
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ice-binding site of surface-bound type III antifreeze protein partially decoupled from water†
Dominique
Verreault
 a,
Sarah
Alamdari
b,
Steven J.
Roeters
a,
Ravindra
Pandey
c,
Jim
Pfaendtner
b and
Tobias
Weidner
*ab
a,
Sarah
Alamdari
b,
Steven J.
Roeters
a,
Ravindra
Pandey
c,
Jim
Pfaendtner
b and
Tobias
Weidner
*ab
aDepartment of Chemistry, Aarhus University, Langelandsgade 140, 8000 Aarhus C, Denmark. E-mail: weidner@chem.au.dk
bDepartment of Chemical Engineering, University of Washington, Seattle, Washington 98195, USA
cDepartment of Chemistry, Indian Institute of Technology, Roorkee 247667, India
First published on 17th September 2018
Abstract
Type III antifreeze proteins (AFP III) have been widely recognized as one class of ice-binding proteins produced by several biological organisms to withstand freezing conditions. Besides their ability to restrict ice growth through their ice-binding site (IBS), AFP III have also been shown to possess a great propensity for hydrophobic surfaces such as the air–water interface. Yet, it is not known whether AFP III adsorb with a specific orientation and how hydrophobic interactions affect the IBS. Molecular insights on the accessibility of the IBS and its interactions with water are important for understanding AFP III action in vivo but also for their application as ice-inhibiting agents for deicing, frozen food storage, as well as for long-term blood and organ cryo-preservation. Here, the orientation of fish AFP III adsorbed at the air–water interface has been studied using a combination of molecular dynamics (MD) simulations and vibrational sum-frequency generation (SFG) spectroscopy together with spectral calculations. The SFG/MD analysis indicated that when AFP III adsorbs at the air–water interface, it mostly retains its native state and orients with a tilt angle of 120° with respect to the surface normal. We found that the IBS is only partially solvated, leaving the pyramidal ice plane binding domain exposed to the vapor phase. These findings suggest that interactions with hydrophobic interfaces (e.g., cell membranes, polymers) could lead to the partial decoupling of the IBS from water and, to some extent, to a loss of AFP III antifreezing activity.
1. Introduction
Antifreeze proteins (AFPs) belong to a class of ice-binding proteins that enable various psychrophilic organisms (bacteria, algae, plants, insects, fishes) to survive in otherwise freezing environments.1–3 These proteins have the remarkable ability to adsorb to ice crystal seeds that form in the extracellular fluids at or below the equilibrium freezing point and to inhibit their further growth.4,5 Type III antifreeze protein (AFP III) is one example of such a protein produced by several species of fish (e.g., eelpouts, wolffish) inhabiting the polar and subpolar seas.6,7 AFP III is a 7 kDa, cusp-shaped, globular protein comprised of a one-turn α-helix and a few short β-strands arranged in β-sheet-like motifs, giving the protein a rigid and compact fold.8–11 Its surface is mostly hydrophobic (∼62% nonpolar residues) with some interspersed hydrophilic patches. More importantly, like other types of AFPs, AFP III possesses a flat and relatively hydrophobic ice-binding site (IBS),12–14 made of two domains that enable binding to distinct ice crystal planes. Through the IBS, AFPs adsorb to the surface of nucleating ice crystals and inhibit their further growth, a process commonly known as the adsorption–inhibition mechanism.4 In order for AFPs to adsorb to growing ice crystals it has been proposed that the IBS first organizes interfacial water molecules into an ice-like lattice that subsequently matches and fuses to the quasi-liquid layer present on the ice surface.15,16 The adsorbed AFPs then create micro-curvatures which, according to the Kelvin effect, prevent further growth of the ice front and result in a reduction of the freezing point below the melting point (the so-called thermal hysteresis gap).17 Even though this mechanism is now widely accepted, molecular details of each step have yet to be fully understood.18,19Because of its overall amphiphilic character and hydrophobic IBS, AFP III is also likely to have a relatively high affinity for hydrophobic interfaces. For instance, it has been shown that even at very low concentrations, AFP III reduces water's surface tension, indicating its strong propensity to be localized at the air–water interface.20,21 More recently, Bakker and co-workers further confirmed the adsorption of AFP III to the air–water interface using surface-sensitive, vibrational sum-frequency generation (SFG) spectroscopy by observing spectral features associated with CH modes (2850–2950 cm−1) of the protein and the absence of the sharp water peak at ∼3700 cm−1 assigned to the dangling OH groups of topmost water molecules.22 Molecular dynamics (MD) simulations have been used to study the water interactions and dynamics with AFP III at the air–water interface.23,24 However, experimentally, the interfacial orientation of AFP III and the extent of conformational changes it might undergo remain to be determined. For instance, it is not known whether AFP III orients itself in such a way as to limit the exposure of the IBS to the aqueous phase and/or to favor interactions with neighboring proteins. In the latter case, it is well known from protein crystallization that AFP III has a tendency to aggregate through protein–protein contacts mediated by the IBS.25 It would thus be interesting to know what orientation AFP III assumes at the air–water interface and whether that orientation would favor interactions with the vapor phase and/or with its neighbours, and if that would cause conformational changes eventually affecting the protein's antifreezing activity. Such information could prove useful, for instance, in applications where AFPs are used in the presence of hydrophobic surfaces.
In this work, we study the conformation of AFP III in the bulk and possible conformational changes induced at the air–water interface using secondary structure analysis from circular dichroism (CD) measurements and molecular dynamics (MD) simulations. We also use a combination of MD simulations, SFG spectroscopy, and spectral calculations to determine the orientation of AFP III at the hydrophobic air–water interface. We further examine the solvation of the IBS, and whether it becomes partially or completely exposed to the vapor phase. Results show that the translocation of AFP III from the bulk to the air–water interface does not significantly alter the protein native conformation. In addition, we found that upon adsorption, AFP III preferentially orients with its long axis at a tilt angle of 120° with respect to the surface normal. This specific orientation results in a partial dehydration of the IBS, with the pyramidal ice binding domain exposed to the vapor phase. We postulate that the partial decoupling of water from the IBS in the presence of hydrophobic interfaces could lead to a partial loss of AFP III antifreezing activity.
