
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bulk phase behavior of lithium imide–metal nitride ammonia decomposition catalysts†‡
Joshua W.
Makepeace
 *ab,
Thomas J.
Wood
b,
Phillip L.
Marks
ab,
Ronald I.
Smith
b,
Claire A.
Murray
c and
William I. F.
David
*ab
*ab,
Thomas J.
Wood
b,
Phillip L.
Marks
ab,
Ronald I.
Smith
b,
Claire A.
Murray
c and
William I. F.
David
*ab
aInorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: josh.makepeace@chem.ox.ac.uk; bill.david@stfc.ac.uk
bISIS Pulsed Neutron and Muon Facility, Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
cDiamond Light Source, Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0DE, UK
First published on 23rd August 2018
Abstract
Lithium imide is a promising new catalyst for the production of hydrogen from ammonia. Its catalytic activity has been reported to be significantly enhanced through its use as a composite with various transition metal nitrides. In this work, two of these composite catalysts (with manganese nitride and iron nitride) were examined using in situ neutron and X-ray powder diffraction experiments in order to explore the bulk phases present during ammonia decomposition. Under such conditions, the iron composite was found to be a mixture of lithium imide and iron metal, while the manganese composite contained lithium imide and manganese nitride at low temperatures, and a mixture of lithium imide and two ternary lithium–manganese nitrides (LixMn2−xN and a small proportion of Li7MnN4) at higher temperatures. The results indicate that the bulk formation of a ternary nitride is not necessary for ammonia decomposition in lithium imide–transition metal catalyst systems.
Introduction
In addition to being the feedstock for fertilisers that produce roughly half the global food supply,1 ammonia has significant potential for use as a sustainable fuel. The combination of its relatively high volumetric energy density and clean combustion emission profile makes ammonia an attractive energy storage choice for transportation and inter-seasonal grid balancing applications. In order to facilitate its use in low-temperature fuel cells, or to promote its combustion,2 ammonia must be either partially or completely cracked into hydrogen and nitrogen. Therefore, the development of highly active catalysts for the decomposition of ammonia is key to its successful application as a fuel.3Catalysts for ammonia cracking have predominantly consisted of transition metal nanoparticles supported on porous carbon or oxide supports. Ruthenium is widely accepted to be the most active transition metal for this reaction, and so research efforts have focussed on the optimisation of ruthenium-based catalysts4 (although iron and nickel catalysts are still of relevance as their low cost allows for higher metal loadings to compensate for their lower intrinsic activity5).
Since initial reports on the promising performance of sodium amide (NaNH2) as an alternative catalyst to traditional transition metal systems,6 lithium amide–imide (LiNH2–Li2NH) has emerged as the most important metal amide/imide ammonia decomposition catalyst, having shown high activity on its own7 and as part of a mixed metal imide.8 In both these cases the reported high-temperature ammonia decomposition activity was superior to supported ruthenium and nickel catalysts.
Very high catalytic activity can also be obtained by creating intimate mixtures of lithium imide with transition metals/metal nitrides,9–11 with lithium imide–manganese nitride mixtures showing superior activity (of around 27 kgNH3 kgcat−1 h−1) to a 5 wt% ruthenium on carbon nanotube catalyst, which is one of the most active ammonia decomposition catalyst formulations. These composites are proposed to function by the cyclical formation and decomposition of ternary nitrides. With manganese nitride, for example, the proposed reactions are as follows:
| 7Li2NH(s) + 2MnN(s) → 2Li7MnN4(s) + 7/2H2(g) + 1/2N2(g) | (1) |
| 2Li7MnN4(s) + 7/3NH3(g) → 7Li2NH(s) + 2MnN(s) + 2/3N2(g) | (2) |
| NH3(g) → 1/2N2(g) + 3/2H2(g) | (3) |
Experimental
Synthesis
All sample manipulations were performed in an argon-filled glove box due to the air and moisture sensitivity of the materials (O2, H2O < 0.1 ppm). Deuterated lithium amide (LiND2, 99% by XRD: ESI,‡ Fig. S1) was prepared by the reaction of lithium nitride (Li3N, 98%, Sigma Aldrich) with 3 bar of deuterated ammonia (ND3, 99%, 99 atom% D, Aldrich) at 300 °C for 3 hours. The powdered nitride was sealed in a cylindrical stainless steel reactor (I.D. 16 mm, height 100 mm) and attached to a gas panel. The reactor was evacuated and refilled with ammonia, and heated at 2 °C min−1 to the desired temperature. After reaction, the reactor was again evacuated, with the product powder extracted in the glove box. The lithium amide product was then hand-ground with one molar equivalent of lithium nitride for 15 minutes and heated in the same reactor under 1 bar of static argon pressure at 2 °C min−1 to 250 °C and held at that temperature for 12 hours to form deuterated lithium imide (Li2ND). The purity of the synthesised material was determined to be greater than 95% (with minor impurities of lithium oxide and lithium nitride) by powder X-ray diffraction (see ESI,‡ Fig. S2).Iron nitride (Fe2−4N, Alfa Aesar) was used as received. Analysis of the XRD pattern (see ESI,‡ Fig. S3) of the material gave an average iron stoichiometry of Fe3.5N from a mixture of Fe3N and Fe4N. Manganese nitride (MnN) was synthesised by the reaction of manganese powder (Mn, 99.95%, Alfa Aesar) with 60 cm3 min−1 flowing ammonia at 1 bar and 480 °C for 60 hours. The powder was placed inside a cylindrical stainless steel reactor (diameter 24.1 mm, height 100 mm) fitted with a gas inlet pipe running from the reactor lid to 10 mm from the base of the reactor. The gas outlet was located in the lid of the reactor, enabling gas flow over the top of the powder. The reactor was heated in a tube furnace (Severn Thermal Solutions) to the required temperature at 5 °C min−1. Analysis of the XRD pattern of the product indicated that the sample was 91 wt% MnN and 9 wt% MnO (see ESI,‡ Fig. S4).
