---|---|---|---|---|
| In vacuum | In PCM | DFT/DFT (ωB97XD) | RM1+D/RM1 | |
| E(CT)–E(GS) or E(L)–E(H) | 2.55 | 2.59 | 5.06 | 4.84 |
| E(S1)–E(CT) or E(L + 1)–E(L) | 0.22 | 0.10 | 0.55 | 0.55 |
The comparison of energy levels offsets shown in Table 4 reveals that the absolute energies of CT state are overestimated (on the order of 5 eV) in the 1-electron treatment in comparison to the TD-DFT results (on the order of 2.5 eV). This likely originated from the neglect of additional electron–hole interaction in the CT state, which is absent in the 1-electron description. This notable discrepancy is irrelevant to our dynamical studies, since we are not aiming to describe the electron–hole recombination. However, should one utilize the 1-electron description, it can be expected that based solely on the energy level energies, the results of the 1-electron description might underestimate the recombination rates. This originates from two reinforcing effects: (a) the twice larger 1-electron gap would mean roughly twice smaller NACs61 and four-fold slower coherent dynamics; (b) overestimated energy gap would exponentially decrease the Franck–Condon factors in comparison to those expected from the TD-DFT gaps.
The energy levels offset between CT and SubPc-localized excited states computed using 1-electron picture are on the order of 0.55 eV both for DFT/DFT and RM1+D/RM1 methods. These values are also about twice as large as those predicted by TD-DFT/CAM-B3LYP (0.1–0.2 eV). However, the difference in the absolute values is not as dramatic as in the case of CT vs. ground state energy gap. The analysis of the transition rates given above still applies, although with smaller overall difference. To reiterate, based solely on the energy levels analysis, the 1-electron picture is expected to give underestimated rates in comparison to the TD-DFT treatment.
The analysis of the amplitudes of the 1-electron transitions in the many-electron TD-DFT states (ESI,† Table S3), reveals that the two dominant 1-electron transitions that constitute the CAM-B3LYP CT state are of HOMO → LUMO type (with two degenerate LUMO orbitals, 1 and 2). Within the present NA-MD framework, this mixing of distinct determinants is accounted for by considering the total populations of individual transitions (e.g. 0 → 1 and 0 → 2). One can regard our 1-electron treatment as a diabatic representation with regard of the electron–hole interaction Hamiltonian. The first bright excited state, S1, involves the dominant 0 → 5 transition. According to Table 2, this corresponds to HOMO → LUMO+1 transition within the 1-electron description. To summarize, we do observe a correspondence of the TD-DFT excited states to the preferential single-electron orbital excitations. Our approach accounts for the possible mixing of similarly-localized excitations by considering the total populations of such individual excitations (microstates).
3.4. Assessment of nonadiabatic dynamics methodologies for CT dynamics at the SubPc/C60 interface
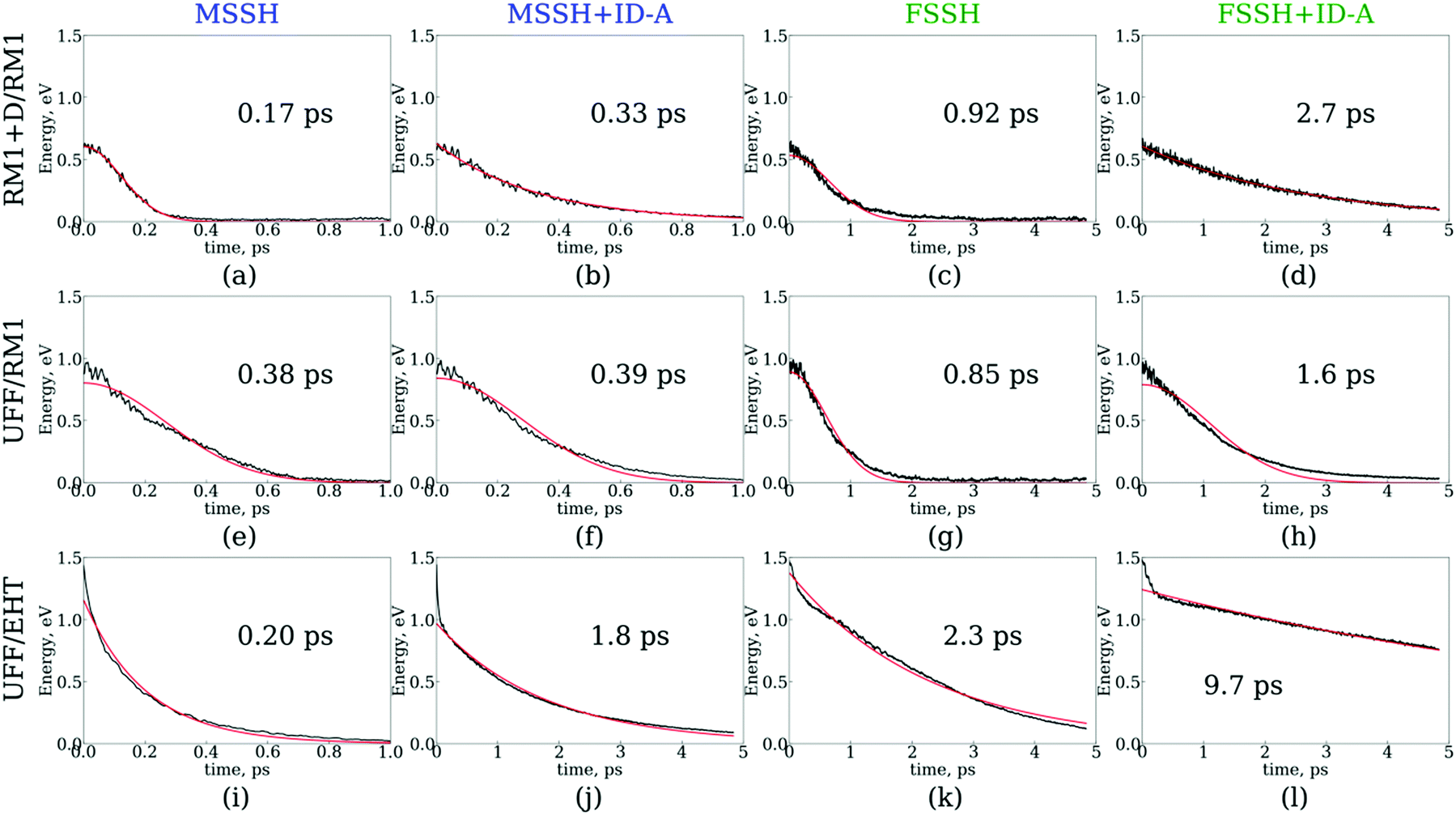
The results of the NA dynamics simulations in the SubPc/C60 dimer computed with systematically varied combinations of TSH, nuclear dynamics, and electronic structure methodologies are summarized in Fig. 6 and 7. In particular, we report the population transfer (Fig. 6) and excitation energy relaxation (Fig. 7) dynamics computed with one of three nuclear dynamics/electronic structure models, RM1+D/RM1, UFF/RM1, and UFF/EHT, and one of four TSH methodologies, MSSH or FSSH, with or without decoherence correction via ID-A.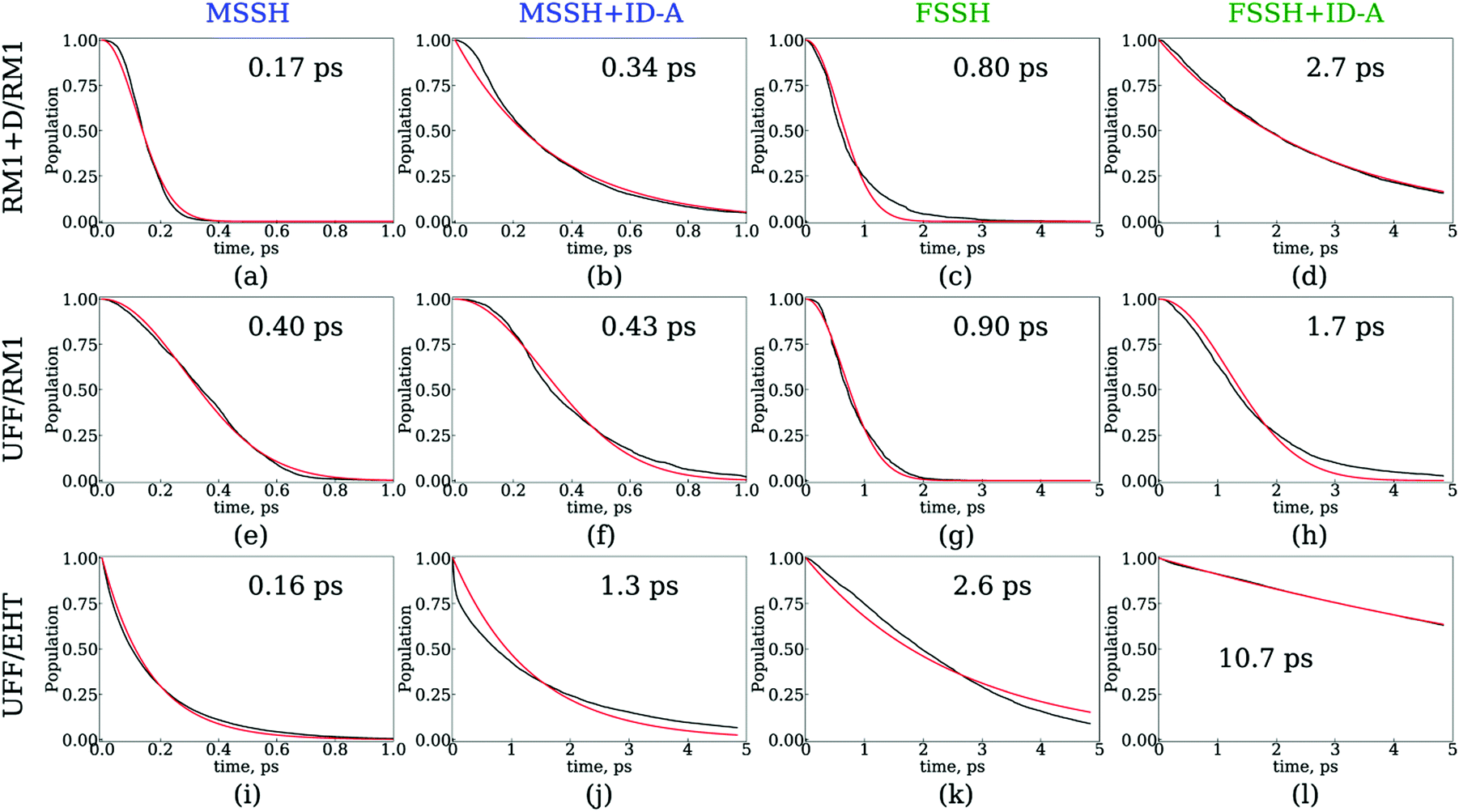 | ||