2. Materials and methods
2.1. Sample preparation
Type III AFP isolated from the ocean pout (Zoarces americanus) was purchased from A/F Protein Inc. (Waltham, MA, USA) and used without further purification. For CD measurements, 5 μM (35 μg mL−1) AFP III solutions were prepared in 5 mM phosphate buffer saline (PBS) at pH 7.5 (Sigma-Aldrich, Taufkirchen, Germany). For surface tension and SFG measurements, 0.18 mM (1.25 mg mL−1) AFP III solutions were prepared in D2O (99.9 atom% D, Sigma-Aldrich).2.2. Surface tensiometry
The surface tension of an AFP III solution has been measured using a single-channel Langmuir tensiometer (DeltaPi, Kibron Inc., Helsinki, Finland). Prior to measurements, the trough was thoroughly cleaned with acetone, ethanol and milli-Q water (18.2 MΩ cm), and dried under a nitrogen stream. The tensiometer was calibrated using pure D2O at ambient temperature (22 °C). The surface tension of the AFP III solution was recorded until an equilibrium value was reached (∼30 min) within the accuracy of the measurement (±0.01 mN m−1). Surface tension measurements of AFP III were repeated at least three times to ensure reproducibility.2.3. Circular dichroism
Far-UV CD spectra of AFP III were recorded on a spectropolarimeter (J-815, JASCO Deutschland GmbH, Pfungstadt, Germany) in the wavelength range from 180 to 250 nm using a path length of 0.02 cm. Each CD spectrum was acquired at a scan speed of 20 nm min−1 and averaged over three scans. A reference spectrum of the buffer (blank) was subtracted from the protein spectra. The CD spectra are expressed in terms of the mean residue ellipticity26| [θ]MRW,λ = 3298ΔAλ/Cmd | (1) |
2.4. MD simulations
The adsorption of AFP III at the air–water interface was simulated with a protein of known crystal structure (AFP III from Zoarces viviparus (European eelpout), PDB reference: 4UR4)11 placed in a vacuum/water box using the GROMACS 5.1.2 engine.29,30 Using 10 random initial orientations, the protein was first equilibrated in a 6.4 nm × 6.4 nm × 6.4 nm water box containing about 7800 water molecules and one chloride ion (for charge neutralization) at 300 K and 1 bar in the NPT ensemble using a Berendsen barostat31 (τ = 1.0 ps) and a stochastic velocity-rescaling thermostat32 (τ = 0.1 ps) for 2 ns. The AMBERff14SB force field33 and TIP3P rigid water model34 were used to model the system. Hydrogen bonds were constrained by the LINCS algorithm35 to a time step of 2 fs. van der Waals interactions were shifted to 0 at 1.1 nm. Electrostatic interactions were calculated with particle-mesh Ewald (PME) summations36 using a 1.2 nm cutoff between the reciprocal and real space domains. To simulate the air–water interface, the simulation box dimensions were doubled in the z-dimension (6.4 nm × 6.4 nm × 12.8 nm) creating a vacuum region. Water molecules in the top 2.5 nm of the interface were translated to the bottom of the simulation box, positioning the protein near the interface. Production runs were carried out in the NVT ensemble for 50 ns. Six simulations of the adsorbed protein were carried out up to 500 ns to assess the protein stability at the interface. These trials were used to assess the stability of each of the two orientations at the interface. An additional trial was simulated in the case were a protein reoriented over the long simulation times, to confirm its stability. The same methodology was also applied for the adsorption of a mutant of AFP III obtained by replacing in silico in the PDB sequence 4UR4 the threonine(T) residue at position 18 by an asparagine(N) (abbreviated, hereafter, as T18N), where T18 is a residue known to be critical in the protein binding affinity to the primary prism ice plane.37Analysis of the trajectories for both AFP III and the T18N mutant was done using the PLUMED 2.4.0 library38 in GROMACS. The orientation of AFP III was determined by monitoring the tilt angle (θ = cos−1(|d|/|dz|)) between a vector (d) along the protein's long axis, defined by the center of mass between residues methionine(M)-1 and isoleucine(I)-14, and its projection along the surface normal (dz) (Fig. 1). The absolute value in the argument takes care of which air–water interface (top (+) or bottom (−)) AFP III was adsorbed to within the periodic unit cell. To compare the secondary structure of AFP III and T18N AFP III, a DSSP analysis based on the α-carbons was performed using the do_dssp tool v1.0 in GROMACS.39,40 The structure, as defined by DSSP, includes α-helices, β-bridges, β-sheets and turns. Fractions were calculated by dividing the number of residues in each secondary structure type by the total number of residues in the sequence.
