
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The magnetic fingerprint of dithiazolyl-based molecule magnets†
Tommaso
Francese
ab,
Jordi
Ribas-Arino
 a,
Juan J.
Novoa
a,
Remco W.A.
Havenith
bcd,
Ria
Broer
b,
Coen
de Graaf
bef and
Mercè
Deumal
*a
a,
Juan J.
Novoa
a,
Remco W.A.
Havenith
bcd,
Ria
Broer
b,
Coen
de Graaf
bef and
Mercè
Deumal
*a
aDept. Ciència de Materials i Química Física, Secció Química Física & IQTCUB, Universitat de Barcelona, Martí i Franquès 1, Barcelona, E-08028, Spain. E-mail: merce.deumal@ub.edu
bTheoretical Chemistry, Zernike Institute for Advance Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
cStratingh Institute for Chemistry, University of Groningen, 9747 AG Groningen, The Netherlands
dGhent Quantum Chemistry Group, Department of Inorganic and Physical Chemistry, Ghent University, Krijgslaan 281 (S3), B-9000 Gent, Belgium
eDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Marcel·lí Domingo 1, 43007 Tarragona, Spain
fICREA, Passeig Lluis Companys 23, 08010, Barcelona, Spain
First published on 12th July 2018
Abstract
Magnetic bistability in organic-radical based materials has attracted significant interest due to its potential application in electronic devices. The first-principles bottom-up study herein presented aims at elucidating the key factors behind the different magnetic response of the low and high temperature phases of four different switchable dithiazolyl (DTA)-based compounds. The drastic change in the magnetic response upon spin transition is always due to the changes in the JAB magnetic interactions between adjacent radicals along the π-stacks of the crystal, which in turn are driven mostly by the changes in the interplanar distance and degree of lateral slippage, according to the interpretation of a series of magneto-structural correlation maps. Furthermore, specific geometrical dispositions have been recognized as a ferromagnetic fingerprint in such correlations. Our results thus show that an appropriate substitution of the chemical skeleton attached to the DTA ring could give rise to new organic materials with dominant ferromagnetic interactions.
Introduction
Theoretical predictions of the magnetic properties of switchable purely organic molecule-based magnets have experienced an incredible development during the last two decades.1–5 Some of these compounds present peculiarities that could not be easily explained. Our attention is paid to dithiazolyl (1,3,2-DTA, see Fig. 1a) compounds because they are one of the most prominent candidates for potential technological applications, like memory storage devices, sensors and quantum computers.6 Therefore, here we focus on why some DTA compounds present not only spin transition from a low (LT) to a high (HT) temperature configuration, but also bistability. In general terms, a bistable system presents two stable phases that can coexist within a reasonably wide temperature range. Often, the phase transition from LT to HT for DTA compounds can be driven by temperature and/or light.7,8 In fact, this is why they can be exploited for technological purposes. Note that the mechanism per se of the phase transition that characterizes these systems is out of the scope of this paper. Instead we are interested in describing the static magneto-response of experimentally well-characterized planar bistable DTA compounds. Let us stress the fact that, in a static study, it is assumed that a molecule-based material can be described by a single crystal structure (usually characterized by X-ray/powder diffraction experiments) over a range of temperatures of interest for simulation purposes, neglecting the effect that, for example, thermal fluctuations might have on these systems.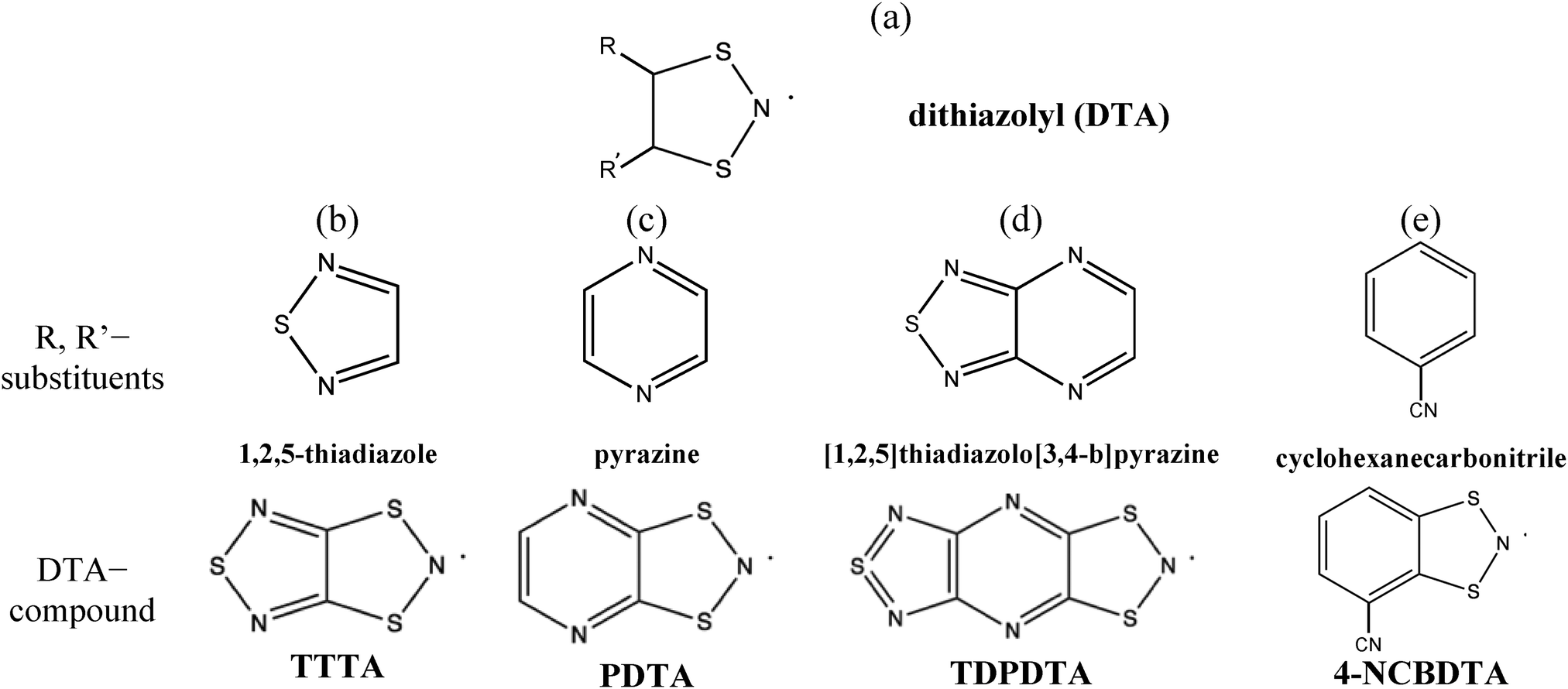 | ||
| Fig. 1 (a) General chemical skeleton of the DTA-ring, with R, R′ substituents. Note that the formal position of the unpaired electron is marked with a dot on the N atom of the DTA-ring. For (b) TTTA, (c) PDTA, (d) TDPDTA, and (e) 4-NCBDTA, the corresponding R, R′ substituents that, together with the DTA moiety, give rise to the DTA-based compounds investigated in this paper are shown. | ||
In particular, the properties of TTTA,9–11 PDTA,12 TDPDTA,13 and 4-NCBDTA14 materials will be investigated. According to magnetic susceptibility χT(T) data, all of them show bistability except 4-NCBDTA, which presents just a spin transition. Specifically, TTTA and PDTA bistabilities encompass room temperature, while for TDPDTA it occurs at much lower temperature. They all share the same dithiazolyl (DTA) chemical skeleton, where the nitrogen formally hosts an unpaired electron (see Fig. 1a–e).
We are interested in understanding how the R, R′ substituents of the DTA-moiety (Fig. 1a) affect the magnetic behavior. The four selected DTA compounds present a common trend in the solid state: the planar DTA radicals pile up forming stacks (see the schematic representation in Fig. 2a–c). Accordingly, the interaction between the π-systems of neighboring DTA-radicals is expected to be responsible for the dominant intermolecular magnetic interactions in these systems, as concluded from a previous First-Principles Bottom-Up (FPBU15) study on the TTTA compound.16 Let us now briefly describe the main features of the compounds under study.
