
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Singlet and triplet energy transfer dynamics in self-assembled axial porphyrin–anthracene complexes: towards supra-molecular structures for photon upconversion†
Victor
Gray
 ,
Betül
Küçüköz
,
Fredrik
Edhborg
,
Maria
Abrahamsson
,
Kasper
Moth-Poulsen
and
Bo
Albinsson
*
,
Betül
Küçüköz
,
Fredrik
Edhborg
,
Maria
Abrahamsson
,
Kasper
Moth-Poulsen
and
Bo
Albinsson
*
Chalmers University of Technology, Department of Chemistry and Chemical Engineering, Gothenburg, Sweden. E-mail: balb@chalmers.se
First published on 12th February 2018
Abstract
Energy and electron transfer reactions are central to many different processes and research fields, from photosynthesis and solar energy harvesting to biological and medical applications. Herein we report a comprehensive study of the singlet and triplet energy transfer dynamics in porphyrin–anthracene coordination complexes. Seven newly synthesized pyridine functionalized anthracene ligands, five with various bridge lengths and two dendrimer structures containing three and seven anthracene units, were prepared. We found that triplet energy transfer from ruthenium octaethylporphyrin to an axially coordinated anthracene is possible, and is in some cases followed by back triplet energy transfer to the porphyrin. The triplet energy transfer follows an exponential distance dependence with an attenuation factor, β, of 0.64 Å−1. Further, singlet energy transfer from anthracene to the ruthenium porphyrin appears to follow a R6 Förster distance dependence. Porphyrin–anthracene complexes are also used as triplet sensitizers for triplet–triplet annihilation (TTA) based photon upconversion, demonstrating their potential for photophysical and photochemical applications. The triplet lifetime of the complex is extended by the anthracene ligands, resulting in a threefold increase in the upconversion efficiency, ΦUC to 4.5%, compared to the corresponding ruthenium porphyrin–pyridine complex. Based on the results herein we discuss the future design of supra-molecular structures for TTA upconversion.
Porphyrins, the heterocyclic 18 π-electron conjugated system common in many natural and biological systems, and their related structures are widely studied. In nature, they are essential for light harvesting, energy and electron transfer reactions, catalysis and oxygen transport.1–3 Accordingly, scientists have studied porphyrin compounds and developed numerous model systems to understand and exploit these versatile and useful compounds. A variety of uses have been explored, such as: light harvesting antennas for artificial photosynthesis and solar energy conversion devices;4–12 singlet oxygen sensitizers for photodynamic therapy;13 wires in molecular electronics;14–17 and triplet sensitizers for triplet–triplet annihilation based photon upconversion,18–21 to name a few. In both the natural and man-made systems, understanding the mechanisms of energy and electron transfer reactions is crucial. For example, in plants and photosynthetic bacteria triplet energy transfer (TET) occurs from the light-absorbing chlorophyll moiety to pendant carotenoids as a way of protecting the biological species against reactive singlet oxygen.22–24 Engineered photosynthetic systems with increased efficiencies will require even greater oxygen protection, motivating further exploration of TET in model porphyrin donor–acceptor systems.24 Furthermore, in the development of supramolecular structures designed for photochemical applications, such as triplet–triplet annihilation based photon upconversion25–27 or photocatalysis28–30 the delicate balance between energy and electron transfer reactions requires careful tuning.
As part of the ongoing studies of energy and electron transfer reactions in porphyrin donor–acceptor system for various photochemical and electrochemical applications,4,5,7,24,25,31–44 we present a detailed photophysical study of a series of new, well-defined, self-assembled, axially coordinated ruthenium porphyrin–anthracene donor–acceptor systems. These systems consist of 2,3,7,8,12,13,17,18-octaethylporpyrin ruthenium(II) carbonyl (RuOEP(CO)) coordinated axially by five different pyridine functionalized anthracene based ligands with various bridge lengths (8–25 Å) or two different dendrimer ligands with 3–7 monomer units. TET from the porphyrin to the anthracene ligand and singlet energy transfer (SET) in the opposite direction are consequently studied as a function of bridge length, and ligand dendrimer size. Transient absorption spectroscopy and time resolved phosphorescence spectroscopy are combined with steady state techniques to study the energy landscape of the complexes. We find that TET to the ligand extends the triplet lifetime of the complex from 20 μs up to 600 μs. With an extended triplet lifetime the sensitization capabilities are improved and we employ the complexes as sensitizers for sensitized triplet–triplet annihilation photon upconversion (TTA-UC). Aiming towards efficient TTA-UC materials in the solid state we further discuss the capabilities of these complexes as sensitizers in supra-molecular TTA-UC systems. As such, this comprehensive study of anthracene–porphyrin complexes unravels important design principles for supra-molecular systems achieving efficient energy transfer.
