
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systematic analysis of electron energy-loss near-edge structures in Li-ion battery materials†
Motofumi
Saitoh‡
 a,
Xiang
Gao‡
a,
Takafumi
Ogawa
a,
Yumi H.
Ikuhara
a,
Shunsuke
Kobayashi
a,
Craig A. J.
Fisher
a,
Akihide
Kuwabara
ab and
Yuichi
Ikuhara
*ac
a,
Xiang
Gao‡
a,
Takafumi
Ogawa
a,
Yumi H.
Ikuhara
a,
Shunsuke
Kobayashi
a,
Craig A. J.
Fisher
a,
Akihide
Kuwabara
ab and
Yuichi
Ikuhara
*ac
aNanostructures Research Laboratory, Japan Fine Ceramics Center, Nagoya 456-8587, Japan
bNational Institute for Materials Science, Tsukuba, Ibaraki 305-0047, Japan
cInstitute of Engineering Innovation, The University of Tokyo, Tokyo 113-8656, Japan. E-mail: ikuhara@sigma.t.u-tokyo.ac.jp
First published on 24th September 2018
Abstract
Electrical conductivity, state of charge and chemical stability of Li-ion battery materials all depend on the electronic states of their component atoms, and tools for measuring these reliably are needed for advanced materials analysis and design. Here we report a systematic investigation of electron energy-loss near-edge structures (ELNES) of Li-K and O-K edges for ten representative Li-ion battery electrodes and solid-state electrolytes obtained by performing transmission electron microscopy with a Wien-filter monochromator-equipped microscope. While the peaks of Li-K edges are positioned at about 62 eV for most of the materials examined, the peak positions of O-K edges vary within a range of about 530 to 540 eV, and the peaks can be categorised into three groups based on their characteristic edge shapes: (i) double peaks, (ii) single sharp peaks, and (iii) single broad peaks. The double peaks of group (i) are attributable to the d0 electronic configuration of their transition metal ions bonded to O atoms. The origin of the different peak shapes of groups (ii) and (iii) is more subtle but insights are gained using density functional theory methods to simulate O-K ELNES edges of group (ii) material LiCoO2 and group (iii) material LiFePO4. Comparison of their densities of states reveals that in LiCoO2 the Co–O hybrid orbitals are separated from Li–O hybrid orbitals, resulting in a sharp peak in the O-K edge, while Fe–O, Li–O and P–O hybrid orbitals in LiFePO4 partially overlap each other and produce a broad peak.
1 Introduction
Li-ion batteries (LIBs) are widely used in a myriad of applications such as portable devices, electric vehicles and renewable energy storage systems. In an effort to develop superior LIBs, many candidate electrode materials have been investigated in terms of energy density, output voltage, charge–discharge rates and cycle lifetimes.1–3 At the same time, efforts to improve battery safety, compactness and stability have led to the development of solid electrolytes that exhibit good Li-ion conductivities and structural integrity after many charge–discharge cycles when used in all-solid-state LIBs.4At the microscopic level, charging and discharging of LIBs involves diffusion of Li atoms, conduction of electrons and changes in valence states of the transition-metal (TM) atoms. Characterisation of the structural and chemical changes that take place during battery operation is thus of fundamental importance in the development of improved LIB materials.
Electrodes and electrolytes in LIBs typically contain TM and Li cations bonded to anions such as O, S and sometimes F. Energy-loss near-edge structure (ELNES) spectra of these elements, obtained by performing electron energy-loss spectroscopy (EELS) within a transmission electron microscope (TEM), can provide useful data about their energetics and coordination environments with a spatial resolution of a few nanometres.5–12 In this work we focus on a number of actively investigated oxide LIB materials, some of whose EELS spectra have not yet been reported.
TM-L2,3 ELNES spectra, ranging over energies between 450 and 900 eV depending on the element in question and its coordination environment,13 are widely used to characterise TM valence states because TM-L spectra change markedly when the valence state changes.14–16 Similarly, the shapes of O-K ELNES at around 530 eV and Li-K ELNES at around 60 eV depend strongly on the local crystal environments of these atoms and the nature of the interatomic bonding.11,17,18 In these cases, ELNES simulations using density functional theory (DFT) are indispensable for identifying the electronic transitions producing the fine structures observed experimentally.7,11,19,20
In the low-energy region (less than 100 eV) in which Li-K edges appear, high energy resolution is necessary to obtain peak shapes accurately. While measurements with resolutions up to 0.3 eV (as determined by the full width half maximum (FWHM) of the zero-loss peak) can be obtained using cold-FE guns,21 to obtain resolutions better than 0.1 eV requires use of monochromators.22–24 The latter allows Li-K edge spectra to be used to measure Li concentrations and distributions quantitatively with nanoscale resolution, as reported for materials LiCoO2,25,26 LiFePO427,28 and Li2MnO3.29 One drawback of this method, however, is that TM-M2,3 (or TM-N2,3) edges lie near to Li-K edges, making their peaks difficult to deconvolute.
Although measurements of Li-K and O-K edges of many LIB materials have been reported,14,15,30–36 to the best of our knowledge, the dependence of the ELNES spectra on the type of TM, the crystal structure, and the bonding environment has not been studied systematically. This lack of systematic comparison has sometimes led to conflicting results in the literature, and hampers deeper understanding of the relationship between electronic structure, chemical environment and electrochemical properties of these materials.
In this study, using a monochromator-equipped TEM, we carried out a systematic comparison of the effect of different TM elements and crystal environments on Li-K and O-K ELNES spectra for ten typical LIB-related materials. The structure types of each material and the coordination numbers (CN), valence states and d-electron configurations of their component TM elements are listed in Table 1, with representative crystal structures depicted in Fig. 1. The materials examined include six cathode materials, two solid electrolyte materials, one anode material, and one non-conductive material (as reference). The cathode materials include two layer-type materials, LiCoO2 and Li2MnO3; two olivine-type materials, LiFePO4 and LiCoPO4; and two spinel-type materials, LiMn2O4 and LiNi0.5Mn1.5O4. LiCoO2,37,38 LiMn2O439,40 and LiFePO441,42 have been widely investigated and are used commercially. Li2MnO3 is a lithium-excess material with superior capacity to LiCoO2; LiCoPO4 exhibits higher output voltage than LiFePO4,43,44 and LiNi0.5Mn1.5O4 also exhibits higher output voltage than LiMn2O4.45,46 The latter three are being actively researched as next-generation cathode materials. The two solid-state electrolytes examined were perovskite-related materials Li0.33La0.56TiO3 and Li0.14La0.29NbO3. Li site occupancy factors in these materials are low compared to the others, so LiNbO3 was also characterised as a reference material because it shares their perovskite-like structure but with fully occupied crystallographic Li sites. The anode material was spinel-type Li4Ti5O12.
