
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the interaction between solid benzene and water using vacuum ultraviolet and infrared spectroscopy
Anita
Dawes
 *a,
Natalia
Pascual
b,
Nigel J.
Mason
a,
Sabrina
Gärtner
c,
Søren V.
Hoffmann
d and
Nykola C.
Jones
d
*a,
Natalia
Pascual
b,
Nigel J.
Mason
a,
Sabrina
Gärtner
c,
Søren V.
Hoffmann
d and
Nykola C.
Jones
d
aSchool of Physical Sciences, The Open University, Walton Hall, Milton Keynes, MK7 6AA, UK. E-mail: Anita.Dawes@open.ac.uk; Fax: +44 (0)1908 654192; Tel: +44 (0)1908 654241
bDepartment of Chemistry and Physics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne 3086, Australia
cISIS Neutron and Muon Source, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Didcot, OX11 0QX, UK
dISA, Centre for Storage Ring Facilities, Department of Physics and Astronomy, Aarhus University, Ny Munkegade 120, DK-8000 Aarhus C, Denmark
First published on 11th May 2018
Abstract
We present results of a combined vacuum ultraviolet (VUV) and infrared (IR) photoabsorption study of amorphous benzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixtures and layers to investigate the benzene–water interaction in the solid phase. VUV spectra of 1:1, 1:10 and 1:100 benzene:water mixtures at 24 K reveal a concentration dependent shift in the energies of the 1B2u, 1B1u and 1E1u electronic states of benzene. All the electronic bands blueshift from pure amorphous benzene towards gas phase energies with increasing water concentration. IR results reveal a strong dOH-π benzene–water interaction via the dangling OH stretch of water with the delocalised π system of the benzene molecule. Although this interaction influences the electronic states of benzene with the benzene–water interaction causing a redshift in the electronic states from that of the free benzene molecule, the benzene–benzene interaction has a more significant effect on the electronic states of benzene. VUV spectra of benzene and water layers show evidence of non-wetting between benzene and water, characterised by Rayleigh scattering tails at wavelengths greater than 220 nm. Our results also show evidence of benzene–water interaction at the benzene–water interface affecting both the benzene and the water electronic states. Annealing the mixtures and layers of benzene and water show that benzene remains trapped in/under water ice until water desorption near 160 K. These first systematic studies of binary amorphous mixtures in the VUV, supported with complementary IR studies, provide a deeper insight into the influence of intermolecular interactions on intramolecular electronic states with significant implications for our understanding of photochemical processes in more realistic astrochemical environments.
:water mixtures and layers to investigate the benzene–water interaction in the solid phase. VUV spectra of 1:1, 1:10 and 1:100 benzene:water mixtures at 24 K reveal a concentration dependent shift in the energies of the 1B2u, 1B1u and 1E1u electronic states of benzene. All the electronic bands blueshift from pure amorphous benzene towards gas phase energies with increasing water concentration. IR results reveal a strong dOH-π benzene–water interaction via the dangling OH stretch of water with the delocalised π system of the benzene molecule. Although this interaction influences the electronic states of benzene with the benzene–water interaction causing a redshift in the electronic states from that of the free benzene molecule, the benzene–benzene interaction has a more significant effect on the electronic states of benzene. VUV spectra of benzene and water layers show evidence of non-wetting between benzene and water, characterised by Rayleigh scattering tails at wavelengths greater than 220 nm. Our results also show evidence of benzene–water interaction at the benzene–water interface affecting both the benzene and the water electronic states. Annealing the mixtures and layers of benzene and water show that benzene remains trapped in/under water ice until water desorption near 160 K. These first systematic studies of binary amorphous mixtures in the VUV, supported with complementary IR studies, provide a deeper insight into the influence of intermolecular interactions on intramolecular electronic states with significant implications for our understanding of photochemical processes in more realistic astrochemical environments.
1 Introduction
Benzene is a prototypical aromatic molecule and a building block of polycyclic aromatic hydrocarbons (PAHs) that are ubiquitous in the interstellar medium and found in great abundance in star forming regions. It is also the basis for many aromatic organic and biological molecules and biopolymers and as such, understanding the interaction between benzene and polar molecules such as water has been of considerable interest in physical chemistry. However much of this work has focused on liquid phases or clusters in the gas phase with far fewer studies in the condensed phase.Water is the simplest hydrogen-bonding molecule and is the most abundant liquid medium and solvent in biological systems, but it is also the most abundant condensed phase constituent of interstellar ices. Though not directly observed yet, evidence suggests that aromatic molecules/PAHs are likely to be present in the water-dominated icy grain mantles in dense molecular clouds, either acting as condensation nuclei for ice mantle growth or incorporated into the ice itself.1 Observational spectra of young stellar objects reveal absorption features that hint towards possible presence of condensed PAHs in icy mantles, e.g. C–H stretching modes2,3 around 3080 cm−1 and C–C stretching modes4 near 1600 cm−1. PAHs are also prevalent in our Solar System, on icy bodies in the outer Solar System,5 in comets6 as well as interplanetary dust particles7 and carbonaceous meteorites.8 Furthermore, PAHs identified in meteoritic and cometary samples show deuterium enrichment that points to their pre-solar dense molecular cloud origins9 where they most likely would have appeared concomitantly with water-rich ices.
The evolution of these aromatic molecules from the pre-stellar molecular cloud to incorporation into planetary systems and our Solar System and their role as precursors for complex organic and biological molecules has prompted numerous laboratory experiments investigating the photochemistry of PAHs in ices via ultraviolet processing under simulated astrophysical conditions. Results of irradiation of PAH–ice mixtures with Lyman-α dominated UV light (121 nm) reveal that PAHs are efficiently ionised in water ice, particularly at low temperatures (<50 K),10 with ionisation energy lowered by up to 2 eV compared to the gas phase11 and PAH cations remaining stable within the ice up to 120 K.12 The efficiency of cation formation is found to be strongly dependent on PAH concentration, with efficiency increasing in very dilute mixtures.13 Irradiation of mixtures of PAHs with water also results in side-group addition reactions forming aromatic alcohols and ketones.14,15 Irradiation of matrix isolated coronene–water mixtures at longer wavelengths (>235 nm) to avoid ionisation with high energy photons, was found to result in the formation of oxygenated photoproducts.16 A laser desorption study of benzene–water ice layers showed compelling evidence of benzene facilitated desorption of water molecules under irradiation with 250 nm photons.17 As water photoabsorption is negligible at this wavelength, the observed desorption of water molecules occurs due to transfer of energy from a neighbouring benzene molecule within the ice matrix following symmetry-forbidden resonant excitation of the 1B2u vibronic state of benzene and subsequent unimolecular decay via1A1g ← 1B2u internal conversion. Furthermore, water molecules were observed to desorb undissociated and with a translational temperature of 450 K. Such photochemical and photodesorption processes would have a significant impact on molecular complexity and gas phase abundances in star forming regions.18
Clearly aromatic molecules display significant photochemical activity via electronic excitation in the condensed phase. As such UV photoabsorption spectroscopy is necessary to better understand and quantify the interactions of UV photons with such species. In our previous paper19 we presented the results of vacuum ultraviolet spectra of pure amorphous and crystalline benzene, as a prototypical aromatic molecule, investigated under conditions relevant to that of ices in the interstellar medium and on planetary surfaces. In this paper we have extended our study to investigate the interaction of benzene and water ice layers and mixtures of different concentrations. While vibrational infrared bands are highly sensitive to the intramolecular shifts in electron densities that are associated with intermolecular interactions, vacuum ultraviolet spectra too can provide invaluable insight into the morphology of molecular solids via their electronic and vibronic structure and scattering. Although it is well known that the electronic spectra of molecules in the solid phases can differ considerably from that of the free molecules in the gas phase, very few experiments have been carried out in the VUV on binary molecular systems. We therefore carried out complementary VUV and IR studies of binary mixtures and layers of benzene and water with the unique capability of performing such investigations in situ using the same portable vacuum sample chamber under completely reproducible experimental conditions.
2 Experimental methods
Both VUV and IR studies in this paper have been carried out using the same Open University Portable Astrochemistry Chamber at two different sites. The VUV photoabsorption measurements were carried out at the ASTRID2 Synchrotron Facility, Aarhus, Denmark by attaching the portable system to the AU-UV beamline.20 This work is a continuation of the work we published in Dawes et al.19 (referred to as AD17 from now on), so a detailed description of the experimental set-up and procedures can be found there. In summary, for the data presented in this paper, the VUV spectra were acquired in the range 112–340 nm with 0.05–1 nm wavelength step size depending on the width of the spectral features to be resolved. With the AU-UV monochromator beamline settings used for the investigations detailed in this paper the measured average photon flux per point was 3.5 × 1010 photons s−1. Absorbance spectra were calculated from the recorded incident and transmitted intensities as a function of wavelength (nm). The infrared studies were carried out at the Open University Molecular Physics Labs using a FTIR (Nicolet Nexus 670) spectrometer with an external MCT detector. A detailed description of this set up and general procedures can be found in ref. 21. Briefly, in these experiments absorbance spectra were collected at 1 cm−1 resolution, by averaging 128 scans in the range 4000–800 cm−1.The experiments were carried out at a base pressure of low 10−9 mbar. Pure benzene (anhydrous, Sigma-Aldrich, 99.8% purity) and water (Sigma, double distilled and deionized water, Molecular Biology Reagent) were separately freeze-pump-thawed (3 times) and vapour deposited onto a cold (24–25 K) transmitting substrate held in a copper holder attached to a closed cycle helium cryostat (Sumitomo). Magnesium fluoride (MgF2) and zinc selenide (ZnSe) (Crystran) windows were used as substrates for VUV and IR transmission respectively. Mixtures of benzene and water were prepared using a dedicated gas line with ratios determined via partial pressures measured with a mass-independent baratron (MKS) prior to deposition.
Film thicknesses were determined by monitoring the sinusoidal variation of the intensity of a helium–neon laser reflected off the substrate during deposition. Deposition rates were between 0.1 and 0.2 nm s−1 with discrepancies arising due the mass dependent ion gauge readings during deposition (with deposition pressures between 5 × 10−8 and 1 × 10−7 mbar) for benzene and water mixtures. The same deposition conditions were strictly maintained in both VUV and IR experiments. Sample thicknesses between 17 nm and 1.7 μm were selected to optimise the signal to noise ratio of specific absorbance peaks under investigation; with thinner samples used to avoid saturation of high intensity bands, and thicker samples used to resolve weak features (e.g. weak IR benzene features in dilute mixtures with water and weak benzene VUV vibronic bands between 230 and 270 nm) in the spectra. In such cases the same experiments were repeated for thinner and thicker samples respectively and spectra were thickness normalised and compiled accordingly. We confirm that no thickness dependent effects (other than scattering effects) were observed in the spectra. In the case of thinner pure layers of benzene or water where it was not possible to perform laser thickness calculations from the recorded laser signal due to insufficient data points to show a sinusoidal trend for a successful fit, thicknesses were determined from the absorbance data using known cross sections of solid benzene from AD1719 and amorphous solid water22 using the rearranged form of Beer–Lambert Law, where the absorbance, A is related to the cross section σ via the following equation:
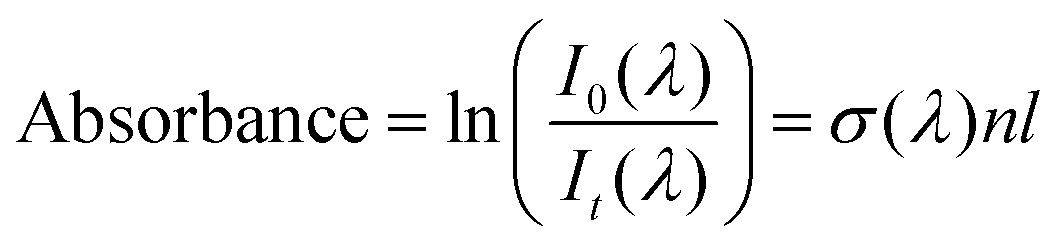 | (1) |
3 Results and discussion
Investigation of the interaction between benzene and water in this study has been carried out via both layered (benzene/water and water/benzene) and mixed (benzene:water ratios of 1:1, 1:10 and 1:100) systems discussed in Sections 3.2 and 3.1 respectively. Both VUV and IR studies were carried out for all systems, under the same experimental conditions (e.g. sample deposition and annealing) in the same Open University Portable Astrochemistry Chamber. A great advantage of using VUV spectroscopy over IR in the study of benzene–water system is the comparatively high cross section of benzene electronic absorption bands relative to water (by almost a factor of ten for the most intense bands). Conversely in the IR, the vibrational bands of benzene are weak relative to water (two orders of magnitude difference in the band strengths of the most intense peaks of benzene and water). This makes like-for-like comparison of they layered system difficult in the infrared, hence most of the discussion of the results in this paper focuses on VUV spectra and IR spectra are only presented for mixtures.