1 Results and discussion
1.1 Ligand design and synthesis
Ligand design is based on our previous work with 9,10-diphenylanthracene (DPA) based ligands and dendrimeric structures.25,45Fig. 1 shows the structures of the ligands: ligand 1 has a meta-pyridine substitution, ligands 2–5 have para-pyridine substitutions with increasing bridge lengths and dendrimeric ligands 6 and 7 have an increasing number of DPA monomer units. Fig. 1 also shows the structure of the triplet sensitizer octaethylporphyrin ruthenium(II) carbonyl (RuOEP(CO)).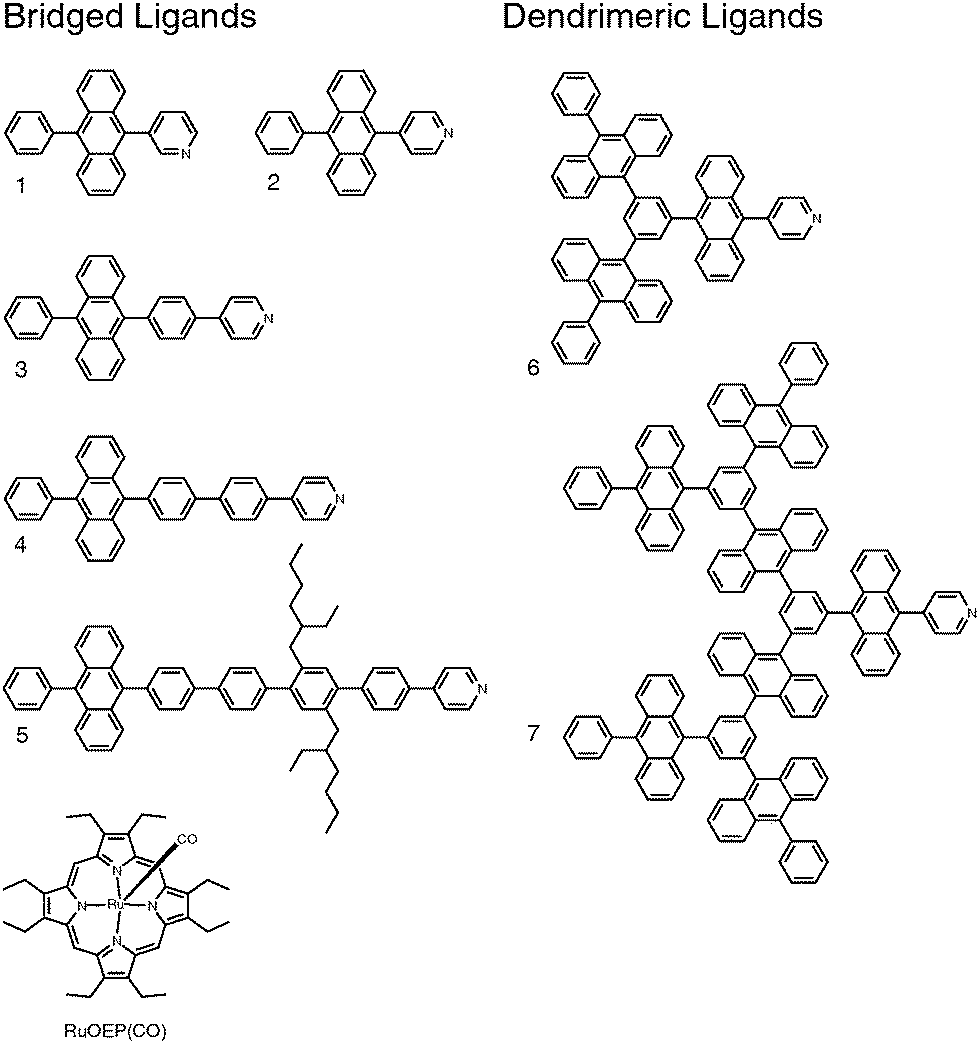 | ||
| Fig. 1 Structures of the studied anthracene ligands 1–7 and the octaethylporphyrin ruthenium(II) carbonyl RuOEP(CO). | ||
The synthesis of ligands 1–5 was reported previously.25 The dendrimer ligands were synthesized by repeating a sequence of three high yielding and simple reactions; bromination followed by borylation followed by a Suzuki-coupling, similar to our previously established route for DPA dendrons and dendrimers.45 The synthetic route is illustrated in Fig. S1 and described in more detail in the ESI.† Similar to what was reported previously RuOEP(CO) was obtained by refluxing the corresponding free-base porphyrin with Ru3CO12.46–48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ‡
‡
1.2 Binding equilibrium
Upon mixing the porphyrin RuOEP(CO) with pyridine (Pyr) or the pyridine containing ligands 1–7, pyridine–porphyrin complexes RuOEP(CO)L (L = 1–7 or Pyr) are formed. The binding is followed using UV/Vis absorption spectroscopy, as seen in Fig. 2 and Fig. S8 (ESI†). Upon binding to pyridine the porphyrin Q-band absorption is red-shifted49,50 and the complex Q-band has absorption peaks at 549 and 518 nm.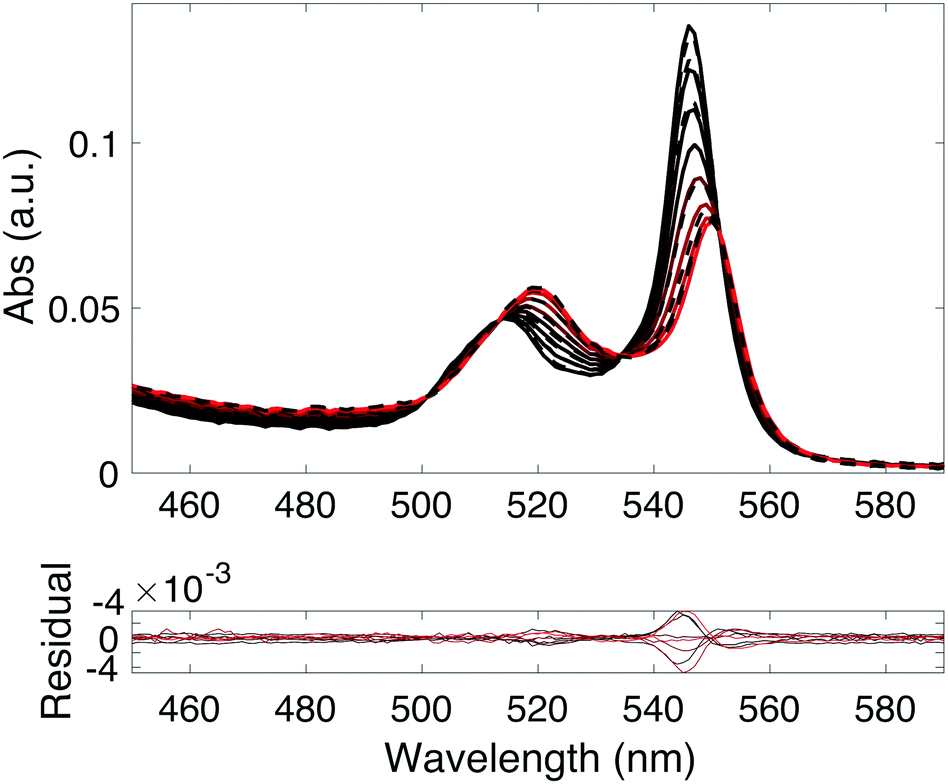 | ||
| Fig. 2 Shift in RuOEP(CO) (0.1 mM) absorption as ligand 1 is added (0–0.1 mM) to form complex RuOEP(CO)1. Measured (dashed) and predicted (solid) spectra with residual below. Titration progression from black to red spectra. | ||
The pyridine–ruthenium porphyrin bond is very strong, with a binding constant in the order of 106–107 M−1 (ESI,† Table S1), to be compared to the pyridine–zinc porphyrin bond of 6000 M−1, reported in our previous work.25 The larger binding constant leads to about 90% of the ligand being bound to the porphyrin at a 1:1 mixture of RuOEP(CO) (total porphyrin concentration is 0.1 mM) and L (total ligand concentration is 0.1 mM). To form significant amounts of the zinc porphyrin–pyridine ligand complexes large excess of the ligand is required. The ruthenium based complexes are therefore better suited for fundamental studies since well-defined complexes can easily be formed.
1.3 Triplet energy transfer and phosphorescence quenching
RuOEP(CO)Pyr is slightly phosphorescent in degassed toluene solutions, Fig. S9 (ESI†), with a quantum yield (ΦP) of 0.68%. From the phosphorescence onset the triplet state energy 3RuOEP(CO)Pyr* is estimated to be 1.90 eV (650 nm), corresponding to a singlet–triplet excited state splitting of 0.35 eV.The triplet energy of the anthracene ligands is expected to be similar to that of DPA, 1.77 eV.51,52 Therefore, a small driving force for TET from the porphyrin to the anthracene ligand exists. A common way of studying TET from phosphorescent porphyrins is by monitoring the phosphorescence quenching of the sensitizer by an acceptor. As can be seen in Table 1 there is only partial quenching of the RuOEP(CO)L phosphorescence compared to RuOEP(CO)Pyr. At first this low degree of quenching and that all compounds show similar phosphorescence quantum yields might seem surprising; its origin will be discussed in the following paragraph.
| Ligand | Φ p (%) | k TET (s−1) | k bTET (s−1) | τ TL (μs) |
|---|---|---|---|---|
| a Phosphorescence quantum yields, ΦP determined in degassed toluene relative to Cresyl violet in methanol. kTET is the triplet energy transfer (TET) rate, kbTET is the rate of back TET and the intrinsic lifetime of the ligand is τTL = kTL−1. | ||||
| Pyr | 0.68 ± 0.10 | — | — | — |
| 1 | 0.29 ± 0.07 | >10 × 109 | >2 × 108 | 680 |
| 2 | 0.29 ± 0.01 | 3.6 × 107 | 7.3 × 105 | 735 |
| 3 | 0.29 ± 0.01 | 7.6 × 105 | 2.1 × 104 | 500 |
| 4 | 0.30 ± 0.05 | 8.4 × 104 | 1.7 × 103 | 190 |
| 5 | 0.42 ± 0.12 | 3.3 × 104 | 0.2 × 103 | 315 |
| 6 | 0.19 ± 0.01 | 3.7 × 107 | 3.4 × 105 | 820 |
| 7 | 0.11 ± 0.01 | 3.8 × 107 | 2.4 × 105 | 610 |
Nanosecond resolved transient absorption (TA) and time resolved phosphorescence measurements were both performed to further study the TET process, and the results are summarized in Table 2. The TA spectra of the RuOEP(CO)L complexes are found in Fig. 3 and Fig. S10 (ESI†). RuOEP(CO)Pyr displays strong T1–Tn excited state absorption (ESA) in the 400–500 nm region and a distinct ground state bleach (GSB) at 520 nm and 550 nm, in accordance with the ground state absorption spectra. Both the ESA and GSB decay with a time constant of 20 μs, in accordance with the phosphorescence decay, vide infra.
