
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeasuring viscosity inside mesoporous silica using protein-bound molecular rotor probe†
Pegah S.
Nabavi Zadeh
 *a,
Milene
Zezzi do Valle Gomes
b,
Maria
Abrahamsson
a,
Anders E. C.
Palmqvist
b and
Björn
Åkerman
a
*a,
Milene
Zezzi do Valle Gomes
b,
Maria
Abrahamsson
a,
Anders E. C.
Palmqvist
b and
Björn
Åkerman
a
aChalmers University of Technology, Department of Chemistry and Chemical Engineering, Physical Chemistry, SE-41296 Gothenburg, Sweden. E-mail: pegah.nabavi@chalmers.se; Tel: +46 317723056
bChalmers University of Technology, Department of Chemistry and Chemical Engineering, Applied Chemistry, SE-41296 Gothenburg, Sweden
First published on 19th June 2018
Abstract
Fluorescence spectroscopy of protein-bound molecular rotors Cy3 and Cy5 is used to monitor the effective viscosity inside the pores of two types of mesoporous silica (SBA-15 and MCF) with pore diameters between 8.9 and 33 nm. The ratio of the peak intensities is used to measure viscosity independently of solvent polarity, and the response of the lipase-bound dyes is calibrated using glycerol/water mixtures (no particles). The two dyes are either attached to the same protein or separate proteins in order to investigate potential effects of energy transfer (FRET) on the fluorescence properties, when using them as reporter dyes. The effective viscosity inside the pores at infinite protein dilution is one order of magnitude higher than in bulk water, and the effect of protein concentration on the measured viscosity indicates a stronger effect of protein–protein interactions in the pores than in similarly concentrated protein solutions without particles. In MCF-particles with octyl-groups covalently attached to the pore walls, a more efficient uptake of the lipase resulted in FRET between the protein-bound dyes even if the two dyes were attached to different proteins. In contrast to the unmodified particles the intensity-ratio method could therefore not be used to measure the viscosity, but the presence of FRET in itself indicates that octyl–protein interactions lead to a non-homogenous protein distribution in the pores. The dye labels also report a less polar pore environment as sensed by the proteins through a redshift in the dye emission. Both observations may help in understanding the higher efficiency of lipase immobilization in octyl-modified particles.
1. Introduction
Enzyme immobilization in confining materials is used to improve the enzyme stability and enzymatic function.1,2 Mesoporous silica particles (MPS) is a common confining material which is used in biocatalytic applications because of the large surface area and narrow pore size distribution of MPS particles as well as a high chemical and mechanical stability of the silica material.3 The porous structure also allows for high enzyme loadings and easy recovery of both product and enzymes.4 Moreover, the MPS material creates a protective environment where enzymes sometimes can tolerate elevated temperature5 and high salt concentration.4From a physical chemistry perspective, several studies have been performed to investigate the behavior of enzymes inside the pores and also to characterize the environment that the enzymes experience during and after the immobilization.6–8 For instance, spectroscopic methods based on fluorescent reporter dyes have been used to measure pH inside pores of the MPS particles, either with the pH probe being bound to the silica walls of the particle pores,9 or covalently attached to the proteins.10 The latter approach has the advantage of monitoring the pH at the actual position of the enzyme in the pore. Protein-bound dye has also been used to monitor the immobilization in real time using the fluorescence of the epicocconone dye which is believed to be sensitive to local viscosity,7,11 moreover, the rotational mobility of enzymes once inside the pores has been studied by fluorescence anisotropy spectroscopy.12,13 The results showed that the rate of both translation into the pores and rotation of the proteins once inside the pores are strongly retarded when the protein and the pore are close in size. Such a dependence on the relative protein/pore-size is expected for simple steric reasons, but since both processes involve the motion of macromolecules in water, their rates will be also sensitive to the local viscosity of the solvent. This possibility, and the hypothesis that the increase in epicocconone fluorescence intensity reflects a higher viscosity inside pores than in bulk water, led us to try to measure directly the effective viscosity of the water experienced by the confined proteins inside the pores. This aspect of the pore environment for pore sizes suitable in enzyme immobilization has to our knowledge not been investigated yet by fluorescence spectroscopy.
In this study, we focus on Mesostructured Cellular Foam (MCF) particles with pore diameters in the range of tens of nm, because of their potential in biocatalytic applications, specifically in co-immobilization of several types of enzymes.14,15 Additionally, Santa Barbara Amorphous-15 (SBA-15) particles were used for comparison with our earlier works.7,12
Following the strategy of our previous studies,7,10 a viscosity-sensitive fluorescent dye was bound covalently to proteins, here lipase, rather than having it attached to the pore wall.16 Therefore, the microviscosity measured is that sensed by the protein at its actual location in the pore. There are several types of dyes whose fluorescence properties are sensitive to the microviscosity of the solvent, often based on the concept of molecular rotors which are slowed down by increasing viscosity.17–20 However, viscosity variations are often accompanied by changes in the polarity of the solvent. Here, we use a ratiometric method for measuring specifically the effective viscosity inside the pores, separately from potential polarity effects. This ratiometric method based on two different fluorescent carbocyanine dyes has been used previously for measuring the viscosity in the cytoplasm of living cells.21
In the present study, sulfo-cyanine3 NHS ester and sulfo-cyanine5 NHS ester (denoted Cy3 and Cy5 respectively; see Fig. 1) were chosen as the two fluorescent dyes since both are sensitive to viscosity as molecular rotors, but to different extents. Because carbocyanines with shorter polymethine chains (such as Cy3) photoisomerize in the excited state more efficiently than the longer analogues (such as Cy5),21–23 the molecular motion is slowed down, and when the viscosity of the solvent increases, the quantum yield of fluorescence increases more for Cy3 than Cy5.
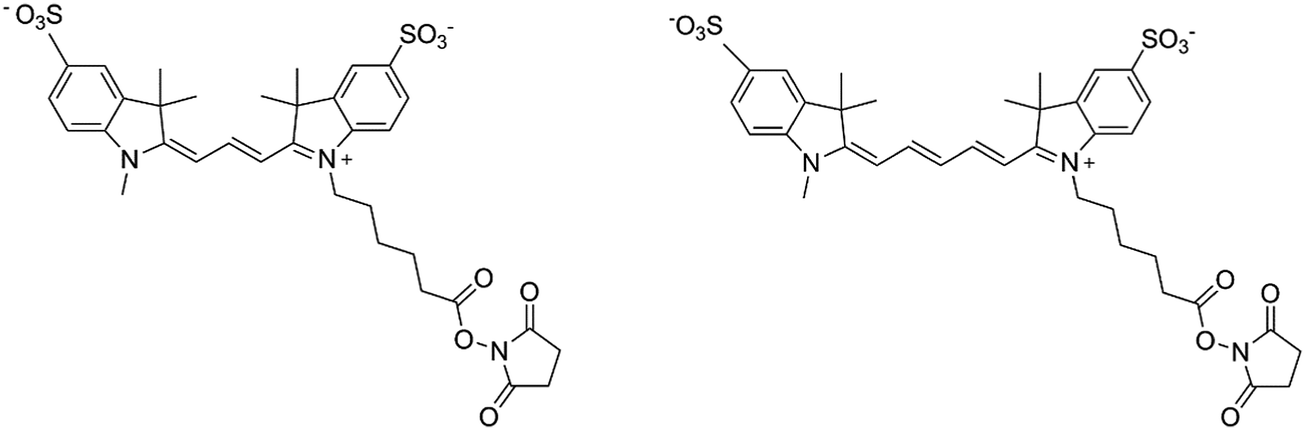 | ||
| Fig. 1 Structure of sulfo-cyanine3 NHS ester (Cy3) (left) and sulfo-cyanine5 NHS ester (Cy5) (right). | ||
These two dyes are suitable for the purpose of this study, since both dyes can be covalently bound to lipase, through amine groups of the enzyme. Secondly both Cy3 and Cy5 are negatively charged and hydrophilic which minimize the possibility of the dye binding to the negatively charged silica pore walls. Finally they are more photostable than epicocconone, thus avoiding the bleaching problem encountered with this dye during prolonged excitation.7
By using the ratio of the emission intensities at the maximum emission wavelength of the two dyes, the effective viscosity can be measured independently of potential polarity effects.21 In this ratiometric approach, each enzyme should ideally carry both dyes, but there is the potential drawback of Förster resonance energy transfer (FRET) between the two dyes, because the enzyme size is on the nanometer length scale where FRET may be efficient and thus may affect the fluorescence spectra of the dye pairs. Two different strategies of dye labeling for the enzyme were therefore investigated. In the first approach (“double labeling”) each enzyme carries both dyes (Cy3 and Cy5), while in the second strategy (“single labeling”) each enzyme carries either Cy3 or Cy5 and a mixture of these two labeled enzymes were used. Notably, the narrow pores of the MPS particles may increase the possibility of FRET between the two dyes even in the single labeling strategy, because the concentration of proteins in the pores can be very high under typical immobilization conditions.12 Therefore, the amount of protein loading in the MPS particles was used as a variable in the present study aiming to avoid energy transfer in the single labeling and also to investigate the protein–protein interaction effect on the viscosity value.
The two silica materials (MCF and SBA-15) present different 3D-structure, pore architecture and pore size. In addition, MCFs with octyl-functionalized surface were also used in this work and compared with unmodified MCFs of the same particle type. This study focused on the effective viscosity in different particle types and enzyme concentrations in the pores and using only one type of protein, here lipase. An earlier study10 showed that at least the pH in the pores of SBA-15 reported by different protein types (including lipase) were the same.