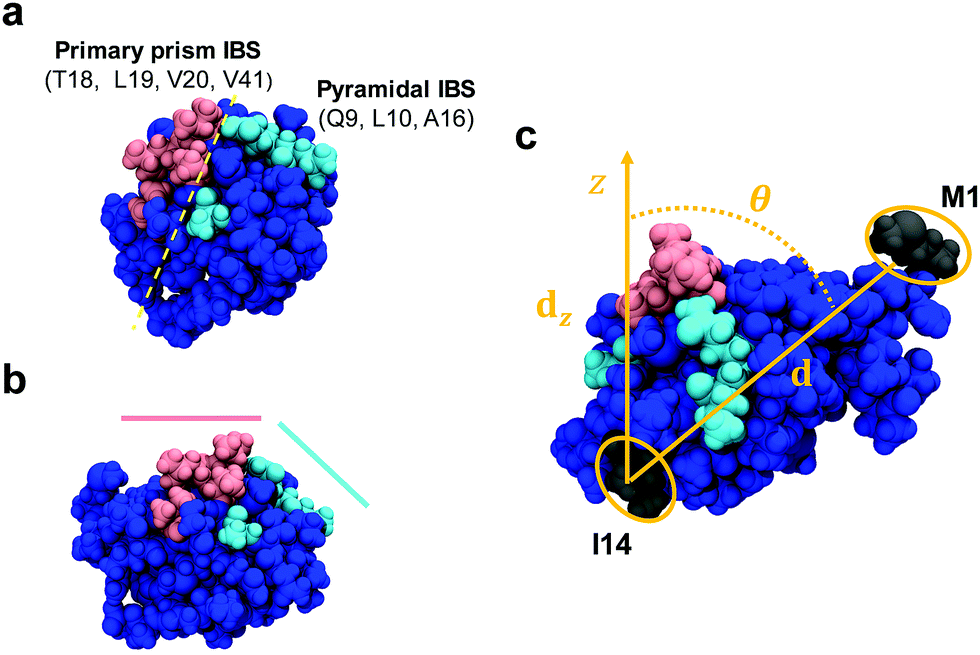 | ||
| Fig. 1 Space-filling model of AFP III (PDB reference: 4UR4) with (a) top and (b) side views. The IBS is divided into two domains that bind to, respectively, the primary prism and pyramidal ice planes. Key residues from these domains are highlighted in magenta and cyan, respectively. The domains form two relatively flat planes inclined to one another by about −30°.37 (c) Definition of the AFP III tilt angle between the vector d along the center of mass between residues M1 and I14 (colored in dark grey) and its projection dz along the surface normal (z-axis). All images in this figure were generated using the program VMD (http://www.ks.uiuc.edu/Research/vmd). | ||
2.5. SFG spectroscopy
The SFG setup was based on a Ti:sapphire femtosecond laser oscillator (Mai Tai, Spectra-Physics, Darmstadt, Germany) coupled to a regenerative amplifier (Spitfire Ace, Spectra-Physics) pumped by a Nd:YLF (neodymium-doped yttrium lithium fluoride) laser (Empower, Spectra-Physics). The amplifier produced a 5 mJ output centered at 800 nm with 40 fs pulse duration and 1 kHz repetition rate. A fraction (1.7 mJ) of the amplified output was used to pump an optical parametric amplifier (TOPAS-C, Spectra-Physics). The signal and idler pulses generated from the parametric amplifier were then mixed in a non-collinear difference frequency generator unit (NDFG, Spectra-Physics), resulting in 4 μJ broadband (full width at half maximum (FWHM) of ∼100 cm−1) infrared (IR) pulses centered at ∼1650 cm−1. The IR beamline (including the sample stage) was constantly purged with dry purified N2 to minimize IR absorption from water vapor. Another fraction (1 mJ) of the amplifier output was directed to an air-spaced Fabry–Perot etalon (SLS Optics, Tromode, Isle of Man) to generate narrowband visible (VIS) pulses (25 μJ; FHWM ∼ 15 cm−1). The VIS and IR beams were directed at incident angles of 36° and 41° (relative to the surface normal), respectively, and spatially and temporally overlapped at the surface of the AFP III solution contained in a custom-built Teflon trough (see Fig. S1, ESI†). The sum-frequency (SF) signal generated in the reflection direction from the surface was collected, filtered, and focused onto a spectrograph (Shamrock SR303i, Andor Technology, Belfast, UK), dispersed by a grating, before being refocused on an electron multiplying charge-coupled device camera (Newton DU-970P-BV, Andor Technology). Each SFG spectrum in the amide I region (1600–1700 cm−1) was recorded for 10 min using the ssp polarization combination (s-polarized SF, s-polarized VIS, and p-polarized IR), background-subtracted (with the signal recorded with the VIS beam blocked) and normalized to a non-resonant reference spectrum from z-cut quartz. The normalized SFG spectra were fitted to a Lorentzian line profile given by41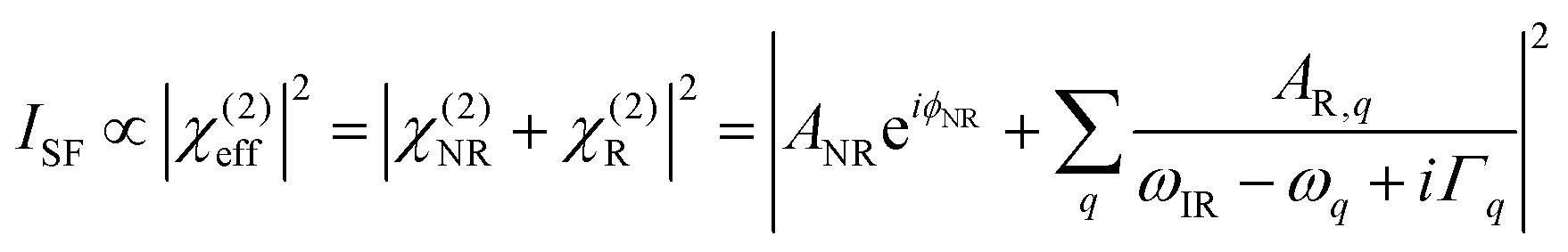 | (2) |
2.6. Spectral amide I SFG calculations
SFG amide I spectra were calculated using the one-exciton approach described previously.42 The diagonal (local-mode) frequencies of the Hamiltonian were assumed to be similar for all amide groups, which would correspond to a situation in which all amide groups are hydrogen-bonded equally (either to water molecules or other amide groups). Only the frequencies of amide groups upstream of proline residues in the amino acid chain are red-shifted by 19 cm−1 with respect to the local-mode frequency of other residues, 1650 cm−1, which is similar to the values used in previous publications.43,44 The off-diagonal elements were calculated with different methods for nearest- and non-nearest neighbors. The nearest-neighbor couplings are dominated by through-bond effects, and thus derived from an ab initio calculated map45 that gives the coupling as a function of the dihedral angle between the neighboring residues.46 The non-nearest neighbor couplings are dominated by through-space effects, and thus determined with the Coulomb-like transition dipole coupling (TDC) model.47,48 The time-independent Schrödinger equation was then solved by diagonalizing the Hamiltonian to obtain the eigenmodes and eigenvalues of the delocalized normal modes. From these, the IR and Raman responses were calculated and, by taking the outer product of the two responses, the SFG response. The spectra were subsequently calculated by convoluting these responses with Lorentzians that have a total half width at half maximum (which is the sum of the half width of the VIS pulse and the homogeneous linewidth) of 19 cm−1.3. Results and discussion