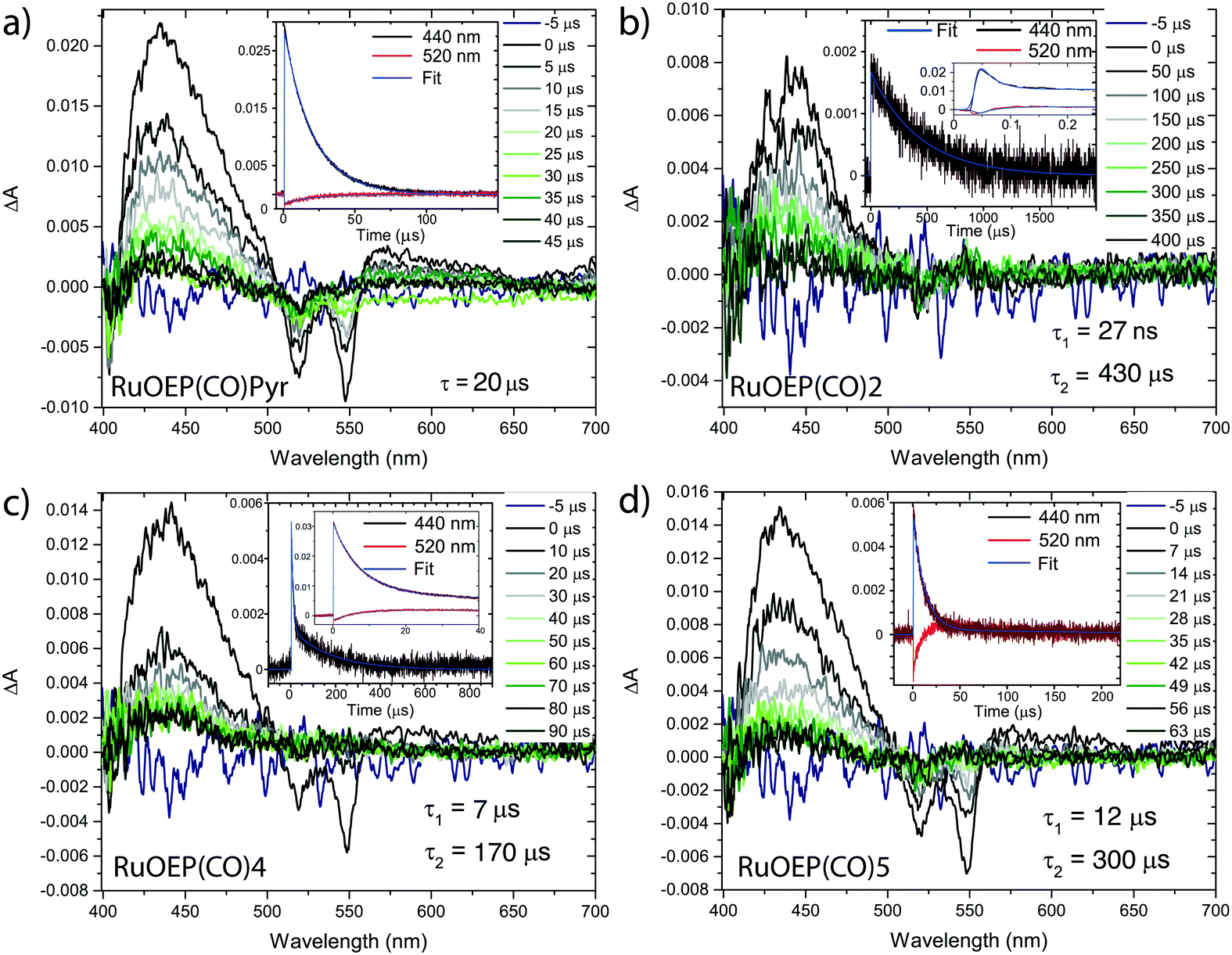 | ||
| Fig. 3 Transient absorption spectra of (a) RuOEP(CO)Pyr, (b) RuOEP(CO)2, (c) RuOEP(CO)4 and (d) RuOEP(CO)5. All samples excited at 550 nm in degassed toluene. | ||
With the pyridine anthracene ligand 2 the ground state bleach decays with τ = 27 ns, Table 2, as does the strong porphyrin T1–Tn ESA, which decays into a longer lived (τ = 430 μs), weaker ESA in the same 400–500 nm region. This weaker ESA feature corresponds to the T1–Tn transition of the anthracene unit. The fast decay of the porphyrin centered triplet followed by the observation of an excited anthracene triplet suggest that there is efficient TET from the porphyrin to the ligand; 3RuOEP*(CO)L → RuOEP(CO)3L*. For anthracene ligands with longer bridges the 3RuOEP*(CO)L triplet decays slower, Fig. 3 and Fig. S10 (ESI†), indicating a slower TET, as expected by the exponential distance dependence of TET.36,37,40
For the larger dendrimeric ligands, 6 and 7, the porphyrin triplet also decays in about 27 ns, the same as for 2, Fig. S10 (ESI†). As the shortest porphyrin–anthracene distance is expected to be the same in these three complexes, the TET rate is expected to be similar. The observed longer decay time increases slightly in the larger ligands, from 430 μs to 600 μs and 520 μs for 2, 6 and 7, respectively. Ligand 1, with a meta-pyridine, displays the fastest decay of the porphyrin triplet, faster than the time-resolution of our TA setup. The expected tilted binding in RuOEP(CO)1 most likely increases the orbital overlap between the porphyrin triplet, located on the ring π-system, and the anthracene unit, explaining the increased TET rate.
Since the TET is only slightly exergonic in the studied systems (∼0.13 eV) it is also possible for TET back (bTET) from the anthracene ligand to the porphyrin to occur; RuOEP(CO)3L* → 3RuOEP*(CO)L. In other words, the two states could be in equilibrium; 3RuOEP*(CO)L ⇌ RuOEP(CO)3L*. To obtain the actual TET rate, both the forward and backward TET must be considered when analysing the biexponential decays obtained from the TA-measurements. Eqn (1) and (2) describe the relationship between the observed decay times τ1 and τ2 (Table 2) with the rate of TET and bTET, respectively:
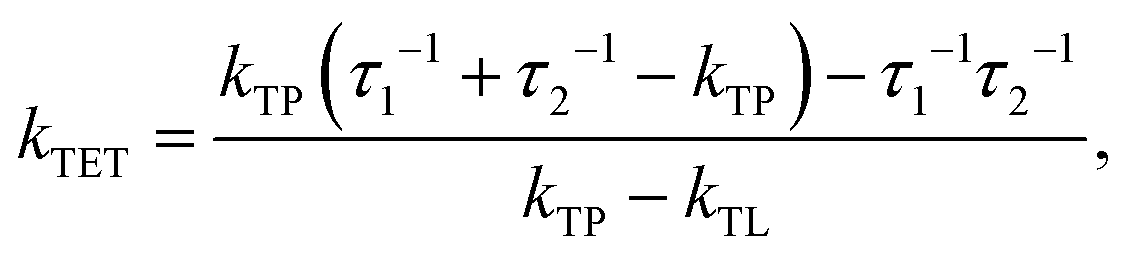 | (1) |
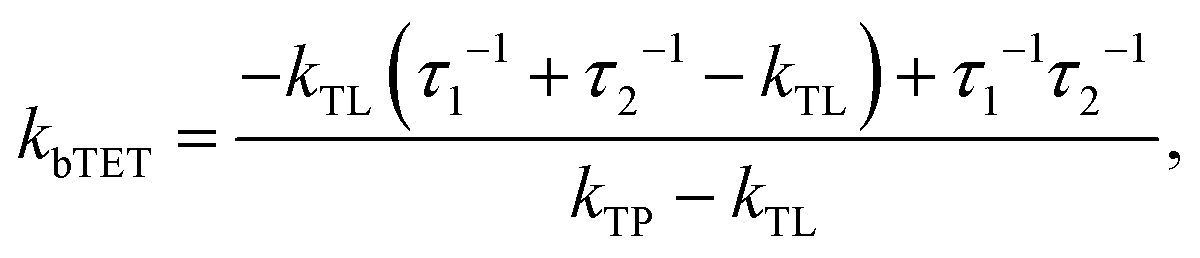 | (2) |
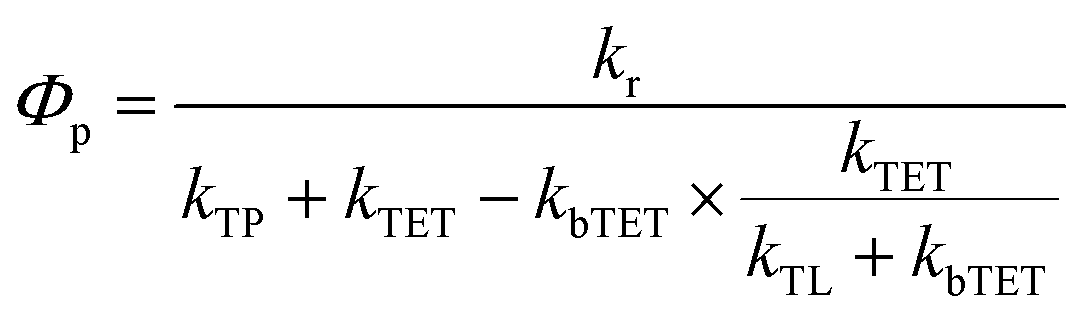 | (3) |
For the shortest bridges the effect of bTET is small and kTET is similar to τ1−1. In complexes with larger donor–acceptor separation the TET and bTET are slower, and the effect of bTET is larger. Due to microscopic reversibility the rate kbTET is related to kTET through the Boltzmann factor as described in eqn (4):
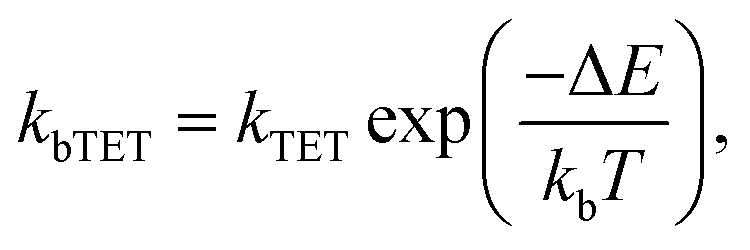 | (4) |
Furthermore, the energy transfer dynamics were also monitored for some of the samples using time-resolved phosphorescence, Table 2 and Fig. S11 (ESI†). In μs emission measurements, the donor only complex, RuOEP(CO)Pyr, displays a phosphorescence decay of 17 μs, comparable to that observed for the triplet decay in the TA measurements, Table 2. In the donor–acceptor complexes where the TET is below the time-resolution of our phosphorescence measurements only a longer lifetime is observed, supporting that bTET occurs resulting in the observed delayed phosphorescence. The dendrimeric ligand complexes RuOEP(CO)6 and RuOEP(CO)7 also show mono exponential delayed phosphorescence lifetimes. In complexes RuOEP(CO)4 and RuOEP(CO)5, however, a biexponential decay is observed and in both cases the shorter of the two lifetimes are in good agreement to the TET rate determined from TA measurements. The longer lifetime is again longer than the donor only complex, further supporting bTET as an explanation to the minor phosphorescence quenching. The magnitude of the long lifetimes agrees well between experiments, there is however some discrepancy between the exact lifetimes in the different experiments. One reason can be that in nanosecond TA spectroscopy the long lived ligand state is directly probed and its decay is accurately monitored. In the phosphorescence measurements, on the other hand, the emissive porphyrin triplet state is monitored and the long lived ligand state is thus monitored indirectly. Therefore, the long lived component is expected to only make up a few percent of the total decay making it difficult to measure with the same accuracy.