| Fig. 6 Dynamics of charge transfer from the SubPc unit to the C60 molecule computed with several surface hopping methods: MSSH (a, e and i), MSSH with the ID-A decoherence correction (b, f and j), FSSH (c, g and k), and FSSH with the ID-A decoherence correction (d, h and l). The charge transfer dynamics were computed with three nuclear dynamics/electronic structure methods: RM1+D/RM1 (a–d), UFF/RM1 (e–h), and UFF/EHT (i–l). The total SH population on orbitals with indices 4 through 8 is plotted. Red lines are fitting results with Gaussian kinetics (a, c, e, f, g, and h) or exponential kinetics (b, d, i, j, k, and l). | ||
 | ||
| Fig. 7 Dynamics of electronic energy relaxation computed using several surface hopping methods: MSSH (a, e, and i), MSSH with the ID-A decoherence correction (b, f, and j), FSSH (c, g, and k), and FSSH with the ID-A decoherence correction (d, h, and l). The dynamics were computed using three nuclear dynamics/electronic structure methods: RM1+D/RM1 (a–d), UFF/RM1 (e–h), and UFF/EHT (i–l). Red lines are fitting results with Gaussian kinetics (a, c, e, f, g, and h) or exponential kinetics (b, d, i, j, k, and l). The zero energy level corresponds to the LUMO energy. | ||
To estimate the CT rates, we computed the total TSH population on the donor states as a function of time. The notable mixing of C60 and SubPc-localized states (especially with the UFF/EHT model) leads to a possibility of state identity changes during the course of the CT dynamics. To make our results invariant with respect to such state changes, we computed the total population of the SubPc-localized states and the energetically close states, which may also be strongly mixed with those localized on SubPc (Fig. 6), 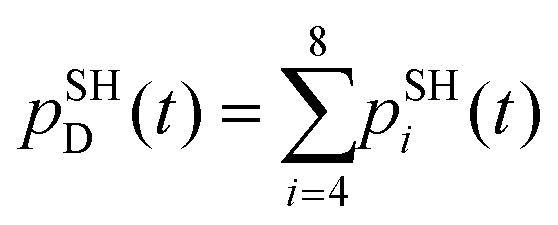 , where pSHi(t) is TSH population on adiabatic state i as a function of time, computed as a fraction of trajectories that are in state i. An alternative approach to make the results invariant (or at least weakly dependent on state identity changes) is to study the excitation energy relaxation dynamics. In particular, Fig. 7 reports the SH-population weighted excitation energy of the system:
, where pSHi(t) is TSH population on adiabatic state i as a function of time, computed as a fraction of trajectories that are in state i. An alternative approach to make the results invariant (or at least weakly dependent on state identity changes) is to study the excitation energy relaxation dynamics. In particular, Fig. 7 reports the SH-population weighted excitation energy of the system: 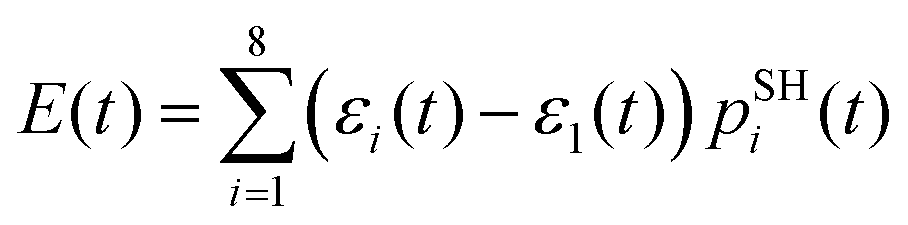 , where εi(t) is an adiabatic energy of state i (orbital energy) at time t. This quantity is zero when the entire population is on the lowest acceptor (index 1) state.
, where εi(t) is an adiabatic energy of state i (orbital energy) at time t. This quantity is zero when the entire population is on the lowest acceptor (index 1) state.
Fig. 6 and 7 suggest that the timescales for population transfer and excitation energy relaxation are consistent with each other. Thus, we compare the dynamics computed via various methodologies without reference to a particular type of processes, keeping in mind both of them. Generally, the dynamics computed with the RM1+D/RM1 and UFF/RM1 models (top two rows) without decoherence correction (panels a, c, e, f, g and h) follow Gaussian kinetics, X(t) = X0exp(−t2/τ2), where X stands for either population, P, or energy, E. Adding the decoherence correction (panels b and d) results in exponential kinetics, X(t) = X0exp(−t/τ), although in the UFF/RM1 case it tends to be a combination of the two types of kinetics. Finally, the UFF/EHT model yields exponential kinetics, whether decoherence correction is performed or not.
