
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the capability of in silico mutation protocols for predicting the finite temperature conformation of amino acids†
Rodrigo
Ochoa
 a,
Miguel A.
Soler
b,
Alessandro
Laio
bc and
Pilar
Cossio
*ad
a,
Miguel A.
Soler
b,
Alessandro
Laio
bc and
Pilar
Cossio
*ad
aBiophysics of Tropical Diseases, Max Planck Tandem Group, University of Antioquia, Medellin, Colombia. E-mail: pilar.cossio@biophys.mpg.de; grupotandem.biotd@udea.edu.co
bInternational School for Advanced Studies (SISSA), Via Bonomea 265, I-34136 Trieste, Italy
cThe Abdus Salam International Centre for Theoretical Physics (ICTP), Strada Costiera 11, 34151 Trieste, Italy
dDepartment of Theoretical Biophysics, Max Planck Institute of Biophysics, 60438 Frankfurt am Main, Germany
First published on 3rd October 2018
Abstract
Mutation protocols are a key tool in computational biophysics for modelling unknown side chain conformations. In particular, these protocols are used to generate the starting structures for molecular dynamics simulations. The accuracy of the initial side chain and backbone placement is crucial to obtain a stable and quickly converging simulation. In this work, we assessed the performance of several mutation protocols in predicting the most probable conformer observed in finite temperature molecular dynamics simulations for a set of protein–peptide crystals differing only by single-point mutations in the peptide sequence. Our results show that several programs which predict well the crystal conformations fail to predict the most probable finite temperature configuration. Methods relying on backbone-dependent rotamer libraries have, in general, a better performance, but even the best protocol fails in predicting approximately 30% of the mutations.
1 Introduction
Mutational protocols have been used successfully for predicting structures of proteins that were not resolved experimentally,1 for determining the interfaces in protein–protein interactions,2,3 and for designing de novo peptides.4–6 One widespread application is the computational scanning of alanine or glycine residues, in order to identify hot spots and key amino acids responsible of the protein–protein stabilizing interactions.7,8 The placement of the mutated residue is crucial to understand the potential effects of the mutation.9,10Most mutation protocols require the backbone or C-alpha position of the amino acid, and then generate a side chain conformation. Some protocols, for example Rosetta fixbb11 and SCWRL412 use rotamer libraries from public databases of experimentally-resolved protein structures to predict the side chain configuration. Other protocols use minimization approaches to find the optimal conformation that minimizes an empirical scoring function.13 Combinatorial approaches have been developed to mutate multiple or single amino acids.14,15
To improve the accuracy of the side chain prediction, a fundamental step is to sample the conformations of the system. This can be performed using stochastic methods such as Monte Carlo, based on movements constrained by dihedrals,16 and classical or enhanced molecular dynamics (MD) simulations.17 However, sampling the rotamer space of the amino acid is time consuming. In protein design applications, where iterative single-point mutations protocols are required for designing novel molecules, running long MD simulations for each mutation is computationally expensive because the number of mutations that have to be explored, even for a small peptide, is extremely large.18 Protocols able to predict rotamers correctly can diminish the required simulation time to explore the conformational space.
In addition, starting an MD simulation from the wrong rotamer can be problematic because it can lead to conformational changes affecting events such as protein folding19 and binding.20 For example, in some cases, the folded conformations of the Trp-cage domain have been simulated with low success using classic and enhanced MD. This has been associated to the erroneous placement of near-native rotamers of the central tryptophan side chain.21 In the study of protein–protein interaction, starting the simulation with key residues at the interface in the wrong conformation can be detrimental. In ESI,† we provided an example involving the protein–protein complex Barnase–Barstar,22 where starting from a single wrong rotamer (compared to the crystal structure) causes a considerable loss of native contacts which are not retrieved along all the MD simulation (see Fig. S1, ESI†). Such effect can, for example, impact the prediction of binding affinities. Therefore, an optimal mutational protocol should predict side chain conformations that are in agreement with the equilibrium distributions from finite temperature simulations.
