
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-touching plasma–liquid interaction – where is aqueous nitric oxide generated?†
Helena
Jablonowski
 *a,
Ansgar
Schmidt-Bleker
b,
Klaus-Dieter
Weltmann
b,
Thomas
von Woedtke
b and
Kristian
Wende
a
*a,
Ansgar
Schmidt-Bleker
b,
Klaus-Dieter
Weltmann
b,
Thomas
von Woedtke
b and
Kristian
Wende
a
aZIK Plasmatis at Leibniz Institute for Plasma Science and Technology (INP Greifswald e.V.), Felix-Hausdorff-Str. 2, 17489 Greifswald, Germany. E-mail: helena.jablonowski@inp-greifswald.de
bLeibniz Institute for Plasma Science and Technology (INP Greifswald e.V.), Felix-Hausdorff-Str. 2, 17489 Greifswald, Germany
First published on 28th September 2018
Abstract
Nitric oxide is a relatively stable free radical and an important signal molecule in plants, animals, and humans with high relevance for biological processes involving inflammatory processes, e.g. wound healing or cancer. The molecule can be detected in the gas phase of non-thermal plasma jets making it a valuable tool for clinical intervention, but transport efficiency from the gas phase into the liquid phase or tissue remains to be clarified. To elucidate this fact, the nitric oxide concentration in buffered solutions is determined using electron paramagnetic resonance spectroscopy. The origin of the nitric oxide in the liquid could be excluded, therefore, potential precursors such as hydroxyl radicals, superoxide anions, atomic hydrogen and stable species (nitrite, nitrate and hydrogen peroxide) were detected and the potential formation pathway as well as ways of enhancing the production of nitric oxide by alteration of the feed gas and the surrounding gas composition during plasma treatment of the liquid have been pointed out.
Introduction
Nitrogen oxides NxOy are major constituents of cold atmospheric pressure discharge, with N2O5, NO2, N2O alongside with HNO2 and HNO3 being frequently observed in the gas phase.1–4 In plasma treated aqueous liquids, nitrite and nitrate ions are easily observed. Interestingly, also the biologically highly relevant nitric oxide (˙NO) can be found in the gas phase and plasma treated liquids, sparking the interest of both fundamental and applied research.5 The molecule is a comparably stable radical and has a key position in diverse biological cascades like vascular and neuronal signal transduction, cell death, or host defense.6–8 The primary targets of ˙NO are specific heme-iron receptors, the soluble or bound guanylate cyclases (GCs), promoting the production of cyclic guanosine monophosphate as the major downstream effector.9 Clinically, blood and tissue ˙NO levels are relevant for blood pressure regulation, platelet aggregation, control of inflammatory processes, and malignant diseases.10,11 Nitric oxide is a double-edged sword, showing biphasic behaviour, e.g. in oncology; low concentrations undesirably promote tumor cell growth while higher concentration above 200 nM mediate tumor cell apoptosis.12,13 Contributing to this are oxidation processes of ˙NO which lead to the formation of N2O3 or nitrogen dioxide (˙NO2) and the reaction with superoxide anion radicals (O2˙−) forming the strong oxidant peroxynitrite (ONOO−).14 In contrast, topical application of ˙NO from chemical sources typically show beneficial effects in wound models.15,16 Application of nitric oxide, especially topically, is challenging, e.g. due to the poor stability of ˙NO donor compounds or the intricate handling of gaseous ˙NO limiting clinical success. In this regard, cold physical plasma is a modern option to deliver ˙NO in a temporal and spatially controllable manner, rendering it interesting for therapeutic applications, e.g. in non-healing chronic wounds. Previous work has indicated the contribution of plasma derived nitrogen species, especially nitric oxide, on experimentally or clinically observed effects.17–19 Further studies showed improved wound healing by cold plasma without specifying the active players but where the role of nitric oxide is discussed.20–22Such an understanding of the deposition and/or generation processes of nitric oxide is key. As the solubility of ˙NO in water is rather low (Hcp(˙NO) = 1.9 × 10−5 M Pa−1), and gas phase densities are low to moderate, a mere gas phase generation of ˙NO and its subsequent solvation must be challenged.23 In opposition, nitric oxide may be generated via chemical reactions between plasma generated reactive nitrogen and oxygen species in the bulk liquid or at the gas–liquid interface. Knowledge of the importance of the latter is growing and several recent publications emphasize its role over that of the bulk liquid.24–29 However, as discussed by Bruggeman et al., transport processes, chemical reactions in the bulk gas, the liquid phase, and at the interface occur simultaneously.30 This makes the segregation of processes challenging, especially with the leak in gas–liquid interface diagnostic approaches. In many systems, the contribution of gas and liquid flow mechanics further escalates complexity ultimately driving research to focus on simulation approaches. Chen et al. modelled the oxygen-based chemistry of a surface helium–oxygen plasma by dividing it into a gas bulk model, a penetration model combined with Henry's law and a reaction chemistry part with the Poisson equation representing the gas–liquid interface, and for the bulk liquid a penetration model with reaction chemistry and the Poisson equation.31 Verlackt et al. modelled the situation for a plasma jet similar to the one used in the presented study.27 In their model, they could not find ˙NO in the bulk liquid, only in the interfacial region. Furthermore, their model predicts that long-lived species are mainly generated in the interface either by direct solvation or by reaction of, for instance, ˙NO originated from the gas phase.27
Here, the mechanism of nitric oxide deposition in physiological liquids by an argon plasma jet (kINPen09) was investigated. Via controlling of the ambient conditions and the working gas composition the possibility to regulate its ˙NO production was tested. Using electron paramagnetic resonance spectroscopy, ion chromatography, and colorimetric assays in combinations with gaseous ˙NO as control, a solvation of plasma derived ˙NO was excluded in favour of its de novo formation in the liquid. Furthermore, several potential gas or liquid phase precursors were ruled out and potentially dominating formation pathways were hypothesised.
Experimental
Plasma treatment
The plasma treatment took place in a 24 well plate using the well-characterized kINPen09 (neoplas GmbH, Greifswald, Germany) – a prior version of the certified medical product, the kINPen® MED (neoplas tools GmbH, Greifswald, Germany) as the plasma source.32–37 The plasma jet was equipped with a gas curtain device to enable working in a controlled environment.38–41 The total feed gas flow of the kINPen09 was 3 standard litre per minute (slm), using argon (argon N50, Air Liquide, Paris, France) as feed gas with addition of 1% molecular gases, oxygen, and/or nitrogen (oxygen N48, nitrogen N50, Air Liquide, Paris, France). For some experiments, humidification of the feed gas was used; therefore, 1% of the feed gas was guided through a bubbler system (final humidity approximately 300 ppm). In the following, this experimental situation will be referred to as ‘humidified feed gas’, whereas the situation without explicit humidification of the feed gas will be referred to as ‘dry feed gas’. The curtain gas device was fed with N2 or synthetic air (nitrogen N50, synthetic air: 20.5 ± 0.5% O2 in N2, Air Liquide, Paris, France). A total curtain gas flow rate of 5 slm was used (see Fig. 1). Details about the effect of the curtain gas can be found in previous publications.1,4,39,40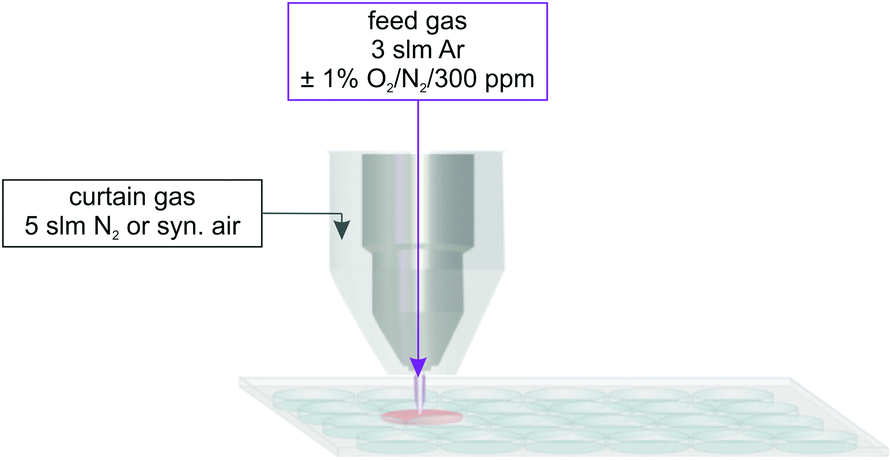 | ||
| Fig. 1 Scheme for the plasma treatment of 750 μL phosphate buffer in a 24 well plate using the kINPen09 with an additional curtain gas device. | ||
In a 24 well plate, 750 μL of phosphate buffer (Na2HPO4/H2NaPO4, Sigma Aldrich; 100 mM, pH 7.3) was treated for 30 s. The distance between the plasma jet nozzle and the liquid surface was kept at 9 mm.1 Furthermore, argon gas with 40 ppm ˙NO admixture was guided through the kINPen without igniting the plasma as a positive control.