| Composition | Structure type | CN | TM | |||
|---|---|---|---|---|---|---|
| O | Li | TM | ion | d config. | ||
| LiFePO4 | Olivine | 4 | 6 | 6 | Fe2+ | d6 (HS) |
| LiCoPO4 | Olivine | 4 | 6 | 6 | Co2+ | d7 (HS) |
| LiCoO2 | Layered rocksalt | 6 | 6 | 6 | Co3+ | d6 (LS) |
| Li2MnO3 | Layered rocksalt | 6 | 6 | 6 | Mn4+ | d3 |
| LiMn2O4 | Spinel | 4 | 4 | 6 | Mn3+ | d4 |
| Mn4+ | d3 | |||||
| LiNi0.5Mn1.5O4 | Spinel | 4 | 4 | 6 | Ni2+ | d8 |
| Mn4+ | d3 | |||||
| Li4Ti5O12 | Spinel | 4 | 4,6 | 6 | Ti4+ | d0 |
| LiNbO3 | Trigonal perovskite | 4 | ∼12 | 6 | Nb5+ | d0 |
| Li0.33La0.56TiO3 | Layered perovskite | 6 | ∼12 | 6 | Ti4+ | d0 |
| Li0.14La0.29NbO3 | Layered perovskite | 6 | ∼12 | 6 | Nb5+ | d0 |
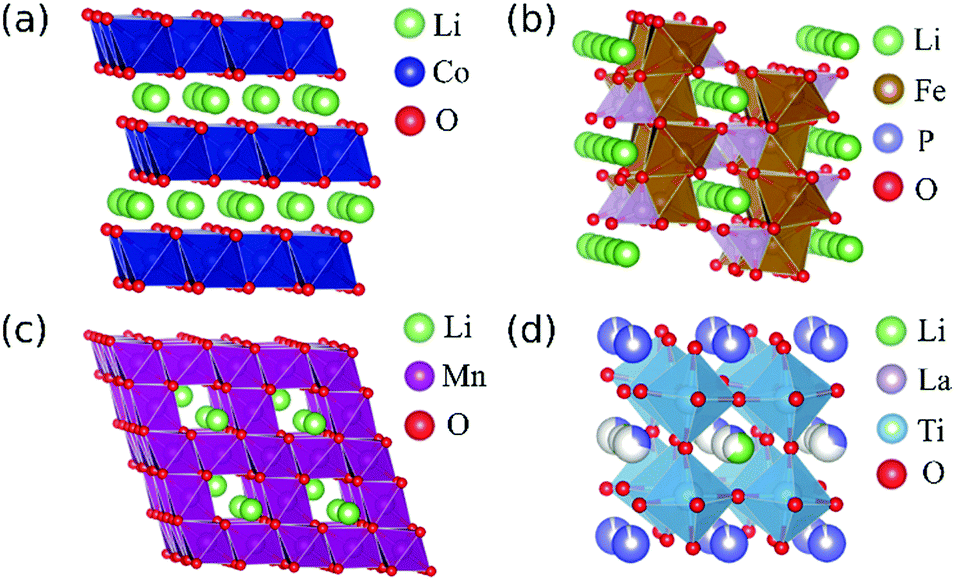 | ||
| Fig. 1 Representative structures of Li-ion battery materials examined in this study: (a) LiCoO2 with layered rocksalt structure, (b) LiFePO4 with olivine structure, (c) LiMn2O4 with spinel structure and (d) LixLa(2−x)/3TiO3 with layered perovskite structure. Multicoloured spheres in (d) represent partially occupied sites. | ||
These materials all exhibit differently shaped EELS spectra, especially in the case of O-K ELNES. To understand the origins of these differences, we carried out DFT-based calculations of LiCoO2 and LiFePO4, which exhibit representative spectrum shapes. Although several theoretical investigations have been reported for both compounds,20,47–49 the reported results are not sufficiently detailed to provide explanations for the differences in ELNES peak positions and shapes. In particular, electronic structures of excited states have not yet been reported, so in this study we performed ELNES simulations of LiCoO2 and LiFePO4 taking into account the electronic structures of the excited state, and compared the results with experimental spectra.
2 Methods
2.1 Specimen preparation
Samples of the ten compounds were prepared by different methods, as described below:X-ray diffraction measurements were used to confirm that materials (1) to (10) were each prepared successfully as a single phase with the appropriate crystal structure under ambient conditions (see Fig. S1 in ESI†).
2.2 EELS measurements
TEM specimens of thin films of LiCoPO4, LiMn2O4 and Li4Ti5O12 were prepared using low-temperature (liquid N2-cooled to below −100 °C) and low-voltage (3.0 kV to 0.2 kV) ion-milling (PIPS, Gatan Inc.). Special care was taken to minimise damage to the samples, and surfaces were cleaned using a plasma cleaner (Solarus, Gatan Inc.) to remove residual amorphous phases. TEM specimens of the other bulk samples were prepared by careful crushing, and the resulting powders dispersed and deposited onto lacey carbon films (above copper substrates).EELS measurements were carried out using a JEM-2400FCS atomic resolution microscope (JEOL Ltd.) equipped with a spherical-aberration corrector (CEOS GmbH), an EELS spectrometer (GIF Tridiem ERS, Gatan Inc.) and a Wien-filter type monochromator. The microscope was installed in a room designed and constructed to minimise AC magnetic fields, floor vibrations, air-flow volume, temperature fluctuations and acoustic vibrations. In this stable environment, a high energy resolution below 30 meV was achieved.24
EELS spectra were recorded with an accelerating voltage of 60 kV to reduce irradiation damage and a convergence/collection semi-angle of 30/35 mrad. The energy resolution, ΔE, specified in terms of the FWHM of the zero-loss peak, was controlled by changing the width of an energy selection slit in the monochromator. To examine the effect of energy resolution on ELNES spectra, we measured Li-K edges of LiFePO4, LiCoPO4, LiCoO2 and Li4Ti5O12 under various slit conditions, viz., no slit, slit of width 4 μm, and slit of width 1 μm, keeping the dispersion value constant at 0.05 eV per pixel. The resulting ΔE were 0.7 eV, 0.35 eV and 0.1 eV, respectively.
Measurements to compare Li-K edges of all materials were performed using a dispersion value, slit width, and measurement exposure time of 0.05 eV per pixel, 1 μm and 0.2 s, respectively. Similarly, for O-K edges, values of 0.1 eV per pixel, 4 μm and 1 s, respectively, were used. The dispersion value of 0.1 eV per pixel used for O-K edge measurements was larger than that for Li-K edges, but is comparable to the natural line width of the O-K edge, 0.12 eV.56 These conditions provided resolutions of ΔE = 0.1 and 0.3 eV for Li-K and O-K edges, respectively. Integration times per measurement for both edges were 10–20 s. Energy alignment was carried out by measuring the zero-loss peak just before and after recording edge spectra. The standard deviation of the zero-loss peak position was 0.03 eV over a time span of 10 minutes (see Fig. S2 in ESI†).
2.3 ELNES simulations
Theoretical O-K ELNES spectra of LiCoO2 and LiFePO4 were generated from first-principles calculations using the DFT+U method including spin polarisation.57 In all calculations, the GGA-PBE functional was used for the exchange-correlation term.58 Values of U of 4.91 eV and 3.71 eV, respectively, were applied to d electrons in Co3+ in LiCoO2 and Fe2+ in LiFePO4.59 In LiFePO4, although the most stable spin configuration is anti-ferromagnetic, the temperature of the anti-ferromagnetic-to-ferromagnetic transition is below room temperature,49 so we assumed LiFePO4 to be in the ferromagnetic state for the calculations.Before generating ELNES spectra, lattice parameters of LiCoO2 and LiFePO4 were determined by performing structural optimisation using the VASP code60 with a planewave cut-off energy of 500 eV. The hexagonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m unit cell (12 atoms) of LiCoO2 was optimised using a 15 × 15 × 3 Brillouin zone k-point mesh, and the orthorhombic Pnma unit cell (28 atoms) of LiFePO4 using a 6 × 9 × 15 k-point mesh.
m unit cell (12 atoms) of LiCoO2 was optimised using a 15 × 15 × 3 Brillouin zone k-point mesh, and the orthorhombic Pnma unit cell (28 atoms) of LiFePO4 using a 6 × 9 × 15 k-point mesh.
O-K ELNES spectra were generated using the core-hole method implemented in the WIEN2k code with an all-electron basis set (APW + lo method).61 In this method, the transition probabilities were calculated using a supercell with an electron removed from the 1s orbital of an O atom (core hole) and placed in the conduction band.62 According to the spin selection rule, we need to consider the probability of each spin state of the excited electron. In the case of an up-spin excited electron, only up-spin orbitals are relevant to the transition calculation, and vice versa for down-spin excited electrons. Because LiCoO2 in its ground state is non-magnetic, the probabilities of up-spin excitation and down-spin excitation are identical, so we only performed calculations for an up-spin excitation. In the case of LiFePO4, although up- and down-spin excitations are not equivalent, our test calculations revealed that the differences between the two excited states are small. The theoretical ELNES spectra of LiFePO4 reported here were generated by averaging contributions of up- and down-spin orbitals from calculation of a down-spin excitation.