The mixed lithium-imide/metal-nitride samples were prepared by mixing the deuterated lithium imide with the appropriate metal nitride in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (the average stoichiometry of the iron nitride sample was used for this calculation). The mixtures were ground by hand for approximately 20 minutes to ensure an intimate mixture of the two reactants. These mixtures were then placed in a tungsten carbide milling jar filled with nine, 10 mm-diameter tungsten-carbide balls, sealed and milled in a planetary ball mill (PM100, Retszch) for 5 hours at 400 rpm, broken up into 15 minute segments. After each segment, the milling was paused for two minutes and the direction of rotation of the mill was reversed. This method was chosen in preference to the reaction of the metal chloride with lithium amide9–11 as it eliminates the need to wash the deuterated samples with deuterated solvent in order to remove the lithium chloride by-product.
:1 molar ratio (the average stoichiometry of the iron nitride sample was used for this calculation). The mixtures were ground by hand for approximately 20 minutes to ensure an intimate mixture of the two reactants. These mixtures were then placed in a tungsten carbide milling jar filled with nine, 10 mm-diameter tungsten-carbide balls, sealed and milled in a planetary ball mill (PM100, Retszch) for 5 hours at 400 rpm, broken up into 15 minute segments. After each segment, the milling was paused for two minutes and the direction of rotation of the mill was reversed. This method was chosen in preference to the reaction of the metal chloride with lithium amide9–11 as it eliminates the need to wash the deuterated samples with deuterated solvent in order to remove the lithium chloride by-product.
Powder diffraction
Time-of-flight neutron powder diffraction data of the two samples were collected using the POLARIS diffractometer14 at the ISIS Pulsed Neutron and Muon Facility, United Kingdom. The experimental setup for these experiments was very similar to that described in previous reports,7,8 with a detailed description given in the ESI.‡ Briefly, the powder sample was loaded into a custom-designed stainless steel ‘flow over’ cell, which itself was housed within a furnace mounted into the diffractometer. Although steel itself can decompose ammonia, quartz reactors are unsuitable given the strong basicity of the lithium imide material, having been shown to react with SiO2 under ammonia to form lithium silicates.15 Previous tests of blank reaction cells have shown that the catalytic performance is significantly lower than with the added catalyst,3 reaching only 39% conversion at 550 °C with a 10 sccm (standard cubic centimetres per minute) flow of ammonia. This is a necessary compromise in order to avoid unwanted reaction of the catalyst. After leak-checking, gas was flowed through the cell (5–15 sccm) using a custom gas panel, and then analysed by mass spectrometry. Neutron powder diffraction data were collected in 100 s segments throughout the experiment. Additional reference diffraction patterns were collected at room temperature from each of the two starting materials, held in vanadium cans (6 mm diameter).The in situ neutron powder diffraction experiment was similar for each of the two samples. Firstly, the sample was heated to 550 °C under flowing argon. Heating under inert gas prior to exposure to ammonia minimises the formation of lithium amide, which will cause melting of the sample in the temperature range ∼370–500 °C with potential blockage of the reaction cell. After equilibrating under argon, the gas flowing over the sample was switched to deuterated ammonia to examine the structure and composition of the samples while decomposing ammonia. The samples were equilibrated under a number of different ammonia flow/temperature regimes in order to explore potential variation in the active form of the catalyst. Finally, the gas flow was switched to natural isotopic abundance ammonia to perform a hydrogen–deuterium isotope exchange experiment.
A second in situ powder diffraction experiment was performed using X-rays at the Diamond Light Source, United Kingdom. A sample of Li2NH + MnN (∼10 mg) was placed into a 1 mm diameter sapphire capillary, held in place by a plug of quartz wool (out of the hot zone to avoid reaction with the catalyst), and was loaded into a custom gas flow cell.16 The cell was leak checked and mounted on the I11 beamline.17 It was placed under a 1 sccm flow of helium gas and heated to 500 °C using a hot air blower before the gas was switched to ammonia (1 sccm). After equilibrating, the ammonia flow rate was increased to 2 sccm in order to examine the effect of variable flow rate. This was followed by a switch back to helium, cooling of the sample to 475 °C, and a second exposure to ammonia (1 sccm) at this lower temperature. Diffraction data were collected in two-minute segments throughout the experiment, recorded using the high-resolution MAC detectors and an X-ray wavelength of 0.826209 Å.
The powder diffraction data were analysed using the TOPAS Academic18 software package. A significant proportion of this analysis was performed using the batch analysis feature, where an initial refinement is performed, with the output then automatically used as the input for the refinement of the next dataset, and so on for all of the datasets. This allows for a large number of datasets with minor differences between them to be processed more efficiently.