Each data point consists of at least three independent samples; each sample was performed in triplicate. The error bars represent the minimal and maximal deviation of the mean value.
EPR measurements
For the detection of the ˙NO, ˙OH, O2˙−, and ˙H, electron paramagnetic resonance (EPR) spectroscopy was performed. An X-band (9.87 GHz) spectrometer (EMXmicro, Bruker BioSpin GmbH, Rheinstetten, Germany) was used with the following instrument parameters for all measurements: modulation frequency of 100 kHz, modulation amplitude of 0.1 mT, microwave power of 5.024 mW, receiver gain of 30 dB, and time constant of 0.01 ms. Depending on the used spin trap, the magnetic field scan was adjusted. A nitronyl nitroxyl radical (NNR) spin trap, carboxy-PTIO (2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, sodium salt), with an initial concentration of 60 μM, was used to detect ˙NO, giving an imino nitroxyl radical (INR) as a spin trap adduct with ˙NO,42 whereas 10 mM BMPO (5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide) was used for the detection of superoxide anion radicals (O2˙−) as well as hydroxyl radicals (˙OH) and hydrogen radicals (˙H). Due to economic reasons only selected data points (0, 0.2, 0.5, 0.8, 1% O2 in Ar, 100% Ar, untreated and gas ctrl.) were measured using BMPO as a spin trap. Both spin traps were supplied from Dojindo Laboratoire (Kumamoto, Japan). For all EPR measurements, the spin trap was added to the phosphate buffer prior to plasma treatment to ensure trapping of all plasma generated radicals in the solution.Prior to plasma treatment, an untreated spin trap solution (spin trap dissolved in phosphate buffer) was measured. Briefly, 50 μL of the treated or control solution was transferred to a borosilicate glass tube (125 mm length, 0.8525 mm inner diameter), and measured with a fixed delay of three minutes. EPR spectra evaluation was performed by using Bruker's Xenon software (Bruker BioSpin, Rheinstetten, Germany). Using the spin counting functionality and by calibrating the spectrometer, the absolute spin number could be determined. More details about the EPR procedure can be found in a previous publication.43
Ion chromatography
The detection of nitrite (NO2−) and nitrate (NO3−) was performed via ion chromatography (ICS-5000, Dionex Corp., Sunnyvale, USA). The ion chromatography measurements of nitrate and nitrite were performed as well only for the selected data points (0, 0.2, 0.5, 0.8, 1% O2 in Ar, 100% Ar, untreated and gas ctrl.), which were investigated using BMPO as a spin trap for the EPR measurements. After treatment, the sample was diluted three fold using ultrapure water (MilliQ, Milli-Q® Merck KGaA, Darmstadt, Germany) before injecting 10 μL onto an IonPac® AS23 anion exchange column (2 × 250 mm, Thermo Fisher Scientific Inc., Waltham, USA). An isocratic mobile phase (4.5 mM Na2CO3/0.8 mM NaHCO3) with 250 μL min−1 flow rate was used. Besides the use of the conductivity detector, data were collected as well from a UV detector (210 nm). The system was calibrated using the Dionex 7-anion standard on a weekly basis.Colorimetric assay
Hydrogen peroxide (H2O2) was detected via a colorimetric reaction with xylenol orange using a commercially available assay (Pierce™ Quantitative Peroxide Assay Kit, Thermo Scientific, Rockford, USA) according to the manufacturer's protocol. The assay is based on the oxidation of ferrous to ferric ion by hydrogen peroxide in the presence of the dye which can be detected photometrically (Tecan Infinite M200 Pro, Tecan Group Ltd, Männedorf, Switzerland) at 595 nm. Each 96 well plate contained a set of standards for calibration (0 to 150 μM, in triplicates) and the samples (n = 4, in triplicate).Results & discussion
In the following, possible reaction mechanisms that can lead to the generation of ˙NO in systems are discussed. In the first paragraph below, the solvation of gaseous ˙NO in the liquid is analysed since it is known from the literature5,44 that the investigated plasma source produced ˙NO in the gas phase. This process could be excluded as the predominant process for the origin of the liquid phase ˙NO. Therefore, in the following paragraph the focus is on the formation pathways yielding liquid phase ˙NO. Starting with a brief review regarding the relevant mechanisms that occur in the gas phase via the interfacial region, the bulk region, furthermore, multi-phase reactions and destruction reactions of ˙NO are discussed.Solvation of gaseous ˙NO
A previous study by Schmidt-Bleker et al. has shown that the generation of NOx in the gas phase by the kINPen operated with an O2/N2 feed gas admixture can be enhanced by humidifying the feed gas.5 That study has also shown that ˙NO can also be detected in the far field (12 mm or more in front of the nozzle5) of the jet, if the feed gas is humidified and a N2 gas curtain is applied. However, it is noted that in the near field (from the nozzle until 12 mm1) gaseous ˙NO can be expected under all conditions to be present to some extent if at least some N2 and O2 or H2O is available. As gaseous ˙NO can quickly be oxidised by various species (including ˙O, O3, HO2˙, ˙OH; section ‘˙NO formation’ reaction (6) and (7)), the detected downstream nitrogen oxides (HxNOy) are an indicator of the presence of ˙NO in the effluent. To investigate the possible solvation of ˙NO in the buffer solution, argon gas with 40 ppm ˙NO admixture guided through the kINPen without igniting the plasma was used as a control. The results of the control compared to the plasma-generated concentration of the ˙NO spin adduct (INR) in the liquid are shown in Fig. 2. For highlighting the difference obtained for ˙NO concentration caused by plasma treatment and by just guiding an Ar + ˙NO gas stream onto the liquid, in Fig. 2 already the plasma generated ˙NO-adduct concentration is given. Details about the plasma treatment data are given in the following sections. Remarkably, to reach a similar INR concentration in the phosphate buffer solution, the pure gas treatment time had to be extended to 600 s, which was 20-fold of the plasma treatment time. Furthermore, the used ˙NO-density of 40 ppm was more than fivefold higher than the estimated amount generated by the kINPen in the gas phase, which is calculated to be less than 8 ppm at maximum according to the following assumption. In order to estimate the maximum possible ˙NO density in the gas phase, all nitrogen oxides measured via FTIR spectroscopy in the gas phase reported in Schmidt-Bleker et al. are added: these include besides ˙NO, also ˙NO2, HNO2, HNO3 and N2O5 which are all likely downstream products of ˙NO.5 From the literature, a density of up to 8 ppm in the maximum of the gaseous phase ˙NO can be estimated and by assuming a linear increase of ˙NO transfer (due to Henry's law) to the liquid from the gas phase4,5 (which is subsequently trapped by the spin trap) in an ideal case, 16 nM INR could originate from solvation of gaseous ˙NO following a 30 s treatment with the kINPen, which is well below the measured concentrations.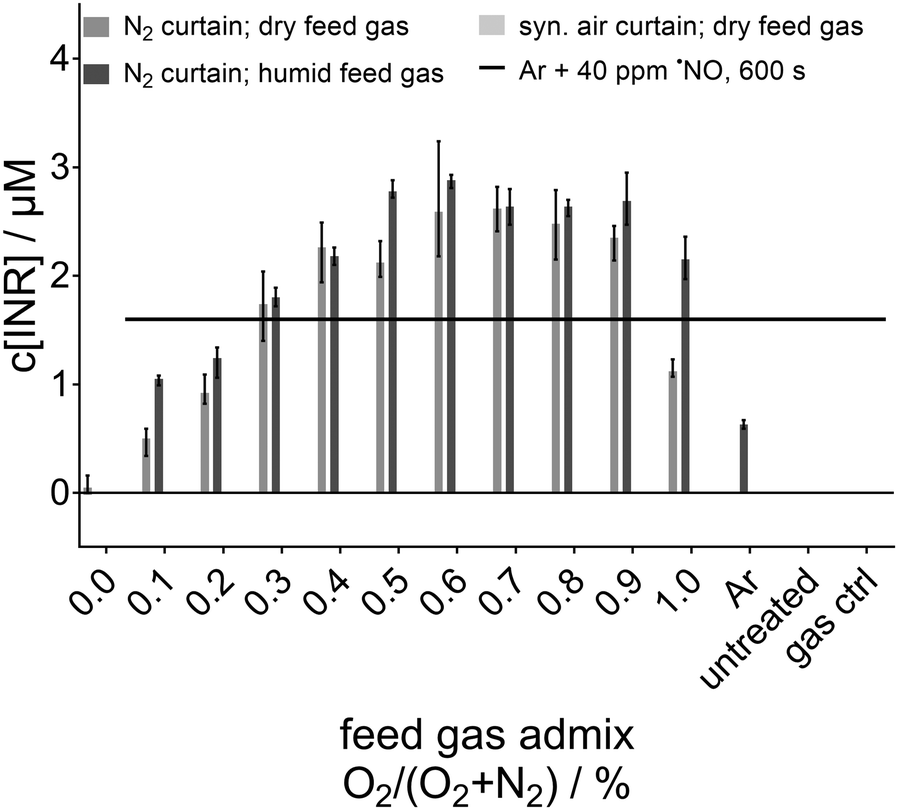 | ||
| Fig. 2 ˙NO spin adduct (INR) concentrations in the 30 s plasma treated phosphate buffer solution for different gas conditions compared to the ˙NO-concentration solvated during 600 s treatment with an Ar + ˙NO gas mixture (40 ppm ˙NO in Ar) as a function of the N2 and O2 content in the feed gas. | ||
Hence, a pure solvation process of gaseous ˙NO can be excluded, and it can be stated that the determined ˙NO in the buffer solution had its origin in the liquid or at the gas/liquid interface region. The aqueous equilibrium concentration of a species at a given partial gas pressure is described by the Henry coefficient. ˙NO (Hcp(˙NO) = 1.9 × 10−5 M Pa−1) is only moderately soluble in water.23,45 Since the solubility of ˙NO2 is also low, (although tenfold higher than the solubility of ˙NO); (Hcp(˙NO2) = 1.2 × 10−4 M Pa−1), solvation of gaseous ˙NO2 is also an unlikely source for the ˙NO detected in plasma-treated liquid, even if all dissolved ˙NO2 was converted to ˙NO.46 It is mentioned that the solubility of both HNO2 and HNO3 is higher by several orders of magnitude (Hcp(HNO2) = 4.8 × 10−1 M Pa−1, Hcp(HNO3) = 2.1 × 103 M Pa−1),46 which are present in the gas phase at densities of up to 1 ppm according to a study of Schmidt-Bleker et al.5
Since the detected liquid phase ˙NO was not mainly generated in the gas phase and transferred into the liquid, other formation pathways have to be taken into account. Possible generation pathways of aqueous ˙NO are discussed in the following.