3.1 Benzene and water mixtures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :water mixtures.
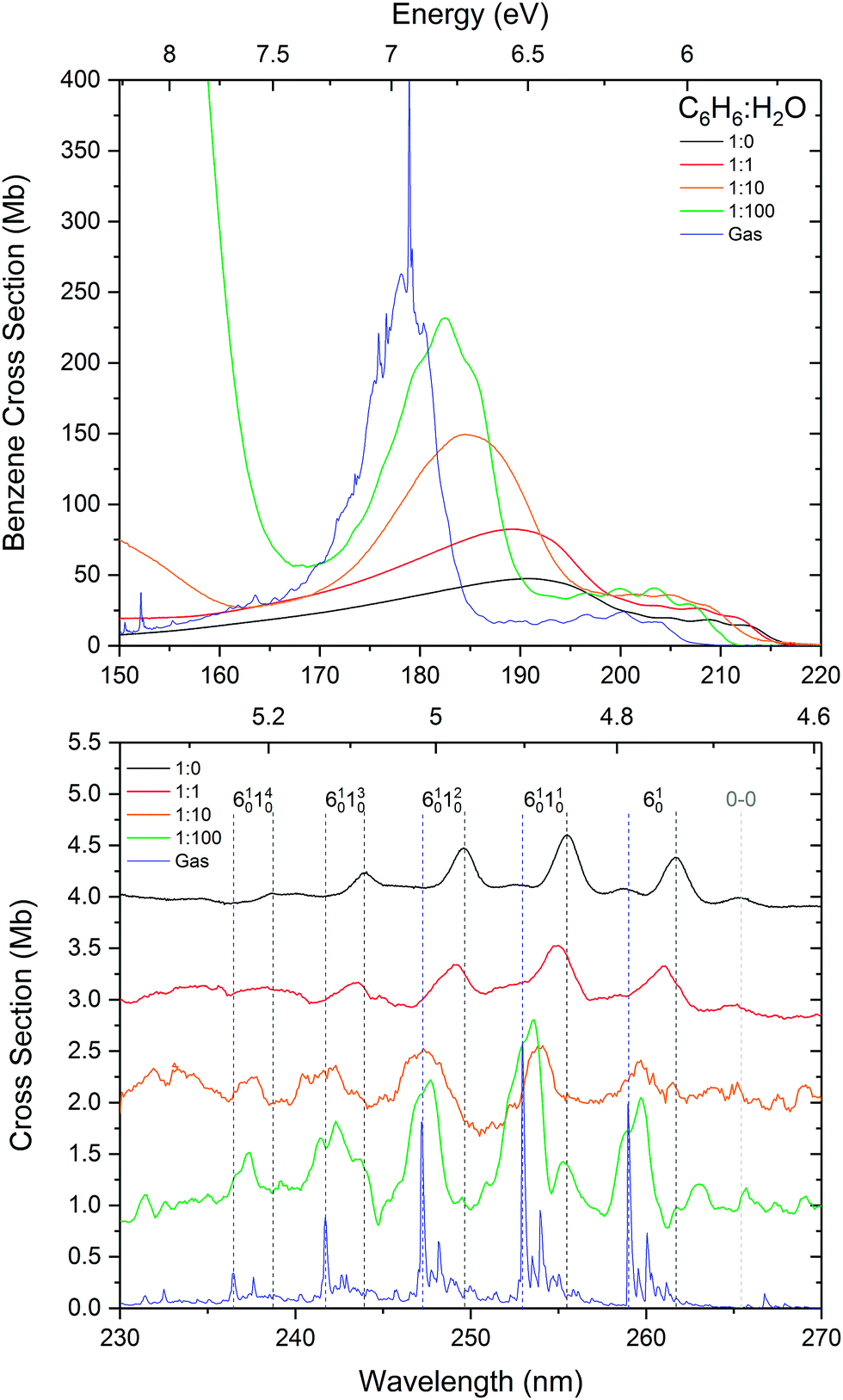
Fig. 1 shows the VUV photoabsorption spectra of 1:1, 1:10 and 1:100 mixtures of benzene and water, compared with the spectra of pure amorphous benzene at 24 K and gas phase benzene from AD17.19 It is difficult to investigate the effect of concentration on the water band around 144 nm (8.61 eV) corresponding to the (4a1: Ã1B1 ← 1b1:
:water mixtures.
Fig. 1 shows the VUV photoabsorption spectra of 1:1, 1:10 and 1:100 mixtures of benzene and water, compared with the spectra of pure amorphous benzene at 24 K and gas phase benzene from AD17.19 It is difficult to investigate the effect of concentration on the water band around 144 nm (8.61 eV) corresponding to the (4a1: Ã1B1 ← 1b1: ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 1A1) transition due to this band overlapping with the tail of an intense benzene band and the much lower (by an order of magnitude) cross section of amorphous solid water compared with solid benzene. The water band is not visible in the 1:1 mixture. It peaks at 144.1 nm in the 1:10 mixture, its position unaffected by mixing at this concentration. In the 1:100 mixture it is blueshifted to 143.2 nm (8.66 eV) as seen in thinner films (not shown in Fig. 1 where it appears saturated due to a thicker sample used to resolve benzene bands).
1A1) transition due to this band overlapping with the tail of an intense benzene band and the much lower (by an order of magnitude) cross section of amorphous solid water compared with solid benzene. The water band is not visible in the 1:1 mixture. It peaks at 144.1 nm in the 1:10 mixture, its position unaffected by mixing at this concentration. In the 1:100 mixture it is blueshifted to 143.2 nm (8.66 eV) as seen in thinner films (not shown in Fig. 1 where it appears saturated due to a thicker sample used to resolve benzene bands).
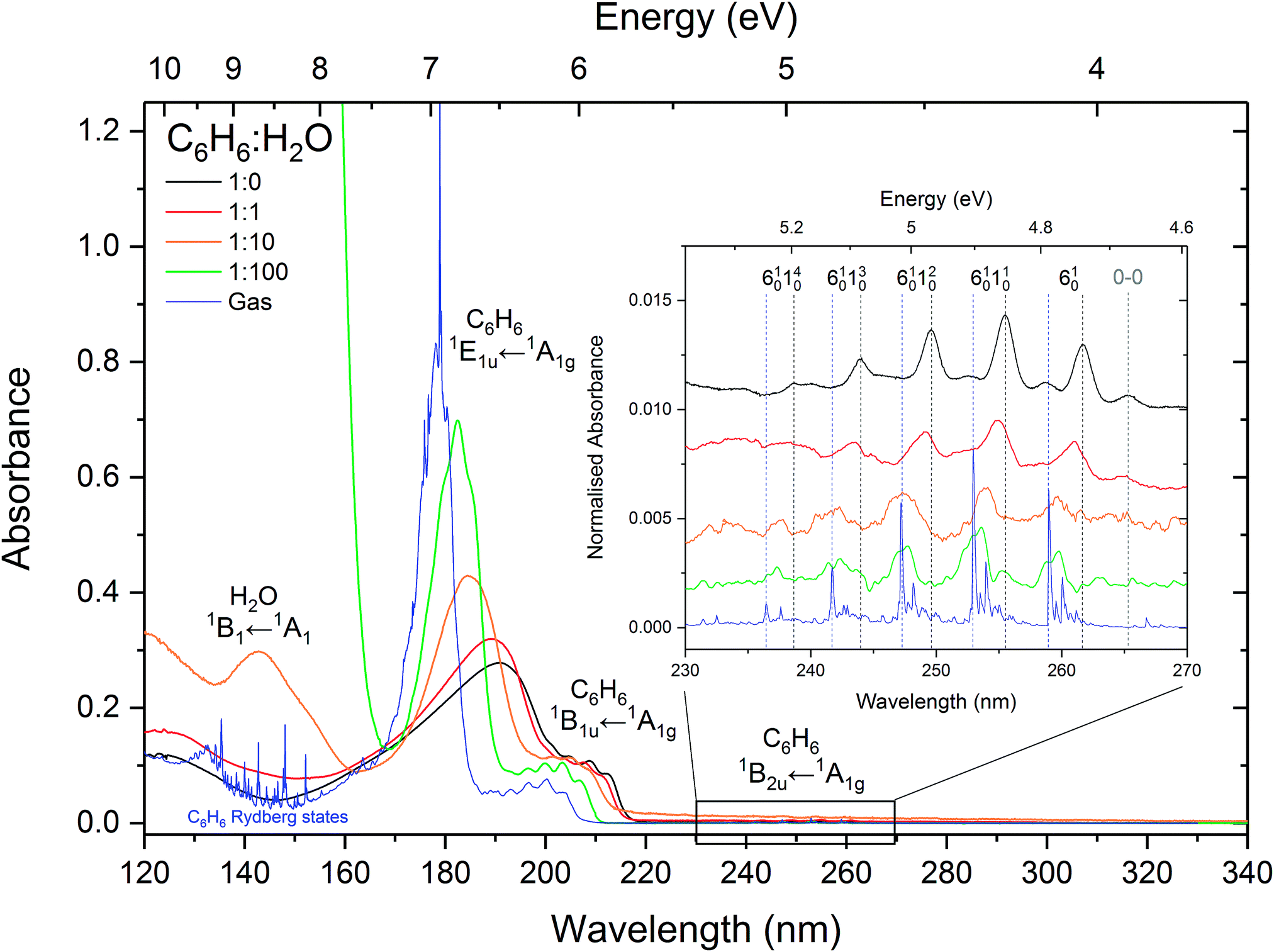 | ||
| Fig. 1 VUV photoabsorption spectra of 1:1, 1:10 and 1:100 benzene:water mixtures deposited at 24 K compared with pure benzene at 24 K and gas phase benzene from ref. 19. The band assignments and positions are summarised in Table 1. The spectra are normalised to the integrated area of the overlapping bands corresponding to the 1E1u ←1A1g and 1B1u ← 1A1g transitions. The inset shows a zoom into the region between 230 and 270 nm to highlight the vibronic bands associated with the 1B2u ← 1A1g transition. These have been baseline corrected for any scattering or sinusoidal thin film effects and offset for clarity. The lower signal-to-noise ratios in the spectra of 1:10 and 1:100 mixtures in this region are due the very weak benzene signal at these concentrations. | ||
It is immediately noticeable from the VUV spectra that there is a concentration dependent shift in the benzene electronic bands, with a general blueshift in all the bands with decreasing benzene concentration. The most intense benzene band in the spectra is due to the transition from the ground state to the third singlet 1E1u state. This band due to the 1E1u ← 1A1g transition shifts from 191.8 nm (6.47 eV) in pure benzene to 189.0 nm (6.56 eV) in 1:1, 184.8 nm (6.71 eV) in 1:10 and 182.7 nm (6.79 eV) in the 1:100 mixture of benzene:water corresponding to approximately a 0.1 eV blueshift with each dilution. As the band blueshifts it also narrows, grows in relative intensity and develops some structure, as can be seen in the spectrum of the 1:100 mixture, that remarkably resembles the profile of the gas phase peak.
The next intense band that appears as a structured red shoulder on the 1E1u ← 1A1g band is due to a transition to the second singlet 1B1u state. This transition is the symmetry forbidden, however it is observed in the optical spectrum via intensity borrowing from the neighbouring intense transition and Herzberg–Teller vibronic coupling giving rise to the vibronic structure. This band due to the 1B1u ←1A1g transition experiences smaller blueshifts in energy with decreasing benzene concentration. The average blueshifts with respect to pure benzene are 0.03, 0.11 and 0.15 eV for the 1:1, 1:10 and 1:100 benzene:water mixtures respectively. Sharpening of the vibronic bands is apparent with increasing benzene dilution.
