
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microsolvated complexes of ibuprofen as revealed by high-resolution rotational spectroscopy†
P.
Pinacho
 a,
A.
Krin
bcd,
C.
Pérez
bcd,
S.
Zinn
bcd,
J. C.
López
a,
S.
Blanco
*a and
M.
Schnell
*bcd
a,
A.
Krin
bcd,
C.
Pérez
bcd,
S.
Zinn
bcd,
J. C.
López
a,
S.
Blanco
*a and
M.
Schnell
*bcd
aDepartamento de Química Física y Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid, 47011 Valladolid, Spain. E-mail: sblanco@qf.uva.es
bDeutsches Elektronen-Synchrotron, Notkestrasse 85, 22607 Hamburg, Germany. E-mail: melanie.schnell@desy.de
cMax-Planck Institut für Struktur und Dynamik der Materie, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: melanie.schnell@mpsd.mpg.de
dChristian-Albrechts-Universität zu Kiel, Institute of Physical Chemistry, Max-Eyth-Strasse 1, 24118 Kiel, Germany
First published on 3rd May 2018
Abstract
Hydrogen-bonded complexes between ibuprofen and water generated in a supersonic expansion were characterized using chirped-pulse Fourier transform microwave spectroscopy in the 2–8 GHz frequency range. Four spectra were observed allowing the determination of their rotational parameters. Comparison with quantum-chemical calculations led to their identification as the lowest energy 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ibuprofen–water complexes. These correspond to the complexes between water and the four different conformers of ibuprofen previously detected in the gas phase, owing to their similar stabilization energies and abundances. Water seems to not change the conformational distribution of ibuprofen.
:1 ibuprofen–water complexes. These correspond to the complexes between water and the four different conformers of ibuprofen previously detected in the gas phase, owing to their similar stabilization energies and abundances. Water seems to not change the conformational distribution of ibuprofen.
1 Introduction
(RS)-2-(4-(2-Methylpropyl)phenyl)propanoic acid, best known as ibuprofen (Scheme 1), is one of the most used non-steroidal anti-inflammatory medicines.1,2 Ibuprofen has a polar end, the carboxylic acid group, which can act both as a hydrogen donor and a hydrogen acceptor establishing moderate to strong interactions with other polar molecules. The other part of ibuprofen is non-polar. Ibuprofen has a high solubility in organic solvents, such as acetone or propanol, but exhibits a low solubility in water.3 It should be noted that ibuprofen has a chiral center (marked atom in Scheme 1), the α-carbon atom, connecting the carboxylic acid and the phenyl ring. It is reported that the S-enantiomer of ibuprofen has a higher pharmacological activity than the R-enantiomer.4 In mammals, an enzyme of the alpha-methylacyl-CoA racemase family transforms in vivo the less-active R to the active S form. This conversion is of importance because it allows ibuprofen to be distributed as a racemate.4,5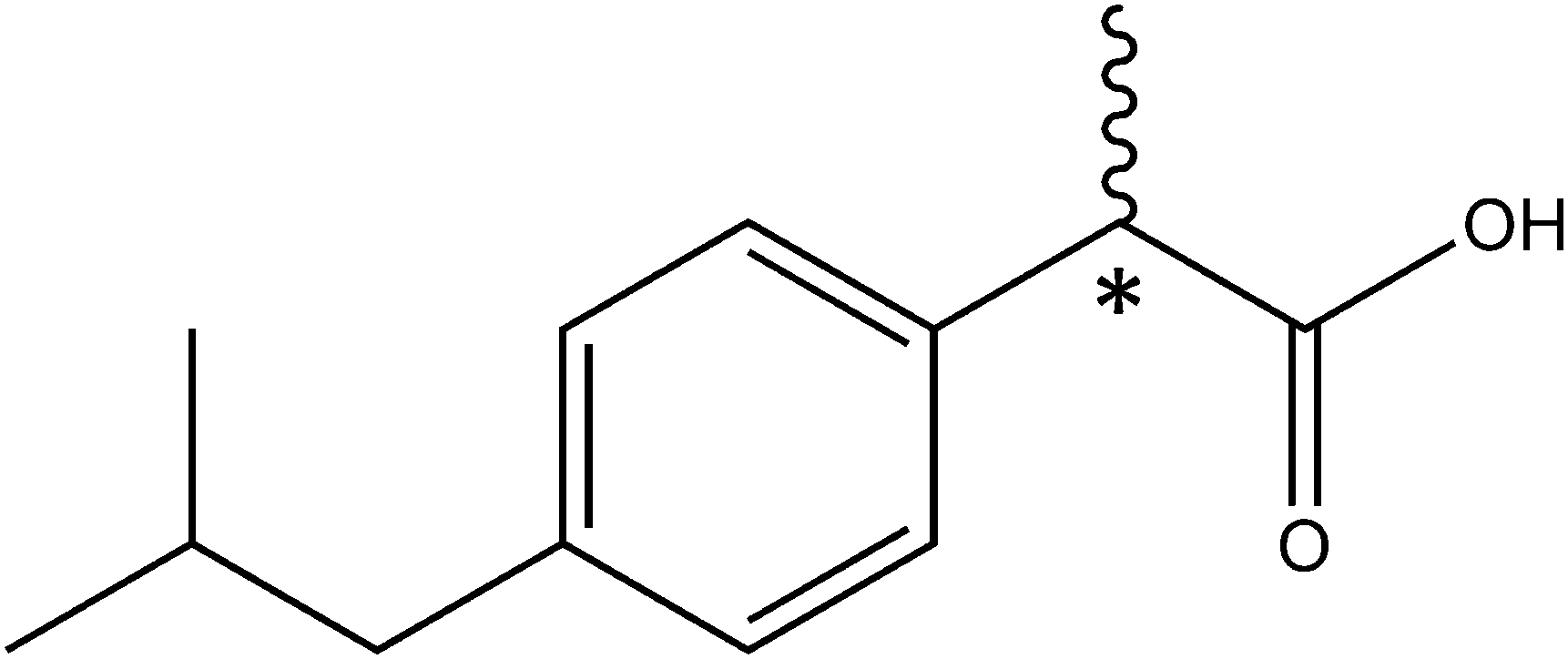 | ||
| Scheme 1 Ibuprofen (IUPAC: (RS)-2-(4-(2-methylpropyl)phenyl)propanoic acid). The asterisk denotes the stereogenic center. | ||