Fig. 4 shows the dependence of the TET rate as a function of donor–acceptor distance, RDA. Since TET processes proceed through an electron exchange mechanism an exponential distance dependence is expected:36,37
| kTET = k0exp(−βRDA) | (5) |
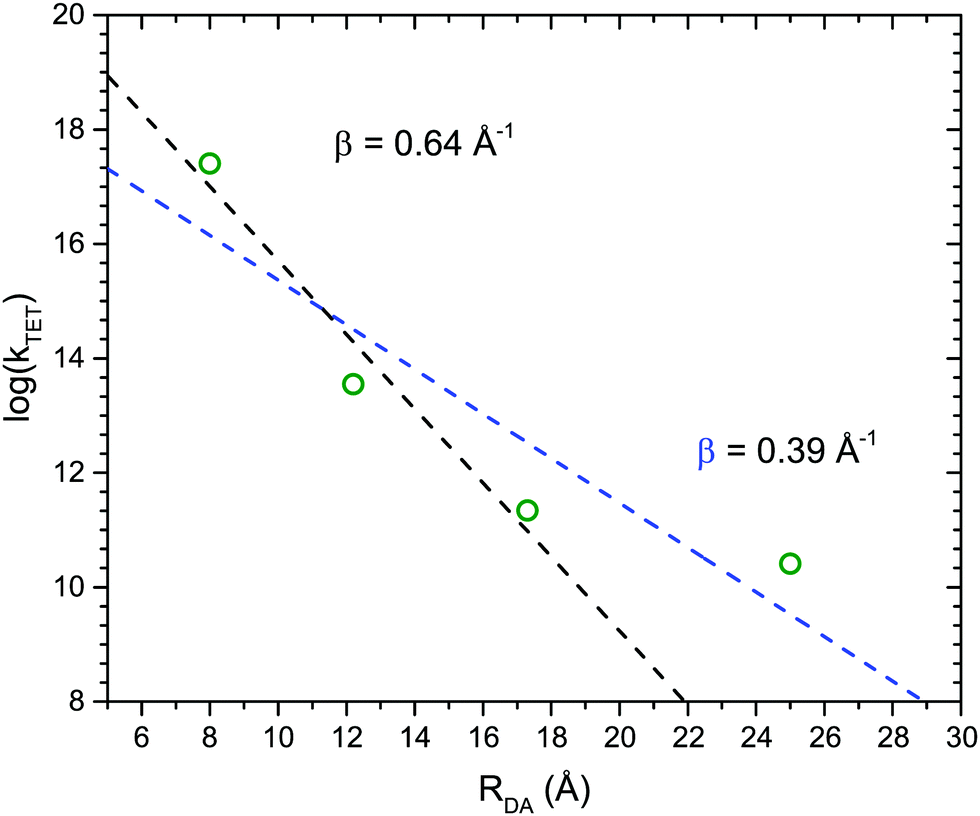 | ||
| Fig. 4 Triplet energy transfer rates (kTET) as a function of donor–acceptor separation (RDA) in toluene. The triplet energy transfer rate decreases exponentially with RDA, with an attenuation factor β = 0.39 or 0.64 Å−1, depending on if the longest bridge (RuOEP(CO)5, RDA = 25 Å) is included in the linear fit or not. | ||
1.4 Singlet energy transfer from anthracene ligands to RuOEP(CO)
Due to the large singlet–triplet splitting in anthracene chromophores, 1.3–1.4 eV, the anthracene ligands function either as triplet acceptors or singlet donors in the studied RuOEP(CO)L complexes. Upon complex formation the prompt anthracene emission is quenched by more than 95%, indicating efficient singlet energy transfer (SET) to the porphyrin. Fig. 5 displays the ps-resolved fluorescence decays of complexes with ligands 3–5 and 7; fluorescence decays of ligands with shorter bridges could not be resolved on the current setup. The decays are biexponential, where the fast component is attributed to SET and the longer component to residual emission from unbound ligand. The fraction of the long component in the decay is not directly related to the fraction of unbound species in the sample, as the lifetime of the free ligand is longer than the repetition rate (80 MHz) a long lived emission signal is built up over time. However, the fact that complexes with ligands 4, 5 or 7 all have similar fractions of long lived components indicate that the amount of free ligand is similar in all samples, as expected. From the binding constants we estimate that <10% free ligand is present in the samples. The much larger fraction of the long lived component of complex RuOEP(CO)3 results from the fact that the fast component is shorter than the pulse width. We can still obtain a range of reasonable values for the short component for RuOEP(CO)3 by expecting a similar fraction of the long lived component and deconvolving the signal with a reasonable pulse width. Further details of the fitting can be found in the ESI.†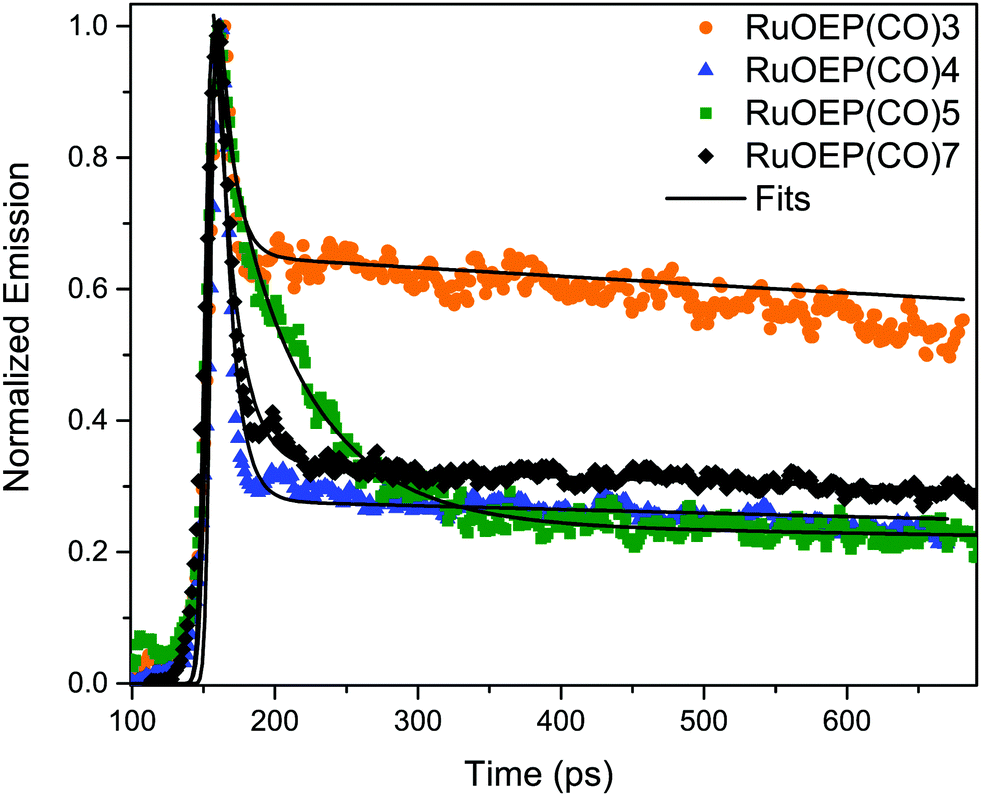 | ||
| Fig. 5 Fluorescence decays of complexes RuOEP(CO)L, where L is 3, 4, 5 or 7. Samples excited at 375 nm and decays are the average emission between 415–440 nm. | ||
The SET in complex RuOEP(CO)5 occurs with τSET = 48 ps, Table 3. As expected, shorter bridges exhibit faster SET, τSET is about 5 ps for complex RuOEP(CO)4. Shortening the bridge further, as in complex RuOEP(CO)3, results in a SET that approaches the limit of our time resolution, observed as an increased fraction of the long lived component in the decay. As discussed above we can still estimate the SET rate to be between 0.5 and 3 ps by assuming a similar fraction of residual fluorescence as observed in complexes RuOEP(CO)4, RuOEP(CO)5 and RuOEP(CO)7. In complexes RuOEP(CO)1, RuOEP(CO)2 and RuOEP(CO)6 SET is much faster than our time resolution and only the long lived component is observed. It should be pointed out, however, that the steady state emission is quenched almost quantitatively in all complexes. Therefore, the long lived component must arise by residual amounts of unbound ligand, also in complexes RuOEP(CO)1, RuOEP(CO)2 and RuOEP(CO)6 where the decay component related to SET is not resolved. An interesting observation is that, in the complex with the largest dendrimeric ligand 7, SET is slowed down compared to the monomer ligand 2 and second generation dendrimer ligand 6. It indicates that on average the excited singlet state is located further away in the larger structures.