Here, we assess the performance of different mutation protocols using as a benchmark MD simulations of a set of protein–peptide complexes that differ only by single-point mutations in the peptide sequence. We compare the side chain dihedral angles, the number of contacts and the number of hydrogen bonds resulting from the mutation protocols to the equilibrium distribution of those quantities. The results suggest a rational pipeline to improve the mutational protocols for efficient MD simulations, and protein or peptide design.
2 Methodology
2.1 Single-point mutation benchmark systems
A set of crystal protein structures of the Oligopeptide Binding Protein (OppA) interacting with a tripeptide of motif K-x-K23 (where x is one out of 11 amino acids) was used as a reference system (Fig. 1A) to test a set of single-point mutation protocols. The structures were obtained from the Protein Data Bank (PDB)24 (PDB ids: 1B4Z, 1B51, 1QKA, 1B3G, 1B32, 1B3F, 1B46, 1B5J, 1QKB, 1B5I, 1B40) and formatted to share the same amino acid number and chain identifiers.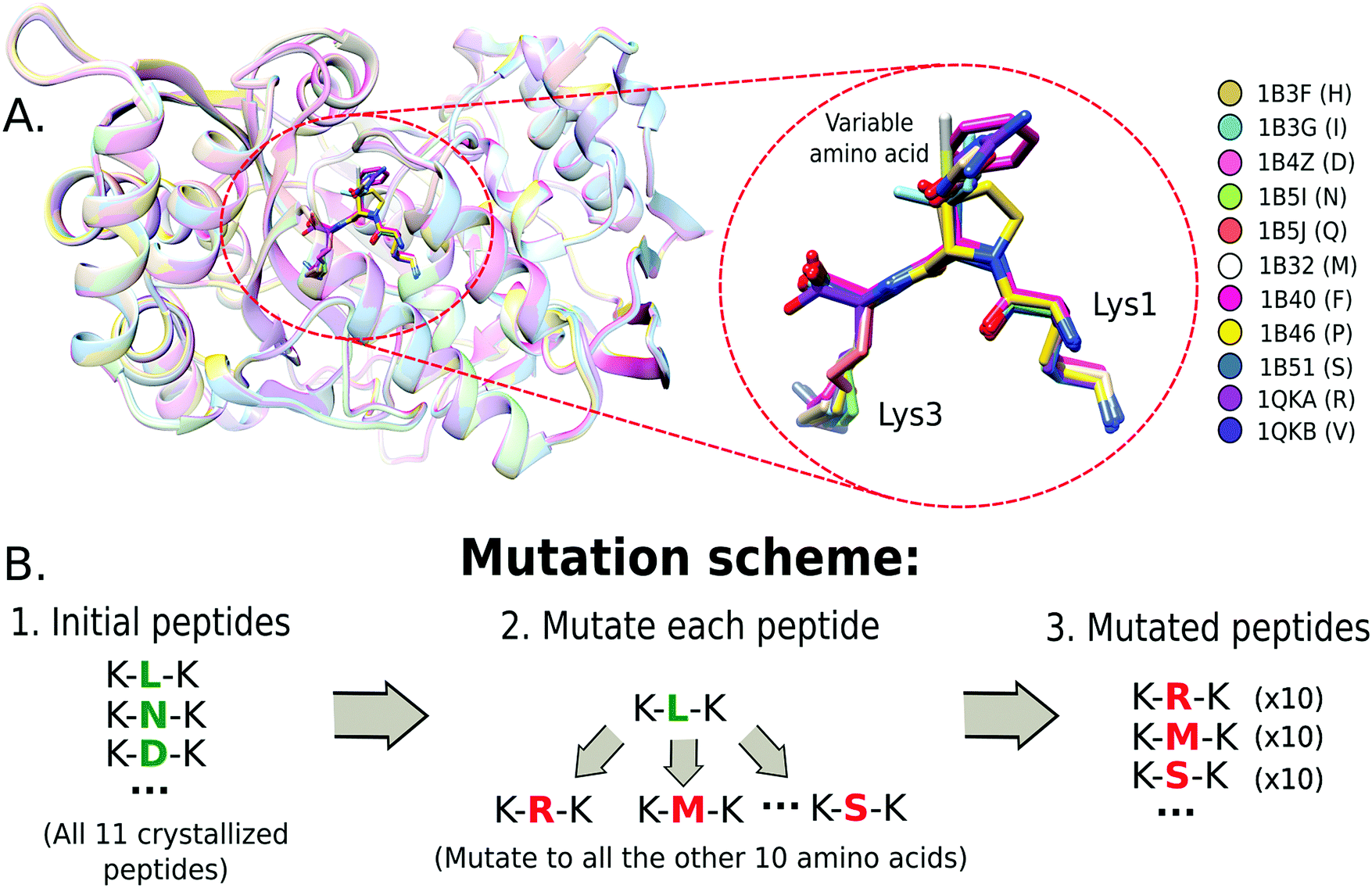 | ||
| Fig. 1 Mutation scheme for the OppA–tripeptide complexes. (A) Multiple structural alignment of the crystals (with PDB codes) containing the tripeptides. (B) Combinatorial scheme used to test the possible single-point mutations among the selected OppA crystals. | ||
We also assessed the protocols with other protein–peptide systems. Two complexes, which had structures differing only by a single amino acid on the peptide sequence, were selected: the HLA class I antigen A-2 alpha chain25 and the MDM4 protein complexed with a 12-mer peptide.26 For the HLA class I complex, four crystals were selected from the PDB differing only by a single peptide amino acid at the 5th position (PDB ids: 3GSO, 3GSU, 3GSV, 3GSR). For MDM4–peptide complex, the variable amino acid is at the peptide's 6th position (PDB ids: 3JZP, 3JZO).
2.2 Mutation scheme
A combinatorial approach was used to construct the mutants. This consists of mutating the variable amino acid to all the other amino acids available in the crystals. In Fig. 1B, we show the mutation scheme for the OppA system, where the variable amino acid from the tripeptide of each crystal is mutated to all other 10 amino acids. Thus, each structure produces 10 different mutations, resulting in 10 different structures for the same peptide sequence. This strategy allowed us to characterize the possible impact of steric and volume constraints due to the starting structure.2.3 Molecular dynamics simulations
Each protein–peptide complex from the benchmark was submitted to 20 nanoseconds (ns) of MD simulations with previous minimization and NVT/NPT equilibration phases. The system was minimized using the steepest descent algorithm, with 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps and a maximum force threshold of 10 kJ mol−1 nm−1. NVT and NPT equilibrations were performed for 100 ps using position restraints on the heavy atoms of the protein to allow for the equilibration of the solvent. GROMACS v5.127 was used to perform the MD simulations. The Amber99SB-ILDN protein force-field28 and TIP3P water model29 were used. The protein was solvated with a cubic box of water with a distance of 8 Å from the furthest atom of the protein. After solvation, counterions of Na+ and Cl− were included in the solvent to make the box neutral. The simulation was run using a modified Berendsen thermostat30 at 330 K temperature-coupling, and the Parrinello–Rahman barostat.31 The electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method with 1.0 nm short-range electrostatic and van der Waals cutoffs.32 The equations of motion were solved with the leap-frog integrator33 using a timestep of 2 fs.
000 steps and a maximum force threshold of 10 kJ mol−1 nm−1. NVT and NPT equilibrations were performed for 100 ps using position restraints on the heavy atoms of the protein to allow for the equilibration of the solvent. GROMACS v5.127 was used to perform the MD simulations. The Amber99SB-ILDN protein force-field28 and TIP3P water model29 were used. The protein was solvated with a cubic box of water with a distance of 8 Å from the furthest atom of the protein. After solvation, counterions of Na+ and Cl− were included in the solvent to make the box neutral. The simulation was run using a modified Berendsen thermostat30 at 330 K temperature-coupling, and the Parrinello–Rahman barostat.31 The electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method with 1.0 nm short-range electrostatic and van der Waals cutoffs.32 The equations of motion were solved with the leap-frog integrator33 using a timestep of 2 fs.
The convergence of the simulations was monitored by computing several observables: the number of hydrogen bonds between the peptide and protein, the number of heavy atom contacts34 made by the mutated amino acid, and the all-atom Root-Mean-Square Deviation (RMSD) of the peptide from the reference crystal. In some cases, the simulations were extended to achieve convergence.
