
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Short-range ion dynamics in methylammonium lead iodide by multinuclear solid state NMR and 127I NQR†
Alessandro
Senocrate
 ,
Igor
Moudrakovski
* and
Joachim
Maier
,
Igor
Moudrakovski
* and
Joachim
Maier
Department of Physical Chemistry of Solids, Max Planck Institute for Solid State Research, Heisenbergstr. 1, 70569, Stuttgart, Germany. E-mail: i.moudrakovski@fkf.mpg.de
First published on 11th July 2018
Abstract
We explore the short-range ion dynamics in methylammonium lead iodide (MAPbI3, the archetypal halide perovskite) by means of solid-state NMR (1H, 13C, 14N, 15N and 207Pb) and Nuclear Quadrupolar Resonance (127I NQR), in combination with molecular dynamics simulations. We focus on the rotational motion of the methylammonium (MA) cation, and on the interaction between MA and the inorganic lattice, since these processes are linked to electronic carrier lifetimes, optical and electronic properties and even structural stability of this promising solar cell material. We show that the motion of the MA cation can be described by a bi-axial rotation, with similar interactions of CH3 and NH3+ groups with the inorganic framework. This motion becomes nearly isotropic above the cubic phase transition, dominating the spin–lattice relaxation of 1H, 13C and 15N through spin-rotational interactions. In addition, we observe strong cross-relaxation between 207Pb and 127I, which fully controls spin–spin and spin–lattice relaxation in 207Pb.
Introduction
The outstanding photo-electrochemical properties of hybrid organic–inorganic halide perovskites have motivated in recent years a massive number of studies on these promising solar cell materials.1–3 Even though these compounds were described nearly 40 years ago,4 the characterization of their properties and application in devices is much more recent.1,5 Despite the momentous effort already spent in characterizing these materials, a significant portion of the physics and chemistry underlying their properties is still missing. As a notable example, the short-range dynamics of the organic cations is still not well understood, despite being of great importance since it is linked to the long carrier lifetimes in these systems.6–8 In addition, the interaction of the organic cations with the inorganic framework through hydrogen bonding9 has been suggested to contribute to the stabilization of the structure,10 while also influencing the optical and electronic properties of the materials.11,12 Therefore, the goal of the present study is to probe such short-range ion dynamics in the archetypal halide perovskite MAPbI3via solid-state NMR (1H, 13C, 14N, 15N and 207Pb) and Nuclear Quadrupolar Resonance (127I NQR), with particular attention to the rotational motion of the methylammonium (MA) cation, as well as to the interaction between MA and the inorganic lattice. Long-range ion dynamics, that gives rise to a significant iodine conductivity in MAPbI3, was studied by our group in a previous work.13Owing to its unsurpassed sensitivity to short-range interactions, NMR spectroscopy is very effective to study dynamic processes,14–18 and its first applications to the investigation of halide perovskites go back as early as 1985.19–22 In these studies, reorientation of the organic moiety was observed in all crystalline phases of MAPbI3 (but also MAPbBr3 and MAPbCl3) through 1H, 2H and 14N spin–lattice relaxation times (T1) measurements.20–22 Even though decreasing the temperature naturally hinders MA reorientation, rotation about the C–N axis in a correlated fashion was observed down to 50 K. The phase transitions (orthorhombic–tetragonal at 162 K and tetragonal–cubic at 327 K)23 are not visible in the T1 measurements of 2H and 14N,21 but can be detected by 1H measurements,22 albeit only the lower temperature transition was investigated. More recently, NMR spectroscopy has been applied to halide perovskites to investigate the effects of degradation,24 extended defects and crystal quality25 and details of Pb–X environment and cation dynamics.13,26–29
In this work, we report on NMR and NQR spectra discussed in light of molecular dynamics simulations and relaxation effects. First we look at the MA cation with 1H, 13C, 14N and 15N NMR as a function of temperature and corresponding relaxation time measurements. Combination of these data will show that the motion of the MA cation in MAPbI3 can be well described by a bi-axial rotation, with similar interactions of CH3 and NH3+ groups with the inorganic framework. This motion develops into a nearly isotropic rotation above the cubic phase transition, becoming a dominant contribution to the spin–lattice relaxation of 1H, 13C and 15N through spin-rotational interactions. We follow by presenting 207Pb NMR and 127I NQR spectroscopic and relaxation data, where we observe strong cross-relaxation between 207Pb and 127I, which fully dominates spin–spin and spin–lattice relaxation in 207Pb.
Experimental part
Synthesis
As a first step, MAI was prepared according to a modified reported procedure.30 Single crystals of MAPbI3 were then obtained as reported by Saidaminov,31 using an equimolar mixture of MAI and commercial PbI2 powders (99.9995%, Alfa Aesar). The synthesis step was carried out in normal laboratory atmosphere. These single crystals were subsequently crushed under inert atmosphere to obtain crystalline powders and directly transferred into the appropriate NMR sample holders, which were sealed to prevent exposure to oxygen and humidity. In order to synthetize 13C- and 15N-labelled MAPbI3, we used commercial 13C,15N methylamine (99% enrichment, Sigma-Aldrich) to obtain labelled MAI, that was used in the single crystal synthesis step to yield labelled MAPbI3 containing 20% 13C and 15N. Powder XRD data on the crushed single crystals are given in the (Fig. S1, ESI†).NMR measurements
The majority of NMR measurements were performed on a Bruker Avance III 400 MHz instrument at magnetic field of 9.4 T. Measurements with stationary samples used a Bruker PE400 double resonance high power probe with a 5 mm solenoid coil. The spectra of 14N and 207Pb were obtained using solid-echo (π/2–τ–π/2) and Hahn-echo (π/2–τ–π) pulse sequences with τ = 20 μs, and the relaxation delays set to provide for a complete relaxation (1s for 14N and 5s for 207Pb at room temperature). Between 100 and 400 scans were accumulated for both nuclei. 1H spectra were acquired with simple Bloch Decay experiment (i.e., pulse-acquisition), with the π/2 pulse of 2.5 μs, and the relaxation delay set long enough for a complete relaxation (10 s at room temperature). 13C and 15N spectra were obtained using Bloch decay experiment accompanied by a composite proton decoupling. The excitation π/2 pulses were 5 ms and 6 ms, and relaxation delays at room temperature of 150 s and 600 s, respectively. For isotopically enriched samples between 16 and 64 scans were commonly accumulated. Unless stated otherwise, spin–lattice relaxation time measurements were performed in stationary conditions, mainly with the saturation-recovery technique32 in order to minimize the experimental time.The experiments with Magic Angle Spinning (MAS) used a Bruker BL4 double resonance MAS probe with 4 mm OD ZrO2 spinners. Dry nitrogen was used as spinning, bearing, and cooling gas. The spinning rate was, when possible, kept between 10 and 14 kHz. The spectra were acquired using ramped Cross Polarization (CP) and also Bloch Decay, both accompanied by composite proton decoupling at the proton RF power of 50 kHz. The temperature in the probes was regulated with a Bruker BVT3000 temperature controller. Actual temperature inside the spinner was calibrated on 207Pb signal of powdered Pb(NO3)2.33 Chemical shifts were referenced externally relative to TMS (1H and 13C, δiso = 0.0 ppm), tetramethyl lead (207Pb, δiso = 0.0 ppm, set with the absolute referencing to TMS), nitromethane (14,15N, δiso = 0.0 ppm, set with the secondary reference of NH4Cl, δiso = −341.2.0 ppm).34
NQR measurements were performed using the same spectrometer console and probes, which were kept at a sufficient distance from the magnet to avoid the effects of stray magnetic fields. All RF-pulses have been calibrated directly on a sample of MAPbI3, with π/2 pulses being adjusted to 2 μs. A solid-echo π/2–τ–π/2 pulse sequence with τ = 20 μs and relaxation delay of 2 ms has been used to acquire the spectra, with the number of accumulations being between 128 and 256. Pure NQR spin–lattice relaxation times have been measured using inversion-recovery method.