3.1. Conformational analysis of AFP III in water
To determine the secondary structure content of unbound AFP III in solution and to study a potential temperature effect on its conformation, far-UV CD spectra of AFP III were measured in water (Fig. 2). The CD spectra of AFP III exhibit a positive band around 220 nm and a large negative band near 195 nm, consistent with spectra of native AFP III reported previously.49,50 Based on the secondary structure analysis of the CD spectra, AFP III consists of unordered structures (∼60–70%), β-strands (∼20%) and turns (∼10%), and has basically no helical content (see Table S1, ESI†). The secondary structure thus inferred is consistent with X-ray and NMR data that reported the presence of two three-stranded β-sheets and little-to-no helical features.8,10,12 In addition, the secondary structure of AFP III in solution was not significantly altered by a change in temperature, even at near-freezing temperatures, which supports previous findings indicating that temperature had only a minimal effect on the crystal structure of fish AFP III, with the exception of minor changes in side-chains conformation.51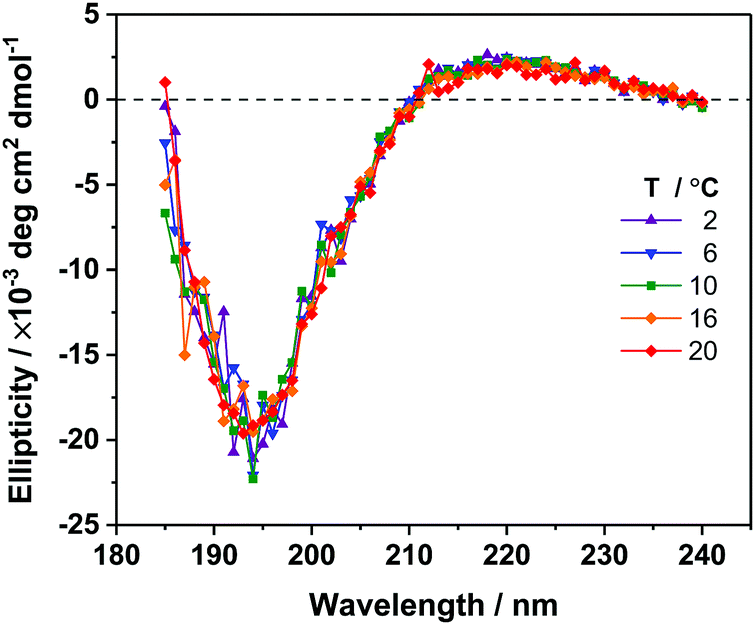 | ||
| Fig. 2 Far-UV CD spectra of AFP III in 5 mM phosphate buffer at pH 7.5, for different temperatures. | ||
3.2. Conformational and orientational analysis of AFP III at the air–water interface
To investigate the adsorption and orientation of AFP III at the air–water interface, SFG experiments were performed in parallel with MD simulations. First, amide I spectra of the interfacial region were obtained using SFG spectroscopy. This second-order nonlinear spectroscopic method has the advantage of being intrinsically surface-specific due to selection rules, which dictate that SFG is only allowed in a medium lacking inversion symmetry (e.g., the interfacial layer between two bulk isotropic media).52–54 SFG spectroscopy can provide information about the interfacial conformation and orientation of peptide and protein secondary structures.55 Surface tension measurements confirmed that AFP III has a strong propensity for the air–water interface. At equilibrium, AFP III decreased the surface tension of pure heavy water (72.3 mN m−1 at 22 °C) to ∼59 mN m−1, a value slightly higher than those reported previously at comparable protein concentrations,20,21 which could be explained by the absence of solution stirring.A representative amide I SFG spectrum of the AFP III layer at the air–water interface is shown in Fig. 3a. The SFG spectrum, together with a fit (for fitting parameters, see Table S2, ESI†), shows a broad backbone amide I band centered at ∼1655 cm−1. Peaks in this frequency range are often assigned to α-helices, but also to random coil structures.55–59 However, this interpretation would be at odds with the crystal structures of AFP III determined by X-ray crystallography and NMR,8–10 where practically no helical features were found. Signal contributions from the β-strands arranged in two triple β-sheets and the predicted 310-helices are ostensibly missing, which means that these structural features may either be too small or flexible to generate significant vibrational couplings that would result in corresponding spectral contributions to the SFG signal, or their respective signals are not clearly discernable in the far field. The latter is more likely because, contrary to linear vibrational spectroscopy, the coherent SFG signal from different secondary structure elements within the protein will interfere based on their relative phase and orientation. Presumably, none of the signal near 1655 cm−1 discussed above is related to helical structures but is the result of interference between sheets and other, non-standard folding motifs. To extract structural and orientation information from complex SFG spectra of multi-domain proteins, a theoretical analysis of the spectral signature and the surface structure can help disentangle spectral contributions from different protein sites. Direct spectral inspection or fitting can lead to significant misinterpretation and uncertainties.