| Ligand | τ SET (ps) | τ 0 (ns) |
|---|---|---|
| a τ SET refers to the quenched lifetime of the anthracene fluorescence in the RuOEP(CO)L complex and τ0 is the unquenched lifetime. b Determined for the ligand in separate TCSPC experiments and fixed in the analysis of the shorter lifetimes τSET. c Lifetimes estimated assuming that the singlet energy transfer is solely governed by FRET, they should therefore be considered as an upper limit. | ||
| 1 | — | 7.1 |
| 2 | — (0.1c) | 7.1 |
| 3 | 0.5–3 (0.8c) | 4.3 |
| 4 | 5 ± 1 (5c) | 3.8 |
| 5 | 48 ± 2 | 4.0 |
| 6 | — | 5.9 |
| 7 | 8 ± 1 | 5.4 |
The rate of SET in complexes RuOEP(CO)L, with L = 3–5, follows a RDA6 dependence, Fig. S12 (ESI†), indicating that SET is governed by FRET with a Förster distance R0 = 50 Å. Based on the FRET mechanism and taking the spectral overlap between ligand emission and porphyrin absorption into account, the lifetimes can be estimated to 0.1 ps for the shortest bridged ligand 2 in the complex, see the ESI† for details about the calculations. As can be seen in Table 3, the estimated lifetimes for complexes RuOEP(CO)3, 0.8 ps, and RuOEP(CO)4, 5 ps, are in good agreement with our experimental observations and also confirm that the expected lifetimes in the shortest bridged complex, RuOEP(CO)2, are well below our time resolution.
R 0 for the RuOEP(CO)L complexes is about 10 Å larger than for the corresponding zinc octaethylporphyrin (ZnOEPL) complexes, resulting in the SET in the current study being faster compared to similar complexes based on ZnOEP.25 For example, the SET in RuOEP(CO)5 is three times faster compared to the corresponding zinc complex ZnOEP5. The faster SET in the ruthenium complex cannot be explained by the difference in the Förster overlap integral (J) as it actually is smaller for the case of the ruthenium complex. One significant difference between the zinc and ruthenium complexes is the ligand binding strength. The Zn–N bond is quite weak (Kbind ∼ 103–104 M−1), resulting in a dynamic ZnOEPL complex. With three orders of magnitude larger binding constant (Kbind ∼ 106–107 M−1) the Ru–N bond should be stronger, making the RuOEP(CO)L complex more stable. Therefore, one possible explanation for the faster SET in the ruthenium complexes could be that the through bond SET is greater in the stronger bonding ruthenium complex.37,60,61 With the longer ligands, however, where the porphyrin anthracene separation is large, close to 25 Å, it is expected that FRET is the dominating SET mechanism. Therefore, considering FRET, the faster SET could imply that the ligand can move away from the 90° binding angle to a larger extent in the ruthenium case, as schematically described in Fig. 6. A greater deviation from the 90° binding angle results in a more favourable geometry for FRET. For a detailed discussion on the angle dependence the reader is referred to ref. 25. In short, an apparent binding angle can be estimated based on the fluorescence quenching experiments and a known spectral overlap.25 For the RuOEP(CO)L complexes we estimate an apparent binding angle of 45°, compared to >70° in ZnOEPL.25 We find this large difference, however, highly unlikely, especially considering the stronger ruthenium–pyridine bond, possibly indicates that another process is also involved.
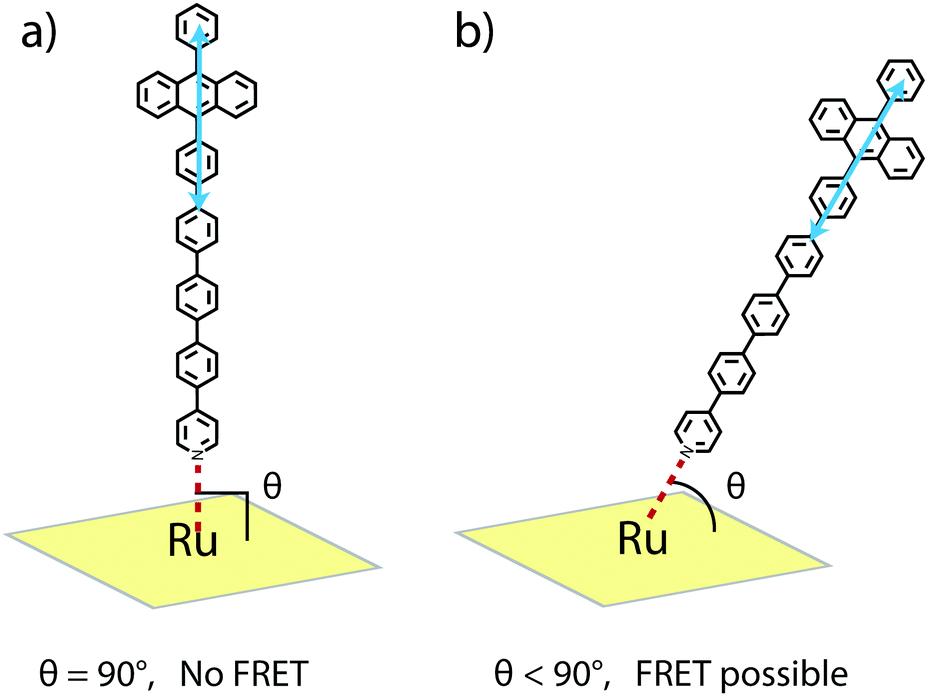 | ||
| Fig. 6 Schematic illustration of an axially coordinated ruthenium–anthracene complex with (a) perpendicular binding and (b) non-perpendicular binding, relative to the porphyrin plane and anthracene transition dipole moment. In the studied systems movement is expected and only an apparent binding angle can be estimated from the FRET measurements. | ||
Based on the experimental results described above we can construct an energy level diagram for the RuOEP(CO)L complexes, as shown in Fig. 7. From the Marcus–Rehm–Weller equation, we estimate the energy of the charge separated state (RuOEP+(CO)L−) to about 2.49 eV, for details of the calculation the reader is referred to the ESI.† The energy is estimated from redox potentials in acetonitrile and should therefore be considered as a lower limit since the current systems are studied in less polar toluene. As can be seen in Fig. 7 the energy of the charge separated state is accessible from the ligand singlet state RuOEP(CO)1L*. If populated, it would also contribute to the singlet quenching. Comparing the experimental quenching rates to a FRET governed distance dependence a reasonable R6 dependence is followed as seen when comparing the slope 6 in the log–log plot in Fig. S12 (ESI†) to the experimental values. However, the best linear fit is obtained for a slightly smaller slope of 5.4, possibly indicating that there is also another factor influencing the singlet quenching.