Firstly, we analyze the effects of the TSH methodology on the CT/energy relaxation rates by comparing the different columns for each row of panels shown in Fig. 6 and 7. In all cases, we consistently observe that MSSH produces faster dynamics in comparison to those of FSSH. This is to be expected because the MSSH method allows for transitions between states that are not coupled directly but whose quantum populations are not negligible. In all cases, the introduction of decoherence correction makes the dynamics notably slower and often changes the kinetics from Gaussian to exponential. This deceleration of the dynamics originates from the fact that decoherence corrections force the states to conserve their purity by frequently collapsing the evolved coherent superposition onto one of the possible energy eigenstates. Such collapses effectively discard the progress of coherent dynamics, reducing the changes for the system to end up in a new state. In the limit case of instantaneous decoherence (zero decoherence time), one approaches the quantum Zeno effect,162–165 according to which no transitions occur from the state being constantly observed. The slowing down of transitions upon introducing the decoherence correction have been previously observed. Thus, this effect is not new. What is new, however, is the estimate of the variation of the CT or energy relaxation timescales that can be obtained using various TSH approaches applied to SubPc/C60 systems. In particular, with the RM1+D/RM1 and UFF/RM1 methods, the timescales vary in the range from 170 fs up to 2.7 ps, spanning an order of magnitude. At a more approximate UFF/EHT level, this variation is as large as two orders of magnitude, varying from 160 fs to 10.7 ps.
Secondly, we analyze the effect of the interaction potential on the CT/energy relaxation rates by comparing the top two rows of panels in each column of Fig. 6 and 7. In particular, we fix the electronic structure methodology to RM1 and focus on the effects of varying the nuclear dynamics (RM1+D vs. UFF). As Table 1 suggests, the bare UFF leads to a geometry in which the boron atom of SubPc is more distant from the closest C atom of C60, as opposed to the RM1+D model. At the same time, the SubPc molecule “wraps around” the C60 to a larger extent when UFF is used as opposed to RM1+D. These geometrical features lead to smaller orbital overlaps and hence to smaller couplings, resulting in the slower dynamics observed via UFF/RM1 in comparison to those observed via RM1+D/RM1.
This order reverses for the FSSH+ID-A method. As Table 1 suggests, the binding energy predicted by UFF is somewhat larger than that predicted by RM1+D, suggesting a more regular intermolecular distance fluctuation with few possibilities for additional noise. To confirm this, the Fourier transform of the autocorrelation function was calculated for the energy gap as shown in Fig. 8. The transforms shown in Fig. 8 represent averaging over the gaps between the orbitals with indices 1, 2, and 3 and the orbitals with indices 4, 5, 6, 7, and 8,
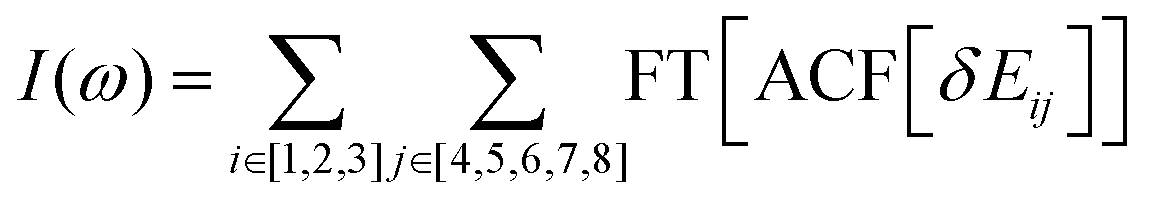
 | ||
| Fig. 8 Fourier transform of the autocorrelation function for the energy gap. This transform is averaged over the gaps between the orbitals with indices 1, 2, and 3, and the orbitals with indices 4, 5, 6, 7, and 8. (a) RM1+D/RM1. (b) UFF/RM1. | ||
In the RM1+D/RM1 case, there is a strong peak at around 1800 cm−1 (Fig. 8a). In the UFF/RM1 case (Fig. 8b), a number of additional low-frequency peaks appear at approximately 600 cm−1 and 1300 cm−1 with the 1800 cm−1 peak weakened. This indicates that the “reactive” coordinate is coupled to a larger number of “environmental” modes when the UFF/RM1 method is used. As recently demonstrated,166 the abundance of such bath modes can facilitate non-adiabatic transitions due to breaking the time reversibility properties of the dynamics. The peak at 1800 cm−1 in Fig. 8a may be attributed to the “breathing” mode, suggested in Section 3.2. To support this hypothesis, the spectra in Fig. 8a are compared to normal mode spectra of the structure optimized at the DFT/ωB97XD level, as shown in Fig. 9. Visualization of the normal modes reveals that those at 840 cm−1, 1000 cm−1, and 1530 cm−1 (pointed by the red arrows in Fig. 9, the displacement vectors shown in Fig. 10) can be regarded as the “breathing modes” of SubPc/C60 structure. Comparison of frequencies in Fig. 8a and 9, suggests that the higher frequency “breathing mode” at 1530 cm−1 in Fig. 9 corresponds to the mode at 1800 cm−1 in Fig. 8a, which is strongly coupled to CT transition. The frequencies differences of the peaks are likely due to differences in interatomic interaction potentials (RM1+D vs. DFT). Thus, we conclude that the “breathing mode” at around 1530 cm−1 may be the one that strongly affects CT in SubPc/C60 system.
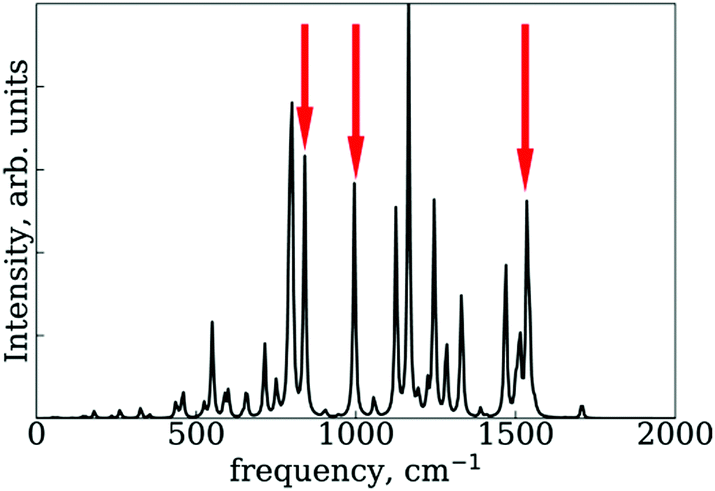 | ||
| Fig. 9 Normal mode spectra of the SubPc/C60 structure optimized by DFT at ωB97XD level. The red arrows indicate peaks of “breathing mode” at SubPc/C60 structure. | ||
 | ||
| Fig. 10 Normal mode displacement vectors of the SubPc/C60 at (a) 840 cm−1, (b) 1000 cm−1, and (c) 1530 cm−1. The displacement vectors are indicated by blue arrows. | ||
Thirdly, we investigate the effect of the electronic structure methodology used. We use trajectories with their dynamics driven by the UFF interaction potential and use either the RM1 or EHT methods to compute the orbitals, NACs, and energies. The induced differences in the NA dynamics can be understood by comparing the bottom two rows of panels in Fig. 6 and 7. The main effect observed is that the dynamics computed at the EHT level are notably slower than those at the RM1 level. This effect likely originates owing to the differences in orbital localization, as apparent from Fig. 4. Indeed, the EHT method induced a notable mixing of SubPc and C60-localized orbitals (e.g., orbitals with indices 6 and 7 in the UFF/EHT column) as opposed to the RM1 method. Because the acceptor orbitals are localized on C60, the matrix elements  will lead to a smaller contribution than
will lead to a smaller contribution than  because of symmetry considerations. The C60-localized orbitals change in time with respect to C60-localized orbitals to a smaller extent than how the SubPc-localized orbitals change in time with respect to C60-localized ones. Thus, the stronger mixing of the donor and acceptor states described by the EHT method leads to smaller couplings and slows down the transition between such states.
because of symmetry considerations. The C60-localized orbitals change in time with respect to C60-localized orbitals to a smaller extent than how the SubPc-localized orbitals change in time with respect to C60-localized ones. Thus, the stronger mixing of the donor and acceptor states described by the EHT method leads to smaller couplings and slows down the transition between such states.
