
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hidden magnetic order in plutonium dioxide nuclear fuel†
James T.
Pegg
 *ab,
Ashley E.
Shields
c,
Mark T.
Storr
b,
Andrew S.
Wills
a,
David O.
Scanlon
ade and
Nora H.
de Leeuw
af
*ab,
Ashley E.
Shields
c,
Mark T.
Storr
b,
Andrew S.
Wills
a,
David O.
Scanlon
ade and
Nora H.
de Leeuw
af
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: pegg.james.t@gmail.com
bAtomic Weapons Establishment (AWE) Plc, Aldermaston, Reading RG7 4PR, UK
cOak Ridge National Laboratory, One Bethel Valley Road, Oak Ridge, Tennessee 37831, USA
dDiamond Light Source Ltd., Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
eThomas Young Centre, University College London, Gower Street, London WC1E 6BT, UK
fCardiff University, School of Chemistry, Main Building, Park Place, Cardiff, CF1D 3AT, UK
First published on 26th July 2018
Abstract
A thorough understanding of the chemistry of PuO2 is critical to the design of next-generation nuclear fuels and the long-term storage of nuclear materials. Despite over 75 years of study, the ground-state magnetic structure of PuO2 remains a matter of much debate. Experimental studies loosely indicate a diamagnetic (DM) ground-state, whereas theoretical methods have proposed either a collinear ferromagnetic (FM) or anti-ferromagnetic (AFM) ground-state, both of which would be expected to cause a distortion from the reported Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry. In this work, we have used accurate calculations based on the density functional theory (DFT) to systematically investigate the magnetic structure of PuO2 to resolve this controversy. We have explicitly considered electron-correlation, spin–orbit interaction and noncollinear magnetic contributions to identify a hereto unknown longitudinal 3k AFM ground-state that retains Fmm crystal symmetry. Given the broad interest in plutonium materials and the inherent experimental difficulties of handling this compound, the results presented in this paper have considerable implications for future computational studies relating to PuO2 and related actinide structures. As the crystal structure is coupled by spin–orbit interactions to the magnetic state, it is imperative to consider relativity when creating computational models.
m symmetry. In this work, we have used accurate calculations based on the density functional theory (DFT) to systematically investigate the magnetic structure of PuO2 to resolve this controversy. We have explicitly considered electron-correlation, spin–orbit interaction and noncollinear magnetic contributions to identify a hereto unknown longitudinal 3k AFM ground-state that retains Fmm crystal symmetry. Given the broad interest in plutonium materials and the inherent experimental difficulties of handling this compound, the results presented in this paper have considerable implications for future computational studies relating to PuO2 and related actinide structures. As the crystal structure is coupled by spin–orbit interactions to the magnetic state, it is imperative to consider relativity when creating computational models.
1 Introduction
In the nuclear fuel cycle, PuO2 plays a significant role as a fissile material and a thorough understanding of its fundamental properties is therefore important. To characterise the nature of condensed matter systems, it is imperative to identify coupled and competing magnetic interactions. An accurate description of the underlying physics, including the magnetic ground-state, is key to the design of reliable computational models. A controversy exists between experimental and theoretical methods,1 which affects the calculation of phonon frequencies, thermodynamic properties and excited electronic states,1,2 and has implications for the validation of basic models of reactivity, the design of surface models and the interaction of adsorbents.3 A singlet diamagnetic (DM) ground-state is inferred by experimental measurements and the one-electron interpretation of the crystal electric field,4–8 whereas a ferromagnetic (FM) or an anti-ferromagnetic (AFM) ground-state is calculated by first-principle methods.2,3,9–21 In this paper, the controversy detailed in the literature is addressed by high-end first-principles methods and, for the first time, a fully relativistic noncollinear investigation of magnetic order in PuO2 has been conducted. As a key computational consideration, the spin–orbit interaction (SOI, which couples the lattice structure to the magnetic state) is fully considered in this study.22 A number of experimental measurements on PuO2 report Fmm (no. 225) crystal symmetry (Fig. 1),23 but suggestions of low-temperature (T < 30.8 K) lattice anomalies (reminiscent of transverse 3k AFM behaviour in UO2 with Pa (no. 205) crystal symmetry) remain uninvestigated.24
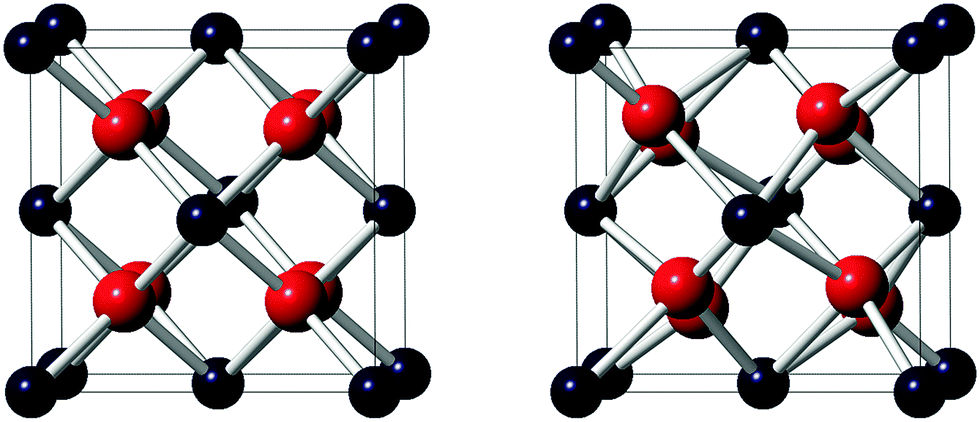 | ||
| Fig. 1 The crystal structure of PuO2 with; (left) Fmm (no. 225) (right) Pa (no. 205) crystal symmetry. The colours in the parentheses indicate the Pu4+ (blue) and O2− (red) ions. Note, in Pa symmetry, the displacement of the O2− ions is exaggerated. | ||