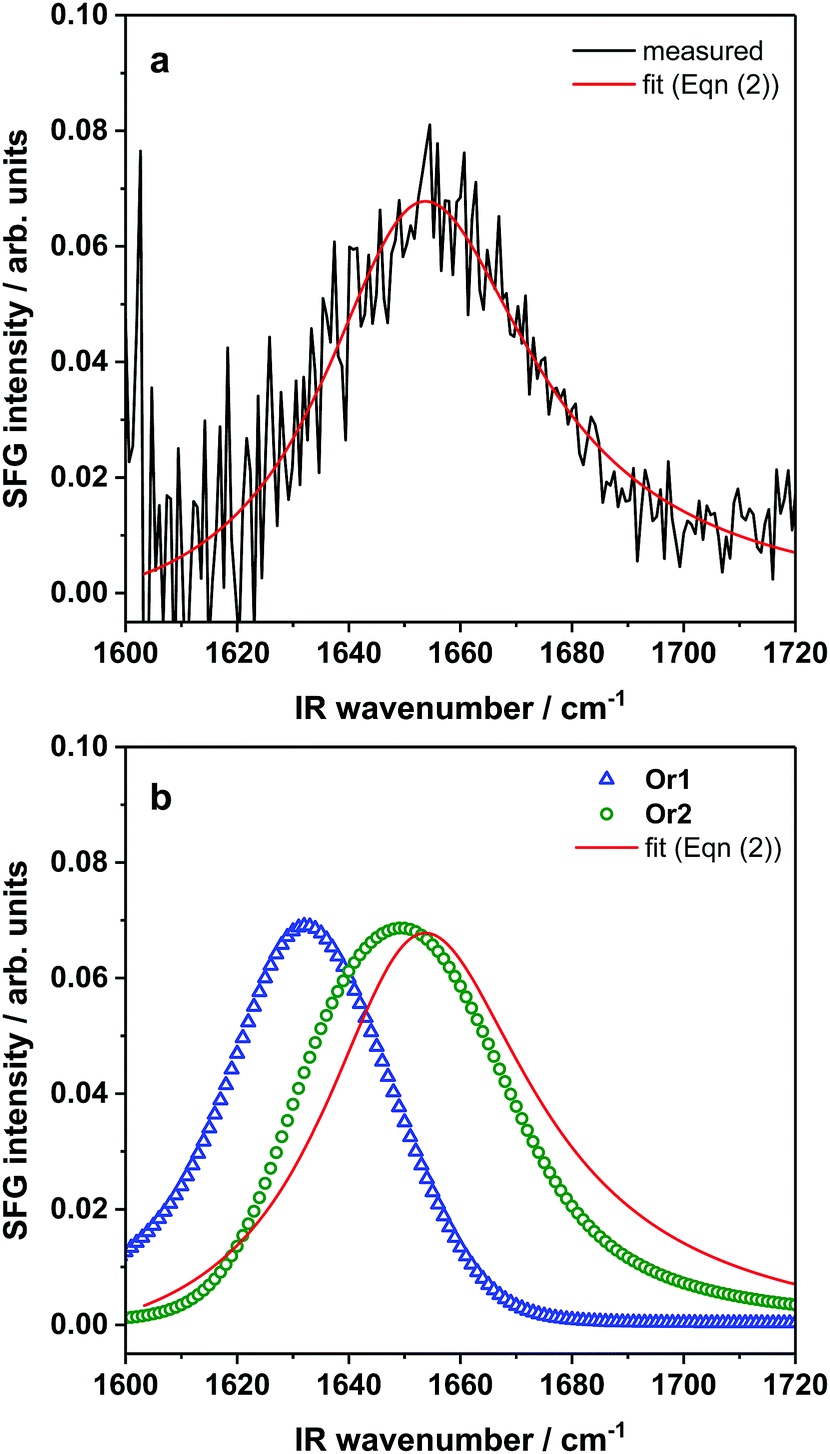 | ||
| Fig. 3 (a) Measured amide I SFG spectrum and fit of AFP III adsorbed at the air–water interface and (b) calculated SFG spectra from MD simulation snapshots of AFP III in Or1 and Or2. The spurious noise observed at low frequencies in the experimental SFG spectrum is due to the normalization procedure. | ||
For a closer look at how the IBS is folded and oriented, the SFG experiments were complemented with MD simulations. As has been demonstrated recently, direct calculations of amide I SFG spectra from MD simulation snapshots through an exciton model, compared with experimental spectra can provide information about the folding and absolute orientation of peptides and proteins.42 Short simulation runs (50 ns) showed that AFP III readily adsorbs to the air–water interface, in good agreement with the surface tension data. In its bound state, AFP III assumes one of two possible orientations (hereafter referred to, respectively, as Or1 and Or2) with respective tilt angles of ∼50° and ∼120° relative to the surface normal (Fig. 4 and see Table S3, ESI†). In both orientations, AFP III has its long axis (defined by the vector d between residues M1 and I14) inclined relative to the water surface, but with the major difference that different domains of the IBS are exposed to the air–water interface (Fig. 5). In Or1, key residues (T18, L19, V20, and V41) involved in binding to the primary prism ice plane37 are predominantly exposed to the vapor phase, whereas in the case of Or2, residues (Q9, L10, A16) belonging to the pyramidal ice binding domain, are facing the vapor phase. Because both domains of the IBS contain several hydrophobic residues, it is difficult to determine which of these has a greater affinity for the vapor phase and therefore which protein orientation would be preferred. For the wild-type AFP III, five of the trials initially adsorbed to Or1 (trials 1, 2, 3, 9, and 10) and the other half adsorbed to Or2. Similarly, four of the mutant structures (trials 1, 2, 3, and 8) adsorbed to Or1, and the remaining adsorbed to Or2. These structures were observed to adsorb and remain stable over at least 40 ns of simulation time.
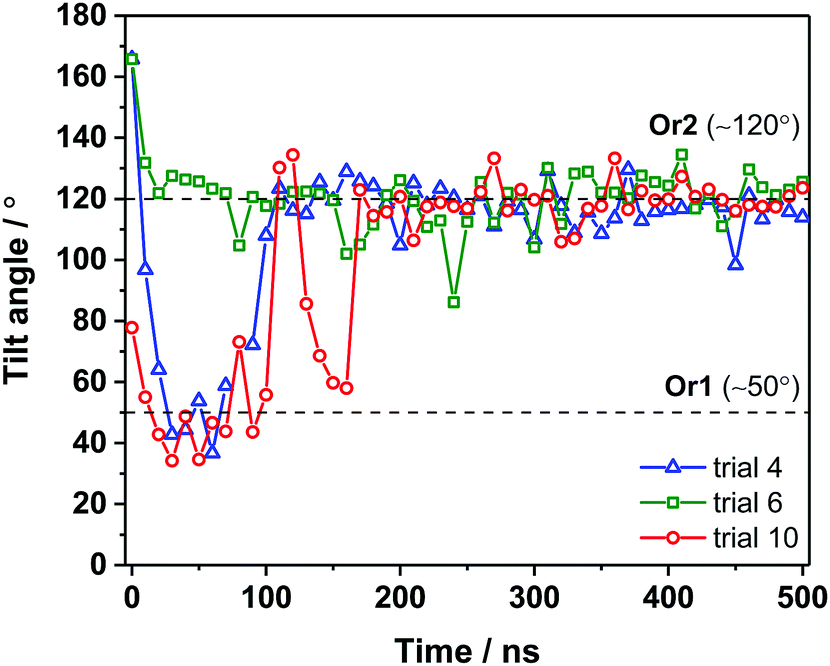 | ||
| Fig. 4 Tilt angle of AFP III adsorbed at the air–water interface over 500 ns simulation runs. For clarity, simulation data is given at every 10 ns step. Breaks in the data indicate that occasionally the protein briefly desorbed such that the tilt angle could not be calculated. | ||
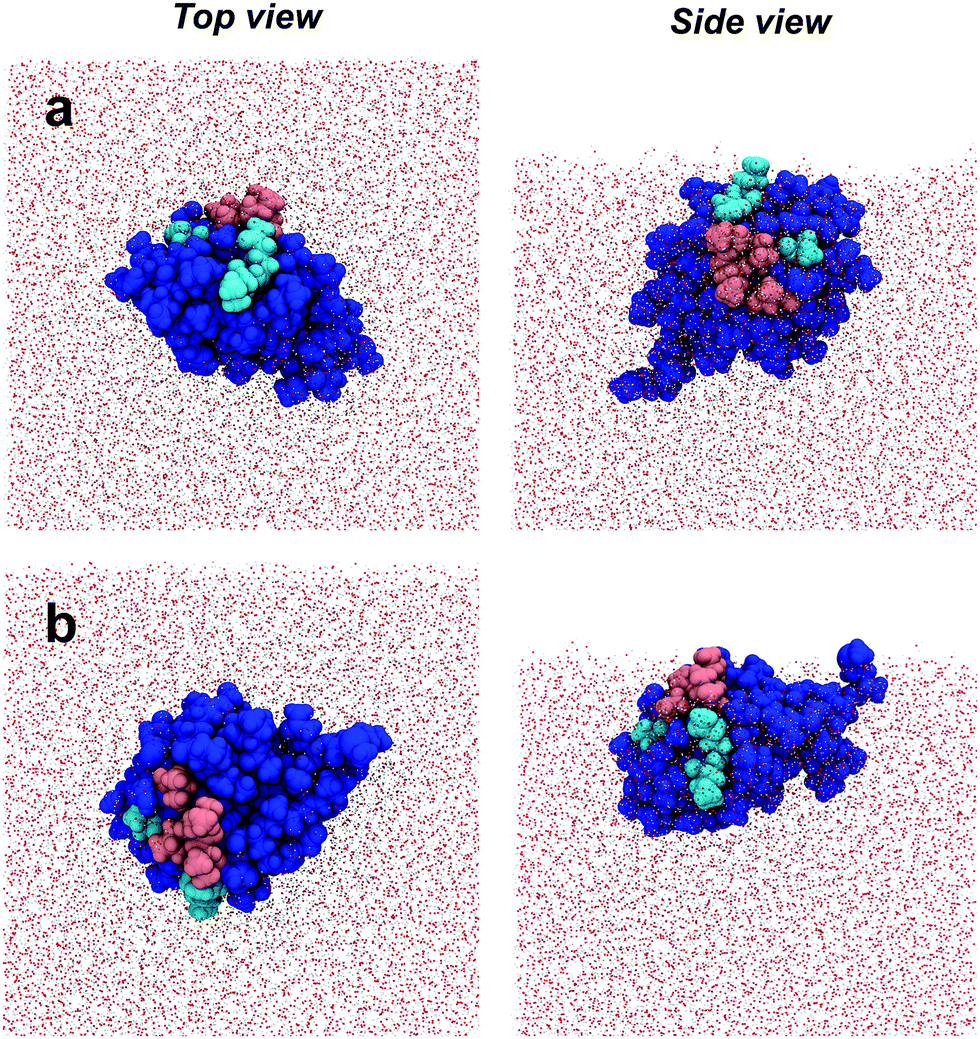 | ||
| Fig. 5 Simulation snapshots of AFP III adsorbed at the air–water interface. Top and side views of (a) orientation 1 (Or1) and (b) orientation 2 (Or2). The IBS residues that bind to the primary prism and pyramidal ice planes are highlighted in magenta and cyan, respectively. | ||