3.5. The role of the SubPc/(C60)n interface in the non-adiabatic dynamics of charge transfer
As illustrated in the previous section, the CT timescale computed using the RM1+D/RM1 method is shorter than the one observed in the experiment.47 The predicted fast CT process may be due to the use of the minimalistic dimer model. In a previous study, one of us suggested that such a model may notably affect the computed timescales and should be used carefully.48 To further assess the role of the interfacial structure, we consider the SubPc/(C60)n (n = 2, 3, and 4) systems. These clusters aim to model surface of the C60 layer in a heterojunction. Thus, the initial structures are constructed to maximize the interfacial area with the given number of C60 molecules. The structures adjust during the thermalization and MD stages, as dictated by the nature of all-atomic interactions. The systems do not reside in the most symmetric geometries all the time and can exhibit small fluctuations during the dynamics, although do not show notable structural reorganization on the timescale of simulations either. The realistic interfaces may be more complex, exhibiting amorphous packing of C60 units at the interface. However, it is very likely that even in such complex structures, the SubPc would either be reside on a “flat” surface of C60 or on a kink/step/apical defect sites. In all situations, the structure may locally resemble that of the utilized minimal clusters. Another option concerns alternative orientation of SubPc unit (as for instance discussed in the work of Wilcox et al.47), but this situation is outside the scope of this work.Unlike in the mentioned study,48 we use the adiabatic states of the entire system. The use of a semiempirical methodology enables us to study systems as large as SubPc/(C60)4 directly. Based on the results discussed above, we choose to focus on the CT dynamics in the extended systems using the FSSH+ID-A method combined with the RM1+D/RM1 interaction potential. The computed timescales are summarized in Table 5. Our calculations indicate that the CT process in SubPc/(C60)n occurs within 2.2–5.0 ps, with a hint of acceleration in larger systems. These timescales are comparable to experimentally determined CT rates (ca. 10 ps).47
| Property | n | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Timescale, ps | 2.7 | 5.0 | 2.2 | 2.2 |
| E D − EA, eV | 0.55 | 0.53 | 0.51 | 0.53 |
| NAC, meV | 1.7 | 0.74 | 1.3 | 1.2 |
| SubPc (B atom)–C60 (closest atom) distance, Å | 4.2 | 4.2, 6.9 | 4.0, 7.0, 7.0 | 4.1, 7.8, 7.8, 8.1 |
In the series of SubPc/C60–SubPc/(C60)3–SubPc/(C60)4, we observe a hint of CT acceleration, which can be rationalized on the basis of Fermi's golden rule. Indeed, the increased density of acceptor (C60) states in SubPc/(C60)4 can facilitate CT, making it faster than in the dimer model, where the density of states is not that high. It is interesting to note that the average magnitude of the NAC between the donor states (localized on SubPc) and the acceptor C60 states steadily decreases in the abovementioned series from 1.7 meV in the minimal dimer model to 1.3 meV in the system with three C60 moieties (Table 3). Thus, based solely on the obtained NAC magnitudes, one would expect a slowing down of the CT process in the larger systems. The fact that such deceleration is not observed indicates that the increased density of states may indeed compensate for the NAC-dependent effects.
The observed decrease of the NACs in the extended systems can be attributed to the delocalization of the acceptor (C60) states over a larger surface. The orbital delocalization may seem counterintuitive considering the relatively large distance between neighboring C60 fragments orbitals. However, due to symmetry, the fragment-localized quasi-diabatic states would be energetically-resonant, which leads to their strong mixing in the adiabatic (whole system) representation. Such mixing-based delocalization of fragment orbitals was reported earlier.48 The symmetry-based delocalization was also discussed earlier for organic dimers.167 Finally, visualizing representative SubPc/(C60)n (n = 2–4) orbitals in Fig. S5–S7 of ESI† directly supports the above hypothesis. It should be noted that since the extent of delocalization depends on the energetics of C60 fragments modulated by their local environment, thermal fluctuation may induce orbital localization and “re-localization” happening from one nuclear configuration to another. Some configurations (more energetical dispersion) may have notably localized orbitals, whereas those with more uniform distribution of the site energies would favor larger extent of orbital delocalization.
Such delocalization leads to the decrease of the time overlaps between the donor (SubPc) and acceptor (C60) states, thereby also decreasing the NAC magnitudes. A similar effect has been discussed in a number of other works. In particular, in the recent work of Biancardi et al.,20 a similar rationalization of the decrease of the CT rates in zinc phthalocyanine/graphene sheets when the number or graphene layers is increased was suggested. The decrease of the NACs in the systems with larger number of C60 molecules also correlates with the increased average distances between the SubPc molecule and the nearby C60 units (Table 5), which also contributes to lowering the overlaps of the donor and acceptor orbitals.
Our calculations also suggest that the CT process in SubPc/(C60)2 is notably slower than that in the other three models with n = 1, 3, and 4. The CT deceleration in this system correlates with the remarkably smaller average NAC magnitude. Although the present results do not directly explain the origin of such anomalous behavior, we proposed a possible explanation based on our experience and general considerations. The decreased couplings observed for SubPc/(C60)2 may be attributed to the symmetry of the system and the interactions existing in it. As we have explained above, the local environment of the acceptor fragments may lead to energetic inhomogeneity or degeneracy and facilitate orbital delocalization or induce orbital trapping. Certain symmetries of the acceptor cites are possible for the systems SubPc/(C60)n with n = 3 and 4. At the same time, the C60 “sites” are clearly distinct in the n = 2 case, because of the asymmetric placement of SubPc unit. Because of this asymmetry, the acceptor states in SubPc/(C60)2 may localize more often on the C60 unit that is more distant from the SubPc. As a consequence, the donor–acceptor overlaps are decreased, leading to smaller (trajectory-averaged) NACs. The increased symmetry of all the other systems may favor the localization of the acceptor states on the C60 unit closest to the SubPc unit, leading to larger orbital overlaps and NACs.
In addition, the symmetry consideration may come into play in the following way. As we have deduced earlier in this work, the “breathing” mode of SubPc that changes the degree of planarity of the molecule is the key mode that drives the CT process in SubPc/C60 systems. The symmetry of all SubPc/(C60)n (n = 1, 3, and 4) systems leads to the compensation of the attraction of the SubPc unit to lateral C60 units such that the boron atom of the SubPc unit stays relatively on top of the closest C60 unit. Such configuration allows for an unhindered “breathing” motion of SubPc, facilitating the CT process. In contrast, there is no compensation of the lateral interactions in the SubPc/(C60)2 system, which offsets the center of SubPc away from the closest C60 molecule. This is reflected in the increased smallest B–C distance of 4.2 Å and the decreased largest B–C distance of 6.9 Å, in comparison to the 4.0 Å and 7.0 Å values for the SubPc/(C60)3 system. The displaced geometry of the SubPc/C60 pair decreases the orbital overlap and hiders the “breathing” motion of SubPc. Both effects decrease the NAC and slow down the CT process, leading to the non-monotonous dependence of the CT rates on the number of C60 units used in the model. A broader implication of this observation is that a broken interfacial symmetry at the SubPc/C60 interface (e.g., at kink sites or step defects in realistic systems) may be responsible for extended CT times longer than those predicted in highly symmetrical idealized models.