The lowest lying 1B2u ← 1A1g electronic transition is also symmetry forbidden. Similarly intensity borrowing from the allowed 1E1u state and vibronic coupling gives rise to weak vibronic bands between 230 and 270 nm (see inset in Fig. 1). These vibronic bands are blueshifted in the solid phase from pure benzene by 0.01, 0.03 and 0.04 eV in the 1:1, 1:10 and 1:100 benzene:water mixtures respectively. It is interesting to note that the 0–0 exciton band origin near 265 nm (pure benzene) is clearly visible in the spectra of all the solid samples (not observed in the gas phase – see AD1719 for discussion on this), blueshifted accordingly as a function of concentration. The vibronic bands in the 1:100 mixture appear to have two components. It may be that we are seeing the second intense 6101n01611 progression that is seen in gas phase benzene19 which is not surprising as benzene molecules are expected to be more or less matrix isolated in the water ice at this concentration.
The observed energy shifts in the three lowest-lying singlet states of benzene as a function of benzene:water concentration in the solid phase is certainly very intriguing. It has been shown from computational and experimental work that there is hydrogen-bonding-like interaction between water and benzene via dipole–quadrupole interaction between water as a proton donor and benzene as a proton acceptor. The most favoured configuration is one with one hydrogen (more stable) or both of the hydrogens (less stable) in the water molecule pointing towards and interacting with the delocalised π cloud above or below the benzene ring in dipole(water)–quadrupole(benzene) interaction.23,24 Theoretically the interaction of water oxygen atoms with the rim of the benzene ring is also possible in cases where water molecules are found beyond the perimeter of the benzene ring.25 This arrangement could occur in an amorphous solid matrix prepared by vapour deposition at 25 K, but is not expected to have much of an influence on the π* ← π electronic transitions of benzene. The progressive shift in energy of the electronic bands from that of pure amorphous solid benzene to benzene diluted in amorphous solid water can therefore be attributed to the quadrupole–dipole hydrogen bonding interaction between benzene and water, as the benzene molecules become surrounded with water molecules disrupting the π electron density of benzene molecules and hence perturbing the energy of the π* ← π valence shell transition. However, the shift to higher energies of all the electronic bands towards that of the gas phase transition energies, and the narrowing and striking similarity in the 1E1u ← 1A1g band profile of the 1:100 mixture with that of the gas phase, also begs the question of whether the benzene molecules are effectively being matrix isolated with increasing dilution in the water matrix. One piece of evidence to strongly support this is the band position and profile in the optical spectra of benzene diluted in inert gas matrices. Band positions of around 182 nm were reported in spectra of benzene matrix isolated in xenon and krypton at 20 K,26 similar to that of 1:100 benzene–water mixture in our spectra. Furthermore, where structure on the 1E1u band is ascribed to Rydberg states in the gas phase, features on this band in the matrix isolated benzene are reportedly assigned to Wannier excitons.
This therefore prompts us to turn the argument around to challenge our initial interpretation of the VUV spectra. What we initially see in our spectra starting with pure solid benzene is a blueshift in electronic states with increasing dilution of benzene in a water ice matrix, giving the impression that the benzene–water interaction causes an increase in the energy of the excited states (observed as a blueshift). But if we turn our argument around and start with a free benzene molecule (gas phase) and surround it in water molecules (1:100 mixture), our results then strongly suggest from the observed gas-to-solid shifts, that there is a redshift in the electronic states of benzene as it interacts with surrounding water molecules. So the benzene–water interaction in fact lowers the excitation energies of benzene by 0.01 eV, 0.11 eV and 0.18 eV for the 1B2u, 1B1u and 1E1u states respectively. Indeed TD-DFT calculations of VUV spectra of benzene–ice clusters by Sharma et al.27 show small redshifts (0.05–0.12 eV) in the bands due to the 1B2u ← 1A1g and 1B1u ←1A1g compared with isolated benzene in agreement with our results. The authors also describe a redshift in the 1E1u ← 1A1g band of between 0.04 and 0.12 eV depending on the nature of the binding site of the ice surface. The further progressive redshift in the benzene electronic states in our spectra can be explained as benzene–benzene interactions coming into play with increasing benzene concentration (1:10 and 1:1 mixture), with a maximum gas-to-solid shift observed in the pure solid benzene spectrum. This implies that the benzene–benzene interaction is in fact stronger than the benzene–water interaction in lowering the energy of the first three observed excited states of the benzene molecule. Indeed this is in agreement with temperature programmed desorption (TPD) studies of benzene from water surfaces reported by Thrower et al.28
The effects of benzene–benzene interaction can clearly be seen when the benzene:water mixtures are annealed as shown in Fig. 2. The films were annealed to 90 K, 140 K and 160 K corresponding to the temperatures at which pure benzene crystallises (75–90 K), benzene desorbs and water begins to crystallise (140 K) and water begins to desorb (160 K). In the 1:1 mixture there is no benzene crystallisation evident at 90 K, however there is a small redshift in the 1E1u band, which is most likely due to some degree of mobility of benzene molecules. With the slight shift being in the direction towards the position of pure benzene indicating rearrangement whereby benzene–benzene interactions are being favoured over benzene–water interaction. The slight increase in intensity of the peak is characteristic of matrix reconstruction and associated changes in the density and optical constants. A further redshift is seen at 140 K in the 1:1 mixture, and interestingly evidence of crystallisation can be seen from the characteristic sharpening of the 1B2u and 1B1u vibronic bands and splitting in the 1E1u peak. Crystal field Davydov splitting can be seen in the 0–0 band of the 1B2u vibronic states (as seen in pure solid benzene in AD1719) suggesting areas of well ordered benzene crystal within the 1:1 benzene:water matrix and possible water-benzene segregation at 140 K. Decrease in the intensity of the 1E1u peak suggests that some of the benzene is desorbing at 140 K, with the rest of the benzene co-desorbing with water between 140 and 160 K in agreement with temperature programmed desorption studies.33 The 1:10 mixture shows no sign of crystallisation at 90 K and only weak splitting in the 1E1u band is seen at 140 K. Unlike the 1:1 mixture, there is no change in peak position seen in the 1:10 mixture suggesting that the benzene matrix is strongly disrupted by water molecules preventing the benzene–benzene crystal field interaction that is expected at these temperatures. As expected, annealing the 1:100 mixture shows no signs of crystallisation and benzene remains trapped in the ice until water begins to desorb at 160 K. There is slight redshift in the 1B2u vibronic bands and broadening of the 1B1u and 1E1u bands. It is not clear what the cause of this is, but may be due to some benzene–benzene interaction as the ice re-orders and desorbs and benzene is very mobile at 160 K which is well above its desorption temperature. The reason why we might be observing this in the 1:100 mixture and not in the 1:10 mixture may be due to the sample thickness. The 1:100 film was much thicker (840 nm) than the 1:10 (90 nm) in order to resolve the benzene peaks in the dilute mixture and was therefore captured while it was still in the process of desorbing, whereas the thinner samples were already completely desorbed.
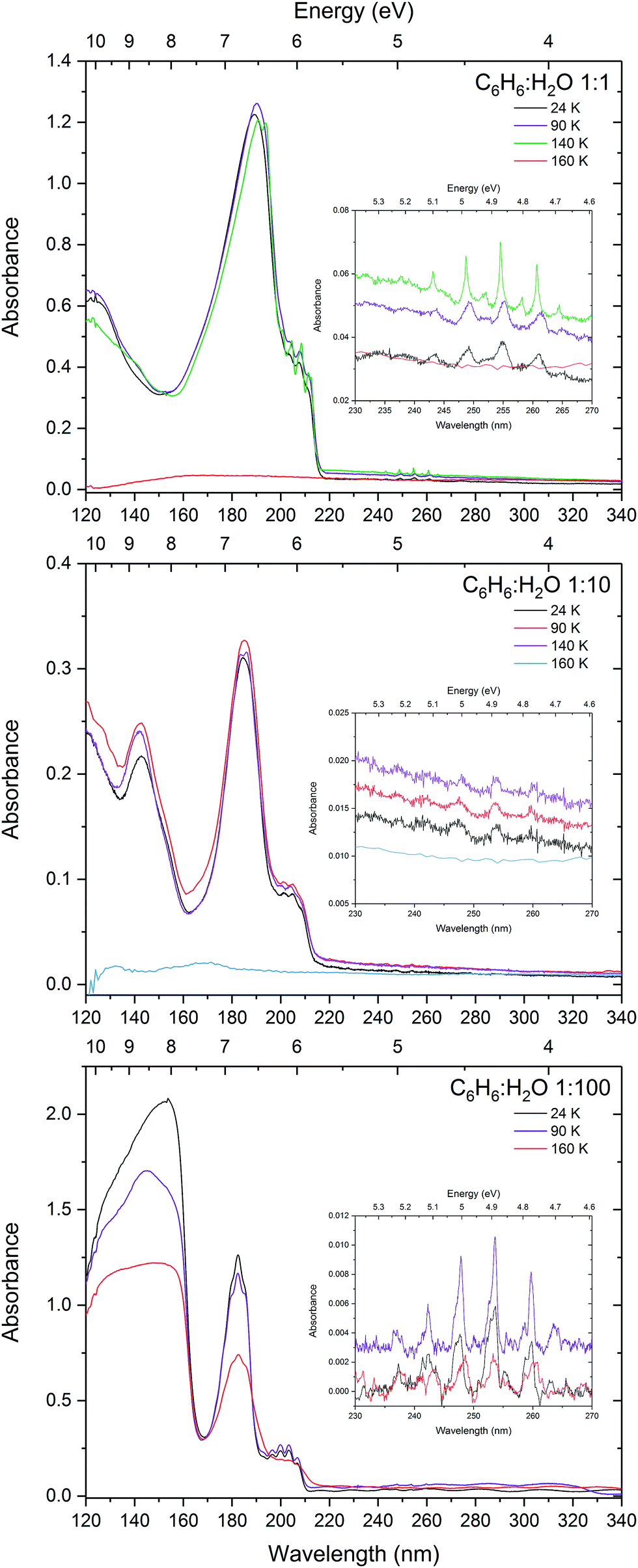 | ||
| Fig. 2 VUV spectra of 1:1, 1:10 and 1:100 mixture of benzene and water annealed to 90 K, 140 K and 160 K with insets showing a zoom into the 1B2u ← 1A1g vibronic bands. | ||
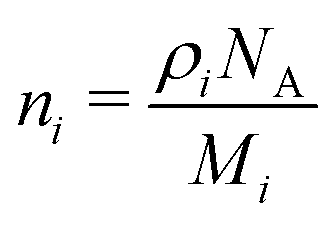 | (2) |
:1, 1:10 and 1:100 mixtures respectively, selected for optimal signal-to-noise ratio and avoiding band saturation. Additionally an 840 nm thick sample was used to resolve the 1B2u ← 1A1g benzene vibronic structure in the 1:100 mixture. The results are shown in Fig. 3. On comparison with the peak area normalised (absorbance scale) data shown in Fig. 1, the trend in the cross section data does appear to be similar. There is a progressive increase in the cross sections of the intense 1E1u ← 1A1g electronic transition with increased dilution in water from that of pure amorphous benzene towards that of the gas phase. There appears to be a slight increase in the cross sections of the vibronic bands associated with the 1B2u ← 1A1g electronic transition with increasing dilution of benzene in water which is coincident with a blueshift towards gas phase energies. It is to be noted that these calculations are approximate with estimated errors of up to 20% in the values of the cross sections which would account for any observed discrepancies between the absorbance area normalised data in Fig. 1 and the cross section data in Fig. 3. There are many factors that are expected to contribute to these errors. We know little about the morphology of the ice mixtures and hence the effect on the density of the ice due to mixing. Although it is assumed that there is homogeneous mixing between benzene and water in the ice film as evidenced by the shifts in the bands as a function of concentration, it is possible that there are areas of inhomogeneity due to the non-wetting interaction between water and benzene. The mixing ratios are determined from the partial pressures of benzene and water vapours that are pre-mixed in the gas line. Experience tells us that there could be discrepancies between the mixing cell ratios and the ratios of the vapour deposited ice films. The blue wing of the intense 1E1u ← 1A1g transition of benzene overlaps with the with the red tail of the water band, adding a concentration dependent error that would be particularly significant in very dilute benzene mixtures. Furthermore the effect of benzene on water cross sections is not known. Further systematic study over a finer range of concentrations with both water in excess and benzene in excess are required to fully ascertain the contribution to the errors in the calculations of the cross sections. Qualitatively however the estimated cross sections are reasonable within experimental error and it is interesting to note that the cross sections tend towards the gas phase values with increasing dilution, consistent with the observed energy shifts.
 | ||
| Fig. 3 Calculated VUV photoabsorption cross sections in megabarns (where 1 Mb = 10−18 cm2) of benzene in 1:1, 1:10 and 1:100 mixtures in water at 24 K, compared with pure amorphous benzene and gas phase benzene cross sections from AD17.19 The 1E1u ← 1A1g and 1B1u ←1A1g electronic transitions are shown in the top panel and the 1B2u ← 1A1g electronic transition is shown in the bottom panel with graphs offset vertically by 0.1 Mb for clarity. | ||
:water mixtures.