The measured viscosity values are compared with the viscosity and dynamics of water in silica porous (channels) materials with different pore sizes (from 100 nm to 2 nm) which have been reported earlier24–27 using other methods such as proton-NMR and quasielastic neutron scattering.
2. Experimental
2.1. Chemicals and materials
Lipase was purchased from Sigma-Aldrich and the enzyme properties can be found in Table 1. The amine-reactive fluorescent probes sulfo-cyanine3 NHS ester (Cy3) and sulfo-cyanine5 NHS ester (Cy5) were used as sodium and potassium salts, respectively, purchased from Lumiprobe, Life Science Solutions. The structures and spectral properties of probes can be seen in Fig. 1 and Table 2, respectively. The properties of the dyes were taken as provided by Lumiprobe. All fluorescence experiments for this study were performed at 25 °C, in 0.1 M phosphate buffer to give pH 6, if not otherwise stated.| Fluorescent dyea | Exb (nm) | Emc (nm) | Q | ε (M−1 cm−1) | CF280f | M w (D) | DOLh |
|---|---|---|---|---|---|---|---|
| a Cy3: Sulfo-cyanine3 NHS ester, Cy5: sulfo-cyanine5 NHS ester. b Excitation wavelength. c Maximum emission wavelength. d Fluorescence quantum yield. e Extinction (molar absorption) coefficient at excitation wavelength. f Correction factor at 280 nm wavelength for calculation degree of labeling. g Molecular weight. h Average of degree of labeling, the number of dyes per each enzyme, the uncertainty is ±0.1 as calculated from the variation between 3 and 4 independent experiments. | |||||||
| Cy3 | 550 | 565 | 0.1 | 162![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.06 | 735.80 | 1.05 |
| Cy5 | 650 | 665 | 0.2 | 271000 |
0.13 | 777.95 | 1.07 |
For the synthesis of MPS particles, Pluronic™ P123 (EO20PO70EO20, Mw = 5800), 1,3,5 trimethylbenzene (TMB, 98%), tetraethylorthosilicate (TEOS, ≥98%), hydrochloric acid (HCl) (37 wt%), ammonium fluoride (NH4F, ≥ 99.9%), octyltriethoxysilane (OC, ≥97.5%) and toluene anhydrous (99.8%) were purchased from Sigma-Aldrich.
Two types of MPS particles were used in this study, Santa Barbara Amorphous-15 (SBA-15) and siliceous Mesostructured Cellular Foams (MCF). Fig. 2 shows schematically the main difference in morphology and structure between SBA-15 and MCF silica particles. SBA-15 particles have parallel hexagonal pore channels, while MCF particles have large spherical pores connected by (smaller) windows.
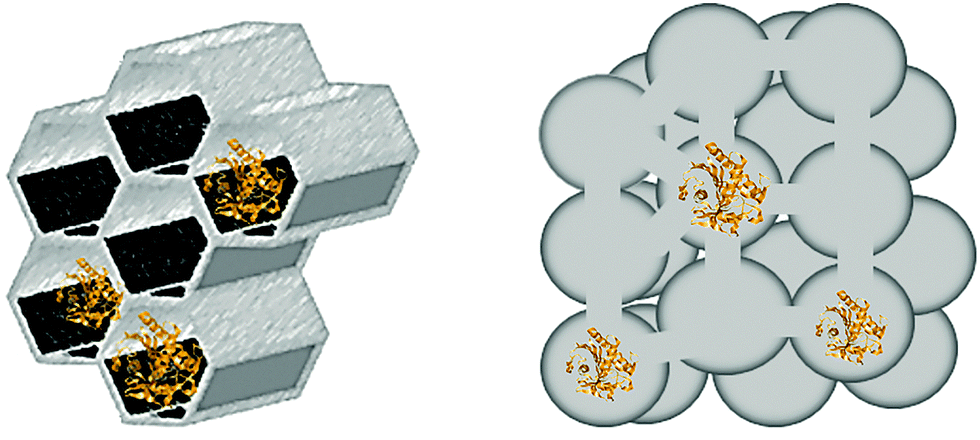 | ||
| Fig. 2 Schematic morphology and pore structure of SBA-15 (left) and MCF (right) particles. | ||
SBA-15 was a gift from Hanna Gustafsson (Applied Chemistry, Chalmers) and was synthesized and characterized as described previously.29 Two MCFs were synthesized according to the procedure developed by Schmidt-Winkel et al.,30 with few modifications aiming at variations in the pore and window sizes. The synthesis was performed using tetraethyl orthosilicate (TEOS) as silica precursor, Pluronic™ P123 (EO20PO70EO20, Mw = 5800) as the structure-directing agent, and 1,3,5 trimethyl benzene (TMB) as a swelling agent. In the synthesis of MCF2, the mass ratio of TEOS to surfactant, and TMB to surfactant used were TMB/P123 = 0.4 and TEOS/P123 = 2.2, respectively, and for MCF12 it was changed to TMB/P123 = 0.5 and TEOS/P123 = 3.0. See ESI,† Section S.1 for a detailed description of the synthesis.
The surface of the two MCFs were functionalized with octyl groups according to the procedure previously described by Russo et al.,31 in which 1 g of each MCF was dried in a vacuum oven during 4 h at 120 °C and then mixed with 15 ml of toluene and 0.7 ml of triethoxy(octyl)silane. The samples were stirred for 10 min at room temperature, and transferred to an autoclave and heated at 100 °C for 24 h. The octyl (OC)-functionalized MCFs were recovered by filtration, washed with toluene and dried in air at 120 °C during 19 h. The final products were denoted as MCF2-OC and MCF12-OC.
The MPS particles were characterized by nitrogen adsorption analysis using TriStar 3000 instrument from Micromeritics Instrument Corporation in order to obtain average pore diameter and window size, specific pore volume and BET surface area. The properties of the used MPS are summarized in Table 3.
| MPSa | Pore size (nm) | Window sized (nm) | BET surface areae (m2 g−1) | V pore (cm3 g−1) |
|---|---|---|---|---|
| a Type of MPS preparation. SBA-15: santa barbara amorphous, MCF: mesostructured cellular foam, OC stands for an octyl-functionalized silica surface. b Average pore diameter obtained by the Barret–Joyner–Halenda method.32 c Average pore diameter obtained by simplified BdB (Broekhoff-de Boer)-FHH (Frenkel–Halsey–Hill) method33 using the adsorption isotherm. d Average window diameter obtained by simplified BdB (Broekhoff-de Boer)-FHH (Frenkel–Halsey–Hill) method33 using the desorption isotherm. e Specific pore surface area obtained by the Brunhauer–Emmett–Teller method.34 f Specific pore volume calculated using a single point adsorption value at the relative pressure of 0.990. | ||||
| SBA-15 | 8.9b | — | 554 | 1.17 |
| MCF2 | 26.5c | 11.2 | 523 | 1.94 |
| MCF2-OC | 24.8c | 11.2 | 453 | 1.67 |
| MCF12 | 33.0c | 13.0 | 375 | 1.84 |
| MCF12-OC | 32.7c | 13.0 | 325 | 1.68 |
2.2. Binding of probes (Cy3 and Cy5) to lipase
Cy3 and Cy5 (Fig. 1) were bound to lipase (here denoted MML) using the reaction of NHS (N-hydroxysuccinimide) ester groups on the probes with amine groups of the enzyme. Two different strategies were used to modify the enzymes with the probes, single labeling where each enzyme is modified with either Cy3 (denoted Cy3-MML) or Cy5 (Cy5-MML), or double labeling where each enzyme is labeled with both Cy3 and Cy5 (Cy3-MML-Cy5).The dye modification in the single labeling approach was performed by mixing the enzyme stock solution (4 mg MML lipase in 900 μl of 0.1 M phosphate buffer, pH 8) with 100 μl of the dye stock solution (0.75 ± 0.02 mg of either Cy3 or Cy5 in 100 μl of Milli-Q water), based on the protocol for mono-labeling provided by Lumiprobe. After vortexing for 1 min, the mixture was kept on ice overnight. The labeled protein was purified on a size-exclusion column (NAP-10, GE Healthcare) in 0.1 M phosphate buffer, pH 8 to remove all non-bound dyes. The number of attached dyes (Cy3 or Cy5) per protein (the degree of labeling DOL in Table 2), was determined based on the absorbance ratio of the enzyme and the dye before and after the binding reaction using the molar absorption coefficient of dyes and the enzyme, see Tables 2 and 3.35 The results showed that for both Cy3 and Cy5 on the average 1 ± 0.1 dye molecule is bound per protein (data not shown), although we do not know to which of the seven lysines in MML36 the dye is attached. Circular dichroism experiments showed that the modification by Cy3 or Cy5 caused no detectable conformational change in the MML proteins. (See ESI,† Section S.2 for CD spectra.)
The double labeling was performed in the same way as with single labeling, except the lipase stock solution was prepared by mixing 4 mg protein in 800 μl of 0.1 M phosphate buffer, pH 8 with 100 μl each of the two dye stock solutions (Cy3 and Cy5). The DOL-values per dye were the same as in the single labeling approach within the experimental uncertainty of 0.1.