2.4 Mutation protocols
We analyzed five mutation protocols. Modeller v9.19,35 which relies on minimization cycles of rotamers derived from homology-based models. SCWRL436 and Rosetta fixbb v2017.26,11 which use backbone-dependent rotamer libraries to predict side chain conformations but with different scoring functions. TLEaP from AmberTools v16,37 used to generate topology files from protein structures for the Amber program.37 FoldX v438 a protocol implemented in protein folding simulations that depends on energy calculations derived from an empirical force field, used also to study the effects of point mutations.These programs were configured to generate single-point mutations of the variable amino acids. The mutations were performed starting from the crystal structure and also from the structure obtained in the last frame of the MD trajectories (see ESI†). The prediction of the mutated side chain was made, for all protocols, with the peptide in complex with the protein.
2.5 Evaluation criteria
To evaluate the performance of the mutation protocols, we compare the dihedral angles and number of contacts predicted by the protocol to the most probable conformations from the MD simulations. The evaluation criteria are exemplified in Fig. 2. The conformational ensemble of protein folds39 and protein–protein complexes40,41 obtained from equilibrated finite temperature simulations has proved to depict accurately the interactions between the involved molecules. | ||
| Fig. 2 Visual summary of the strategies used to evaluate the mutation protocols. The first strategy compares the side chain dihedral angle of the mutated peptide to structures obtained in the MD trajectory. The second strategy assesses the conservation of the heavy atom contacts. | ||
Backbone dihedrals (ϕ, ψ) and side chain dihedrals χ1 and χ2 were calculated for each mutated amino acid. For the side chain dihedrals, the distribution from the MD simulations was used to verify that the predicted rotamers are located within the conformations visited in the MD. The χ1 ([0,360] deg) and χ2 ([0,360] deg) 2D dihedral angle space was binned using a 50 × 50 grid. Thus, along each dihedral direction a bin size of 7.2 deg was taken, resulting in a 7.2 × 7.2 deg2 for the 2D bin size. To indicate if a side chain prediction is visited in the trajectory, we looked at the 2D bin corresponding to the rotamer prediction and compared its population to that of the most populated bin. We count a rotamer as visited during the MD trajectory if the rotamer falls within a bin which has an MD population that is at least 5% of that of the most populated bin. We assume that if the mutated conformation is sampled in the trajectory then it is possible to reach equilibrium in a similar or shorter time. The threshold allows us to compute a success rate for the mutation protocols that can assess which protocols perform better.
Another evaluation criterion was defined using the heavy-atom contacts between the mutated amino acid and the protein. For this purpose, we used the information from the bins previously established in the side chain dihedral analysis. Specifically, each mutated complex, which is located in a bin from the χ1 and χ2 grid, was compared to the MD structures belonging to the same bin. The heavy atom contacts were monitored using the contact matrix42 with different distance thresholds d0, including 4 Å, 3.5 Å and 3 Å. The results of the latter are included in the main text, and the others are available in the ESI.† The contact matrix is defined as:

where the i sum runs over the peptide atoms and the j sum runs over the protein atoms. The result is a number from 0 to 1, where 1 is the most successful scenario (i.e., all the contacts predicted by the mutation are also present in the MD structure). The average and standard deviation of the SMR for each protocol and each mutated amino acid were obtained by averaging over five structures from the corresponding dihedral bin.
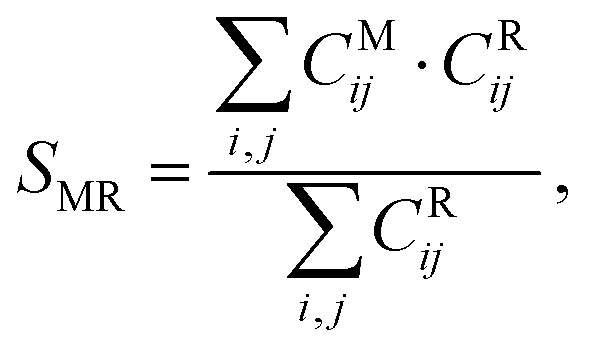
2.6 Rosetta mutation-protocol modifications
The Rosetta Commons project (http://www.rosettacommons.org) has available Monte Carlo approaches to optimize both backbone and side chain dihedrals of protein structures. These methods can be used to refine the system before or after performing a single-point mutation. We implemented the following protocols: relaxing with rigid backbone,43 prepacking of interface side chains,44 refinement of the system with FlexPepDock45 and inclusion of backbone flexibility for both protein and peptide using the BackRub protocol.9 The differences between these protocols are related to the type of molecular movements, the computational exhaustiveness and the internal constraints used to obtain the lowest energy conformations of the selected amino acids. The performance of these modifications was also evaluated using the previously described criteria.3 Results