Results and discussion
Li2ND + FexN
The results of the neutron powder diffraction experiment for the Li2ND/FexN composite sample are shown in Fig. 1. During the initial heating under argon flow, a large release of N2 and a smaller release of D2 were recorded, beginning at around 380 °C. The diffraction data indicate that this event correlates with the sequential denitriding of the Fe3−xN (through a range of nitrogen content as evidenced by the smooth change in the lattice parameter during denitriding) and Fe4N phases to form Fe metal (the intense peak at 2.05 Å), according to reactions 4 and 5:| Fe3N(s) → 3Fe(s) + 1/2N2(g) | (4) |
| Fe4N(s) → 4Fe(s) + 1/2N2(g) | (5) |
| 3Li2ND(s) + Fe4N(s) → 2Li3FeN2(s) + 2Fe(s) + 3/2D2(g) | (6) |
| 2Li3FeN2(s) + ND3(g) → 3Li2ND(s) + 2Fe(s) + N2(g) | (7) |
 | ||
| Fig. 1 Results of the neutron powder diffraction experiment on lithium imide–iron nitride. The panels show: (a) the temperature of the sample and gas flow rate and composition, (b) the molar gas fractions of the various gas species monitored in the experiment, (c) a contour plot of the neutron powder diffraction with regions used for analysis of summed diffraction data indicated with numbered, white lines and (d) a plot of the molar composition of the sample obtained from Rietveld analysis of the diffraction data. | ||
Examination of the remainder of the diffraction data shows that, aside from small variations in stoichiometry which will be discussed in the following text, the sample remains as a mixture of Li2ND and Fe throughout the range of ammonia decomposition reaction conditions explored. Only once the reaction is cooled under ND3 can the reformation of a small amount of Fe4N be observed together with release of D2 (at approximately 27.5 h), along with the conversion of Li2ND to LiND2 through a melt. This suggests that if the ammonia decomposition mechanism in this system relies on the formation of the ternary nitride, then it must only exist as a short-lived intermediate. Given the large particle sizes indicated by the sharp Bragg peaks observed for the crystalline phases, it seems unlikely that the bulk of the sample is being rapidly converted between the ternary nitride and the Li2ND–Fe composite. Thus, if the ternary nitride is indeed the active species, the ammonia decomposition reaction must only take place on the interface of Li2ND and Fe particles. The ammonia fraction observed for this sample at 550 °C and 10 sccm ND3 flow is approximately the same as was observed for Li2ND on its own in a similar experimental setup.7
After equilibrating under ND3, the sample was then exposed to a number of different ND3 flow rate and temperature regimes. Visual inspection of the diffraction data presented in Fig. 1c indicates that no significant changes in the phase composition of the sample were observed under the conditions used, despite the ammonia gas fraction clearly changing with changing conditions, i.e. a higher ammonia gas fraction at higher ND3 flow rates and lower temperatures, as indicated in Fig. 1b.
A more detailed analysis of the sample was performed by Rietveld analysis of summed sections of the diffraction data where the ammonia gas fraction was stable for each flow/temperature regime (see ESI,‡ Fig. S6 for an illustration of the summed regions). Of particular interest are potential changes in the stoichiometry of Li2ND towards LiND2 (forming Li1+xND2−x), which have previously been observed in the context of ammonia decomposition reactions.7Fig. 2 shows the refined lattice parameter and lithium occupancy of the Li2ND phase from each summed diffraction dataset under the various reaction conditions of the experiment, with the lattice parameter of the Fe phase included as a reference (fits to each summed dataset are given in the ESI,‡ Fig. S7–S17).
 | ||
| Fig. 2 Refined lattice parameter and lithium occupancy for the Li1+xND2−x phase in the Li2ND + FexN sample under the various temperature/ND3 flow segments of the in situ neutron diffraction experiment. Error bars represent one standard deviation. | ||
Attempts to directly refine the stoichiometry of the sample using the summed diffraction data (see the lithium occupancy in Fig. 2) did not show any significant changes over the different segments; the analysis was hindered by the relatively poor signal-to-noise ratio in the peaks associated with the Li2ND phase. However, the lattice parameter data provide a more robust measure of the subtle changes in the sample under the various reaction conditions. While the variation in the Fe lattice parameter is consistent with normal thermal expansion behaviour, the variation in Li2ND lattice parameter is more complex.
Changes in the lattice parameter of lithium imide have been associated with variation in the sample stoichiometry in previous ammonia decomposition7 and hydrogen storage23in situ diffraction measurements. In general, these studies have shown that a larger lattice parameter is associated with a more hydrogen-rich phase (although a recent study on the room-temperature structure of a number of lithium amide–imide non-stoichiometric phases synthesised by reaction of lithium nitride with lithium amide showed the opposite trend24).
The Li2ND lattice parameter decreased dramatically on initial exposure of the sample to ND3 (segment 1 → 2), with a further decrease in segment 3 despite an increase in the ND3 flow rate. Both of these segments would be expected to show in an increase in the lattice parameter associated with a shift toward amide stoichiometry, based on the previous results. This may indicate that the Li2ND which remained after the formation of the ternary nitride has a different stoichiometry or defect structure than the Li2ND formed by decomposition of the ternary nitride on exposure to ND3. A plot of the variation in the lattice parameter of Li2ND in each individual run throughout the experiment (ESI,‡ Fig. S18) shows that on exposure to ND3, an initial exponential decrease in the lattice parameter is followed by a much slower decrease (fit here with a linear function). This process appears to not have completed before the flow was changed in segment 3. Therefore, the lattice parameter continues to decrease, though at a slower rate, which may be as a result of the higher ammonia flow rate.
Segments 4–7 are an isothermal region where the ND3 flow rate was sequentially increased between 5 sccm and 15 sccm. The lattice parameter increases with the increasing flow, which is consistent with increasing hydrogen (deuterium) content through absorption of ammonia:
| (1 + x)Li2ND(s) + (1−x)ND3(g) → 2Li1+xND2−x(s) | (8) |
In order to probe these data, the thermal expansion of the Li2ND lattice during the heating under argon was calculated. The 150–400 °C section was used, as the heating ramp rate was almost constant in that range. The thermal expansion coefficient from the linear fit of these data (ESI,‡ Fig. S19) was then used to predict the expected change of the Li2ND lattice parameter with the temperature changes in segments 7–10. These predicted values are compared with the measured values in Fig. 3, showing the measured lattice parameter is significantly larger than expected from the thermal expansion data, again reflecting absorption of ammonia. Comparison of these with the lattice parameters for increasing ND3 flow highlights the strong influence of temperature on the stoichiometry of lithium imide under ammonia.