Finally, we have extended our assessment of the surface hopping methods (MSSH and FSSH and their decoherence-corrected versions) using the data obtained for the extended systems (Table 6). In all four types of calculations, we observe slower CT dynamics in SubPc/(C60)2, and thus the non-monotonic behavior is a consequence of the intrinsic properties of the system and is not TSH-methodology dependent. The other timescales correlate with the FSSH+ID-A timescales discussed above. It is the absolute values of these numbers that differ across the methods. We consistently observe an overestimation of the CT rate when using the MSSH method, similar to the effects we discussed in details for the minimal system. The FSSH method predicts up to five times larger timescales in comparison to the MSSH method. Both numbers increase when the decoherence correction is applied. In general, the trends follow those observed in the smaller molecular model. To recapitulate, the present calculations suggest that the FSSH+ID-A method is the most reasonable scheme among the four we tested, and in fact it predicts timescales (2.2–5.0 ps) closest to the experimental values (10 ps).
| System | MSSH | MSSH+ID-A | FSSH | FSSH+ID-A |
|---|---|---|---|---|
| SubPc/C60 | 0.17 | 0.33 | 0.92 | 2.7 |
| SubPc/(C60)2 | 0.22 | 0.94 | 1.0 | 5.0 |
| SubPc/(C60)3 | 0.14 | 0.39 | 0.57 | 2.2 |
| SubPc/(C60)4 | 0.13 | 0.36 | 0.50 | 2.2 |
4. Conclusions
In this work, we have reported the development of the open-source Libra-X software for atomistic NA-MD simulations within the NBRA and beyond. This package is freely available on the Internet viahttps://github.com/Quantum-Dynamics-Hub/Libra-X. Our code relies on the Libra library for NA dynamics and on external electronic structure packages, namely GAMESS, Quantum Espresso, or Gaussian09. The presently available functionality enables simulations of molecular and solid-state system of various sizes and at various levels of theory. In particular, we demonstrate the utility of the proposed software to study CT rates in extended models of SubPc/C60 interfaces.We have assessed a number of interaction potentials for the modelling of the binding, geometry, and electronic structure of the SubPc/C60 dimer. We have found that the bare RM1 method is incapable of accurately describing the intermolecular interactions in SubPc/C60 systems. By introducing the attractive dispersion interactions of the Lennard-Jones type with scaled UFF parameters provides an improved description of the system's geometry and the binding energies, as compared with the reference DFT values. The resulting RM1+D (scaled) approach preserves the conical (nearly flat) structure of SubPc. At the same time, the over-scaled UFF vdW parameters can overstabilize the SubPc/C60 system and distort the conical structure of the SubPc molecule.
We have analyzed several calculation protocols to obtain an electronic structure and a geometry of SubPc/C60 which are consistent with those computed via the DFT/DFT protocol. We found that, among the tested semiempirical approaches, the RM1+D/RM1 method is the most suitable for modelling such systems, because the MO characters (localization and degeneracies) obtained with this method are consistent with those found via the DFT/DFT calculations. The other tested methods tend to distort the orbital localization or their ordering as well as lead to a mixing of the donor and acceptor states. To summarize our observations, orbital localization is strongly controlled by the degree of the conical distortion of the SubPc molecule, with the “flatter” base favoring electron localization on SubPc, whereas an out-of-plane bent, and “wrapped around” structure favors charge localization on C60. We suggest that the “breathing” motion of SubPc is likely the mode that drives the CT process in the SubPc/C60 system. We propose that this mode could be controlled via photo- or chemical activation, which could be used to create nanoscale charge transfer switches based on SubPc/C60 systems.
We have investigated the lower excited states of SubPc/C60 system using TD-DFT. We have found that the excitation character of these states strongly depends on the density functional chosen. Among ωB97XD, LC-ωPBE, CAM-B3LYP functionals, only CAM-B3LYP predicts the first excited state to have the CT character and energy around 2.5–2.6 eV, with the SubPc-localized exciton to be 0.1–0.2 eV higher in energy. Both ωB97XD and LC-ωPBE functionals predict the inverse order of CT and SubPc exciton. The polarization effects due to the solid-state environment modelled with PCM (dielectric constant of 4.2) have relatively negligible effect, changing the state energies by no more than 0.05 eV. The environment tends to stabilize the SubPc exciton and destabilize the CT state.
Using the NA-MD implementation in Libra-X, we have conducted a comprehensive investigation of the dependence of computed CT rates on the NA-MD methodologies used. We have demonstrated that the computed timescales can vary by almost two orders of magnitude: from 160 fs to ca. 10 ps. We conclude that the decoherence-corrected FSSH method is a suitable TSH approach, whereas the MSSH method tends to overestimate the CT rates. The inclusion of the decoherence correction is critical for getting correct order of magnitude in the computed CT rates. Among the tested methods, the RM1+D/RM1 protocol seems to be a suitable methodology for obtaining the nuclear trajectories used in NA-MD simulations.
With the help of the semiempirical methods available in GAMESS and Libra, we have been able to simulate the several-picosecond dynamics in SubPc/(C60)n systems with n up to 4. We observed a decrease of the NACs in systems with a larger number of C60 molecules, which we attribute to a decreased overlap of the localized donor and delocalized acceptor states. Such a monotonic decrease of NACs is observed only in relatively symmetric structures, such as SubPc/(C60)n with n = 1, 3, and 4. The SubPc/(C60)2 structure breaks this monotonic relationship and shows a remarkably smaller NAC. We attribute such anomalous value to the reduced donor–acceptor overlap due to distorted symmetry of the interactions. We also hypothesize that such asymmetry can hinder the “breathing” mode of SubPc, further slowing down the CT process.
Finally, we conclude that the best of the tested methodologies (FSSH+ID-A with RM1+D/RM1) and the molecular models (SubPc/(C60)n, all n = 2–4) predict the CT timescales to be in the 2.2–5.0 ps range, which is comparable to the experimentally reported timescale of 10 ps. The larger timescale is associated with the locally asymmetric interfacial structures, such as SubPc/(C60)2. Thus, as suggested earlier, the use of non-ideal, extended molecular models is important for explaining the experimental timescales for the CT process in SubPc/C60 heterojunctions. We hypothesize that, in experimental samples, the non-ideal motifs may be accommodated via various surface defects, such as kink sites or step edges. The SubPc molecules absorbed at such sites are expected to show CT dynamics inhibited with respect to an ideally positioned SubPc, as in the minimal dimer model.
The working examples of computational protocols used in this work, the key input (including execution Python files, scripting, and plotting files) and output files, as well as important structural data are available online at https://github.com/AkimovLab/Project_Libra-GAMESS. The repository also provides the digital equivalents of some figures shown in the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
KS is grateful to Dr. Ryosuke Jinnouchi, Dr Nobuko Ohba, Dr. Soichi Shirai, and Prof. Oleg Prezhdo for the fruitful discussions. AVA acknowledges the financial support of the SUNY RF, award number 57333. The computational work has been supported by the Center for Computational Research, University at Buffalo.References
- A. Mishra, K. R. Fischer Markus and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS PubMed.
- H. Tian, Z. Yu, A. Hagfeldt, L. Kloo and L. Sun, J. Am. Chem. Soc., 2011, 133, 9413–9422 CrossRef CAS PubMed.
- T. Liu, L. Huo, X. Sun, B. Fan, Y. Cai, T. Kim, Y. Kim Jin, H. Choi and Y. Sun, Adv. Energy Mater., 2015, 6, 1502109 CrossRef.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- Z. Zheng, M. Awartani Omar, B. Gautam, D. Liu, Y. Qin, W. Li, A. Bataller, K. Gundogdu, H. Ade and J. Hou, Adv. Mater., 2016, 29, 1604241 CrossRef PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- A. Tang, C. Zhan, J. Yao and E. Zhou, Adv. Mater., 2016, 29, 1600013 CrossRef PubMed.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Nat. Commun., 2016, 7, 13740 CrossRef CAS PubMed.