The low-temperature characterisation of the PuO2 magnetic ground-state is challenging, due to: nucleonic radioactive decay,25 the toxicity of the metals,26 and the inhomogeneity of samples.7 A number of inconsistencies in the literature have been recorded.27 For example, a singlet Γ1 DM ground-state (in accordance with the one-electron crystal-field model) has been inferred by: magnetic susceptibility (T = 4 K),4 inelastic neutron scattering (INS) (T > 30 K),5,6 and nuclear magnetic resonance (NMR) (T > 4 K)7,8 measurements. Although a number of inconsistencies in the literature have been recorded.27 A singlet Γ1 ground-state has been inferred from the temperature-independent magnetic susceptibility of PuO2 (5.4 × 10−4 cm3 mol−1).1 In contrast, the temperature-dependent magnetic susceptibility of Th1−xPuxO2 (∼1.1 × 10−3 cm3 mol−1) indicates a magnetic response that is inconsistent with earlier results.28 INS measurements of PuO2 indicate a singlet Γ1 to triplet Γ4 transition of 123 meV, which corresponds to a temperature-dependent magnetic susceptibility of ∼1 × 10−3 cm3 mol−1 when T > 400 K. The magnetic coupling of proximal cations by intermediary DM anions is described by magnetic superexchange.29 The full width at half maximum (FWHM) INS peak results of 25 meV is indicative of AFM superexchange.1,30 In addition, magnetic susceptibility measurements of U1−xPuxO2 indicate AFM superexchange with high concentrations of plutonium.31 Finally, the low-temperature 17O NMR measurements of PuO2 result in an apparent DM state at 6 K and it is conceivable that even lower temperatures may result in a different magnetic ground-state. For instance, low temperature measurements of U1−xPuxO2 indicate a Néel temperature under 5 K when x = 0.5.31 To compensate for experimental issues (thermal energy released by the decay of actinide nuclei), theoretical methods offer an alternative and complementary means of investigation.
A computational investigation of the PuO2 systems is highly complicated. This is due to exchange–correlation effects, relativistic influences and magnetic behaviour. It is now well recognised that in highly correlated f-electron systems, conventional DFT32,33 methods fail to reproduce the correct electronic structure. For instance, identifying finding well-known AnO2 insulators as metallic by underestimating the band-gap.17,21 To calculate highly-correlated materials, a number of methods offer a correction to the self-interaction error. These include the self-interaction correction (SIC) method,34 modified density functional theory (DFT+U),32,33,35–37 dynamic mean field theory (DMFT),38,39 and hybrid density functionals.12,40,41 In condensed matter physics, the DFT+U method offers a computationally tractable means of study, where the on-site Coulomb repulsion of the f-electrons is treated by tuneable U and J modifiers. In contrast, although the use of hybrid functionals are computationally expensive, they represent one of the more accurate methods available.
DFT studies have consistently reported either a collinear ferromagnetic (FM) or a collinear antiferromagnetic (AFM) ground-state.3 The ordered magnetic states have been attributed to approximations inherent to DFT methods.42 In response to the highly-controversial ordered magnetic structures, a number of investigations have focused on the DM solution.42,43 As a combination of the exact diagonalization of the Anderson impurity model and LDA+U (U = 6.5 eV), correlated band theory yielded a DM solution with an f-shell occupation of ∼4.5 at the Pu atoms. The fully spherical symmetric Coulomb interaction, crystal field, and SOI were included in the multiorbital impurity model.43 The authors found an exited triplet state at 126 meV; however, a complete investigation of ordered magnetic states is needed.
In cases where an ordered magnetic state has been incorporated within the DFT methodology, the investigators have limited the discussion to FM(001) and collinear 1k AFM states.2,3,9–21 As the actinide elements are heavy-fermion systems, SOI is imperative for the computational treatment of nuclear materials.44 A limited number of studies have included SOI (due to the high computational cost) in their calculations.18–21,42,43 The inclusion of noncollinear magnetic behaviour PuO2 has been ignored.
In addition to general complications associated with highly-correlated systems, the magnetic ground-state of the AnO2 is influenced by superexchange, multipolar and noncollinear interactions.1,27 Other methods have been used to model PuO2 systems. The complete active space (CAS) wavefunctions to correct the exchange–correlation errors common to DFT can be used to achieve a DM ground-state.45 A limitation of the embedded cluster method, however, is that the quantum cluster may not be sufficiently large enough to capture the superexchange interactions in PuO2, and therefore the findings are incomplete. In addition, the ground-state electronic structure and crystal-field levels of PuO2 have been calculated by DFT in terms of the total energy differences.30 The total energy of the crystal-field states are calculated by the Clebsch–Gordon method from a standard model of rare-earth elements and as a function of their single-electron components. The calculated ground Γ1 state-excited Γ4 state transition of 99 meV compares with the measured INS result of 123 meV.
In the current study, the magnetic structure of PuO2 has been investigated by noncollinear relativistic calculations. We have implemented three levels of theory. These include density functional theory (PBEsol),32,33 DFT with the Hubbard correction (PBEsol+U),32,33,35–37 and hybrid (HSE06) functionals.12,40,41,46 To date, noncollinear contributions to the magnetic ground-state have remained unexplored, and we have therefore investigated the noncollinear interactions illustrated in Fig. 2. To highlight the importance of SOI, we have compared with non-SOI HSE06 calculations (see additional information, ESI†).