The anti-inflammatory properties of ibuprofen arise from inhibiting the formation of prostaglandins, the molecules responsible for several pathological processes such as cellular inflammation or pain sensitization, by blocking the catalytic active site of cyclooxygenase (COX) enzymes6 that convert arachidonic acid into prostaglandin PGH2,7 the precursor of various other prostaglandins. This blocking occurs through the interaction of the carboxyl group of ibuprofen with the amino acid residues inside the enzymatic channel.
It is well known that water molecules form a part of the structures of biological macromolecules,8 playing a decisive role in many biological processes such as protein folding,9 conformational10 or tautomeric11 equilibria among others. Water molecules are also present inside the cyclooxygenase channels and seem to play an important structural role through water-mediated interactions between different amino acid residues.7 However, the extent to which water molecules contribute to the inhibition on COX enzymes has not been shown. Furthermore, addition of water might influence the structures of the respective solute molecules.12 This makes it interesting to study the interactions between ibuprofen and water.
A better knowledge of the solute–solvent interactions can be achieved by studying the microsolvated solute complex in the isolated environment of a supersonic jet, where other interactions occurring in condensed phases are not present. Those complexes can be considered as the first steps of the solvation process. Microsolvated complexes with different degrees of hydration formed in a supersonic expansion can be characterized using high-resolution rotational spectroscopy.
Complexes of different kinds of biologically important molecules with water have been already studied, contributing relevant information about solute–solvent interactions in those systems.13 For example, it has been recently shown that microsolvation may affect not only the structure of the solute,12 but also its conformational preferences as observed for the 12C4 crown ether.13d
The ibuprofen monomer has already been studied in the gas phase using high-resolution chirped-pulse Fourier transform microwave (CP-FTMW) spectroscopy14 to elucidate its structure.15 Four conformers were found which differ in the arrangement of the flexible isobutyl group. Their structures are closely related and share the same kind of stabilization forces, which explains why the spectra of the four conformers showed similar intensities, pointing to comparable populations in the molecular beam.
In the present work, we studied the microwave spectrum of ibuprofen in order to characterize the microsolvated complexes generated in the supersonic expansion and analyze the stabilization forces involved in their formation. Different possibilities for the interaction between ibuprofen and water have been explored, which may serve as a model for the hydrogen bond interactions taking place inside the COX enzymes. An important aspect in this case is the possible influence of water on the conformational landscape of ibuprofen.