DFT calculations
All calculations were performed with the Castep code35,36 integrated within the Biovia Materials Studio 2017 suite. The computations use the generalized gradient approximation (GCA) and Perdew–Burke–Ernzerhof (PBE) functional,37 with the core–valence interactions described by ultra-soft pseudopotentials.28 A Monkhorst–Pack grid38 was used to realize integrations over the Brillouin zone, with k point spacings generally being less than 0.04 Å−1. The convergence of the calculated NMR parameters was tested for both the size of a Monkhorst–Pack k-grid and a basis set cut-off energy, with cut-off energies being in the range of 550–610 eV. To account for relativistic effects in such heavy elements as lead and iodine, all calculations were performed including the zeroth-order regular approximation (ZORA)39,40 through scalar-relativistic pseudopotentials.41 Geometry optimization calculations were performed using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm, with the same functional, k-grid spacings and cut-off energies as in the single-point energy calculations. Convergence tolerance parameters for geometry optimization were as follows: maximum energy 2 × 10−5 eV per atom, maximum force 0.05 eV Å−1, maximum stress 0.1 GPa and maximum displacement 0.002 Å. Crystallographic data used in the calculations were taken from the literature.514N NMR parameters in methylammonium cation were calculated in a 3 × 3 × 3 supercell of MAPbI3 after a molecular dynamic simulation run for 1 ps 270 K. During the DFT MD simulation only the cations move, while the lattice parameters of the inorganic framework composed by heavy atoms of iodine and lead are fixed. Fig. S8 (ESI†) shows the structure before and after the simulations.Spectral simulations and fitting
Analytical simulations of the experimental spectra were carried out with the DMFit42 simulation package and the SOLAGuide module in TopSpin 3.2 processing software from Bruker. 14N NMR line shapes affected by the motion of the MA-cation were simulated with NMR-WEBLAB,43 and EXPRESS44 computational packages.Results and discussion
1H solid state NMR
The isotope 1H offers the highest sensitivity among all NMR active nuclei in MAPbI3. As shown in Fig. 1a, 1H spectra obtained with 10–15 kHz Magic Angle Spinning (MAS) show two well-resolved signals at 3.4 ppm (CH3) and 6.4 ppm (NH3+), the position of which remain nearly constant between 183 and 373 K (Fig. S2, ESI†). On few other occasions, we have observed some minor signals near 0.9 ppm likely due to residual solvent from the synthesis (DMF), while no peaks assignable to water have ever been detected. Although the MAS offers a considerable improvement in spectral resolution, the majority of our measurements have been performed in stationary conditions, since having a consistent spinning of the glass-sealed samples (required to avoid oxygen- and moisture-induced degradation) in the broad range of studied temperatures was rather difficult. A further argument against the MAS measurements was the influence of the spinning on the spin–lattice relaxation time measurements (Fig. S5a, ESI†), which is also found in other materials.45,46 In contrast to spinning samples, the 1H stationary spectra show a broadened, unresolved signal with a ∼8 kHz FWHM and a line shape well represented by a Gaussian (Fig. 1a).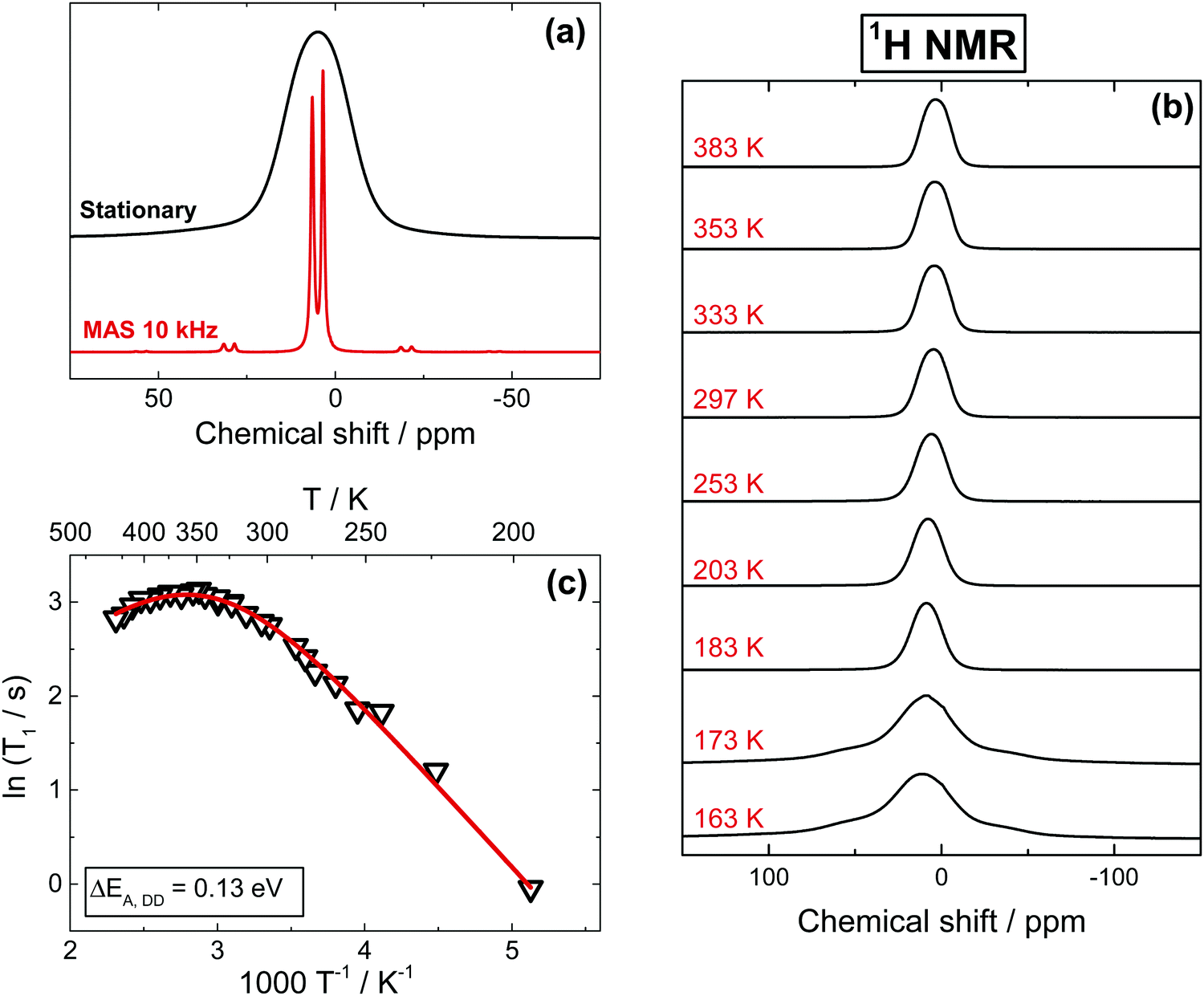 | ||
| Fig. 1 (a) Stationary and MAS 1H NMR spectra of MAPbI3 recorded at 297 K. (b) Stationary 1H spectra as a function of temperature. (c) 1H T1 relaxation times obtained from stationary spectra. Extracted activation energy values for the dipole–dipole relaxation mechanism dominating at low temperatures is ΔEA,DD = 0.13 eV. A value for the spin-rotational contribution of ΔEA,SR= −0.1 eV is obtained after fitting the entire curve using fixed values initially extracted from the low temperature part. Due to a possibility of sample degradation, only a limited temperature range is available, making the spin-rotational behavior in T1 challenging to accurately fit and the resulting activation energy intrinsically only indicative. | ||
Before looking further at the experimental results, it can be helpful to discuss the factors affecting the shape and width of NMR signals. The broadening of 1H NMR signals in a stationary MAPbI3 sample is primarily due to the homonuclear dipole–dipole (DD) interaction between protons in the MA cation. Additional contributions come from the heteronuclear interaction between protons and 14N of the amine group and, to a lesser extent, between protons and the atoms of the inorganic lattice. DD interactions in solids can be quantified by the so-called second moment M2 of the spectrum, which can be calculated for 1H in MA based on the geometry of the system (Section 3, ESI†). We note a substantial deviation of more than 1 order of magnitude between the M2 values calculated in a static environment and the experimental values, indicating that the MA cation, along with rotation around the C–N axis, must be involved in another reorientation with a symmetry axis higher than 2-fold (for the detailed discussion, see Section 3, ESI†). Since the methyl and amino groups composing MA have similar masses, such a reorientation is likely a rotational oscillation about an axis passing near the center of the C–N bond, tilted relative to the bond at angle θ (schematics below in Fig. 5). This composite rotation will give a reduction of M2 by a factor ρ = 1/16·(3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos2θ − 1)2.20,47 Since 180° flips (θ = 180°) are not the dominant motion reducing M2 we speculate that a precessional motion of the C–N bond tilted at θ ≤ 60° is responsible for the small observed M2 value, a hypothesis that is in very good agreement with the 14N NMR results discussed below. The absence of 180° flips was observed by neutron scattering experiments48 and it is also indicated by the quadrupolar splitting visible in our 14N NMR spectra (given below in Fig. 4a). Note that DD interactions are not completely averaged neither between the protons within the cation, nor to other atoms (127I, 207Pb) or to protons from neighboring cells. As a result, both line shape and FWHM of 1H stationary signals change very little from 503 K down to 183 K (Fig. 1b). Both the temperature independence of 1H FWHM and the small magnitude of M2 were previously reported in the tetragonal phases of MAPbCl3 and MAPbBr3,20 along with a sharp increase of the second moment after transition into the orthorhombic phase that was assigned to a diminished C3 reorientation in the MA cation (i.e., around the C–N axis). Similarly, we observe that at T ≤ 173 K the signal undergoes a drastic transformation, morphing into a broad, poorly resolved triplet with separation between the components of ∼18 kHz (Fig. 1b). Such a line shape is expected for a system of three protons at the corners of an equilateral triangle,49,50 with separation between the peaks being a function of both distance and mobility of the protons.
cos2θ − 1)2.20,47 Since 180° flips (θ = 180°) are not the dominant motion reducing M2 we speculate that a precessional motion of the C–N bond tilted at θ ≤ 60° is responsible for the small observed M2 value, a hypothesis that is in very good agreement with the 14N NMR results discussed below. The absence of 180° flips was observed by neutron scattering experiments48 and it is also indicated by the quadrupolar splitting visible in our 14N NMR spectra (given below in Fig. 4a). Note that DD interactions are not completely averaged neither between the protons within the cation, nor to other atoms (127I, 207Pb) or to protons from neighboring cells. As a result, both line shape and FWHM of 1H stationary signals change very little from 503 K down to 183 K (Fig. 1b). Both the temperature independence of 1H FWHM and the small magnitude of M2 were previously reported in the tetragonal phases of MAPbCl3 and MAPbBr3,20 along with a sharp increase of the second moment after transition into the orthorhombic phase that was assigned to a diminished C3 reorientation in the MA cation (i.e., around the C–N axis). Similarly, we observe that at T ≤ 173 K the signal undergoes a drastic transformation, morphing into a broad, poorly resolved triplet with separation between the components of ∼18 kHz (Fig. 1b). Such a line shape is expected for a system of three protons at the corners of an equilateral triangle,49,50 with separation between the peaks being a function of both distance and mobility of the protons.