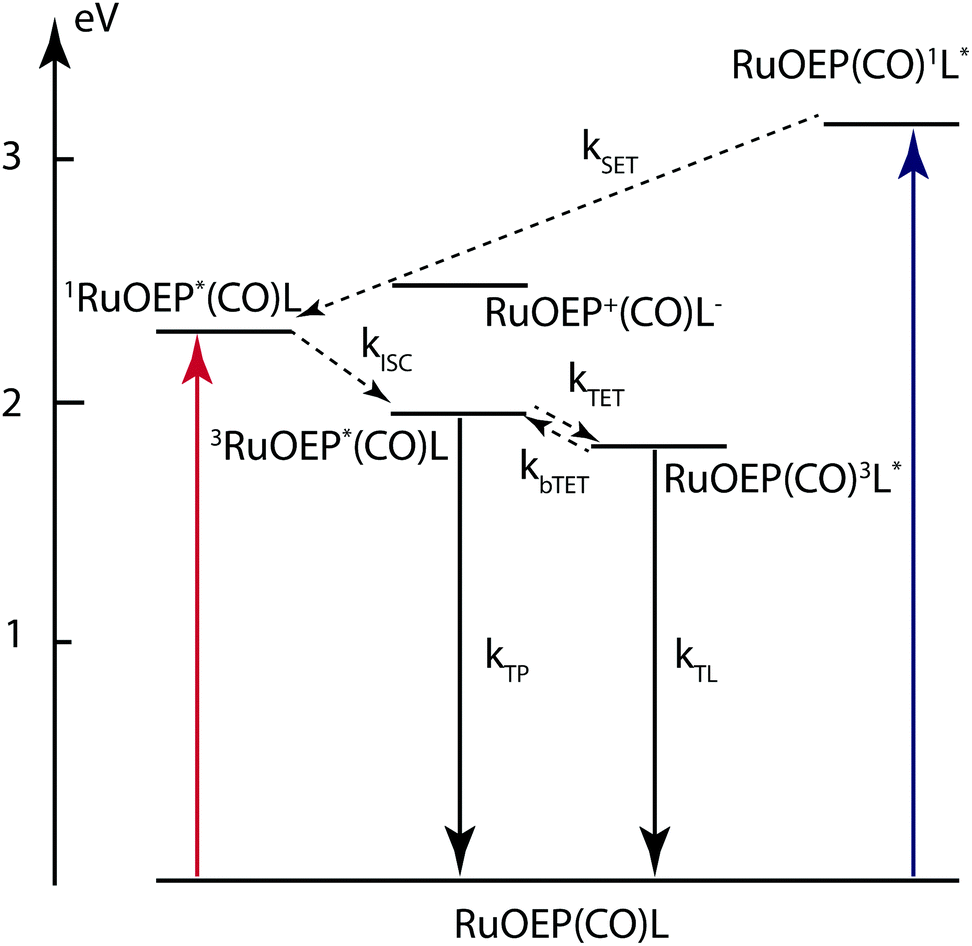 | ||
| Fig. 7 Energy level diagram of RuOEP(CO)L complexes. The charge separated state RuOEP+(CO)L− at 2.49 eV is estimated from the redox potentials of the free components in acetonitrile and should therefore be considered a lower limit. | ||
1.5 RuOEP(CO)–ligand complexes applied to triplet–triplet annihilation photon upconversion
Recently, many metallo-porphyrins have been used in sensitized triplet–triplet annihilation based photon upconversion (TTA-UC) systems. In TTA-UC systems the porphyrin triplet sensitizes an annihilator molecule through TET, the triplet excited annihilator can then undergo triplet–triplet annihilation (TTA) with another triplet excited annihilator forming a singlet excited annihilator which can emit a high energy photon. The most popular porphyrins for TTA-UC are based on palladium and platinum,20 probably due to their high intersystem crossing (ISC) yield, phosphorescent nature and stable structure. To the best of our knowledge, no ruthenium based porphyrins have been used as sensitizers for TTA-UC, even though they also have high ISC yields.54One of the main challenges faced in the field of TTA-UC is the development of solid state materials that maintain the high efficiencies observed in the liquid systems. The difficulty arises from the diffusion limited TET and TTA processes which are reduced in most solid systems. One approach is to develop large supra-molecular structures where TET and TTA can occur intra-molecularly, overcoming the diffusion limit.26 In such a supra-molecular system the sensitizer must be in close proximity to an annihilator. One advantage of ruthenium porphyrins, compared to the palladium and platinum analogues, is that pyridine ligands can coordinate to the ruthenium atom. By designing ligands that can accept the triplet energy, as described herein, the triplet energy transfer can be enhanced. We therefore used the RuOEP(CO)L complexes as sensitizers for TTA-UC with free DPA as an annihilator in toluene. It should be pointed out that the RuOEP(CO)L complexes are specifically designed to study the TET, thus TTA still must occur between two free annihilators, sensitized by two distinct complexes for upconversion to occur. As such the studied systems are only one part of a potential supra-molecular structure also incorporating intra-molecular TTA.62
As can be seen in Fig. 8, when RuOEP(CO)2 is used as a sensitizer, the upconversion quantum yield, ΦUC, is increased about 3 times to about 4.5%,¶ compared to RuOEP(CO)Pyr which has ΦUC = 1.5%. The quantum yields are determined in the strong annihilation limit (linear regime), Fig. S13 (ESI†). The increase in ΦUC can be understood in terms of the extended triplet lifetime of the RuOEP(CO)2 complex. Fig. S14 and the corresponding section in the ESI† describe the effect on the TET efficiency and ΦUC by extending the triplet lifetime of the sensitizer. With the current annihilator concentration it is expected that RuOEP(CO)2 is approximately 1.5–2 times more efficient due to the increased TET efficiency, in line with our experimental observations.
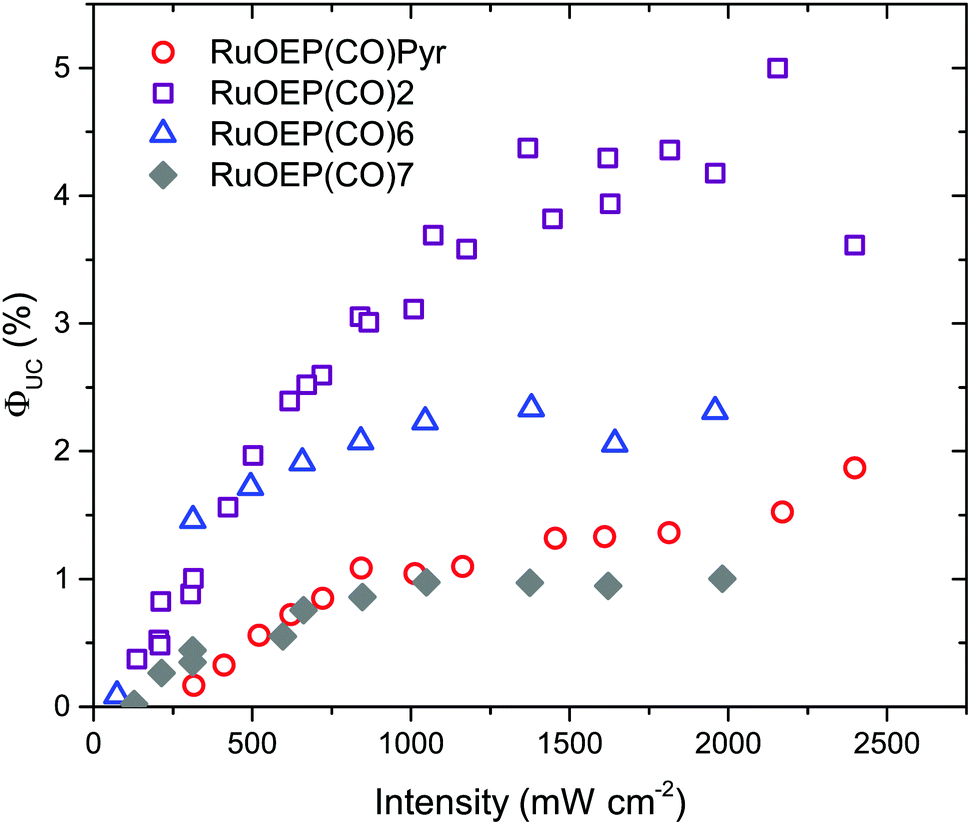 | ||
| Fig. 8 Photon upconversion quantum yield, ΦUC, as a function of excitation intensity. Purple squares represent samples containing RuOEP(CO)2, blue triangles RuOEP(CO)6, grey diamonds RuOEP(CO)7 and red circles RuOEP(CO)Pyr as the sensitizer complex (19 μM, porphyrin/ligand 1:1), 9,10-diphenylanthracene (DPA, 0.1 mM) is added as the annihilator. All samples in toluene, prepared in a glovebox (<0.1 ppm O2 and excited at 532 nm). | ||
Interestingly the effect of the extended triplet lifetime is not observed to the same extent for the complexes with dendrimeric ligands, RuOEP(CO)6 (ΦUC = 2.3%) and RuOEP(CO)7 (ΦUC = 1.0%), Fig. 8. ΦUC decreases more for the larger dendrimers. One possible explanation, in line with the observed decrease in ΦUC, is a steric effect on the probability of free DPA colliding with the triplet excited monomer in the dendrimer ligand. This is a result of the current upconversion system still relying on diffusional TET and TTA. In fully supra-molecular structures where an annihilator dendrimer is coordinated to multiple sensitizers, allowing for both TTA and TET to occur intra-molecularly we envision that such problems can be circumvented.