2.3. Protein immobilization
The double-labeled protein Cy3-MML-Cy5 (each enzyme carrying both probes) was found to be less suitable for our present purposes due to FRET (see Results, Section 3.1, ESI†). Therefore, particle–protein complexes were only prepared by immobilizing mixture of single-labeled proteins (Cy3-MML + Cy5-MML), at a 1:1 ratio if nothing else is stated. Aqueous stock solutions of the MPS were prepared by dispersing 5 mg of dry mesoporous silica particles in 1 ml phosphate buffer, 0.1 M, pH 6, using vortexing for 10 min at 10 rpm followed by sonication (ultrasonic cleaner model CD-4800 at a power of 70 W) for 20 min in order to dissolve any particle aggregates, and a final step of vortexing for 5 min.
Protein–particle samples were prepared by adding 5–10 μl of the mixture of single-labeled lipase (Cy3-MML + Cy5-MML in a 1:1-ratio, 4 mg ml−1 total enzyme concentration) with 200 μl of MPS solutions (5 mg ml−1) diluted to a final volume of 500 μl with phosphate buffer. By varying the amount of added volume of the single-labeled protein mixture, samples were prepared containing 20, 40 or 60 μg of added enzyme per mg of MPS. For control experiments, only Cy3 labeled protein was added to the same final total protein concentration in 1 mg of the MPS particles.
The protein–particle samples were incubated at 25 °C for 24 h during gentle stirring, and then centrifuged for 6 min. The pelleted protein–particles complexes were re-suspended, and washed three times with 100 μl of phosphate buffer at pH 6 by repeated centrifugation and resuspension. The loaded MPS particles with the immobilized proteins were finally re-suspended by adding 500 μl of buffer and vortexing for a few minutes until homogenous samples were obtained for the spectroscopic measurements.
The fraction of the added protein which is associated with the particles (degree of immobilization; DOI) was calculated from the difference between the total added amount of enzyme and the amount of enzyme remaining in the supernatant after particle washing, which was measured by UV absorption at 280 nm using a Varian Cary50 spectrophotometer. The protein loading PLD (μg protein per mg particle) was then obtained from the dry mass of added particles. The amount of immobilized protein was also expressed as the pore filling (Pf), the volume fraction of protein in the pores, which was calculated from the PLD value as described previously.12,35 (See ESI,† Section S.3 for more details). The relative loading of the co-immobilized Cy3-MML and Cy5-MML in the single labeling approach was estimated by measuring the Cy3 and Cy5 absorbance in the supernatant, and converting them into amounts of respective protein remaining in the supernatant by using the degree of labeling in Table 2.
2.4. Spectroscopic measurements
:1 ratio) were recorded in aqueous solutions with the concentration of glycerol varying from 0 to 70% in phosphate buffer pH 6, at 25 °C with 15 different viscosity values between 0.92–18.38 cP. The entire emission spectrum for each sample was obtained using a Cary Eclipse fluorimeter (Varian) with a temperature-controlled sample chamber (±0.1 °C) using an excitation wavelength of 550 nm for Cy3-MML and 650 nm for Cy5-MML. The excitation and emission slits were set at 5 nm. The steady state emission intensity maxima Imax were used to calculate the intensity ratio R = Imax(565 nm)/Imax(665 nm) between Cy3-MML and Cy5-MML. The ratios were corrected for the optical density of the samples in order to make the calibration curve independent of protein concentration, and were then plotted versus the known viscosity values of water/glycerol-mixtures at 25 °C.37 The reported ratios are the mean values from triplicate samples prepared independently, and the experimental error of ±0.01 corresponds to half of the maximum variation between the three experiments.
The validity of the calibration curve was tested by measuring the intensity ratio R for the mixture of single-labeled Cy3-MML + Cy5-MML in a solvent containing 40% mass fraction of ethanol in phosphate buffer pH 6 (no glycerol). An aqueous solution with 40% ethanol has very similar viscosity (η = 2.34 cP) but distinctly different dielectric constant (εr = 63.3) compared to a solution of 30% glycerol in aqueous solution (η = 2.26 cP, εr = 71.4).21,37,38 Therefore, these two samples (30% glycerol and 40% ethanol) should give the same R-values if the ratiometric method indeed reflects the viscosity of the solvent independently of its polarity.
000 counts had been collected in the peak channel. The emitted light was detected at magic angle to the excitation light through a monochromator tuned to the maximum emission wavelength of the sample. The photons were collected by a microchannel plate photomultiplier tube from Hammamatsu and fed into a multichannel analyzer with 4096 channels.
3. Results and discussion
3.1. Labeling strategy
In order to use lipase-bound dyes to measure the local viscosity in the pores, the Cy3 and Cy5 as a ratio pair should ideally be located as close to each other as possible, so as to probe the same environment. One strategy is to attach both dyes to the same protein, the double labeling strategy, denoted Cy3-MML-Cy5. However, the potential of FRET between Cy3 and Cy5 in this approach led us to compare it to the alternative strategy to use a 1:1 mixture of single-labeled lipase Cy3-MML and Cy5-MML, denoted Cy3-MML + Cy5-MML. Both strategies were investigated since the single labeling also has a potential drawback, which is a possibility of uneven protein loading, if the two dyes (Cy3 and Cy5) affect the lipase immobilization to different degrees. Fig. 3 shows the emission spectra of free lipase-dyes (no particles) for the double labeled sample (Fig. 3a) and the mixed sample of single labeled (Fig. 3b), which both are compared to the individual emission spectra of Cy3-MML and Cy5-MML.
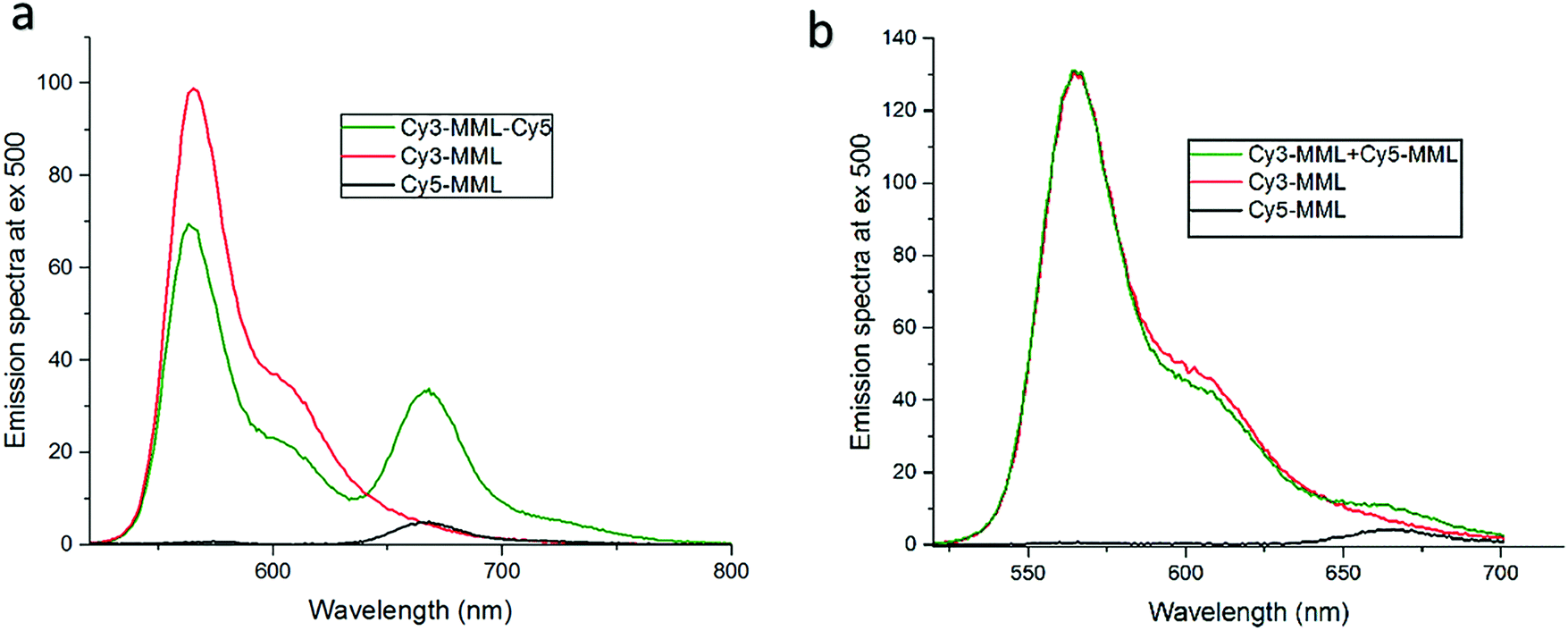 | ||
| Fig. 3 Steady state Förster resonance energy transfer for the two different strategies of labeling, compared to individual spectra for labeled proteins Cy3-MML (red) and Cy5-MML (black). (a) Double-labeled lipase (Cy3-MML-Cy5; green curve) and (b) a 1:1-mixture of single-labeled lipase (Cy3-MML + Cy5-MML; green curve). Phosphate buffer, pH 6 and particle free solution. Excitation at 483 nm, emission at 565 nm and 665 nm indicate Cy3 and Cy5, respectively. | ||
It is seen in Fig. 3a that the emission spectrum of the double-labeled sample (green curve) clearly differs from the sum of the spectra of the individually labeled proteins. The emission of the Cy5-MML (centered at 665 nm in the green curve) is strongly enhanced compared to the Cy5-MML by itself (black curve). This enhancement is accompanied by a significantly reduced intensity of the Cy3-MML in the double labeled sample (centered at 565 nm in the green curve) compared to the emission of Cy3-MML by itself (red curve). Taken together, these two observations strongly indicate energy transfer from Cy3 to Cy5 when the dyes are bound to the same protein. These results show that the average distance between Cy3 and Cy5 attached to the same enzyme in double labeling is too small for the two dyes to act as independent reporter dyes as required in the ratiometric method. Furthermore, the observation of FRET is consistent with that the diameter of the lipase (2RH = 4.5 nm) is comparable to the Förster distance (Ro) of the Cy3/Cy5-pair (Ro = 6 nm; see ESI,† Section S.5).39,40
By contrast, Fig. 3b shows that the emission spectrum of the mixture of Cy3-MML and Cy5-MML is almost identical to the sum of the spectra of the individually labeled lipase. This observation and time-resolved fluorescence measurements of Cy3-MML and the mixed sample (see ESI,† Section S.5) support that FRET is negligible in the single labeling approach, at least at the relatively low protein concentrations used in the particle-free solution measurements. Importantly, the situation may be different after immobilization in the MPS particles due to an enhanced local protein concentrations in the pores.