To get further insight into AFP III orientation and to assess potential conformational changes (or even denaturation) at the air–water interface, longer simulation runs (500 ns) were also performed on AFP III adsorbed in Or1 and Or2 (Fig. 4). Changes in backbone RMSD over 500 ns of simulation time were less than 0.2 nm for both the wild-type and mutant AFP III. This is within the thermal fluctuations of a protein backbone, indicating no large conformational changes to the secondary structure. It turns out that at longer time scales, AFP III initially found in Or2 remained in the same orientation, whereas proteins in Or1 ultimately converged to Or2 after about 200 ns (e.g., see trial 10 in Fig. 4), thus indicating that the latter orientation is likely the most stable. After sufficiently long times (>300 ns), AFP III stabilizes at the air–water interface with a tilt angle that follows a narrow angular distribution (119° ± 7°). Secondary structure analysis on one of the long MD trajectories (trial 10) further revealed that AFP III mostly maintains its native fold at the air–water interface (Fig. S6 and see also Fig. S2a, ESI†), even though conformational changes are observed, mainly during the adsorption process. The secondary structure analysis of unbound AFP III (up to 150 ns) showed that AFP III consists of β-sheet-like structures (51%), turns (∼43%), as well as some helical features (∼7%). Apart from the presence of helical content, these fraction values agree relatively well with those obtained from the CD spectra analysis (see Table S1, ESI†), thus validating the choice of crystal structure in the MD simulations as a representative of the protein structure in solution.
To test whether the MD simulations are in agreement with the SFG results, theoretical SFG spectra were directly calculated from snapshots of the simulation. The calculated SFG spectrum of AFP III in Or2 is in good agreement with the experimental data for the AFP III monolayer (Fig. 3b). It reproduces well the band position and spectral shape of the experimental spectrum. In contrast, the Or1 state results in a calculated spectrum that is significantly shifted with respect to the experimental spectrum. The spectral differences are almost exclusively related to differences in orientation (see Fig. S5, ESI† for additional spectra for different orientation as reference). SFG and MD results therefore indicate that AFP III most likely favors the Or2 state where it adsorbs to the air–water interface with its long axis oriented about 120° to the surface. The large spectral difference observed for the two orientations highlights the sensitivity of spectra calculations to orientational changes. The calculations also enabled to determine the spectral responses of the three types of secondary structures, namely α-helices, β-sheets and coils, for AFP III in Or2 (see Fig. S5, ESI†). As expected, the response from α-helices appears as a relatively narrow band centered at ∼1642 cm−1, whereas β-sheets display their typical band splitting with intensity at 1635 and 1670 cm−1 corresponding, respectively, to antiparallel and parallel β-sheet arrangements.55 Coil structures, on the other hand, gave a rather broad response across the whole amide I region, which taken together with the contributions of the other secondary structures, results in an overall broad SFG spectrum.