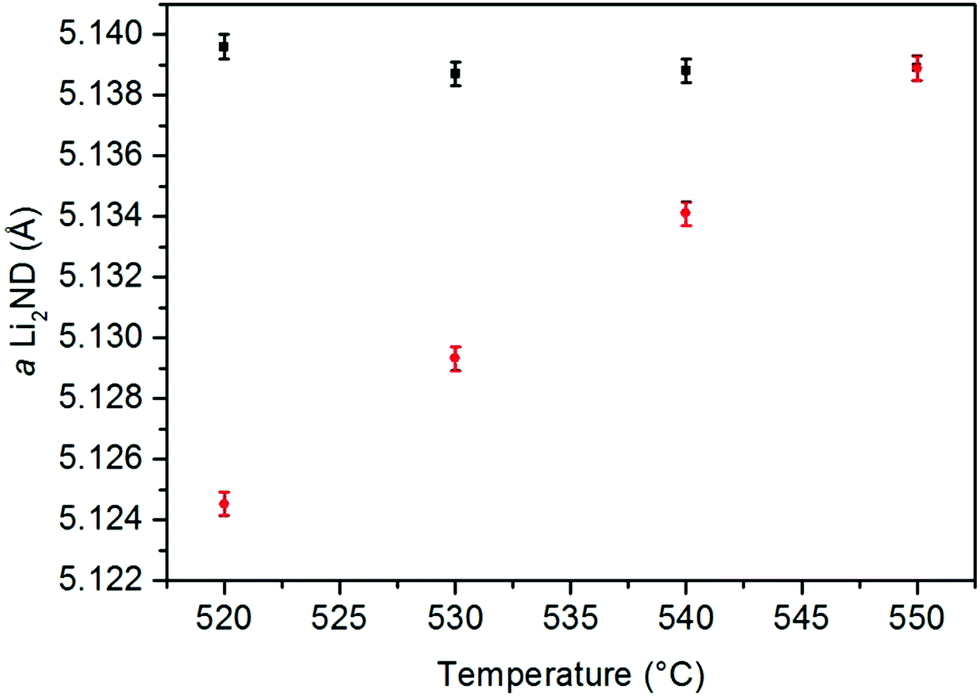 | ||
| Fig. 3 Comparison of the lattice parameter of Li2ND at various temperatures extracted from the Rietveld analysis of the measured diffraction data (black squares) with the value predicted from the thermal expansion coefficient (red circles). Error bars represent one standard deviation. | ||
The increase in temperature back to 550 °C in segment 11 results in a larger lattice parameter than was observed under the same conditions in segment 7. This is due to the slow kinetics of the reverse reaction of reaction (8).
The final stage of the experiment, beginning at 21.8 h, was a hydrogen–deuterium isotope exchange experiment where natural abundance ammonia gas was introduced in place of ND3. In the section of the diffraction data following this change, the background intensity can be seen to increase, along with a significant decrease in the intensity (and relative intensities) of the Bragg peaks of the Li2ND phase. The intensities of the Fe peaks were unaffected. This is as a result of the incoherent neutron scattering of hydrogen as it is incorporated into the lithium imide structure. Rietveld analysis of the diffraction data in this section shows the structure changing from completely deuterated to completely hydrogenated, with a concomitant decrease in the lattice parameter (ESI,‡ Fig. S20). Reintroduction of ND3 shows the exchange to be reversible.
Under ammonia, this sample showed similar behaviour to that observed for lithium imide in isolation in the previous in situ study, with the addition of the iron phase. While the formation of the ternary imide Li3FeN2 was observed under inert gas, no ternary phases were observed under ND3 in this measurement of bulk structure. This may indicate a very short lifetime for this intermediate or that the formation of the ternary phase is confined to the surface, which cannot be effectively measured in this experiment.
Li2ND + MnN
The results of the neutron diffraction experiment on the lithium imide–manganese nitride composite are summarised in Fig. 4. The experiment proceeded similarly to the iron composite sample. During heating under argon, two distinct gas release events were observed with peaks at 483 °C and 550 °C. The higher relative amount of D2 in the gas release events compared with the iron sample reflects more complete conversion of the sample to ternary lithium–manganese nitrides. Li2ND was completely consumed and converted into two ternary nitrides: Li7MnN4 and LixMn2−xN.| 7Li2ND(s) + 2MnN(s) → 2Li7MnN4(s) + 7/2D2(g) + 1/2N2(g) | (9) |
| Li2ND(s) + 4MnN(s) → 3Li0.67Mn1.33N(s) + N2(g) + 1/2D2(g) | (10) |
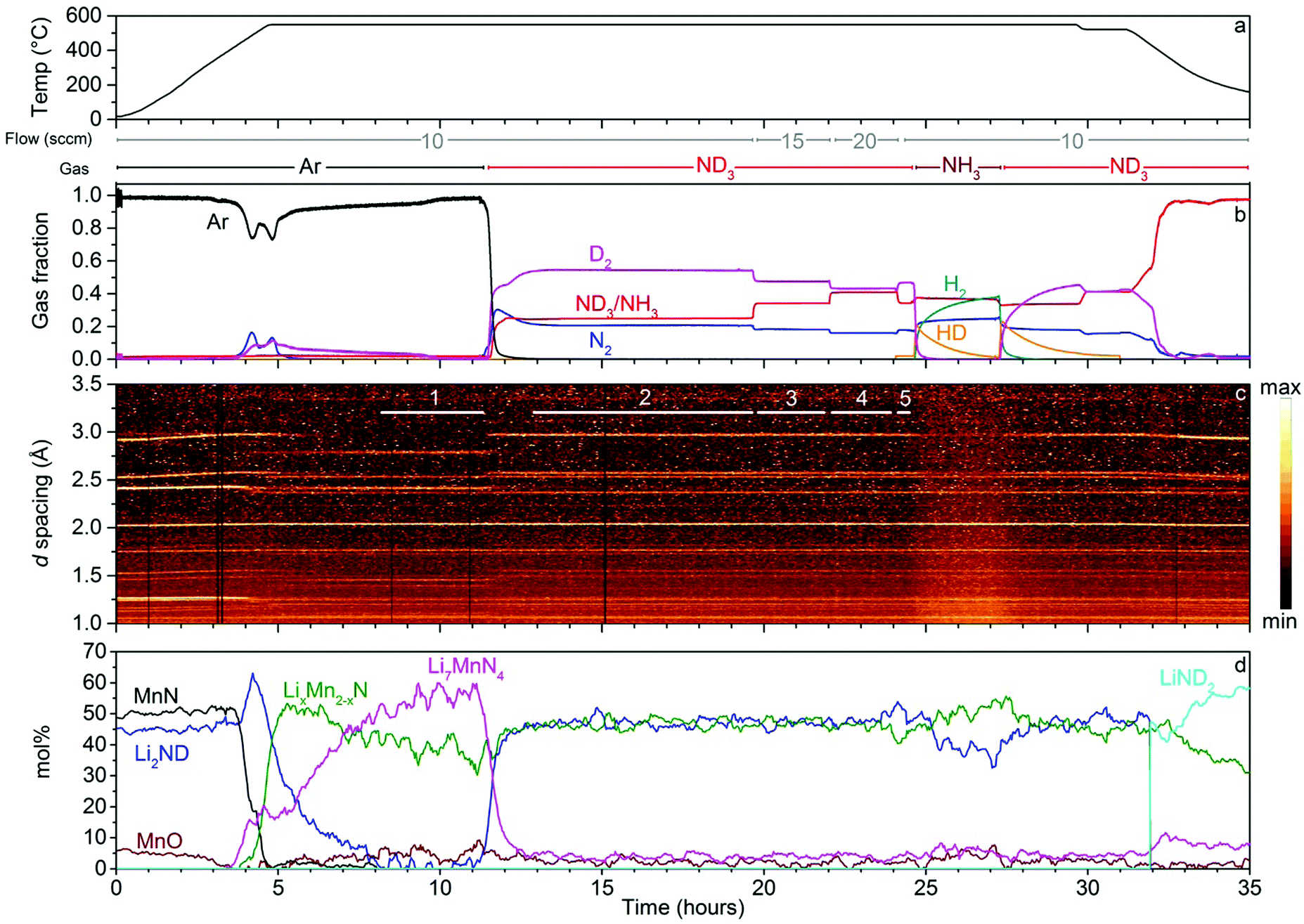 | ||