The effect of intermolecular interactions between benzene and water is clearly evident from the infrared spectra of C6H6:H2O mixtures. Fig. 4 shows the infrared spectra of pure water, benzene and 1:1, 1:10 and 1:100 mixtures of C6H6:H2O. What is immediately noticeable is an intense broad (60 cm−1 FWHM) feature at 3616 cm−1 in the OH stretching region of H2O in the spectrum of the 1:1 mixture of benzene and water. This is the signature region in the IR spectrum of water corresponding to ‘dangling’ O–H stretches of incompletely hydrogen-bonded water molecules. The position of the band matches well with that of matrix isolated benzene–water at 17 K, reported by Engdahl et al.31 who observe a concentration dependent structured band between 3620 and 3612 cm−1 due to the benzene–water complex. Silva et al.30 report a broad (35 cm−1 FWHM) band at 3585 cm−1 in dilute mixtures of benzene and water at 12–15 K. This band is assigned to the π hydrogen-bonded OH benzene–water complex,38 dOH-π. A benzene-induced apparent redshift of the ‘dOD’ band was observed by Bahr et al.33 in D2O films exposed to various coverages of benzene at 124 K. The authors report that as benzene coverage was increased, the surface dangling OD band of amorphous deuterated water (at 2728 cm−1) decreased in intensity, whilst a broad ‘dOD*’ band emerged, redshifted by 75 cm−1 relative to dOD. The authors report the same effect in co-deposited benzene and D2O and attributed the ‘dOD*’ band to the formation of benzene–D2O complexes. Similarly shifts in the ring modes of benzene adsorbed on solid H2O have been reported by Thrower,39 and Marchione.40,41
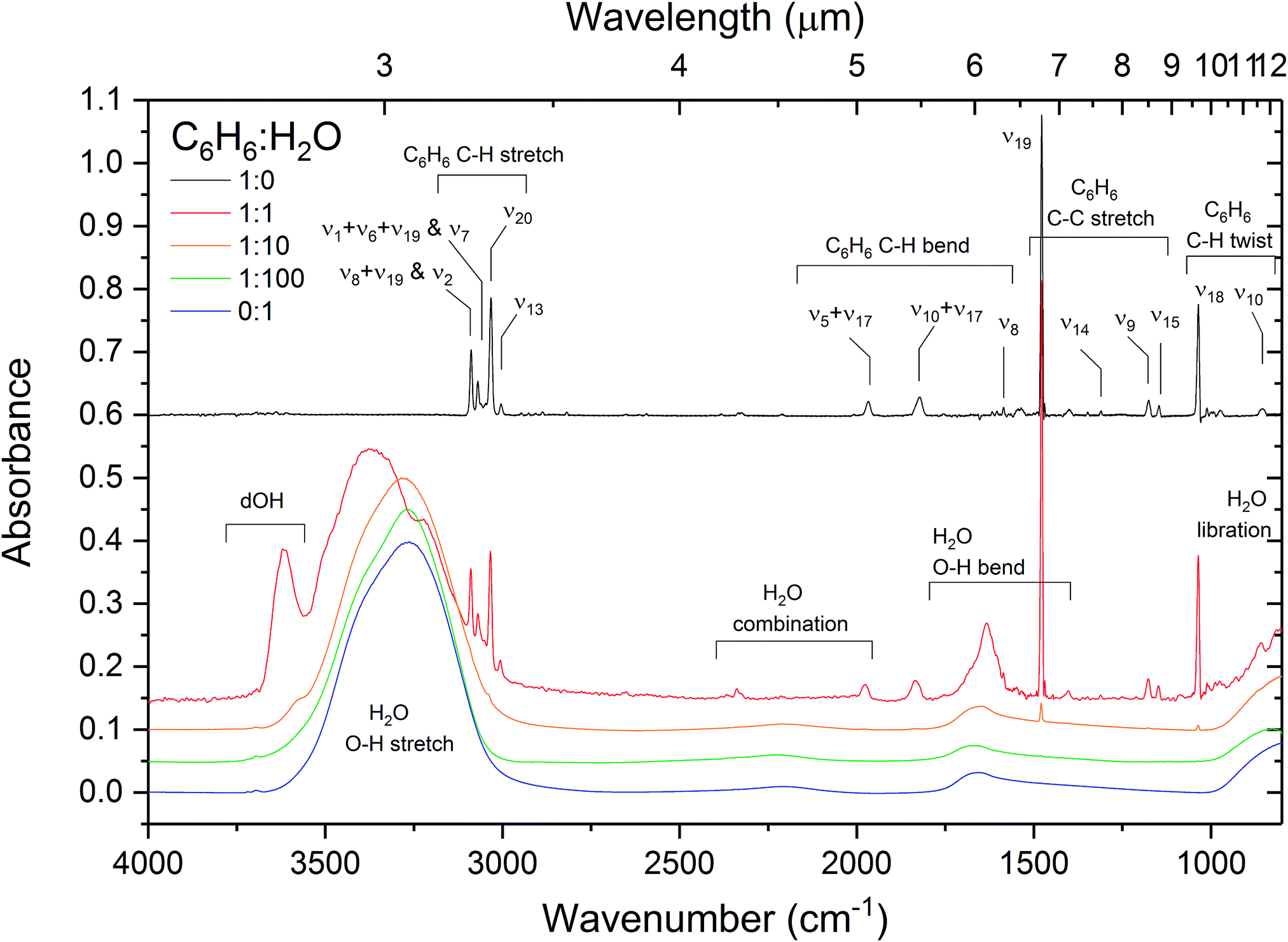 | ||
| Fig. 4 Infrared absorption spectra of benzene, water and 1:1, 1:10 and 1:100 benzene:water mixtures deposited at 24 K with assignments given for the most prominent bands. The band assignments and positions are summarised in Table 2. The spectra of benzene:water mixtures are normalised to the OH stretch band area for comparison. Spectra are offset vertically for clarity. | ||
This dOH-π band, shown in Fig. 5, is intense and broad in the 1:1 mixture centered about 3616 cm−1 and appears to be composed of at least two components. Splitting in the π-bonded OH band is also reported by Gruenloh et al.38 in the interaction between benzene and the water octamer. Silva and Devlin30 suggest that their observed dOH-π bands in diluted mixtures of benzene and water fits well when resolved into two components at 3595 and 3575 cm−1 in the temperature range from 12 to 125 K. By comparing the similarities between the two components of this band with that of the pure dOH bands, the authors assign them to benzene bonded to 2-coordinated (higher wavenumber) and 3-coordinated (lower wavenumber) dOH respectively. This band appears diminished in intensity and redshifted to 3568 cm−1 in our 1:10 mixture, shifting towards the reported positions of the antisymmetric ‘double-donor OH-stretches (3568–3543 cm−1) in the interaction of benzene with the water octamer’38 or the ‘dO mode’ (3549 cm−1) in amorphous solid water.42 We can therefore attribute the ‘redshift’ of the dOH-π band in the 1:10 mixture of benzene to the fact that less of the water ice matrix is disrupted by benzene molecules, giving rise to more higher coordinated water bonds.
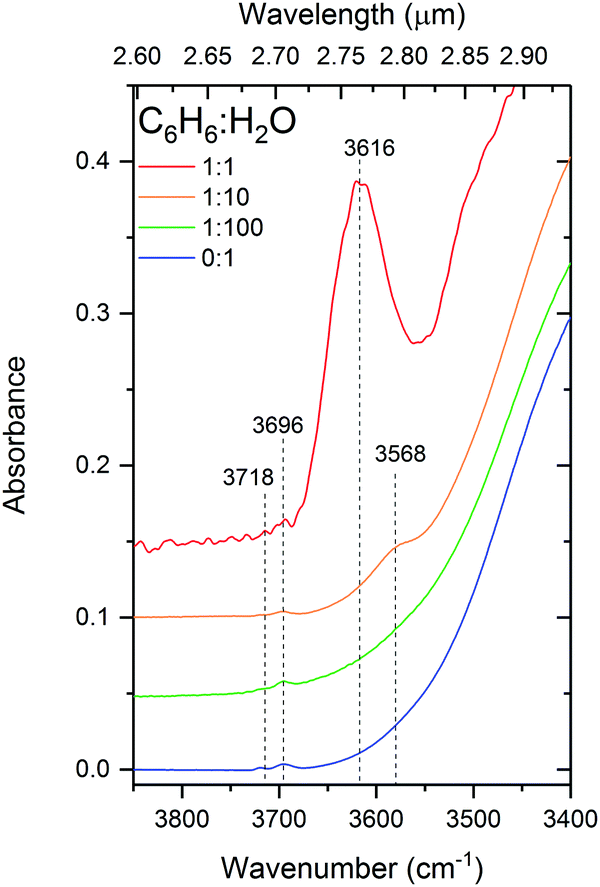 | ||
| Fig. 5 Infrared spectra in the dangling-OH region for 1:1, 1:10 and 1:100 C6H6:H2O mixtures deposited at 24 K, compared with d-OH bands of pure H2O. | ||
On annealing the 1:1 mixture of benzene and water there is a redshift in the dOH-π band from 3616 cm−1 at 24 K to 3599 cm−1 at 90 K and 3593 cm−1 at 140 K. There is little or no evidence of the dOH-π band at 160 K as most of the benzene is desorbing with water crystallisation. This is consistent with benzene desorption and subsequent increasing coordination of the water bonds with crystallisation.
All of the water features are affected in the IR spectra with higher concentrations of benzene, particularly in the 1:1 mixture. The 1:1 system presents an interesting case in that stoichiometrically it is not possible to have a homogeneous 1:1 benzene–water mixture at this ratio. The OH stretch band of water is slightly blueshifted and broadened compared to that of pure water ice at low benzene concentrations. Splitting of the symmetric (ν1) and asymmetric (ν3) OH stretching modes is clearly seen in the band in the 1:1 mixture with considerable enhancement in the asymmetric mode. This can certainly be attributed to the influence of the water(dipole)–benzene(quadrupole) hydrogen bonding interaction on the water molecules. Evidence of dOH-π bonding discussed above combined with computational studies23–25 reveal preferred orientations involving one or both of the water OH bonds pointing towards the plane of the benzene ring. As stoichiometrically one water molecule could only bind to one benzene ring in a 1:1 mixture, an enhanced asymmetric stretch mode would suggest that the preferred orientation is one with one of the OH bonds pointing towards the benzene ring thus breaking the symmetry. There is also an observed redshift in the position of the water bending mode with increasing benzene concentration. Since strong hydrogen bonding is associated with a blueshift in the ν2 bending mode of H2O,21 the observed redshift with respect to pure amorphous solid water confirms that the strength of benzene–water interaction is weaker than that of the water–water hydrogen bonding interaction24 and is a result of benzene molecules disrupting the water hydrogen bonding network. This is in agreement with results from the VUV spectra (Section 3.1.1) and temperature programmed desorption results of Thrower et al.28
The influence of dilution of benzene in water on the IR bands of benzene is small, resulting only in slight blueshifts in the bands (see Table 2) relative to pure benzene and no visible broadening. It is to be noted that the IR band strengths of benzene are relatively low compared to H2O making interpretation of concentration dependent changes in the benzene band profiles difficult.