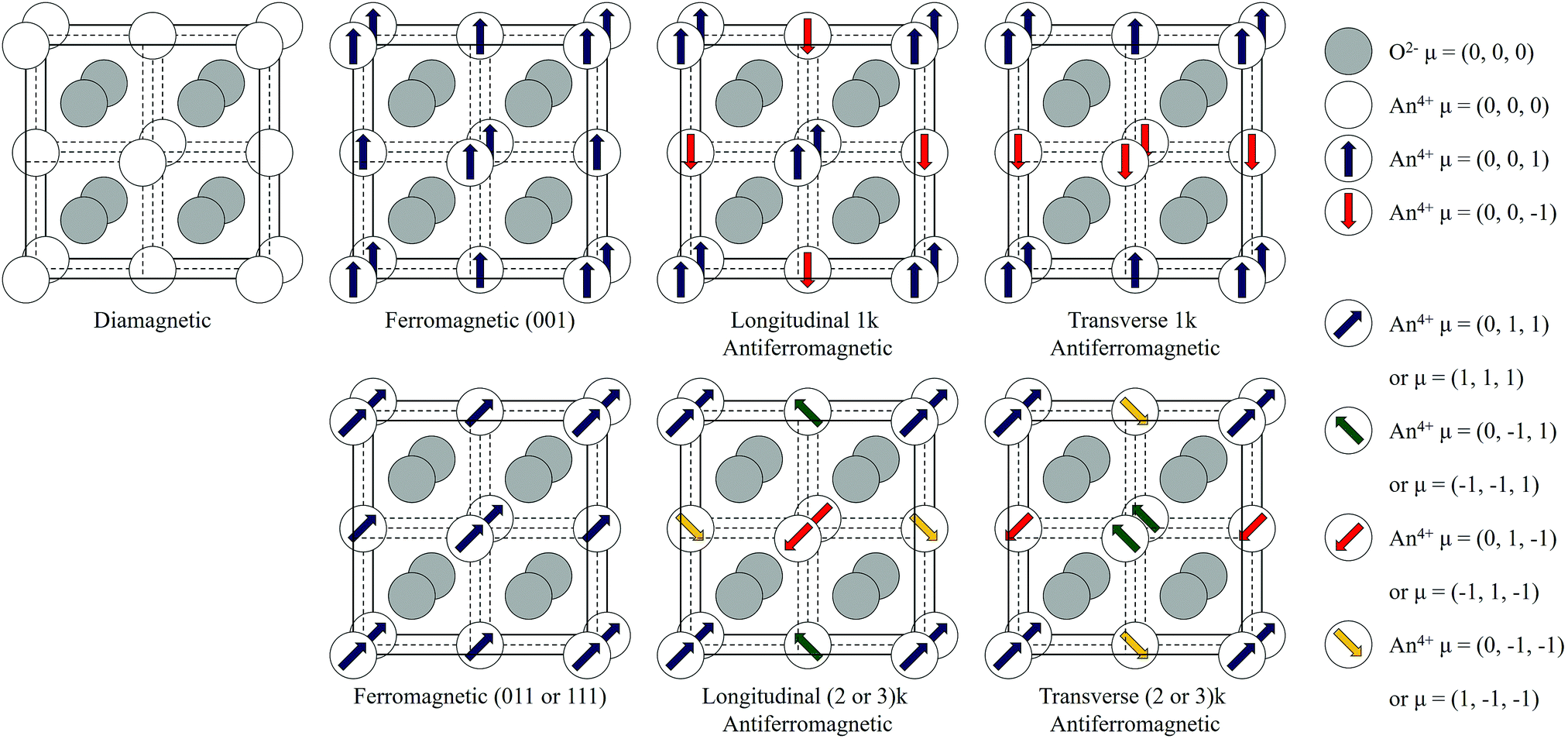 | ||
| Fig. 2 The magnetic structure of PuO2 for the diamagnetic (DM), ferromagnetic (FM) and antiferromagnetic (AFM) states. The direction of the effective magnetic moment (μ) of each ion is described by the μ = (a, b, c) wave-vector matrix. Here, transverse (T) and longitudinal (L) domains are considered for the AFM state. The number of independent wave vectors is denoted by the k prefix. | ||
2 Computational methodology
A noncollinear relativistic computational study of the PuO2 magnetic structure by the Vienna Ab initio Simulation Package (VASP) code has been conducted.34,38,47 The code employs relativistic effective core potentials (ECPs), the frozen-core projector-augmented wave (PAW) method and a planewave basis set. The cut-off energy of the planewave basis set is 500 eV. The cut-off energy and k-point mesh have been checked for convergence. The following explicit electrons for each atomic species are considered: O (1s22s22p2) and Pu (5f56s26p66d17s2). The ion–electron interaction was described using the projector-augmented wave (PAW)41,48 approximation, considering spin–orbit interactions (SOI)49 and noncollinear magnetic wave vectors. The spin quantisation axis is defined by (0, 0, 1) plane, from which magnetic and spinor-like values are calculated. Ionic forces have been evaluated using the Hellmann–Feynman theorem50 and conjugate gradient algorithm.51 The space group has been evaluated to 10−5 Å based on a symmetry analysis of the unit cell.As a computationally intensive method, hybrid functionals incorporate Hartree–Fock (HF) exchange energy into the DFT formulism. In this study, the hybrid Heyd–Scuseria–Ernzerhof (HSE06) functional is employed.12,40,41,46 The functional offers improved: thermochemical data, band-gaps (for low-medium semi-conductors) and lattice constants.