Additional information about the overall dynamics and specific modes of motion of MA can be obtained from the 1H spin–lattice relaxation time (T1) measurements as a function of temperature given in Fig. 1c. Between 183 K and 310 K, the relaxation is characteristic of the fast motion limit of the Bloembergen–Purcell–Pound (BPP) model,51 and is being governed by DD interactions with other protons (Section 4, ESI†). Assuming an Arrhenius dependence of the motional correlation time (ln(τρ) ∝ 1/T1), we found an activation barrier (ΔEA,DD) of 0.13 eV for this temperature range, well within the expected range for rotational motion of the MA cation inside the Pb–I framework and in good agreement with earlier reports.22 We note that the here reported T1 values are an average of the proton relaxations in CH3 and NH3+ groups, with the respective contributions likely being equal, or at least very similar, since the observed T1 is mono-exponential in the whole temperature range, with only a slight deviation near the orthorhombic phase transition (Fig. S3, ESI†). This mono-exponential dependence clearly suggests that CH3 and NH3+ groups interact similarly with the inorganic framework. In contrast, in a previous report,261H T1 relaxation times measured under MAS conditions were used to claim a preferential interaction of NH3+ groups with the inorganic framework, albeit the difference in T1 between the two groups was rather small and likely within experimental error. We have also measured 1H T1 relaxation times under MAS conditions (Fig. S5a, ESI†), and observed similarly minor differences between the relaxation times of the two groups. We believe that this very minor discrepancy, in conjunction with the strong influence of MAS conditions on T1 relaxation times,45,46 makes the above claims of preferential interactions unwarranted.
As expected, the relaxation times observed for 1H in pure MAPbI3 and in 13C,15N-labelled samples are identical within experimental error; a similar activation energy is also found for MAPbBr3 (Fig. S4, ESI†). Interestingly, as seen in Fig. 1c, above room temperature T1 makes a downward turn, that is characteristic of spin-rotational (SR) interaction. As we will show below, the same behavior is present for 13C and 15N in MAPbI3, and was reported for 1H in MAPbBr3 (also Fig. S4, ESI†) and MAPbCl3.20 The relaxation rate is therefore defined by a combination of two processes, namely by a dipole–dipole and a spin-rotational mechanism, the latter becoming dominant in the vicinity of the tetragonal to cubic phase transition where the increased free space in the inorganic cage allows for a less hindered (and thus faster) MA rotation. As expected, in the case of 1H T1 measurements in MAPbBr3 (Fig. S4, ESI†), the inversion behavior takes place at lower temperatures, albeit notably not as low as the phase transition into the cubic phase (237 K).23 This observation is consistent with previous reports.20 Lastly, we note that while we can accurately extract an activation energy for DD relaxation (given above), the experimentally accessible temperature range in which SR contributions dominate is rather narrow, making the estimate of such activation energy inaccurate (see caption of Fig. 1).
13C NMR
The 13C spectra, as shown in Fig. 2a, give a better spectral resolution than 1H NMR, though at the expense of substantially longer acquisition times due to the low natural abundance of the isotope (1.1%). To overcome this issue, we use a 13C-labelled sample (20% enrichment). Both Cross-Polarization (CP) from protons and direct excitation (Bloch Decay accumulation) combined with the high power proton decoupling (HPDEC) sequence can be successfully used. While in many organic materials CP usually offers a better sensitivity because of the shorter 1H relaxation times and effective CP dynamics, this is not the case for MAPbI3. Slow proton magnetization recovery and a very fast relaxation in the rotating frame T1ρ make CP very ineffective above 250 K (Fig. S5b, ESI†). In contrast, the direct excitation is less affected by the MA dynamics and here the relative intensities of the signals reflect quantitatively the concentration of different sites, making this our approach of choice. MAS has also been applied in order to obtain better resolved spectra (Fig. 2a and Fig. S7a, ESI†). However, since our preliminary results indicated an influence of MAS on 13C T1 relaxation times (Section 5, see also Fig. S6, ESI†), as also reported for other materials,45,46,52 all T1 measurements were performed in stationary conditions.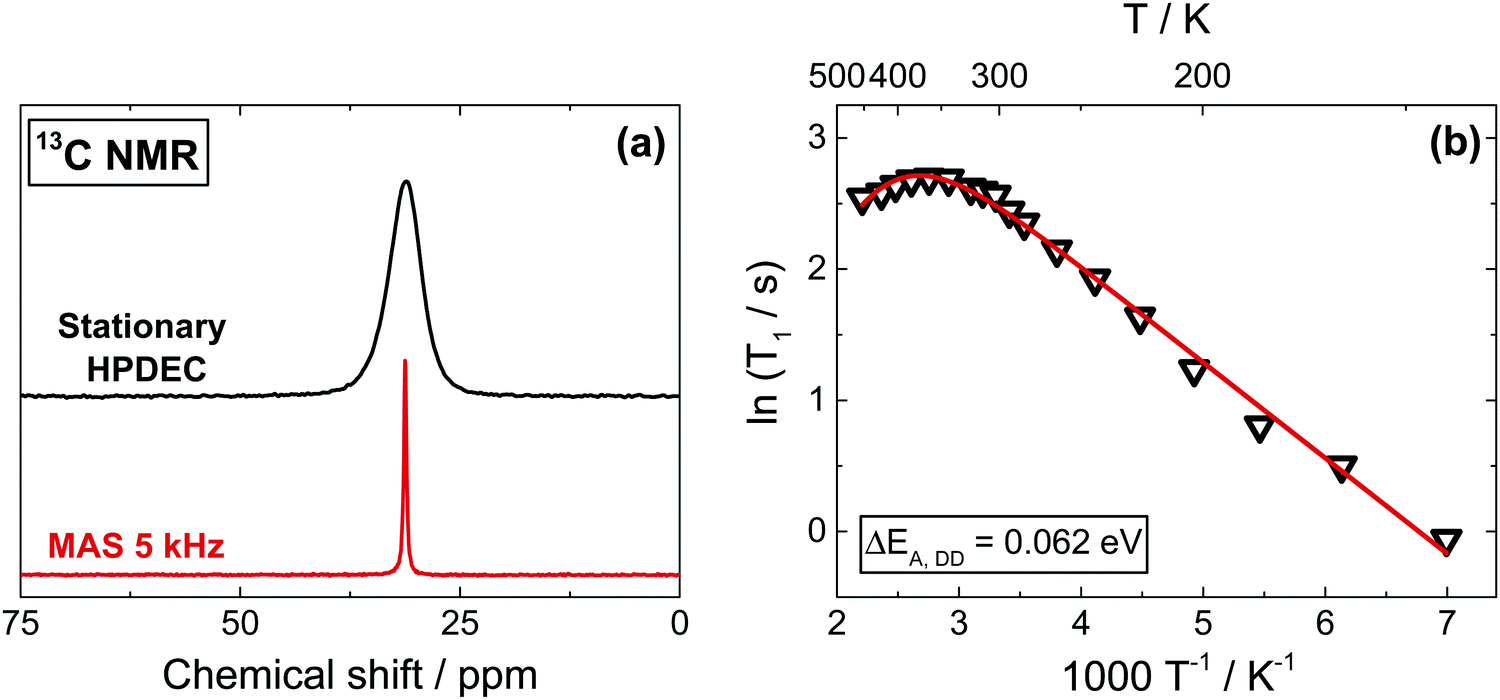 | ||
| Fig. 2 (a) 13C stationary and MAS NMR spectra in (13C, 15N)-labelled MAPbI3. (b) Spin–lattice relaxation time extracted from stationary 13C NMR spectra as a function of temperature. The activation energy (ΔEA,DD = 0.062 eV) was extracted from the linear low temperature part, and the obtained value was used to generate the complete fit, yielding ΔEA,SR = −0.38 eV. Again, the narrow range accessible at high temperatures makes the activation energy for SR highly inaccurate. | ||
13C spin–lattice relaxation times can be composed of contributions from three primary relaxations mechanisms,53–55 such as: (i) heteronuclear DD interactions with protons, (ii) relaxation due to the chemical shift anisotropy (CSA) and (iii) relaxation due to SR interactions (as in protons). We leave the full expressions of the relaxation mechanism and of the related contributions to the ESI,† (Section 5); here it suffices to say that the nearly symmetrical signal shape even at very low temperatures suggest a negligible contribution of CSA in 13C spectra. This is in agreement with ab initio DFT calculations on the tetragonal phase of MAPbI3 yielding a very small value (38 ppm) for the shielding anisotropy (Table S1, ESI†). As shown in Fig. 2b, 13C T1 measurements as a function of temperature present a linear trend in the Arrhenius dependence below 340 K, as expected from DD interactions dominating the relaxation (ΔEA,DD = 0.062 eV). Analogously to 1H and 15N (below), above room temperature we observe an inversion of the trend, corresponding to a change in dominant relaxation mechanism from DD to a SR interaction. Again, the trend inverts close to the tetragonal to cubic phase transition, relating it to the increased free space of the inorganic framework giving a reduced obstruction to the rotation of the MA cation.