:1, 1:10 and 1:100 benzene:water mixtures at 24 K, compared with pure solid benzene at 24 K and and gas phase benzene from our previous work.19 The gas to solid energy shifts (in eV) are shown under headings ‘g–s’
| Band assignment | Benzene(solid)19 | 1:1 C6H6:H2O |
1:10 C6H6:H2O |
1:100 C6H6:H2O |
Benzene(gas)19 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nm | eV | g–s/eV | nm | eV | g–s/eV | nm | eV | g–s/eV | nm | eV | g–s/eV | nm | eV | |
| 1E1u ← 1A1g | ||||||||||||||
| 191.8 | 6.47 | 0.50 | 189.0 | 6.56 | 0.41 | 184.8 | 6.71 | 0.26 | 182.7 | 6.79 | 0.18 | 178.0 | 6.97 | |
| 1B1u ←1A1g | ||||||||||||||
| 610 | 212.7 | 5.83 | 0.24 | 211.3 | 5.87 | 0.20 | 208.6 | 5.94 | 0.13 | 207.0 | 5.99 | 0.08 | 204.2 | 6.07 |
| 610110 | 208.8 | 5.94 | 0.25 | 207.6 | 5.97 | 0.22 | 204.8 | 6.05 | 0.14 | 203.6 | 6.09 | 0.10 | 200.3 | 6.19 |
| 610120 | 204.9 | 6.05 | 0.26 | 203.8 | 6.08 | 0.22 | 201.6 | 6.15 | 0.16 | 200.1 | 6.20 | 0.11 | 196.6 | 6.31 |
| 610130 | 201.2 | 6.16 | 0.26 | 200.2 | 6.19 | 0.23 | 198.0 | 6.26 | 0.16 | 196.5 | 6.31 | 0.11 | 193.0 | 6.42 |
| 610140 | 193.3 | 6.41 | 0.12 | 189.8 | 6.53 | |||||||||
| 1B2u ← 1A1g | ||||||||||||||
| 610 | 261.6 | 4.74 | 0.05 | 261.2 | 4.75 | 0.04 | 259.9 | 4.77 | 0.02 | 259.6 | 4.78 | 0.01 | 258.9 | 4.79 |
| 610110 | 255.7 | 4.85 | 0.05 | 255.2 | 4.86 | 0.04 | 253.9 | 4.88 | 0.02 | 253.5 | 4.89 | 0.01 | 252.9 | 4.90 |
| 610120 | 249.8 | 4.96 | 0.05 | 249.3 | 4.97 | 0.04 | 248.0 | 5.00 | 0.02 | 247.6 | 5.01 | 0.01 | 247.2 | 5.02 |
| 610130 | 244.2 | 5.08 | 0.05 | 243.6 | 5.09 | 0.04 | 242.5 | 5.11 | 0.02 | 242.2 | 5.12 | 0.01 | 241.7 | 5.13 |
| 610140 | 238.8 | 5.19 | 0.05 | 238.3 | 5.20 | 0.04 | 237.5 | 5.22 | 0.02 | 237.3 | 5.23 | 0.02 | 236.4 | 5.25 |
:water mixtures, pure benzene and water deposited at 24 K, shown in Fig. 4, and compared with experimental values from literature with details given in the table footnotes
| Molecule | Band | Assignment | Pure H2O/CH3OH | C6H6:H2O mixtures |
Literature | |||
|---|---|---|---|---|---|---|---|---|
| 1:1 |
1:10 |
1:100 |
2:1 (10 K, ref. (a)) |
Various (see footnote) | ||||
|
a Hagen et al.,29 2:1 C6H6:H2O mixture (10 K).
b *Engdahl et al.,31 C6H6–H2O complex in Ar (17 K).
c *Bahr et al.33
d Bernstein et al.35
e Silva and Devlin,30 3:97 C6H6:H2O(13 K).
f Kearley et al.,32 calculated C6H6 crystal.
g Chandrasekaran et al.,34 benzene dimers. *Assignments converted from Herzberg to Wilson notation.36 |
||||||||
| C6H6 | C–H stretch | ν 8 + ν19, ν2f,g | 3088 | 3089 | 3092 | Weak | 3090 | 3042b |
| ν 1 + ν6 + ν19, ν7f,g | 3069 | 3070 | Weak | 3069 | 3057g | |||
| ν 20 , | 3032 | 3034 | 3038 | 3034 | 3117f, 3043g | |||
| ν 13 , | 3003 | 3006 | Weak | 3013g | ||||
| C–H bend | ν 5 + ν17b | 1967 | 1974 | 1977 | 1973 | 1964b | ||
| ν 10 + ν17b | 1822 | 1834 | 1836 | 1831 | 1821b | |||
| ν 8 , | 1586 | 1586 | 1585 | |||||
| C–C stretch | ν 19 , , | 1477 | 1478 | 1479 | 1480 | 1478 | 1480b, 1459f | |
| ν 14 , | 1311 | 1312 | 1305 | |||||
| ν 9 , | 1176 | 1177 | Weak | 1176 | 1154f | |||
| ν 15 , , | 1146 | 1148 | 1148 | 1131f | ||||
| Twist | ν 18 , , | 1034 | 1035 | 1036 | Weak | 1035 | 1036b | |
| ν 12 , | 1010 | 1011 | 1012 | |||||
| ν 17 , | 990 | 989 | 977 | |||||
| ν 10 , | 855 | 858 | 860 | |||||
| H2O | dOH | 3696, 3718 | 3695 | 3695 | 3696, 3718e | |||
| OH stretch | ν 1, ν3 | 3270 | 3215, 3352 | 3288 | 3279 | 3206, 3370 | 3638, 3734b | |
| OH bend | ν 2 | 1655 | 1631 | 1650 | 1653 | 1630 | 1594–1609b | |
| C6H6:H2O | dOH-πf | 3616 | 3568 | 3626 | 3616b, 3585e | |||
There are four bands visible in the C–H stretching region of the IR spectrum of pure benzene between 3100 and 3000 cm−1. The same bands are seen in the 1:1 mixture, redshifted by 1–3 cm−1. These bands are too weak to be seen in the more diluted mixtures. The free benzene molecule exhibits four C–H fundamental modes: ν2(A1g), ν7(E2g), ν20(E1u) and ν13(B1u) (in Wilson notation). However, only the ν20 fundamental is IR allowed and in the gas phase the ν1 + ν6 + ν19 and ν8 + ν19 combination bands are observed.43 Due to the breakdown in symmetry in the solid phase due to intermolecular interactions between the benzene molecules the IR forbidden modes are likely to be allowed, however they are weak and overlap with the combination bands.34
It can clearly be seen on annealing the benzene film that the bands near 3090 cm−1 and 3003 cm−1 split (Fig. 6). This occurs as a result of crystallisation and ordering of the benzene molecules breaking the degeneracy of the vibrational modes. The splitting is also observed in the 1:1 mixture albeit at a higher temperature, consistent with crystallisation observed at higher temperature in the VUV spectra. Benzene C–H stretching features are too weak to be resolved in the 1:10 and 1:100 mixtures. The splitting in the C–H stretch, ν19 C–C stretch and ν18 C–H twist modes in the 1:1 mixture at 140 K is consistent with that of pure benzene at 90 K and indicates little disruption by the presence of water molecules. Furthermore at this temperature the water O–H stretch band resembles the profile of pure water crystallisation, suggesting segregation of benzene and water as both become mobile and benzene begins to desorb.
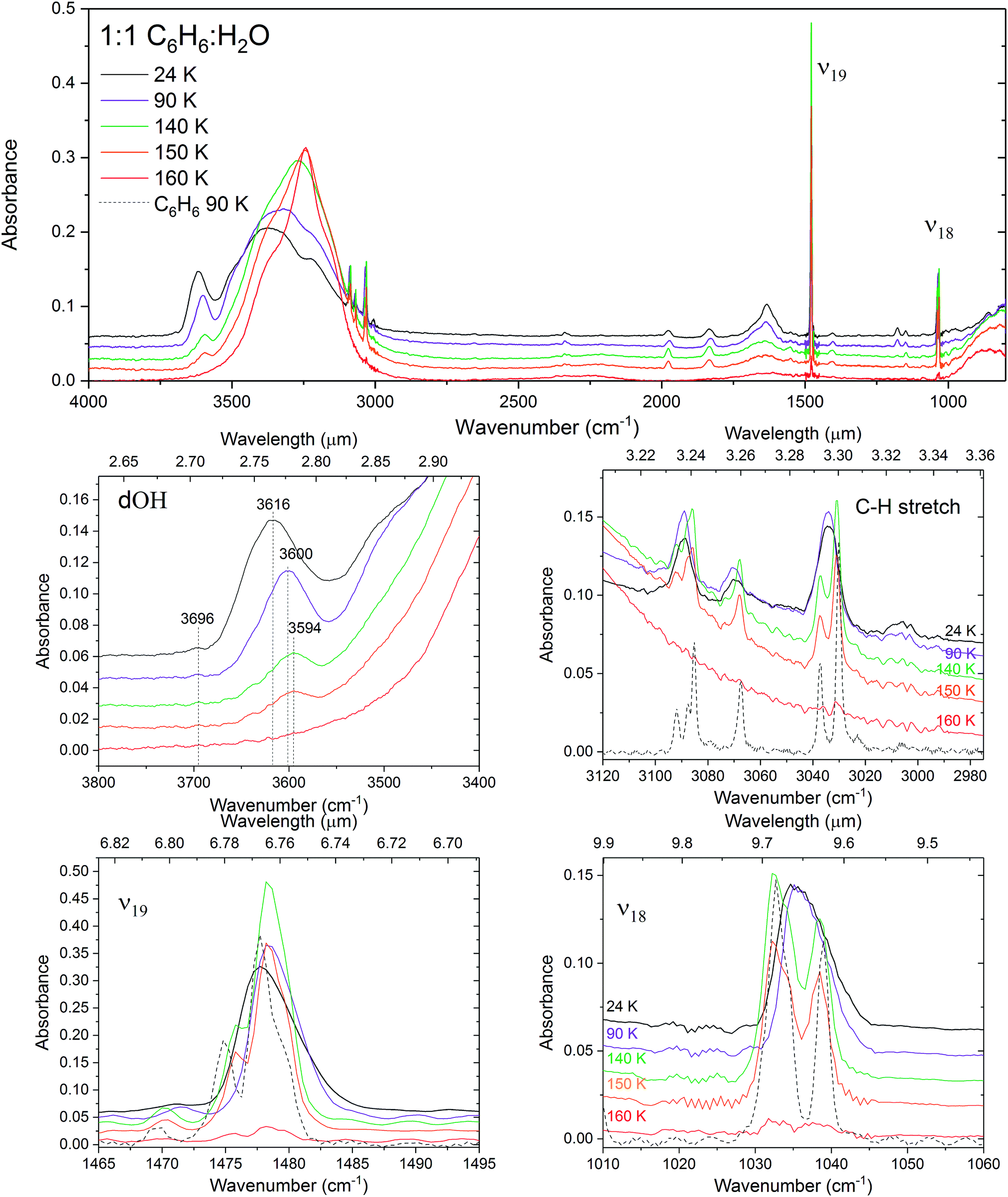 | ||
| Fig. 6 Infrared spectra of the 1:1 mixture of C6H6:H2O deposited at 24 K and annealed to 90, 140, 150 and 160 K. The insets show zooms into the dOH-π band region and the main benzene bands: the C–H stretches, ν19 C–C stretch and ν18 antisymmetric C–H twist and compared with the spectrum of pure crystalline benzene at 90 K represented with a dashed black line. The spectra are slightly offset vertically for clarity. | ||
The fact that very small changes are seen in the infrared band profiles of benzene in the 1:1 benzene:water mixture further supports our interpretation that the concentration dependent energy shifts in the VUV spectra are due to benzene molecules becoming matrix isolated in water and the electronic bands shifting towards the gas phase energies with increasing dilution. Little change is seen in the vibrational bands of benzene and this is consistent with the negligible changes in the spacings of the vibronic bands in the VUV spectra. Intermolecular interactions between benzene and water strongly influence the water molecules as can be seen from the striking changes in the vibrational spectra of the O–H stretch and O–H bending modes of water and the presence of the dOH-π feature.