In summary, the lack of energy transfer in the mixture of single-labeled enzyme (Cy3-MML + Cy5-MML) makes this approach promising for our attempt to measure the local viscosity using the ratiometric method, whereas the double labeling strategy (Cy3-MML-Cy5) is not suitable for our purposes. Luby-Phelps et al. have used the same single-labeling approach to measure the viscosity of the cytoplasm,21 but the possibility of double labeling of the used Ficoll carrier polymer was not investigated. We find that the double labeled sample can serve as a useful positive control for FRET. For instance, from the demonstrated lack of FRET in Fig. 3b, we conclude there is no interaction (association) between MML-enzymes in particle-free solution. Therefore, any potential FRET after immobilization is most likely due to an accidental proximity between two proteins in the narrow pores. All results presented below are based on the strategy of a mixture of single-labeled enzymes, in which case the reporter dyes can be considered to act independently as long as there is no FRET.
3.2. Viscosity calibration curve for protein-bound Cy3 and Cy5
The attachment of Cy3 and Cy5 to lipase may affect the fluorescence response of the dyes to solvent properties, because the local environment for the dyes may be different when they are bound to the protein compared to non-bound (free) dyes. Therefore, the ratiometric method needs to be calibrated with the mixture sample of Cy3-MML + Cy5-MML. Indeed, fluorescence anisotropy results (ESI,† Section S.6) indicate that the attached Cy3 partly interacts with the lipase, which may affect how Cy3 senses viscosity as a fluorescence-based molecular rotor. However, this interaction between Cy3 and MML is not altered when the labeled lipase is immobilized in any of the five particle types of this study. (See ESI,† Section S.6). This observation strongly supports that the viscosity calibration curve measured for protein-bound dyes in free solution (no particles) can be applied to intensity ratios measured when the labeled lipase are inside the MPS pores.For the calibration experiments in free solution (no particles), the viscosity of the phosphate buffer is increased by adding glycerol. Fig. 4 shows the fluorescence emission spectra of the Cy3-MML + Cy5-MML mixture in different aqueous mixtures with glycerol at mass fractions in the range 0–70% in phosphate buffer. The two curves with the lowest peak intensity correspond to 0% glycerol, with increasing of the peak intensities to the highest values which correspond to 70% glycerol. It is seen that the emission intensity increases monotonously with increasing glycerol concentration for both dyes which is consistent with a viscosity increase, although the relative increase is larger for Cy3 as expected. Moreover, there is a red shift in the peak intensity of both dyes when the glycerol concentration increases. This observation indicates that both dyes are sensitive to other solvent effects as well, probably the polarity of the water/glycerol mixtures.41 However, the red shift is larger for Cy5 (from 665 nm to 672 nm) compared to Cy3 (from 565 nm to 568 nm). The difference in response between Cy3 and Cy5 regarding intensity and redshift is the basis of using the ratiometric method to separate viscosity from polarity effects.
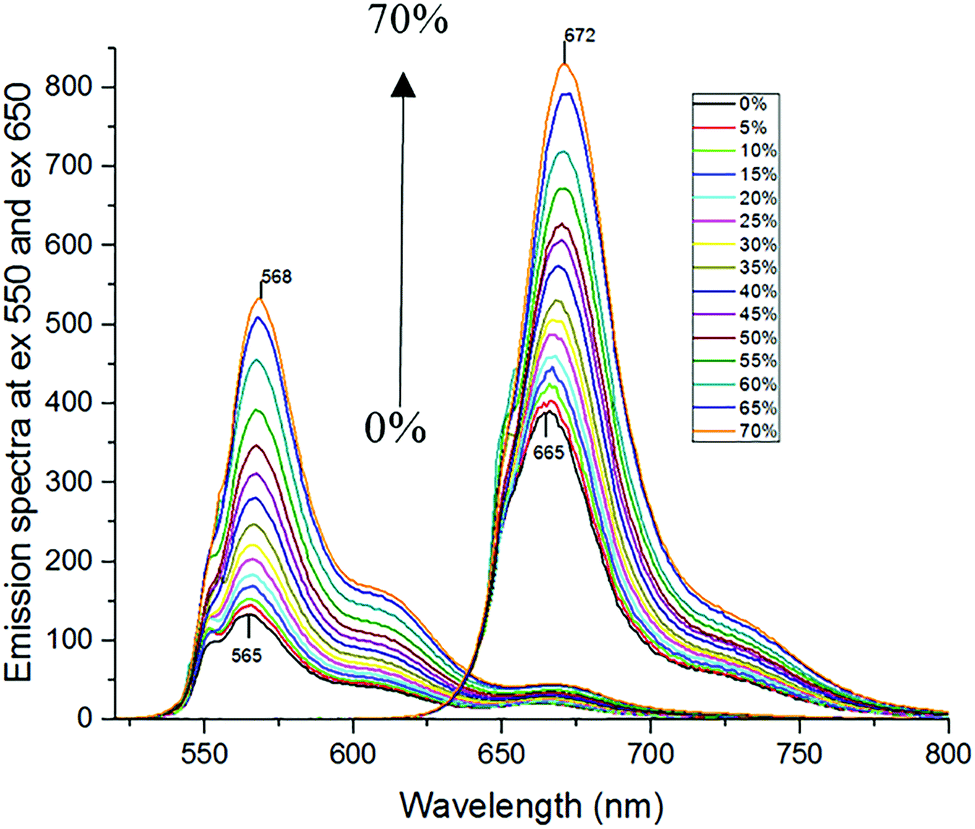 | ||
| Fig. 4 Fluorescence intensities of the mixture of single-labeled lipase (Cy3-MML + Cy5-MML) in different concentration of glycerol (0–70% mass fraction of glycerol mixed with phosphate buffer). The Cy3 intensity maximum is at about 565 nm, and for Cy5 at about 665 nm. For both dyes the curve with lowest maximum corresponds to 0% glycerol (pure phosphate buffer), and both intensity maxima increases monotonously as the mass fraction of glycerol increases to 70%. Excitation wavelengths were 550 nm for Cy3-MML and 650 nm for Cy5-MML, respectively. | ||
When the concentration of glycerol is increased from 0 to 70% in phosphate buffer, the viscosity at 25 °C increases from 0.92 to 18.38 cP.37Fig. 5 shows that the fluorescence intensity ratio R = Imax(565 nm)/Imax(665 nm) of the Cy3-MML + Cy5-MML mixture increases more than 2.3-fold in this viscosity range. The R-values fall essentially on a single master curve, which can be used as a calibration curve.