| EHSEXC = (a)EHF,SRX(μ) + (1 − a)EPBE,SRX(μ) + EPBE,LRX(μ) + EPBEC. | (1) |
As a computationally more tractable means of investigation, the DFT+U32,33,35–37 method addresses the on-site Columbic repulsion of the Pu f-electrons. The correction of the electron self-interaction error (SIE) implicit in DFT calculations, is treated by the rotationally invariant Liechtenstein et al.36 formulism. In the rotationally invariant Liechtenstein et al.36 formulism, the on-site Coulombic (U) and exchange (J) modifiers are treated as independent variables.
 | (2) |
In the past, the influence of J on noncollinear magnetic materials has been investigated.44,54 The introduction of J increased the anisotropic nature of the f-states.42 As such, in the current study, the J modifier is kept at a constant value of 0.00 eV.44 By comparison, when J = 0.00 eV, the Dudarev et al.35 formulism and Liechtenstein et al.36 formulism are equivalent.54 The integration of reciprocal space was completed with a Γ-centred k-point (5·5·5) mesh52 by Blöchl tetrahedron method.53 The iteration threshold for electronic and ionic convergence is 1 × 10−8 eV and 1 × 10−3 eV Å−1, respectively. The DFT32,33 and DFT+U35–37 calculations are performed with PBE revised for solids (PBEsol)55 exchange–correlation functional. In the calculation of actinide materials, the functional has shown improved results.44 For the optical absorbance calculation, the k-point mesh is 15 × 15 × 15. For the band structure calculations, the Fmm k-point pathway of high symmetry points in the Brillion zone is defined as Γ → L → W → X → Γ. The density of states and band structure have been illustrated using the SUMO code, a command-line plotting tool for ab initio calculations.56
2.1 Noncollinear magnetic behaviour
In the high-temperature PM state, the AnO2 crystallises in the calcium fluorite (CaF2) motif with Fmm (no. 225) cubic symmetry. In this structure, the An4+ cations occupy octahedral (4a) sites; whereas, the O2− anions occupy tetrahedral (8c) sites. As the magnetic planes are indistinct, the implicit PM disorder stabilizes the high symmetry Fmm crystal structure. At low-temperature, the AnO2 exhibit either DM or AFM behaviour. In addition, multiple first-principle methods calculate a FM ground-state under certain constraints. In the DM and FM states, the implications of noncollinearity are irrelevant. In the AFM state, noncolinear contributions result in 1k (one wave-vector), 2k (two independent wave-vectors) or 3k (three independent wave-vectors) configurations. These configurations are further differentiated when considering longitudinal and transverse domains (Table 1).
| Ion | Ferromagnetic | Antiferromagnetic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Longitudinal domain | Transverse domain | ||||||||
| (001) | (011) | (111) | 1k | 2k | 3k | 1k | 2k | 3k | |
| (0, 0, 0) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) |

|
(0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, 1) | (0, ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , 1) , 1) |
(, , 1) |
(0, 0, ) |
(0, , ) |
(1, , ) |

|
(0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, ) |
(0, 1, ) |
(, 1, ) |
(0, 0, 1) | (0, , 1) |
(, , 1) |

|
(0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, ) |
(0, , ) |
(1, , ) |
(0, 0, ) |
(0, 1, ) |
(, 1, ) |
In the Russell–Saunders coupling scheme, under the cubic crystal-field, the degeneracy of the 5I4 multiplet is split into ascending Γ1 (1), Γ4 (3), Γ3 (2), and Γ5 (3) crystal-field levels (the number in the parentheses denotes the degeneracy).30,42,43 The calculated singlet Γ1 ground-state to triplet Γ4 excited state transition is 123 meV as measured by neutron inelastic scattering.6 The crystal-field model is technically only valid for the one-electron case; nevertheless, the DM state inferred by experiment is often justified by crystal-field theory.
The majority of studies on the AnO2 are often limited by the computational expense of the systems and restricted to discussions of collinear 1k magnetism.2,3,9–21,57 In general, the actinide dioxides (AnO2) exhibit low-temperature noncollinear 3k AFM or PM behaviour (Table S1, ESI†).7,24,58–64 The low-temperature magnetic state can result in a distortion of the crystal structure.1,27,44,65 For instance, in uranium dioxide (UO2) transverse 3k AFM order results in Pa symmetry; whereas, in neptunium dioxide (NpO2) longitudinal 3k AFM order preserves Fmm symmetry. As the crystal structure is coupled by spin–orbit interactions (SOI) to the magnetic state, it is imperative to incorporate relativistic influences when creating computational models.