˙NO formation
By the use of electron paramagnetic resonance spectroscopy and the nitronyl nitroxyl radical (NNR) as a spin trap, the ˙NO adduct, imino nitroxyl radical (INR), was determined. This determined concentration of the ˙NO adduct (INR) is given in Fig. 3, whereby in Fig. 3a the concentration for humidified feed gas and in Fig. 3b for dry feed gas is shown as a function of the O2 content in the feed gas. The feed gas composition was not always argon with 1% admixture (varied from 1% N2 and 0% O2 to 0% N2 and 1% O2) also pure argon (Ar) was used. With increasing O2 content in the feed gas the INR concentration was raised until a maximal concentration of almost 3 μM for both humidified and dry feed gas. This maximal concentration was reached for an intermediate amount of O2 in the feed gas before it decreased again. The controls, untreated buffer solution and gas treated (only feed gas without plasma ignition) yielded no INR signals. Ar treatment instead resulted in a formation of INR, whereby it differed for humid and dry feed gas. Only with humidity added to the feed gas is the concentration detectable. In addition, the maximum in INR formation was at different intermediate O2 contents in the feed gas depending on whether humid or dry feed gas was applied. For the dry condition, the maximum was shifted more to a higher O2 content (0.6% to 0.8% O2) whereas for humidified feed gas it was around 0.5% to 0.6% O2. The shift is only minor and by taking the error bars into account the maximum was for the humid and for the dry feed gas condition in the range between 0.5 or 0.4 to 0.9% O2, respectively. In the humid condition, there was also one plasma treatment condition where no INR was determined, more specifically, for a feed gas composition of 1% N2 and 99% Ar. This was not the case for dry conditions, although the INR concentration for that feed gas admixture was by far the lowest detected. Even though there were differences observed for humid and dry feed gas, these were not so remarkable that it could be stated that one of these two condition yielded a surplus in INR, and therefore, ˙NO-formation. In contrast, the observed dynamic of the INR concentration due to the feed gas admixtures of O2 and N2 have an important impact onto the ˙NO-formation. Besides the ˙NO formation via INR, the formed concentrations of NO2− (Fig. 3c and d) and NO3− (Fig. 3e and f) are given as well as a function of the O2 content in the humidified (Fig. 3c and e) or dry feed gas (Fig. 3d and f). Other than INR, NO2− showed a remarkably different behaviour between the humid and dry feed gas; with humidified feed gas, the resultant concentrations were several fold higher than for dry feed gas. Furthermore, the maximal yielded NO2− concentration was found for 0.2% O2 and 0.8% N2 in the feed gas for both the humid and dry feed gas condition. NO3− did not alter much, although it was slightly more produced for 0%, 0.2%, and 1% O2 in the humid feed gas than in the dry condition.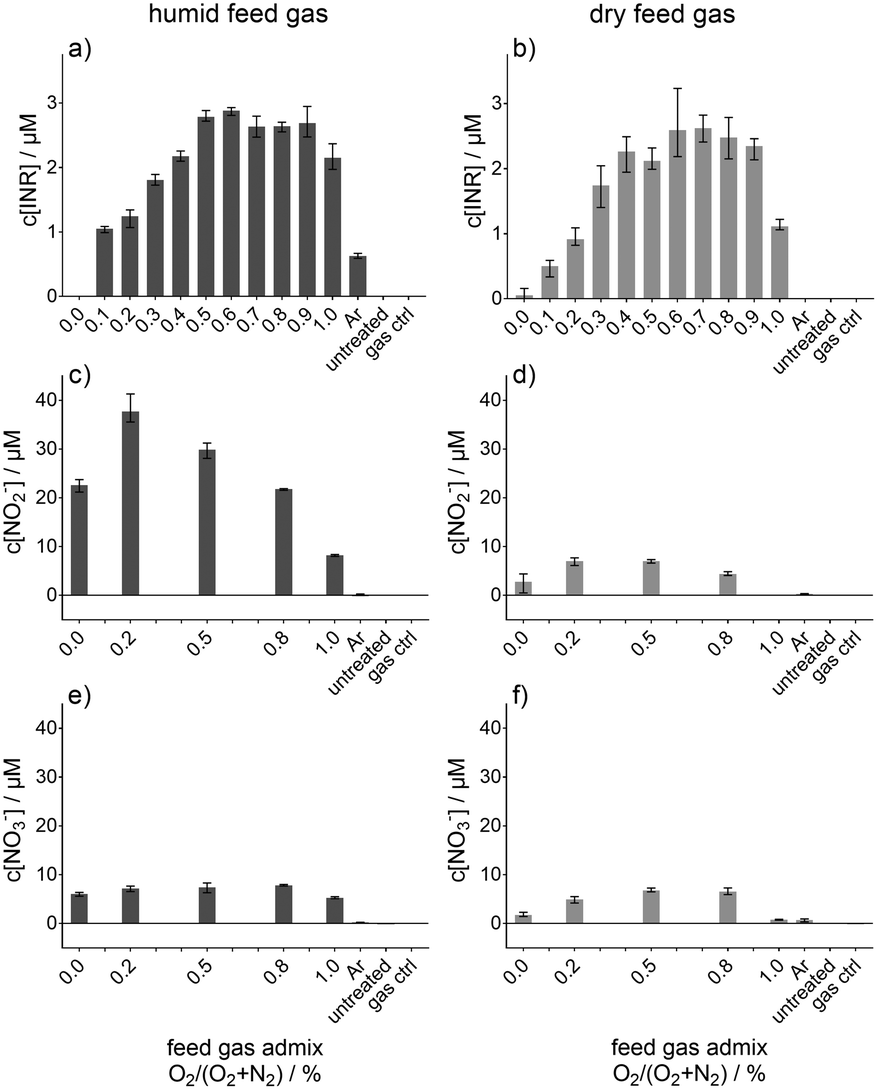 | ||
| Fig. 3 Concentration of nitric oxide spin trap adduct (a) and (b), nitrite (c) and (d), and nitrate (e) and (f). Each column represented either humid (left column) or dry (right column) feed gas for a N2 curtain gas. The concentrations were obtained after 30 s plasma treatment of phosphate buffer and given as function of the O2 content (%) in the feed gas argon (Ar). “Ar” indicates treatment with pure argon without the O2/N2 admixture; “gas ctrl” indicates treatment with pure gas mixture without plasma ignition; “untreated” indicates pure phosphate buffer without any treatment. | ||
Beside these nitrogen species also some potentially relevant oxygen species, O2˙−, ˙OH, and H2O2 as well as atomic hydrogen (˙H) were investigated under the same conditions (see Fig. 4). The radical adducts of O2˙− (Fig. 4a and b), ˙OH (Fig. 4c and d), and ˙H (Fig. 4e and f) were determined by EPR using BMPO as a spin trap. The concentration of H2O2 is given in Fig. 4g and h. The behaviour of O2˙− (BMPO˙-OOH, Fig. 4a and b) showed only slight differences for humid or dry feed gas. Under the humid condition, an O2 content of 0.2% yielded the highest concentration, whereas the maximum was reached for dry feed gas for 1% followed by the 0% O2 admixture. For 0% O2 content in the humid feed gas instead, the lowest BMPO˙-OOH concentration was determined. The difference between the humid and dry feed gas was clearer for the ˙OH (BMPO˙-OH, Fig. 4c and d). There, the O2 free conditions, pure Ar and Ar + 1% N2 as dry feed gas, resulted in almost twice as high concentrations as for the humid feed gas. The other feed gas compositions did not vary between humid and dry. The difference between humid and dry was also observed for the same two pronounced feed gas compositions for ˙H (BMPO˙-H, Fig. 4e and f). Under the O2 containing feed gas conditions no BMPO˙-H was detected. A strong relation of the H2O2 concentration to humid feed gas was observed (Fig. 4g), whereby pure Ar yielded the maximum. For dry feed gas (Fig. 4h), pure Ar resulted as well in the highest concentration, although it was less than half of the one of the humid feed gas. A further difference was observed for Ar + 1% N2, a condition which resulted in higher H2O2 concentration than all other dry feed gas compositions, whose concentrations were much lower.