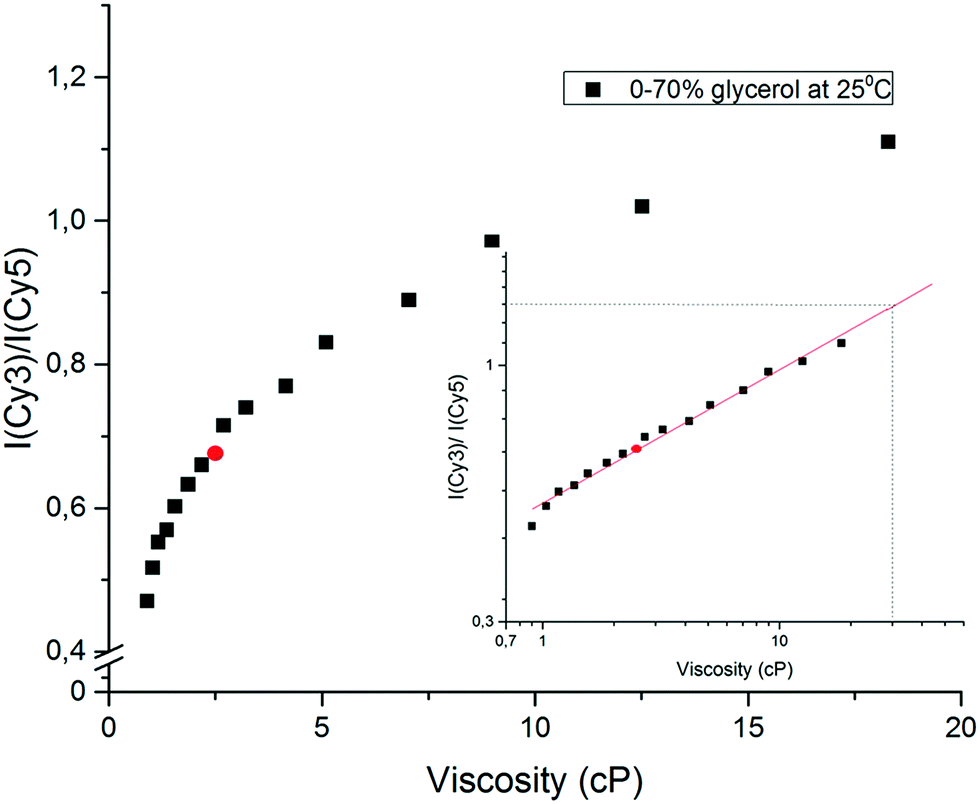 | ||
| Fig. 5 Calibration curve for intensity ratio R of the Cy3-MML + Cy5-MML sample vs. viscosity (no particles). Black squares shows intensity ratio vs. viscosity in aqueous solutions of glycerol between 0 and 70% in phosphate buffer. Red circle shows the intensity ratio in a control solvent with 40% ethanol in phosphate buffer (no glycerol; see main text). Inset shows log–log plot of the same data (black squares). | ||
To check the validity of the calibration curve in Fig. 5, the Cy3-MML + Cy5-MML sample was investigated in a control solvent of 40% ethanol in phosphate buffer (no glycerol) which has the same macroscopic viscosity as 30% glycerol whereas the dielectric constant of these two samples are significantly different (Table 4). The measured intensity R-value for this solvent (red circle in Fig. 5) falls well on the calibration curve, which strongly supports that the ratiometric method can be used to measure viscosity independently of solvent polarity, also when the Cy3 and Cy5 dyes are attached to lipase. Similarly, the solvent with 70% glycerol in water has nearly the same dielectric constant as the 40% ethanol control solvent, but the viscosity values in these two solvents differ by a factor of almost 8 (see Table 4).
In a second control experiment using circular dichroism, there are no significant differences in the CD spectra of Cy3-MML + Cy5-MML in phosphate buffer compared to the 70% glycerol/buffer mixture (see ESI,† Section S.2). The result indicates that the enzyme retains its secondary structures even at the highest glycerol concentration (70% mass fraction). This observation is consistent with other studies of the effect of glycerol on protein folding and unfolding,42–44 and it means that the fluorescence responses of the dyes in Fig. 4 underlying the calibration curve are not affected by a glycerol-induced change in lipase structure.
The calibration curve in Fig. 5 is markedly non-linear which shows that the sensitivity is higher when the viscosity of the solvent is closer to pure water viscosity. A well-established format20,45,46 for using viscosity calibration curves is the double logarithmic plot shown in the inset of Fig. 5, where the approximately linear form of the plot allows for extrapolation at high viscosities, if necessary. The viscosity values derived from Fig. 5 for the immobilized samples will be denoted effective viscosities (see below), shorthand for the microviscosity that the enzyme-attached probes senses in an aqueous solution of glycerol that has the same macroscopic viscosity.
3.3. Degree of protein loading and pore filling in the MPS particles
The 1:1 mixture of Cy3-MML + Cy5-MML was immobilized into five different mesoporous silica particles with different structure, pore size and surface modifications (see Table 3). Table 5 shows the measured protein loading (PLD) in each porous material using three different added total amounts of protein (per mg of particles) in the immobilization step. The stated values of PLD correspond to the total amount of immobilized protein, i.e. the sum of Cy3-MML and Cy5-MML. It is seen that the total protein loading increases in each particle type when the amount of added protein is increased. The relative degree of loading of the two separate proteins could be evaluated using the known degree of labeling for Cy3 and Cy5, and the absorbance of each of the reporter dyes linked to MML remaining in the supernatant after the immobilization (see Section 2.3). The results indicated that the 1:1 mixing ratio of Cy3-MML and Cy5-MML is retained in the pores within 2% (data not shown). Therefore, Cy3-MML and Cy5-MML immobilize to the same extent at a given total amount of added protein which suggests that the two dyes are not affecting the degree of immobilization.
| Added proteinb (μg mg−1) | Protein loading, PLDa (μg mg−1) | ||||
|---|---|---|---|---|---|
| SBA-15 (8.9 nm)c | MCF2 (26.5 nm)c | MCF12 (33.0 nm)c | MCF2-OC (24.8 nm)c | MCF12-OC (32.7 nm)c | |
| a Total amount of immobilized Cy3-MML and Cy5-MML per mg of silica particles. b Total amount of added protein Cy3-MML and Cy5-MML per mg of silica particles. c Pore diameter of each silica particle (see Table 3). | |||||
| 20 | 4.8 ± 0.5 | 7.9 ± 0.5 | 8.5 ± 0.5 | 15.1 ± 0.5 | 16.5 ± 0.5 |
| 40 | 12.1 ± 0.5 | 13.6 ± 0.5 | 14.5 ± 0.5 | 26.5 ± 0.5 | 30.3 ± 0.5 |
| 60 | 15.3 ± 0.5 | 19.8 ± 0.5 | 25.6 ± 0.5 | 37.2 ± 0.5 | 44.5 ± 0.5 |
Table 5 also shows that the PLD value increases at a given total amount of added protein in the order SBA-15, MCF-2 and MCF-12, i.e. in the order of increasing pore size. It is noteworthy that MCF particles modified with octyl groups have consistently higher protein loading than the unmodified particles of the same type (at the same amount of added protein), even though the measured pore size is slightly smaller after the OC-modification (see Table 3). Smaller effective pore sizes can be expected due to the presence of the additional carbon chains on the silica surface. The higher protein loading in OC-modified particles suggests that the hydrophobic surface from the octyl groups provide a favorable interaction for the immobilization of lipase, rather than sterically counteracting it. This observation is consistent with interfacial activation of lipase,47–49 as discussed further in Section 3.6.
In a previous study12 we have shown that enzyme immobilization under commonly used conditions may lead to high protein concentrations in the pores, with protein volume fractions of 20% or higher. Such a high protein concentration may lead to FRET between Cy3 and Cy5 even in the single labeling strategy, which would prevent the use of the ratiometric method. The relatively low amounts of added protein used in this study were chosen to avoid such energy transfer after protein immobilization. Table 6 shows the pore filling Pf (the fraction of the pore volume occupied by proteins) in the samples, as calculated from the PLD values in Table 5 (see ESI,† Section S.3).
| Added proteinb | Pore filling, Pf (%) | ||||
|---|---|---|---|---|---|
| SBA-15 (8.9 nm)c | MCF2 (26.5 nm)c | MCF12 (33.0 nm)c | MCF2-OC (24.8 nm)c | MCF12-OC (32.7 nm)c | |
| a Volume fraction protein (in %) in the pores, with ±0.05 percentage uncertainty. b Total amount of added protein per mg of silica particles. c Pore diameter of each silica particle (see Table 3). | |||||
| 20 μg mg−1 | 0.37 | 0.37 | 0.41 | 0.81 | 0.88 |
| 40 μg mg−1 | 0.93 | 0.63 | 0.71 | 1.43 | 1.60 |
| 60 μg mg−1 | 1.18 | 0.92 | 1.25 | 2.00 | 2.36 |