| Fig. 4 Results of the neutron powder diffraction experiment on lithium imide–manganese nitride. The panels show (a) the temperature of the sample and gas flow rate and composition, (b) the molar gas fractions of the various gas species monitored in the experiment, (c) a contour plot of the neutron powder diffraction with regions used for analysis of summed diffraction data indicated with numbered, white lines and (d) the molar composition of the sample obtained from Rietveld analysis of the diffraction data. | ||
The LixMn2−xN phase forms rapidly, while the kinetics of formation of the Li7MnN4 phase are relatively slow, with D2 production observed until just before 10 h. At the end of the period under argon, these two phases account for all of the Bragg peaks in the diffraction data (excluding the steel from the reaction cell).
On introduction of ND3, the Li2ND peaks reappeared with the almost complete decomposition of the Li7MnN4, in a similar fashion to Li3FeN2, with a broader nitrogen release observed in the mass spectrometry data between 11.5 and 13 h:
| 6Li7MnN4(s) + 7ND3(g) → 21Li2ND(s) + 6Mn(s) + 5N2(g) | (11) |
The relatively low intensity of the manganese metal Bragg peaks and their partial overlap with the LixMn2−xN phase made the batch refinement unstable (ESI,‡ Fig. S21). As a result it was not included in the main analysis. Additionally, extra weak Bragg peaks associated with an unidentified phase(s) were observed in the summed data under ND3. It is possible that these peaks are associated with either a nitride of manganese or another ternary lithium–manganese nitride. All known phases in these two categories were checked against the peaks, but no satisfactory fits were obtained. Given the poor signal-to-noise ratio on these peaks and their limited number, it was not possible to index them and their origin remains unknown.
Unlike the iron-based sample, the ternary nitride phases remained present for this sample under ammonia decomposition conditions. A small amount of Li7MnN4 (∼5 mol%) persisted in the sample, while the scale factor for the LixMn2−xN phase remained almost unchanged upon introduction of ND3. Under a reducing gas environment such as is generated in ammonia decomposition conditions, the lower formal (1–2+) oxidation state of manganese in this nitride may be more stable than the Mn5+ present in Li7MnN4, which may explain the relative stabilities of the two nitride phases.
As with the Li2ND/FexN sample, the ND3 flow rate was varied in order to study the active form of the catalyst over a range of reaction conditions. Increasing the ND3 flow rate to 15 sccm and then 20 sccm resulted in slightly lower ammonia decomposition levels as expected, and again resulted in little change to the observed diffraction patterns. Refinement of the Li2ND phase in summed diffraction datasets for these regions (ESI,‡ Fig. S23–S28) produced similar results to the iron-containing sample; and while a significant difference in the lithium occupancy was not detected, the change in ND3 flow rate did result in a slight increase in the lattice parameter associated with a shift toward amide stoichiometry (Fig. 5).