2 Experimental and theoretical methods
Broadband Fourier transform microwave spectroscopy
The study of the microsolvated complexes of ibuprofen was carried out using the Hamburg broadband chirped-pulse Fourier transform microwave (CP-FTMW) spectrometer COMPACT.16 Ibuprofen was purchased (Sigma-Aldrich 98% chemical purity, melting point 76 °C) and used without further purification. Ibuprofen was held in a reservoir and heated to 115 °C. Its vapour was mixed with the carrier gas (Ne, 3 bar stagnation pressure) before the supersonic expansion into the vacuum chamber. No water was added to the experiment, which lets us assume that the ibuprofen–water complexes were formed from water molecules remaining in the carrier gas and the gas tubes. Another source might be the crystallization water present in the commercial sample.The complexes were excited using a 4 μs long linear frequency chirp covering the range from 2 to 8 GHz (300000 acquisitions). The chirp was amplified by a TWT amplifier (300 W power) and broadcast into the vacuum chamber using a horn antenna. The free induction decay (FID) emission signal was recorded for 40 μs and Fourier transformed to the frequency domain, resulting in a frequency resolution of 25 kHz. The fast-frame option was applied.17
Quantum-chemical calculations
Prior to the analysis of the microwave spectrum, the potential energy surface (PES) for the microsolvation of ibuprofen was explored using quantum-chemical calculations18 at the B3LYP-D3/aug-cc-pVDZ level of theory.19,20 Grimme's empirical dispersion correction (GD3)21 has been used to obtain more accurate predictions. Based on the resulting structures, further optimizations were performed at the MP2/6-311++G(d,p) level of theory.22,23 From the PES study, optimized geometries for several ibuprofen–water complexes were obtained and used to predict their spectra on the basis of the calculated rotational constants and electric dipole moment components.3 Results and discussion
Ibuprofen–water complexes potential energy surface
As mentioned previously, the carboxyl group is the only polar group present in ibuprofen. It can present stabilizing hydrogen bond interactions with the water molecule. For each of the observed conformers of ibuprofen (C1, C2, C3 and C4)15 different possibilities for the interactions with one water molecule have been explored (Fig. 1 and Fig. S2–S4, ESI†). Table 1 summarizes the rotational parameters obtained at the B3LYP-D3/aug-cc-pVDZ level of theory. Additional calculated rotational parameters are presented in Table S2 (ESI†). The results of the MP2/6-311++G(d,p) calculations are given in Table S3 (ESI†). Both levels of theory led to comparable results with respect to the relative stability of the complexes and their structures. The results from the B3LYP-D3/aug-cc-pVDZ level of theory are given here since this level requires less computational resources and seems to match slightly better with the experimental rotational parameters than the MP2/6-311++G(d,p).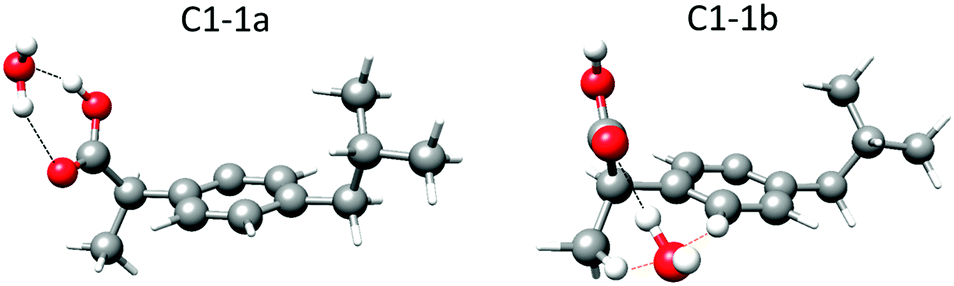 | ||