- M. Guérette, A. Najari, J. Maltais, J. R. Pouliot, S. Dufresne, M. Simoneau, S. Besner, P. Charest and M. Leclerc, Adv. Energy Mater., 2016, 6, 1502094 CrossRef.
- B. J. Tremolet de Villers, K. A. O’Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Chem. Mater., 2016, 28, 876–884 CrossRef CAS.
- G. Zhang, K. Zhang, Q. Yin, X.-F. Jiang, Z. Wang, J. Xin, W. Ma, H. Yan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2017, 139, 2387–2395 CrossRef CAS PubMed.
- K. Sen, R. Crespo-Otero, W. Thiel and M. Barbatti, Comput. Theor. Chem., 2014, 1040-1041, 237–242 CrossRef CAS.
- M. H. Lee, E. Geva and B. D. Dunietz, J. Phys. Chem. A, 2016, 120, 2970–2975 CrossRef CAS PubMed.
- X. Ji, J. Wang, L. Mei, W. Tao, A. Barrett, Z. Su, S. Wang, G. Ma, J. Shi and S. Zhang, Adv. Funct. Mater., 2017, 28, 1705083 CrossRef.
- M. Yamamoto, J. Föhlinger, J. Petersson, L. Hammarström and H. Imahori, Angew. Chem., Int. Ed., 2017, 56, 3329–3333 CrossRef CAS PubMed.
- M. C. Staniford, M. M. Lezhnina and U. H. Kynast, RSC Adv., 2015, 5, 3974–3977 RSC.
- M. V. Martinez-Diaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- A. Biancardi, C. Caraiani, W.-L. Chan and M. Caricato, J. Phys. Chem. Lett., 2017, 8, 1365–1370 CrossRef CAS PubMed.
- H. M. Rhoda, M. P. Kayser, Y. Wang, A. Y. Nazarenko, R. V. Belosludov, P. Kiprof, D. A. Blank and V. N. Nemykin, Inorg. Chem., 2016, 55, 9549–9563 CrossRef CAS PubMed.
- L. G. Kaake, A. Jailaubekov, K. J. Williams and X.-Y. Zhu, Appl. Phys. Lett., 2011, 99, 083307 CrossRef.
- I. Radivojevic, G. Bazzan, B. P. Burton-Pye, K. Ithisuphalap, R. Saleh, M. F. Durstock, L. C. Francesconi and C. M. Drain, J. Phys. Chem. C, 2012, 116, 15867–15877 CrossRef CAS PubMed.
- G. Mattioli, C. Melis, G. Malloci, F. Filippone, P. Alippi, P. Giannozzi, A. Mattoni and A. Amore Bonapasta, J. Phys. Chem. C, 2012, 116, 15439–15448 CrossRef CAS.
- A. Varotto, C.-Y. Nam, I. Radivojevic, J. P. C. Tome, J. A. S. Cavaleiro, C. T. Black and C. M. Drain, J. Am. Chem. Soc., 2010, 132, 2552–2554 CrossRef CAS PubMed.
- B. C. O'Regan, I. López-Duarte, M. V. Martínez-Díaz, A. Forneli, J. Albero, A. Morandeira, E. Palomares, T. Torres and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 2906–2907 CrossRef PubMed.
- J.-J. Cid, A. Kahnt, P. Vázquez, D. M. Guldi and T. Torres, J. Inorg. Biochem., 2012, 108, 216–224 CrossRef CAS PubMed.
- K. L. Mutolo, E. I. Mayo, B. P. Rand, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2006, 128, 8108–8109 CrossRef CAS PubMed.
- H. Gommans, D. Cheyns, T. Aernouts, C. Girotto, J. Poortmans and P. Heremans, Adv. Funct. Mater., 2007, 17, 2653–2658 CrossRef CAS.
- R. R. Lunt, N. C. Giebink, A. A. Belak, J. B. Benziger and S. R. Forrest, J. Appl. Phys., 2009, 105, 053711 CrossRef.
- X. Tong, B. E. Lassiter and S. R. Forrest, Org. Electron., 2010, 11, 705–709 CrossRef CAS.
- J. Kim and S. Yim, Appl. Phys. Lett., 2011, 99, 193303 CrossRef.
- R. Pandey, A. Gunawan Aloysius, K. A. Mkhoyan and J. Holmes Russell, Adv. Funct. Mater., 2012, 22, 617–624 CrossRef CAS.
- P. Sullivan, A. Duraud, L. Hancox, N. Beaumont, G. Mirri, J. H. Tucker, R. A. Hatton, M. Shipman and T. S. Jones, Adv. Energy Mater., 2011, 1, 352–355 CrossRef CAS.
- R. A. Kipp, J. A. Simon, M. Beggs, H. E. Ensley and R. H. Schmehl, J. Phys. Chem. A, 1998, 102, 5659–5664 CrossRef CAS.
- C. G. Claessens, D. González-Rodríguez and T. Torres, Chem. Rev., 2002, 102, 835–854 CrossRef CAS PubMed.
- D. González-Rodríguez, T. Torres, D. M. Guldi, J. Rivera, M. Á. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 6301–6313 CrossRef PubMed.
- C. C. Mattheus, W. Michaelis, C. Kelting, W. S. Durfee, D. Wöhrle and D. Schlettwein, Synth. Met., 2004, 146, 335–339 CrossRef CAS.
- D. González-Rodríguez, G. Claessens Christian, T. Torres, S. Liu, L. Echegoyen, N. Vila and S. Nonell, Chem. – Eur. J., 2005, 11, 3881–3893 CrossRef PubMed.
- M. E. El-Khouly, S. H. Shim, Y. Araki, O. Ito and K.-Y. Kay, J. Phys. Chem. B, 2008, 112, 3910–3917 CrossRef CAS PubMed.
- R. Pandey, Y. Zou and R. J. Holmes, Appl. Phys. Lett., 2012, 101, 033308 CrossRef.
- D. M. Guldi, Chem. Commun., 2000, 321–327, 10.1039/A907807J.
- J. Xue, B. P. Rand, S. Uchida and S. R. Forrest, J. Appl. Phys., 2005, 98, 124903 CrossRef.
- R. Pandey and R. J. Holmes, IEEE J. Sel. Top. Quantum. Elec., 2010, 16, 1537–1543 CAS.
- T. Liu and A. Troisi, Adv. Mater., 2012, 25, 1038–1041 CrossRef PubMed.
- M. H. Lee, E. Geva and B. D. Dunietz, J. Phys. Chem. C, 2014, 118, 9780–9789 CrossRef CAS.
- D. E. Wilcox, M. H. Lee, M. E. Sykes, A. Niedringhaus, E. Geva, B. D. Dunietz, M. Shtein and J. P. Ogilvie, J. Phys. Chem. Lett., 2015, 6, 569–575 CrossRef CAS PubMed.
- A. V. Akimov, J. Chem. Theory Comput., 2016, 12, 5719–5736 CrossRef CAS PubMed.
- Y. Zhao and W. Liang, Chem. Soc. Rev., 2012, 41, 1075–1087 RSC.
- M. H. Lee, B. D. Dunietz and E. Geva, J. Phys. Chem. C, 2013, 117, 23391–23401 CrossRef CAS.
- M. H. Lee, B. D. Dunietz and E. Geva, J. Phys. Chem. Lett., 2014, 5, 3810–3816 CrossRef CAS PubMed.
- A. K. Manna and B. D. Dunietz, J. Chem. Phys., 2014, 141, 121102 CrossRef PubMed.
- A. K. Manna, D. Balamurugan, M. S. Cheung and B. D. Dunietz, J. Phys. Chem. Lett., 2015, 6, 1231–1237 CrossRef CAS PubMed.