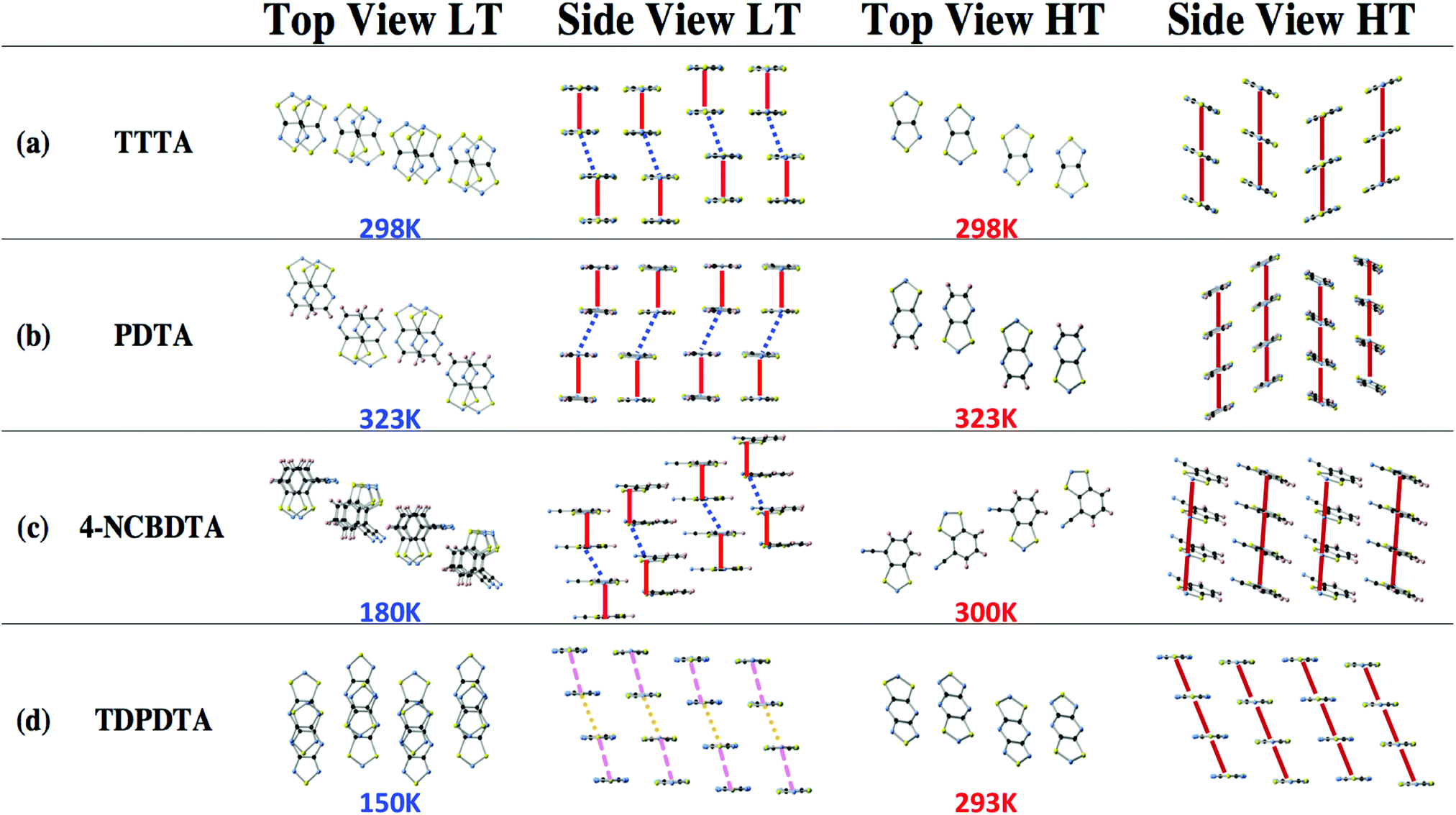 | ||
| Fig. 2 Schematic representation of the crystal packing showing the top and side views of the common pattern of the DTA stacking for the low temperature (LT) and high temperature (HT) phases of TTTA, PDTA, 4-NCBDTA and TDPDTA (a–d, respectively). In the LT phases, a common crystal packing trend is followed by TTTA, PDTA and 4-NCBDTA where the eclipsed-π dimers (red line) are alternated with π-slipped dimers (dashed blue line). For the (d) LT-TDPDTA phase, the packing differs from the other three compounds, presenting an almost uniform arrangement (dashed purple and orange lines represent two different distances between radicals within the same column). In the HT configurations instead, all four materials follow the same regular π-stack configuration. | ||
TTTA exhibits bistability at room temperature (namely, TC↓ = 220 K to TC↑ = 315 K). It has been extensively studied both from the structural and the electronic perspectives.9–11 The structure has been experimentally resolved at different temperatures (150 K, 225 K, 250 K, 300 K and 310 K). The system undergoes a first-order phase transition between the LT and HT phases. The LT-TTTA diamagnetic phase belongs to the triclinic P1 space group. The HT-TTTA paramagnetic phase instead is monoclinic with space group P21/c. The columns of radicals in the LT phase are distorted π-stacks consisting of slipped pairs of nearly-eclipsed radicals (Fig. 2a-LT). In contrast, the columns of the HT phase at room temperature are regular π-stacks of radicals, where each single TTTA molecule exhibits a tilted overlap with its two adjacent TTTA molecules along the stacking direction (Fig. 2a-HT).
The PDTA system presents many similarities compared to TTTA. It is a planar bistable system, whose susceptibility curve encompasses room temperature (namely, TC↓ = 297 K to TC↑ = 343 K). The compound has been experimentally characterized at 95 K, 293 K and 323 K.12 As TTTA, it experiences a first-order phase transition. The LT-PDTA motif presents eclipsed π-pairs alternated with π-shifted pairs (Fig. 2b-LT); whereas the HT-PDTA phase presents a uniform π-stack pair pattern (Fig. 2b-HT). The LT-PDTA crystal structure belongs to the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , consisting of centrosymmetric pairs of PDTA dimers, while the HT-PDTA phase belongs to the monoclinic space group C2/c.
, consisting of centrosymmetric pairs of PDTA dimers, while the HT-PDTA phase belongs to the monoclinic space group C2/c.
The 4-NCBDTA system is not bistable but presents spin transition. The 4-NCBDTA structures studied were resolved at 180 K (LT) and 300 K (HT) by X-ray powder diffraction.14 Both the LT-4-NCBDTA and the HT-4-NCBDTA phases belong to the monoclinic space group P21/c. For the LT-4-NCBDTA phase, there is an alternation between eclipsed π-pairs and π-shifted pairs (Fig. 2c-LT). In contrast, for the HT-4-NCBDTA structure, the packing is uniform (Fig. 2c-HT).
The TDPDTA radical presents three fused rings, i.e. two five membered rings coupled by a six-membered ring. This has a direct effect on the crystal packing. Both the LT-TDPDTA and HT-TDPDTA phases belong to the triclinic space group P1.13 The system shows a hysteretic behavior, with the temperature range between TC↓ = 50 K and TC↑ = 200 K. The HT phase displays the common uniform packing of ribbons of the TDPDTA radicals packed in a slipped π-stack arrangement, as the other three crystals (Fig. 2d-HT). However, the LT phase presents alternate layers of TDPDTA shifted laterally every two radicals within a given column, giving rise to almost uniform arrays of dimers (see Fig. 2d-LT).
To sum up, the current study of TTTA, PDTA, TDPDTA, and 4-NCBDTA addresses three main issues from a static perspective. Our first goal is to study by means of the FPBU15 approach the magnetic interactions between pairs of radicals to identify the magnetic topology of the molecule-based DTA crystals. Next, we aim at providing a magneto-structural correlation map as a function of the substituents of the DTA-moiety to highlight the ferromagnetic fingerprint region in DTA-based materials. At this point, we would like to stress that this magneto-structural map could be used as a practical tool to help experimentalists to design more stable and efficient purely organic radicals with ferromagnetic properties in the solid state. Finally, our objective is to assess whether structural (geometrical) as well as electronic (DTA-chemical skeleton, interactions between substituents) factors affect or not the magnitude of the significant radical···radical JAB magnetic couplings.