15N NMR
Due to very low natural abundance (0.4%), 15N spectra are barely detectable in non-labelled MAPbI3. However, since 15N NMR can produce highly resolved spectra, especially when combined with CP and proton decoupling, we use a 15N-labelled sample (20% enrichment) to record the spectra (Fig. 3a). MAS conditions have also been applied in order to obtain better resolved spectra (Fig. 3a and Fig. S7b, ESI†). The relaxation mechanisms expecting to contribute into 15N T1 are the same as those discussed for 13C and described in the previous section (and in Section 5, ESI†). Also here, we can reliably rule out any significant contribution of the CSA to the relaxation process based on our ab initio DFT calculations, which give a low value of 10.2 ppm (Table S1, ESI†) for the 15N shielding anisotropy, which is by at least two orders of magnitude too small to produce any appreciable contribution to T1.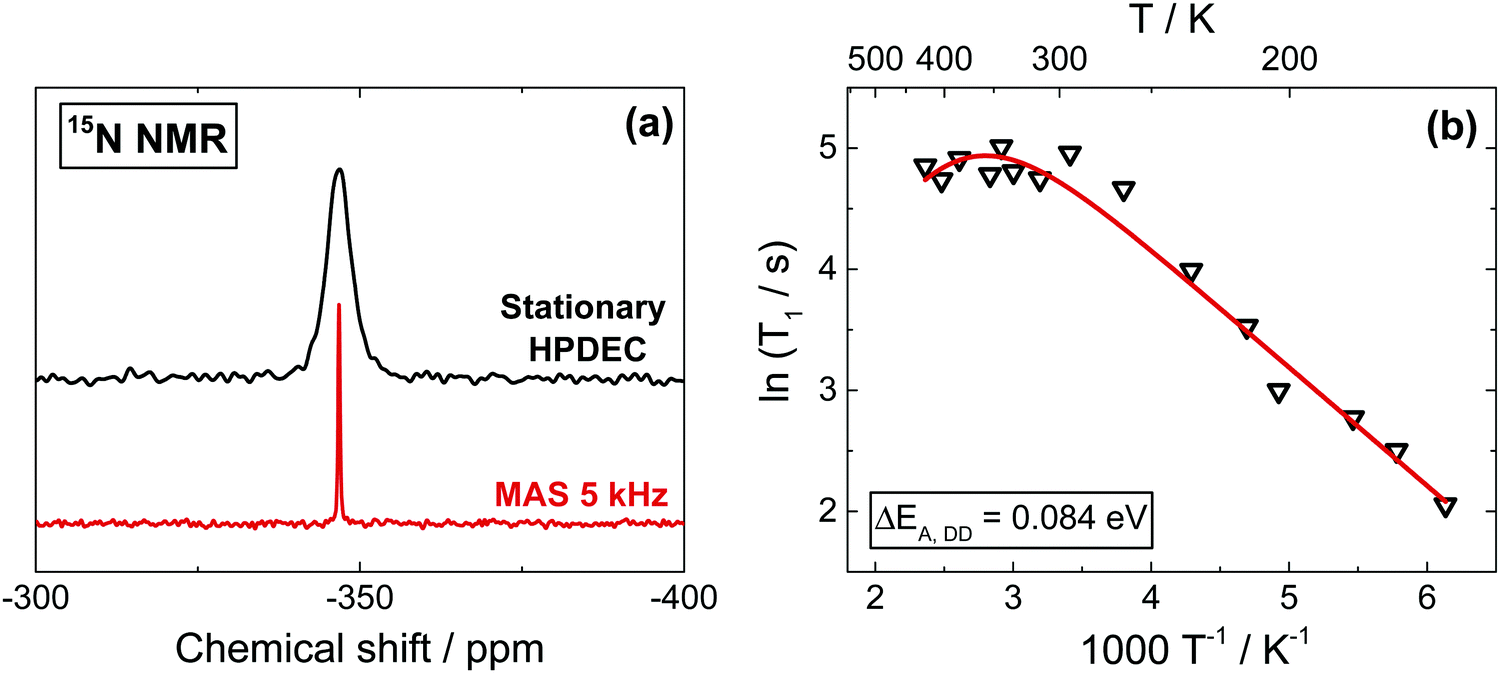 | ||
| Fig. 3 (a) 15N stationary and MAS spectra (13C, 15N)-enriched MAPbI3. (b) 15N Spin–lattice relaxation time T1 extracted from stationary spectra. The activation energy (ΔEA,DD = 0.084 eV) was extracted from the linear low temperature part, and the obtained value was subsequently used to generate the complete fit, yielding ΔEA,SR = −0.17 eV. Also here, the narrow range accessible at high temperatures yields a highly inaccurate activation energy value for the SR contribution. | ||
Also here, T1 demonstrates a linear trend in the tetragonal phase, as expected for internuclear proton–nitrogen DD relaxation in the fast motion limit (ΔEA,DD = 0.084 eV), which turns into a negative temperature dependence in the cubic phase due to the increasingly dominating SR contribution. The rather small difference between the activation barriers of the DD relaxation in 13C and 15N is expected for an almost symmetrical rotor such as CH3–NH3+. In addition, the activation energy extracted from 1H T1 measurements is comparable with the ones of 13C and 15N, as expected since they refer to the same motion (and to the same type of relaxation process).
14N NMR
Fig. 4a shows 14N NMR spectra of MAPbI3 in the tetragonal and cubic phases.13 In the tetragonal phase, the spectrum is dominated by quadrupolar interactions, demonstrating a typical spin-1 powder pattern with axial symmetry of the electric field gradient (EFG), while in the cubic phase the quadrupolar interactions are averaged, and therefore the splitting absent. Spin–lattice relaxation time T1 becomes longer with the temperature (Fig. 4b), indicating fast motion limit for the dominant relaxation mechanism in the whole studied range (183 K to 503 K). For a spin-1 nucleus such as 14N, the dominating mechanism of spin–lattice relaxation is due to quadrupolar interactions. In the fast motion limit, such quadrupolar relaxation can be expressed as:14,15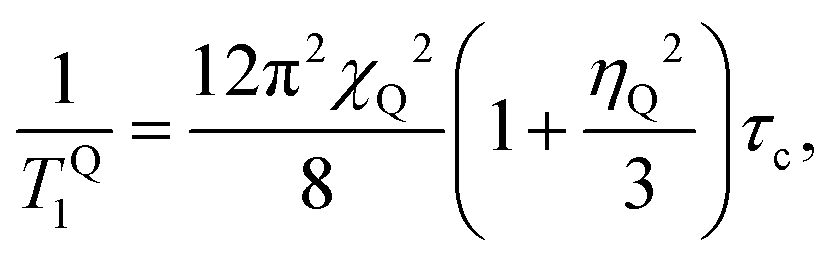 | (1) |
 | ||
| Fig. 4 (a) Variable temperature 14N NMR spectra in MAPbI3, collected using a solid echo pulse sequence. No difference except in the overall intensity is observed between pure and partially labelled samples. (b) Arrhenius plot of 14N T1 relaxation times in MAPbI3 and (c) 14N FWHM as a function of temperature in MAPbI3. For the signals (tetragonal phase) demonstrating the quadrupolar splitting, the value of the splitting has been used in place of the FWHM. | ||
The T1 temperature dependences (in Arrhenius coordinates, Fig. 4b) are linear in both cubic and tetragonal phases, producing activation barriers of 0.1 eV and 0.16 eV, respectively. These values cannot be directly compared with the one extracted in the 15N case (Fig. 3b), as the relaxation mechanisms involved are different. Nevertheless, they are fully in agreement with the ones extracted from other 14N NMR measurements.56 The observed decrease of the activation barrier across the phase transition implies a less hindered rotation of MA in the cubic phase, where the 14N signal displays a Lorentzian line shape with FWHM of only 710 Hz at 333 K, decreasing slowly as the temperature increases to 453 K (Fig. 4c). Such a small and steady change in the signal is consistent with a faster local motion of MA cations.