- R. D. Coalson, D. G. Evans and A. Nitzan, J. Chem. Phys., 1994, 101, 436 CrossRef CAS.
- R. Hoffmann, J. Chem. Phys., 1963, 39, 1397–1412 CrossRef CAS.
- J. H. Ammeter, H. B. Bürgi, J. C. Thibeault and R. Hoffmann, J. Am. Chem. Soc., 1978, 100, 3686–3692 CrossRef CAS.
- G. Calzaferri, L. Forss and I. Kamber, J. Chem. Phys., 1989, 93, 5366–5371 CrossRef CAS.
- L. Li, J. C. Wong and Y. Kanai, J. Chem. Theory Comput., 2017, 13, 2634–2641 CrossRef CAS PubMed.
- R. Crespo-Otero and M. Barbatti, Chem. Rev., 2018, 118, 7026–7068 CrossRef CAS PubMed.
- M. Barbatti, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 620–633 CAS.
- Y. Lin and A. V. Akimov, J. Phys. Chem. A, 2016, 120, 9028–9041 CrossRef CAS PubMed.
- A. V. Akimov, J. Comput. Chem., 2016, 37, 1626–1649 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. S. Gordon and W. S. Michael, Advances in electronic structure theory: GAMESS a decade later, Elsevier, Amsterdam, 2005, pp. 1167–1189 Search PubMed.
- J. C. Tully, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef CAS.
- D. S. Kilin, O. V. Prezhdo and M. Schreiber, J. Phys. Chem. A, 2007, 111, 10212–10219 CrossRef CAS PubMed.
- D. S. Kilin, K. Tsemekhman, O. V. Prezhdo, E. I. Zenkevich and C. von Borczyskowski, J. Photochem. Photobiol., Chem., 2007, 190, 342–351 CrossRef CAS.
- D. S. Kilin, K. L. Tsemekhman, S. V. Kilina, A. V. Balatsky and O. V. Prezhdo, J. Phys. Chem. A, 2009, 113, 4549–4556 CrossRef CAS PubMed.
- A. V. Akimov and O. V. Prezhdo, J. Chem. Theory Comput., 2013, 9, 4959–4972 CrossRef CAS PubMed.
- A. V. Akimov and O. V. Prezhdo, J. Am. Chem. Soc., 2014, 136, 1599–1608 CrossRef CAS PubMed.
- A. V. Akimov, R. Asahi, R. Jinnouchi and O. V. Prezhdo, J. Am. Chem. Soc., 2015, 137, 11517–11525 CrossRef CAS PubMed.
- J. Blumberger, Chem. Rev., 2015, 115, 11191–11238 CrossRef CAS PubMed.
- S. Jensen and D. Kilin, Int. J. Quantum Chem., 2012, 112, 3874 CrossRef CAS.
- D. S. Kilin and D. A. Micha, J. Phys. Chem. C, 2009, 113, 3530–3542 CrossRef CAS.
- D. S. Kilin and D. A. Micha, J. Phys. Chem. C, 2011, 115, 770–775 CrossRef CAS.
- S. Kilina, N. Dandu, E. R. Batista, A. Saxena, R. L. Martin, D. L. Smith and S. Tretiak, J. Phys. Chem. Lett., 2013, 4, 1453–1459 CrossRef CAS PubMed.
- S. Kilina, S. Ivanov and S. Tretiak, J. Am. Chem. Soc., 2009, 131, 7717–7726 CrossRef CAS PubMed.
- S. Kilina, D. Kilin and S. Tretiak, Chem. Rev., 2015, 115, 5929–5978 CrossRef CAS PubMed.
- S. V. Kilina, C. F. Craig, D. S. Kilin and O. V. Prezhdo, J. Phys. Chem. C, 2007, 111, 4871–4878 CrossRef CAS.
- S. V. Kilina, D. S. Kilin and O. V. Prezhdo, ACS Nano, 2009, 3, 93–99 CrossRef CAS PubMed.
- J. Liu, S. V. Kilina, S. Tretiak and O. V. Prezhdo, ACS Nano, 2015, 9, 9106–9116 CrossRef CAS PubMed.
- J. Spencer, L. Scalfi, A. Carof and J. Blumberger, Faraday Discuss., 2016, 195, 215–236 RSC.
- A. Carof, S. Giannini and J. Blumberger, J. Chem. Phys., 2017, 147, 214113 CrossRef.
- C. Craig, W. Duncan and O. Prezhdo, Phys. Rev. Lett., 2005, 95, 163001 CrossRef PubMed.
- R. D. Senanayake, A. V. Akimov and C. M. Aikens, J. Phys. Chem. C, 2016, 121, 10653–10662 CrossRef.
- A. Nijamudheen and A. V. Akimov, J. Phys. Chem. C, 2017, 121, 6520–6532 CrossRef CAS.
- G. B. Rocha, R. O. Freire, A. M. Simas and J. J. Stewart, J. Comput. Chem., 2006, 27, 1101–1111 CrossRef CAS PubMed.
- M. J. Dewar and W. Thiel, J. Am. Chem. Soc., 1977, 99, 4899–4907 CrossRef CAS.
- M. J. Dewar, E. G. Zoebisch, E. F. Healy and J. J. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- J. J. Stewart, J. Comput. Chem., 1989, 10, 209–220 CrossRef CAS.
- J. J. Stewart, J. Comput. Chem., 1989, 10, 221–264 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. A. Pople, D. P. Santry and G. A. Segal, J. Chem. Phys., 1965, 43, S129–S135 CrossRef CAS.
- T. Raeker and B. Hartke, J. Phys. Chem. A, 2017, 121, 5967–5977 CrossRef CAS PubMed.
- E. Fabiano, T. W. Keal and W. Thiel, Chem. Phys., 2008, 349, 334–347 CrossRef CAS.
- T. Nelson, S. Fernandez-Alberti, V. Chernyak, A. E. Roitberg and S. Tretiak, J. Phys. Chem. B, 2011, 115, 5402–5414 CrossRef CAS PubMed.
- T. Nelson, S. Fernandez-Alberti, A. E. Roitberg and S. Tretiak, Acc. Chem. Res., 2014, 47, 1155–1164 CrossRef CAS PubMed.
- L. G. C. Rego and V. S. Batista, J. Am. Chem. Soc., 2003, 125, 7989–7997 CrossRef CAS PubMed.
- R. da Silva, L. G. C. Rego, J. A. Freire, J. Rodriguez, D. Laria and V. S. Batista, J. Phys. Chem. C, 2010, 114, 19433–19442 CrossRef CAS.
- L. G. C. Rego, B. C. Hames, K. T. Mazon and J.-O. Joswig, J. Phys. Chem. C, 2014, 118, 126–134 CrossRef CAS.
- R. da Silva, D. A. Hoff and L. G. C. Rego, J. Phys.: Condens. Matter, 2015, 27, 134206 CrossRef PubMed.
- A. Torres, R. S. Oliboni and L. G. C. Rego, J. Phys. Chem. Lett., 2015, 6, 4927–4935 CrossRef CAS PubMed.
- A. V. Akimov and O. V. Prezhdo, J. Chem. Theory Comput., 2014, 10, 789–804 CrossRef CAS PubMed.
- S. Hammes-Schiffer and J. C. Tully, J. Chem. Phys., 1994, 101, 4657–4667 CrossRef CAS.
- W. R. Duncan, C. F. Craig and O. V. Prezhdo, J. Am. Chem. Soc., 2007, 129, 8528–8543 CrossRef CAS PubMed.
- C. M. Isborn, X. Li and J. C. Tully, J. Chem. Phys., 2007, 126, 134307 CrossRef PubMed.
- S. V. Kilina, A. J. Neukirch, B. F. Habenicht, D. S. Kilin and O. V. Prezhdo, Phys. Rev. Lett., 2013, 110, 180404 CrossRef PubMed.