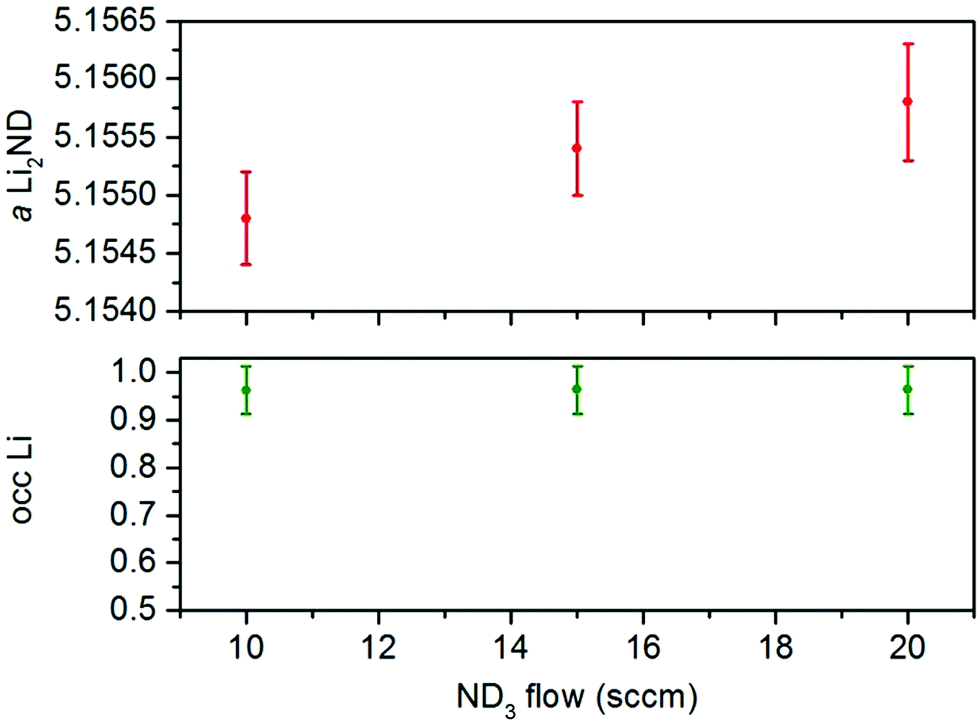 | ||
| Fig. 5 Variation of the lattice parameter and lithium occupancy of the Li2ND phase in the Li2ND + MnN composite under different ND3 flow rates. Error bars represent one standard deviation. | ||
Refinement of the Li2ND lattice parameter and deuterium occupancy during the H–D isotope exchange experiment also showed almost complete and completely-reversible conversion of Li2ND to Li2NH (ESI,‡ Fig. S29), again demonstrating the bulk interaction of lithium amide–imide with ammonia.
The presence of both Li2ND and LixMn2−xN under ammonia decomposition conditions in this experiment raises the question of whether the formation of the ternary nitride LixMn2−xN is necessary for ammonia decomposition. Fig. 6 details the results of an in situ synchrotron X-ray powder diffraction experiment on the Li2NH + MnN system. This experiment was conducted at the lower temperature of 500 °C to avoid oxidation of the sample through reaction with the sapphire capillary. In this experiment, no Li–Mn–N phases were observed, under helium gas or ammonia. MnN was found to persist in the sample throughout the experiment, undergoing a tetragonal to cubic phase transition at 460 °C (this is a somewhat higher temperature than has been reported from DSC measurements,28 which may indicate a temperature difference between the set point of the system and the actual temperature of the sample inside the capillary). When the gas flow was switched to ammonia, the cubic MnN phase was unchanged, while a step increase in the size of the Li2NH lattice can be seen (Fig. 6c, 135 min), consistent with a shift towards amide stoichiometry as explained previously. The increase in the ammonia flow rate from 1 sccm to 2 sccm (Fig. 6c, 150 min) shows a further, smaller increase in the lattice parameter, indicating a further shift towards amide stoichiometry. The return to helium flow (Fig. 6c, 175 min) resulted in a gradual decrease in the Li2NH lattice parameter, consistent with the return towards imide stoichiometry.
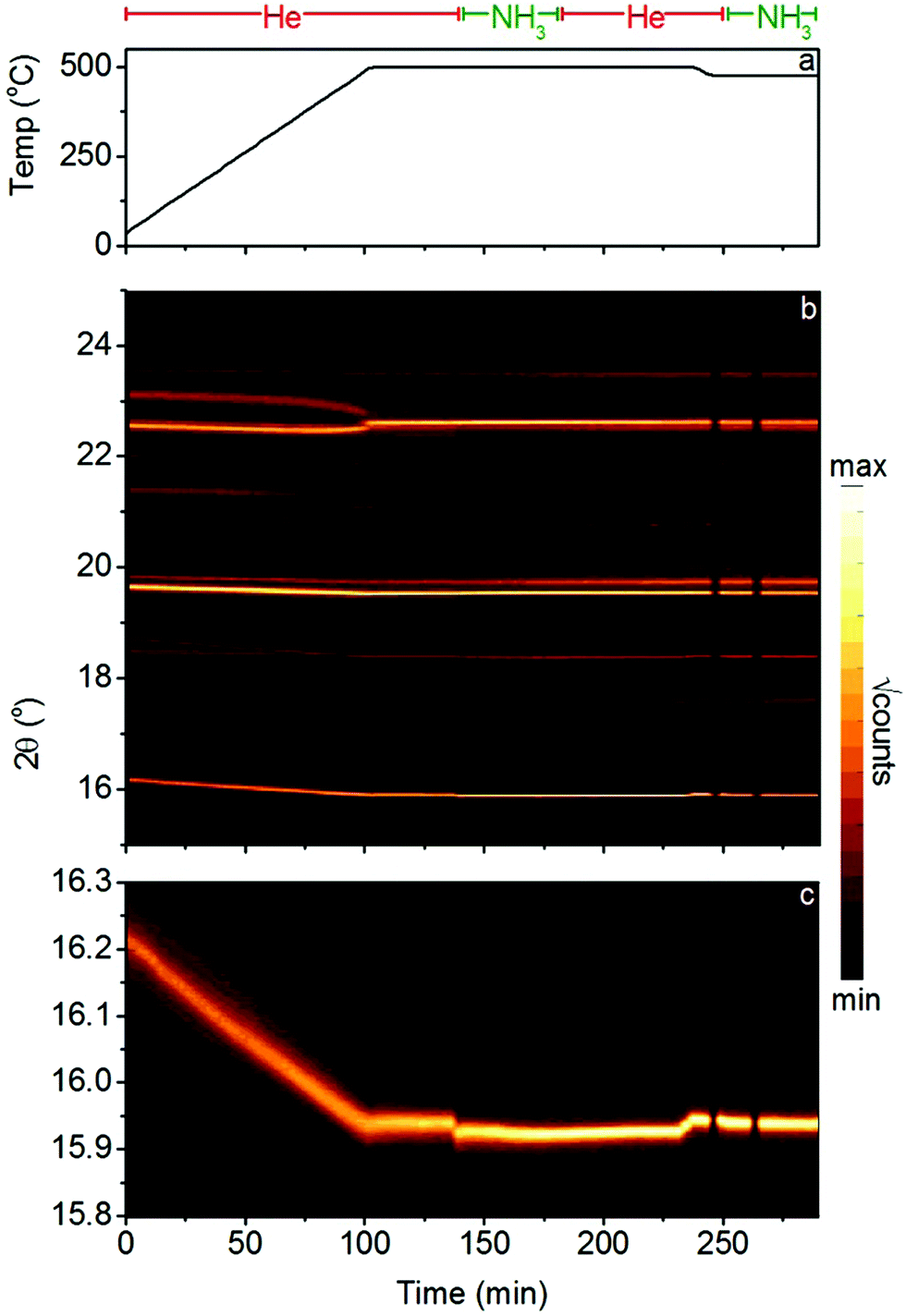 | ||
| Fig. 6 In situ synchrotron X-ray diffraction on the ammonia decomposition reaction by the lithium imide–manganese nitride composite, showing (a) the gas species (1 sccm constant flow) and sample temperature, (b) contour plot of the diffraction data and (c) a contour plot of the Li2ND(111) region of the diffraction pattern. | ||
The absence of any Li–Mn–N phases in this experiment, while still in a temperature-flow regime where significant ammonia decomposition activity is expected,10 suggests that the presence of such a phase in the bulk is not a prerequisite for ammonia decomposition. There was no evidence in the diffraction data for broad features which might be associated with nanoparticles or amorphous features, which suggests that the Rietveld analysis should accurately reflect the bulk phase composition of these samples. While this does not preclude the possibility that the ternary phase is present as a short-lived or surface/interface intermediate, as discussed previously, or that both Li2ND and LixMn2−xN are active ammonia decomposition catalysts, it does suggest that these composites may be best described as promoted lithium imide catalysts.
Conclusions