Methodological details
The First-Principles Bottom-Up (FPBU) working strategy has been extensively used to study molecule-based magnets.15–17 First, one selects all possible magnetically relevant pairs of radicals in the crystal by analysis of the crystal packing from the X-ray resolved experimental structure. As for the DTA-based crystals, although the spin density of a DTA radical is delocalized over the atoms of the entire DTA-ring (see the ESI† Section 1), the pairs of radicals have been chosen based on the N*⋯N* distance, where N* refers to the nitrogen atom that formally holds the unpaired electron (Fig. 1a). The N*⋯N* threshold distance was set to 10.0 Å to select di pairs of radicals because it is known that the spin-coupling interaction exponentially decays with distance.18Next, one calculates the radical⋯radical JAB spin-coupling interaction for each pair of radicals selected in the crystal. The microscopic JAB magnetic interaction is evaluated in terms of energy differences. Therefore, for the energy calculations, the neutral environment of any given DTA-radical must be well described. Direct observation of the crystal hints at using a two-radical cluster as a model. From the general Heisenberg Hamiltonian ( ) for an A–B pair of S = 1/2 radicals, the JAB value can be defined as JAB = [EBS − ET],19 where ET and EBS are the triplet and singlet broken symmetry20 energies of the dimer cluster model, respectively. All energy calculations were performed at the DFT/UB3LYP21 level as implemented in the Gaussian09 package.22 The standard 6-31+G(d) basis set23 was used in all energy calculations.
) for an A–B pair of S = 1/2 radicals, the JAB value can be defined as JAB = [EBS − ET],19 where ET and EBS are the triplet and singlet broken symmetry20 energies of the dimer cluster model, respectively. All energy calculations were performed at the DFT/UB3LYP21 level as implemented in the Gaussian09 package.22 The standard 6-31+G(d) basis set23 was used in all energy calculations.
Once all JAB exchange couplings have been computed, one must propose the magnetic topology of the crystal in terms of the non-negligible JAB magnetic interactions. This step is extremely important because it enables us to visualize how the microscopic magnetic interactions propagate. The use of Statistical Mechanics to calculate the macroscopic magnetic properties of the DTA-based crystals is bound to the selection of a magnetic model, whose extension along (a, b, c) crystallographic axes regenerates the whole magnetic topology.
Finally, having chosen the magnetic models, one constructs the matrix representation that contains all JAB values required to appropriately parameterize the Heisenberg Hamiltonian. The energy eigenvalues and corresponding spin quantum numbers that result from the full diagonalization of the adequate matrix on the space of spin functions of the magnetic models are used to calculate the magnetic susceptibility χT(T) data for each magnetic model we select using the corresponding expression provided by Statistical Mechanics.24 Finally, the simulated data for the systems studied by means of the FPBU working strategy are compared to the experimental data.
Results and discussion
The static analysis of the four DTA-based materials studied in this work is reported in three different sections. In the first section, the compounds are analyzed by means of the FPBU approach, providing a unique view of the driving spin-coupling interactions and how they propagate within the experimentally X-ray resolved crystal. The second section, instead, deals with the study of geometrical factors and their correlation with magnetic coupling interactions. In this case, we will make use of a set of representative structural models of each system to explore the different magneto-structural correlations displayed by each DTA-based material. The aim of this study is to uncover the intermolecular arrangement that most likely favors ferromagnetic (FM) interactions. Finally, the third section will quantify the contribution of electronic and structural factors to the overall JAB exchange coupling interactions.1. Magnetic susceptibility curves: calculated vs. experimental
Previous studies on TTTA16,25 and 4-NCBDTA26 have already reported their magnetic topology and their corresponding simulated magnetic susceptibility curves. For both compounds, good agreement was accomplished with respect to the experimental data. Notice that the magnetic topology is defined in terms of non-negligible JAB exchange interactions between radicals.27 Therefore, the magnetic topology embraces all the most important JAB spin coupling interactions (see the ESI,† Section 2, for a list of the atomic coordinates of the magnetically dominant pairs of radicals). For the LT-TTTA phase, the basic magnetic motif can be described as an antiferromagnetic (AFM) 1D chain formed by alternating eclipsed and slipped dimers. There are large AFM interactions (JLT-TTTAeclipsed![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) dimer = −1755 cm−1) that are responsible for the overall diamagnetic behavior of this phase, which are then connected by weaker JLT-TTTAslippeddimer coupling interactions (−50 cm−1) along the π-stack (see Fig. 2a).16 This basic magnetic motif is then interconnected in a three-dimensional (3D) network of much weaker coupling interactions that can be neglected for simulation purposes. The HT-TTTA magnetic motif consists instead of regular AFM 1D chains (JHT-TTTAregulardimer = −135 cm−1, see Fig. 2b), which explains the experimentally observed weak paramagnetism. Likewise, the 4-NCBDTA basic magnetic motifs resemble those for TTTA, presenting similar magnetic exchange coupling values, namely JLT-4NCBDTAeclipseddimer = −1700 cm−1 and JLT-4NCBDTAslippeddimer = −80 cm−1 for the LT phase, and JHT-4NCBDTAregulardimer = −340 cm−1 for the HT phase.26
dimer = −1755 cm−1) that are responsible for the overall diamagnetic behavior of this phase, which are then connected by weaker JLT-TTTAslippeddimer coupling interactions (−50 cm−1) along the π-stack (see Fig. 2a).16 This basic magnetic motif is then interconnected in a three-dimensional (3D) network of much weaker coupling interactions that can be neglected for simulation purposes. The HT-TTTA magnetic motif consists instead of regular AFM 1D chains (JHT-TTTAregulardimer = −135 cm−1, see Fig. 2b), which explains the experimentally observed weak paramagnetism. Likewise, the 4-NCBDTA basic magnetic motifs resemble those for TTTA, presenting similar magnetic exchange coupling values, namely JLT-4NCBDTAeclipseddimer = −1700 cm−1 and JLT-4NCBDTAslippeddimer = −80 cm−1 for the LT phase, and JHT-4NCBDTAregulardimer = −340 cm−1 for the HT phase.26
Here we report the magnetic topology for both HT- and LT-phases of the PDTA and TDPDTA systems (see Fig. 3a and c for HT and Fig. 3b and d for LT). We can clearly see how the main JAB magnetic interaction propagates along the DTA π-stacking direction of the crystal structure for all phases (see the strongest JAB couplings highlighted in red in tables inserted in Fig. 3). For PDTA (Fig. 3a and b), the alternating eclipsed radical pair and slipped dimer pattern in the LT-phase become a regular π-stacking in the HT-phase, in accordance with the general tendency hitherto observed for DTA-based systems (see TTTA in Fig. 2a and b). One can realize that the magnetic topologies are three-dimensional (3D). Yet, the inter-π-stacking JAB magnetic interactions are at least one order of magnitude smaller that the strongest JAB π-stacking couplings (see tables inserted in Fig. 3). Consequently, while we are dealing with 3D magnetic topologies, for simulation purposes, the basic magnetic motif can be considered to be one-dimensional (1D) (see the ESI,† Section 3). Comparison between PDTA (Fig. 3a and b) and TDPDTA (Fig. 3c and d) shows that, although the inter-π-stacking JAB magnetic interactions have the same order of magnitude, the intra-π-stacking JAB magnetic couplings for TDPDTA are weaker than for PDTA. This is not an issue for the LT-TDPDTA phase, which can still be taken to be 1D for simulation purposes. However, for the HT-TDPDTA phase the intra- and inter-π-stacking JAB values might appear to be comparable. Thus, the 3D magnetic topology of the HT-TDPDTA phase has been analyzed to conclude that a 1D magnetic model can be nevertheless used for the calculation of the magnetic properties (see the ESI,† Section 3, for a detailed discussion on the magnetic models for PDTA and TDPDTA).
