
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anharmonicity in a double hydrogen transfer reaction studied in a single porphycene molecule on a Cu(110) surface
S.
Liu†
a,
D.
Baugh†‡
a,
K.
Motobayashi
b,
X.
Zhao
c,
S. V.
Levchenko
cd,
S.
Gawinkowski
e,
J.
Waluk
 ef,
L.
Grill
g,
M.
Persson
h and
T.
Kumagai
*a
ef,
L.
Grill
g,
M.
Persson
h and
T.
Kumagai
*a
aDepartment of Physical Chemistry, Fritz-Haber Institute of the Max-Planck Society, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: kuma@fhi-berlin.mpg.de
bDepartment of Physical Science and Engineering, Nagoya Institute of Technology, Gokiso, Showa, Nagoya 466-8555, Japan
cTheory Department, Fritz-Haber Institute of the Max-Planck Society, Faradayweg 4-6, 14195 Berlin, Germany
dLaboratory of Modelling and Development of New Materials, NUST MISIS, Leninskiy prospekt 4, Moscow 119049, Russia
eInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, Warsaw 01-224, Poland
fFaculty of Mathematics and Natural Sciences, College of Science, Cardinal Stefan Wyszyński University, Wóycickiego 1/3, 01-938 Warsaw, Poland
gDepartment of Physical Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria
hSurface Science Research Centre and Department of Chemistry, University of Liverpool, Liverpool L69 3BX, UK
First published on 29th March 2018
Abstract
Anharmonicity plays a crucial role in hydrogen transfer reactions in hydrogen-bonding systems, which leads to a peculiar spectral line shape of the hydrogen stretching mode as well as highly complex intra/intermolecular vibrational energy relaxation. Single-molecule study with a well-defined model is necessary to elucidate a fundamental mechanism. Recent low-temperature scanning tunnelling microscopy (STM) experiments revealed that the cis ↔ cis tautomerization in a single porphycene molecule on Cu(110) at 5 K can be induced by vibrational excitation via an inelastic electron tunnelling process and the N–H(D) stretching mode couples with the tautomerization coordinate [Kumagai et al. Phys. Rev. Lett. 2013, 111, 246101]. Here we discuss a pronounced anharmonicity of the N–H stretching mode observed in the STM action spectra and the conductance spectra. Density functional theory calculations find a strong intermode coupling of the N–H stretching with an in-plane bending mode within porphycene on Cu(110).
Hydrogen (H) transfer reactions are of fundamental importance in nature.1 The H transfer dynamics is complicated by anharmonic potential energy surfaces caused by the hydrogen bond which acts as a pathway of H atoms. The anharmonicity results in a strong intermode coupling of A–H stretching (A is an electronegative atom) with other lower frequency modes, which leads to highly complex spectral features in vibrational spectra, particularly in the A–H stretching band.2 Although anharmonicity in H-bonding systems has been studied extensively using vibrational spectroscopy and quantum chemical calculations,3 an accurate and quantitative description of the peculiar vibrational spectral features as well as the H-transfer dynamics remains a challenging topic. Since the anharmonic nature is hidden by inhomogeneous effects in bulk samples, it is essential to study a well-defined model and employ local spectroscopy down to the single-molecule level. Intramolecular H transfer reactions, so-called tautomerization, have served as an important model for studying a fundamental mechanism of H-transfer dynamics.4 Porphycene has been investigated as a model of a double H transfer that occurs in a shallow and strongly anharmonic potential energy surface resulting from strong intramolecular hydrogen bonds.5–8 Recent low-temperature scanning tunnelling microscopy (STM) experiments revealed that the cis ↔ cis tautomerization of a single porphycene molecule on Cu(110) can be induced by vibrational excitation via inelastic electron tunnelling and that the N–H stretching mode couples with the tautomerization coordinate.9 DFT calculations were used to determine the adsorption structure and also proposed that the cis–cis tautomerization occurs by a stepwise mechanism in which the meta-stable trans configuration is involved as an intermediate state along the reaction pathway.9–11 Here we provide an in-depth analysis of single-molecule vibrational spectroscopy of STM and discuss a pronounced anharmonicity of the N–H stretching mode.