Muffin tin radii (RMT) for Li, O, P, Co and Fe were 1.70, 1.60 (LiCoO2)/1.45 (LiFePO4), 1.45, 2.00 and 2.10 bohr, respectively. The cutoff parameter that determines the maximum wave number of plane waves, Rkmax, was set at 7.0, and O-1s states were treated as core levels for both LiCoO2 and LiFePO4. A 4 × 4 × 1 (192 atom) supercell was constructed from the LiCoO2 unit cell and a 1 × 2 × 2 (112 atom) supercell from the LiFePO4 unit cell, with 3 × 3 × 2 and 4 × 3 × 3 k-point meshes, respectively, for integrating over the Brillouin zone. In LiFePO4, as there are three non-equivalent O sites, we calculated the contribution of each type of O separately and took the weighted average when calculating the projected density of states (PDOS) of O-p orbitals and ELNES spectra. The core level of the most distant O atom from the excited O atom in each supercell was used as reference to adjust the PDOS position. The transition energy obtained from the difference in total energies between ground and excited states was used to adjust ELNES edge positions.
3 Results and discussion
3.1 Effect of energy resolution on Li-K ELNES
The effect of different energy resolutions, i.e., ΔE = 0.7, 0.35 and 0.1 eV, on EELS spectra in the low-loss region around Li-K edges was examined for LiFePO4, LiCoPO4, LiCoO2 and Li4Ti5O12 (Fig. 2). As can be seen in Fig. 2(c) and (d), the positions and shapes of the strongest peaks change little with ΔE in the cases of Li4Ti5O12 and LiCoO2. In the case of LiCoPO4, when ΔE = 0.35 eV, a small trough appears at higher energy than the largest peak, around 64 eV, and when ΔE = 0.1 eV, small peaks appear on both sides of this trough as indicated by the blue arrow in Fig. 2(b). The splitting of the peak on the left side of the trough from the largest peak is accompanied by a shift of the largest peak to slightly lower energy, allowing the edge position to be determined with higher accuracy. In the case of LiFePO4, the single peak at 58.2 eV apparent when ΔE = 0.7 eV changes into a main peak and shoulder peak at 57.6 and 58.3 eV, respectively, when ΔE = 0.1 eV, as indicated by the red arrows in Fig. 2(a).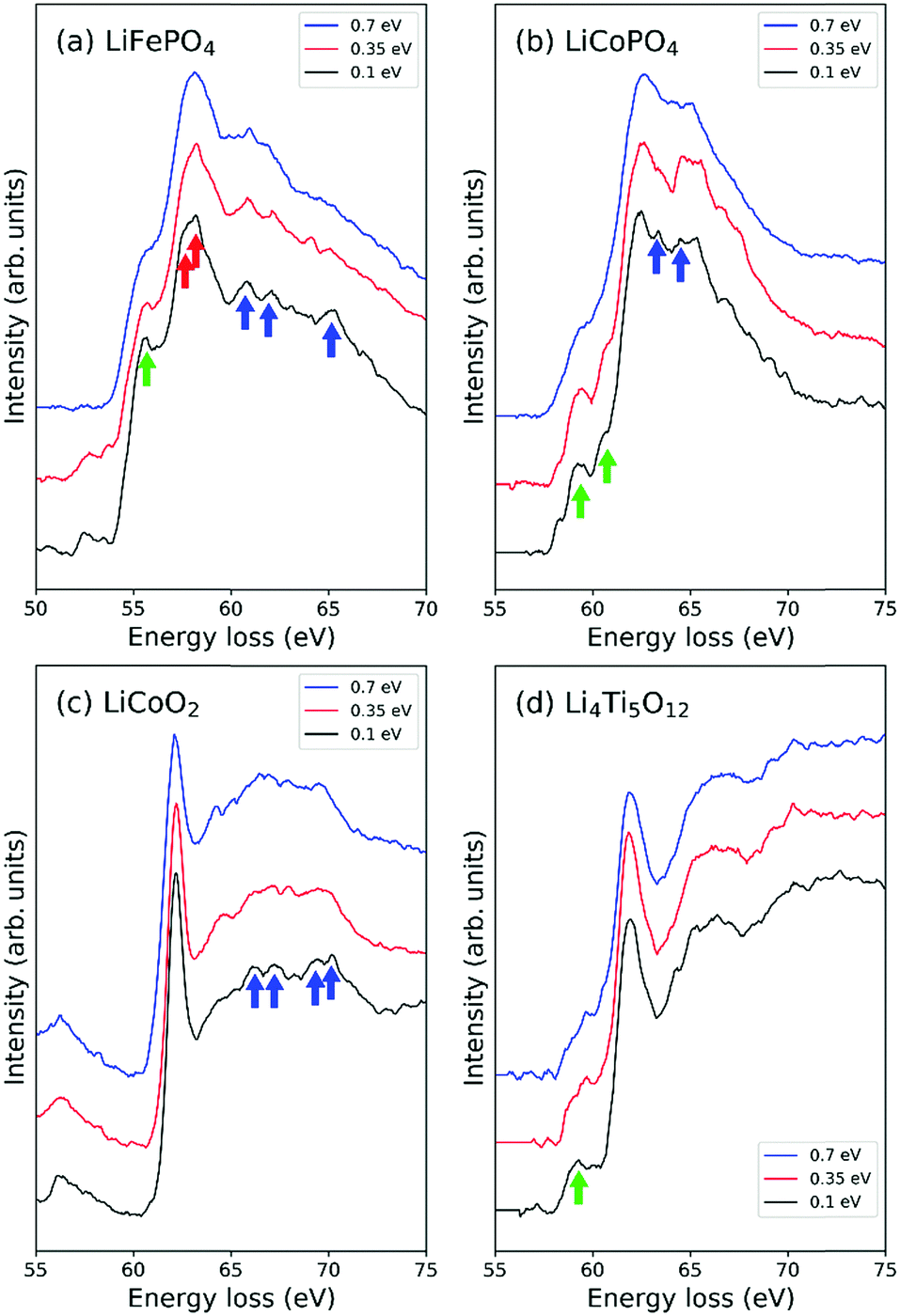 | ||
| Fig. 2 Li-K edges of EELS spectra using energy resolutions, ΔE, of 0.7, 0.35 and 0.1 eV for (a) LiFePO4, (b) LiCoPO4, (c) LiCoO2 and (d) Li4Ti5O12. Fine structures emerging when an energy resolution of 0.1 eV is used are indicated by arrows. Green arrows indicate pre-edge peaks, blue arrows indicate post-edge peaks and red arrows indicate splitting of main edges. | ||
In the cases of LiFePO4, LiCoPO4 and Li4Ti5O12, pre-edge peaks become more distinct with decreasing ΔE as indicated by green arrows in Fig. 2(a), (b) and (d). In LiCoO2 and LiFePO4 small bumps appear in the tails of the largest peaks at higher energies when ΔE ≤ 0.35 eV, as indicated by blue arrows in Fig. 2(a) and (c). Although such peaks are small, they are useful for checking the accuracy of calculated spectra. For example, the spectral shape including tail peaks of LiCoO2 has been verified by DFT calculations using the Z + 1 method by Kikkawa et al.30 In the case of LiFePO4, while the largest peak in Fig. 2(a) at around 58 eV corresponds to the Fe-M2,3 edge, the peaks above 60 eV indicated by the blue arrows are thought to be components of the Li-K edge because it has been reported that the intensities of these peaks decrease with decreasing Li content.32 In this case, performing EELS measurements with high resolution becomes critically important as such features may otherwise be missed.