| Fig. 1 Lower energy complexes predicted at the B3LYP-D3/aug-cc-pVDZ level of theory for 1:1 ibuprofen–water complexes in the C1 conformation of the ibuprofen molecule showing the hydrogen bonds (black dashed lines) and the secondary weak interactions (orange dashed lines). The hydrogen atoms from the water molecule and those involved in long-range interactions are displayed as balls. The other hydrogen atoms are displayed as sticks. The corresponding parameters are given in Table 1 and Table S2 in the ESI.† | ||
:1 ibuprofen–water complexes
| Parametera | C1-1a | C2-1a | C3-1a | C4-1a | C1-1b | C2-1b | C3-1b | C4-1b |
|---|---|---|---|---|---|---|---|---|
| a A, B and C are the rotational constants. κ is the asymmetry parameter derived from the rotational constants; κ = (2B − A − C)/(A − C). μα (α = a, b or c) are the electric dipole moment components, 1 D ≈ 3.33 × 10−30 C m. ΔE is the energy relative to the most stable complex. De is the dissociation energy calculated using BSSE corrections.27 | ||||||||
| A/MHz | 851.9 | 865.2 | 1148.3 | 1135.2 | 860.9 | 880.0 | 966.4 | 954.5 |
| B/MHz | 223.4 | 225.0 | 195.1 | 196.6 | 232.3 | 229.5 | 224.4 | 223.2 |
| C/MHz | 200.0 | 198.6 | 188.1 | 186.8 | 219.2 | 219.0 | 204.4 | 205.7 |
| κ | −0.93 | −0.92 | −0.98 | −0.98 | −0.96 | −0.97 | −0.95 | −0.95 |
| μ a/D | −1.88 | −1.92 | −1.88 | −1.84 | 0.51 | 0.59 | 0.09 | 0.07 |
| μ b/D | −0.25 | −0.19 | 0.51 | 0.57 | 1.67 | 1.62 | −1.94 | −1.63 |
| μ c/D | 0.16 | −0.03 | 0.09 | 0.05 | 1.06 | 1.40 | −0.52 | −0.87 |
| ΔE/cm−1 | 0 | 27 | 32 | 9 | 1165 | 1175 | 1146 | 1167 |
| D e/cm−1 | −4016 | −4020 | −4018 | −4017 | −2722 | −2708 | −2706 | −2721 |
| D e/kJ mol−1 | −48 | −48 | −48 | −48 | −33 | −32 | −32 | −33 |
In the complexes labeled as 1a, the water molecule closes a sequential cycle with the carboxyl group acting both simultaneously as hydrogen donors and acceptors and thus forming two O–H⋯O hydrogen bonds. This is the most stable arrangement for the interaction between an acid group and one water molecule, as reported before in detail.24 The calculated relative energies for the four complexes are predicted to be in a narrow range (within 32 cm−1 ≈ 0.4 kJ mol−1), similar to the monomer conformers.15 However, the calculated stability ordering of the complexes is not the same as that calculated for the corresponding monomer forms. Altogether, this makes it impossible to discriminate whether one of the complexes is more stable than the others (see discussion on conformer population below).
In the complexes labeled as 1b, the water molecule establishes an O–H⋯O hydrogen bond to the carbonyl oxygen, which acts as a hydrogen bond acceptor, an O⋯H–C secondary weak interaction with the hydrogen atom in the methyl group in the β-position, and an O⋯H–C secondary weak interaction with the closest hydrogen atom in the phenyl ring (Fig. 1). The calculated energy difference between the four 1b complexes is within 30 cm−1 or 0.35 kJ mol−1. The energy difference between 1a and 1b conformers is approximately 1150 cm−1 or 14 kJ mol−1 (Table 1).