3.2 Benzene and water layers
In our previous paper, AD17,19 results of VUV spectra of pure benzene vapour deposited at 25 K showed that thin layers of benzene did not ‘wet’ the MgF2 substrate resulting in the formation of 3D amorphous ‘clumps’. This was evident from thickness dependent scattering tails observed in the VUV spectra at wavelengths greater than 220 nm. At first instance we will look at the interaction of benzene deposited on an amorphous water surface and compare it with that of benzene deposited on a MgF2 substrate: the ‘benzene on water’ layered system. Following that we will look at the effect that water deposition may have on a thin ‘clumpy’ benzene layer.The scattering tails in the absorbance spectra of benzene in the region 220–340 nm (avoiding the onset of the strong absorption band near 220 nm) were fitted with a function of the form
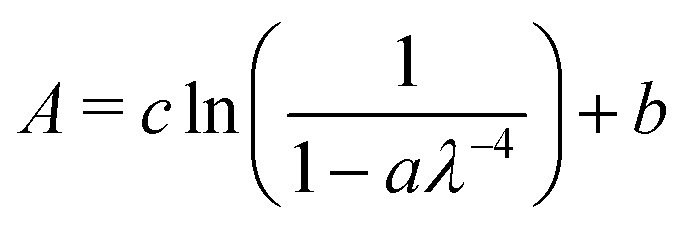 | (3) |
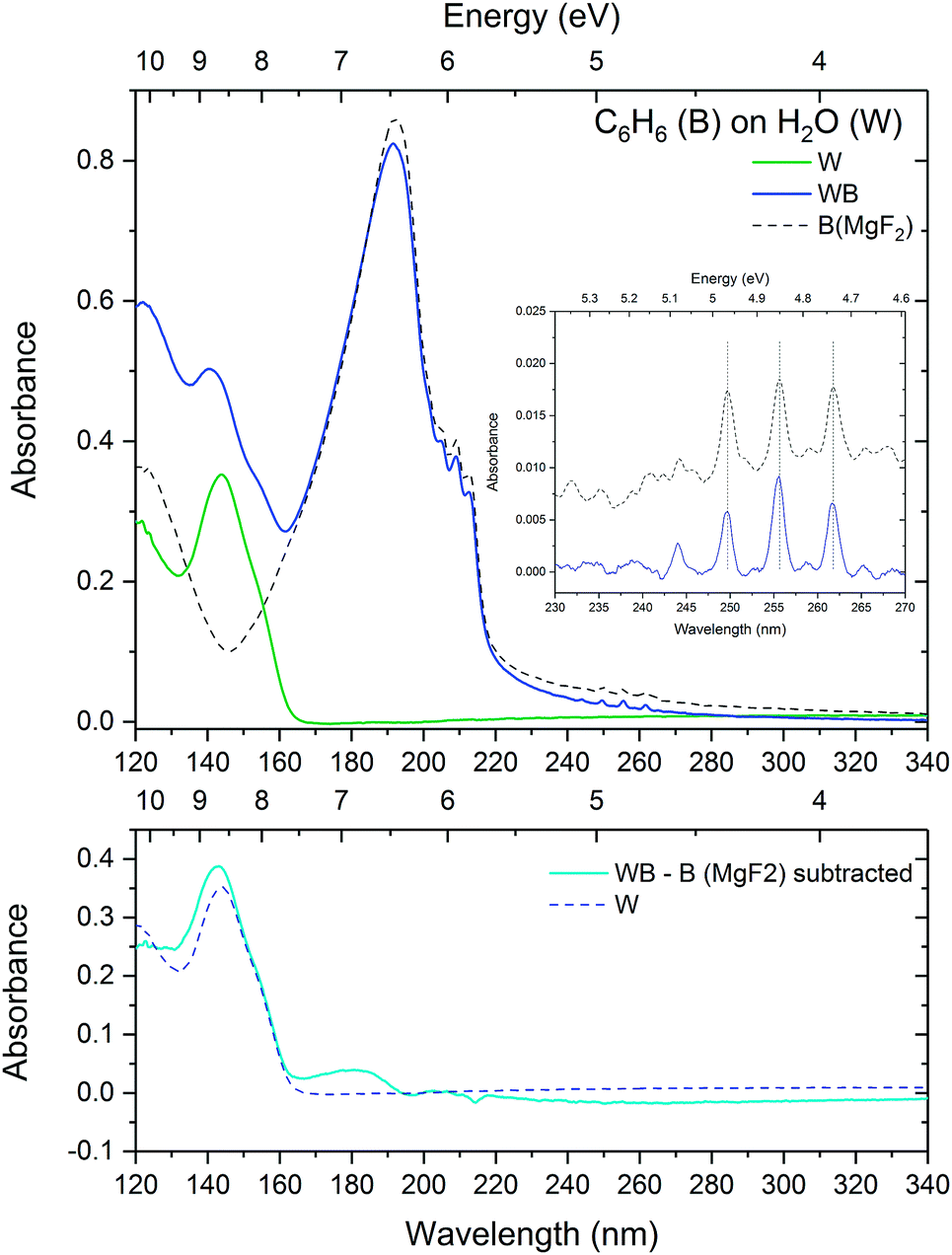 | ||
| Fig. 7 Top: VUV spectra of a layer of water (25 nm) deposited on a MgF2 substrate (green) at 25 K with a layer of benzene (24 nm) on top (blue) compared with benzene deposited on MgF2. The inset shows a zoom into the region of vibronic structure between 230 and 270 nm, baseline corrected for Rayleigh scattering. Bottom: Residual spectrum (solid line) after subtracting benzene on MgF2 from the layered spectrum together with the initially deposited water spectrum (dashed line). | ||
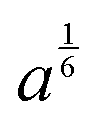 , proportional to the size of scatterers are given in the last column
, proportional to the size of scatterers are given in the last column
| Layer | Thickness (nm) | c | a (×109) |
|
|---|---|---|---|---|
| B | 22 | 0.0446 ± 0.0005 | 1.926 ± 0.009 | 35.3 |
| BW1 | 22(+17) | 0.0326 ± 0.0011 | 1.549 ± 0.030 | 34.0 |
| BW2 | 22(+17+33) | 0.0043 ± 0.0001 | 2.341 ± 0.001 | 36.4 |
| BW2 90 K | 0.0066 ± 0.0001 | 2.339 ± 0.001 | 36.4 | |
| BW2 160 K | 0.0486 ± 0.0016 | 1.484 ± 0.029 | 33.8 | |
| WB | 24 (+25) | 0.0368 ± 0.0002 | 2.173 ± 0.002 | 36.0 |
| WB 90 K | 0.0560 ± 0.0002 | 2.084 ± 0.003 | 35.7 | |
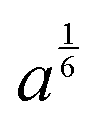
The vibronic bands of benzene deposited on water are slightly blueshifted by 0.1 nm with respect to benzene deposited on MgF2. As seen in Section 3.1.1, a blueshift in the vibronic bands is consistent with concentration dependent benzene–water mixing, suggesting strong evidence for benzene–water interaction at the benzene–water interface disrupting the benzene–benzene bonds. The bottom panel of Fig. 7 shows the residual spectrum after subtraction of a spectrum of benzene deposited on MgF2 together with the initially deposited water spectrum. The negative baseline above 220 nm is due to scattering effects. However what is also apparent is negative features corresponding to the vibronic bands of the 1B1u state. The positions of the negative features are redshifted with respect to the layered spectrum. This negative redshifted component translates to an enhanced blueshifted component of the vibronic bands and suggests an effect due to the benzene–water interaction consistent with the discussion in Section 3.1.1 where the benzene–benzene interaction results in a resdhift of the bands while the condensed phase benzene–water interaction causes a blueshift in the electronic states of benzene. The position of the positive feature near 180 nm in the residual is consistent with the energy of the 1E1u state in the dilute 1:100 benzene–water matrix. All these arguments point to strong evidence of a benzene–water interaction at the interface of the benzene–water layer. What is also interesting to note is the slightly blueshifted position of the water band 4a1: Ã1B1 ← 1b1: 1A1 transition from 143.8 nm as deposited to 142.2 nm in the residual. This is consistent with the blueshift in the water band observed in the 1:100 mixture of benzene:water (Section 3.1.1) relative to pure water and may hence be indicative of benzene–water interaction.
On annealing the layered benzene-on-water system (Fig. 8), benzene behaves in much the same way as benzene on a MgF2 substrate, crystallising by 90 K and desorbing between 140 and 150 K leaving the underlying layer of water behind. On crystallising at 90 K, the scattering results show little change in the size of the ‘clumps’ on the water surface.