3 Results & discussions
3.1 Magnetic structure
In the current study, the magnetic structure of PuO2 has been investigated by first-principle methods. The magnetic state clearly influences: relative energetics, band structure, magnetic moment, and crystal symmetry. In our HSE06 and PBEsol+U calculations, the DM state of PuO2 is the highest energy magnetic configuration. In each instance, a longitudinal 3k AFM ground-state with Fmm cubic symmetry has been calculated. In the absence of SOI, a degenerate FM(011)–FM(111) ground-state has been calculated (see additional information, Table A1, ESI†). The result highlights the need (when modelling PuO2) to consider: relativistic effects, exchange–correlation, and noncollinear magnetism. In our hybrid (HSE06) calculations (compared to the DM configuration) the ordered FM and AFM configurations are ∼1.01 eV per F.U. (formula unit) lower in energy (Table 2). The finding is inconsistent with the DM ground-state indicated by current experimental data.4–8
| Magnetic configuration | Relative energy (eV per F.U.) | Band-gap (eV) | Magnetic moment (μB/Pu ion) | Lattice volume (Å3) | Space group (number) | |
|---|---|---|---|---|---|---|
| Diamagnetic | 1.069 | 2.92 | 0.00 | 155.37 |
Fmm (no. 225) |
|
| Ferromagnetic | (001) | 0.074 | 1.91 | 3.66 | 155.88 | I4/mmm (no. 139) |
| (011) | 0.037 | 1.79 | 3.68 | 156.03 | Immm (no. 71) | |
| (111) | 0.027 | 1.68 | 3.68 | 155.99 |
Rm (no. 166) |
|
| Antiferromagnetic | ||||||
| Longitudinal | 1k | 0.058 | 2.83 | 3.63 | 155.69 | I4/mmm (no. 139) |
| 2k | 0.011 | 3.05 | 3.65 | 155.65 | I4/mmm (no. 139) | |
| 3k | 0.000 | 3.04 | 3.65 | 155.65 |
Fmm (no. 225) |
|
| Transverse | 1k | 0.062 | 2.50 | 3.64 | 155.64 | Fmmm (no. 69) |
| 2k | 0.020 | 2.93 | 3.66 | 155.73 | Pbca (no. 61) | |
| 3k | 0.009 | 2.68 | 3.66 | 155.72 |
Pa (no. 205) |
|
| Experimental | — | 2.8066 | 0.004–8 | 157.2523 |
Fmm (225)23 |
|
It is noted that the experimental optical band-gap (2.80 eV) has been measured by epitaxial thin film absorbance.66 As the band-gap is influenced by the magnetic state, the experimental PM (high-temperature, T ∼ 298 K) result and computational longitudinal 3k AFM result (low-temperature, T ∼ 0 K) cannot be directly compared. In addition, the magnetic structure is coupled by SOI to the crystal symmetry. In our calculations, the DM and longitudinal 3k AFM states retain Fmm crystal symmetry, only. The collinear 1k AFM states (used in past studies) calculate an incorrect crystal structure. A tetragonal I4/mmm (no. 139) (longitudinal 1k AFM) or orthorhombic Fmmm (no. 69) (transverse 1k AFM) crystallographic distortion results.2,3,9–21 This inconsistent with experimental results. In addition, transverse 3k AFM order results in Pa crystal symmetry (as observed in UO2 and AmO2).44
As a more computationally tractable method of theory (compared with the hybrid functionals), the results obtained from PBEsol+U are reported for different values of U (Fig. 3). In an identical manner to hybrid (HSE06) calculations, the magnetic state influences the electronic and crystallographic structure. PBEsol (U = 0 eV) calculates a degenerate metallic FM (111), (011), (001) ground-state; however, the metallic ground-state is inconsistent with the known insulating nature. The result exemplifies the failure of conventional DFT methods with highly-correlated systems. The band-gap is only replicated by the AFM and DM states when U = 5–7 eV.66 In particular, the band-gap of the experimental DM state and theoretical longitudinal 3k AFM ground-state is reproduced when U = 6.35 eV and U = 6.00 eV, respectively. In contrast, when calculating the band-gap for the FM states, at every value of U, the band-gap is underestimated relative to experimental values.
 | ||
| Fig. 3 The relative ground-state energies, band-gaps, and effective magnetic moments against the Coulomb modifier (U) for diamagnetic (DM), ferromagnetic (FM), and antiferromagnetic (AFM) states of PuO2, calculated with PBEsol (U = 0 eV) and PBEsol+U. The antiferromagnetic transverse (T) and longitudinal (L) domains are additionally represented. The k-prefix denotes the number of independent wave vectors: (upper row) the calculated energy of magnetic states relative to the longitudinal 3k antiferromagnetic ground-state; (middle row) the direct band-gap; (lower row) the effective Pu magnetic moment. The DM (yellow), FM (red), longitudinal AFM (green) and transverse AFM (blue) states are denoted by the colours in the parentheses. | ||
In experimental studies, DM order has been inferred from the absence of an effective Pu4+ magnetic moment.4–8 In this study, the effective magnetic Pu4+ moment of the ordered FM and AFM states is reported. In the FM states, the effective magnetic moment decreases from 3.85 to 3.75 μB/Pu ion in the U = 0–2 eV range; however, increases from 3.75 μB/Pu ion to 3.83 μB/Pu ion when U = 3–7 eV. In contrast, the effective magnetic moment of the AFM states continually increases from 3.55 μB/Pu ion to 3.80 μB/Pu ion over the range of U = 0–7 eV. However, when U = 6 eV, the effective magnetic moment converges at 3.80 μB/Pu ion for all magnetic states.