3.2 Comparison of Li-K ELNES
Fig. 3(a) shows experimentally obtained ELNES spectra for all materials measured with a resolution of ΔE = 0.1 eV. The main peak positions of TM-M2,3 (or N2,3 for Nb) edges and Li-K edges are indicated by grey and black inverted triangles, respectively. The weak peaks assumed to be Li-K edges in the spectra of Li0.14La0.29NbO3 and Li0.33La0.56TiO3 are indicated by open inverted triangles. Fig. 3(b) shows a magnified view of the energy region around the Li-K edges after background subtraction. TM-M2,3 (or N2,3 for Nb) edges have also been subtracted, except for the Fe-M2,3 edge for LiFePO4. Table 2 compares these peak positions with data available in the literature.13,14,30,32–34,36 Agreement to within 1 eV was obtained in all cases.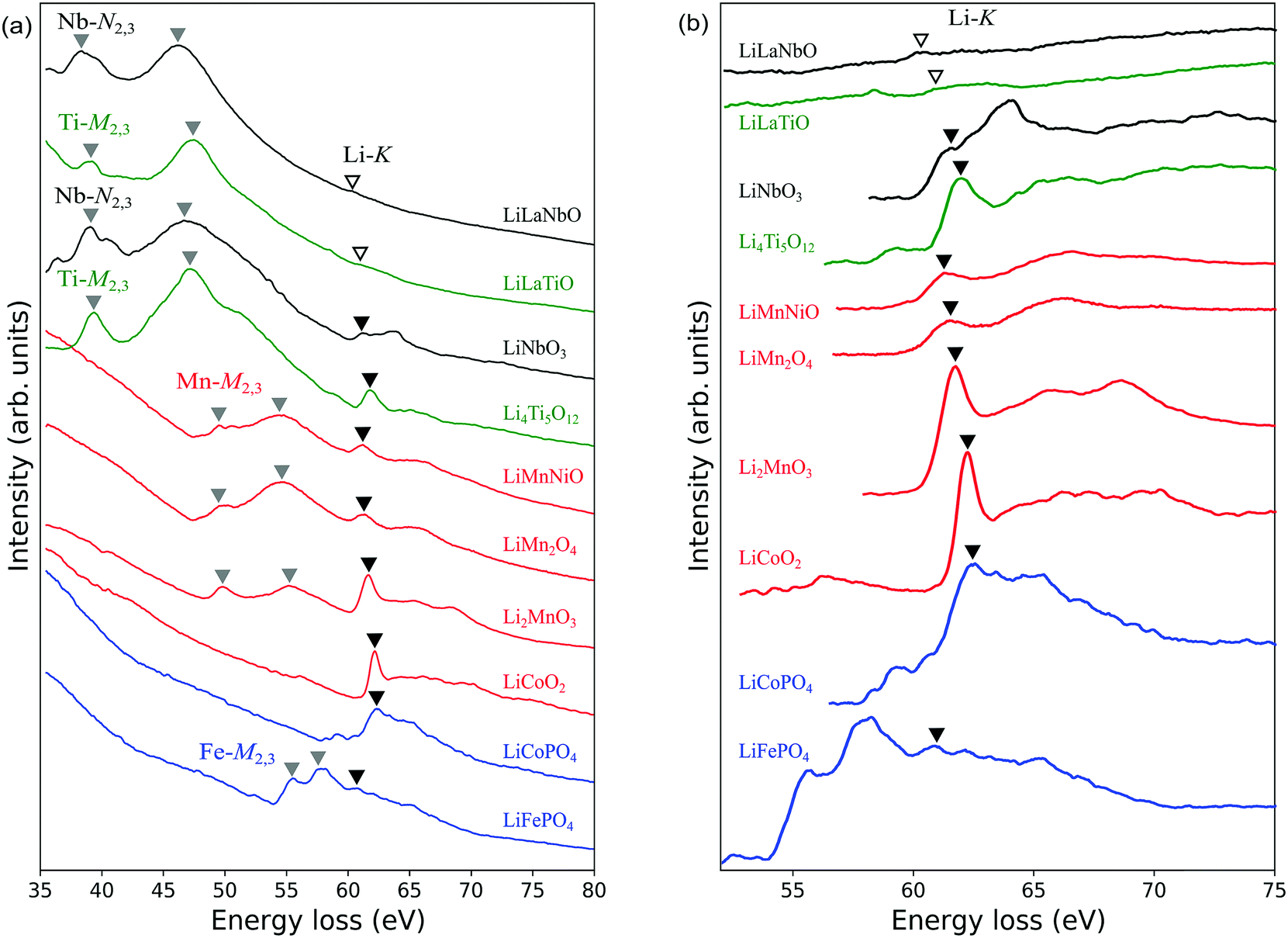 | ||
| Fig. 3 EELS spectra of Li-ion battery materials: (a) comparison of total spectra from 10 materials; (b) magnified view of Li-K edges in the energy range 52 to 75 eV after background subtraction. LiLaNbO, LiLaTiO and LiMnNiO stand for Li0.14La0.29NbO3, Li0.33La0.56TiO3 and LiMn1.5Ni0.5O4, respectively. Peaks of the TM edges (M2,3 for Mn, Fe, Co, Ni and Ti; N2,3 for Nb) and the main peaks of Li-K edges are indicated by grey and black inverted triangles, respectively. Open inverted triangles in the case of LLNO and LLTO indicate weak peaks assumed to be Li-K edges. The tails of Mn/Fe-M2,3 edges overlap Li-K edges, and Co/Ni-M2,3 edges merge completely with Li-K edges. | ||
| Composition | TM-M,N edges (eV) | Li-K edge (eV) | ||
|---|---|---|---|---|
| This work | Previous work | This work | Previous work | |
| a Ref. 20. b Ref. 36. c Ref. 14. d Ref. 33. e Ref. 51. f Ref. 30. g Ref. 34. | ||||
| LiFePO4 | 55.9, 58.3 | 55.5, 57.4a | 60.9 | 60.3a |
| LiCoPO4 | — | 62.4 | 63.0e | |
| LiCoO2 | — | 62.2 | 62.0f | |
| Li2MnO3 | 50.0, 55.5 | 50.4, 55.4b | 61.7 | 62.6b |
| LiMn2O4 | 50.2, 54.9 | 49.5, 54.5c | 61.4 | 60.6c |
| LiNi0.5Mn1.5O4 | 49.7, 54.6 | 61.2 | ||
| Li4Ti5O12 | 39.3, 47.3 | 39.5, 47.3d | 61.8 | 61.6d |
| LiNbO3 | 39.0, 46.6 | 61.3 | ||
| Li0.33La0.56TiO3 | 39.2, 47.4 | (58.8) | 59.1g | |
| Li0.14La0.29NbO3 | 38.4, 46.4 | (60.4) | ||
TM-M2,3 (Nb-N2,3) edges are known to correspond to transitions from 3p3/2,1/2 (or 4p3/2,1/2) core states to the unoccupied conduction bands. A systematic comparison of the spectra reveals peak positions shift to higher energies as the atomic number increases. This trend reflects the shift to lower core energy levels with increasing atomic number.
In LiNi0.5Mn1.5O4, LiFePO4, LiCoPO4 and LiCoO2, the M2,3 edges of Ni, Fe and Co are close to and partially overlap the Li-K edges, complicating detection of the latter. As mentioned in the previous section, in the case of LiFePO4, we cannot assign the weak protrusion at around 62 eV to the Li-K edge without comparison with measurements from delithiated samples. Similarly, the Li-K edge in LiCoO2 has been identified by comparison with spectra from delithiated samples.30 In the case of LiCoPO4, no such data are available, so we here assume the peak at around 62 eV to be the Li-K edge, similar to the case for LiCoO2. The Ni-M2,3 edge of LiNi0.5Mn1.5O4 is assumed to occur at energies higher than the Li-K edge by several electron-volts because in Ni metal it appears at 68 eV.13 Based on this, the small protrusion at 61.9 eV to the right of the Mn-M2,3 edges was assigned to the Li-K edge.
In the magnified spectra of Li0.14La0.29NbO3 and Li0.33La0.56TiO3, very slight peaks are visible at around 60 eV. However, it is difficult to distinguish these Li-K edges from Nb-N1 and Ti-M1 edges which also exist at around 60 eV,34 so it is difficult to identify the Li-K edges with certainty. The chief reason for this is the lower Li concentrations in these solid electrolytes compared to the other materials; the volume densities of Li atoms in these two materials are approximately 2.3 and 5.7 atoms per nm3, respectively, while those of the other materials are between 13 and 40 atoms per nm3.
Previous studies of other Li-containing materials by ELNES or XANES showed that the positions of Li-K edges can vary by more than 5 eV depending on the particular chemical environment of the Li atoms, as reflected in the Mulliken electronegativities or oxidation numbers of neighbouring atoms.18,63 In our work, the main Li-K peaks were observed to lie at around 61–62 eV for all materials except Li0.33La0.56TiO3 and Li0.14La0.29NbO3. This suggests that, although there are differences between the compounds, such as the CN of Li, Li–O bond lengths, and the types of second nearest neighbour cations, the positions of the main peaks are only slightly affected by the local environments around Li.
For some materials, the fine structures of Li-K edges contain small pre-edge peaks and/or sub-peaks above the main peak. These structures are expected to contain valuable information about the chemical bonding states in these compounds. It remains difficult, however, to extract these details because of the overlap with TM-M2,3 edges mentioned above. Furthermore, although DFT calculations can help explicate these fine structures, simulation of a Li-K edge requires a calculation scheme that includes electron-hole coupling effects, e.g., the Bethe-Salpeter equation (BSE) method,64 which is beyond the scope of the present work.