Microwave spectrum
The recorded broadband microwave spectrum is very dense and dominated by the rotational transitions of the four monomer conformers,15 which show the characteristic patterns of R-branch μa-type transitions (Fig. 2a). Once we excluded the monomer lines, new sets of R-branch μa-type transitions could be identified (Fig. 2b). Detailed analysis allowed the assignment25 of four different species (labeled I, II, III and IV). No μb- or μc-type lines associated with these species were observed. The spectra were fit using a semirigid rotor Hamiltonian in the asymmetric reduction and in the Ir representation.26 The obtained rotational parameters are given in Table 2. The complete list of measured frequencies for these complexes is collected in Tables S6–S9 in the ESI.† In accordance with the previous work,15 the lines do not present splittings attributable to the internal rotation of the methyl groups of ibuprofen, or to tunneling associated to water motions.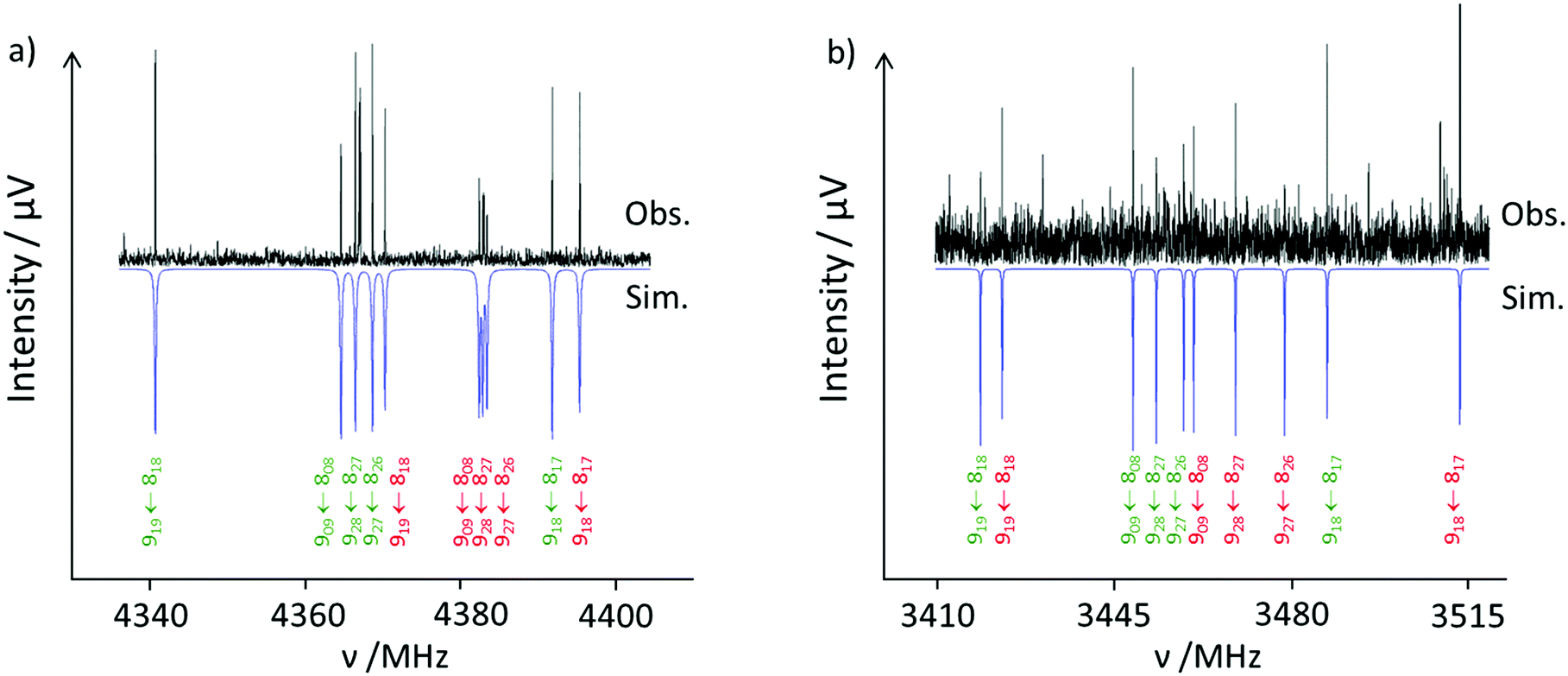 | ||
| Fig. 2 Excerpt of the ibuprofen microwave spectrum. (a) Observed (upwards) and simulated (downwards) spectra showing the rotational transitions 919 ← 818; 909 ← 808; 928 ← 827; 927 ← 826 and 918 ← 817 for the ibuprofen monomer conformers C3 (green) and C4 (red). (b) Observed (upwards) and calculated (downwards) spectra showing the rotational transitions 919 ← 818; 909 ← 808; 928 ← 827; 927 ← 826 and 918 ← 817 for ibuprofen–water complexes C3-1a (green) and C4-1a (red). The relative intensity between the monomer and the complex spectra is more than 20:1. | ||
:1 ibuprofen–water complexes observed
| Fitted parametersa | I (C1-1a) | II (C2-1a) | III (C3-1a) | IV (C4-1a) |
|---|---|---|---|---|
| a A, B and C are the rotational constants. ΔJ and ΔJK are quartic centrifugal distortion constants. N is the number of rotational transitions fitted. σ is the rms deviation of the fit. b Standard errors are given in parentheses in units of the last digits. | ||||
| A/MHz | 851.363(28)b | 872.598(39) | 1156.72(18) | 1129.39(11) |
| B/MHz | 225.08507(24) | 225.15642(29) | 195.75673(18) | 197.90286(19) |
| C/MHz | 200.49914(23) | 198.35992(35) | 188.10105(17) | 187.77533(22) |
| Δ J/kHz | 0.01832(73) | 0.02003(98) | 0.00732(48) | 0.00661(57) |
| Δ JK/kHz | −0.115(18) | −0.096(14) | −0.0650(66) | −0.0696(67) |
| N | 47 | 39 | 50 | 55 |
| σ/kHz | 6.5 | 6.7 | 4.4 | 5.2 |
A plausible identification of the complexes giving rise to each observed rotational spectrum can be achieved by analyzing the experimental rotational parameters and comparing them with those predicted for the different conformers based on quantum-chemical calculations. The observation of only μa-type spectra allowed excluding the 1b complexes since they are predicted to have high μb values (Table 1) and thus should have sizable b-type transitions. This is also plausible because of their high energy difference of 14 kJ mol−1.