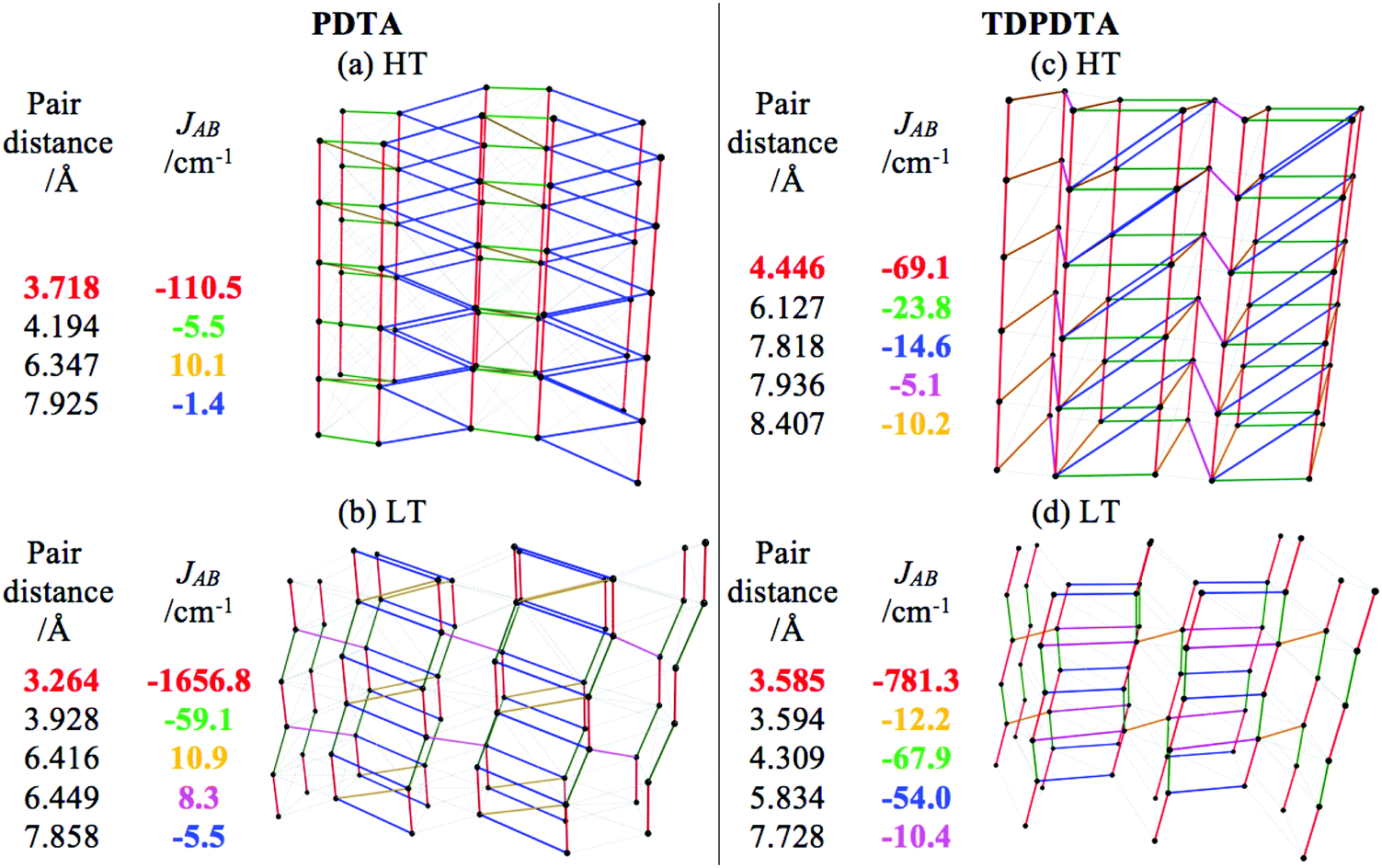 | ||
| Fig. 3 Magnetic topology for HT- and LT-phases of (a and b) PDTA and (c and d) TDPDTA crystals, respectively. Note that the N*⋯N* inter radical distance (in Å) and the corresponding JAB (in cm−1) are also given in a table next to the corresponding magnetic topology. |JAB| < 1 cm−1 are represented by light grey lines. | ||
The experimental and computed magnetic susceptibility χT(T) data for PDTA and TDPDTA show qualitative agreement with experiment (see Fig. 4). For the LT-PDTA phase, the shape of the calculated curve almost perfectly overlaps the measured χT(T) values (Fig. 4a). The simulated LT-PDTA phase behaves as experimentally measured because the radicals form nearly eclipsed pairs and there is an extremely strong AFM calculated JAB value (ca. −1650 cm−1). The LT-PDTA phase thus becomes magnetically silent, similarly to TTTA and 4-NCBDTA. Note that the large AFM value of JAB comes from the dominant magnetic interaction propagating along the DTA π-stacking direction in all the analyzed crystals (see Fig. 2a). Yet the experimental and calculated χT(T) values do not show a perfect correspondence for either the LT-TDPDTA or HT data of both crystals. The LT-TDPDTA and HT magnetic responses are qualitatively consistent with the general experimental shape. However, they do not numerically reproduce the measured χT(T) values. Although enlarging the magnetic model improves the computed χT(T) data (see the discussion in the ESI,† Section 3),25 the experimental values cannot be fully reproduced. Therefore, the study of PDTA and TDPDTA proves one more time that a static study of the magnetism of DTA-based systems has its limitations. For DTA-based materials, this flaw is due to the fact that a single geometry has been disclosed not to be representative enough of the system due to the thermal fluctuations that the crystal experiences.26,28 Despite being aware of the fact that molecular dynamics studies are needed to address this issue (and will soon be performed), the static results provide a rationale for the different magnetic responses of the two phases of PDTA and TDPDTA. The resulting data are in agreement with previous studies on TTTA and 4-NCBDTA. In all these four DTA-derivatives, the change in the magnetic response upon phase transition is due to different J(di) magnetic radical⋯radical interactions along the π-stacking direction. Therefore, one can safely conclude that the different magnetic behavior of the phases of bistable DTA-compounds is ruled by the same physical principles.
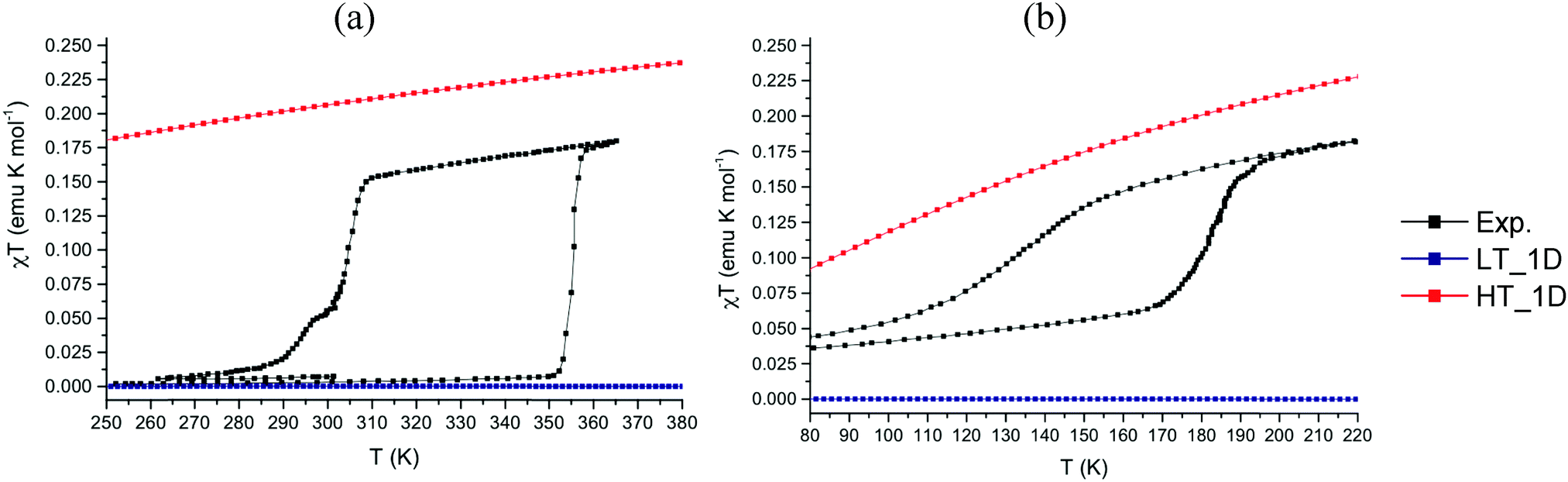 | ||
| Fig. 4 Comparison between experimental χT(T) data (in black) and computed magnetic susceptibility χT(T) for (a) PDTA and (b) TDPDTA using the most representative 1D minimal magnetic models that better reproduce the data for LT (in blue) and HT (in red) phases. | ||
2. Evaluating the nature of the magnetic interactions in DTA-based materials: magneto-structural correlation maps
We complemented our FPBU investigation screening the JAB values for a multiple set of DTA-based dimers as a function of two geometrical variables: namely, the interplanar distance (dIP) and the degree of slippage (dSL) between the two radicals. These two geometrical variables have been found to be of general application for a series of organic radicals in order to establish magneto-structural correlations.29 For all DTA-based materials studied here, the HT phase is the most interesting because it is not diamagnetic. Therefore, the study that we hereafter present aims at rationalizing the different experimental data of the magnetic susceptibility displayed by the HT phases of TTTA, PDTA, 4-NCBDTA and TDPDTA using magneto-structural correlation maps.To this aim, we selected one DTA-radical from the HT phase of each single crystal (TTTA, PDTA, TDPDTA, and 4-NCBDTA) as a reference, keeping the bond distances as resolved experimentally. This radical was then oriented in the xy-plane, and then duplicated along the z-axis to create the model used to screen the JAB values (see Fig. 5a for Model I of TTTA).