We tested various protocols to perform single point mutations on peptides bound to protein targets. We used all-atom MD simulations with explicit solvent to sample the conformational space and compare to the results from the mutation protocols. First, we present the results for the OppA–tripeptide complex using the evaluation criteria, and then we show the results for the HLA class I and the MDM4 complexes (see Methods).3.1 Convergence of the molecular dynamics simulations
We used the equilibrium ensemble from MD simulations as a test to evaluate the performance of the mutation protocols. The benchmark system of the 11 OppA crystals presented stable observables, such as the number of hydrogen bonds and all-atom RMSD, during the 20 ns of simulation (see Methods and Fig. S2, ESI†). We used the complete 20 ns trajectory to calculate the equilibrium distributions. We tracked the distribution of the backbone and side chain dihedrals angles of the mutated amino acid during the MD trajectories. We found that the backbone dihedrals remain quite stable during all the MD trajectories (see Fig. S3, ESI†).For the side chain dihedrals (χ1 and χ2), we checked that the most frequent conformations are categorized in three on-rotamer groups: gauche(+), trans and gauche(−), centered in 300, 180° and 60° respectively.46 In general, for both χ1 and χ2 most of the side chain conformations were classified as gauche(+), which is in fact the most abundant conformation in the PDB, with the gamma side chain pointing in an opposite direction to the main chain nitrogen (see Fig. S4, ESI†).47
3.2 Side chain dihedral prediction
After calculating the distributions from the converged trajectories, we compared the predicted χ1 and χ2 dihedrals of each mutation protocol to the distribution from the MD simulations (see Methods for details). We first evaluated the prediction of χ1 dihedral for all the 11 amino acids. The results for serine and valine are presented in Fig. 3. We also report for each amino acid the percentage of success in predicting the dihedral with a conformation located in a histogram bin with a population larger than 5% of the most populated rotamer bin (Table 1). If only χ1 is considered, Rosetta fixbb and FoldX are the methods with highest success rates.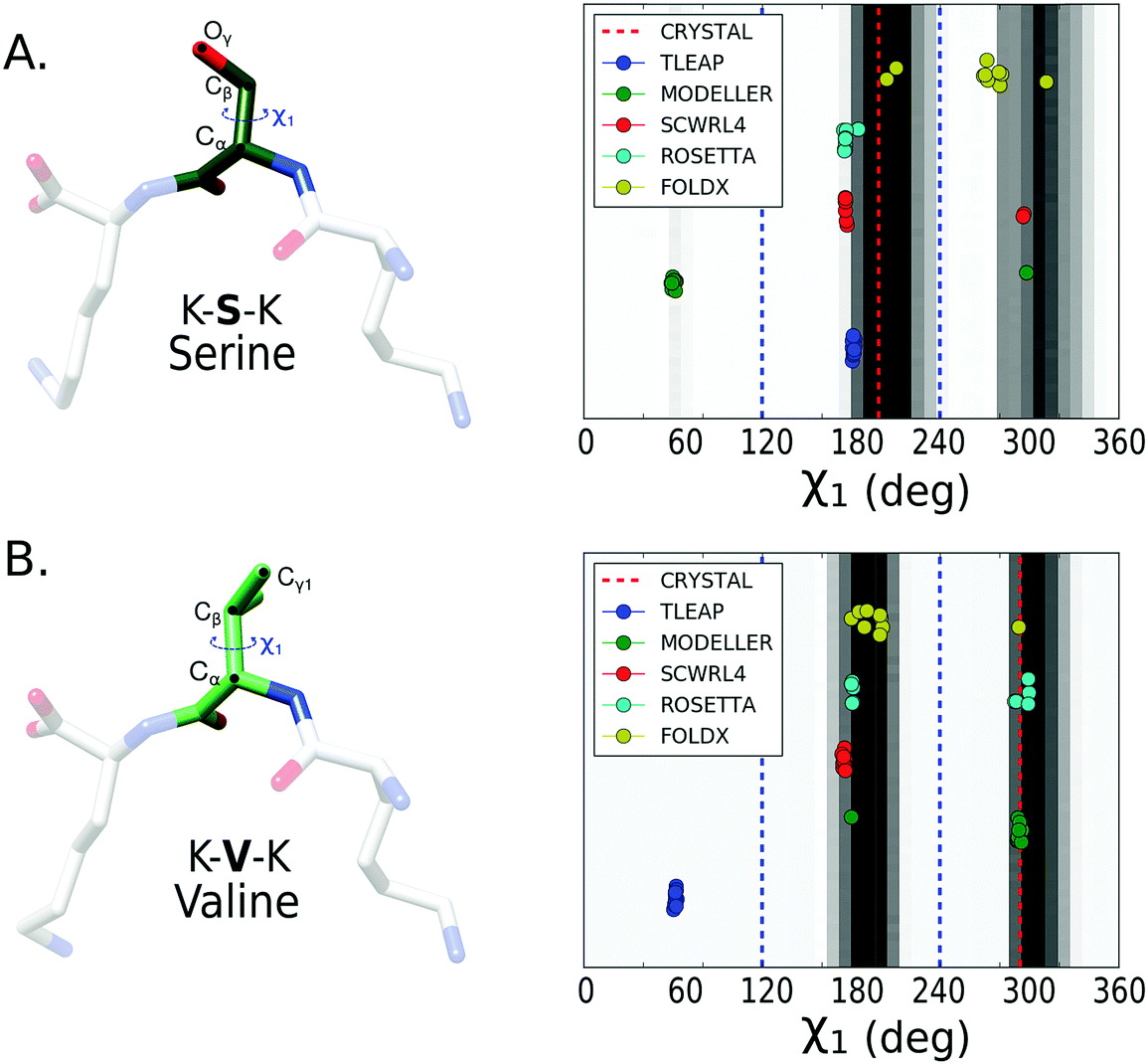 | ||
| Fig. 3 1D histogram of χ1 dihedral for two amino acids serine (A) and valine (B) mutated over the tripeptide in complex with the OppA protein. The black regions represent the χ1 dihedrals most frequently explored during the MD simulation of the complex containing the amino acid of interest. Each mutation protocol prediction is represented by a circle, with 10 circles per protocol given the combinatorial approach proposed for the OppA system (Methods). The main side-chain groups are split by dashed blue lines in the three main on-rotamer regions: gauche(+), trans and gauche(−), centered in 300°, 180° and 60° respectively. | ||
| AA | Modeller | Scwrl4 | TLEaP | Rosetta | Foldx |
|---|---|---|---|---|---|
| ARG | 90 | 90 | 0 | 100 | 70 |
| ASN | 60 | 0 | 100 | 80 | 80 |
| ASP | 20 | 0 | 0 | 30 | 100 |
| GLN | 80 | 80 | 0 | 100 | 80 |
| HIS | 100 | 100 | 0 | 100 | 100 |
| ILE | 100 | 100 | 100 | 100 | 100 |
| MET | 90 | 100 | 0 | 90 | 80 |
| PHE | 70 | 100 | 0 | 100 | 80 |
| PRO | 100 | 100 | 100 | 100 | 100 |
| SER | 10 | 100 | 100 | 100 | 80 |
| VAL | 100 | 100 | 0 | 100 | 100 |
| Average | 74.5 | 79.1 | 36.4 | 90.9 | 88.2 |
To better assess the side chain dihedral prediction, we performed a similar analysis taking into account both the χ1 and χ2 dihedrals. In Fig. 4, we show the results for the mutated amino acids isoleucine (I), arginine (R) and asparagine (N) (see Fig. S5 and S6 for other amino acids, ESI†). In Table 2, we report the percentage of mutations that succeeded, for each protocol, in predicting both the χ1 and χ2 dihedrals using a 5% bin-threshold (see Methods). A similar table using a 30% bin-threshold was calculated (Table S1, ESI†), which despite being stricter over the conformational space shows the same tendencies as reported in Table 2. The results show that SCWRL4, Rosetta fixbb and FoldX (see Methods) are the most successful protocols for predicting both the χ1 and χ2 dihedral angles. However, the success rate decreases for all protocols in comparison to the results for only χ1. The good performance of SCWRL4 and Rosetta fixbb can be related to the fact that both use backbone-dependent rotamer libraries as the basis for rotamer selection.