A first look at the rotational parameters in Table 2 allows classifying the complexes into two families corresponding to the different values of the A rotational constant for the complexes. The first family comprises species I and II with A values around 850–870 MHz and very similar values of B and C rotational constants. This family can be correlated with the predicted conformers C1-1a and C2-1a in which the isobutyl group of ibuprofen is on the same side of the phenyl ring as the carboxyl group (see Fig. 3), i.e., in a cis arrangement. The values of the A rotational constant for rotamers I and II are sufficiently distinct to assign species I to C1-1a and species II to C2-1a, respectively.
 | ||
| Fig. 3 Observed 1:1 ibuprofen–water complexes showing the different orientations of the isobutyl group that arise from the four different monomer conformers. | ||
The second family comprises species III and IV with A values between 1120 and 1160 MHz and again with very similar values of B and C rotational constants. They can be related to the forms C3-1a and C4-1a, having the isobutyl and the carboxyl groups on opposite sides of the phenyl ring (see Fig. 3), i.e., in a trans arrangement. Again, the difference in the A rotational constant seems to correlate rotamers III and IV to forms C3-1a and C4-1a, respectively. The differences in orientation of the isobutyl group from conformer C1-1a to C2-1a and from C3-1a to C4-1a cause small shifts in the values of the rotational constants, which can be used to further identify the rotamers.
Another corroboration of the assignment can be obtained by attending to the planar moments of inertia, derived from the inertial moments [Pαα = (Iββ + Iγγ − Iαα)/2 (α, β, γ = a, b or c)]. These provide information about the mass extension out of a given inertial plane. For example, Pcc reflects the mass extension out of the ab inertial plane. Table 3 compares the experimental and calculated values of the planar moments of inertia. This comparison corroborates the identification done based on the rotational constants. The trends in the variation of the experimental planar moment values for species I and II or for species III and IV are well reproduced by the theoretical values.
:1 ibuprofen–water complexes (I–IV) compared with the predicted values calculated at the B3LYP-D3/aug-cc-pVDZ level of theory
| Experimental | I | II | III | IV |
|---|---|---|---|---|
| P aa/μÅ2 | 2086.136(20) | 2106.595(26) | 2415.751(68) | 2398.797(44) |
| P bb/μÅ2 | 434.468(20) | 441.192(26) | 270.990(68) | 292.605(44) |
| P cc/μÅ2 | 159.143(20) | 137.973(26) | 165.916(68) | 154.874(44) |
| B3LYP-D3 | C1-1a | C2-1a | C3-1a | C4-1a |
|---|---|---|---|---|
| P aa/μÅ2 | 2101.17 | 2103.35 | 2418.50 | 2415.43 |
| P bb/μÅ2 | 428.19 | 441.34 | 268.25 | 290.02 |
| P cc/μÅ2 | 164.25 | 142.76 | 171.85 | 155.16 |
No isotopic information on the ibuprofen–water complexes has been obtained, since the signal to noise (S/N) level for the complexes was approximately 4:1. Thus, it was not possible to acquire further information on their structures. However, given the agreement between the observed and the theoretical rotational constants, the predicted structures can be taken as a reasonable description of the geometries of the observed species.
The main difference between the detected forms of ibuprofen–water complexes is the orientation of the isobutyl group of the ibuprofen molecule with respect to the phenyl ring, similar to the corresponding monomers, and not in the arrangement of the water molecule. As mentioned before, the interaction occurs through the two hydrogen bonds between the carboxyl group of ibuprofen and the water molecule (Fig. 2). The calculated distances and angles for the hydrogen bonds are very similar for the four complexes and are of the same order as those reported for other complexes between an acid group and one water molecule (Table S4 in ESI†).24 The interaction between water and the acid group can thus be interpreted to be nearly identical for the four conformers.
Theoretical calculations predict that in the case of the four observed conformers, the formation of the complex does not drive any significant conformational or structural change. In fact, the rotational constants of the monomer and those calculated assigning zero mass to water in the complex are almost identical (Table S5 in ESI†).