 | ||
| Fig. 5 (a) Model I used for sampling the respective dIP (in purple) and dSL (in green) distances to compute the magneto-structural correlation maps. Note that the DTA-radical shown is TTTA. (b) Schematic representation of the correlation maps. The x-axis provides the dIP contribution, whereas the y-axis provides the dSL contribution. The calculated JAB interactions are displayed along the z-axis, and also projected onto the xy-plane. | ||
Analyzing the geometry of the DTA-radicals within the experimentally resolved crystal, it is observed that the shortest and longest dIP interplanar distances belong to TDPDTA (3.305 Å) and 4-NCBDTA (3.524 Å) systems, respectively. The dSL degree of slippage has, instead, the shortest value for 4-NCBDTA (1.007 Å), whereas the largest is found in TDPDTA (2.974 Å). Accordingly, we have selected dIP interplanar distances ranging from 2.5 to 3.8 Å and dSL degrees of slippage ranging from 1.0 to 3.5 Å to obtain the correlation maps. Note that this distance range is large enough not only to explore those already known geometrical arrangements but also other dispositions that might be magnetically important. Therefore, the configurations created extend from an almost completely eclipsed dimer (dSL = 1.0) to the completely detached pair (dSL = 3.5) (see Fig. 5b). Specifically, the number of configurations along dIP is 130, while the number along dSL is 25. Each single magneto-structural correlation map is thus obtained by analyzing 3250 dimers, and computing the JAB coupling value for each pair of radicals.
Fig. 6 displays the calculated JAB interactions as dIP and dSL vary, and the corresponding projection onto the xy-plane. One can easily realize that our results show that we can locate not only regions with the expected highly AFM interactions, but also FM regions which have not yet been experimentally met. The JAB range of interest considered goes from −100 cm−1 up to +150 cm−1, as highlighted in the color scale legend. AFM coupling values beyond 100 cm−1 (JAB ≪ −100 cm−1) are represented in black, because such strong JAB spin coupling actually corresponds to a diamagnetic pair of radicals, which is magnetically silent. Thus, there is no possibility of exploiting the magnetic properties of the systems. In contrast, screening the different regions from JAB = −100 cm−1 up to the strongest FM area allows us to identify the radical pairs with geometrical configurations that favor FM interactions. Let us remark here that the slipped geometries of other organic radicals displaying FM interactions have also been predicted in the literature.29a,c
 | ||
| Fig. 6 Magneto-structural correlation maps for (a) TTTA (bistable), (b) PDTA (bistable), (c) 4-NCBDTA (spin switch), and (d) TDPDTA (bistable). | ||
The four DTA-based systems present very different magneto-structural correlation maps (see Fig. 6). As a consequence, the extension of the FM areas varies significantly. Indeed, direct observation of the correlation maps reveals that whether the compounds are bistable or just spin switchable does not depend on JAB. Contrarily, a correlation can be inferred between the number of fused rings of the DTA-radical and the tendency of the JAB magnetic coupling to show AFM values. Moving from two fused ring systems (TTTA, 4-NCBDTA, and PDTA) to three fused ring radicals (TDPDTA), the probability of orbital overlap to be efficient increases and, thus, the FM coupling is quenched.
The compound presenting the widest FM area is the prototypical TTTA system (Fig. 6a). The FM zone ranges from (dIP = 2.50 Å, dSL = 1.49 Å) to (dIP = 3.24 Å, dSL = 2.40 Å). The JAB values vary from 0 cm−1 (orange border line) to +87 cm−1 (brownish area). Between these two limiting values, we have a gradient of FM spin coupling values. Each depicted distinctive zone contains a set of radical pairs whose arrangement enhances the FM coupling. Since all JAB values have been computed at the UB3LYP20,21 level, our next goal is to prove the good description of the magnetic interaction between TTTAs (and DTA-radicals in general) provided by DFT.
First, the JAB magnetic coupling interactions computed at the DFT/UB3LYP level were benchmarked with the Difference Dedicated Configuration Interaction (DDCI3)30 method for our system. Therefore, a series of FM configurations from the TTTA correlation map were selected and their JAB values compared to the outcomes of the DDCI calculations (see A–C models in the ESI,† Section 4, for a detailed discussion). The corresponding JAB values calculated at the DDCI level confirm the existence of FM interactions associated with certain geometrical configurations (e.g. JDDCImodelA = 30.6 cm−1vs. JUB3LYPmodelA = 7.9 cm−1 in the ESI,† Section 4). In this sense, DFT, although less accurate than DDCI3, offers a cheap and consistent method to rely on for predicting FM/AFM coupling. Therefore, DFT can be trusted to evaluate FM and AFM tendencies, but not the absolute value of the JAB coupling per se.
A similar scenario to TTTA is also reported for PDTA (Fig. 6b) and 4-NCBDTA (Fig. 6c) compounds, although a contraction of the FM region is observed, i.e. there are fewer geometrical arrangements presenting FM coupling. Unexpectedly, a dissimilar magneto-structural correlation map is obtained by analyzing the TDPDTA compound (Fig. 6d). Herein, there is no presence of a FM area at all. To further investigate this trend, we have employed the orbital analysis as defined by Kahn's model.31 Accordingly, the FM JAB interactions are favored when the orbital overlap (OO) between the SOMO of the two radicals tends to be zero. Fig. 7 reports the orbital overlap analysis carried out for TTTA and TDPDTA compounds. Note that, for each magneto-structural correlation map, we analyzed four slices along dSL at fixed chosen dIP distances, using as reference TTTA. The dIP values (2.5, 3.1, 3.22 and 3.5 Å) were chosen to sample the most FM region, the FM boundary (JAB = 0 cm−1), the moderate AFM region, and the region where the experimentally resolved dimer should be located.
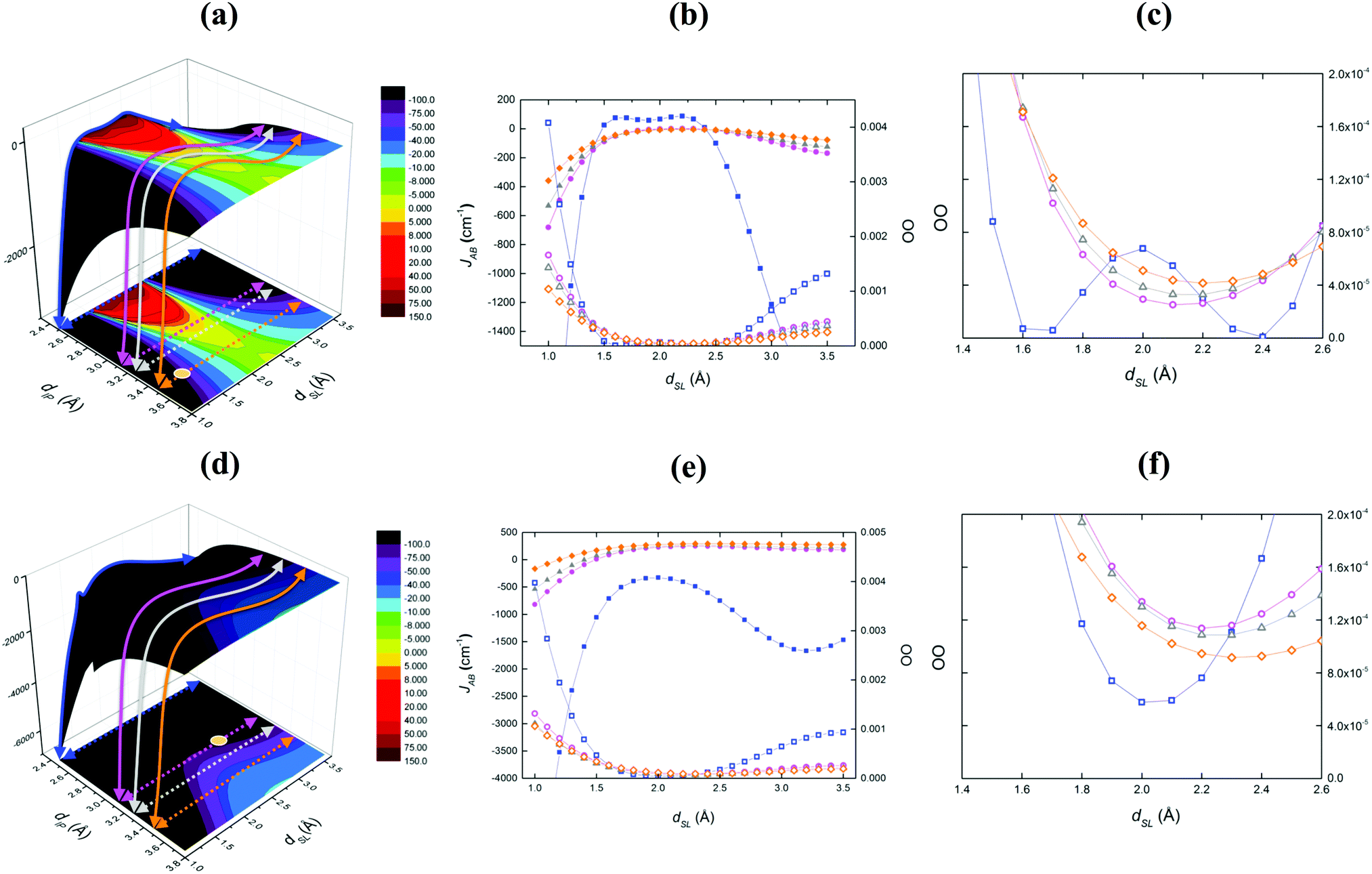 | ||
| Fig. 7 Orbital overlap (OO) analysis graphs for TTTA (a–c) and TDPDTA (d–f). (a) Fixed dIP values sampled at 2.5, 3.1, 3.22 and 3.5 Å in order to analyze the most FM region (blue line), the FM boundary (JAB = 0 cm−1, purple line), the moderate AFM region (gray line), and the region where the experimentally resolved TTTA dimer should be located (orange line and the corresponding orange dot in the projected map). (d) Same regions are analyzed for TDPDTA for comparison reasons. Note that the TDPDTA experimental geometry lies in the FM boundary (purple line). (b and e) Effective orbital analysis (OO) compared to the JAB values for the four slices in (a and d). The empty symbols refer to the orbital overlap OO data, while the full symbols refer to JAB data. (c and f) Zoom of the region with the smallest orbital overlap OO, ranging from 1.4 Å and 2.6 Å. | ||