We can now focus on the analysis of the line shape of 14N spectra, in order to gain information on the modes of motion involving MA. In general, line shapes in 14N NMR depend on the interaction between the quadrupole moment of the nuclear spin and the EFG tensor surrounding the nucleus. In the specific case of a MA cation, this quadrupolar coupling is primarily of intramolecular origin, and is therefore strongly affected by the type and the rate of the MA molecular motions. Interestingly, the very large frequency spread of the experimental spectra (Fig. 4a) means that it is possible to probe molecular motion over a very broad frequency range. To help us identify the possible modes of motion involving MA, we performed 14N spectral simulations. In the general case of non-axial symmetry, the NMR frequencies (ωQ) in a 14N spectrum can be expressed as:
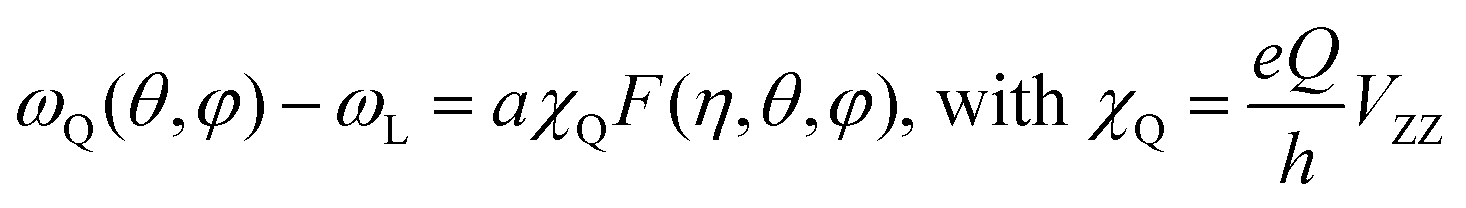 | (2) |
As shown in Fig. 5, the 14N spectra are strongly and diversely influenced by the different internal motions considered, offering a way of discriminating among them. Looking at Fig. 5a, it is clear that a fast (>10−6 s) continuous rotation around the C–N axis (or equivalently fast ≥3-sites symmetrical jumps) is necessary in order to produce the spectra experimentally observed in the tetragonal phase (Fig. 4a). Also, as shown in Fig. 5b, by adding to a fast rotation around the C–N bond a precessional motion with increasing angle θ (schematized in Fig. 5c), the quadrupolar splitting in the spectra reduces steadily until the angle reaches 54.7°, when it virtually disappears indicating nearly complete averaging of the quadrupolar interactions forming the line shape. This behavior is experimentally observed across the phase transition into the cubic phase (Fig. 4a and 5b), and corresponds to a pseudo-isotropic motion of MA cations within the inorganic framework. We recognize also that, in the cubic phase, identical spectra would be obtained with the presence of a fast enough isotropic motion, for example due to a translational diffusion of MA cations or to the presence of 180° flips involving MA molecules. Both the former and the latter motion were clearly ruled out from previous electrochemical13 and neutron scattering experiments48 respectively.
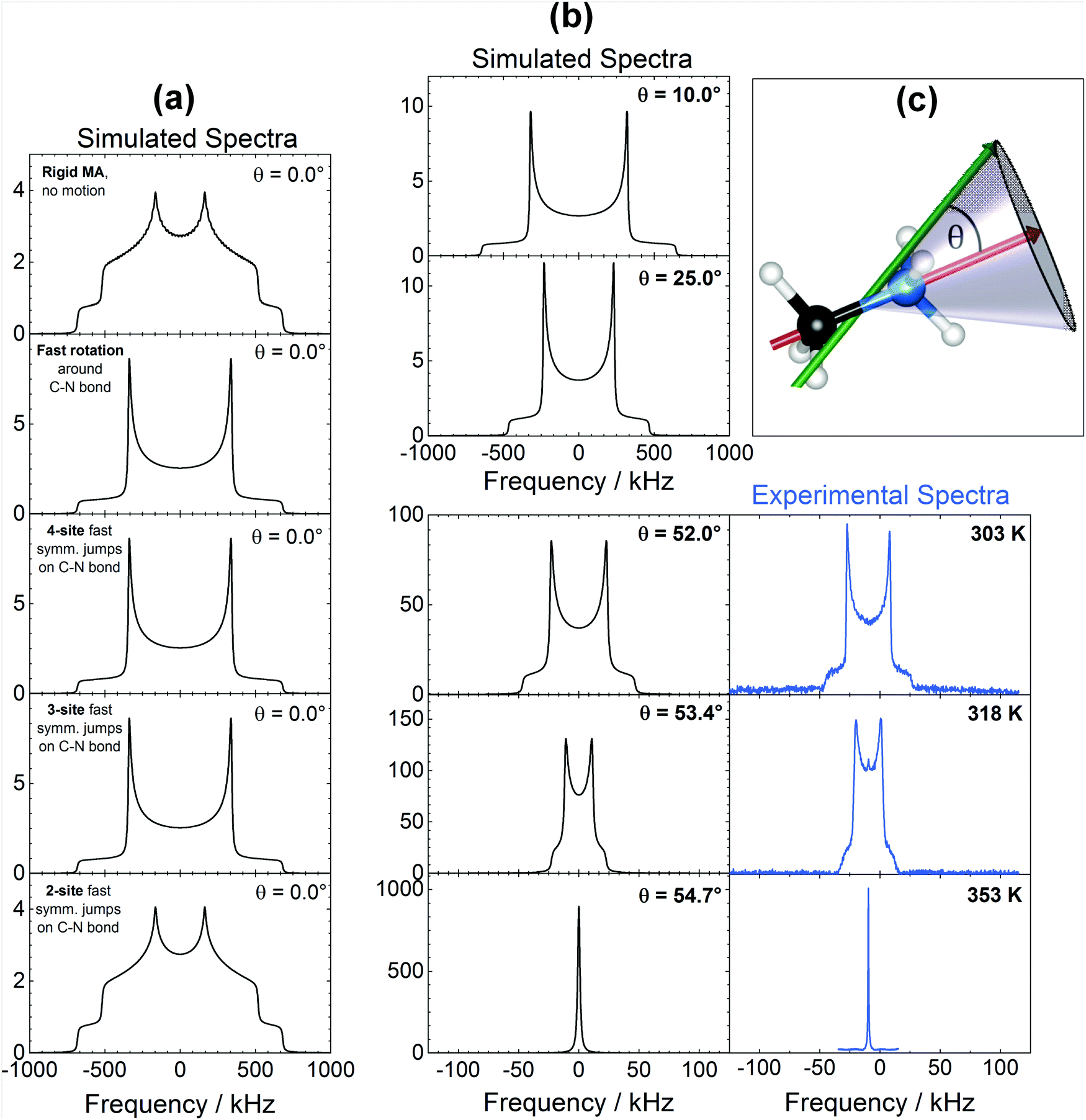 | ||
| Fig. 5 14N NMR spectral simulations assuming different MA modes of motion. (a) Simulated spectra in different dynamic conditions. (b) Simulated spectra obtained by modeling fast rotation around C–N axis together with precession in conical fashion at different angles θ. The data are compared with experimental spectra across the cubic phase transition. (c) Sketch of the modeled conical precessional motion. | ||
207Pb NMR
207Pb is a moderately sensitive spin-1/2 nucleus showing a remarkably broad range of chemical shifts (>8000 ppm) depending on its oxidation state and coordination environment, so much so that an unusually large difference (∼1500 ppm) is found between the chemical shifts of MAPbI3 and PbI2. In order to rationalize this peculiar chemical shift behavior, we performed extensive DFT calculations of the 207Pb shielding in a series of lead compounds of known environment (results are summarized in Table S2, ESI†). As shown in Fig. 6, the calculations correctly capture the general trend between the experimental chemical shifts and the different chemical environments, confirming the experimental observations. We recognize that the extracted slope in the shielding-shift trend (Fig. 6) has a value of almost 2, while it would commonly not strongly deviate from 1. This semi-quantitative agreement, however, is likely due to the incomplete accounting for relativistic effects in Pb nuclei stemming from the use of pseudopotentials instead of the full electron wave functions and to the lack of spin–orbit effects in the valence wave functions. | ||
| Fig. 6 Correlation between calculated isotropic shielding and experimental 207Pb isotropic chemical shifts in various diamagnetic compounds of lead. All experimental data are taken from the literature, with their corresponding ICSD numbers given in Table S2 (ESI†). The chemical shift of lead is referenced to lead tetramethyl (δiso = 0 ppm). | ||
The detection of 207Pb NMR in the solid state, particularly in iodine surroundings, presents a substantial challenge. As shown in Fig. 7a, the iodine environment around Pb nuclei causes a very short spin–spin relaxation time T2 (∼20 μs) and a subsequent formidable broadening of the signals at temperatures above 250 K (Fig. 7b). The 207Pb linewidth remains nearly constant (∼16.5 kHz) in the 433–253 K range, while below 253 K the line shape starts showing distinct asymmetry, consistently with the chemical shift anisotropy, and even significantly broadens at 173 K. Lowering temperature also increases T2 (Fig. 7b), reaching a peculiar 3-fold enhancement at 183 K, even though the value falls back to 20 μs at 173 K (near the orthorhombic phase transition), in agreement with previous reports.25 This unusual behavior, which is also found in T1 measurements (Fig. 7b), cannot be ascribed to standard relaxation mechanisms and common spectral density functions, but can be easily understood by taking into account strong spin–spin interactions between the spin-1/2 207Pb nucleus and the quadrupolar 127I nucleus. In this case, the 207Pb T2 is defined primarily by cross-relaxation to 127I, which presents very short relaxation times (Fig. 8c). Reducing the temperature slows down the cross-relaxation rate, yielding an increase in T2 relaxation times. The transition to the orthorhombic phase appears to alter the dynamics of the spin-exchange, resulting again in a shorter T2.