3.3 O-K ELNES
Fig. 4 shows O-K ELNES spectra of the LIB materials examined in this study obtained with ΔE = 0.3 eV. The peak positions occur at energies between 530 eV and 550 eV. Based on the peak shapes, the materials can be categorised into the following three groups: (i) Li0.14La0.29NbO3, Li0.33La0.56TiO3, LiNbO3 and Li4Ti5O12, with double peaks split by a few electron-volts (indicated by inverted open triangles); (ii) LiCoO2, Li2MnO3, LiMn2O4 and LiNi0.5Mn1.5O4, with a single sharp, strong peak at the edge position (indicated by black inverted triangles); and (iii) LiCoPO4 and LiFePO4, with a single broad peak (indicated by grey inverted triangles). In Table 3, the peak positions are listed for all ten materials and compared with available reported values. In all cases, differences in energy are less than 2 eV.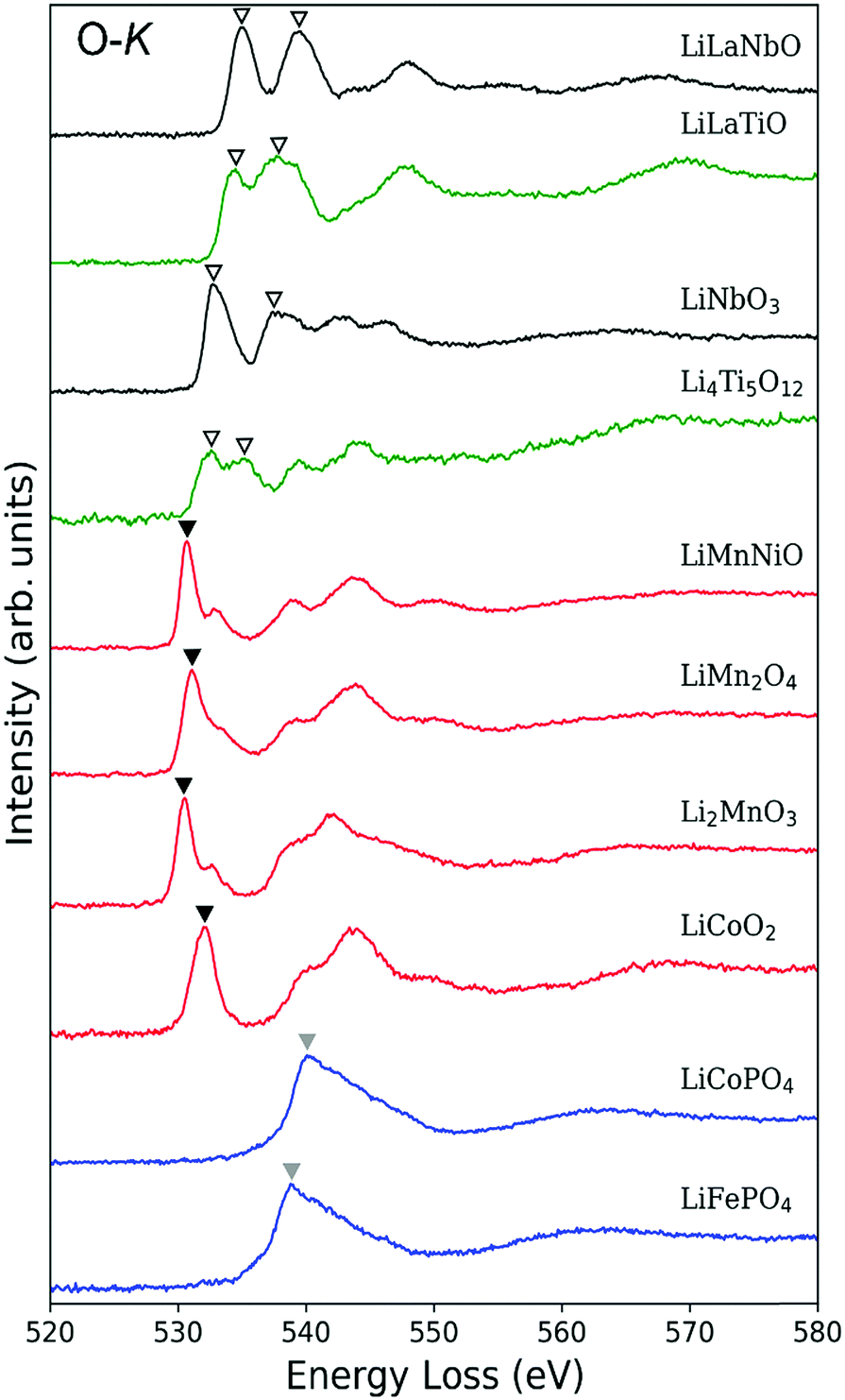 | ||
| Fig. 4 EELS spectra around O-K edges of the Li-ion battery materials. LiLaNbO, LiLaTiO and LiMnNiO stand for Li0.14La0.29NbO3, Li0.33La0.56TiO3 and LiMn1.5Ni0.5O4, respectively. Spectra can be classified into three groups according to edge shape: (i) double peaks of similar intensity (open inverted triangles); (ii) single sharp peak (black inverted triangles) with relatively weak sub-peak; and (iii) a single broad peak (grey inverted triangles). | ||
| Composition | O-K edge (eV) | Shape | |
|---|---|---|---|
| This work | Previous work | ||
| a Ref. 35. b Ref. 51. c Ref. 30. d Ref. 36. e Ref. 14. f Ref. 33. g Ref. 34. | |||
| LiFePO4 | 538.7 | 537.0a | Broad single |
| LiCoPO4 | 540.1 | 540.4b | Broad single |
| LiCoO2 | 532.0 | 530.5c | Sharp single |
| Li2MnO3 | 530.5 | 530.9d | Sharp single |
| LiMn2O4 | 531.1 | 529.5e | Sharp single |
| LiNi0.5Mn1.5O4 | 530.7 | Sharp single | |
| Li4Ti5O12 | 532.6, 535.2 | 532.7, 535.4f | Double |
| LiNbO3 | 532.7, 537.8 | Double | |
| Li0.33La0.56TiO3 | 534.5, 537.7 | 532.9, 536.1g | Double |
| Li0.14La0.29NbO3 | 535.1, 539.6 | Double | |
The observed trends can be partly interpreted in terms of the effect of TM-d orbitals overlapping with neighbouring O-2p orbitals. As reported in Table 1, TM atoms in group (i) materials have a d0 configuration, while TM atoms in the other two groups have partially occupied d orbitals. Since all TM atoms of the materials examined here are in octahedral coordination environments, their d orbitals are split into t2g and eg bands.
In group (i) materials, the t2g and eg bands hybridise with unoccupied O-2p orbitals to form part of the conduction band. Such hybridisation results in splitting of the O-2p states into which an O-1s electron is excited, resulting in the double peak in the O-K ELNES spectra.
In the case of group (ii) materials, the origin of the single sharp peak and small sub-peaks can also be explained in terms of the TM-d orbitals, with d orbitals in the conduction band splitting by different amounts as a result of their partial occupation (for details see Section S3 in ESI†).
O-K ELNES spectra of group (iii) materials contain only one broad peak, and are distinctly different to those of group (ii) materials despite also containing partially occupied TM-d orbitals. In addition, the peak positions of group (iii) materials are energetically higher than those of the other groups. These differences cannot be explained satisfactorily using a simple model of the electronic structure, so we simulated the ELNES of a group (ii) material, LiCoO2, and a group (iii) material, LiFePO4, using the core-hole method within the framework of DFT, analysing and comparing their electronic structures in detail.