For TTTA, the corresponding OO values are nearly zero when the JAB interaction between the radicals becomes FM (see the blue line for dIP = 2.5 Å in Fig. 7a). In fact, the zero-orbital overlap correlates with the interstitial disposition of the π orbitals of the two radicals within the dimer (see blue squares in Fig. 7b and c). This structural arrangement enhances the FM coupling between TTTA radicals. Contrarily, the same slice at 2.5 Å for TDPDTA displays OO values clearly different from zero (see blue squares in Fig. 7e and f). Thus, it agrees with JAB being AFM in all dSL ranges of sampled values (see the blue line in Fig. 7d). Indeed, one can realize that, in the case of TDPDTA, the degree of delocalization of the spin density on the fused rings to the DTA-moiety is larger than in the other three compounds. As a result, non-zero orbital overlap (OO) within the dimer is present in a much wider geometrical range and, in turn, any possibility for FM coupling is quenched.
The second cross-section of the TTTA material at dIP = 3.20 Å and dSL = 1.0–3.5 Å (see Fig. 7a, purple line) explores the boundary of the FM area. It clearly shows the tendency of OO to present non-negligible values (purple circles in Fig. 7b and c). The corresponding JAB interactions, instead, still display some ferromagnetism, in agreement with our assumption that the OO rather than being accurate is only one of the contributions (direct exchange and spin polarization are other factors that determine the total coupling). For the sake of comparison with TTTA, the same slice of interest is taken in the TDPDTA case (purple circles in Fig. 7e and f). Correspondingly, the OO analysis exhibits significant values, describing AFM spin couplings. Last but not least, always using the TTTA compound as our reference, we have also considered the cross-sections just outside the FM area (dIP = 3.30 Å, dSL = 1.0–3.5 Å) and in accordance with the position of the experimentally X-ray resolved TTTA dimer (dIP = 3.47 Å, dSL = 1.32 Å) (see grey and orange lines in Fig. 7a). Additionally, the position of the experimental structure of the TDPDTA dimer was also considered (dIP = 3.305 Å, dSL = 2.97 Å in Fig. 7d). The representation of the zoom in Fig. 7c and f shows the clear tendency of the orbital overlap towards larger values, both in TTTA (grey triangles) and TDPDTA (orange diamonds) cases, and hence towards larger AFM interactions.
For TDPDTA, a further analysis was carried out. The geometry of the Model I dimer (JAB = −89 cm−1) that most resembles the experimental structure (JAB = −70 cm−1) was selected. Then, we applied successive longitudinal translations (dLG = 0.1 Å) between both TDPDTA monomers, generating 10 different configurations (see the ESI,† Section 5). Specifically, one of the generated configurations (Conf. #4) presents dIP = 3.28 Å, dSL = 3.05 Å and JUB3LYPConf#4 = −69.6 cm−1, which are very close to the experimental available data (see Table 1), although internal distortions and degrees of freedom have been neglected in these newly generated models. Strikingly, the longitudinal translation accomplishes TDPDTA radical arrangements that show FM coupling (JUB3LYPConf#9 = 5.0 cm−1 and JUB3LYPConf#10 = 15.6 cm−1). This suggests that the magneto-structural correlation map of TDPDTA must also account for longitudinal translation (dLG) as well as interplanar distance (dIP) and degree of slippage (dSL) to capture the complete behavior.
| Parameters | TTTA (298 K) | J AB/cm−1 | TDPDTA (293 K) | |
|---|---|---|---|---|
| PDTA (323 K) | 4-NCBDTA (300 K) | |||
| Experimental data | ||||
| d IP-X-ray (Å) | 3.46 | 3.44 | 3.52 | 3.31 |
| d SL-X-ray (Å) | 1.34 | 1.40 | 1.01 | 2.97 |
| N*–N* (Å) | 3.71 | 3.72 | 3.67 | 4.45 |
| J AB (cm−1) | −135 | −111 | −342 | −69 |
| Model I | ||||
| d IP – TM (Å) | 3.46 | 3.43 | 3.52 | 3.27 |
| d SL – TM (Å) | 1.31 | 1.43 | 1.01 | 3.03 |
| N*–N* (Å) | 3.69 | 3.71 | 3.66 | 4.46 |
| J AB (cm−1) | −142 | −147 | −295 | −89 |
| Error % | 5.44 | 32.57 | 13.73 | 27.14 |
| Model II | ||||
| d IP – TM (Å) | 3.46 | 3.46 | 3.46 | |
| d SL – TM (Å) | 1.34 | 1.34 | 1.34 | |
| N*–N* (Å) | 3.71 | 3.71 | 3.71 | |
| J AB (cm−1) | −135 | −125 | −135 | |
| Error % | 0.16 | 7.46 | 0.04 | |
| Model III | ||||
| d IP – TM (Å) | 3.43 | 3.51 | 3.32 | |
| d SL – TM (Å) | 1.43 | 1.03 | 2.92 | |
| N*–N* (Å) | 3.71 | 3.66 | 4.42 | |
| J AB (cm−1) | −99 | −295 | −72 | |
| Error % | 10.67 | 13.83 | 4.42 | |
Although the concept of orbital overlap is not new32 and has inspired many groups,33 the qualitative description obtained by the orbital overlap analysis portraits remarkably well the behavior of PDTA and TDPDTA systems, and their magnetic variability as a function of the geometrical configuration. All these results are consistent with Kahn's qualitative model, and agree with other recent studies.34 It thus follows that these conclusions can be exploited to drive the synthesis of compounds whose crystal packing avoids the orbital overlap and favors the FM coupling.
The previous magneto-structural correlation maps for TTTA, PDTA and 4-NCBDTA undeniably show a series of radical arrangements resulting in FM coupling. Why is it that there are very few examples of DTA-based ferromagnets in nature? The answer turned out to be very simple since it relies on the energetic cost for reaching the FM area. The Interaction Energy Map (IEM) was computed for the four DTA systems (see the ESI,† Section 6). Note that the contour of the FM and weak AFM regions (−5 cm−1 < JAB < 0 cm−1) is delimited by dashed black lines on each IEM. The only exception is the IEM for TDPDTA, since it does not present any FM sector of interest (due to the lack of screening longitudinal translations).
The IEM for TTTA shows that the most FM region (+50 cm−1 < JAB < +150 cm−1) has a prohibitive energetic cost, which explains why TTTA experimentally shows no large FM coupling interactions. In fact, more than 20 kcal mol−1 are needed to reach this specific area. The energetically forbidden region involves an interplanar distance dIP < 3.0 Å, while the energetically reachable region encompasses 3.25 Å < dIP < 3.00 Å and 2.3 Å < dSL < 1.8 Å. The latter region partly contains the FM region of interest (0 cm−1 < JAB < +10 cm−1), which means that the adequate DTA-radical under certain conditions and specific geometrical conformation can result in a FM configuration.