Conformer line intensities
The intensities of the observed spectra of the ibuprofen–water complexes are approximately equal. Those intensities are assumed to be proportional to Ni·μi,α2, with Ni being the number density of species i in the supersonic jet, and μi,α the corresponding electric dipole moment component, in this case μa. Since this dipole moment component is predicted to have similar values (μa ≈ −1.9 D, Table 1) for the four complexes, their population in the supersonic jet can be deduced to be similar. Complex formation takes place in the expansion region close to the nozzle orifice where the rate of three-body collisions is high enough. Once formed, the complexes are cooled down to their ground vibrational state as the supersonic expansion progresses. It can be reasonably assumed that all the ibuprofen molecules existing in the jet have the same probability to form the corresponding 1a complexes with water. As observed previously,15 all four monomer conformers are present in the jet with similar populations. Thus, our observation that the four 1a complexes have similar relative populations and thus almost equal intensities is a consistent result. Furthermore, given that in all the complexes the interaction with water is the same, it is possible to assume the same relative energy for them as for the monomer conformers. This result is not in contrast with the different energy order predicted by theoretical calculations. The predicted energy differences are smaller than the accuracy limits of common theoretical methods.Dissociation energies
Table 1 gives the dissociation energies of the complexes calculated using the basis set superposition error (BSSE) correction.27 There is practically no difference between the dissociation energies obtained for the four 1a complexes, which is another confirmation that the hydrogen bonds established between water and ibuprofen have the same energies for the four complexes. The same behavior can be seen for the four 1b complexes with almost equal dissociation energies. There is a difference in the dissociation energy between the 1a and 1b complexes of ca. 15.5 kJ mol−1, however, implying that the 1a complexes are more tightly bound. If the dissociation energy of a complex is related to its survival probability in the first stages of the supersonic expansion, this difference could explain why only the 1a complexes have been detected.4 Conclusions
The microwave spectra of four different complexes between ibuprofen and water formed in a supersonic jet were observed using CP-FTMW spectroscopy. The good agreement between quantum-chemically predicted and experimentally determined rotational parameters led to the identification of the observed species as the four lower energy forms of the complex between ibuprofen and water. In the four cases, the water molecule closes a sequential cycle with the carboxyl group establishing two hydrogen bonds. The main structural difference between the observed forms lies in the conformation of ibuprofen that corresponds to those structures reported before.15 Water does not drive any conformational change in the ibuprofen molecule and essentially does not modify energetically the distribution of conformers from the monomer. In other words, the interaction of the carboxyl group with water does not alter the conformational landscape of ibuprofen, which solely arises from the different orientations of the isobutyl group. This result might be important with respect to transporting the molecule to the COX enzyme and its molecular recognition. The conservation of the ibuprofen flexibility when the carboxyl group of ibuprofen interacts with other polar groups resulting in a cyclic interaction structure might be essential in the recognition process.Conflicts of interest
Authors declare that there are no conflicts of interest.Acknowledgements
J. C. L., S. B., and P. P. acknowledge the Ministerio de Economía y Competitividad (Grants CTQ2016-75253-P and CTQ2013-40717-P) and the Junta de Castilla y León (Grant VA334U14) for financial support. P. P. acknowledges the University of Valladolid for a mobility grant. Open Access funding provided by the Max Planck Society.Notes and references
- K. D. Rainsford, Int. J. Clin. Pract., 2013, 67, 9–20 CrossRef PubMed.
- World Health Organization model list of essential medicines: http://www.who.int/medicines/publications/essentialmedicines/en/.
- M. A. Filippa and E. I. Gasull, Fluid Phase Equilib., 2013, 354, 185–190 CrossRef.
- T. J. Woodman, P. J. Wood, A. S. Thompson, T. J. Hutchings, G. R. Steel, P. Jiao, M. D. Threadgill and M. D. Lloyd, Chem. Commun., 2011, 47, 7332–7334 RSC.
- (a) K. D. Rainsford, Inflammopharmacology, 2009, 17, 275–342 CrossRef PubMed; (b) G. Q. Lin, Q. D. You and J. F. Cheng, Chiral drugs: Chemistry and Biological Action, John Wiley & Sons, Hoboken, New Jersey, 2011 Search PubMed.
- (a) G. Dannhardt and W. Kiefer, Eur. J. Med. Chem., 2001, 36, 109–126 CrossRef PubMed; (b) V. Limongelli, M. Bonomi, L. Marinelli, F. L. Gervasio, A. Cavalli, E. Novellino and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5411–5416 CrossRef PubMed.