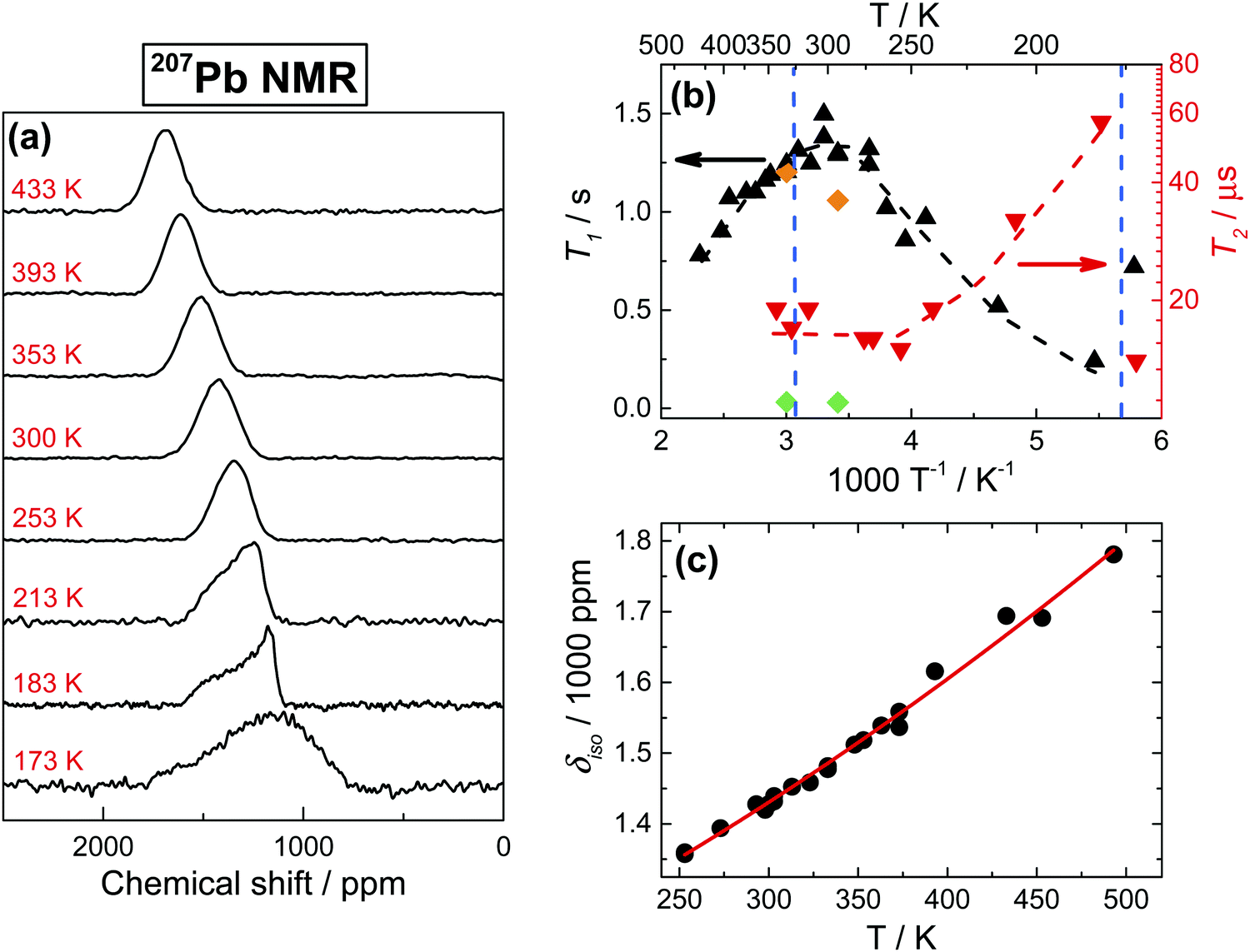 | ||
Fig. 7 (a) 207Pb NMR spectra of MAPbI3 as a function of temperature. (b) Temperature dependence of 207Pb spin–lattice and spin–spin relaxation times T1 and T2. Dashed lines are only to guide the eye. Vertical lines indicate the phase transitions. Orange ( ) and green ( ) and green ( ) points are T1 relaxation times collected at 21.1 T in stationary and 8 kHz MAS conditions respectively. Note that the higher field has no influence on T1 for stationary samples, while spinning greatly reduces it down to 20–30 ms. (c) Isotropic chemical shift of 207Pb signal as a function of temperature, fitted with a quadratic equation δiso = 1.04 + 0.96T + 0.001T2. ) points are T1 relaxation times collected at 21.1 T in stationary and 8 kHz MAS conditions respectively. Note that the higher field has no influence on T1 for stationary samples, while spinning greatly reduces it down to 20–30 ms. (c) Isotropic chemical shift of 207Pb signal as a function of temperature, fitted with a quadratic equation δiso = 1.04 + 0.96T + 0.001T2. | ||
 | ||
| Fig. 8 (a) 127I NQR signals in MAPbI3 at 300 K (tetragonal phase) and at 340 K (cubic phase). (b) Temperature dependence of the 127I quadrupolar coupling constant χQ and of the asymmetry parameter η. Phase transition is visible at 327 K (blue line). (c) Quadrupolar spin–lattice relaxation time T1Q as a function of temperature. | ||
Focusing now on spin–lattice relaxation, the magnitude of 207Pb T1 in MAPbI3 is similar to that previously reported for PbI2.59 As shown in Fig. 7b, T1 is practically independent of the magnetic field and depends only marginally on the temperature. Also, T1 temperature dependences in 127I and 207Pb appear rather similar (Fig. 8c and 7b), clearly demonstrating the large influence of cross-relaxation to 127I on the 207Pb spin–lattice relaxation process. An additional proof of such coupling is that T1 is strongly affected by MAS, decreasing by more than 1 order of magnitude under spinning conditions (Fig. 7b, details in the caption). This effect was recently reported and discussed in detail for lead and mercury halides,60 where in the specific case of PbI2 MAS conditions were required to induce the Pb–I coupling; unsurprisingly, thanks to the much stronger quadrupolar interactions, in MAPbI3 the coupling is present already in stationary conditions (Fig. 7a).
As a last point, we discuss the temperature dependence of the 207Pb isotropic chemical shift (δiso). As shown in Fig. 7c, δiso exhibits a distinct temperature dependence, as commonly observed in lead compounds61–64 and also in MAPbCl3,29 even though our experimental data show a small but detectable deviation from the normally reported linear dependences. For MAPbI3, this temperature dependence can be fully attributed to changes in the lattice parameters,25 as it is usually the case in Pb compounds.61–64 We note that this temperature dependence shows no clear sign of the cubic phase transition.
127I NQR
127I is a quadrupolar spin-5/2 nucleus with 100% natural abundance and reasonably high Larmor frequency, and thus could be considered a high receptivity nucleus. However, due to its very high quadrupole moment (−69.6 fm2),65127I NMR must be restricted to systems with a relatively high symmetry66 (for MAPbI3, preliminary 127I NMR spectra have been reported in ref. 13). In MAPbI3, however, a significant EFG at the iodine sites (χQ > 550 MHz) provides a good sensitivity in NQR spectroscopy, offering an alternative method to probe the I-environment in this material. As shown in Fig. 8a, two pairs of transitions in 127I NQR are detected for the two distinct iodine sites present in the tetragonal phase, while in the cubic phase only 2 signals are observed, corresponding to conversion into a single iodine site. The resonance frequencies of the signals, along with the quadrupolar constant χQ, show appreciable temperature dependence (Fig. 8b and Fig. S9, ESI†), a behavior that is commonly attributed to a planar-torsional oscillation of a resonant nucleus (I) about another atom to which it is bonded (Pb).The temperature dependence of the 127I quadrupole resonance frequency in MAPbI3 can be estimated based on the Bayer theory67 of torsional oscillations of atoms in the lattice.68 In this model the nuclei are agitated by the torsional motion at a rate that is very fast compared to their NQR frequencies, and they are therefore affected by an averaged temperature-dependent EFG that is different from the stationary situation (details in Section 8 and Fig. S9, ESI†). Thanks to this approach,68 it is possible to fit the experimental data and extract a torsional frequency νt ≈ 1.15 THz, corresponding to 38 cm−1. For a simple comparison, the reported Raman frequencies for Pb–I contributions in MAPbI3 were calculated to be between 20–50 cm−1, with a perceptible coupling to MA-modes.69,70 Another work, instead, experimentally observed Pb–I vibrations at higher frequencies (75–125 cm−1), also here with contributions coming from MA-modes.71 In addition, as shown in Fig. 8c, we record the quadrupolar spin–lattice relaxation time (T1Q) for 127I as a function of temperature. Again according to the Bayer theory,67 this temperature dependence can be used to calculate an accurate estimate of the correlation time τa of the torsional vibrations, albeit a closed solution for a spin 5/2 nucleus does not exist.72 Under certain assumptions, a partial solution can still be found for our system, but only in a very narrow temperature range (290–340 K), making the obtained τa values of 2 × 10−10 s (290 K) and 4 × 10−10 s (340 K) only indicative. We refer the reader to the ESI,† (Section 8) for the complete treatment and related assumptions.