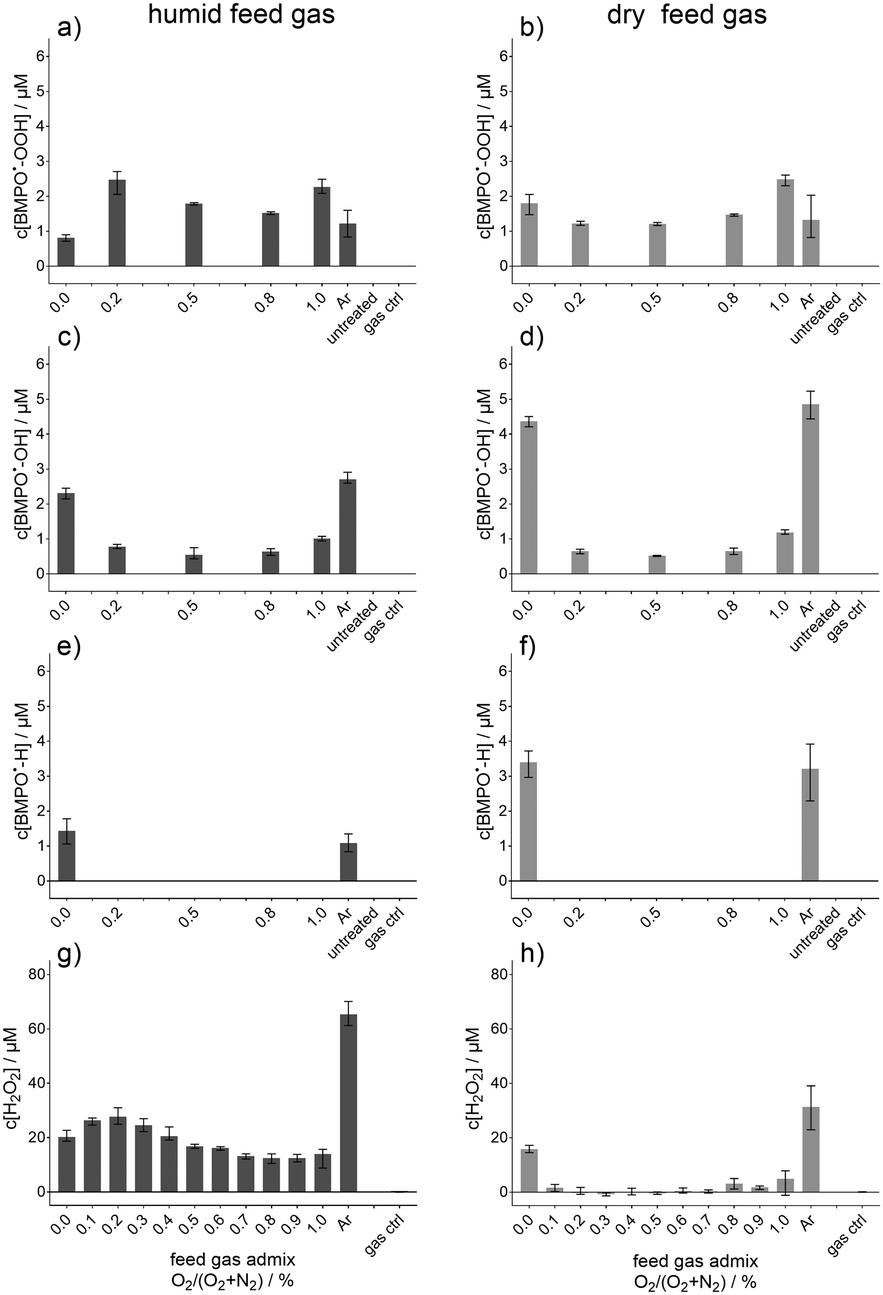 | ||
| Fig. 4 Concentration of O2˙− spin trap adduct (a) and (b), ˙OH spin trap adduct (c) and (d), ˙H spin trap adduct (e) and (f), and H2O2 (g) and (h). Each column represented either humid (left column) or dry (right column) feed gas for a N2 curtain gas. The concentrations were obtained after 30 s plasma treatment of phosphate buffer and given as a function of the O2 content (%) in the feed gas argon (Ar). “Ar” indicates treatment with pure argon without O2/N2 admixture; “gas ctrl” indicates treatment with pure gas mixture without plasma ignition; “untreated” indicates pure phosphate buffer without any treatment. | ||
In the following, other potential formation pathways than solvation are discussed and weighted in consideration of the above described measurement results.
Although the ˙NO in the liquid phase was not predominantly generated in the gas phase and solvated in the liquid, at least some of the precursors of the liquid phase ˙NO had their origin in the gaseous phase. Hence, in the following subsection a more detailed insight into the formation of several precursors in the gas and plasma phase is briefly reviewed. Following the gaseous phase reactions, the gas–liquid interface and the bulk liquid reaction involved in either precursor or direct ˙NO formation are discussed. Furthermore, in the subsection ‘multi-phase reactions’ reactions which occur in more than one of the discussed phases are taken into account for the formation of ˙NO in the liquid or at its interface.
| Ar* + N2 → 2N + Ar1 | (1) |
A numerical study of the kINPen operated with O2/N2 feed gas admixtures suggests that gaseous ˙NO is mainly formed through the reaction (2).
| N2* + ˙O → ˙NO + N47 | (2) |
| N2 + ˙O → ˙NO + N | (3) |
| O2 + N → ˙NO + ˙O | (4) |
| N + ˙OH → ˙NO + ˙H | (5) |
The latter reaction (5) also implies that generation of N or N2* as a precursor for the generation of ˙NO in the liquid (reactions (2)–(4)) is unlikely to occur in the liquid itself, since reactive species with sufficient energy are most likely to dissociate water (ESI,† reaction (S2)). Hence, it is most likely that ˙NO is either transferred from the gaseous to the liquid phase (discussed in paragraph ‘Solvation of gaseous ˙NO’), or generated though secondary reactions from further nitrogen oxides generated in the core plasma, or at the liquid interface. The dominant nitrogen oxides that can be expected in the effluent of the kINPen comprise ˙NO2, which can be generated from ˙NO by various oxidizing agents Y (such as O3, ˙O, HO2˙) in reactions of the form of reaction (6) and HNO2 and HNO3, which are mainly generated in reactions such as reaction (7).47
| Y + ˙NO → ˙NO2 + products | (6) |
| ˙NOx + ˙OH → HNOx+1 | (7) |
| N2O3 ↔ ˙NO2 + ˙NO | (8) |
Another mechanism that is well-known in the context of plasma-treated liquids is the generation of peroxynitrous acid (ONOOH) in the reaction (9) whereas ONOOH partially (3 to 30%) decomposes to form ˙NO2 and ˙OH, where ONOOH could act as precursors for the generation of ˙OH.53
| HNO2 + H2O2 → ONOOH + H2O → ˙NO2 + ˙OH + H2O | (9) |
Reaction (10) could also occur at the surface of the liquid, where the pH is low enough for relevant amounts of HNO2 to exist.
| HNO2 + ˙H → ˙NO + H2O54 | (10) |
| ˙H + NO2− → ˙NO + OH−55,56 | (11) |
| ˙H + ˙NO2 → ˙NO + ˙OH57 | (12) |
A relevant mechanism could be the generation of ˙NO from ˙NO2 through reaction (13).
| ˙NO2 + ˙O → ˙NO + O258 | (13) |
The promoting effect of a N2 curtain gas could have a double impact; on one hand the N2 could be an additional source of the N-containing precursors as was discussed during the prior processes. On the other hand, it avoids the formation of huge amounts of gaseous O3 which inhibits the formation of aqueous ˙NO by destroying precursors necessary for its generation.59 For instance, NO2− could effectively be removed from the liquid via O3 (ESI† reaction (S4)).