3.4 O-K ELNES simulations of LiCoO2 and LiFePO4
Fig. 5(a) shows experimental and simulated O-K ELNES spectra of LiCoO2 when ΔE = 0.3 eV. The simulated spectrum matches the shape of the experimental spectrum well, as well as the calculated O-K X-ray absorption near edge structure.47,48Fig. 5(b) shows PDOSs of O-p, Co-d and Li-p orbitals, with ground-state and excited-state PDOSs plotted as solid and dashed lines, respectively. The excited-state PDOSs are from the O atom containing the core hole in the case of the O-p orbital, and from Co and Li atoms neighbouring the excited O atom in the case of Co-d and Li-p orbitals, respectively.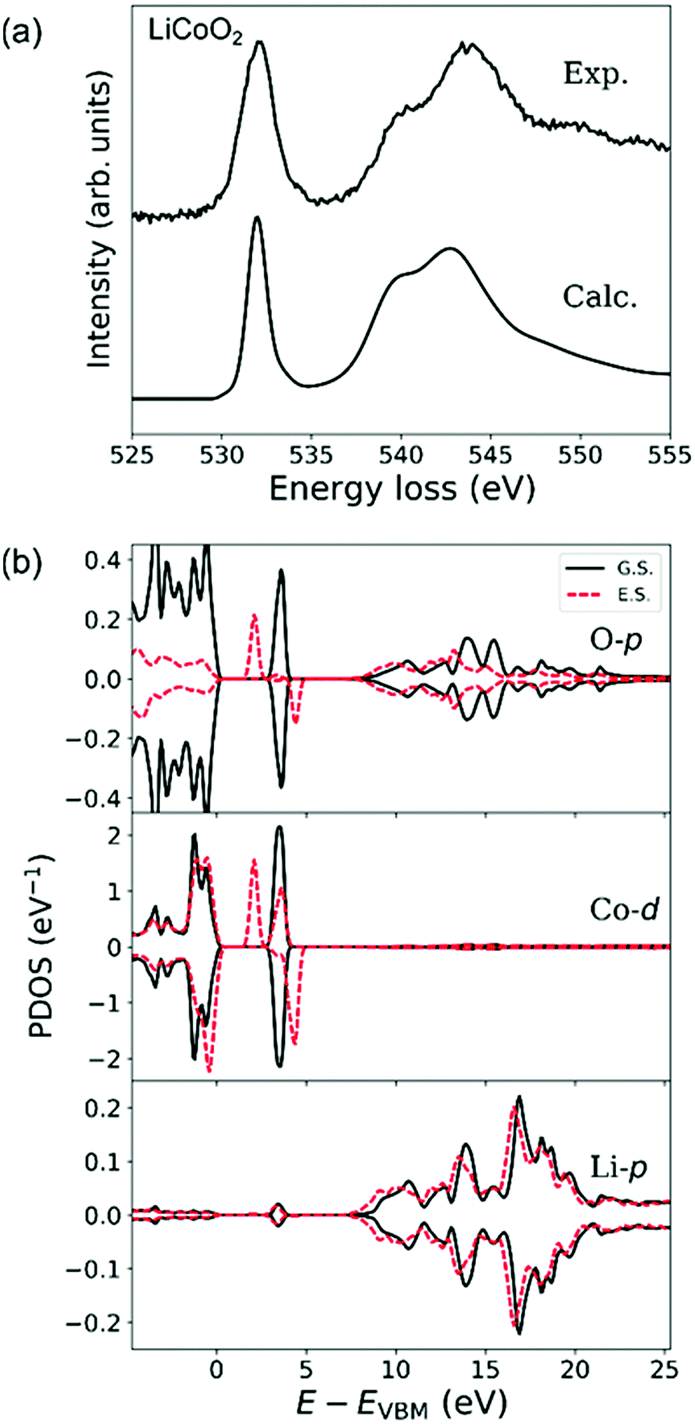 | ||
| Fig. 5 (a) Experimental and calculated ELNES spectra of the O-K edge of LiCoO2 and (b) projected densities of states (PDOSs) of O-p, Co-d and Li-p orbitals in LiCoO2. Solid lines are PDOSs for the ground state (G.S.) and dashed lines are for the excited state (E.S.). PDOSs of Co and Li in the excited state were extracted from atoms neighbouring the excited O atom. | ||
In ground-state LiCoO2, the valence band consists of Co-3d (t2g) and O-2p orbitals and the conduction band of Co-3d (eg), O-2p and Li-2p orbitals.30,47 The octahedral environment of the Co atom causes Co-d orbitals to split into fully occupied t2g states and unoccupied eg states. The calculated band gap of 2.5 eV is consistent with previous DFT calculations.48
Formation of an O-1s hole reduces the symmetry reduction of CoO6 octahedra resulting in splitting of Co-d (eg) orbitals hybridised with 2p orbitals of the excited O atom. This splitting lowers the conduction band minimum by about 1.5 eV, resulting in a lowering of the energy of the O-K edge, and produces the very small shoulder-like sub-peak in the tail of the main peak in the ELNES. Other changes in the shape of the broad hybridisation band between O-2p and Li-2p orbitals at energies higher than 7 eV in the excited state are also noticeable in the PDOS plots in Fig. 5(b). Since the O-K ELNES spectrum is produced by the transition of an O-1s electron to unoccupied O-p states, Fig. 5(b) shows that the single sharp ELNES peak at around 532 eV and the broad peak at energies higher than 536 eV originate from hybridisation of O-p orbitals with Co-d and Li-p orbitals, respectively.
Experimental and calculated O-K ELNES spectra of LiFePO4 using ΔE = 0.3 eV are compared in Fig. 6(a). Fig. 6(b) shows PDOSs of O-p, Fe-d, P-p and Li-p orbitals in ground and excited states in the same manner as for LiCoO2 in Fig. 5(b) where PDOSs of the excited O atom and Fe, P and Li atoms neighbouring it have been averaged over contributions from crystallographically independent sites.
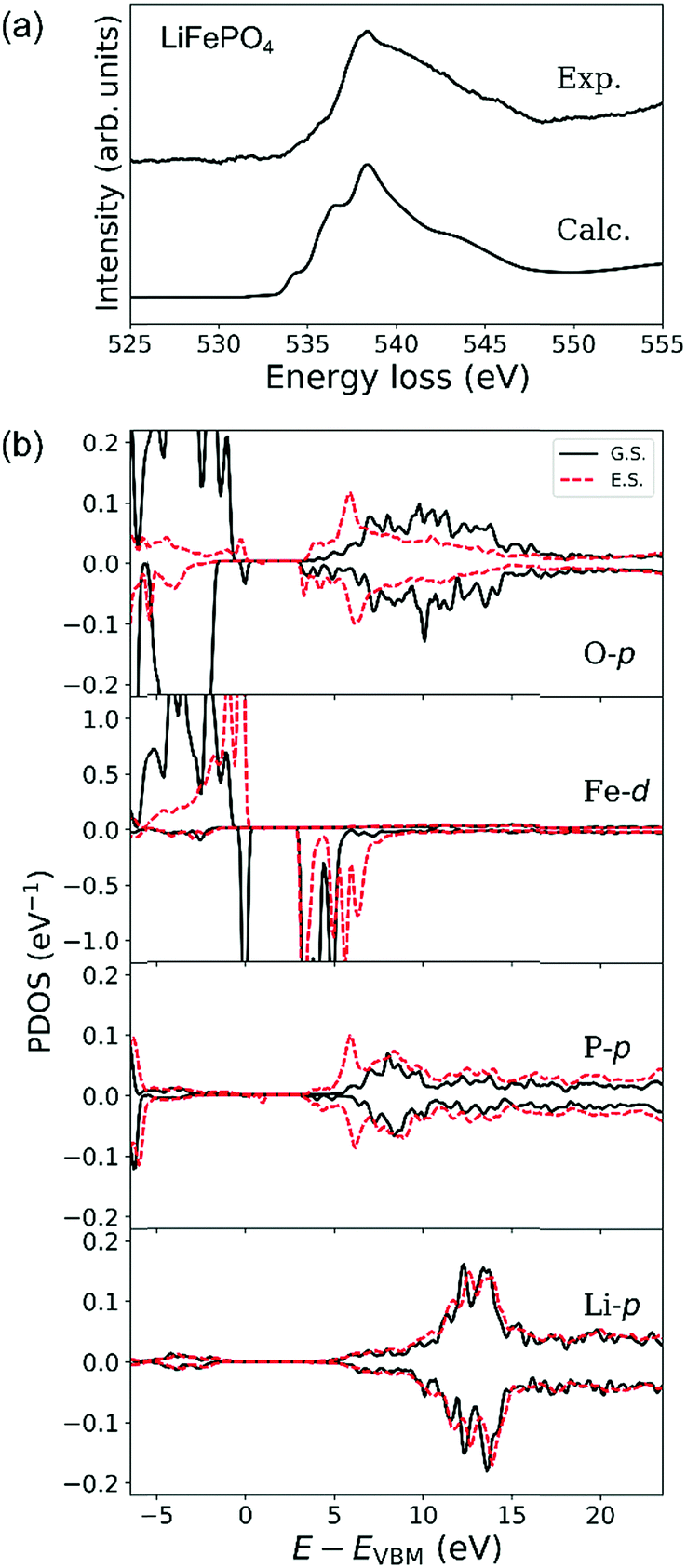 | ||
| Fig. 6 (a) Experimental and calculated ELNES spectra of the O-K edge of LiFePO4 and (b) projected densities of states (PDOSs) of O-p, Fe-d, P-p and Li-p orbitals in LiFePO4. Solid lines are PDOSs for the ground state (G.S.) and dashed lines are for the excited state (E.S.). PDOSs of Fe, P and Li in the excited state were extracted from atoms neighbouring the excited O atom. Calculated O-p PDOSs and ELNES spectra are averages of data from O atoms on the three non-equivalent crystallographic sites in the olivine-type structure. | ||
The calculated O-K ELNES spectrum in Fig. 6(a) has a single broad peak and appears more similar to the experimental spectrum compared to those previously calculated using the Z + 1 method19 or from dielectric function calculations.20 This result demonstrates the efficacy of using the core–hole method to simulate the ELNES spectra with reasonable accuracy.62
In the ground state, while the top of the valence band of LiFePO4 consists of O-2p and Fe-3d orbitals, in the energy range from 3 to 5 eV above the valence band maximum (VBM), the conduction band mainly consists of O-2p and Fe-3d orbitals and the region with energies higher than 5 eV consists of O-p, P-p and Li-p orbitals (Fig. 6(b)). The relatively large differences between up- and down-spin contributions in PDOSs of Fe-d and O-p orbitals reflect the high-spin states of Fe2+ ions with a d6 electronic configuration, i.e., (t2g)4(eg)2. The observed broad distribution of the O-p PDOS in the conduction band is a result of hybridisation between O-p orbitals and neighbouring Fe, P and Li orbitals. This overlapping of several orbitals in the conduction band is characteristic of LiFePO4, in contrast to the clear separation between hybrid O–Co and Li–O orbitals in the conduction band of LiCoO2.