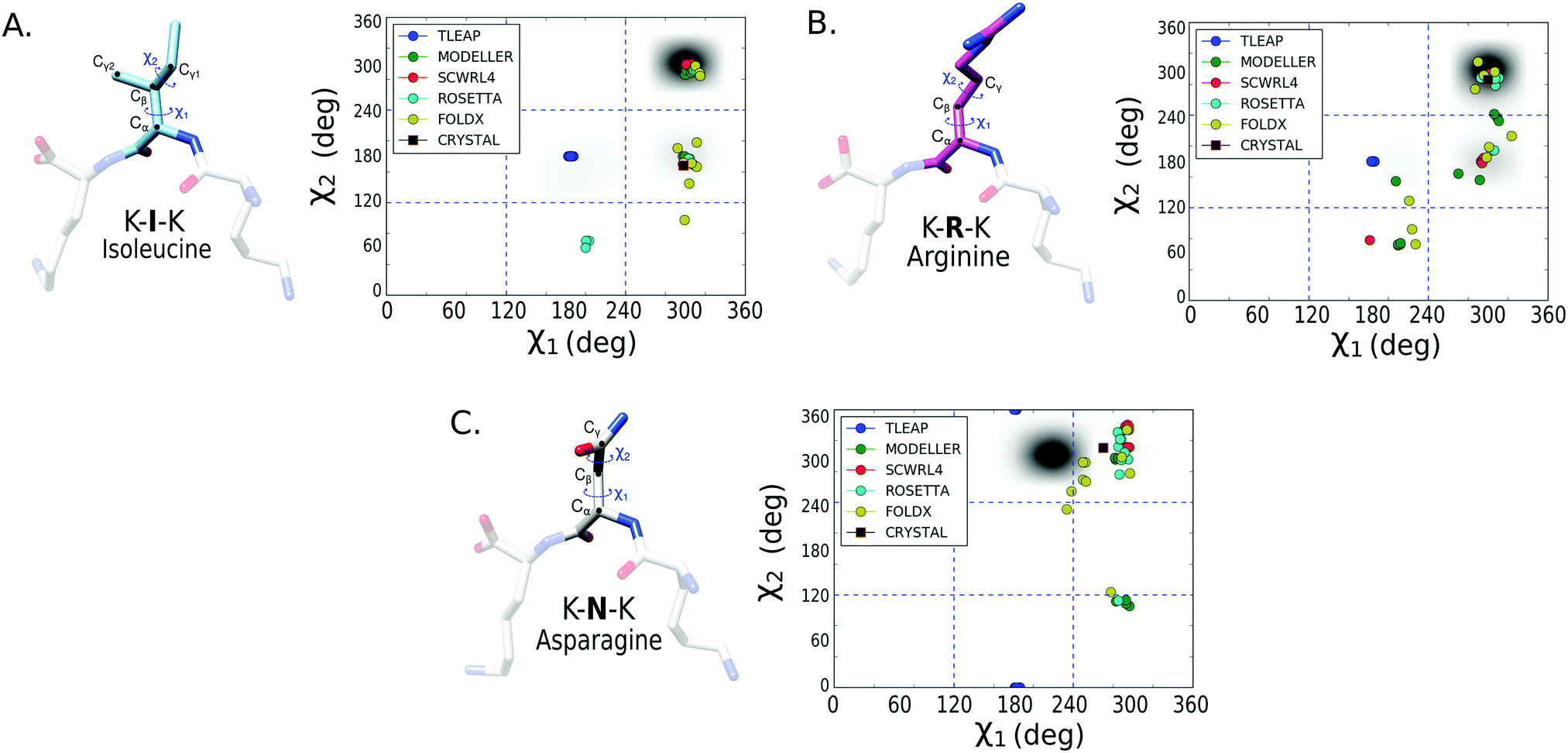 | ||
| Fig. 4 2D histogram of χ1vs. χ2 for three mutated amino acids: isoleucine (A), arginine (B) and asparagine (C) on the tripeptide of the OppA complex. The black zones represent the dihedrals most frequently explored during the MD simulation of the complex containing the amino acid of interest. Each mutation-protocol prediction is represented by circles, with 10 circles per protocol given the combinatorial approach proposed for the OppA system. The main side-chain conformations are split by dashed blue lines in 9 regions based on the possible χ1 and χ2 combinations. | ||
| AA | Modeller | Scwrl4 | TLEaP | Rosetta | Foldx |
|---|---|---|---|---|---|
| ARG | 0 | 90 | 0 | 100 | 40 |
| ASN | 50 | 0 | 0 | 30 | 40 |
| ASP | 20 | 0 | 0 | 20 | 90 |
| GLN | 50 | 80 | 0 | 100 | 20 |
| HIS | 60 | 10 | 0 | 80 | 60 |
| ILE | 100 | 10 | 0 | 70 | 70 |
| MET | 50 | 60 | 0 | 80 | 30 |
| PHE | 60 | 100 | 0 | 90 | 0 |
| PRO | 20 | 100 | 100 | 100 | 100 |
| Average | 45.6 | 50.0 | 11.1 | 74.4 | 50.0 |
A similar analysis was also performed for mutations created from the last MD frame (instead of using the crystal structure), and we observed similar trends for the χ1 and χ2 dihedrals prediction (see Table S2, ESI†).
In addition, we analyzed the performance from the perspective of the starting amino acid that was after mutated. This analysis elucidates if the starting amino acid produces an effect, e.g., due to its size or side-chain orientation, over the mutation protocol performance. We find that for the OppA crystals, the results for each protocol are similar for all the amino acids (see Table S3, ESI†). This can be explained by the side chain orientation, which is always pointing to the same direction (Fig. 1A). This implies that the starting amino acid has small impact on the mutation protocols.