2 Conclusion
Herein we report a comprehensive study of the energy transfer processes of seven new ruthenium porphyrin–anthracene coordination complexes, RuOEP(CO)L. The design and synthesis of pyridine containing anthracene ligands with various bridge lengths (8–25 Å), consisting of 1–7 anthracene units, is described. The anthracene ligands bind strongly to the ruthenium atom resulting in close to quantitative binding for a 1:1 mixing ratio. Upon excitation of RuOEP(CO) the formed triplet is quickly transferred to the axially coordinated anthracene derivative. By separating the anthracene and porphyrin with phenyl bridges we observe a decrease in the triplet energy transfer rate, and an attenuation factor β of 0.64 Å−1 is determined. We further study the singlet energy transfer from the anthracene ligand to RuOEP(CO) using ps-resolved fluorescence lifetime measurements. The singlet anthracene is readily quenched by the porphyrin. By employing large dendrimeric structures or longer phenyl bridge separators the quenching is reduced. The singlet quenching follows a Förster type R6 distance dependence, even though the transition dipole moments are close to perpendicularly oriented.
The current study is part of our broader investigation of supra-molecular structures for TTA based photon upconversion. Here we demonstrate that by effectively extending the triplet lifetime of the ruthenium complex by coordination of the anthracene ligands, the upconversion quantum yield is increased threefold to 4.5% with complex RuOEP(CO)2. We have previously studied the properties of DPA based dendrimers and oligomers45,62 and shown that intra-molecular TTA is possible in such structures. Combined with the current study, where efficient intra-molecular TET is achieved, we are en route to developing fully functioning, well defined, supramolecular TTA upconversion systems, with broad implications spanning solar energy harvesting devices to bioimaging and drug targeting.
3 Experimental
Synthetic procedures for the new ligands 1, 6 and 7, and NMR spectra are found in the ESI.†3.1 Photophysical characterization
Steady-state absorption was recorded on a Cary 5000 spectrophotometer and steady state emission measurements were carried out on a Spex Flurolog 3 spectrofluorimeter (JY Horiba). Phosphorescence decays were carried out on the mentioned spectrofluorimeter. Unquenched fluorescence lifetimes were determined on a time correlated single photon counting (TCSPC) setup using a 377 nm laser diode (PicoQuant) and a MCP-PMT detector (10000 counts in the top channel, 2048 channels). Quenched fluorescence lifetimes were determined by time resolved emission measurements which were taken with a streak camera system; excitation pulses were generated using a Tsunami Ti:sapphire laser (Spectra-Physics) that was pumped by a Millennia Pro X laser (Spectra-Physics). The Tsunami laser output was set to 740 nm and frequency doubled to 370 nm using a frequency doubler/tripler (GWU, Spectra Physics) to excite the anthracene ligands. The emitted photons were passed through a spectrometer (Acton SP2300, Princeton Instruments) and were detected using a streak camera (C5680, Hamamatsu) with a synchroscan sweep unit (M5675, Hamamatsu).
Nanosecond transient absorptions measurements were performed on a home built system with a Surelite Continuum Nd:YAG laser equipped with an Surelite Continuum OPO generating a 7 ns pump beam. A quartz-halogen lamp with a monochromator was used for the probe light. Either a CCD camera (iStar, Andor Technology) or a monochromator (Oriel Cornerstone 130, Newport) together with a 5 stage PMT (Applied Photophysics) coupled to an oscilloscope (TDS 2022, Tektronix) was used for recording the transient spectra or decay signal, respectively.
Upconversion samples were excited at 532 nm using a frequency doubled cw Nd:YAG laser (Millenia V, Spectra-Physics), the excitation intensity was varied using a neutral density filter and the intensity was measured using a power-meter from Starlite Ophir. The laser spot diameter was determined to be 2.5 mm using a laser alignment paper. The emission was recorded at a 90° angle by out-coupling the emission with an optical fibre to an AvaSpec 2048 (Avantes) USB fibre spectrometer with a 532 nm notch filter between the sample and the fibre to protect the spectrometer from the intense scattered excitation light.
All photophysical measurements were carried out in toluene using quartz cuvettes. Samples were prepared in a glovebox from Innovative Technologies (<0.1 ppm O2) under a nitrogen atmosphere. RuOEP(CO)L phospohorescence quantum yields were determined using relative Cresyl violet in methanol (Φ = 54%). Upconversion quantum yields, ΦUC, were also determined using relative Cresyl violet in methanol employing the standard equation for relative fluorescence quantum yield determination:
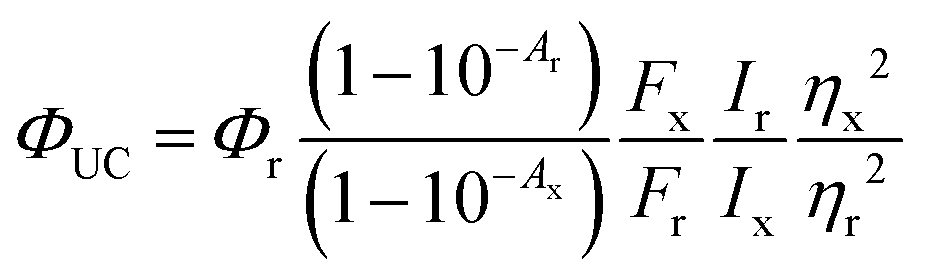 | (6) |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge funding from the Swedish Energy Agency, the Swedish Research Council, the Swedish Strategic Research Foundation and Knut & Alice Wallenberg Foundation. Dr Martyn Jevric is acknowledged for providing compound 2.References
- M. F. Perutz, H. Muirhead, J. M. Cox and L. C. G. Goaman, Nature, 1968, 219, 131–139 CrossRef CAS PubMed.
- G. McDermott, S. M. Prince, A. A. Freer, A. M. Hawthonthwaite-Lawless, M. Z. Papiz, R. J. Cogdell and N. W. Isaacs, Nature, 1995, 374, 517–521 CrossRef CAS.
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. van Grondelle and G. D. Scholes, Chem. Rev., 2017, 117, 249–293 CrossRef CAS PubMed.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198–205 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS PubMed.
- G. Kodis, P. A. Liddell, L. D. Garza, P. C. Clausen, J. S. Lindsey, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. A, 2002, 106, 2036–2048 CrossRef CAS.
- Y. Nakamura, I. W. Hwang, N. Aratani, T. K. Ahn, D. M. Ko, A. Takagi, T. Kawai, T. Matsumoto, D. Kim and A. Osuka, J. Am. Chem. Soc., 2005, 127, 236–246 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russel, R. A. J. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282–7285 CrossRef CAS PubMed.
- S. Mathew, N. A. Astani, B. F. E. Curchod, J. H. Delcamp, M. Marszalek, J. Frey, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, J. Mater. Chem. A, 2016, 4, 2332–2339 CAS.
- P. Qin, P. Sanghyun, M. I. Dar, K. Rakstys, H. Elbatal, S. A. Al-muhtaseb, C. Ludwig and M. K. Nazeeruddin, Adv. Funct. Mater., 2016, 26, 5550–5559 CrossRef CAS.
- J. Schmitt, V. Heitz, A. Sour, F. Bolze, H. Ftouni, J.-F. Nicoud, L. Flamigni and B. Ventura, Angew. Chem., Int. Ed., 2015, 54, 169–173 CrossRef CAS PubMed.