As can be seen from Table 6, the pore fillings are less than 3%, which is considerably smaller than in our previous study,12 as was intended by reducing the amount of added protein. The pore fillings of the unmodified particles are comparatively insensitive to the particle type even though the specific pore volume (Vpore) of the SBA-15 particles is 60% smaller compared to the MCF particles (see Table 3). The Pf values in the OC-modified particles are approximately twice as high as for the corresponding non-modified particle type at the same amount of added protein, which reflects the more efficient protein uptake in OC-modified particles in terms of pore occupancy. Notably, the useful parameter Nprot, the number of proteins per MCF-particle, could not be calculated as was done previously for SBA-15 particles,12,35 since the morphology of the MCF-particles are not uniform30 in the way the SBA-15 particles are.29
3.4. Fluorescence intensity ratio (R) inside the MPS particle pores
In order to evaluate the effective viscosity inside the pores from the fluorescence intensity ratio of the immobilized Cy3-MML + Cy5-MML sample, the emission spectra for Cy3 and Cy5 were recorded in the range 551–800 nm (excitation at 550 nm for Cy3 and at 650 nm for Cy5). Fig. 6 shows the emission spectrum in the SBA-15 particles at two different values of total added protein, after correction for light scattering by the particles (see Methods, Section 2.4.3).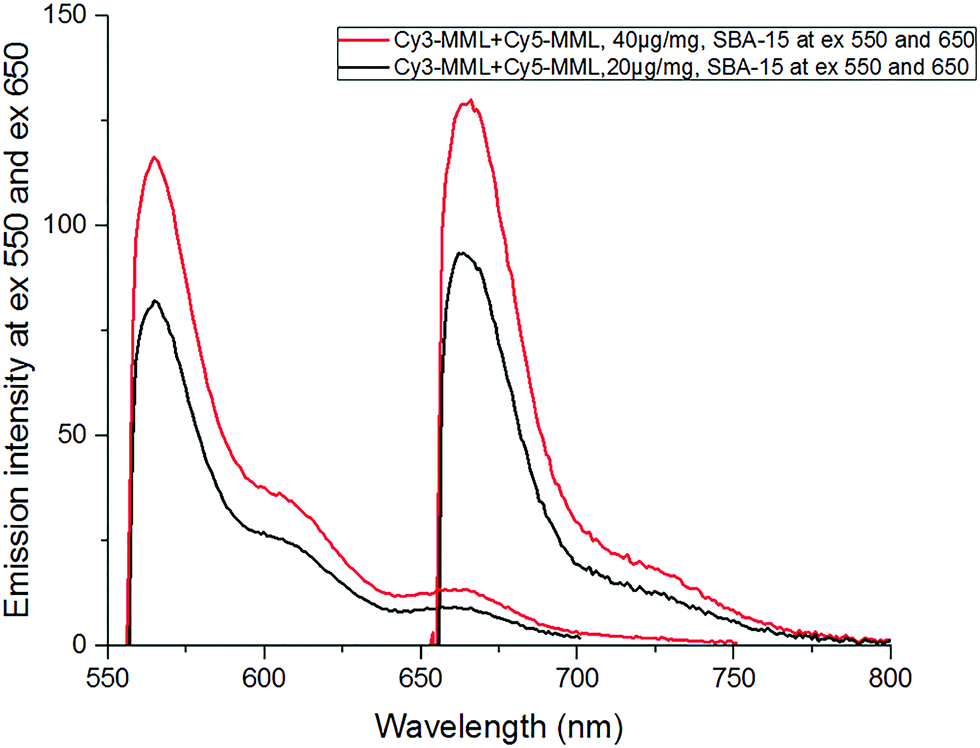 | ||
| Fig. 6 Emission spectra of the 1:1 mixture of Cy3-MML and Cy5-MML immobilized in SBA-15, at two different amounts of added protein (20μg (black) and 40 μg (red) total protein per mg of particles). The spectra have been corrected for light scattering by the particles. Excitation wavelengths were 550 nm for Cy3 and 650 nm for Cy5. | ||
Importantly, time-resolved fluorescence measurements on the same particle–protein samples (see ESI,† Section S.5) show that there is no FRET between the two dyes after immobilization of the Cy3-MML + Cy5-MML mixture in SBA-15 particles. This lack of FRET indicates that (at the relatively low levels of protein loading used in this study) the individual proteins are not packed close enough in the pores to bring the attached Cy3 and Cy5 within Förster distance. Importantly time-resolved anisotropy measurements (see ESI,† Section S.6) also show that the Cy3 has no detectable interaction with pore walls during the excited state life time, which otherwise may have interfered with the rotor-based sensing of the viscosity. Taken together, these two observations support that the calibration curve for the mixture of single-labeled lipase measured in particle-free solution (Fig. 5) can be applied to the results obtained for the same lipase mixture when immobilized in the particles. Notably, the actual dye absorbance of each sample (because of differences in the protein loading PLD) must be taken into account when the ratio of emission intensities (R) are calculated (see Section 2.4.1). The R-values for 20 μg and 40 μg added total protein in SBA-15 are 1.33 and 1.39, respectively, see Table 7, which both are slightly higher than the highest R value in calibration curve in Fig. 5 (at 70% glycerol). Therefore, a minor extrapolation is necessary to evaluate the effective viscosities for each R value.
| Added proteinb | SBA-15 (8.9 nm)c | MCF2 (26.5 nm)c | MCF12 (33.0 nm)c | |||
|---|---|---|---|---|---|---|
| R | Viscosity (cP) | R | Viscosity (cP) | R | Viscosity (cP) | |
| a Intensity ratio (R) and effective viscosity inside the different mesoporous silica particles, with ±0.01 and ±0.2 cP uncertainty, respectively. b Total amount of added protein per mg of silica particles. c Pore diameter of each silica particle (see Table 3). | ||||||
| 20 μg mg−1 | 1.33 | 31.3 | 1.23 | 23.4 | 1.20 | 21.4 |
| 40 μg mg−1 | 1.39 | 36.1 | 1.27 | 26.3 | 1.22 | 22.7 |
| 60 μg mg−1 | 1.48 | 46.4 | 1.35 | 32.4 | 1.30 | 28.2 |
The R value of the (Cy3-MML + Cy5-MML) sample immobilized in both unmodified and OC-modified MCF particles were measured by recording the emission spectra of Cy3 and Cy5 and correcting them in the same way as described for the SBA-15 particles above (see ESI,† Section S.7). Time-resolved intensity measurements of immobilized Cy3-MML + Cy5-MML show that there is no FRET in unmodified MCF2 and MCF12 particles even at the highest protein loading used here. However, in the OC-modified MCF-particles, FRET is observed at all protein loadings (see ESI,† and Section S.5) and because of this observation, the R value of the OC-modified MCF particles cannot be reported properly. First we will discuss the viscosity results in the unmodified particles and then turn to the results in the MCF-OC particles.
3.5. Effective viscosity and protein concentration in unmodified particles
Table 7 shows the collected data on the R-values and the derived effective viscosities in the unmodified SBA-15 and MCF-particles at the three different amounts of added protein used in the immobilization (see Table 6).As can be seen in Table 7, the effective viscosity values in the unmodified SBA-15, MCF2 and MCF12 particle pores are considerably higher (between 30–40 times) than pure water viscosity at the same temperature (0.89 cP at 25 °C). The effective viscosity decreases in the order of increasing pore size (for a given amount of added protein), which indicates that confining the water in smaller pores increases its effective viscosity.
These observations are summarized in Fig. 7, which shows the effective viscosity in the three particle types plotted versus the pore filling values (Pf) by combining the data of Tables 6 and 7. It is seen that in all three particle types, the effective viscosity decreases with decreasing pore filling (decreasing immobilized protein concentration), revealing a protein–protein interaction effect on the measured viscosity.
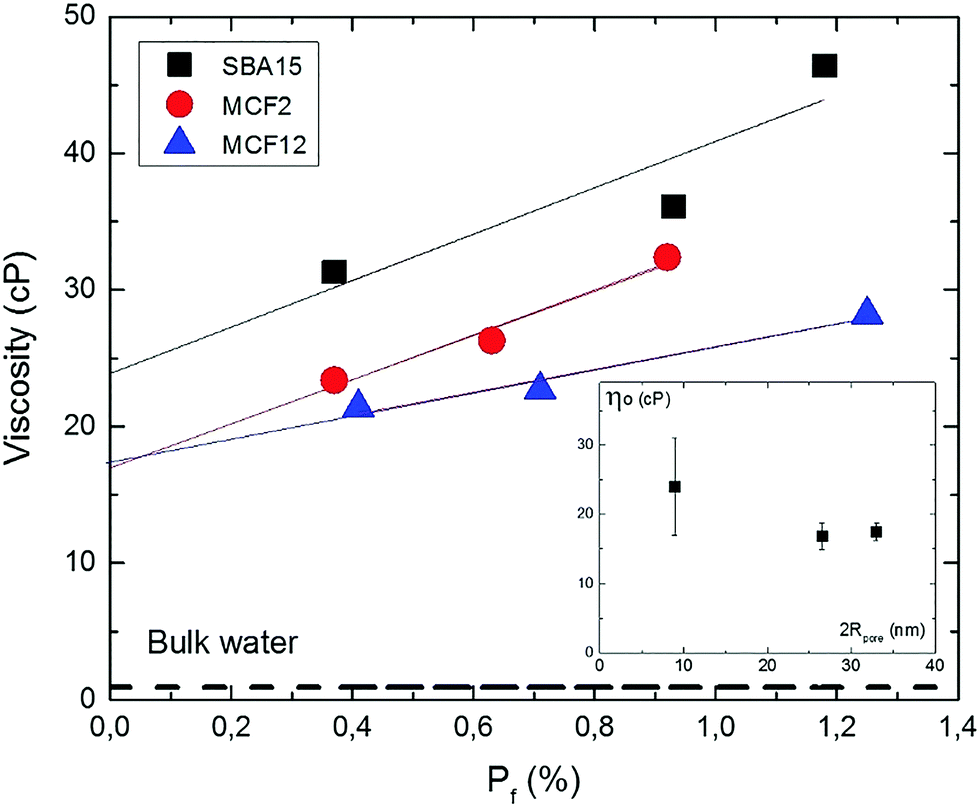 | ||
| Fig. 7 Effect of protein concentration on the effective viscosity in the pores. Plot of the viscosity data for the three particle types (Table 7) vs. pore filling Pf (Table 6) for SBA15 (black squares), MCF2 (red circles) and MCF12 (blue triangles). Experimental uncertainties are indicated by symbol size. Dashed line corresponds to the viscosity of bulk water at 25 °C. Straight lines are least square fits. The intercepts ηo (in cP) at Pf = 0 are 24 ± 7 cP (SBA15), 16.8 ± 1.9 cP (MCF2) and 17.5 ± 1.3 cP (MCF12). The inset shows ηo plotted vs. pore diameter (Table 3), with the uncertainties obtained from the linear fits. | ||
The viscosity ηo in the limit of infinite protein dilution in the pores (Pf = 0), obtained as the intercepts of the fitted straight lines in Fig. 7, are 24 ± 7, 16.8 ± 1.9 and 17.5 ± 1.3 cP for SBA15, MCF2 and MCF12, respectively. These values correspond to the effective viscosity sensed by a single protein in the pores since the effect of protein–protein interactions on the effective viscosity is removed by extrapolation to zero protein concentration.