3.3 Conservation of contacts
For the second evaluation criteria, we calculated the average and standard deviation of the conserved contacts for each selected protocol and each predicted amino acid (Methods). For each variable amino acid, the comparison was made between the 10 mutated structures of the OppA complex and five selected structures from the trajectory with similar dihedral angles (see Methods for details), giving a total of 50 comparisons per amino acid per protocol. The results using a contact threshold of 3 Å are shown in Table 3 (see Tables S4 and S5 for other thresholds, ESI†). Similarly as for the dihedral analysis, we found that methods using backbone-dependent rotamer libraries are better in predicting the contacts observed in the MD simulations, demonstrating the capabilities of the Rosetta fixbb and SCWRL packages to perform single point-mutations in peptides bound to proteins.| AA | Modeller | Scwrl4 | TLEaP | Rosetta | Foldx |
|---|---|---|---|---|---|
| ARG | 0 | 0.39 ± 0.03 | 0 | 0.51 ± 0.02 | 0.19 ± 0.04 |
| ASN | 0.36 ± 0.06 | 0 | 0 | 0.06 ± 0.02 | 0.05 ± 0.02 |
| ASP | 0.08 ± 0.03 | 0 | 0 | 0.05 ± 0.02 | 0.53 ± 0.05 |
| GLN | 0.27 ± 0.05 | 0.56 ± 0.06 | 0 | 0.72 ± 0.05 | 0.16 ± 0.05 |
| HIS | 0.49 ± 0.07 | 0.70 ± 0.05 | 0 | 0.53 ± 0.06 | 0.46 ± 0.07 |
| ILE | 0.70 ± 0.06 | 0.81 ± 0.05 | 0 | 0.62 ± 0.07 | 0.65 ± 0.07 |
| MET | 0.48 ± 0.07 | 0.52 ± 0.07 | 0 | 0.76 ± 0.06 | 0.22 ± 0.06 |
| PHE | 0.51 ± 0.07 | 0.73 ± 0.06 | 0 | 0.62 ± 0.07 | 0 |
| PRO | 0.14 ± 0.05 | 0.20 ± 0.06 | 0.24 ± 0.06 | 0.48 ± 0.07 | 0.22 ± 0.06 |
| SER | 0.10 ± 0.04 | 0.32 ± 0.06 | 0.93 ± 0.03 | 0.10 ± 0.04 | 0.60 ± 0.07 |
| VAL | 0.89 ± 0.04 | 0.93 ± 0.02 | 0 | 0.91 ± 0.04 | 0.92 ± 0.04 |
| Average | 0.37 ± 0.08 | 0.47 ± 0.09 | 0.11 ± 0.08 | 0.49 ± 0.09 | 0.36 ± 0.09 |
3.4 Performance of the modifications to the Rosetta mutation protocol
Based on the previous analysis, Rosetta fixbb has one of the best performances. We studied if it can be improved based on some available protocols9,43–45 to move the backbone and side chains atoms. We tested five Rosetta protocols. One protocol (pre-Relax) was applied before the mutation, and the other four were made after the mutation. The results of the dihedral analysis and contact conservation using these protocols are described in Table 4. These results indicate that relaxing the complex with a fixed (Post-Relax) or flexible (BackRub) backbone, after the mutation, slightly improves both the dihedral rotamer prediction and the conservation of contacts. The protocols designed for docking (Pre-Pack and FlexPepDock) were not able to improve the rotamer prediction.| Measured average | fixbb | preR | postR | preP | FPD | BR |
|---|---|---|---|---|---|---|
| χ 1, χ2% prediction | 74.4 | 65.6 | 81.1 | 38.9 | 64.4 | 82.2 |
| Contact conservation | 0.48 | 0.45 | 0.49 | 0.21 | 0.25 | 0.45 |
3.5 Dihedral analysis for other protein–peptide complexes
We tested the protocols over two additional systems. The first one is the MDM4 protein in complex with a 12-mer peptide.26 The peptide has an alpha helix conformation, and the initial amino acid tyrosine (Y, PDB:3JZO) has a similar orientation as the mutated tryptophan (W, PDB:3JZP) (see Fig. S7A, ESI†). The complex was stable during the MD simulation, as shown by monitoring the RMSD (Fig. S7C, ESI†).The second system is the HLA class I (see Fig. S7B, ESI†) in complex with a 9-mer peptide.25 The 5th position of the peptide was variable, having four possible amino acids: methionine (M, PDB:3GSO), threonine (T, PDB:3GSU), valine (V, PDB:3GSR) and glutamine (Q, PDB:3GSV) (Fig. S7B, ESI†). Similar to the MDM4 case, the complex is stable during the simulations (Fig. S7D, ESI†).
Both systems were submitted to single-point mutations using a combinatorial scheme similar to that described in the methods but according to the number of available structures. Using the same assessment as for the OppA system, in Table 5, we describe for each mutation if the mutation protocol was able to predict both the χ1 and χ2 dihedrals correctly. For the case of the MDM4 complex, all protocols, with the exception of TLEaP, were capable to predict the rotamers explored by the MD trajectory (see Fig. S8, ESI†). For the HLA class I the performance was different. The best performing protocols were Modeller, SCWRL4 and Rosetta fixbb after relaxing the complex. However, none of the protocols were able to put the methionine side chains as explored by the MD simulations. This might be influenced by the intrinsic flexibility of the amino acids that are not creating hydrogen bonds with the pockets of the HLA class I α chain.48
| PDB ID | Mutation | Modeller | Scwrl4 | TLEaP | Rosetta | Ros-postR | Foldx |
|---|---|---|---|---|---|---|---|
| MDM4 | |||||||
| 3JZP | W → Y | Yes | Yes | No | Yes | Yes | Yes |
| 3JZO | Y → W | Yes | Yes | No | Yes | Yes | Yes |
| HLA class I | |||||||
| 3GSO | M → Q | Yes | Yes | No | Yes | Yes | No |
| 3GSU | T → Q | Yes | Yes | No | Yes | Yes | No |
| 3GSR | V → Q | Yes | Yes | No | No | Yes | No |
| 3GSV | Q → M | No | No | No | No | No | No |
| 3GSU | T → M | No | No | No | No | No | No |
| 3GSR | V → M | No | No | No | No | No | No |
| Total (%) | 62.5 | 62.5 | 0 | 50 | 62.5 | 37.5 | |
4 Discussion
For comparing the performance of the mutational protocols it was necessary to use a robust benchmark system with various crystals differing only by a single mutation. We found the OppA system ideal for these purposes due to the wide availability of crystal structures. Moreover, working with tripeptides allowed the system to reach equilibrium in a short computational time (∼20 ns), which is not guaranteed for other systems where proteins are usually longer.49 We analyzed two additional systems to assess the performance with longer and structurally different peptide chains. We found that the mutation protocol performances are consistent for all test sets.By comparing the predicted side chain dihedral angles and contacts to the equilibrium distributions from finite temperature simulations, we found that the protocols based on rotamer libraries derived from protein structures50 perform better. In this context, SCWRL4 and Rosetta fixbb are suitable methods for performing single-point mutations. Both mutation protocols use backbone-dependent rotamer libraries but differ in how each rotamer is scored. SCWRL4 uses a scoring scheme based on single and pairwise rotamer energies computed from attractive and repulsive hydrogen bond and van der Waals terms.51 Rosetta, in addition, includes weighted terms related to statistical energies derived from distance-dependent pair potentials and solvation energies.52
The results for other mutation protocols were in some cases comparable or better than Rosetta and SCWRL4, but on average their performance was worst. One of the closest in performance to SCWRL4 and Rosetta fixbb is Modeller, which relies on cycles of minimization with the inclusion of homology-derived dihedral angle restraints, but without using rotamer libraries.35 This may be a factor that hinders its performance. TLEaP, the worst performing program, is used by AMBER to generate topology files for MD simulations, with the option to add missing atoms based on force field information.53 The main issue with TLEaP is that most of the dihedrals are predicted as trans/trans conformation without optimizating the dihedrals based on environment interactions. For FoldX,54 we obtained variable performances, showing successful results in cases where other protocols were unable to predict correctly the conformation, for example for aspartic acid. However, it is important to remark that neither the protocols that use rotamer-libraries, nor those that do not, are able to consistently predict the side chain conformations sampled in the simulations.