O3 is a double-edged sword as well; it can also contribute to the formation of ˙NO2via reaction with the gas phase ˙NO (reaction (14)). Although the optimum for O3 production was not under N2 curtain gas, there will be still a relevant amount produced in the gas phase with an N2 curtain, especially for higher admixtures of O2.5 There, also the three body reaction of ˙O with O2 and a third body (M) takes place (reaction (15)).
| O3 + ˙NO → ˙NO2 + O260,61 | (14) |
| ˙O + O2 + M → O3 + M58 | (15) |
At first sight, an ˙O-related process cannot explain the measured INR density in the case using the N2 curtain with the 1% N2 feed gas admixture and dry feed gas (Fig. 3b), where no ˙O can be expected. There, the only way to create ˙O in the liquid under this condition is the complete dissociation of H2O into ˙H and ˙OH in the first step and in the second step ˙OH into ˙O and ˙H. With a bond dissociation energy of 5.17 eV (first step) and 4.44 eV (second step), this is in principle possible under these conditions.35 For dry feed gas, the dissociation took place in the gas phase, whereas for the N2 curtain with the 1% N2 feed gas admixture, the process occurred in the interface. In the literature, the generation via a reaction of NO2− with ˙H has been mentioned in an aqueous system (see reaction (11)).55
˙H has recently been measured in the effluent of an Ar-operated plasma operated with an Ar gas curtain jet.62 Additionally, ˙H can be generated from solvated electrons e−via the reaction with H+ (see ESI,† reaction (S5)).
However, this process is unlikely in these experiments, since the effluent of the plasma jet does not directly touch the liquid surface.
In Fig. 3c–f, the generated concentrations of NO2− and NO3− after plasma treatment are given over the O2 feed gas admixture, whereby, the concentrations given in Fig. 3c and e represented the humid and Fig. 3d and f the dry feed gas.
Interestingly, there was a strong difference between these two; humidification of the feed gas yielded a much more pronounced formation of NO2− compared to NO3−. NO3− was detected in similar amounts for the humid and dry feed gas. In the latter case, also the NO2− was in the same concentration range, up to 8 μM. The difference in these two cases suggests that a NO2−-related mechanism may not be the predominant pathway of the liquid phase ˙NO formation because the trend and amounts of ˙NO formation are similar in the humid and dry feed gas (see Fig. 3a and b). The observed trends for NO2−, NO3− and ˙NO for dry feed gas are similar. In contrast, the trends for humid feed gas are not alike. Hence, in that case, the detected NO2− and NO3− is predominantly produced via a different pathway than via ˙NO for the humid and dry feed gas condition; for instance, the solvation of gaseous HNO2 and HNO3 could be highly relevant.
| ˙NO2 + ˙O → ˙NO + O258 | (16) |
| ˙N + ˙OH → ˙NO + H˙63,64 | (17) |
| ˙NO2 + ˙OH → HO2˙ + ˙NO65 | (18) |
| ˙NO2 + ˙N → 2˙NO66 | (19) |
| ˙N + ˙O → ˙NO67 | (20) |
The concentration of the generated spin trap adduct for O2˙−/HO2˙, ˙OH, and ˙H was determined using BMPO as a spin trapping agent as it is able to distinguish between the O2˙−- and ˙OH-adduct.43,68
In Fig. 4, the resulting concentrations for the trapped O2˙− (a), ˙OH (c), and ˙H (e) are shown in the case of humidified feed gas admixtures and N2 as the curtain gas. Interestingly, under plasma treatment conditions (with O2 in the feed gas), where higher amounts of ˙NO were detected (see Fig. 3a), the concentration of ˙OH was lower (see Fig. 4c). In contrast, high ˙OH-adduct concentrations were observed – pure Ar or Ar + 1% N2 as feed gas – for conditions where no or just minor amounts of ˙NO were measured after plasma treatment. Without the admixture of O2, the ˙OH-adduct concentration was maximal (see Fig. 4c and d). Moreover, when O2 was present in the humidified feed gas, the BMPO˙-OH concentration was reduced by 50% or more. This dependence on the feed gas humidity indicated no direct linkage of the ˙OH with the formation of ˙NO. It could be also excluded that ˙OH is consumed by the formation of ˙NO, as there were treatment conditions where different concentrations of the ˙OH adduct were detected, for instance the N2 curtain gas with 0.2% or 0.8% O2 admixture to the feed gas. Here, the determined concentrations of the ˙NO spin probe adduct differed a lot whereas those of the ˙OH adduct were similar. A similar situation was observable for humidified feed gas for the 0.2% and 1% O2 feed gas admixture.
For ˙H, the observations were almost the same, besides that in the case of O2 present in the humid feed gas no BMPO˙-H adduct was detectable, presumably because gaseous ˙H quickly reacts with O2, leading to the generation of HO2˙ (ESI,† reaction (S6)).
This would also imply that solvation from the gas phase is the predominant source of ˙H detected in the liquid or at least the formation in the bulk liquid or at the interface was hindered by presence of O2 in the feed gas. Furthermore, ˙H can also quite likely be generated at the liquid surface by dissociation of water; under O2-free conditions VUV radiation can reach the liquid surface as mentioned above.35 For the kINPen09, the argon excimer radiation at 126 nm is present and is able to dissociate H2O.35
As already discussed when considering the NO2−-formation, also ˙H does probably not predominantly contribute to the production of ˙NO viareaction (11).
The O2˙−-adduct behaved slightly differently; the lowest BMPO˙-OOH concentrations reached for these humidified plasma treatments were observed when only N2 was added to the feed gas. When N2 was used as the curtain gas, no diffusion of O2 from the environment took place. Hence, the results for O2˙− were as expected when the O2˙− mainly originated from the gas phase. There, HO2˙ will be generated and can be transferred to the liquid.1,5 In general, the production of O2˙− adducts in all other conditions, Ar and Ar with O2 addition, was more pronounced when the feed gas was humidified. Due to the pH value of 7.3 of the used phosphate buffer, the HO2˙ will be present in its deprotonated form as O2˙−.45 The highest concentration was detected for the 0.2% and 1% O2 admixture in the humid feed gas. The reason for the maximum at the 1% O2 admixture could be the simultaneous formation of ˙OH and O3 in the gas phase. If both species react with each other, HO2˙ could be formed (see ESI,† reaction (S7)). For 0.2% O2 in the feed gas, one reaction, which enhanced the O2˙−/HO2˙ formation could be ˙O with H2O2 (see ESI,† reaction (S8)), as by addition of humidity to the feed gas the formation of H2O2 will be enhanced.33,69 This was also shown here under these specific conditions. This will be discussed in more detail later on.
For dry feed gas and N2 curtain gas, the BMPO-adducts of O2˙−, ˙OH, and ˙H are given in Fig. 4b, d and f. For O2-free conditions, the measured concentrations for BMPO˙-OH and BMPO˙-H were about twice the one of the humidified feed gas plasma treatment. This indicated again that also the formation of ˙NO via ˙OH and ˙H was not likely to be the predominant process.
In contrast, the feed gas humidity did not influence the O2˙− adduct concentration as much as the ˙OH adduct concentration; the O2˙− adduct concentrations were in the same range for both dry and humid feed gas. Interestingly, for the dry feed gas, the two highest conditions were obtained for the 1% O2 and 1% N2 feed gas. In particular the admixture of only N2 was in contrast to the humidified case, as there the lowest BMPO˙-OOH concentration was observed. Due to the absence of O2 in the gas phase, the high energetic species could reach the interfacial region and the liquid surface yielding reduction of the liquid phase O2 to O2˙−. In the case of the 1% O2 admixture, the HO2˙ could also be mainly produced in the plasma/gas phase as was the case for humidified feed gas, although, direct production in the liquid itself was likely as well. For instance, reactions of ˙OH, ˙O, O3, and H2O2 (see ESI,† reactions (S7)–(S10)) could result in an increase of O2˙−/HO2˙ in the solution.