When an electron is elevated from an O-1s shell in LiFePO4, the PDOSs of O-p, Fe-d and P-p orbitals at the bottom of the conduction band (around 3 to 7 eV above the VBM) change dramatically, while their PDOSs at higher energies undergo relatively little change. Introduction of the core hole shifts the bottom of the conduction band, which in the ground state consists mainly of Fe-d electrons between 3 and 5 eV above the VBM, higher by about 1.5 eV. The PDOS of P-p orbitals between 5 and 7 eV above the Fermi level also increases dramatically, a consequence of the strong covalent bonding between P and O atoms. The band gap, however, remains essentially unchanged because Fe-d orbitals at the top of the valence band in the ground state are elevated to the bottom of the conduction band in the excited state. These changes enhance the energetic overlap between the Fe–O, Li–O and P–O hybrid orbitals, resulting in the characteristic broad peak in the O-K ELNES of LiFePO4, in contrast to that of LiCoO2. The broad single peaks observed for group (iii) materials (LiFePO4 and LiCoPO4) can thus be attributed to the presence of tetrahedral PO4 units in addition to the TM–O polyhedra. Group (ii) materials, which lack PO4 tetrahedral units, exhibit single, sharp O-K edges regardless of the overall crystal structure, e.g., layered-rocksalt in the case of LiCoO2 or spinel in the case of LiMn2O4.
Although the band gaps of LiCoO2 and LiFePO4 are almost the same, the peak positions of O-K edges in LiCoO2 and LiFePO4 differ by about 6.7 eV. Our ELNES simulations also demonstrated a similar trend, with a calculated difference in peak positions of 4.9 eV. Analysis of the electronic structures showed that this difference arises from the difference in changes in the conduction bands in the excited state. As mentioned above, the peak in the O-K edge of LiCoO2 corresponds to the conduction band minimum composed of hybrid O–Co orbitals, and decreases by about 1.5 eV upon introduction of a core hole. In the case of LiFePO4, the conduction band minimum is not decreased when a core hole is introduced, and neither are there isolated hybrid O–TM orbitals. This difference provides one explanation for why the main peak of the O-K edge in LiFePO4 appears at higher energy than in LiCoO2.
4 Conclusions
Li-K and O-K ELNES spectra of ten different representative LIB materials were systematically investigated by EELS using an aberration-corrected TEM equipped with a monochromator. Measurements with a high energy resolution of ΔE = 0.1 eV revealed fine structures in Li-K ELNES spectra that can otherwise remain hidden. The peak positions of Li-K edges were found to be about 62 eV for most of the LIB materials because of the similar coordination environments of their Li ions. In contrast, the peak positions of O-K edges varied over a range of about 10 eV.The shapes of O-K edges could be divided into three groups. Group (i) materials contained octahedrally coordinated TM ions with empty d orbitals, and these materials produced ELNES with double peaks at the O-K edges because of splitting of TM-d orbitals into t2g and eg levels. Group (ii) and (iii) materials both contained TM ions with partially filled d orbitals, but the shapes of their O-K edges were found to be distinctly different, the former with a single sharp peak and the latter with a single broad peak.
ELNES simulations based on DFT revealed that the single sharp peak in the O-K edge of group (ii) material LiCoO2 results from hybridisation of O-p and Co-d eg orbitals with energies close to the conduction band minimum. In contrast, the broad peak of the O-K edge of group (iii) material LiFePO4 results from hybridisation between O-p orbitals with P-p, Fe-d and Li-p orbitals in the conduction band at similar energy levels.
Our systematic analysis of EELS spectra demonstrates how a combination of high-resolution ELNES measurements and simulations based on DFT calculations provides useful insights into the electronic structures and coordination environments of atoms in LIB materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Research & Development Initiative for Scientific Innovation of New Generation Batteries 2 (RISING 2) project from the New Energy and Industrial Technology Development Organization (NEDO), Japan. The authors thank Toyota Motor Corporation for providing some of the samples.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef.
- P. E. Batson, Ultramicroscopy, 1992, 47, 133–144 CrossRef.
- K. Suenaga, Science, 2000, 290, 2280–2282 CrossRef PubMed.
- S. Muto and K. Tatsumi, Microscopy, 2016, 1–11 Search PubMed.
- K. J. Carroll, D. Qian, C. Fell, S. Calvin, G. M. Veith, M. Chi, L. Baggetto and Y. S. Meng, Phys. Chem. Chem. Phys., 2013, 15, 11128–11138 RSC.
- R. Senga and K. Suenaga, Nat. Commun., 2015, 6, 7943 CrossRef PubMed.
- W. Zhou, M. P. Oxley, A. R. Lupini, O. L. Krivanek, S. J. Pennycook and J.-C. Idrobo, Microsc. Microanal., 2012, 18, 1342–1354 CrossRef PubMed.
- T. Mizoguchi, T. Sasaki, S. Tanaka, K. Matsunaga, T. Yamamoto, M. Kohyama and Y. Ikuhara, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 1–10 CrossRef.
- R. Huang, Y. H. Ikuhara, T. Mizoguchi, S. D. Findlay, A. Kuwabara, C. A. J. Fisher, H. Moriwake, H. Oki, T. Hirayama and Y. Ikuhara, Angew. Chem., Int. Ed., 2011, 50, 3053–3057 CrossRef PubMed.
- R. F. Egerton, Electron Energy-Loss Spectroscopy in the Electron Microscope, Springer, US, Boston, MA, 3rd edn, 2011 Search PubMed.
- X. Gao, Y. H. Ikuhara, C. A. J. Fisher, H. Moriwake, A. Kuwabara, H. Oki, K. Kohama, R. Yoshida, R. Huang and Y. Ikuhara, Adv. Mater. Interfaces, 2014, 1, 1400143 CrossRef.