- G. Sedghi, K. Sawada, L. J. Esdaile, M. Hoffmann, H. L. Anderson, D. Bethell, W. Haiss, S. J. Higgins and R. J. Nichols, J. Am. Chem. Soc., 2008, 120, 8582–8583 CrossRef PubMed.
- G. Sedghi, V. M. García-Suárez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martín, D. Bethell, S. J. Higgins, M. Elliot, N. Bennett, J. E. Macdonald and R. J. Nichols, Nat. Nanotechnol., 2011, 6, 517–523 CrossRef CAS PubMed.
- Z. Li, T.-H. Park, J. Rawson, M. J. Therien and E. Borguet, Nano Lett., 2012, 12, 2722–2727 CrossRef CAS PubMed.
- G. Sedghi, L. J. Esdaile, H. Anderson, S. Martin, D. Bethell, S. J. Higgins and R. J. Nichols, Adv. Mater., 2012, 24, 653–657 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937–950 RSC.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
- C. E. McCusker and F. N. Castellano, Top. Curr. Chem., 2016, 374, 19 CrossRef PubMed.
- T. G. Monger, R. J. Cogdell and W. W. Parson, Biochim. Biophys. Acta, 1976, 449, 136–153 CrossRef CAS.
- A. Gall, R. Berera, M. T. A. Alexandre, A. A. Pascal, L. Bordes, M. M. Mendes-pinto, S. Andrianambinintsoa, K. V. Stoitchkova, A. Marin, L. Valkunas, P. Horton, J. T. M. Kennis, R. V. Grondelle, A. Ruban and B. Robert, Biophys. J., 2011, 101, 934–942 CrossRef CAS PubMed.
- J. Ho, E. Kish, D. D. Méndez-Hernández, K. WongCarter, S. Pillai, G. Kodis, J. Niklas, O. G. Poluektov, D. Gust, T. A. Moore, A. L. Moore, V. S. Batista and B. Robert, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5513–E5521 CrossRef CAS PubMed.
- V. Gray, K. Börjesson, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Phys. Chem. C, 2016, 120, 19018–19026 CAS.
- C. Fan, W. Wu, J. J. Chruma, J. Zhao and C. Yang, J. Am. Chem. Soc., 2016, 138, 15405–15412 CrossRef CAS PubMed.
- N. Yanai and N. Kimizuka, Chem. Commun., 2016, 52, 5354–5370 RSC.
- F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem., Int. Ed., 2012, 51, 2417–2420 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- V. Bandi, F. P. D. Souza, H. B. Gobeze and F. D'Souza, Chem. – Eur. J., 2015, 21, 2669–2679 CrossRef CAS PubMed.
- K. Kilså, J. Kajanus, S. Larsson, A. N. Macpherson, J. Mårtensson and B. Albinsson, Chem. – Eur. J., 2001, 7, 2122–2133 CrossRef.
- D. Holten, D. F. Bocian and J. S. Lindsey, Acc. Chem. Res., 2002, 35, 57–69 CrossRef CAS PubMed.
- A. Prodi, C. Chiorboli, F. Scandola, E. Iengo, E. Alessio, R. Dobrawa and F. Würtner, J. Am. Chem. Soc., 2005, 127, 1454–1462 CrossRef CAS PubMed.
- D. M. Guldi, G. M. A. Rahman, V. Sgobba and C. Ehli, Chem. Soc. Rev., 2006, 35, 471–487 RSC.
- Y. Nakamura, N. Aratani and A. Osuka, Chem. Soc. Rev., 2007, 36, 831–845 RSC.
- B. Albinsson, M. P. Eng, K. Pettersson and M. U. Winters, Phys. Chem. Chem. Phys., 2007, 9, 5847–5864 RSC.
- B. Albinsson and J. Mårtensson, J. Photochem. Photobiol., C, 2008, 9, 138–155 CrossRef CAS.
- T. K. Khan, M. Bröring, S. Mathur and M. Ravikanth, Coord. Chem. Rev., 2013, 257, 2348–2387 CrossRef CAS.
- J. G. Woller, J. K. Hannestad and B. Albinsson, J. Am. Chem. Soc., 2013, 135, 2759–2768 CrossRef CAS PubMed.
- M. Gilbert and B. Albinsson, Chem. Soc. Rev., 2015, 44, 845–862 RSC.
- A. Antoniuk-Pablant, G. Kodis, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. B, 2016, 120, 6697 CrossRef PubMed.
- H. Dekkiche, A. Buisson, A. Langlois, P.-L. Karsenti, L. Ruhlmann, P. D. Harvey and R. Ruppert, Inorg. Chem., 2016, 55, 10329–10336 CrossRef CAS PubMed.
- D. R. Martir, G. J. Hedley, D. B. Cordes, A. M. Z. Slawin, D. Escudero, D. Jacquemin, T. Kosikova, D. Philp, D. M. Dawson, S. E. Ashbrook, I. D. W. Samuel and E. Zysman-colman, Dalton Trans., 2016, 45, 17195–17205 RSC.
- Y. Hu, M. B. Thomas, R. G. W. Jinadasa, H. Wang and F. D'Souza, Chem. – Eur. J., 2017, 23, 12805–12814 CrossRef CAS PubMed.
- K. Börjesson, M. Gilbert, D. Dzebo, B. Albinsson and K. Moth-Poulsen, RSC Adv., 2014, 4, 19846 RSC.
- M. Tsutsui, D. Ostfeld and L. M. Hoffman, J. Am. Chem. Soc., 1971, 93, 1820–1823 CrossRef CAS.
- A. Antipas, J. W. Buchler, M. Gouterman and P. D. Smith, J. Am. Chem. Soc., 1978, 100, 3015–3024 CrossRef CAS.
- D. P. Rillema, J. K. Nagle, L. F. Barringer Jr and T. J. Meyer, J. Am. Chem. Soc., 1981, 103, 56–62 CrossRef CAS.
- K. M. Kadish, D. J. Leggett and D. Chang, Inorg. Chem., 1982, 21, 3618–3622 CrossRef CAS.
- K. M. Kadish and D. Chang, Inorg. Chem., 1982, 21, 3614–3618 CrossRef CAS.
- S. Chattopadhyay, C. V. Kumar and P. Das, Chem. Phys. Lett., 1983, 98, 250–254 CrossRef CAS.
- V. Gray, D. Dzebo, A. Lundin, J. Alborzpour, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Mater. Chem. C, 2015, 3, 11111–11121 RSC.
- J. Andraos, J. Chem. Educ., 1999, 76, 1578–1583 CrossRef CAS.
- L. M. A. Levine and D. Holten, J. Phys. Chem., 1988, 92, 714–720 CrossRef CAS.
- J. Vura-Weis, S. H. Abdelwahed, R. Shukla, R. Rathore, M. A. Ratner and M. R. Wasielewski, Science, 2010, 328, 1547–1550 CrossRef CAS PubMed.
- N. I. Nijegorodov, W. S. Downey and M. B. Danailov, Spectrochim. Acta, Part A, 2000, 56, 783–795 CrossRef CAS.
- E. A. Weiss, M. J. Ahrens, L. E. Sinks, A. V. Gusev, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 5577–5584 CrossRef CAS PubMed.
- E. A. Weiss, M. J. Tauber, R. F. Kelley, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2005, 127, 11842–11850 CrossRef CAS PubMed.
- T. Mani, D. M. Niedzwiedzki and S. A. Vinogradov, J. Phys. Chem. A, 2012, 116, 3598–3610 CrossRef CAS PubMed.
- K. Kilså, J. Kajanus, J. Mårtensson and B. Albinsson, J. Phys. Chem. B, 1999, 103, 7329–7339 CrossRef.
- K. Pettersson, A. Kyrychenko, E. Ro and T. Ljungdahl, J. Phys. Chem. A, 2006, 110, 310–318 CrossRef CAS PubMed.
- D. Dzebo, K. Börjesson, V. Gray, K. Moth-Poulsen and B. Albinsson, J. Phys. Chem. C, 2016, 120, 23397–23406 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and NMR spectra of the new ligands, description of the fitting procedures and the corresponding photophysical data, time resolved and steady state phosphorescence data, transient absorption spectra. See DOI: 10.1039/c8cp00884a |
| ‡ RuOEP(CO) is a low-spin d-6 complex, with a singlet ground-state.47 |
| § Optimized structures of the RuOEP(CO)L coordination complexes were modelled with PM3 as implemented in the HyperChem 8.0 computational chemistry package. |
| ¶ All reported upconversion quantum yields are on the basis of a maximum of 50%, two absorbed low energy photons can at most produce one high energy photon. |
| This journal is © the Owner Societies 2018 |