The fact that humidity yields no surplus for the formation of oxygen radicals in liquids is known from the literature, although, in that study only humidity but no O2 and N2 was added.69
As mentioned above, the H2O2 concentration in strongly linked to the feed gas humidity. Concentrations in Fig. 4g were detected after plasma treatment with humidified feed gas. The highest concentration, more than threefold higher than that of the others, was obtained for pure Ar. Interestingly, low amounts of O2, up to 0.2% yielded slightly higher H2O2 concentrations than those for only N2 or N2 with 0.3% O2 or more for humidified feed gas. For BMPO-OOH a similar increase was observed for the humidified feed gas and it is known from the literature that HO2˙/O2˙− could contribute to the formation of H2O2 as well.31,45 This explained this slight increase. The other conditions all resulted in almost the same concentration, although the oxygen radical concentrations inside the liquid differed. This underlined the predominant formation of H2O2 in the gas phase if humidity was added to the feed gas.69
In contrast to the humidified feed gas, the dry feed gas yielded much lower H2O2 concentrations for all cases of feed gas admixtures of N2 and/or O2 (Fig. 4h). Between the 0.1% and 0.7% O2 admixture the determined concentrations were in the range of the lower detection limit of the assay used, which was 1 μM according to the data sheet of the manufacturer. For higher amounts of O2, there was a slight increase observed, but not in the range of what was measured for the 1% N2 admixture or for pure argon. Under these two conditions (pure Ar or Ar + 1% N2), high energetic radiation or species such as V(UV) and metastable nitrogen were able to reach the liquid and dissociate water to ˙OH (see subsection ‘Bulk reactions’), yielding via recombination H2O2.35,44,70
Looking at all results together, it can be stated that it was most likely that the aqueous ˙NO production was linked to reactions containing ˙N/NO2˙ and ˙O/O3.
| ˙NO + O2˙− → ONOO−64 | (21) |
| ˙NO + ˙OH → NO2− + H+71 | (22) |
| ˙NO + ˙H → HNO72 | (23) |
| ˙NO + O3 → ˙NO2 + O273 | (24) |
| 4˙NO + O2 + 2H2O → 4H+ + 4NO2− | (25) |
| 2˙NO + O2 → 2˙NO274 | (26) |
| ˙NO3 + ˙NO → 2˙NO258 | (27) |
| ˙NO2 + ˙NO → N2O375 | (28) |
| N2O3 + 2OH− → 2NO2− + H2O75 | (29) |
As relevant amounts of BMPO-OOH have also been detected in the liquid (see Fig. 3a and b), it was likely that some ˙NO is removed by O2˙− or HO2˙ in reactions (21) and (30).
| HO2˙ + ˙NO → ONOOH76 | (30) |
While up to 30% of the generated ONOOH could be regenerated to ˙NO253 and subsequently ˙NO via various pathways, HO2˙ still is expected to lead to a net reduction of the ˙NO content in the liquid. If not stabilized by a spin trap, HO2˙ is also likely to quickly vanish by reaction with ˙OH (ESI,† reaction (S11)) or with another HO2˙ molecule (ESI,† reaction (S12)).
In the previous study concerning the reactive species output of the kINPen in the gas phase, NOx were produced in similar amounts either with N2 curtain gas or with the synthetic air curtain.5 However, in the present study, the synthetic air environment around the plasma plume completely inhibits the formation of aqueous ˙NO (no INR was detected in this case; data not shown, the corresponding NNR signal is shown in Fig. 5).
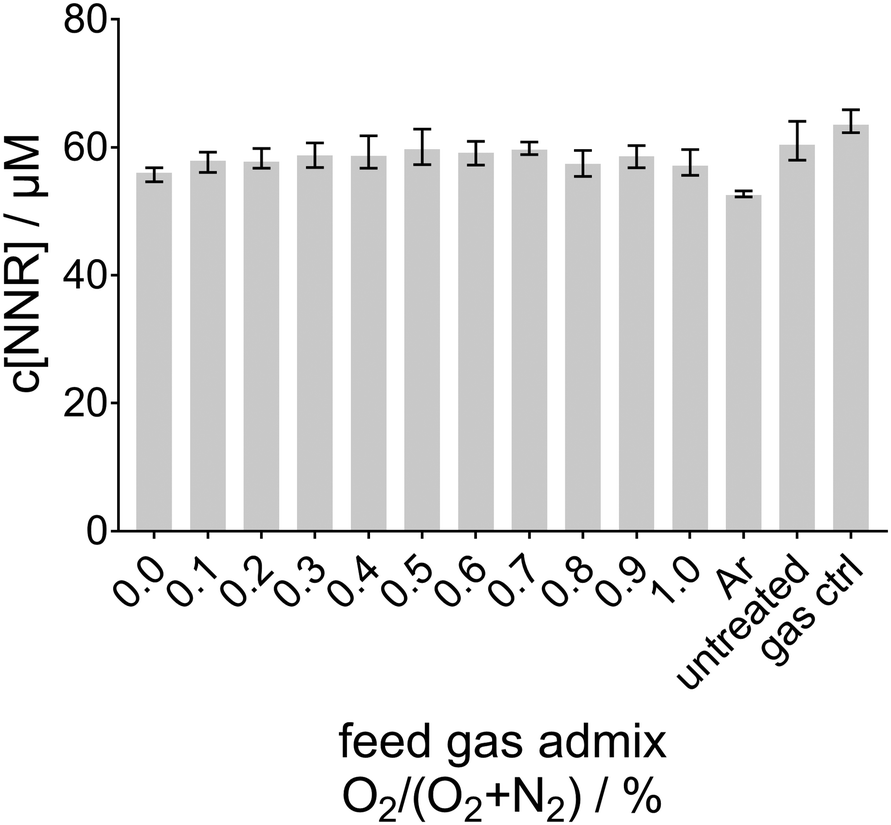 | ||
| Fig. 5 The nitronyl nitroxyl radical concentration for a synthetic air curtain gas and dry molecular feed gas admixtures obtained after 30 s plasma treatment as a function of the O2 content in the feed gas. | ||
Based on the mechanisms discussed in the literature, it is assumed that the exclusion of ambient O2 will mainly have two effects: first, when ambient O2 is excluded (while O2 is still admixed to the feed gas), ˙O is more likely and O3 less likely to reach the liquid surface, as less ˙O is converted to O3 in the gas phase by reaction (15).1,5
In a prior study, among others, the effect of the curtain gas on the oxygen species production was investigated.77 There, also the suppression of the ˙O concentration in the liquid in the presence of synthetic air surroundings was determined. In that study, increasing amounts of O2 up to 1%, but no N2, were added to the feed gas. By the use of TEMPD-HCl as a spin probe ˙O, 1O2, or O3 were detected after plasma treatment. Due to the gaseous formation processes, it could be stated that for direct treatment, as was the case in the here-presented study as well, the N2 curtain gas mainly leads to the formation of aqueous ˙O, whereas an extended treatment distance or a synthetic air curtain gas composition was dominated by the formation of O3.77 This leads to the conclusion that ˙O is an important precursor for aqueous ˙NO, and in the presence of O2, O3 is formed, which prevents the formation of aqueous ˙NO. Therefore, using synthetic air as the curtain gas, it was expected that less or no ˙NO-adducts are detectable in the plasma treated phosphate buffer. In contrast to the measurements using N2 curtain gas using either humid (see Fig. 3a) or dry feed gas (see Fig. 3b), for synthetic air as the curtain gas no INR was detected. In Fig. 5, the nitronyl nitroxyl radical (NNR) concentration is plotted as no INR was detected after plasma treatment. This was the expected result, as due to the presence of O2 in the surroundings mainly O3 is formed. Interestingly, the spin trap concentration from initially 60 μM was slightly reduced due to the plasma treatment. This destruction of the compound was most prominent in the case of pure argon as feed gas. Under this condition, only diffusing species from the surroundings into the plasma plume were excited, therefore, more highly energetic argon species reached the liquid and effected the spin trap therein. Besides the highly energetic argon species, also other radicals present in the liquid such as ˙OH, ˙O, and O2˙− could react with the spin trap compound yielding non-paramagnetic products. This point is discussed in more detail in the paragraph ‘˙NO formation’. Second, ˙H can be expected to reach the liquid surface when ambient O2 is excluded, which quickly forms HO2˙ by reaction with a third body (M) in the reaction (S6) (ESI†). As the rate for the gas phase reaction (S6) (ESI†) is fast (5.65 × 10−32 cm6 s−1), already small amounts of O2 from feed gas admixtures inhibit the transport of gaseous ˙H to the liquid surface. As shown in Fig. 4e and f, ˙H was only detected in the liquid if no O2 is available in the ambient or the feed gas. More details about the role of ˙H in the formation processes of ˙NO can be found in the subsection ‘Bulk reactions’.