Amino acids such as aspartic acid and asparagine were more complex to predict, possibly because the hydrogen bonding capacity of their side chains, which impacts the χ2 dihedral.55 Another aspect to take into account is the chosen force field. The Amberff99SB-ILDN force field has been parameterized to explore the conformational space of proteins in long MD simulations, using quantum mechanics and experimental parameter data.28 Interestingly, the equilibrium distributions for aspartic acid and asparagine for this force field differed the most from expectations based on the Protein Data Bank statistics.24
Computational time is also a relevant consideration for selecting a mutation protocol. Both the time required to predict the rotamer and the computational time of the MD simulation to reach a stable conformation are important variables. All five mutation protocols tested are able to predict the mutated side chain in just a few seconds. When refinements are added to the Rosetta fixbb prediction, the computational time increases to a few minutes, with the exception of the BackRub method that is approximately 10 times longer than the standard Rosetta fixbb protocol. For the case of the MD simulation time, the goal of this work is to select the protocols able to predict the most probable rotamers explored by MD simulations. Consequently, the best performing mutation protocols will reduce the computational sampling time, and consequently the time required to reach a stable conformation. Importantly, we note that very wrong rotamer predictions could destabilize so much the structure that it might be very difficult to obtain a converged MD simulation.
Finally, we found that refining the protein–peptide complex after the mutation can improve the side chain prediction.56 The modularity of the Rosetta protocols to perform the refinement was useful at this scope. We found that relaxing the side chain without having to move the backbone improves the performance whilst maintaining a reasonable computational time.
5 Conclusions
The assessment of the single-point mutation protocols to predict side chains from equilibrium distributions has shown that, although some protocols are able to predict the most probable rotamer explored in MD, there is still large room for improvements. This is of key importance to the MD community, which highly relies on homology modelling and rotamer prediction. In addition, these protocols are also essential for peptide design, where filtering mutations in a random or guided way can contribute dramatically to the design protocol efficiency.57Our work sets a basis to assess, and to further improve and optimize, the mutation protocols to be in accordance with finite temperature simulations. Previously, all strategies have been optimized to predict crystal-structure rotamers. We propose to use also MD simulations as training sets to reparameterize and optimize the mutation algorithms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
RO and PC were supported by Colciencias, University of Antioquia, Colombia, and the Max Planck Society, Germany. The computations were performed in a local server with an NVDIA Titan X GPU. PC gratefully acknowledges the support of NVIDIA Corporation for the donation of this GPU. Open Access funding provided by the Max Planck Society.References
- R. Guerois, J. E. Nielsen and L. Serrano, J. Mol. Biol., 2002, 320, 369–387 CrossRef CAS PubMed.
- I. H. Moal and J. Fernández-Recio, Bioinformatics, 2012, 28, 2600–2607 CrossRef CAS PubMed.
- J. Yang, A. Roy and Y. Zhang, Nucleic Acids Res., 2013, 41, 1096–1103 CrossRef PubMed.
- V. D. Sood and D. Baker, J. Mol. Biol., 2006, 357, 917–927 CrossRef CAS PubMed.
- I. Gladich, A. Rodriguez, R. P. Hong Enriquez, F. Guida, F. Berti and A. Laio, J. Phys. Chem. B, 2015, 119, 12963–12969 CrossRef CAS PubMed.
- M. A. Soler, A. Rodriguez, A. Russo, A. F. Adedeji, C. J. Dongmo Foumthuim, C. Cantarutti, E. Ambrosetti, L. Casalis, A. Corazza, G. Scoles, D. Marasco, A. Laio and S. Fortuna, Phys. Chem. Chem. Phys., 2017, 19, 2740–2748 RSC.
- V. Ramadoss, F. Dehez and C. Chipot, J. Chem. Inf. Model., 2016, 56, 1122–1126 CrossRef CAS PubMed.
- Y. Yan, M. Yang, C. G. Ji and J. Z. Zhang, J. Chem. Inf. Model., 2017, 57, 1112–1122 CrossRef CAS PubMed.
- C. A. Smith and T. Kortemme, J. Mol. Biol., 2008, 380, 742–756 CrossRef CAS PubMed.
- M. Petukh, M. Li and E. Alexov, PLoS Comput. Biol., 2015, 11, 1–23 CrossRef PubMed.
- P. Loffler, S. Schmitz, E. Hupfeld, R. Sterner, R. Merkl and M. Hughes, PLoS Comput. Biol., 2017, 13, e1005600 CrossRef PubMed.
- L. X. Peterson, X. Kang and D. Kihara, Proteins: Struct., Funct., Bioinf., 2014, 82, 1971–1984 CrossRef CAS PubMed.
- E. Feyfant, A. Sali and A. Fiser, Protein Sci., 2007, 16, 2030–2041 CrossRef CAS PubMed.
- Y. Dehouck, J. M. Kwasigroch, M. Rooman and D. Gilis, Nucleic Acids Res., 2013, 41, 333–339 CrossRef PubMed.