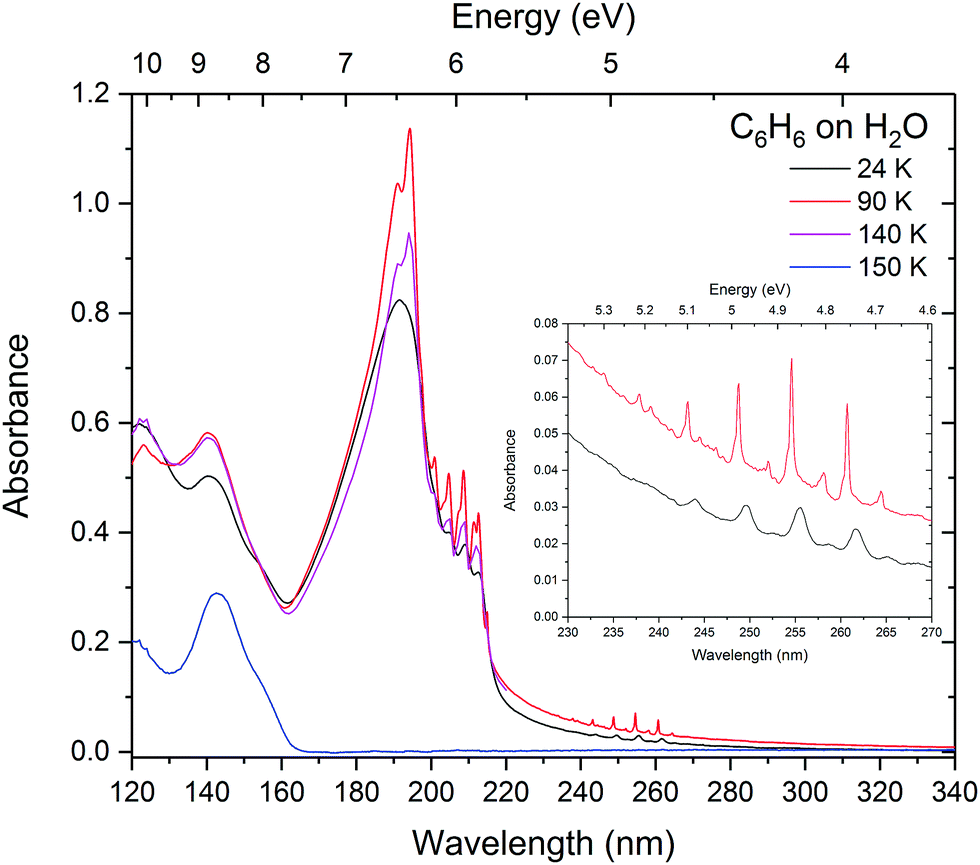 | ||
| Fig. 8 Layers of benzene on water annealed to 90, 140 and 150 K showing that benzene is completely desorbed by 150 K. | ||
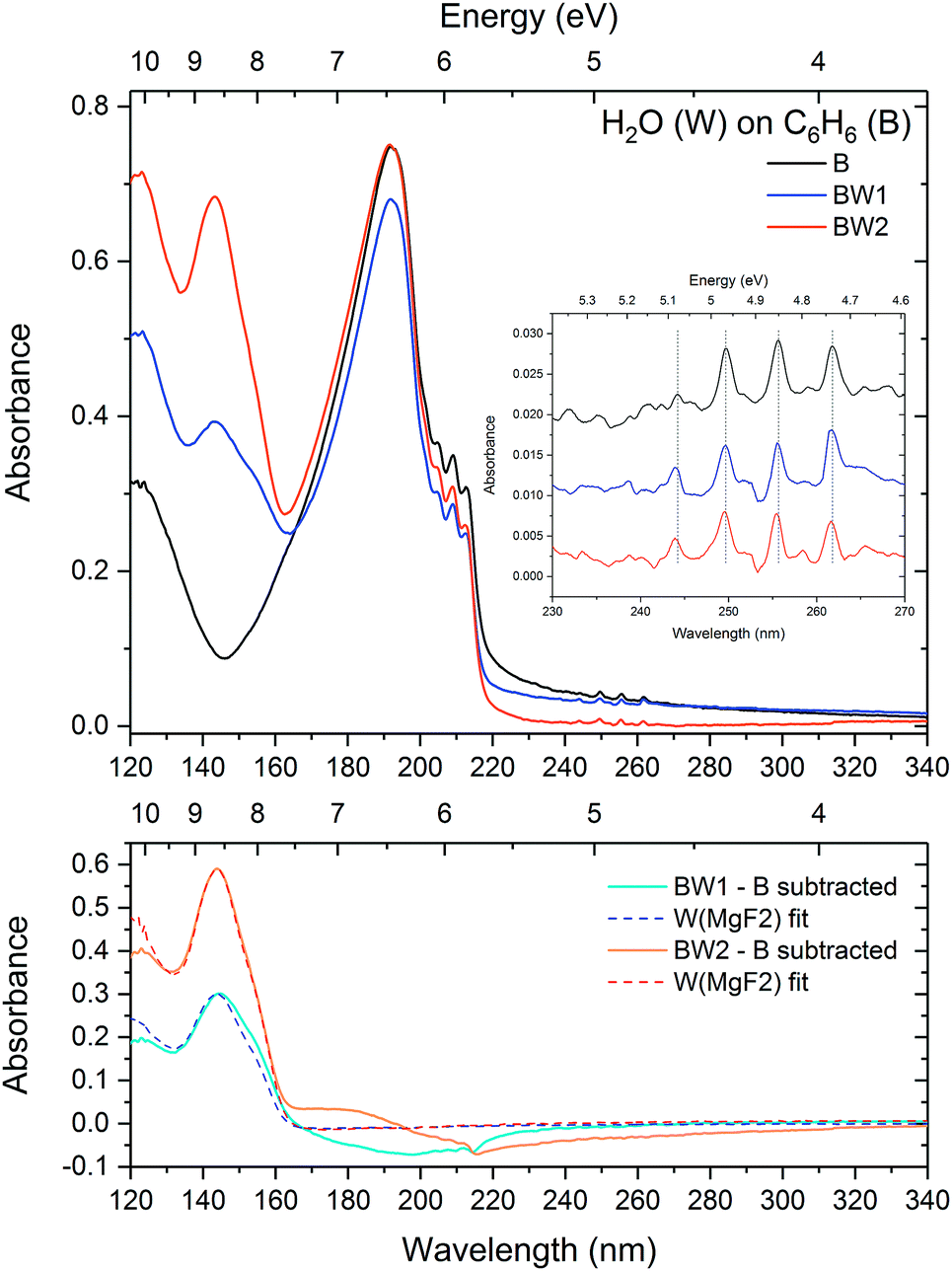 | ||
| Fig. 9 Top: VUV spectra of a layer of benzene (22 nm) deposited on a MgF2 substrate (black) at 25 K with two successive layers (17 and 33 nm) of water deposited on top (blue, orange). The inset shows a zoom into the region of benzene vibronic structure between 230 and 270 nm, baseline corrected for Rayleigh scattering for accurate peak position comparison. Bottom: Residual spectra (solid lines) after subtracting the initially deposited benzene on MgF2 spectrum, together with fits of water deposited on MgF2. | ||
The spectrum of pure benzene clearly exhibits a scattering tail at wavelengths greater than 220 nm, consistent with thickness dependent studies in AD17. It can clearly be seen that the first layer of water influences the scattering tail, reducing the scattering effect, and interestingly seems to reduce the intensity of the benzene peak. This could simply be an apparent effect due to lowering of the scattering baseline with the addition of the water overlayer. From the results of the scattering fits (Table 3) it can be seen that there is an apparent decrease in the clump size with the deposition of the first ‘sub-benzene’ layer of water. With deposition of the second layer of water the ‘clump’ size appears to increase, while the benzene peak intensity in the VUV spectrum remains unchanged. As little or no scattering is visible in the spectra of water deposited on MgF2, the water ice layer itself does not contribute to scattering in this spectral region, thereby suggesting the possibility that there may be some surface reconstruction effects taking place, changing the underlying benzene ‘clumps’ as water molecules cover and fill the gaps between the clumps. Alternatively, the apparent reduced ‘clump’ size in the ‘sub-benzene’ thickness regime could simply be due to water molecules ‘filling the gaps’ between the benzene ‘clumps’ exposing only the tops of the clumps in an iceberg effect. The clump size appears to increase again once the benzene clumps are completely covered with the second water deposition. This would suggest that on deposition water molecules prefer to stick between the benzene clumps rather than on top of them, suggesting a repulsive effect which is consistent with non-wetting in the case of benzene deposited on water.
Evidence of benzene–water interaction at the interface can clearly be seen in the residual spectra where the initial benzene spectrum has been subtracted from the benzene–water layered spectra as shown in the bottom of Fig. 9. As in the water-on-benzene system, aside from the negative Rayleigh scattering tails above 220 nm which is as a result of decrease in scattering with the introduction of the water layer, there are benzene negative and positive features visible in the region 160 to 215 nm. The negative peaks in the residual due to the 1B1u vibronic states are redshifted with respect to the original peaks in the layered spectra suggesting a loss in a low energy component, corresponding to an enhancement in a higher energy component. In fact a very slight blueshift can be discerned in the bands in the layered spectrum relative to the initially deposited benzene layer. Furthermore, there is also a slight blueshift of 0.1 and 0.2 nm seen in the 1B2u vibronic bands between 230 and 270 nm (inset of Fig. 9) after the first and second water deposition respectively. This is consistent with observed shifts in the benzene-on-water layered system discussed in Section 3.2.1 as well as our discussion of benzene:water mixtures (Section 3.1.1), where a blueshift in the bands with increasing water concentration is indicative of water molecules disrupting the benzene–benzene intermolecular interactions.
Though analysis of the VUV water band is not be possible due to the overlap of the water band with that of benzene, it is to be noted in the case of the subtracted spectrum for the first water overlayer (BW1) in Fig. 9 that there is also a slight change in the width (broadening) and a redshift of the water band compared to that of pure water ice deposited on MgF2. Interestingly the shift in this case is in the opposite direction to the blueshift observed in the 1:10 mixture (Section 3.1.1) and the benzene-on-water layered system (Section 3.2.1). It is unclear without a systematic investigation of the water band what the reason for this effect is. One suggestion may be that in the case of benzene on water, the water matrix structure is already established prior to benzene deposition following which the seemingly repulsive non-wetting benzene–water interaction (relative to water–water interaction) at the interface strengthens the bonds due to the water–water interaction, resulting in a blueshift (shifting away from the gas phase energy), raising the energy of the electronic state of water. In contrast, in the case of water on benzene the water molecules bind weakly to the surface benzene molecules during deposition, the weaker benzene–water interaction results in a redshift (shifting towards gas phase energy). As the water layer thickens the bulk water–water interaction dominates as evidenced by the shape and position of the water band in the residual spectrum returning to that of pure water ice with the second water deposition.
The layered samples were subsequently annealed to 90, 145 and 160 K (Fig. 10). Expected shifts and sharpening were observed in the benzene bands as for crystallisation of pure benzene at 90 K. Distinctive Davydov splitting and appearance of an exciton peak are observed indicating bulk crystallisation of benzene. Little change is observed in the scattering tail on annealing to 145 K. A decrease in intensity of both the main benzene peak and the water peak indicate that both benzene and water are beginning to desorb at 145 K as water begins to crystallise. Clearly benzene is trapped under the water ice, but also interestingly this suggests either that there is incomplete coverage of benzene with the water overlayer or that ‘gaps’ in the water layer have opened up as water molecules become mobile allowing some of the underlying benzene molecules to escape. What is more surprising is that both water and benzene continue to desorb at 160 K clearly indicating that some of the benzene still remains trapped under the water. There is also increased scattering with benzene ‘clumps’ becoming exposed as the water overlayer desorbs. There is no evidence of benzene and water mixing during this process.
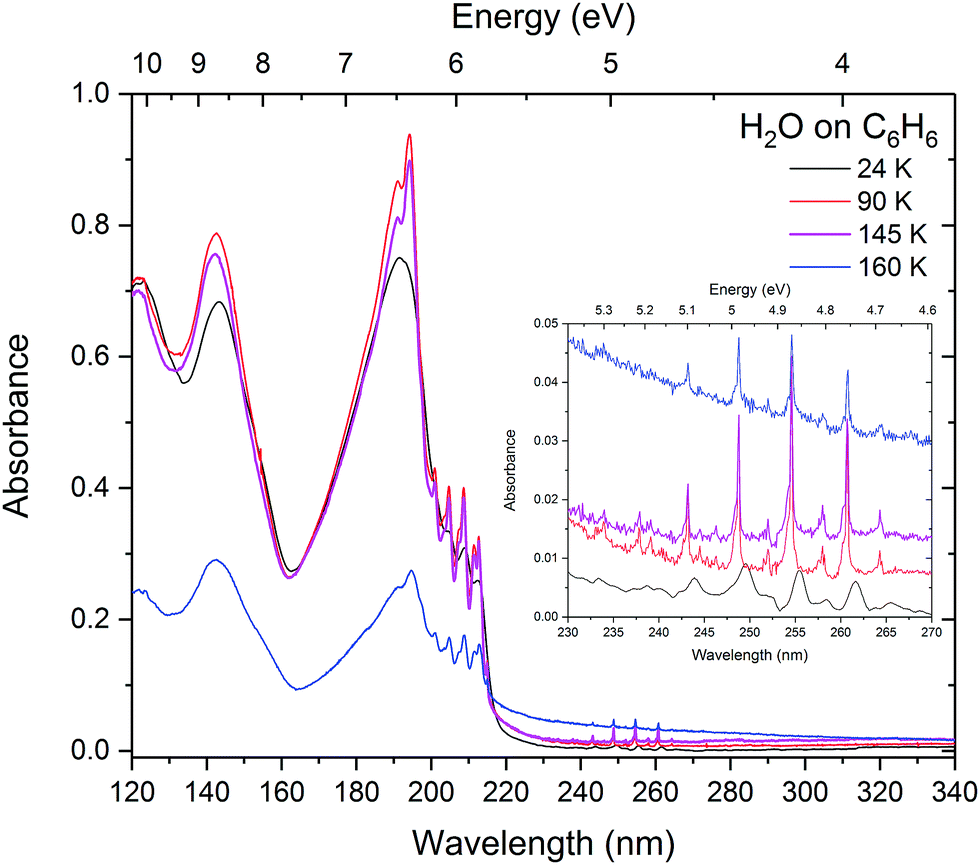 | ||
| Fig. 10 Layers of water on benzene annealed to 90, 145 and 160 K showing that benzene is trapped under water until water and benzene desorb together at 160 K. | ||
4 Summary and conclusions
We have investigated the interaction between benzene and water using vacuum ultraviolet and infrared spectroscopy of benzene:water mixtures and benzene/water layers. Our results suggest that though there is evidence of benzene(quadrupole)–water(dipole) hydrogen bonding interaction, the benzene–benzene and water–water interactions are stronger. Benzene concentration has a strong influence on the IR spectrum of water by disrupting the water hydrogen-bonding matrix, whilst water has little influence on the IR spectrum of benzene, though measurements with other ratios (with benzene in excess) and thicker samples to better resolve the benzene bands are needed to verify this. VUV spectra reveal that the benzene molecules behave more like free benzene molecules when diluted in water. Although there may be a small influence on the electronic states of benzene due to the benzene(quadrupole)–water(dipole) interaction, resulting in the redshift of the electronic bands of benzene with respect to the free benzene molecule, the effect of the benzene–benzene interaction is more pronounced with greater redshifts observed with increasing benzene concentration.