The agreement between calculations and experiments lends additional credence to the MD results. Therefore, the interfacial structure of AFP III, as determined with MD simulations, was analyzed in more detail, especially with respect to the orientation of the IBS domains and their exposure to water. This is important because exposure of the IBS to water is a critical assumption made in spectroscopic and functional studies of AFPs at hydrophobic surfaces. The analysis shows that the pyramidal ice domain of the IBS is mostly exposed to the vapor phase and is decoupled from water. The primary prism ice domain, on the other hand, remains partially submerged in the aqueous phase (Fig. 5). It has been shown that, in bulk water, AFP III does not aggregate, even at physiological concentrations, and remains surrounded by liquid water.21 However, contrary to the primary prism ice domain of the IBS, water molecules in contact with the pyramidal ice domain are known to be rather disordered and dispersed over the surface, therefore not promoting the formation of an ice-like water lattice.25 It is possible that, by localizing at the air–water interface, AFP III could “shake off” the water molecules from the pyramidal ice domain more easily, thereby exposing the largest area of its hydrophobic IBS to the vapor phase. The increased hydrophobic interactions with the vapor phase could thus explain, at least partly, the preferential orientation of AFP III at the air–water interface. Although AFP III does not aggregate in bulk water, the presence of protein–protein interactions in a protein layer adsorbed at the air–water interface cannot be ruled out. For instance, it is well known that AFP III tends to crystallize by interacting with neighboring proteins through the IBS.25 It is therefore possible that, in a densely packed film at high protein concentrations, the primary prism ice domain will also be decoupled from water due to protein–protein interactions. This might also explain the concentration-dependent changes of the water structure near AFP III monolayers reported by Bakker and co-workers.22 In view of the laterally oriented prism ice domain, this observation may be explained by an increasing exclusion of water molecules from the IBS due to protein–protein contacts.
To test whether the orientation with respect to hydrophobic surfaces is related to the antifreeze activity of AFP III, MD simulations were run on the T18N AFP III mutant. The results show that the orientation of T18N AFP III also converges towards Or2 on longer time scales, although it takes slightly more time (300 ns) than native AFP III to reach this orientation (see e.g., trials 1 and 3 in Fig. S3, ESI†). The orientation of the T18N mutant at the air–water interface has been discussed in previous works. On the one side it has been hypothesized, that the mutation does not affect AFP III orientation and surface propensity, probably because AFP III and the mutant are comparable in terms of conformation and hydrophobicity.22 At the same time a very recent simulation study has concluded that the differences in water interaction for wild type and T18N AFP III could be related to differences in the distance between residue 18 and the interface, i.e. the orientation. However, the latter study reported shorter simulation times and fewer trials than in the present study and the simulations reported here show that after 300 ns the orientation and the location of residue 18 with respect to the interface is largely unaffected by the T18N mutation (see Fig. S8–S10, ESI† for location of residue 18).23 The angular distribution of T18N AFP III closely resembles that of AFP III with an average tilt angle of 119° ± 10°. Similar to native AFP III, the pyramidal ice binding domain of the mutant AFP III becomes exposed to the vapor phase in Or2 (see Fig. S4b, ESI†). Furthermore, secondary structure analysis of T18N AFP III adsorbed at the air–water interface reveals that the conformation of AFP III remains unaffected by the mutation as the fractions of secondary structure of T18N AFP III are highly similar to that of the native protein (compare, for example, fraction percentages at 500 ns in Fig. S6, ESI†).
4. Conclusion
We have examined the conformation and orientation of AFP III at the hydrophobic air–water interface. Combined SFG/MD analysis indicates that AFP III remains in a native state when bound to the air–water interface and predominantly adopts an orientation with the protein's long axis at a tilt angle of 120° relative to the surface normal. In this particular orientation the IBS is only partially solvated, leaving the pyramidal ice binding domain exposed to the vapor phase. We argue that the water molecules are more easily displaced from this domain (because they are known to be less ordered and dispersed) in favor of hydrophobic interactions between the exposed residues and the vapor phase. More generally, these results suggest that the orientation of AFP III adsorbed to other hydrophobic interfaces such as lipid and artificial membranes should cause a similar partial decoupling of the IBS from water molecules, which could lead to the loss of its antifreezing activity. This will be important to consider when designing experiments to study the freezing mechanisms of antifreeze proteins at interfaces. It will also be an important factor for the development of protein-based freeze/antifreeze applications and in strategies for biomimetic organs and blood cryo-preservation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. V. and T. W. gratefully acknowledge the Aarhus University Research Foundation (AUFF) for financial support. S. A. acknowledges the NSF (Grant No. DGE-1633216). T. W. is grateful for financial support by the Deutsche Forschungsgemeinschaft (WE 4478/4-1).References
- M. Bar Dolev, I. Braslavsky and P. L. Davies, Annu. Rev. Phys. Chem., 2016, 85, 515–542 CAS.
- R. C. F. Cheung, T. B. Ng and J. H. Wong, Curr. Protein Pept. Sci., 2017, 18, 262–283 CrossRef CAS PubMed.
- P. L. Davies, Trends Biochem. Sci., 2014, 39, 548–555 CrossRef CAS PubMed.
- J. A. Raymond and A. L. DeVries, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 2589–2593 CrossRef CAS.
- J. A. Raymond, P. Wilson and A. L. DeVries, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 881–885 CrossRef CAS.
- A. L. DeVries and C.-H. C. Cheng, in Fish Physiology, ed. J. Steffensen, Elsevier, 2005, ch. 4, vol. 22, pp. 155–201 Search PubMed.
- Fish Antifreeze Proteins, ed. K. V. Ewart and C. L. Hew, Molecular Aspects of Fish & Marine Biology, vol. 1, World Scientific, Singapore, 2012 Search PubMed.
- Z. Jia, C. I. DeLuca and P. L. Davies, Nature, 1996, 384, 285–288 CrossRef CAS PubMed.
- F. D. Sönnichsen, C. I. DeLuca, P. L. Davies and B. D. Sykes, Structure, 1996, 4, 1325–1337 CrossRef.
- F. D. Sönnichsen, B. D. Sykes, H. Chao and P. L. Davies, Science, 1993, 259, 1154–1157 CrossRef.
- C. Wilkens, J. N. Poulsen, H. Ramlov and L. Lo Leggio, Cryobiology, 2014, 69, 163–168 CrossRef CAS PubMed.