Catalyst composites based on light metal amide and imide catalysts are promising low-cost systems for the generation of high-purity hydrogen from ammonia, showing improved performance over state-of-the-art ruthenium-based catalysts. In situ powder diffraction measurements of ammonia decomposition reactions catalysed by composites of lithium imide with manganese nitride and iron nitride have given insight into the active forms of these catalysts. While both systems were found to form ternary nitrides (Li3FeN2, LixMn2−xN and Li7MnN4) at elevated temperatures under inert gas, only LixMn2−xN was found to persist as a significant proportion of the sample under ammonia decomposition conditions. Li3FeN2 completely disappeared, while Li7MnN4 only remained as a minor component of the sample. Lithium imide was present under active conditions in both samples, and demonstrated stoichiometry variation consistent with previous reports on the structure of isolated lithium imide during ammonia decomposition. These results suggest that bulk formation of the ternary nitride is not a complete explanation of the reported enhancements in catalytic activity reported for these imide–nitride composites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Science and Technology Facilities Council for access to neutron beam time at ISIS (RB1520479), and Diamond Light Source for access to synchrotron beam time (EE15226-1). The authors are grateful for the technical and experimental support from Chris Goodway, Paul McIntyre, Adam Sears, Mark Kibble, Hazel Hunter, James Taylor and Jonathan Potter. JWM thanks St John's College, Oxford for financial support. TJW and WIFD acknowledge the EPSRC for financial support (grant number EP/M014371/1).References
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, How a Century of Ammonia Synthesis Changed the World, Nat. Geosci., 2008, 1, 636–639 CrossRef.
- M. Comotti and S. Frigo, Hydrogen Generation System for Ammonia-Hydrogen Fuelled Internal Combustion Engines, Int. J. Hydrogen Energy, 2015, 40, 10673–10686 CrossRef.
- S. Mukherjee, S. V. Devaguptapu, A. Sviripa, C. R. F. Lund and G. Wu, Low-Temperature Ammonia Decomposition Catalysts for Hydrogen Generation, Appl. Catal., B, 2018, 226, 162–181 CrossRef.
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, A Mini-Review on Ammonia Decomposition Catalysts for on-Site Generation of Hydrogen for Fuel Cell Applications, Appl. Catal., A, 2004, 277(1–2), 1–9 CrossRef.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Ammonia as a Possible Element in an Energy Infrastructure: Catalysts for Ammonia Decomposition, Energy Environ. Sci., 2012, 5(4), 6278 RSC.
- W. I. F. David, J. W. Makepeace, S. K. Callear, H. M. A. Hunter, J. D. Taylor, T. J. Wood and M. O. Jones, Hydrogen Production from Ammonia Using Sodium Amide, J. Am. Chem. Soc., 2014, 136(38), 13082–13085 CrossRef PubMed.
- J. W. Makepeace, T. J. Wood, H. M. A. Hunter, M. O. Jones and W. I. F. David, Ammonia Decomposition Catalysis Using Non-Stoichiometric Lithium Imide, Chem. Sci., 2015, 6, 3805–3815 RSC.
- J. W. Makepeace, H. M. A. Hunter, T. J. Wood, R. I. Smith, C. A. Murray and W. I. F. David, Ammonia Decomposition Catalysis Using Lithium–Calcium Imide, Faraday Discuss., 2016, 188, 525–544 RSC.