Benzene is a prototypical aromatic molecule and a building block of PAHs. The inclusion of aromatic molecules in astrochemical ices would influence their photochemistry due to intermolecular interactions. We have investigated the interaction of UV photons with pure benzene and benzene in mixtures with water. Clearly the intermolecular interaction between benzene and water and benzene and benzene has a significant effect on the excitation energies of benzene electronic states as seen from our concentration dependent VUV spectra. The high benzene cross section means that even in small quantities in the ice matrix benzene can have a significant influence on the photochemistry. For instance, the effect of benzene-facilitated desorption17 would be very much concentration dependent. Furthermore, it can be seen from comparison of VUV and IR spectra of benzene and water that even small amounts of benzene (1:100) that are virtually undetectable in the IR spectra, and are unlikely to be detected in observational spectra of ices in dense molecular clouds, have significant VUV cross sections and can contribute to photochemistry.
We calculated the cross sections of benzene for different concentrations of benzene and water ice mixtures. These values may suffer from errors of up to 20%, not unlike the infrared integrated absorption coefficients, or A-values, for molecular vibrational bands which suffer form uncertainties of up to 30%21 in the values quoted in the literature. Nevertheless, the results show a significant enhancement of the peak cross section intensity at lower concentrations, tending towards gas phase cross sections of benzene, with a peak gas phase cross section of 270 Mb and 225 Mb, 150 Mb and 75 Mb for 1:100, 1:10 and 1:1 solid phase mixtures of benzene and water respectively, compared with 47 Mb for pure solid benzene. Given the peak cross sections of smaller molecular constituents of grain mantles such as H2O, CO, NH3 and CO2 are below 10 Mb for most intense electronic transitions occurring below 6.2 eV (200 nm),22 the significance of inclusion of aromatic molecules such as benzene in ice mantles therefore cannot be neglected any less than the significant role gas phase PAHs play in photon dominated regions at the edges of dense molecular clouds. Inclusion and dilution of benzene and PAHs in the ice mantles would be of greater significance than such molecules acting as condensation nuclei since the cross sections tend towards the generous gas phase cross sections, enhancing the probability of photon absorption by one or two orders of magnitude over the surrounding matrix molecules. This would have significant implications for photochemical networks that would certainly need to include molecules such as benzene directly as well as consider the indirect processes such as benzene-facilitated desorption. Benzene benefits from UV active low energy (>200 nm) symmetry forbidden singlet–singlet electronic transitions due to spin–orbit coupling and intensity borrowing from a higher energy allowed 1E1u state. The lowest lying symmetry forbidden 1B2u state and its associated vibronic bands have substantial cross sections that are of similar order of magnitude to the other common grain mantle constituents. It has been demonstrated by Thrower et al. that resonant excitation of one of the vibronic bands at 250 nm and subsequent intersystem crossing and decay to the ground state transfers sufficient energy to the neighbouring water molecules causing them to desorb with high translational energy (450 K). These experiments were carried out using a layered benzene–water system, however it would be of great interest to investigate this effect in mixtures as a function of concentration. Our results show an enhancement in peak cross section of the vibronic bands at lower concentrations, increasing the probability of photon absorption by benzene in a region where most other small molecules have negligible cross sections. Further studies are required to assess whether there is any flux dependence when considering experimental work involving photon irradiation of ices to investigate photodesorption or photochemistry. For instance the benzene-mediated photodesorption studies of Thrower et al. were carried out using a photon flux of 4.5 × 1018 photons cm−2 s−1.
Our results from the layered benzene–water system reveal evidence of non-wetting at the benzene water interface in layered films. We see evidence of clumping and 3D island growth of benzene on water even at temperatures as low as 24 K where the mobility of molecules is low. Although the mechanism for clumping and island formation is not fully understood, the effect may have significant consequences on grain mantle growth on carbonaceous grains that on a molecular level may contain aromatic units with delocalised π electron clouds.44 The effect of benzene–water interaction at the benzene–water interface can clearly be seen in the spectra of layered samples, affecting both the benzene and the water electronic states. This effect would be significant at low coverages and may influence the binding and mobility of surrounding molecules. It can be seen in our results that benzene remains trapped in water ice at temperatures well above benzene desorption, co-desorbing with water molecules. Delayed desorption may allow benzene to be involved in further photochemical processing at higher temperatures.
Our combined VUV and IR results demonstrate the importance of systematic experimental work of binary mixtures in the condensed phase to elucidate the effect of intermolecular interactions on the intramolecular electronic states in order to fully understand photochemical processes in more realistic astrochemical ice environments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. P. and S. G. acknowledge support from The Open University. A. D. gratefully acknowledges the Daphne Jackson Trust for a Fellowship co-sponsored by Science and Technology Facilities Council and The Open University. A. D. also acknowledges the Leverhulme Trust for continuation of this work as part of her Early Career Fellowship. We kindly acknowledge the support of the European Community CALIPSO Infrastructure Programme for access to ASTRID2 Synchrotron Facility, Aarhus, Denmark.Notes and references
- V. Geers, E. Van Dishoeck, K. Pontoppidan, F. Lahuis, A. Crapsi, C. Dullemond and G. Blake, Astron. Astrophys., 2009, 495, 837–846 CrossRef.
- R. G. Smith, K. Sellgren and A. T. Tokunaga, Astrophys. J., 1989, 344, 413–426 CrossRef.
- T. Brooke, K. Sellgren and T. Geballe, Astrophys. J., 1999, 517, 883 CrossRef.
- J. Keane, A. Tielens, A. Boogert, W. Schutte and D. Whittet, Astron. Astrophys., 2001, 376, 254–270 CrossRef.
- D. P. Cruikshank, C. M. Dalle Ore, R. N. Clark and Y. J. Pendleton, Icarus, 2014, 233, 306–315 CrossRef.
- A. Li, Deep Impact as a World Observatory Event: Synergies in Space, Time, and Wavelength, Springer, 2009, pp. 161–175 Search PubMed.
- S. J. Clemett, C. R. Maechling, R. N. Zare, P. D. Swan and R. M. Walker, Science, 1993, 262, 721–725 Search PubMed.
- J. H. Hahn, R. Zenobi, J. L. Bada and R. N. Zare, Science, 1988, 239, 1523–1525 Search PubMed.
- S. A. Sandford, Planet. Space Sci., 2002, 50, 1145–1154 CrossRef.
- J. Bouwman, H. M. Cuppen, A. Bakker, L. J. Allamandola and H. Linnartz, Astron. Astrophys., 2010, 511, A33 CrossRef.
- M. S. Gudipati and L. J. Allamandola, Astrophys. J., Lett., 2004, 615, L177 CrossRef.
- M. S. Gudipati and L. J. Allamandola, Astrophys. J., 2006, 638, 286 CrossRef.
- S. H. Cuylle, L. J. Allamandola and H. Linnartz, Astron. Astrophys., 2014, 562, A22 CrossRef.
- J. Bouwman, D. Paardekooper, H. Cuppen, H. Linnartz and L. Allamandola, Astrophys. J., 2009, 700, 56 CrossRef.
- A. de Barros, A. Mattioda, A. Ricca, G. Cruz-Diaz and L. Allamandola, Astrophys. J., 2017, 848, 112 CrossRef PubMed.
- J. Noble, C. Jouvet, C. Aupetit, A. Moudens and J. Mascetti, Astron. Astrophys., 2017, 599, A124 CrossRef.
- J. Thrower, D. Burke, M. Collings, A. Dawes, P. Holtom, F. Jamme, P. Kendall, W. Brown, I. Clark and H. Fraser, et al. , Astrophys. J., 2008, 673, 1233 CrossRef.
- S. Viti, M. P. Collings, J. W. Dever, M. R. McCoustra and D. A. Williams, Mon. Not. R. Astron. Soc., 2004, 354, 1141–1145 CrossRef.
- A. Dawes, N. Pascual, S. V. Hoffmann, N. C. Jones and N. J. Mason, Phys. Chem. Chem. Phys., 2017, 19, 27544–27555 RSC.
- M. H. Palmer, T. Ridley, S. V. Hoffmann, N. C. Jones, M. Coreno, M. De Simone, C. Grazioli, M. Biczysko, A. Baiardi and P. Limão-Vieira, J. Chem. Phys., 2015, 142, 134302 CrossRef PubMed.
- A. Dawes, N. J. Mason and H. J. Fraser, Phys. Chem. Chem. Phys., 2016, 18, 1245–1257 RSC.
- N. J. Mason, A. Dawes, P. D. Holtom, R. J. Mukerji, M. P. Davis, B. Sivaraman, R. I. Kaiser, S. V. Hoffmann and D. A. Shaw, Faraday Discuss., 2006, 133, 311–329 RSC.
- K. S. Kim, J. Y. Lee, H. S. Choi, J. Kim and J. H. Jang, Chem. Phys. Lett., 1997, 265, 497–502 CrossRef.
- D. Feller, J. Phys. Chem. A, 1999, 103, 7558–7561 CrossRef.
- S. Li, V. R. Cooper, T. Thonhauser, A. Puzder and D. C. Langreth, J. Phys. Chem. A, 2008, 112, 9031–9036 CrossRef PubMed.
- M. B. Robin, in Higher Excited States of Polyatomic Molecules, ed. M. B. Robin, Academic Press, 1975, pp. 209–268 Search PubMed.
- D. Sharma, W. Sameera, S. Andersson, G. Nyman and M. J. Paterson, ChemPhysChem, 2016, 17, 4079–4089 CrossRef PubMed.
- J. Thrower, M. Collings, F. Rutten and M. McCoustra, J. Chem. Phys., 2009, 131, 244711 CrossRef PubMed.
- W. Hagen, A. Tielens and J. Greenberg, Astron. Astrophys., Suppl. Ser., 1983, 51, 389–416 Search PubMed.
- S. Silva and J. P. Devlin, J. Phys. Chem., 1994, 98, 10847–10852 CrossRef.
- A. Engdahl and B. Nelander, J. Phys. Chem., 1985, 89, 2860–2864 CrossRef.
- G. Kearley, M. Johnson and J. Tomkinson, J. Chem. Phys., 2006, 124, 044514 CrossRef PubMed.
- S. Bahr and V. Kempter, J. Chem. Phys., 2007, 127, 074707 CrossRef PubMed.
- V. Chandrasekaran, L. Biennier, E. Arunan, D. Talbi and R. Georges, J. Phys. Chem. A, 2011, 115, 11263–11268 CrossRef PubMed.
- E. R. Bernstein, S. Colson, R. Kopelman and G. Robinson, J. Chem. Phys., 1968, 48, 5596–5610 CrossRef.
- A. Bernhardsson, N. Forsberg, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, J. Chem. Phys., 2000, 112, 2798–2809 CrossRef.
- S. D. Colson and E. R. Bernstein, J. Chem. Phys., 1965, 43, 2661–2669 CrossRef.
- C. J. Gruenloh, J. R. Carney, C. A. Arrington, T. S. Zwier, S. Y. Fredericks and K. D. Jordan, Science, 1997, 276, 1678–1681 CrossRef.
- J. D. Thrower, et al., PhD thesis, Citeseer, 2009.
- D. Marchione, PhD thesis, Heriot-Watt University, 2015.
- D. Marchione and M. R. McCoustra, Phys. Chem. Chem. Phys., 2016, 18, 20790–20801 RSC.
- J. A. Noble, C. Martin, H. J. Fraser, P. Roubin and S. Coussan, J. Phys. Lett., 2014, 5, 826–829 Search PubMed.
- U. Erlekam, M. Frankowski, G. Meijer and G. von Helden, J. Chem. Phys., 2006, 124, 171101 CrossRef PubMed.
- S. Kwok, Nature, 2004, 430, 985 CrossRef PubMed.
| This journal is © the Owner Societies 2018 |