The effective limiting viscosities (ηo) in Fig. 7 are higher than bulk water viscosity (dashed line in Fig. 7) by a factor of approximately 27 in the SBA15 pores and 19 in both MCF-particle types. These enhancement factors indicate that confinement in the silica pores confers a considerable increase in the effective water viscosity, by approximately one order of magnitude. The inset of Fig. 7 shows the values of ηo plotted versus pore diameter. In spite of the substantial uncertainties, the plot indicates that the enhancement of the effective water viscosity is higher in more narrow pores, and levels off at a value of about 15 cP if the pores are larger than 20 nm in diameter. Fukatsu et al.50 have used proton NMR to investigate the mobility of water in the pores of mesoporous silica particles, in the same range of pore diameters as investigated here. Interestingly, they have reported a strongly hindered water motion (as monitored by 1H-NMR relaxation time) below 10 nm pore size, and a levelling off between 15 and 30 nm to an approximately constant level of water dynamics about 10 times slower than in bulk water. Such a reduction in water mobility several nanometers away from the pore wall has been also detected in a study based on the optical Kerr effect.51 In contrast to pore sizes a few tens of nm studied here, the water dynamics in both larger and smaller pores have been studied extensively. The viscosity of pressure-driven flows in 200 nm wide channels is higher with a factor 2.5 than in bulk water.24 The dynamic properties of water molecules in silica particles with pore size in a few nm range (Vycor, MCM-41, SBA-15) has been studied by a wide range of techniques. Quasielastic neutron scattering (QENS) studies25,26,52 indicate that the translational diffusion coefficient of water at room temperature is about a factor two lower than in bulk water, as supported by detailed molecular dynamics simulations.53,54 However, water translation retardation factors as high as 30 have been reported in 1 nm pores by QENS27 and proton-NMR.55 Strong retardation of water rotation (by a factor of about 8) in 2 nm pores has also been observed through the optical Kerr effect.51
To summarize, the effective viscosity values that we report here, are higher than in the 200 nm silica channels as expected, but also larger than most of the reports which involve narrower pore sizes than this study. However, the molecular rotor which is attached to MML does not measure the viscosity of pure water in the pore, but the effective viscosity which the protein-bound probe experiences in the presence of the (single) protein. The effect of the protein itself on the water mobility in the pore may be substantial, because the protein is comparable in size to the pore radius and may perturb the flow-pattern of the water. Therefore, the average effective solvent viscosity experienced by the protein is better measured by a viscosity probe attached to protein itself rather than to the pore wall, since the protein can probe the whole pore lumen.
The high value of ηo in the SBA-particles (Fig. 7) supports our observation from a fluorescence anisotropy study of lipase and BSA in such particles12 that protein–wall interaction are more important rather than protein–protein hydrodynamic interactions in the retardation of protein rotation in the pore, as was concluded from a comparison with theory13,56 of experimental data at considerably higher protein concentrations than studied here. A stronger effect of protein–wall interactions is also consistent with that the average protein–protein distance (approximately 25 nm) at the highest volume fraction in SBA-15 (1.18%, see Table 6) is considerably larger than the maximum protein–wall distance (2 nm) at the corresponding pore diameter (8.9 nm, see Table 3).
The protein–protein interaction revealed by increasing viscosity for higher Pf in Fig. 7 is consistent with the reported decrease in protein diffusion coefficient in concentrated solutions of free BSA proteins (no particles),57 which was ascribed to an increase in protein–protein hydrodynamic interactions. Moreover, our results suggest that the protein–protein interaction effect is stronger in the pores than in bulk protein solution. In qualitative terms, the effective viscosity at 1.4% pore filling (Pf) of MML in the SBA-15 pores is doubled, to almost 50 cP (Fig. 7) compared to ηo (24 ± 7 cP), while the diffusion rate in the BSA experiments57 is reduced by a factor of 2 compared to diffusion coefficient in dilute solution (Do) only when the volume fraction of BSA has reached 10%. A systematic comparison with theory in this regard will require more experiments with MCF particles.
3.6. Effects of hydrophobic modification on modified MCF particles
The reason for including the octyl functionalized particles in this study was to alter the surface chemistry of the pore walls that may enhance enzyme efficiency, and to investigate how it can affect the effective viscosity. Tables 5 and 6 show that the hydrophobic surface modification with octyl groups increases the enzyme uptake in the MCF-particles by approximately a factor of 2 for a given amount of added protein. This observation is consistent with other studies regarding interaction between lipase and hydrophobic surfaces.47,58–60 Lipases are fairly hydrophilic and soluble in water,48,61,62 but there is typically a hydrophobic pocket containing their active site. In homogenous media the active site is covered by a surface, which isolates it from reaction medium (closed form). In an unusual mechanism of action called interfacial activation, in the presence of a hydrophobic surface the enzyme becomes adsorbed to it, leading to a new conformation, called open, where the active site is fully exposed.59 That the enzymes do experience a less polar pore environment in the OC-functionalized particles is supported by the observation that the emission peaks redshift (almost 3 nm)63 compared to MCFs with no modification (see ESI,† Section S.7).It has been also reported previously for alcohol dehydrogenase that the immobilization in MCF functionalized with OC resulted in higher specific activity in comparison to the enzyme immobilized in unmodified MCF.14 The authors suggested that the interactions between the alcohol dehydrogenase and the hydrophobic octyl groups on the surface of the MCF may have increased the enzyme stability and/or the enzyme became oriented inside the pores in a way that the active site is more available to the substrate.14
Many studies have shown that activity and stability of the immobilized enzyme increases,47,59 when immobilized on hydrophobic surfaces accompanied by a decrease in the mobility of lipases.60,64 It is therefore interesting to investigate if an enhanced effective viscosity in the OC-modified particles may lead to a similar protein retardation in the MCF-pores. However, the ratio-method could not be used to evaluate the effective viscosity inside the OC-modified particles because time-resolved fluorescence measurements clearly show FRET between Cy3-MML and Cy5-MML in the OC-modified particles even at the lowest added protein concentrations (see ESI,† Section S.5 and Fig. S.4). Interestingly, in a recent study Kubánková et al. reported that the fluorescence life time of Cy3 itself (no Cy5) can be used to estimate the solvent viscosity, if an appropriate calibration curve is established.65 We have not measured such a calibration curve for our system, but a qualitative comparison between OC-modified and non-modified MCF-particles using the lifetimes of immobilized Cy3-MML suggests that the viscosity might be higher after OC-modification (see ESI,† Section S.8)
Finally, the presence of FRET would seem to be consistent with the higher pore filling in the OC-modified particles compared to unmodified particles (Table 6), because shorter distance between two enzymes leads to higher FRET efficiencies. However, the pore filling in some cases of unmodified particles (e.g. 1.81% in SBA-15 at 60 μg mg−1) is higher, and still there is no FRET. Clearly, the presence of FRET does not only depend on the average protein concentration in the pores. One possibility is that the interaction with the (hydrophobic) pore wall that leads to more efficient uptake (Table 6) also leads to a less random spatial distribution of the enzymes (for instance concentrated along the pore walls) which decreases their average distances and therefore increases the FRET-efficiency. In addition to this potential of using FRET to monitor protein distribution in the pores, our results in the OC-modified particles also demonstrate the importance of confirming the absence of FRET if the ratio-method is to be used for viscosity measurements.
4. Concluding remarks
Cy3 and Cy5 is a good pair of protein-bound dyes to measure viscosity independently of other solvent effects such as polarity, if the two dyes are attached to different proteins. The use of the fluorescence lifetime of Cy3 itself (no Cy5) to measure viscosity65 may be less sensitive to light scattering by the particles, but intensity decays at a single wavelength can to our knowledge not distinguish between viscosity and polarity effects. Spectral shifts in the emission spectrum compared to free protein in water can be used to monitor the polarity of the pore environment.It is important to check for potential FRET if the labeled enzymes are studied in heterogeneous environments where protein–protein distances are potentially small, and the double-labeled proteins is then a useful positive controls for FRET in the actual experimental system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Hanna Gustafsson for the synthesis and characterization of SBA-15 silica particle and Dr Sandra Rocha for all valuable inputs on CD measurements. Funding for this project from the Swedish Research Council (VR) is acknowledged as a part of the Linnaeus Centre for Bio-inspired Supramolecular Function and Design – SUPRA.References
- C. Lei, T. A. Soares, Y. Shin, J. Liu and E. J. Ackerman, Nanotechnology, 2008, 19, 125102 CrossRef PubMed.
- C. Lei, Y. Shin, J. Liu and E. J. Ackerman, J. Am. Chem. Soc., 2002, 124, 11242–11243 CrossRef PubMed.
- S. Hudson, E. Magner, J. Cooney and B. K. Hodnett, J. Phys. Chem. B, 2005, 109, 19496–19506 CrossRef PubMed.
- S. Hudson, J. Cooney and E. Magner, Angew. Chem., Int. Ed., 2008, 47, 8582–8594 CrossRef PubMed.
- B. Chen, W. Qi, X. Li, C. Lei and J. Liu, Small, 2013, 9, 2228–2232 CrossRef PubMed.
- M. Radhakrishna, J. Grimaldi, G. Belfort and S. K. Kumar, Langmuir, 2013, 29, 8922–8928 CrossRef PubMed.