The main result obtained from the OO analysis coupled with the magneto-structural correlation maps is the fact that geometrical rather than electronic factors are apparently responsible for the planar DTA-based materials to exhibit FM coupling interactions. The larger and more extended the structure is, the higher the probability is to have π–π interactions and, in turn, the higher the possibility of non-zero orbital overlap and quenching of the FM interactions. This important result is also confirmed in the next section by means of a set of in silico experiments performed to discriminate the role of the geometrical and electronic facets of the DTA-based compounds.
3. Electronic versus structural contributions
Three independent computational experiments are conducted aiming at distinguishing between the structural and the electronic contribution of the different DTA-substituents. Prior to these computations, the magnetically most important JAB data obtained using the corresponding dimers of the experimentally X-ray resolved HT phases were collected. From each DTA-radical dimer, the interplanar distance (dIP), degree of slippage (dSL), and nitrogen⋯nitrogen distance (N*⋯N*) were extracted, and the JAB interaction computed (see data under ‘Experimental data’ in Table 1). Note that the HT-4-NCBDTA material is the one presenting the strongest AFM coupling (JAB = −342 cm−1, at 300 K), while HT-TDPDTA is the one showing the weakest AFM interaction (JAB = −69 cm−1, at 293 K).For the first computational experiment, Model I was built as explained in Section 2, with dIP, dSL, and N*–N* distance that best resemble the experimentally X-ray resolved pairs of DTA-radicals for TTTA, PDTA, 4-NCBDTA and TDPDTA (note that it was selected among 3250 different radical pair configurations). In order to prove the adequacy of these in silico models, we calculated the JAB values (see data referred to as ‘Model I’ in Table 1). Direct comparison between the JAB values calculated using the experimental X-ray data and the models in the Model I section shows good agreement for each Model I system. Since we are using an isolated pair of radicals, neglecting the inner degrees of freedom of the crystal introduces a certain error in Model I, but the results are reassuring in order to be confident about the conclusions we have previously drawn for DTA-based magnets from the computed magneto-structural correlation maps.
The goal of the second and third computational tests is to discretize the electronic influence of the DTA-substituent with respect to the geometrical configuration of the dimer.
The second set of models is designed to determine the electronic effect on the magnetic coupling. For this purpose, we used the DTA-skeleton of the X-ray HT-TTTA pair of radicals (first column in Table 1 under ‘Experimental data’), and replaced the 1,2,5-thiadiazole substituent by the corresponding substituents of the PDTA, 4-NCBDTA and TDPDTA systems (see Fig. 1c and d). We refer to these TTTA-modified models as Model II. The resulting JAB spin coupling interactions (see ‘Model II’ in Table 1) are practically the same as the JAB value computed using the TTTA dimer itself extracted directly from the X-ray data at 298 K (JAB = −135 cm−1). This points out that the DTA-substituents do not directly influence the coupling between the radicals, and that the differences observed for the four systems have another origin.
Finally, the third model isolates the geometrical factor in the magnetic coupling of the pair of radicals. Among the 3250 configurations generated in the magneto-structural correlation map for the TTTA system, we select the four arrangements of two TTTA radicals that are geometrically closest to the experimental X-ray crystal structure of PDTA, 4-NCBDTA and TDPDTA (see geometrical data in Table 1 for ‘Model III’). The JAB value for the four clusters without any further change, i.e. maintaining the 1,2,5-thiadiazole substituent, is then calculated. In contrast to the calculations for Model II, we do observe important changes in the magnetic coupling. In fact the four JAB interactions calculated using Model III are in rather good agreement with those for the four systems extracted from experiment. Hence, it can be concluded that the substituent-induced crystal packing effects are responsible for the different magnetic behavior of the four systems.
Conclusions
The characterization of a subset of four compounds of the DTA-based family is used to evaluate the nature of the spin coupling and the magnetic properties of bistable and spin switchable systems. TTTA, PDTA, and TDPDTA combine the spin transition with the crystallographic phase transition, leading to a bistable system in which two stable phases (namely LT and HT) coexist within the same range of temperatures. For comparison purposes, we also studied the 4-NCBDTA system, which just presents spin transition.The four DTA-based radicals present a common planar geometry, and have the tendency to dimerize at low temperature, quenching the possibility for FM intermolecular interactions in the LT-phase. Yet the corresponding HT-phases are all paramagnetic, providing a more interesting scenario since there is possibility of FM spin coupling. Although the magnetic topology of all four DTA-based compounds is 3D, for simulation purposes they can be considered to form 1D isolated π-stacking AFM chains because the largest and dominant JABs extend along the π-stacking direction of the planar DTA-radicals. The simulated χT(T) magnetic susceptibility curves of PDTA and TDPDTA show that the LT-phases are magnetically silent, whereas the HT-phases are paramagnetic, in agreement with experiment.12,13 Some numerical discrepancies between the experimental and calculated χT(T) data are believed to be due to the presence of thermal fluctuations, in analogy to what was established in previous studies on TTTA and 4-NCBDTA compounds.25,28
The distinctive macroscopic magnetic response of the HT-phase of each DTA-based compound originates in the different microscopic intra π-stacking JAB radical interactions, which are found to be driven mostly by the changes in interplanar distance and degree of lateral slippage, according to the interpretation of a series of magneto-structural correlation maps. Thus, for TTTA, PDTA and 4-NCBDTA, it has been possible to predict the region where the FM interactions should appear, i.e. whose geometrical disposition enhances FM spin coupling. The presence of the FM regions and their location on the magneto-structural correlation maps has been qualitatively explained by the orbital overlap analysis based on Kahn's model.31 We concluded that only an interstitial orbital arrangement can prevent the orbitals of the two radicals from overlap, and favor FM coupling. Apparently, large fused-ring DTA-compounds (e.g. TDPDTA) are not suitable candidates to display ferromagnetism, because they increase the probability of having π–π orbital overlap at any reasonable relative orientation of the two radicals and, thus, preclude FM coupling. Besides, from our study it is clear that the electronic component introduced by the different DTA-substituents does not influence the value of the JAB spin coupling interaction itself. Instead, the DTA-substituents structurally affect the radical packing and, in turn, the JAB magnetic coupling. These magneto-structural maps could no doubt become a practical tool to help experimentalists to design more stable and efficient purely organic radicals with ferromagnetic properties in the solid state.
The magneto-structural correlation maps as a function of the substituents of the DTA-moiety enable the static FM fingerprint region to be highlighted, which is observed to be very expensive in terms of interaction energy. Indeed, this is the reason why there are very few examples of DTA-based ferromagnets in nature. All these results give further insight into the behavior of the DTA-radical-based magnets, as a step forward for the experimental counterpart in this research field to be able to design compounds with tailored properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MD, JRA and JJN acknowledge “Ministerio de Economia, Industria y Competitividad” (MICINN) for projects MAT2014-54025-P and CTQ2017-87773-P/AEI/FEDER, UE, and Catalan DURSI for projects 2014SGR1422 and 2017SGR348. CdG acknowledges CTQ2017-83566-P/AEI/FEDER, UE (MICINN), 2017SGR629 (DURSI) and COST Action ECOST-Bio CM1305 (EU). TF acknowledges funding from the European Union's Horizon 2020 research and innovation programme under the Marie Curie Skłodowska-Curie grant agreement no. 642294.References
- J. S. Miller, Adv. Mater., 2002, 14, 1105–1110 CrossRef.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- Stable radicals: fundamentals and applied aspects of odd-electron compounds, ed. R. G. Hicks, John Wiley & Sons, 2011 Search PubMed.
- P. M. Lahti, Adv. Phys. Org. Chem., 2011, 45, 93–169 CrossRef.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- (a) J. M. Rawson and J. J. Hayward in Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, Wiley, Hoboken, 2013, pp. 225–237 Search PubMed; (b) D. Bates, C. M. Robertson, A. A. Leitch, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2018, 140, 3846–3849 CrossRef PubMed; (c) K. E. Preuss, Coord. Chem. Rev., 2015, 289–290, 49–61 CrossRef; (d) J. M. Rawson, A. Alberola and A. Whalley, J. Mater. Chem., 2006, 16, 2560–2575 RSC; (e) K. Lekin, S. M. Winter, L. E. Downie, X. Bao, J. S. Tse, S. Desgreniers, R. A. Secco, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2010, 132, 16212–16224 CrossRef PubMed; (f) H. Phan, K. Lekin, S. M. Winter, R. T. Oakley and M. Shatruk, J. Am. Chem. Soc., 2013, 135, 15674–15677 CrossRef PubMed.