Results and discussion
Fig. 1a shows STM images before and after the cis ↔ cis tautomerization of a single porphycene on Cu(110) at 5 K.9,10 The electron-induced tautomerization of porphycene was investigated using STM action spectroscopy (STM-AS) which has emerged as a powerful method to study single-molecule reactions on surfaces.12,13 We measured STM-AS for normal (h-porphycene) and deuterated porphycene (d2-porphycene in which only the inner H atoms are replaced by D)9 and spectral fitting analysis was carried out using an established method.12,13 This fitting analysis enables us to extract the vibrational properties of molecular adsorbates which are associated with the reaction. The reaction yield Y at a specific bias voltage V is defined by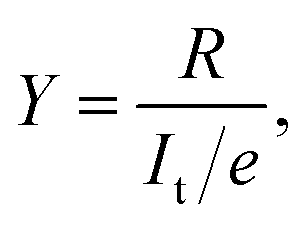 | (1) |
| R ∝ INt, | (2) |
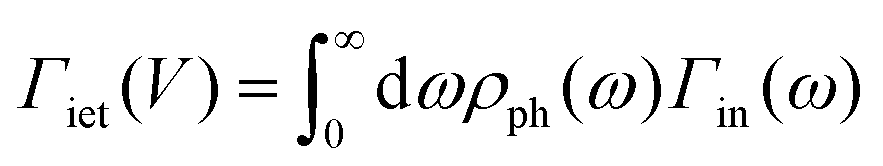 , as derived in ref. 15. Y(V) can be expressed using double integration of the vibrational DOS as
, as derived in ref. 15. Y(V) can be expressed using double integration of the vibrational DOS as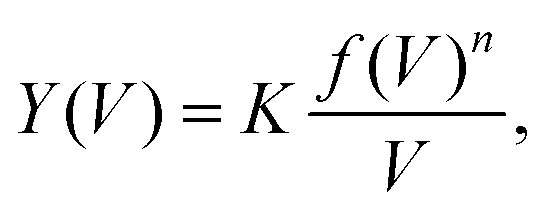 | (3) |
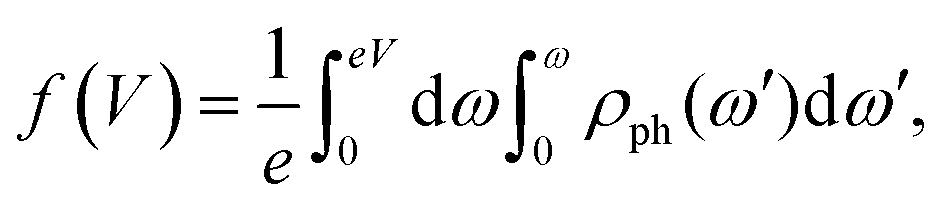 | (4) |
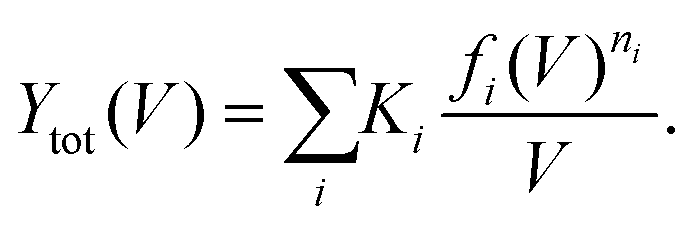 | (5) |
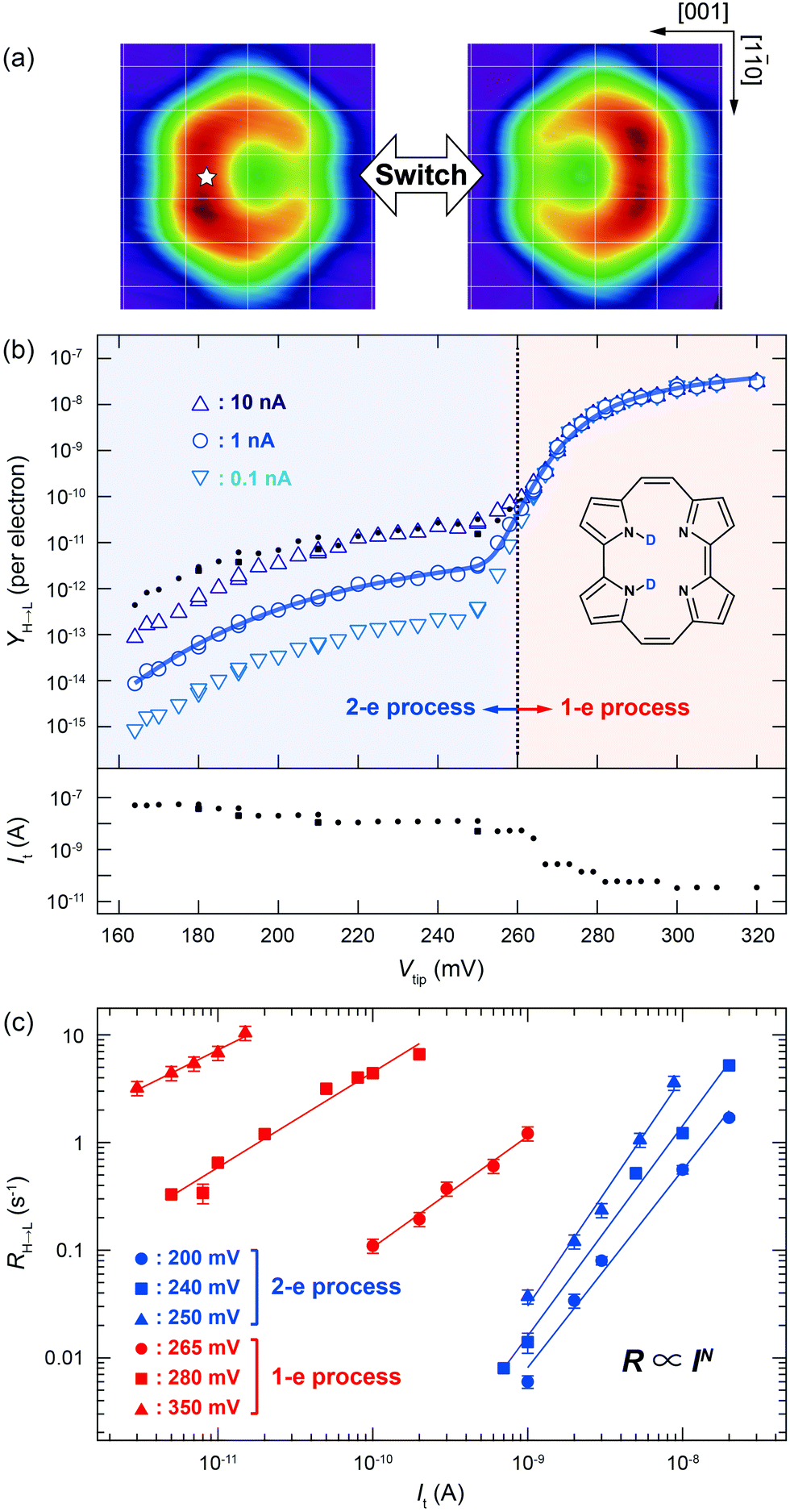 | ||