Conclusions
In conclusions, we have shown that much information on halide perovskites can be collected by means of NMR spectroscopy. Specifically, we studied the short-range ion dynamics in MAPbI3 in a broad temperature range using 1H, 13C, 14,15N, 207Pb multinuclear solid-state NMR and 127I NQR. We found that the motion of the MA cation is ascribable to that of a symmetrical rotor, with similar interactions of CH3 and NH3+ groups with the inorganic lattice. Specifically, the predominant mode of motion for the MA cation is identified as a bi-axial rotation, as supported by 14N NMR and related spectral simulations. This rotation becomes nearly isotropic above the cubic phase transition, and sufficiently fast to dominate the relaxation mechanism through spin-rotational interactions. Regarding the inorganic lattice, 207Pb NMR shows strong cross-relaxation to quadrupolar 127I, which ultimately fully dominates the 207Pb spin–lattice relaxation times T1 and severely influences the linewidth (T2). The temperature behavior of 127I NQR spectra is indicative of a torsional oscillation of I about Pb atoms, where the frequency of oscillations can be estimated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank V. Terskikh (University of Ottawa, Canada) for help with the UHF NMR experiments. Access to the 900 MHz NMR spectrometer was provided by the National Ultrahigh Field NMR Facility for Solids managed by University of Ottawa, Ottawa, Canada (http://www.nmr900.ca). Dr Helga Hoier is thanked for performing the XRD measurements. Open Access funding provided by the Max Planck Society.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells, J. Am. Chem. Soc., 2009, 131(17), 6050–6051, DOI:10.1021/ja809598r.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl and A. Marchioro, et al., Lead Iodide Perovskite Sensitized All-Solid-State Submicron Thin Film Mesoscopic Solar Cell with Efficiency Exceeding 9%, Sci. Rep., 2012, 2(1), 591 CrossRef PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Efficient Hybrid Solar Cells Based on Meso-Superstructured Organometal Halide Perovskites, Science, 2012, 338(6107), 643–647 CrossRef PubMed.
- D. Weber, CH3NH3PbX3, ein Pb(II)-System mit kubischer Perowskitstruktur/CH3NH3PbX3, a Pb(II)-System with Cubic Perovskite Structure, Zeitschrift für Naturforsch B., 1978, 33(12), 1443–1445 Search PubMed.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties, Inorg. Chem., 2013, 52(15), 9019–9038, DOI:10.1021/ic401215x.
- J. Ma and L.-W. Wang, The Nature of Electron Mobility in Hybrid Perovskite CH3NH3PbI3, Nano Lett., 2017, 17(6), 3646–3654, DOI:10.1021/acs.nanolett.7b00832.
- C. Motta, F. El-Mellouhi, S. Kais, N. Tabet, F. Alharbi and S. Sanvito, Revealing the role of organic cations in hybrid halide perovskite CH3NH3PbI3, Nat. Commun., 2015, 6(1), 7026 CrossRef PubMed.
- J. Gong, M. Yang, X. Ma, R. D. Schaller, G. Liu and L. Kong, et al., Electron–Rotor Interaction in Organic–Inorganic Lead Iodide Perovskites Discovered by Isotope Effects, J. Phys. Chem. Lett., 2016, 7(15), 2879–2887 CrossRef PubMed.
- J. H. Lee, J.-H. Lee, E.-H. Kong and H. M. Jang, The nature of hydrogen-bonding interaction in the prototypic hybrid halide perovskite, tetragonal CH3NH3PbI3, Sci. Rep., 2016, 6(1), 21687 CrossRef PubMed.
- E. Mosconi, A. Amat, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, First-Principles Modeling of Mixed Halide Organometal Perovskites for Photovoltaic Applications, J. Phys. Chem. C, 2013, 117(27), 13902–13913 CrossRef.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari and M. K. Nazeeruddin, et al., Cation-Induced Band-Gap Tuning in Organohalide Perovskites: Interplay of Spin–Orbit Coupling and Octahedra Tilting, Nano Lett., 2014, 14(6), 3608–3616, DOI:10.1021/nl5012992.
- C. Quarti, E. Mosconi and F. De Angelis, Interplay of Orientational Order and Electronic Structure in Methylammonium Lead Iodide: Implications for Solar Cell Operation, Chem. Mater., 2014, 26(22), 6557–6569, DOI:10.1021/cm5032046.
- A. Senocrate, I. Moudrakovski, G. Y. Kim, T.-Y. Yang, G. Gregori and M. Grätzel, et al., The Nature of Ion Conduction in Methylammonium Lead Iodide: A Multimethod Approach, Angew. Chem., Int. Ed., 2017, 56(27), 7755–7759 CrossRef PubMed.
- C. P. Slichter, Principles of Magnetic Resonance, Springer-Verlag Berlin Heidelberg, 1990, p. 658 Search PubMed.
- A. Abragam, The principles of nuclear magnetism, Oxford University Press, 1961 Search PubMed.
- K. J. D. MacKenzie and M. E. Smith, in Multinuclear Solid-State NMR of Inorganic Materials, ed. MacKenzie K. J. D. and Smith M. E., Pergamon Materials Series, Pergamon, 2002, vol. 6 Search PubMed.
- M. J. Duer, Introduction to Solid-State NMR Spectroscopy, Blackweell Publishing, Oxford, 2004 Search PubMed.
- V. I. Bakhmutov, Solid State NMR in Materials Science, CRC Press, 2011, p. 280 Search PubMed.
- R. E. Wasylishen, O. Knop and J. B. Macdonald, Cation rotation in methylammonium lead halides, Solid State Commun., 1985, 56(7), 581–582 CrossRef.
- Y. Furukawa and D. Nakamura, Cation Dynamics in the Crystalline Phases of (CH3NH3)PbX3 (X: Cl, Br) as Studied by Proton Magnetic Resonance Techniques, Z. Naturforsch., 1989, 44a(1989), 1122–1126 Search PubMed.
- O. Knop, R. E. Wasylishen, M. A. White, T. S. Cameron and M. J. M. Van Oort, Alkylammonium lead halides. Part 2. CH3NH3PbX3 (X = Cl, Br, I) perovskites: cuboctahedral halide cages with isotropic cation reorientation, Can. J. Chem., 1990, 68(3), 412–422, DOI:10.1139/v90-063.
- Q. Xu, T. Eguchi, H. Nakayama, N. Nakamura and M. Kishita, Molecular Motions and Phase Transitions in Solid CH3NH3PbX3 (X = Cl, Br, I) as Studied by NMR and NQR, Zeitschrift für Naturforsch A., 1991, 46(3), 240–246 Search PubMed.
- A. Poglitsch and D. Weber, Dynamic disorder in methylammoniumtrihalogenoplumbates(II) observed by millimeter wave spectroscopy, J. Chem. Phys., 1987, 87, 11 CrossRef.
- A. M. Askar, G. M. Bernard, B. Wiltshire, K. Shankar and V. K. Michaelis, Multinuclear Magnetic Resonance Tracking of Hydro, Thermal, and Hydrothermal Decomposition of CH3NH3PbI3, J. Phys. Chem. C, 2017, 121(2), 1013–1024 CrossRef.
- W. M. J. Franssen, S. G. D. van Es, R. Dervişoğlu, G. A. de Wijs and A. P. M. Kentgens, Symmetry, Dynamics, and Defects in Methylammonium Lead Halide Perovskites, J. Phys. Chem. Lett., 2017, 8(1), 61–66, DOI:10.1021/acs.jpclett.6b02542.
- T. Baikie, N. S. Barrow, Y. Fang, P. J. Keenan, P. R. Slater and R. O. Piltz, et al., A combined single crystal neutron/X-ray diffraction and solid-state nuclear magnetic resonance study of the hybrid perovskites CH3NH3PbX3 (X = I, Br and Cl), J. Mater. Chem. A, 2015, 3(17), 9298–9307, 10.1039/C5TA01125F.
- C. Roiland, G. Trippé-Allard, K. Jemli, B. Alonso, J.-C. Ameline and R. Gautier, et al., Multinuclear NMR as a tool for studying local order and dynamics in CH3NH3PbX3 (X = Cl, Br, I) hybrid perovskites, Phys. Chem. Chem. Phys., 2016, 18(39), 27133–27142, 10.1039/C6CP02947G.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, P. Péchy, S. M. Zakeeruddin and M. Grätzel, et al., Cation Dynamics in Mixed-Cation (MA)x(FA)1–xPbI3 Hybrid Perovskites from Solid-State NMR, J. Am. Chem. Soc., 2017, 139(29), 10055–10061, DOI:10.1021/jacs.7b04930.
- G. M. Bernard, A. Goyal, M. Miskolzie, R. McKay, Q. Wu and R. E. Wasylishen, et al., Methylammonium lead chloride: A sensitive sample for an accurate NMR thermometer, J. Magn. Reson., 2017, 283, 14–21 CrossRef PubMed.
- J.-H. Im, C.-R. Lee, J.-W. Lee, S.-W. Park and N.-G. Park, 6.5% efficient perovskite quantum-dot-sensitized solar cell, Nanoscale, 2011, 3(10), 4088–4093, 10.1039/C1NR10867K.
- M. I. Saidaminov, A. L. Abdelhady, B. Murali, E. Alarousu, V. M. Burlakov and W. Peng, et al., High-quality bulk hybrid perovskite single crystals within minutes by inverse temperature crystallization, Nat. Commun., 2015, 6, 7586 CrossRef PubMed.
- E. Fukushima and S. B. W. Roeder, Experimental Pulse NMR: A nuts and bolts approach, Addison-Wesley, 1981, p. 539 Search PubMed.