- (a) B. S. Selinsky, K. Gupta, C. T. Sharkey and P. J. Loll, Biochemistry, 2001, 40, 5172–5180 CrossRef PubMed; (b) A. L. Blobaum and L. J. Marnett, J. Med. Chem., 2007, 50, 1425–1441 CrossRef PubMed.
- (a) E. N. Baker and R. E. Hubbard, Prog. Biophys. Mol. Biol., 1984, 44, 97–179 CrossRef PubMed; (b) W. Saenger, Annu. Rev. Biophys. Biophys. Chem., 1987, 16, 93–114 CrossRef PubMed; (c) P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef PubMed.
- (a) G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, New York, 1991 Search PubMed; (b) G. A. Jeffrey, Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed; (c) M. F. Jarrold, Acc. Chem. Res., 1999, 32, 360–367 CrossRef.
- (a) M. Schmitt, M. Böhm, C. Ratzer, C. Vu, I. Kalkman and W. L. Meerts, J. Am. Chem. Soc., 2005, 127, 10356–10364 CrossRef PubMed; (b) W. Caminati, J. C. López, S. Blanco, S. Mata and J. L. Alonso, Phys. Chem. Chem. Phys., 2010, 12, 10230–10234 RSC.
- (a) A. Maris, P. Ottaviani and W. Caminati, Chem. Phys. Lett., 2002, 360, 155–160 CrossRef; (b) S. Mata, V. Cortijo, W. Caminati, J. L. Alonso, M. E. Sanz, J. C. López and S. Blanco, J. Phys. Chem. A, 2010, 114, 11393–11398 CrossRef PubMed.
- (a) S. Blanco, P. Pinacho and J. C. López, Angew. Chem., Int. Ed., 2016, 55, 9331–9335 CrossRef PubMed; (b) S. Blanco, P. Pinacho and J. C. López, J. Phys. Chem. Lett., 2017, 8, 6060–6066 CrossRef PubMed.
- (a) C. Pérez, J. L. Neil, M. T. Muckle, D. P. Zaleski, I. Peña, J. C. López, J. L. Alonso and B. H. Pate, Angew. Chem., Int. Ed., 2015, 54, 979–982 CrossRef PubMed; (b) S. Blanco, J. C. López, A. Lesarri and J. L. Alonso, J. Am. Chem. Soc., 2006, 128, 12111–12121 CrossRef PubMed; (c) C. Pérez, A. Krin, A. L. Steber, J. C. López, Z. Kisiel and M. Schnell, J. Phys. Chem. Lett., 2016, 7, 154–160 CrossRef PubMed; (d) C. Pérez, J. C. López, S. Blanco and M. Schnell, J. Phys. Chem. Lett., 2016, 7, 4053–4058 CrossRef PubMed.
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer, S. T. Shipman and B. H. Pate, Rev. Sci. Instrum., 2008, 79, 053103 CrossRef PubMed.
- T. Betz, S. Zinn and M. Schnell, Phys. Chem. Chem. Phys., 2015, 17, 4538–4541 RSC.
- D. Schmitz, V. A. Shubert, T. Betz and M. Schnell, J. Mol. Spectrosc., 2012, 280, 77–84 CrossRef.
- C. Pérez, S. Lobsiger, N. A. Seifert, D. P. Zaleski, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Chem. Phys. Lett., 2013, 571, 1–15 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef.
- (a) T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef; (b) R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154101 CrossRef PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef.
- (a) M. Canagaratna, J. A. Phillips, M. E. Ott and K. R. Leopold, J. Phys. Chem. A, 1998, 102, 1489–1497 CrossRef; (b) D. Priem, T.-K. Ha and A. Bauder, J. Chem. Phys., 2000, 113, 169–175 CrossRef; (c) J. L. Alonso, E. J. Cocinero, A. Lesarri, M. E. Sanz and J. C. López, Angew. Chem., Int. Ed., 2006, 45, 3471–3474 CrossRef PubMed; (d) E. G. Schnitzler and W. Jäger, Phys. Chem. Chem. Phys., 2014, 16, 2305–2314 RSC.
- H. M. Pickett, J. Mol. Spectrosc., 1991, 148, 371–377 CrossRef.
- J. K. G. Watson, in Vibrational Spectra and Structure a Series of Advances, ed. J. R. Durig, Elsevier, Amsterdam, 1977, vol. 6, pp. 1–89 Search PubMed.
- (a) S. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef; (b) S. S. Xantheas, J. Chem. Phys., 1996, 21, 8821–8824 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp01088a |
| This journal is © the Owner Societies 2018 |