- J. Chen, A. Schmitz, T. Inerbaev, Q. Meng, S. Kilina, S. Tretiak and D. S. Kilin, J. Phys. Chem. Lett., 2013, 4, 2906–2913 CrossRef CAS.
- M. E.-A. Madjet, A. V. Akimov, F. El-Mellouhi, G. R. Berdiyorov, S. Ashhab, N. Tabet and S. Kais, Phys. Chem. Chem. Phys., 2016, 18, 5219–5231 RSC.
- R. Long, W. Fang and A. V. Akimov, J. Phys. Chem. Lett., 2016, 7, 653–659 CrossRef CAS PubMed.
- S. Pal, D. J. Trivedi, A. V. Akimov, B. Aradi, T. Frauenheim and O. V. Prezhdo, J. Chem. Theory Comput., 2016, 12, 1436–1448 CrossRef CAS PubMed.
- A. Nijamudheen and A. V. Akimov, J. Phys. Chem. Lett., 2018, 9, 248–257 CrossRef CAS PubMed.
- M. E. Madjet, G. R. Berdiyorov, F. El-Mellouhi, F. H. Alharbi, A. V. Akimov and S. Kais, J. Phys. Chem. Lett., 2017, 8, 4439–4445 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- A. I. Skoulidas and D. S. Sholl, J. Phys. Chem. B, 2005, 109, 15760–15768 CrossRef CAS PubMed.
- S. S. Konyukhov, I. V. Kupchenko, A. A. Moskovsky, A. V. Nemukhin, A. V. Akimov and A. B. Kolomeisky, J. Chem. Theory Comput., 2010, 6, 2581–2590 CrossRef CAS PubMed.
- C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Nat. Chem., 2011, 4, 83–89 CrossRef PubMed.
- A. V. Akimov and A. B. Kolomeisky, J. Phys. Chem. C, 2011, 115, 13584–13591 CrossRef CAS.
- A. L. Dzubak, L.-C. Lin, J. Kim, J. A. Swisher, R. Poloni, S. N. Maximoff, B. Smit and L. Gagliardi, Nat. Chem., 2012, 4, 810–816 CrossRef CAS PubMed.
- A. V. Akimov and A. B. Kolomeisky, J. Phys. Chem. C, 2012, 116, 22595–22601 CrossRef CAS.
- A. V. Akimov, C. Williams and A. B. Kolomeisky, J. Phys. Chem. C, 2012, 116, 13816–13826 CrossRef CAS.
- W. Bury, D. Fairen-Jimenez, M. B. Lalonde, R. Q. Snurr, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 739–744 CrossRef CAS.
- N. Höft and J. Horbach, J. Am. Chem. Soc., 2015, 137, 10199–10204 CrossRef PubMed.
- K. Gundertofte, T. Liljefors, P. O. Norrby and I. Pettersson, J. Comput. Chem., 1998, 17, 429–449 CrossRef.
- R. Krishna and J. M. van Baten, Phys. Chem. Chem. Phys., 2011, 13, 10593–10616 RSC.
- A. V. Akimov, D. Trivedi, L. Wang and O. V. Prezhdo, J. Phys. Soc. Jpn., 2015, 84, 094002 CrossRef.
- B. R. Landry and J. E. Subotnik, J. Chem. Phys., 2011, 135, 191101 CrossRef PubMed.
- H. M. Jaeger, S. Fischer and O. V. Prezhdo, J. Chem. Phys., 2012, 137, 22A545 CrossRef PubMed.
- A. V. Akimov, R. Long and O. V. Prezhdo, J. Chem. Phys., 2014, 140, 194107 CrossRef PubMed.
- A. V. Akimov and O. V. Prezhdo, ACS Symp. Ser., 2015, 1196, 189–200 CrossRef CAS.
- T. Nelson, S. Fernandez-Alberti, A. E. Roitberg and S. Tretiak, J. Chem. Phys., 2013, 138, 224111 CrossRef PubMed.
- F. Jensen, An Introduction to Computational Chemistry, John Wiley & Sons, 1989 Search PubMed.
- Python Programming Language, http://www.python.org, accessed May 10, 2018.
- The Boost C++ Libraries, http://www.boost.org/, accessed 10 May, 2018.
- D. Abrahams and R. W. Grosse-Kunstleve, C/C++ Users Journal, 2003, http://www.boost.org/doc/libs/1_31_30/libs/python/doc/PyConDC_2003/bpl.html.
- P. Gianozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, 2009, 9096580 Search PubMed.
- A. Farazdel, M. Dupuis, E. Clementi and A. Aviram, J. Am. Chem. Soc., 1990, 112, 4206–4214 CrossRef CAS.
- IQmol Molecular Viewer, http://iqmol.org/, accessed May 10, 2018.
- S. Nose, J. Chem. Phys., 1984, 81, 511–519 CrossRef CAS.
- S. Nose and M. L. Klein, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 339–342 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1989, 40, 2814–2815 CrossRef.
- H. Kamberaj, R. J. Low and M. P. Neal, J. Chem. Phys., 2005, 122, 224114 CrossRef CAS PubMed.
- D. S. Kleinerman, C. Czaplewski, A. Liwo and H. A. Scheraga, J. Chem. Phys., 2008, 128, 245103 CrossRef PubMed.
- O. A. Vydrov, J. Heyd, A. V. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 074106 CrossRef PubMed.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- O. A. Vydrov, G. E. Scuseria and J. P. Perdew, J. Chem. Phys., 2007, 126, 154109 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- A. F. Hebard, R. C. Haddon, R. M. Fleming and A. R. Kortan, Appl. Phys. Lett., 1991, 59, 2109 CrossRef CAS.
- B. Pevzner, A. F. Hebard and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 16439–16449 CrossRef CAS.
- G. J. Dutton and S. W. Robey, J. Phys. Chem. C, 2012, 116, 19173–19181 CrossRef CAS.
- J. A. Pople, J. Chem. Phys., 1967, 47, 2026–2033 CrossRef CAS.
- A. Solovyeva, M. Pavanello and J. Neugebauer, J. Chem. Phys., 2014, 140, 164103 CrossRef PubMed.
- R. Baer and D. Neuhauser, Phys. Rev. Lett., 2005, 94, 043002 CrossRef PubMed.
- E. Livshits and R. Baer, Phys. Chem. Chem. Phys., 2007, 9, 2932–2941 RSC.
- T. Stein, H. Eisenberg, L. Kronik and R. Baer, Phys. Rev. Lett., 2010, 105, 266802 CrossRef PubMed.
- T. Stein, L. Kronik and R. Baer, J. Am. Chem. Soc., 2009, 131, 2818–2820 CrossRef CAS PubMed.
- T. Stein, L. Kronik and R. Baer, J. Chem. Phys., 2009, 131, 244119 CrossRef PubMed.
- S. Zheng, H. Phillips, E. Geva and B. D. Dunietz, J. Am. Chem. Soc., 2012, 134, 6944–6947 CrossRef CAS PubMed.
- B. Misra and E. C. G. Sudarshan, J. Math. Phys., 1977, 18, 756 CrossRef.
- T. Petrosky, S. Tasaki and I. Prigogine, Phys. Lett. A, 1990, 151, 109–113 CrossRef.
- T. Petrosky, S. Tasaki and I. Prigogine, Phys. A, 1991, 170, 306–325 CrossRef.
- O. V. Prezhdo, Phys. Rev. Lett., 2000, 85, 4413–4417 CrossRef CAS PubMed.
- A. V. Akimov, J. Phys. Chem. Lett., 2017, 8, 5190–5195 CrossRef CAS PubMed.
- A. V. Akimov and O. V. Prezhdo, J. Phys. Chem. Lett., 2013, 4, 3857–3864 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03841d |
| This journal is © the Owner Societies 2018 |