- T. M. Barclay, A. W. Cordes, N. A. George, R. C. Haddon, M. E. Itkis, M. S. Mashuta, R. T. Oakley, G. W. Patenaude, R. W. Reed, J. F. Richardson and H. Zhang, J. Am. Chem. Soc., 1998, 120, 352 CrossRef.
- H. Matsuzaki, W. Fujita, K. Awaga and H. Okamoto, Phys. Rev. Lett., 2003, 91, 017403 CrossRef PubMed.
- G. Wolmershäuser and R. Johann, Angew. Chem., Int. Ed., 1989, 28, 920–921 CrossRef.
- G. D. McManus, J. M. Rawson, N. Feeder, J. van Dujin, E. J. L. McInnes, J. J. Novoa, R. Burriel, F. Palacio and P. Oliete, J. Mater. Chem., 2001, 11, 1992 RSC.
- W. Fujita and K. Awaga, Science, 1999, 286, 261 CrossRef PubMed.
- J. L. Brusso, O. P. Clements, R. C. Haddon, M. E. Itkis, A. A. Leitch, R. T. Oakley, R. W. Reed and J. F. Richardson, J. Am. Chem. Soc., 2004, 126, 8256–8265 CrossRef PubMed.
- T. M. Barclay, A. W. Cordes, N. A. George, R. C. Haddon, M. E. Itkis, M. S. Mashuta, R. T. Oakley, G. W. Patenaude, R. W. Reed, J. F. Richardson and H. Zhang, J. Am. Chem. Soc., 1998, 120, 352–360 CrossRef.
- A. Alberola, R. J. Collis, S. M. Humphrey, R. J. Less and J. M. Rawson, Inorg. Chem., 2006, 45, 1903–1905 CrossRef PubMed.
- M. Deumal, M. J. Bearpark, J. J. Novoa and M. A. Robb, J. Phys. Chem. A, 2002, 106, 1299–1315 CrossRef.
- C. S. Clarke, J. Jornet-Somoza, F. Mota, J. J. Novoa and M. Deumal, J. Am. Chem. Soc., 2010, 132, 17817–17830 CrossRef PubMed.
- (a) M. Deumal, F. Mota, M. J. Bearpark, M. A. Robb and J. J. Novoa, Mol. Phys., 2006, 104, 857–873 CrossRef; (b) S. Vela, A. Sopena, J. Ribas-Arino, J. J. Novoa and M. Deumal, Chem. – Eur. J., 2014, 20, 7083–7090 CrossRef PubMed; (c) S. Vela, M. Deumal, J. Ribas-Arino and J. J. Novoa, Inorg. Chem., 2012, 51, 8646–8648 CrossRef PubMed; (d) M. Fumanal and M. Deumal, Phys. Chem. Chem. Phys., 2016, 18, 20738–20749 RSC; (e) S. Vela, M. Fumanal and M. Deumal, Dalton Trans., 2015, 44, 608–614 RSC.
- C. Herring in Direct Exchange between Well-Separated Atoms, In Magnetism, ed. G. T. Rado and H. Suhl, Academic Press, New York, 1966, vol. IIB, p. 5 Search PubMed.
- From the general Heisenberg Hamiltonian
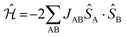 for a pair of A and B radicals, the JAB value is computed as the energy difference between biradical open-shell singlet S and triplet T states, ΔES−T = ES − ET = 2JAB. Open-shell singlet systems can localize alpha spin density and beta spin density on different radicals. In our case, within the DFT framework, once the broken symmetry approximation (BS) is applied, the energy difference can be expressed as: ES − ET = 2(ESBS − ET)/(1 + Sab). The resulting Sab overlap between the alpha (a) SOMO and the beta (b) SOMO is very small, which means that the orbitals are localized on each of the two radicals. This leads to Sab ≈ 0. As a conclusion JAB = ESBS − ET.
for a pair of A and B radicals, the JAB value is computed as the energy difference between biradical open-shell singlet S and triplet T states, ΔES−T = ES − ET = 2JAB. Open-shell singlet systems can localize alpha spin density and beta spin density on different radicals. In our case, within the DFT framework, once the broken symmetry approximation (BS) is applied, the energy difference can be expressed as: ES − ET = 2(ESBS − ET)/(1 + Sab). The resulting Sab overlap between the alpha (a) SOMO and the beta (b) SOMO is very small, which means that the orbitals are localized on each of the two radicals. This leads to Sab ≈ 0. As a conclusion JAB = ESBS − ET. - (a) L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef; (b) L. Noodleman and E. R. Davidson, Chem. Phys., 1986, 109, 131 CrossRef.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef; (b) M. M. Francl, W. J. Petro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef.
- For more information, see: (a) R. L. Carlin, Magnetochemistry, Springer Science & Business Media, 2012 Search PubMed; (b) R. Boca, Theoretical Foundations of Molecular Magnetism; Current Methods in Inorganic Chemistry, Elsevier Science S.A., The Netherlands, 1999, vol. 1 Search PubMed.
- S. Vela, F. Mota, M. Deumal, R. Suizu, Y. Shuku, A. Mizuno, K. Awaga, M. Shiga, J. J. Novoa and J. Ribas-Arino, Nat. Commun., 2014, 5, 4411 CrossRef PubMed.
- S. Vela, M. B. Reardon, C. E. Jakobsche, M. M. Turnbull, J. Ribas-Arino and J. J. Novoa, Chem. – Eur. J., 2017, 23, 3479–3489 CrossRef PubMed.
- J. J. Novoa, M. Deumal and J. Jornet-Somoza, Chem. Soc. Rev., 2011, 40, 3182–3212 RSC.
- S. Vela, M. Deumal, M. Shiga, J. J. Novoa and J. Ribas-Arino, Chem. Sci., 2015, 6, 2371–2381 RSC.
- (a) A. A. Leitch, X. Yu, S. M. Winter, R. A. Secco, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2009, 131, 7112–7125 CrossRef PubMed; (b) J. B. Rota, B. Le Guennic and V. Robert, Adv. Inorg. Chem., 2010, 49, 1230–1237 CrossRef PubMed; (c) M. Verot, N. Brefuel, J. Pecaut, C. Train and V. Robert, Chem. – Asian J., 2012, 7, 380–386 CrossRef PubMed.
- (a) J. Miralles, J. P. Daudey and R. Caballol, Chem. Phys. Lett., 1992, 198, 555 CrossRef; (b) J. Miralles, O. Castell, R. Caballol and J. P. Malrieu, Chem. Phys., 1993, 172, 33–43 CrossRef; (c) C. de Graaf and R. Broer, Magnetic interactions in molecules and solids, Springer, 2016 CrossRef.
- O. Kahn, Molecular magnetism, VCH Publishers, Inc., USA, 1993, p. 393 Search PubMed.
- (a) P. Hay, C. Jeffrey, J. Thibeault and R. Hoffmann, J. Am. Chem. Soc., 1975, 97, 4884–4899 CrossRef; (b) O. Kahn and B. Briat, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 268–281 RSC.
- For a historical review see: N. Onofrio and M. Jean-Marie, J. Phys. Chem. A, 2010, 114, 6149–6156 CrossRef PubMed.
- (a) J. Ribas-Arino, J. J. Novoa and J. S. Miller, J. Mater. Chem., 2006, 16, 2600–2611 RSC; (b) S. C. C. van der Lubbe and C. F. Guerra, Chem. – Eur. J., 2017, 23, 10249–10253 CrossRef PubMed; (c) J. Zapata-Rivera, D. Maynau and C. J. Calzado, Chem. Mater., 2017, 29, 4317–4329 CrossRef.

Footnote |
| † Electronic supplementary information (ESI) available: 1. Spin density of TTTA, PDTA, 4-NCBDTA and TDPDTA; 2. atomic coordinates of the LT and HT magnetically dominant pairs of radicals for TTTA, PDTA, TDPDTA, and 4-NCBDTA; 3. selection of magnetic models of PDTA and TDPDTA; 4. benchmarking the JAB magnetic coupling interactions computed at the DFT/UB3LYP level with the Difference Dedicated Configuration Interaction (DDCI-3) method; 5. TDPDTA shifted configurations; and 6. interaction energy maps of TTTA, PDTA, 4-NCBDTA, and TDPDTA. See DOI: 10.1039/c8cp03173h |
| This journal is © the Owner Societies 2018 |