3.2 Longitudinal 3k antiferromagnetic ground-state
In this paper, a theoretical longitudinal 3k AFM ground-state for PuO2 has been calculated by PBEsol+U (U = 6.00 eV) and hybrid (HSE06) functionals. The noncollinear magnetic ground-state of UO2, NpO2 (established by experimental means) and PuO2 are highly comparable. The optical absorbance, band structure and density of states for the longitudinal 3k AFM ground-state have been calculated by PBEsol+U (U = 6 eV) (Fig. 4 and Table 3). | ||
| Fig. 4 The electronic structure of the longitudinal 3k antiferromagnetic ground-state of PuO2 calculated by PBEsol+U where U = 6 eV; (left) band structure, (right) density of states. The colours in the parentheses indicate the Pu f- (blue), Pu d- (green) and O p- (red) states. | ||
| DFT+U | Band-gap (eV) | Lattice constant (Å) | Bulk modulus (GPa) | Magnetic moment (μB/Pu ion) | Crystal symmetry | Magnetic configuration | |
|---|---|---|---|---|---|---|---|
| Fundamental | Optical | ||||||
| 6.35 | 2.81 | 2.82 | 5.411 | 217 | 0.00 |
Fmm (225) |
DM44 |
| 6.00 | 2.80 | 2.97 | 5.415 | 215 | 3.80 |
Fmm (225) |
Longitudinal 3k AFM |
| — | — | 2.8066 | 5.395–5.39823 | 178–379 | 0.00 |
Fmm (225) |
Exp. |
A charge-transfer insulator with an indirect bandgap of 2.80 eV has been calculated; this contrasts against the Mott-insulator calculated by the fully relativistic linear combinations of Gaussian-type orbitals-fitting function (LCGTO-FF) method.19 The respective X–Γ valence band maximum (VBM) to L conduction band minimum (CBM) is comprised predominately of O p- and Pu f-states. The degeneracy of the bands in the longitudinal 3k AFM ground-state is perturbed by the presence of magnetic order. This key finding was made possible only through the inclusion of noncollinear and spin–orbit interactions.
The fundamental band-gap (the difference between the ionisation potential and the electron affinity, the valence band maximum (VBM) to conduction band minimum (CBM) transition) and optical band-gap (transitions are restricted by orbital symmetry) are influenced by the magnetic state. In our calculations, the longitudinal 3k AFM electronic structure is in excellent agreement with optical absorbance measurements.66 The direct and optical band-gaps differ by 0.17 eV, although both are in excellent agreement with the experimental band-gap of 2.80 eV (Table 3).66
3.3 Antiferromagnetic superexchange & small-moment systems
The experimental low-temperature structure of PuO2 (not necessarily indicative of DM order) is characterised by the absence of a lattice distortion and of a magnetic moment. The ambient PM, experimental DM ground-state, and theoretical longitudinal 3k AFM ground-state retain Fmm crystal symmetry. The absence of any change in crystal structure between the PM and the longitudinal 3k AFM phase is presumed to have helped the ground-state elude detection. A complete analysis can be conducted by comparing other f-electron materials. The results are rationalised by considering magnetic anomalies in NpO2, URu2Si2, UPt3, and UPd3, which are small-moment materials.1
In the absence of an ordered moment and of noticeable lattice anomalies, a phase-transition could be driven by an ordered parameter no longer invariant under time-reversal symmetry.1 The interaction leading to a phase-transition would have to arise from a purely electronic mechanism, as phonons may only carry interactions between time-reversal-invariant multipoles. A magnetic multipole (for which Kramers' theorem would no longer apply) by AFM superexchange is a candidate for an ordered mechanism.1,27,67 As an indication of superexchange, the magnetic susceptibility measurements for PuO2 and Th1−xPuxO2 are inconsistent.4,28,31 The interaction of the Pu4+ ions in Th1−xPuxO2 is reduced by dilution; this suppresses the superexchange mechanism and increases the magnetic susceptibility.1 In addition, anomalies in the INS measurements of PuO2 have been attributed to Jahn–Teller distortions and superexchange interactions.5,6 As a comparison, a superexchange interaction is theorised to occur in NpO2 where a magnetic octupole has been established.68
In another interpretation, PuO2 may be a small-moment system. This is observed in other actinide materials where the AFM response of URu2Si2 (TN = 17.5 K), UPt3 (TN = 5 K) and UPd3 (TN = 4.5 K) is 0.02–0.03 μB/U ion.1 Indeed, a small-moment magnetic state has been theorised for NpO2 where muon spin rotation measurements of the magnetic moment is 0.06–0.15 μB/Np ion.1,69 The nature of small-moment systems in unclear.1 The moment of a system may be reduced by competing magnetic interactions. In the localised PuO2 f-electron system, the interaction may be Rudermann, Kittel, Kasuya, Yoshida (RKKY) or Kondo in nature. If a small-moment in the PuO2 system exists, establishing the magnetic ground-state is a considerable challenge. In our results, the magnetic moment of the longitudinal 3k AFM ground-state is ∼3.65 μB/Pu ion. In this regard, first-principle DFT methods are known to overestimate small-moment actinide materials.1 It is also conceivable that PuO2 undergoes a second low-temperature phase transition, which is known to occur in other actinide compounds. For instance, the specific heat capacity of UPt3 has two distinct anomalies at TC1 = 0.49 K and TC2 = 0.44 K indicative of changing magnetic order. A detailed low-temperature experimental study of magnetic order in PuO2 is needed.
4 Conclusions
In conclusion, the magnetic structure of PuO2 has been investigated by computational means. As a highly-correlated and heavy-fermion system, the electronic structure of PuO2 is notoriously difficult to calculate. Investigations of surface reactivity and bonding often rely on exact calculations of the electronic structure. A key finding is the impact of transverse and longitudinal domains on the band structure. In the case of collinear 1k AFM order, the electronic structure is significantly different depending on the domain employed, which is important as most investigations on AnO2 do not differentiate between collinear magnetic structurers.In our calculations, the metastable DM state (in contrast with the longitudinal 3k AFM ground-state) is inconsistent with current experimental information. In this paper, we have established a longitudinal 3k AFM ground-state with Fmm crystal symmetry. The fact that this magnetic ground-state has not been experimentally observed may be explained in two possible ways. First, PuO2 may be a small-moment material for which current experimental resolution is insufficient to identify an ordered magnetic state. If this is the case, dynamic mean field-theory (DMFT) may be needed to resolve the magnetic moment. Second, an AFM-DM phase transition may occur outside of the temperature range typically studied in the available literature. In theory a superexchange mechanism might result in a hidden magnetic phase transition. This paper calls for further experimental studies to resolve outstanding issues, in particular low-temperature specific heat capacity and muon spin relaxation measurements are needed.