- R. Huang, T. Hitosugi, C. A. J. Fisher, Y. H. Ikuhara, H. Moriwake, H. Oki and Y. Ikuhara, Mater. Chem. Phys., 2012, 133, 1101–1107 CrossRef.
- L. Laffont, C. Delacourt, P. Gibot, M. Y. Wu, P. Kooyman, C. Masquelier and J. M. Tarascon, Chem. Mater., 2006, 18, 5520–5529 CrossRef.
- T. Mizoguchi, J. P. Buban, K. Matsunaga, T. Yamamoto and Y. Ikuhara, Ultramicroscopy, 2006, 106, 92–104 CrossRef PubMed.
- F. Wang, J. Graetz, M. S. Moreno, C. Ma, L. Wu, V. Volkov and Y. Zhu, ACS Nano, 2011, 5, 1190–1197 CrossRef PubMed.
- S. C. Nagpure, S. S. S. Babu, B. Bhushan, A. Kumar, R. Mishra, W. Windl, L. Kovarik and M. Mills, Acta Mater., 2011, 59, 6917–6926 CrossRef.
- M. K. Kinyanjui, P. Axmann, M. Wohlfahrt-Mehrens, P. Moreau, F. Boucher and U. Kaiser, J. Phys.: Condens. Matter, 2010, 22, 275501 CrossRef PubMed.
- K. Kimoto and Y. Matsui, J. Microsc., 2002, 208, 224–228 CrossRef PubMed.
- P. C. Tiemeijer, Inst. Phys. Conf. Ser., 1999, 161, 191 Search PubMed.
- M. Mukai, J. S. Kim, K. Omoto, H. Sawada, A. Kimura, A. Ikeda, J. Zhou, T. Kaneyama, N. P. Young, J. H. Warner, P. D. Nellist and A. I. Kirkland, Ultramicroscopy, 2014, 140, 37–43 CrossRef PubMed.
- M. Mukai, E. Okunishi, M. Ashino, K. Omoto, T. Fukuda, A. Ikeda, K. Somehara, T. Kaneyama, T. Saitoh, T. Hirayama and Y. Ikuhara, Microscopy, 2015, 64, 151–158 CrossRef PubMed.
- Z. Wang, D. Santhanagopalan, W. Zhang, F. Wang, H. L. Xin, K. He, J. Li, N. Dudney and Y. S. Meng, Nano Lett., 2016, 16, 3760–3767 CrossRef PubMed.
- N. Taguchi, H. Sakaebe, K. Tatsumi and T. Akita, e-J. Surf. Sci. Nanotechnol., 2015, 13, 284–288 CrossRef.
- X. Mu, A. Kobler, D. Wang, V. S. K. K. Chakravadhanula, S. Schlabach, D. V. Szabó, P. Norby and C. Kübel, Ultramicroscopy, 2016, 170, 10–18 CrossRef PubMed.
- S. Kobayashi, C. A. J. Fisher, T. Kato, Y. Ukyo, T. Hirayama and Y. Ikuhara, Nano Lett., 2016, 16, 5409–5414 CrossRef PubMed.
- P. Yan, L. Xiao, J. Zheng, Y. Zhou, Y. He, X. Zu, S. X. Mao, J. Xiao, F. Gao, J.-G. Zhang and C.-M. Wang, Chem. Mater., 2015, 27, 975–982 CrossRef.
- J. Kikkawa, S. Terada, A. Gunji, M. Haruta, T. Nagai, K. Kurashima and K. Kimoto, Appl. Phys. Lett., 2014, 104, 114105 CrossRef.
- J. Graetz, A. Hightower, C. C. Ahn, R. Yazami, P. Rez and B. Fultz, J. Phys. Chem. B, 2002, 106, 1286–1289 CrossRef.
- P. Moreau and F. Boucher, Micron, 2012, 43, 16–21 CrossRef PubMed.
- M. Kitta, T. Akita, S. Tanaka and M. Kohyama, J. Power Sources, 2013, 237, 26–32 CrossRef.
- C. Ma, K. Chen, C. Liang, C.-W. Nan, R. Ishikawa, K. More and M. Chi, Energy Environ. Sci., 2014, 7, 1638 RSC.
- Y. Honda, S. Muto, K. Tatsumi, H. Kondo, K. Horibuchi, T. Kobayashi and T. Sasaki, J. Power Sources, 2015, 291, 85–94 CrossRef.
- J. Kikkawa, T. Akita, E. Hosono, H. Zhou and M. Kohyama, J. Phys. Chem. C, 2010, 114, 18358–18365 CrossRef.
- K. Mizushima, P. Jones, P. Wiseman and J. Goodenough, Solid State Ionics, 1981, 3–4, 171–174 CrossRef.
- A. K. Padhi, K. S. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461–472 CrossRef.
- G. G. Amatucci, N. Pereira, T. Zheng and J.-M. Tarascon, J. Electrochem. Soc., 2001, 148, A171 CrossRef.
- A. Yamada, S. C. Chung and K. Hinokuma, J. Electrochem. Soc., 2001, 148, A224 CrossRef.
- L.-X. Yuan, Z.-H. Wang, W.-X. Zhang, X.-L. Hu, J.-T. Chen, Y.-H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269–284 RSC.
- J. M. Lloris, C. Pérez Vicente and J. L. Tirado, Electrochem. Solid-State Lett., 2002, 5, A234 CrossRef.
- F. Zhou, M. Cococcioni, K. Kang and G. Ceder, Electrochem. Commun., 2004, 6, 1144–1148 CrossRef.
- Q. Zhong, A. Bonakdarpour, M. Zhang, Y. Gao, J. R. Dahn, A. Bonakclarpour, M. Zhang, Y. Gao and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 205 CrossRef.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- M. T. Czyzyk, R. Potze and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 3729–3735 CrossRef.
- A. Juhin, F. de Groot, G. Vankó, M. Calandra and C. Brouder, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 1–12 CrossRef.
- G. Rousse, J. Rodriguez-Carvajal, S. Patoux and C. Masquelier, Chem. Mater., 2003, 15, 4082–4090 CrossRef.
- A. Nakamura, S. Furutsuki, S. Nishimura, T. Tohei, Y. Sato, N. Shibata, A. Yamada and Y. Ikuhara, Chem. Mater., 2014, 26, 6178–6184 CrossRef.
- Y. H. Ikuhara, X. Gao, C. A. J. Fisher, A. Kuwabara, H. Moriwake, K. Kohama, H. Iba and Y. Ikuhara, J. Mater. Chem. A, 2017, 5, 9329–9338 RSC.
- Y. H. Ikuhara, X. Gao, R. Huang, C. A. J. Fisher, A. Kuwabara, H. Moriwake and K. Kohama, J. Phys. Chem. C, 2014, 118, 19540–19547 CrossRef.
- A. Kumatani, S. Shiraki, Y. Takagi, T. Suzuki, T. Ohsawa, X. Gao, Y. Ikuhara and T. Hitosugi, Jpn. J. Appl. Phys., 2014, 53, 058001 CrossRef.
- Y. Fujiwara, K. Hoshikawa and K. Kohama, J. Cryst. Growth, 2016, 433, 48–53 CrossRef.
- X. Gao, C. A. J. Fisher, T. Kimura, Y. H. Ikuhara, H. Moriwake, A. Kuwabara, H. Oki, T. Tojigamori, R. Huang and Y. Ikuhara, Chem. Mater., 2013, 25, 1607–1614 CrossRef.
- M. O. Krause, J. Phys. Chem. Ref. Data, 1979, 8, 307–327 CrossRef.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235121 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef.
- G. K. H. H. Madsen, P. Blaha, K. Schwarz, E. Sjöstedt and L. Nordström, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 195134 CrossRef.
- T. Mizoguchi, W. Olovsson, H. Ikeno and I. Tanaka, Micron, 2010, 41, 695–709 CrossRef PubMed.
- J. Tsuji, H. Nakamatsu, T. Mukoyama, K. Kojima, S. Ikeda and K. Taniguchi, X-Ray Spectrom., 2002, 31, 319–326 CrossRef.
- W. Olovsson, I. Tanaka, T. Mizoguchi, P. Puschnig and C. Ambrosch-Draxl, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 041102 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03390k |
| ‡ Authors contributed equally. |
| This journal is © the Owner Societies 2018 |