- A. A. Antson, D. J. Smith, D. I. Roper, S. Lewis, L. S. D. Caves, C. S. Verma, S. L. Buckley, P. J. Lillford and R. E. Hubbard, J. Mol. Biol., 2001, 305, 875–889 CrossRef CAS PubMed.
- G. Chen and Z. Jia, Biophys. J., 1999, 77, 1602–1608 CrossRef CAS PubMed.
- D. S. Yang, W. C. Hon, S. Bubanko, Y. Xue, J. Seetharaman, C. L. Hew and F. Sicheri, Biophys. J., 1998, 74, 2142–2151 CrossRef CAS PubMed.
- C. P. Garnham, R. L. Campbell and P. L. Davies, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7363–7367 CrossRef CAS PubMed.
- D. R. Nutt and J. C. Smith, J. Am. Chem. Soc., 2008, 130, 13066–13073 CrossRef CAS PubMed.
- Y. Yeh and R. E. Feeney, Chem. Rev., 1996, 96, 601–618 CrossRef CAS PubMed.
- H. Nada and Y. Furokawa, Polym. J., 2012, 44, 690–698 CrossRef CAS.
- M. Schauperl, M. Podewitz, T. S. Ortner, F. Waibl, A. Thoeny, T. Loerting and K. R. Liedl, Sci. Rep., 2017, 7, 11901 CrossRef PubMed.
- N. Du, X. Y. Liu and C. L. Hew, J. Phys. Chem. B, 2006, 110, 20562–20567 CrossRef CAS PubMed.
- A. G. Salvay, F. Gabel, B. Pucci, J. Santos, E. I. Howard and C. Ebel, Biophys. J., 2010, 99, 609–618 CrossRef CAS PubMed.
- K. Meister, S. Strazdaite, A. L. DeVries, S. Lotze, L. L. C. Olijve, I. K. Voets and H. J. Bakker, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17732–17736 CrossRef CAS PubMed.
- Z. F. Brotzakis, I. K. Voets, H. J. Bakker and P. G. Bolhuis, Phys. Chem. Chem. Phys., 2018, 20, 6996–7006 RSC.
- Y. Xu, A. Bäumer, K. Meister, C. G. Bischak, A. L. DeVries, D. M. Leitner and M. Havenith, Chem. Phys. Lett., 2016, 647, 1–6 CrossRef CAS.
- T. Sun, S. Gauthier, R. L. Campbell and P. L. Davies, J. Phys. Chem. B, 2015, 119, 12808–12815 CrossRef CAS PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed.
- L. Whitmore and B. A. Wallace, Nucleic Acids Res., 2004, 32, W668–W673 CrossRef CAS PubMed.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1-2, 19–25 CrossRef.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parinello, J. Chem. Phys., 2007, 126, 14101 CrossRef PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- M. W. Mahoney and W. L. Jorgensen, J. Chem. Phys., 2000, 112, 8910–8922 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- C. P. Garnham, A. Natarajan, A. J. Middleton, M. J. Kuiper, I. Braslavsky and P. L. Davies, Biochemistry, 2010, 49, 9063–9071 CrossRef CAS PubMed.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camiloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia and M. Parinello, Comput. Phys. Commun., 2008, 180, 1961–1972 CrossRef.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- W. G. Touw, C. Baakman, J. Black, T. A. H. te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2015, 43, D364–D368 CrossRef CAS PubMed.
- H.-F. Wang, W. Gan, R. Lu, Y. Rao and B.-H. Wu, Int. Rev. Phys. Chem., 2005, 24, 191–256 Search PubMed.
- S. J. Roeters, C. N. van Dijk, A. Torres-Knoop, E. H. G. Backus, R. K. Campen, M. Bonn and S. Woutersen, J. Phys. Chem. A, 2013, 117, 6311–6322 CrossRef CAS PubMed.
- H. Lu, H. Lutz, S. J. Roeters, M. A. Hood, A. Schafer, R. Muñoz-Espí, R. Berger, M. Bonn and T. Weidner, J. Am. Chem. Soc., 2018, 140, 2793–2798 CrossRef CAS PubMed.
- L. Schmüser, S. Roeters, H. Lutz, S. Woutersen, M. Bonn and T. Weidner, J. Phys. Chem. Lett., 2017, 8, 3101–3105 CrossRef PubMed.
- R. D. Gorbunov, D. S. Kosov and G. Stock, J. Chem. Phys., 2005, 122, 224904 CrossRef PubMed.
- P. Hamm and M. Zanni, Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, New York, 2011 Search PubMed.
- S. Krimm and Y. Abe, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2788–2792 CrossRef CAS.
- W. H. Moore and S. Krimm, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 4933–4935 CrossRef CAS.
- V. S. Ananthanarayanan, D. Slaughter and C. L. Hew, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1986, 870, 154–159 CrossRef CAS.
- A. G. Salvay, J. Santos and E. I. Howard, J. Biol. Phys., 2007, 33, 389–397 CrossRef CAS PubMed.
- Q. Ye, E. Leinala and Z. Jia, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 700–702 CrossRef CAS.
- Y. R. Shen, Appl. Phys. B: Lasers Opt., 1999, 68, 295–300 CrossRef CAS.
- Y. R. Shen, Fundamentals of Sum-Frequency Spectroscopy, Cambridge University Press, New York, 2016 Search PubMed.
- X. Wei, S. C. Hong, A. I. Lvovsky, H. Held and Y. R. Shen, J. Phys. Chem. B, 2000, 104, 3349–3354 CrossRef CAS.
- S. Ye, F. Wei, H. Li, K. Tian and Y. Luo, Adv. Protein Chem. Struct. Biol., 2013, 93, 213–255 CAS.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS PubMed.
- L. K. Tamm and S. A. Tatulian, Q. Rev. Biophys., 1997, 30, 365–429 CrossRef CAS PubMed.
- K. T. Nguyen, S. Le Clair, S. J. Ye and Z. Chen, J. Phys. Chem. B, 2009, 113, 12169–12180 CrossRef CAS PubMed.
- S. Krimm and J. Bandekar, Adv. Protein Chem., 1986, 38, 181–364 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03382j |
| This journal is © the Owner Societies 2018 |