- M. Li, F. L. Simonetti, A. Goncearenco and A. R. Panchenko, Nucleic Acids Res., 2016, 44, W494–W501 CrossRef CAS PubMed.
- J. D. Chodera and F. Noé, Curr. Opin. Struct. Biol., 2014, 25, 135–144 CrossRef CAS PubMed.
- E. Verschueren, P. Vanhee, F. Rousseau, J. Schymkowitz and L. Serrano, Structure, 2013, 21, 789–797 CrossRef CAS PubMed.
- S. Piana, J. L. Klepeis and D. E. Shaw, Curr. Opin. Struct. Biol., 2014, 24, 98–105 CrossRef CAS PubMed.
- K. Misura and D. Baker, Proteins: Struct., Funct., Bioinf., 2005, 59, 15–29 CrossRef CAS PubMed.
- C. J. Camacho, Proteins: Struct., Funct., Bioinf., 2005, 60, 245–251 CrossRef CAS PubMed.
- S. Kannan and M. Zacharias, PLoS One, 2014, 9, e88383 CrossRef PubMed.
- C. K. Vaughan, A. M. Buckle and A. R. Fersht, J. Mol. Biol., 1999, 286, 1487–1506 CrossRef CAS PubMed.
- S. H. Sleigh, P. R. Seavers, A. J. Wilkinson, J. E. Ladbury and J. R. Tame, J. Mol. Biol., 1999, 291, 393–415 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- S. Gras, X. Saulquin, J.-B. Reiser, E. Debeaupuis, K. Echasserieau, A. Kissenpfennig, F. Legoux, A. Chouquet, M. Le Gorrec, P. Machillot, B. Neveu, N. Thielens, B. Malissen, M. Bonneville and D. Housset, J. Immunol., 2009, 183, 430–437 CrossRef CAS PubMed.
- J. Phan, Z. Li, A. Kasprzak, B. Li, S. Sebti, W. Guida, E. Schönbrunn and J. Chen, J. Biol. Chem., 2010, 285, 2174–2183 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, Phys. Rev. Lett., 1980, 45, 1196–1199 CrossRef CAS.
- M. Di Pierro, R. Elber and B. Leimkuhler, J. Chem. Theory Comput., 2015, 11, 5624–5637 CrossRef CAS PubMed.
- D. Janežič and F. Merzel, J. Chem. Inf. Comput. Sci., 1995, 35, 321–326 CrossRef.
- P. Cossio, A. Laio and F. Pietrucci, Phys. Chem. Chem. Phys., 2011, 13, 10421 RSC.
- M. A. Marti-Renom, A. C. Stuart, R. Sanchez, F. Melo and A. Sali, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325 CrossRef CAS PubMed.
- G. G. Krivov, M. V. Shapovalov and R. L. Dunbrack, Proteins: Struct., Funct., Bioinf., 2009, 77, 778–795 CrossRef CAS PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 CAS.
- J. Schymkowitz, J. Borg, F. Stricher, R. Nys, F. Rousseau and L. Serrano, Nucleic Acids Res., 2005, 33, 382–388 CrossRef PubMed.
- P. Cossio, D. Granata, A. Laio, F. Seno and A. Trovato, Sci. Rep., 2012, 2, 351 CrossRef.
- E. Sarti, I. Gladich, S. Zamuner, B. E. Correia and A. Laio, Proteins: Struct., Funct., Bioinf., 2016, 84, 1312–1320 CrossRef CAS PubMed.
- M. A. Soler, S. Fortuna, A. de Marco and A. Laio, Phys. Chem. Chem. Phys., 2018, 20, 3438–3444 RSC.
- L. Holm and C. Sander, J. Mol. Biol., 1993, 233, 123–138 CrossRef CAS PubMed.
- P. Conway, M. D. Tyka, F. DiMaio, D. E. Konerding and D. Baker, Protein Sci., 2014, 23, 47–55 CrossRef CAS PubMed.
- C. H. U. Wang and O. R. a. Schueler-furman, Protein Sci., 2005, 14, 1328–1339 CrossRef CAS PubMed.
- B. Raveh, N. London and O. Schueler-Furman, Proteins: Struct., Funct., Bioinf., 2010, 78, 2029–2040 CAS.
- S. Wolfe, Acc. Chem. Res., 1972, 5, 102–111 CrossRef CAS.
- R. L. Dunbrack, Curr. Opin. Struct. Biol., 2002, 12, 431–440 CrossRef CAS PubMed.
- C. M. Ayres, S. A. Corcelli and B. M. Baker, Front. Immunol., 2017, 8, 935 CrossRef PubMed.
- I. Antes, Proteins: Struct., Funct., Bioinf., 2010, 78, 1084–1104 CrossRef CAS PubMed.
- R. L. Dunbrack and M. Karplus, Nat. Struct. Biol., 1994, 1, 334–340 CrossRef CAS PubMed.
- P. Wang, J. Sidney, C. Dow, B. Mothé, A. Sette and B. Peters, PLoS Comput. Biol., 2008, 4, e1000048 CrossRef PubMed.
- R. F. Alford, A. Leaver-Fay, J. R. Jeliazkov, M. J. O’Meara, F. P. DiMaio, H. Park, M. V. Shapovalov, P. D. Renfrew, V. K. Mulligan, K. Kappel, J. W. Labonte, M. S. Pacella, R. Bonneau, P. Bradley, R. L. Dunbrack, R. Das, D. Baker, B. Kuhlman, T. Kortemme and J. J. Gray, J. Chem. Theory Comput., 2017, 13, 3031–3048 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- N. Tokuriki, F. Stricher, J. Schymkowitz, L. Serrano and D. S. Tawfik, J. Mol. Biol., 2007, 369, 1318–1332 CrossRef CAS PubMed.
- J. M. Word, S. C. Lovell, J. S. Richardson and D. C. Richardson, J. Mol. Biol., 1999, 285, 1735–1747 CrossRef CAS PubMed.
- N. Alam, L. Zimmerman, N. A. Wolfson, C. G. Joseph, C. A. Fierke and O. Schueler-Furman, Structure, 2016, 24, 458–468 CrossRef CAS PubMed.
- P. Vlieghe, V. Lisowski, J. Martinez and M. Khrestchatisky, Drug Discovery Today, 2010, 15, 40–56 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03826k |
| This journal is © the Owner Societies 2018 |