Conclusions
In the present study, the origin of ˙NO was investigated. It was known from previous studies that different reactive species induced by the plasma jet have various origins. Here, it could be shown for the first time that liquid phase ˙NO mainly originated from the bulk and interfacial region and not via solvation from the gas phase. This knowledge is of high importance for the impact of plasma treatment on targets, no matter whether they are biological targets or not. Increasing knowledge about the ongoing chemical reactions are essential for matching the treatment condition and the aimed application.The predominant formation and destruction reactions are given in Scheme 1, where the most relevant reactions occurring in the different phases – gas phase, interface, and liquid phase – are put together. By combination of the measurements with knowledge about the gas phase chemistry of the plasma jet from the literature and theoretical considerations of possible formation reactions, the most likely reactions for ˙NO production were determined: this pointed to the liquid in the direction of reactions of nitrogen species with ˙O/O3, which are highlighted by red frames in Scheme 1. The elucidation of the relevant formation reaction added to the overall understanding of the induced chemistry by the kINPen09. Besides this, the elucidated optimum for the formation of ˙NO by kINPen09 enables a regulation of the ˙NO formation as desired in the application, for instance, for tailored therapeutic treatment in cancer medicine or wound healing.
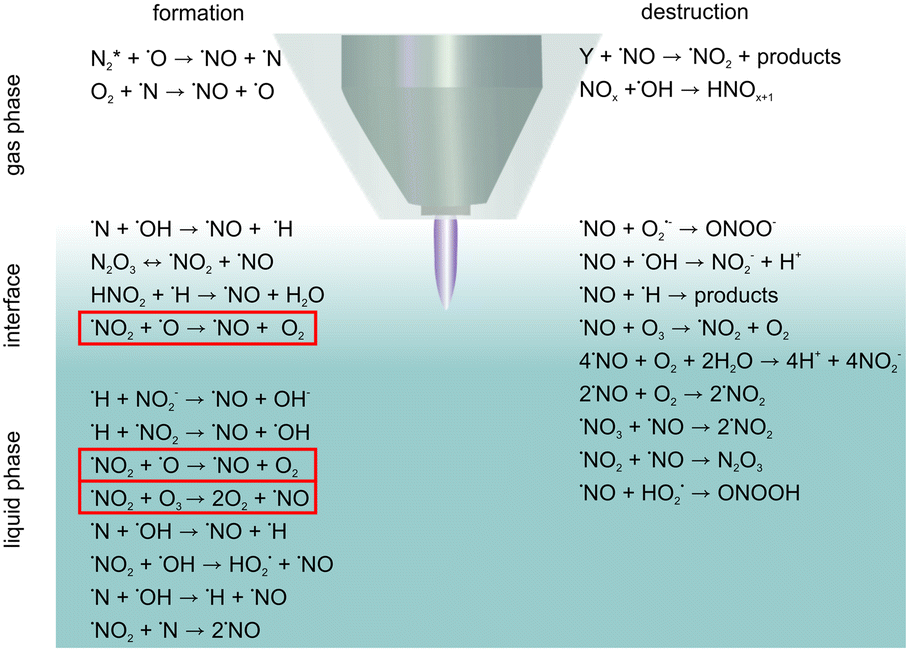 | ||
| Scheme 1 A detailed look into the relevant formation and destruction reactions of ˙NO in the gas phase, interface, and the liquid phase. Red frames indicate the most likely formation reactions for liquid phase ˙NO. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr Jörn Winter for his help. This work is funded by the German Federal Ministry of Education and Research (BMBF) (Grant No. 03Z22DN12).References
- A. Schmidt-Bleker, J. Winter, A. Bösel, S. Reuter and K.-D. Weltmann, Plasma Sources Sci. Technol., 2015, 25, 015005 CrossRef.
- T. Moiseev, N. Misra, S. Patil, P. Cullen, P. Bourke, K. Keener and J. Mosnier, Plasma Sources Sci. Technol., 2014, 23, 065033 CrossRef CAS.
- S. Iseni, S. Reuter and K. D. Weltmann, J. Phys. D: Appl. Phys., 2014, 47, 075203 CrossRef.
- A. V. Pipa, S. Reuter, R. Foest and K. D. Weltmann, J. Phys. D: Appl. Phys., 2012, 45, 085201 CrossRef.
- A. Schmidt-Bleker, R. Bansemer, S. Reuter and K.-D. Weltmann, Plasma Process Polym., 2016, 13, 1120–1127 CrossRef.
- O. I. Aruoma, J. Am. Oil Chem. Soc., 1998, 75, 199–212 CrossRef CAS.
- A. J. Gow and H. Ischiropoulos, J. Cell. Physiol., 2001, 187, 277–282 CrossRef CAS PubMed.
- J. R. Hickok, D. Vasudevan, K. Jablonski and D. D. Thomas, Redox Biol., 2013, 1, 203–209 CrossRef CAS PubMed.
- D. A. Fitzpatrick, D. M. O'Halloran and A. M. Burnell, BMC Evol. Biol., 2006, 6, 26 CrossRef PubMed.
- Y. C. Luiking and N. E. Deutz, Curr. Opin. Clin. Nutr. Metab. Care, 2003, 6, 103–108 CrossRef CAS PubMed.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- S. Huerta, Future Sci. OA, 2015, 1, FSO44 Search PubMed.
- W. Xu, L. Z. Liu, M. Loizidou, M. Ahmed and I. G. Charles, Cell Res., 2002, 12, 311–320 CrossRef PubMed.
- K. Kobayashi, M. Miki and S. Tagawa, J. Chem. Soc., Dalton Trans., 1995, 2885–2889 RSC.
- L. R. Martinez, G. Han, M. Chacko, M. R. Mihu, M. Jacobson, P. Gialanella, A. J. Friedman, J. D. Nosanchuk and J. M. Friedman, J. Invest. Dermatol., 2009, 129, 2463–2469 CrossRef CAS PubMed.
- M. Neidrauer, U. K. Ercan, A. Bhattacharyya, J. Samuels, J. Sedlak, R. Trikha, K. A. Barbee, M. S. Weingarten and S. G. Joshi, J. Med. Microbiol., 2014, 63, 203–209 CrossRef CAS PubMed.
- C. V. Suschek and C. Opländer, Clinical Plasma Medicine, 2016, 4, 1–8 CrossRef.
- A. B. Shekhter, V. A. Serezhenkov, T. G. Rudenko, A. V. Pekshev and A. F. Vanin, Nitric oxide, 2005, 12, 210–219 CrossRef CAS PubMed.
- V. N. Vasilets, A. B. Shekhter, A. E. Guller and A. V. Pekshev, Clinical Plasma Medicine, 2015, 3, 32–39 CrossRef.
- G. Isbary, J. L. Zimmermann, T. Shimizu, Y. F. Li, G. E. Morfill, H. M. Thomas, B. Steffes, J. Heinlin, S. Karrer and W. Stolz, Clinical Plasma Medicine, 2013, 1, 19–23 CrossRef.
- G. Isbary, J. L. Zimmermann, T. Shimizu, G. E. Morfill, A. Mitra, H. Thomas, T. G. Kaempfl, J. Koeritzer, V. Boxhammer, J. Schlegel and W. Stolz, Plasma Medicine, 2012, 2, 85–96 CrossRef.
- A. Schmidt, S. Bekeschus, K. Wende, B. Vollmar and T. von Woedtke, Exp. Dermatol., 2017, 26, 156–162 CrossRef CAS PubMed.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- A. D. Lindsay, D. B. Graves and S. C. Shannon, J. Phys. D: Appl. Phys., 2016, 49, 235204 CrossRef.
- K. Wende, P. Williams, J. Dalluge, W. V. Gaens, H. Aboubakr, J. Bischof, T. von Woedtke, S. M. Goyal, K. D. Weltmann, A. Bogaerts, K. Masur and P. J. Bruggeman, Biointerphases, 2015, 10, 029518 CrossRef PubMed.
- C. E. Anderson, N. R. Cha, A. D. Lindsay, D. S. Clark and D. B. Graves, Plasma Chem. Plasma Process., 2016, 36, 1393–1415 CrossRef CAS.
- C. C. W. Verlackt, W. Van Boxem and A. Bogaerts, Phys. Chem. Chem. Phys., 2018, 20, 6845–6859 RSC.
- N. Kurake, H. Tanaka, K. Ishikawa, K. Takeda, H. Hashizume, K. Nakamura, H. Kajiyama, T. Kondo, F. Kikkawa, M. Mizuno and M. Hori, J. Phys. D: Appl. Phys., 2017, 50, 155202 CrossRef.
- Y. Y. Zhao, T. Wang, M. P. Wilson, S. J. MacGregor, I. V. Timoshkin and Q. C. Ren, IEEE Trans. Plasma Sci., 2016, 44, 2084–2091 CAS.