- P. S. Nabavi Zadeh, K. A. Mallak, N. Carlsson and B. Åkerman, Anal. Biochem., 2015, 476, 51–58 CrossRef PubMed.
- J. Grimaldi, M. Radhakrishna, S. K. Kumar and G. Belfort, Langmuir, 2015, 31, 1005–1010 CrossRef PubMed.
- A. Yamaguchi, M. Namekawa, T. Kamijo, T. Itoh and N. Teramae, Anal. Chem., 2011, 83, 2939–2946 CrossRef PubMed.
- C. Thörn, N. Carlsson, H. Gustafsson, K. Holmberg, B. Åkerman and L. Olsson, Microporous Mesoporous Mater., 2013, 165, 240–246 CrossRef.
- D. Panda, S. Khatua and A. Datta, J. Phys. Chem. B, 2007, 111, 1648–1656 CrossRef PubMed.
- P. S. Nabavi Zadeh and B. Åkerman, J. Phys. Chem. B, 2017, 121, 2575–2583 CrossRef PubMed.
- R. B. Jones, J. Chem. Phys., 2005, 123, 164705 CrossRef PubMed.
- M. Zezzi do Valle Gomes and A. E. C. Palmqvist, New J. Chem., 2017, 41, 11391–11397 RSC.
- M. Zezzi do Valle Gomes and A. E. C. Palmqvist, Colloids Surf., B, 2018, 163, 41–46 CrossRef PubMed.
- A. Yamaguchi, M. Namekawa, T. Itoh and N. Teramae, Anal. Sci., 2012, 28, 1065–1070 CrossRef PubMed.
- G. Weber, M. Shinitzky, A. C. Dianoux and C. Gitler, Biochemistry, 1971, 10, 2106–2113 CrossRef.
- F. Perrin, Ann. Phys., 1929, 10, 169–275 Search PubMed.
- M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669–1678 RSC.
- M. K. Kuimova, G. Yahioglu, J. A. Levitt and K. Suhling, J. Am. Chem. Soc., 2008, 130, 6672–6673 CrossRef PubMed.
- K. Luby-Phelps, S. Mujumdar, R. B. Mujumdar, L. A. Ernst, W. Galbraith and A. S. Waggoner, Biophys. J., 1993, 65, 236–242 CrossRef PubMed.
- E. Lerner, E. Ploetz, J. Hohlbein, T. Cordes and S. Weiss, J. Phys. Chem. B, 2016, 120, 6401–6410 CrossRef PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Baltimor, USA, 3rd edn, 2006 Search PubMed.
- L. Li, Y. Kazoe, K. Mawatari, Y. Sugii and T. Kitamori, J. Phys. Lett., 2012, 3, 2447–2452 Search PubMed.
- S. Takahara, N. Sumiyama, S. Kittaka, T. Yamaguchi and M.-C. Bellissent-Funel, J. Phys. Chem. B, 2005, 109, 11231–11239 CrossRef PubMed.
- S. Takahara, M. Nakano, S. Kittaka, Y. Kuroda, T. Mori, H. Hamano and T. Yamaguchi, J. Phys. Chem. B, 1999, 103, 5814–5819 CrossRef.
- J. Swenson, H. Jansson, W. S. Howells and S. Longeville, J. Chem. Phys., 2005, 122, 084505 CrossRef PubMed.
- X. Y. Wu, S. Jaaskelainen and Y. Y. Linko, Appl. Biochem. Biotechnol., 1996, 59, 145–158 CrossRef.
- H. Gustafsson, C. Thörn and K. Holmberg, Colloids Surf., B, 2011, 87, 464–471 CrossRef PubMed.
- P. Schmidt-Winkel, W. W. Lukens, P. Yang, D. I. Margolese, J. S. Lettow, J. Y. Ying and G. D. Stucky, Chem. Mater., 2000, 12, 686–696 CrossRef.
- P. A. Russo, M. M. L. Ribeiro Carrott, P. A. M. Mourão and P. J. M. Carrott, Colloids Surf., A, 2011, 386, 25–35 CrossRef.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef.
- W. W. Lukens, P. Schmidt-Winkel, D. Zhao, J. Feng and G. D. Stucky, Langmuir, 1999, 15, 5403–5409 CrossRef.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef.
- N. Carlsson, H. Gustafsson, C. Thörn, L. Olsson, K. Holmberg and B. Åkerman, Adv. Colloid Interface Sci., 2014, 205, 339–360 CrossRef PubMed.
- E. Boel, B. Huge-Jensen, M. Christensen, L. Thim and N. P. Fiil, Lipids, 1988, 23, 701–706 CrossRef PubMed.
- J. B. S. A. H. E. Oberstak, Ind. Eng. Chem., 1951, 43, 2117–2120 CrossRef.
- J. Timmermans, The Physico-chemical Constants of Binary Systems in Concentrated Solutions, Interscience, 1960.
- S. Hohng, C. Joo and T. Ha, Biophys. J., 2004, 87, 1328–1337 CrossRef PubMed.
- Y. Sun, S. I. Shopova, C.-S. Wu, S. Arnold and X. Fan, Proceedings of the National Academy of Sciences, 2010, 107, 16039–16042 CrossRef PubMed.
- H. Lee, M. Y. Berezin, M. Henary, L. Strekowski and S. Achilefu, J. Photochem. Photobiol., A, 2008, 200, 438–444 CrossRef PubMed.
- H. Giudicelli and J. Boyer, J. Lipid Res., 1973, 14, 592–595 Search PubMed.
- W. Ou, Y. Park, F. Meng and H. Zhou, Tsinghua Sci. Technol., 2002, 7, 352–362 Search PubMed.
- R. V. Rariy and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13520–13523 CrossRef.
- M. K. Kuimova, S. W. Botchway, A. W. Parker, M. Balaz, H. A. Collins, H. L. Anderson, K. Suhling and P. R. Ogilby, Nat. Chem., 2009, 1, 69–73 CrossRef PubMed.
- M. A. Haidekker, T. P. Brady, D. Lichlyter and E. A. Theodorakis, J. Am. Chem. Soc., 2006, 128, 398–399 CrossRef PubMed.
- R. Fernandez-Lafuente, P. Armisén, P. Sabuquillo, G. Fernández-Lorente and J. M. Guisán, Chem. Phys. Lipids, 1998, 93, 185–197 CrossRef PubMed.
- A. M. Brzozowski, U. Derewenda, Z. S. Derewenda, G. G. Dodson, D. M. Lawson, J. P. Turkenburg, F. Bjorkling, B. Huge-Jensen, S. A. Patkar and L. Thim, Nature, 1991, 351, 491–494 CrossRef PubMed.
- P. Skagerlind, M. Jansson, B. Bergenståhl and K. Hult, Colloids Surf., B, 1995, 4, 129–135 CrossRef.
- Y. Fukatsu, K. Morikawa, Y. Ikeda and T. Tsukahara, Anal. Sci., 2017, 33, 903–909 CrossRef PubMed.
- A. Scodinu and J. T. Fourkas, J. Phys. Chem. B, 2002, 106, 10292–10295 CrossRef.
- A. Faraone, L. Liu, C.-Y. Mou, P.-C. Shih, J. R. D. Copley and S.-H. Chen, J. Chem. Phys., 2003, 119, 3963–3971 CrossRef.
- S. Romero-Vargas Castrillón, N. Giovambattista, I. A. Aksay and P. G. Debenedetti, J. Phys. Chem. B, 2009, 113, 1438–1446 CrossRef PubMed.
- A. A. Milischuk and B. M. Ladanyi, J. Chem. Phys., 2011, 135, 174709 CrossRef PubMed.
- E. Walther Hansen, R. Schmidt, M. Stöcker and D. Akporiaye, Microporous Mater., 1995, 5, 143–150 CrossRef.
- V. Degiorgio, R. Piazza and R. B. Jones, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1995, 52, 2707–2717 CrossRef.
- F. Roosen-Runge, M. Hennig, F. Zhang, R. M. Jacobs, M. Sztucki, H. Schober, T. Seydel and F. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11815–11820 CrossRef PubMed.
- D. I. Fried, D. Bednarski, M. Dreifke, F. J. Brieler, M. Thommes and M. Froba, J. Mater. Chem. B, 2015, 3, 2341–2349 RSC.
- E. A. Manoel, J. C. S. dos Santos, D. M. G. Freire, N. Rueda and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2015, 71, 53–57 CrossRef PubMed.
- A. W. Sonesson, U. M. Elofsson, H. Brismar and T. H. Callisen, Langmuir, 2006, 22, 5810–5817 CrossRef PubMed.
- K. K. Kim, H. K. Song, D. H. Shin, K. Y. Hwang and S. W. Suh, Structure, 1997, 5, 173–185 CrossRef PubMed.
- M. Cygler and J. D. Schrag, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1999, 1441, 205–214 CrossRef.
- N. Carlsson, A.-S. Winge, S. Engström and B. Åkerman, J. Phys. Chem. B, 2005, 109, 18628–18636 CrossRef PubMed.
- A. W. Sonesson, H. Brismar, T. H. Callisen and U. M. Elofsson, Langmuir, 2007, 23, 2706–2713 CrossRef PubMed.
- M. Kubánková, I. López-Duarte, J. A. Bull, D. M. Vadukul, L. C. Serpell, M. de Saint Victor, E. Stride and M. K. Kuimova, Biomaterials, 2017, 139, 195–201 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp01063c |
| This journal is © the Owner Societies 2018 |