- J. Guo, P. Wang, G. Wu, A. Wu, D. Hu, Z. Xiong, J. Wang, P. Yu, F. Chang and Z. Chen, et al., Lithium Imide Synergy with 3d Transition-Metal Nitrides Leading to Unprecedented Catalytic Activities for Ammonia Decomposition, Angew. Chem., Int. Ed., 2015, 127(10), 2993–2997 CrossRef.
- J. Guo, F. Chang, P. Wang, D. Hu, P. Yu, G. Wu, Z. Xiong and P. Chen, Highly Active MnN–Li2NH Composite Catalyst for Producing COx-Free Hydrogen, ACS Catal., 2015, 5(5), 2708–2713 CrossRef.
- J. Guo, Z. Chen, A. Wu, F. Chang, P. Wang, D. Hu, G. Wu, Z. Xiong, P. Yu and P. Chen, Electronic Promoter or Reacting Species? The Role of LiNH2 on Ru in Catalyzing NH3 Decomposition, Chem. Commun., 2015, 51(82), 15161–15164 RSC.
- T. J. Wood, J. W. Makepeace and W. I. F. David, Isotopic Studies of the Ammonia Decomposition Reaction Using Lithium Imide Catalyst, Phys. Chem. Chem. Phys., 2017, 19, 4719–4724 RSC.
- T. J. Wood, J. W. Makepeace, H. M. A. Hunter, M. O. Jones and W. I. F. David, Isotopic Studies of the Ammonia Decomposition Reaction Mediated by Sodium Amide, Phys. Chem. Chem. Phys., 2015, 17(35), 22999–23006 RSC.
- S. Hull, R. I. Smith, W. I. F. David, A. C. Hannon, J. Mayers and R. Cywinski, The Polaris Powder Diffractometer at ISIS, Phys. B, 1992, 181, 1000–1002 CrossRef.
- T. J. Wood and J. W. Makepeace, Assessing Potential Supports for Lithium Amide-Imide Ammonia Decomposition Catalysts, ACS Appl. Energy Mater., 2018, 1, 2657–2663 CrossRef.
- J. E. Parker, J. Potter, S. P. Thompson, A. R. Lennie and C. C. Tang, In Situ Gas Supply System on the Powder Diffraction Beamline I11 at Diamond Light Source, Mater. Sci. Forum, 2012, 706–709, 1707–1712 Search PubMed.
- S. P. Thompson, J. E. Parker, J. Potter, T. P. Hill, A. Birt, T. M. Cobb, F. Yuan and C. C. Tang, Beamline I11 at Diamond: A New Instrument for High Resolution Powder Diffraction, Rev. Sci. Instrum., 2009, 80(7), 075107 CrossRef PubMed.
- A. Coelho, TOPAS-Academic, Version 6, http://www.topas-academic.net/.
- M. Widenmeyer, T. C. Hansen, E. Meissner and R. Niewa, Formation and Decomposition of Iron Nitrides Observed by In Situ Powder Neutron Diffraction and Thermal Analysis, Z. Anorg. Allg. Chem., 2014, 640(7), 1265–1274 CrossRef.
- W. Peikun, J. Guo and P. Chen, The Interactions of Li3FeN2 with H2 and NH3 Science Direct, Int. J. Hydrogen Energy, 2016, 1–7 Search PubMed.
- T. J. Wood, J. W. Makepeace and W. I. F. David, Neutron Diffraction and Gravimetric Study of the Iron Nitriding Reaction under Ammonia Decomposition Conditions, Phys. Chem. Chem. Phys., 2017, 19, 27859–27865 RSC.
- B. J. Kooi, M. A. J. Somers and E. J. Mittemeijer, An Evaluation of the Fe-N Phase Diagram Considering Long-Range Order of N Atoms in Γ′-Fe4N1−x and ε-Fe2N1−z, Metall. Mater. Trans. A, 1996, 27(4), 1063–1071 CrossRef.
- J. W. Makepeace, M. O. Jones, S. K. Callear, P. P. Edwards and W. I. F. David, In Situ X-Ray Powder Diffraction Studies of Hydrogen Storage and Release in the Li–N–H System, Phys. Chem. Chem. Phys., 2014, 16, 4061–4070 RSC.
- J. W. Makepeace and W. I. F. David, Structural Insights into the Lithium Amide–Imide Solid Solution, J. Phys. Chem. C, 2017, 121, 12010–12017 CrossRef.
- R. Niewa, F. DiSalvo, D.-K. Yang, D. Zax, H. Luo and W. Yelon, Synthesis, Crystal Structure and Properties of a Lithium Manganese Nitride, (Li, Mn)2N, J. Alloys Compd., 1998, 266, 32–38 CrossRef.
- M. Widenmeyer, T. C. Hansen, A. Leineweber, A. Weidenkaff and R. Niewa, Nitrogen Transfer between Solid Phases in the System Mn–N Detected via in Situ Neutron Diffraction, Z. Anorg. Allg. Chem., 2017, 643(23), 1929–1938 CrossRef.
- T. J. Wood, J. W. Makepeace and W. I. F. David, Neutron Diffraction and Gravimetric Study of the Manganese Nitriding Reaction under Ammonia Decomposition Conditions, Phys. Chem. Chem. Phys., 2018, 20, 8547–8553 RSC.
- A. Leineweber, R. Niewa, H. Jacobs and W. Kockelmann, The Manganese Nitrides η-Mn3N2 and ΘMn6N5 + x: Nuclear and Magnetic Structures, J. Mater. Chem., 2000, 10(12), 2827–2834 RSC.
Footnotes |
| † The research materials supporting this publication can be accessed by contacting Joshua Makepeace. Neutron diffraction data included in this publication will be publicly available at https://data.isis.stfc.ac.uk/ from 23/2/2019. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp02824a |
| This journal is © the Owner Societies 2018 |