- A. Bielecki and D. P. Burum, Temperature Dependence of 207Pb MAS Spectra of Solid Lead Nitrate. An Accurate, Sensitive Thermometer for Variable-Temperature MAS, J. Magn. Reson., Ser. A, 1995, 116(2), 215–220 CrossRef.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001), Pure Appl. Chem., 2001, 73(11), 1795–1818 Search PubMed.
- M. D. Segall, P. J. D. Lindan, M. I. J. Probert, C. J. Pickard, P. J. Hasnip and S. J. Clark, et al., First-principles simulation: ideas, illustrations and the CASTEP code, J. Phys.: Condens. Matter, 2002, 14(11), 2717–2744 CrossRef.
- J. R. Yates, C. J. Pickard and F. Mauri, Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76(2), 24401, DOI:10.1103/PhysRevB.76.024401.
- J. P. Perdew, K. Burke and Y. Wang, Generalized gradient approximation for the exchange-correlation hole of a many-electron system, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(23), 16533–16539 CrossRef.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13(12), 5188–5192, DOI:10.1103/PhysRevB.13.5188.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, Relativistic total energy using regular approximations, J. Chem. Phys., 1994, 101(11), 9783–9792, DOI:10.1063/1.467943.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, The zero-order regular approximation for relativistic effects: the effect of spin–orbit coupling in closed shell molecules, J. Chem. Phys., 1996, 105(15), 6505–6516, DOI:10.1063/1.472460.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint and T. Azaïs, et al., First-Principles Calculation of NMR Parameters Using the Gauge Including Projector Augmented Wave Method: A Chemist's Point of View, Chem. Rev., 2012, 112(11), 5733–5779, DOI:10.1021/cr300108a.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé and B. Alonso, et al., Modelling one- and two-dimensional solid-state NMR spectra, Magn. Reson. Chem., 2002, 40(1), 70–76 CrossRef.
- V. Macho, L. Brombacher and H. W. Spiess, The NMR-WEBLAB: an internet approach to NMR lineshape analysis, Appl. Magn. Reson., 2001, 20(3), 405–432, DOI:10.1007/BF03162288.
- R. L. Vold and G. L. Hoatson, Effects of jump dynamics on solid state nuclear magnetic resonance line shapes and spin relaxation times, J. Magn. Reson., 2009, 198(1), 57–72 CrossRef PubMed.
- A. M. Gil and E. Alberti, The effect of Magic Angle Spinning on proton spin–lattice relaxation times in some organic solids, Solid State Nucl. Magn. Reson., 1998, 11(3–4), 203–209 CrossRef PubMed.
- S. Hayashi, Effects of magic-angle spinning on spin-lattice relaxations in talc, Solid State Nucl. Magn. Reson., 1994, 3(6), 323–330 CrossRef PubMed.
- J. G. Powles and H. S. Gutowsky, Proton Magnetic Resonance of the CH3 Group. II. Solid Solutions of t-Butyl Chloride in Carbon Tetrachloride, J. Chem. Phys., 1953, 21(10), 1704–1709, DOI:10.1063/1.1698646.
- A. M. Leguy, J. M. Frost, A. P. McMahon, V. G. Sakai, W. Kochelmann and C. Law, et al., The dynamics of methylammonium ions in hybrid organic–inorganic perovskite solar cells, Nat. Commun., 2015, 6, 7124 CrossRef PubMed.
- E. R. Andrew and R. Bersohn, Nuclear Magnetic Resonance Line Shape for a Triangular Configuration of Nuclei, J. Chem. Phys., 1950, 18(2), 159–561, DOI:10.1063/1.1747579.
- H. S. Gutowsky and G. E. Pake, Structural Investigations by Means of Nuclear Magnetism. II. Hindered Rotation in Solids, J. Chem. Phys., 1950, 18(2), 162–170, DOI:10.1063/1.1747580.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Relaxation effects in nuclear magnetic resonance absorption, Phys. Rev., 1948, 73(7), 679–712, DOI:10.1103/PhysRev.73.679.
- A. M. Panich, N. A. Sergeev and M. Olszewski, Effect of magic angle spinning on 13C spin–lattice and spin–spin relaxation in nanodiamonds, J. Phys.: Condens. Matter, 2015, 27(36), 365302 CrossRef PubMed.
- J. L. Sudmeier, S. E. Anderson and J. S. Frye, Calculation of Nuclear Spin Relaxation Times, Concepts Magn. Reson., 1990, 2(4), 197–212, DOI:10.1002/cmr.1820020403.
- R. Ikeda and C. A. McDowell, Spin-rotational relaxation for protons in solid, NH4 ClO4, Chem. Phys. Lett., 1972, 14(4), 389–392 CrossRef.
- V. I. Bakhmutov, Practical NMR Relaxation for Chemists, John Wiley & Sons Ltd, Chichester, UK, 2004, DOI:10.1002/0470094486.
- G. M. Bernard, R. E. Wasylishen, C. I. Ratcliffe, V. Terskikh, Q. Wu and J. M. Buriak, et al., Methylammonium Cation Dynamics in Methylammonium Lead Halide Perovskites: A Solid-State NMR Perspective, J. Phys. Chem. A, 2018, 122(6), 1560–1573, DOI:10.1021/acs.jpca.7b11558.
- P. Polatin, T. Barbara and B. P. Dailey, The determination of the 14N nuclear quadrupole coupling constant for the methylammonium ion in a liquid-crystal solution, J. Magn. Reson., 1982, 47(1), 148–150 Search PubMed.
- D. T. Edmonds, M. J. Hunt and A. L. Mackay, Pure quadrupole resonance of 14N in a tetrahedral environment, J. Magn. Reson., 1973, 9(1), 66–74 Search PubMed.
- R. E. Taylor, P. A. Beckmann, S. Bai and C. Dybowski, 127I and 207Pb Solid-State NMR Spectroscopy and Nuclear Spin Relaxation in PbI2: A Preliminary Study, J. Phys. Chem. C, 2014, 118(17), 9143–9153, DOI:10.1021/jp5023423.
- A. A. Shmyreva, M. Safdari, I. Furó and S. V. Dvinskikh, NMR longitudinal relaxation enhancement in metal halides by heteronuclear polarization exchange during magic-angle spinning, J. Chem. Phys., 2016, 144(22), 224201 CrossRef PubMed.
- T. Mildner, H. Ernst and D. Freude, 207Pb NMR detection of spinning-induced temperature gradients in MAS rotors, Solid State Nucl. Magn. Reson., 1995, 5(3), 269–271 CrossRef PubMed.
- P. A. Beckmann and C. Dybowski, A Thermometer for Nonspinning Solid-State NMR Spectroscopy, J. Magn. Reson., 2000, 146(2), 379–380 CrossRef PubMed.
- T. Takahashi, H. Kawashima, H. Sugisawa and T. Baba, 207Pb chemical shift thermometer at high temperature for magic angle spinning experiments, Solid State Nucl. Magn. Reson., 1999, 15(2), 119–123 CrossRef PubMed.
- L. C. M. van Gorkom, J. M. Hook, M. B. Logan, J. V. Hanna and R. E. Wasylishen, Solid-state lead-207 NMR of lead(II) nitrate: localized heating effects at high magic angle spinning speeds, Magn. Reson. Chem., 1995, 33(10), 791–795, DOI:10.1002/mrc.1260331005.
- P. Pyykko, Year-2008 nuclear quadrupole moments, Mol. Phys., 2008, 106(16–18), 1965–1974 CrossRef.
- D. L. Bryce, C. M. Widdifield, R. P. Chapman and R. J. Attrell, Chlorine, Bromine, and Iodine Solid-State NMR, eMagRes, John Wiley & Sons, Ltd, 2007, DOI:10.1002/9780470034590.emrstm1214.
- H. Bayer, Zur Theorie der Spin-Gitterrelaxation in Molekülkristallen, Zeitschrift für Phys., 1951, 130(2), 227–238, DOI:10.1007/BF01337696.
- J. Ramakrishna, Temperature Dependence of the Iodine Pure Quadrupole Resonance Frequency in Methyl Iodide, Proc. Phys. Soc., 1962, 79(5), 1069 CrossRef . Available from: http://stacks.iop.org/0370-1328/79/i=5/a=320.
- F. Brivio, J. M. Frost, J. M. Skelton, A. J. Jackson, O. J. Weber and M. T. Weller, et al., Lattice dynamics and vibrational spectra of the orthorhombic, tetragonal, and cubic phases of methylammonium lead iodide, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92(14), 144308, DOI:10.1103/PhysRevB.92.144308.
- T. Hata, G. Giorgi and K. Yamashita, The Effects of the Organic-Inorganic Interactions on the Thermal Transport Properties of CH3NH3PbI3, Nano Lett., 2016, 16(4), 2749–2753 CrossRef PubMed.
- P. Pistor, A. Ruiz, A. Cabot and V. Izquierdo-Roca, Advanced Raman Spectroscopy of Methylammonium Lead Iodide: Development of a Non-destructive Characterisation Methodology, Sci. Rep., 2016, 6, 35973 CrossRef PubMed.
- A. C. Daniel and W. G. Moulton, Temperature Dependence of Pure Nuclear Quadrupole Spin—Lattice Relaxation in SnI4, J. Chem. Phys., 1964, 41(6), 1833–1846 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp01535j |
| This journal is © the Owner Societies 2018 |