- P. J. Bruggeman, M. J. Kushner, B. R. Locke, J. G. E. Gardeniers, W. G. Graham, D. B. Graves, R. C. H. M. Hofman-Caris, D. Maric, J. P. Reid, E. Ceriani, D. F. Rivas, J. E. Foster, S. C. Garrick, Y. Gorbanev, S. Hamaguchi, F. Iza, H. Jablonowski, E. Klimova, J. Kolb, F. Krcma, P. Lukes, Z. Machala, I. Marinov, D. Mariotti, S. M. Thagard, D. Minakata, E. C. Neyts, J. Pawlat, Z. L. Petrovic, R. Pflieger, S. Reuter, D. C. Schram, S. Schroter, M. Shiraiwa, B. Tarabova, P. A. Tsai, J. R. R. Verlet, T. von Woedtke, K. R. Wilson, K. Yasui and G. Zvereva, Plasma Sources Sci. Technol., 2016, 25, 053002 CrossRef.
- C. Chen, D. X. Liu, Z. C. Liu, A. J. Yang, H. L. Chen, G. Shama and M. G. Kong, Plasma Chem. Plasma Process., 2014, 34, 403–441 CrossRef CAS.
- S. Reuter, J. Winter, S. Iseni, S. Peters, A. Schmidt-Bleker, M. Dunnbier, J. Schafer, R. Foest and K. D. Weltmann, Plasma Sources Sci. Technol., 2012, 21, 034015 CrossRef.
- J. Winter, K. Wende, K. Masur, S. Iseni, M. Dunnbier, M. U. Hammer, H. Tresp, K. D. Weltmann and S. Reuter, J. Phys. D: Appl. Phys., 2013, 46, 295401 CrossRef.
- A. Schmidt-Bleker, S. A. Norberg, J. Winter, E. Johnsen, S. Reuter, K. D. Weltmann and M. J. Kushner, Plasma Sources Sci. Technol., 2015, 24, 035022 CrossRef.
- H. Jablonowski, R. Bussiahn, M. U. Hammer, K. D. Weltmann, T. von Woedtke and S. Reuter, Phys. Plasmas, 2015, 22, 122008 CrossRef.
- S. Bekeschus, A. Schmidt, K.-D. Weltmann and T. von Woedtke, Clinical Plasma Medicine, 2016, 4, 19–28 CrossRef.
- K. D. Weltmann, E. Kindel, R. Brandenburg, C. Meyer, R. Bussiahn, C. Wilke and T. von Woedtke, Contrib. Plasma Phys., 2009, 49, 631–640 CrossRef.
- S. Reuter, J. Winter, A. Schmidt-Bleker, H. Tresp, M. U. Hammer and K. D. Weltmann, IEEE Trans. Plasma Sci., 2012, 40, 2788–2794 CAS.
- S. Reuter, H. Tresp, K. Wende, M. U. Hammer, J. Winter, K. Masur, A. Schmidt-Bleker and K. D. Weltmann, IEEE Trans. Plasma Sci., 2012, 40, 2986–2993 CAS.
- H. Tresp, M. U. Hammer, K.-D. Weltmann and S. Reuter, Plasma Medicine, 2013, 3, 45–55 CrossRef.
- H. Jablonowski, M. A. Haensch, M. Duennbier, K. Wende, M. U. Hammer, K. D. Weltmann, S. Reuter and T. von Woedtke, Biointerphases, 2015, 10, 029506 CrossRef PubMed.
- N. Hogg, Free Radical Biol. Med., 2010, 49, 122–129 CrossRef CAS PubMed.
- H. Tresp, M. U. Hammer, J. Winter, K. D. Weltmann and S. Reuter, J. Phys. D: Appl. Phys., 2013, 46, 435401 CrossRef.
- S. Iseni, P. J. Bruggeman, K.-D. Weltmann and S. Reuter, Appl. Phys. Lett., 2016, 108, 184101 CrossRef.
- B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine, Oxford University Press, Oxford, 2007 Search PubMed.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- W. V. Gaens, S. Iseni, A. Schmidt-Bleker, K. D. Weltmann, S. Reuter and A. Bogaerts, New J. Phys., 2015, 17, 033003 CrossRef.
- J. Zeldovich, Eur. Phys. J. A, 1946, 21, 577–628 Search PubMed.
- M. H. Stans, NIST Special Publication 1, 1970, p. 58.
- H. Kalka, http://www.aqion.dehttp://www.aqion.de/.
- E. Kaiser and C. Wu, J. Phys. Chem., 1977, 81, 1701–1706 CrossRef CAS.
- W. Tian, A. M. Lietz and M. J. Kushner, Plasma Sources Sci. Technol., 2016, 25, 055020 CrossRef.
- P. Lukes, E. Dolezalova, I. Sisrova and M. Clupek, Plasma Sources Sci. Technol., 2014, 23, 015019 CrossRef.
- C. C. Hsu, M. C. Lin, A. M. Mebel and C. F. Melius, J. Phys. Chem. A, 1997, 101, 60–66 CrossRef CAS.
- B. Smaller, E. C. Avery and J. R. Remko, J. Chem. Phys., 1971, 55, 2414–2418 CrossRef CAS.
- W. Tian and M. J. Kushner, J. Phys. D: Appl. Phys., 2014, 47, 165201 CrossRef.
- M. C. Su, S. S. Kumaran, K. P. Lim, J. V. Michael, A. F. Wagner, L. B. Harding and D. C. Fang, J. Phys. Chem. A, 2002, 106, 8261–8270 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2004, 4, 1461–1738 CrossRef CAS.
- Y. Gorbanev, N. Stehling, D. O'Connell and V. Chechik, Plasma Sources Sci. Technol., 2016, 25, 055017 CrossRef.
- B. T. J. van Ham, S. Hofmann, R. Brandenburg and P. J. Bruggeman, J. Phys. D: Appl. Phys., 2014, 47, 224013 CrossRef.
- F. Girard, M. Peret, N. Dumont, V. Badets, S. Blanc, K. Gazeli, C. Noel, T. Belmonte, L. Marlin, J. P. Cambus, G. Simon, N. Sojic, B. Held, S. Arbault and F. Clement, Phys. Chem. Chem. Phys., 2018, 20, 9198–9210 RSC.
- S. Yatom, Y. C. Luo, Q. Xiong and P. J. Bruggeman, J. Phys. D: Appl. Phys., 2017, 50, 415204 CrossRef.
- I. W. Smith and D. W. Stewart, J. Chem. Soc., Faraday Trans., 1994, 90, 3221–3227 RSC.
- V. G. Kharitonov, A. R. Sundquist and V. S. Sharma, J. Biol. Chem., 1994, 269, 5881–5883 CAS.
- D. Chakraborty, J. Park and M. Lin, Chem. Phys., 1998, 231, 39–49 CrossRef CAS.
- L. F. Phillips and H. I. Schiff, J. Chem. Phys., 1965, 42, 3171–3174 CrossRef CAS.
- K. Niemi, D. O'Connell, N. de Oliveira, D. Joyeux, L. Nahon, J. P. Booth and T. Gans, Appl. Phys. Lett., 2013, 103, 034102 CrossRef.
- F. A. Villamena and J. L. Zweier, J. Chem. Soc., Perkin Trans. 2, 2002, 1340–1344, 10.1039/b201734b.
- J. Winter, H. Tresp, M. U. Hammer, S. Iseni, S. Kupsch, A. Schmidt-Bleker, K. Wende, M. Dunnbier, K. Masur, K. D. Weltmannan and S. Reuter, J. Phys. D: Appl. Phys., 2014, 47, 285401 CrossRef.
- S. Bekeschus, S. Iseni, S. Reuter, K. Masur and K. D. Weltmann, IEEE Trans. Plasma Sci., 2015, 43, 776–781 CAS.
- W. A. Seddon, J. W. Fletcher and F. Sopchysh, Can. J. Chem., 1973, 51, 1123–1130 CrossRef CAS.
- R. Knight and H. Sutton, Trans. Faraday Soc., 1967, 63, 2628–2639 RSC.
- J. Hoigne, H. Bader, W. R. Haag and J. Staehelin, Water Res., 1985, 19, 993–1004 CrossRef CAS.
- J. S. Beckman and W. H. Koppenol, Am. J. Physiol., 1996, 271, C1424–C1437 CrossRef CAS PubMed.
- M. Grätzel, S. Taniguchi and A. Henglein, Ber. Bunsen-Ges., 1970, 74, 488–492 CrossRef.
- S. Goldstein and G. Czapski, Free Radical Biol. Med., 1995, 19, 505–510 CrossRef CAS PubMed.
- H. Jablonowski, J. S. Sousa, K.-D. Weltmann, K. Wende and S. Reuter, Sci. Rep., 2018, 8, 12195 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp02412j |
| This journal is © the Owner Societies 2018 |