Furthermore, we have shown how the crystal structure is influenced by the magnetic state. For instance, transverse 3k AFM behaviour results in Pa crystal symmetry,22,44 synonymous with calculations of UO2 and AmO2 when SOI are considered. Finally, the electronic structure of PuO2 is highly influenced by the magnetic state employed. These results have profound implications for investigations of bonding, reactivity, and surface structure. The re-orientation of magnetic vectors in surface calculations is often neglected (the direction of magnetism is usually defined by the z-axis), resulting in incomparable magnetic configurations between low-index surfaces. As the magnetic structure has a clear impact of the energetics, it is imperative that the re-orientation of magnetic vector is accounted for in computational surface investigations.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This research was supported by the UK Engineering & Physical Science Research Council (EPSRC) (grant no. EP/G036675 and EP/K016288) and the Atomic Weapons Establishment (AWE). AES gratefully acknowledges the United States Department of Homeland Security (DHS), Domestic Nuclear Detection Office (DNDO), National Technical Nuclear Forensics Centre (NTNFC) for a Postdoctoral Research Fellowship. NHdL thanks AWE for a William Penney Fellowship. This work made use of the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk), via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202).Notes and references
- P. Santini, R. Lémanski and P. Erdős, Adv. Phys., 1999, 48, 537–653 CrossRef.
- G. Jomard and F. Bottin, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195469 CrossRef.
- D. Gryaznov, E. Heifets and D. Sedmidubsky, Phys. Chem. Chem. Phys., 2010, 12, 12273–12278 RSC.
- G. Raphael and R. Lallement, Solid State Commun., 1968, 6, 383–385 CrossRef.
- S. Kern, C. K. Loong, G. L. Goodman, B. Cort and G. H. Lander, J. Phys.: Condens. Matter, 1990, 2, 1933 CrossRef.
- S. Kern, R. Robinson, H. Nakotte, G. Lander, B. Cort, P. Watson and F. Vigil, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 104 CrossRef.
- Y. Tokunaga, H. Sakai, T. Fujimoto, S. Kambe, R. E. Walstedt, K. Ikushima, H. Yasuoka, D. Aoki, Y. Homma, Y. Haga, T. D. Matsuda, S. Ikeda, E. Yamamoto, A. Nakamura, Y. Shiokawa, K. Nakajima, Y. Arai and Y. Ōnuki, J. Alloys Compd., 2007, 444–445, 241–245 CrossRef.
- H. Yasuoka, G. Koutroulakis, H. Chudo, S. Richmond, D. K. Veirs, A. I. Smith, E. D. Bauer, J. D. Thompson, G. D. Jarvinen and D. L. Clark, Science, 2012, 336, 901–904 CrossRef PubMed.
- X.-D. Wen, R. L. Martin, G. E. Scuseria, S. P. Rudin and E. R. Batista, J. Phys. Chem. C, 2013, 117, 13122–13128 CrossRef.
- I. D. Prodan, G. E. Scuseria and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045104 CrossRef.
- D. A. Andersson, J. Lezama, B. P. Uberuaga, C. Deo and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 024110 CrossRef.
- I. D. Prodan, G. E. Scuseria and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 033101 CrossRef.
- Y. Yang, B. Wang and P. Zhang, J. Nucl. Mater., 2013, 433, 345–350 CrossRef.
- B. Sun, H. Liu, H. Song, G. Zhang, H. Zheng, X. Zhao and P. Zhang, J. Nucl. Mater., 2012, 426, 139–147 CrossRef.
- P. Zhang, B.-T. Wang and X.-G. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 144110 CrossRef.
- D. Gryaznov, S. Rashkeev, E. Kotomin, E. Heifets and Y. Zhukovskii, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 3090–3094 CrossRef.
- I. D. Prodan, G. E. Scuseria, J. A. Sordo, K. N. Kudin and R. L. Martin, J. Chem. Phys., 2005, 123, 014703 CrossRef PubMed.
- X.-D. Wen, R. L. Martin, T. M. Henderson and G. E. Scuseria, Chem. Rev., 2013, 113, 1063–1096 CrossRef PubMed.
- J. Boettger and A. Ray, Int. J. Quantum Chem., 2002, 90, 1470–1477 CrossRef.
- S. A. Moten, R. Atta-Fynn, A. K. Ray and M. N. Huda, J. Nucl. Mater., 2016, 468, 37–45 CrossRef.
- H. Wang and K. Konashi, J. Alloys Compd., 2012, 533, 53–57 CrossRef.
- J. T. Pegg, A. E. Shields, M. T. Storr, A. S. Wills, D. O. Scanlon and N. H. de Leeuw, 2018.
- J. M. Haschke, T. H. Allen and L. A. Morales, Science, 2000, 287, 285–287 CrossRef PubMed.
- L. Desgranges, Y. Ma, P. Garcia, G. Baldinozzi, D. Siméone and H. E. Fischer, Inorg. Chem., 2016, 56, 321–326 CrossRef PubMed.
- M. Noe and J. Fuger, Inorg. Nucl. Chem. Lett., 1974, 10, 7–19 CrossRef.
- E. Ansoborlo, O. Prat, P. Moisy, C. Den Auwer, P. Guilbaud, M. Carriere, B. Gouget, J. Duffield, D. Doizi, T. Vercouter, C. Moulin and V. Moulin, Biochimie, 2006, 88, 1605–1618 CrossRef PubMed , https://chemrxiv.org/articles/Magnetic_Structure_of_UO2_and_NpO2_by_First-Principle_Methods/6859940.
- P. Santini, S. Carretta, G. Amoretti, R. Caciuffo, N. Magnani and G. H. Lander, Rev. Mod. Phys., 2009, 81, 807–863 CrossRef.
- G. A. Candela, C. A. Hutchison and W. B. Lewis, J. Chem. Phys., 1959, 30, 246–250 CrossRef.
- P. W. Anderson, Phys. Rev., 1950, 79, 350–356 CrossRef.
- M. Colarieti-Tosti, O. Eriksson, L. Nordström, J. Wills and M. S. S. Brooks, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 195102 CrossRef.
- D. Kolberg, F. Wastin, J. Rebizant, P. Boulet, G. H. Lander and J. Schoenes, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 214418 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467–R5470 CrossRef.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef.
- A. Georges, G. Kotliar, W. Krauth and M. J. Rozenberg, Rev. Mod. Phys., 1996, 68, 13 CrossRef.
- Q. Yin, A. Kutepov, K. Haule, G. Kotliar, S. Y. Savrasov and W. E. Pickett, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195111 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef.
- M. T. Suzuki, N. Magnani and P. M. Oppeneer, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 195146 CrossRef.
- A. Shick, J. Kolorenč, L. Havela, T. Gouder and R. Caciuffo, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 041109 CrossRef.
- J. T. Pegg, X. Aparicio-Anglès, M. Storr and N. H. de Leeuw, J. Nucl. Mater., 2017, 492, 269–278 CrossRef.
- F. Gendron and J. Autschbach, J. Phys. Chem. Lett., 2017, 673–678, DOI:10.1021/acs.jpclett.6b02968.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- V. I. Anisimov, Strong coulomb correlations in electronic structure calculations, CRC Press, 2000 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- S. Steiner, S. Khmelevskyi, M. Marsmann and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 224425 CrossRef.
- R. P. Feynman, Phys. Rev., 1939, 56, 340–343 CrossRef.
- W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes3rd Edition: The Art of Scientific Computing, Cambridge University Press, 2007 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223 CrossRef.
- E. Bousquet and N. Spaldin, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 220402 CrossRef.
- G. I. Csonka, J. P. Perdew, A. Ruzsinszky, P. H. Philipsen, S. Lebègue, J. Paier, O. A. Vydrov and J. G. Ángyán, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 155107 CrossRef.
- A. M. Ganose, A. J. Jackson and D. O. Scanlon, Sumo: Command-line tools for plotting and analysis of *ab initio* calculations, 2018, https://raw.githubusercontent.com/SMTG-UCL/sumo/master/paper.md.
- G. Jomard, B. Amadon, F. Bottin and M. Torrent, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 075125 CrossRef.
- L. Asprey, F. Ellinger, S. Fried and W. Zachariasen, J. Am. Chem. Soc., 1955, 77, 1707–1708 CrossRef.
- K. Ikushima, S. Tsutsui, Y. Haga, H. Yasuoka, R. E. Walstedt, N. M. Masaki, A. Nakamura, S. Nasu and Y. Ōnuki, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 104404 CrossRef.
- Y. Tokunaga, Y. Homma, S. Kambe, D. Aoki, H. Sakai, E. Yamamoto, A. Nakamura, Y. Shiokawa, R. E. Walstedt and H. Yasuoka, Phys. Rev. Lett., 2005, 94, 137209 CrossRef PubMed.
- Y. Tokunaga, T. Nishi, S. Kambe, M. Nakada, A. Itoh, Y. Homma, H. Sakai and H. Chudo, J. Phys. Soc. Jpn., 2010, 79, 053705 CrossRef.
- J. Faber and G. H. Lander, Phys. Rev. B: Solid State, 1976, 14, 1151–1164 CrossRef.
- J. Faber, G. H. Lander and B. R. Cooper, Phys. Rev. Lett., 1975, 35, 1770–1773 CrossRef.
- D. Mannix, G. H. Lander, J. Rebizant, R. Caciuffo, N. Bernhoeft, E. Lidström and C. Vettier, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 15187–15193 CrossRef.
- R. Caciuffo, G. Amoretti, P. Santini, G. H. Lander, J. Kulda and P. D. V. Du Plessis, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 13892–13900 CrossRef.
- T. M. McCleskey, E. Bauer, Q. Jia, A. K. Burrell, B. L. Scott, S. D. Conradson, A. Mueller, L. Roy, X. Wen and G. E. Scuseria, J. Appl. Phys., 2013, 113, 013515 CrossRef.
- P. Santini and G. Amoretti, Phys. Rev. Lett., 2000, 85, 2188–2191 CrossRef PubMed.
- R. Caciuffo, J. A. Paixão, C. Detlefs, M. J. Longfield, P. Santini, N. Bernhoeft, J. Rebizant and G. H. Lander, J. Phys.: Condens. Matter, 2003, 15, S2287 CrossRef.
- W. Kopmann, F. J. Litterst, H. H. Klauß, M. Hillberg, W. Wagener, G. M. Kalvius, E. Schreier, F. J. Burghart, J. Rebizant and G. H. Lander, J. Alloys Compd., 1998, 271–273, 463–466 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03583k |
| This journal is © the Owner Societies 2018 |