| Fig. 1 (a) STM images of a single porphycene molecule on Cu(110) at 5 K (Vtip = −100 mV, It = 10 nA, size: 1.49 × 1.42 nm2).10 The white star indicates the tip position during the STM-AS and conductance measurements. The white grid lines represent the surface lattice of Cu(110). (b) STM-AS for d2-porphycene on Cu(110) measured at 5 K. The small closed circles and squares in the upper panel indicate the raw data obtained at the tunnelling current shown in the lower panel. The open markers in the upper panel are the calculated Y(V) normalized to a constant current indicated in the figure. The solid curve represents the best-fitting result of Y(V) for It = 1 nA to eqn (5) with the parameters listed in Table 1. (c) Current dependence of the tautomerization rates (RH→L) measured at various bias voltages. The slopes (N) are determined to be 1.83(±0.11), 2.12(±0.13), 1.95(±0.08), 2.12(±0.13), 1.03(±0.07), 0.88(±0.06) and 0.71(±0.05) at 200, 240, 250, 265, 280, and 350 mV, respectively. | ||
When the reaction is induced by simultaneous excitation of several different vibrational modes, the total reaction order (N) involves contribution from each excitation. Fitting analysis of Y(V) using eqn (5) has been employed to extract the energy and broadening factor of active vibration modes.17–21 Here we use a Gaussian function 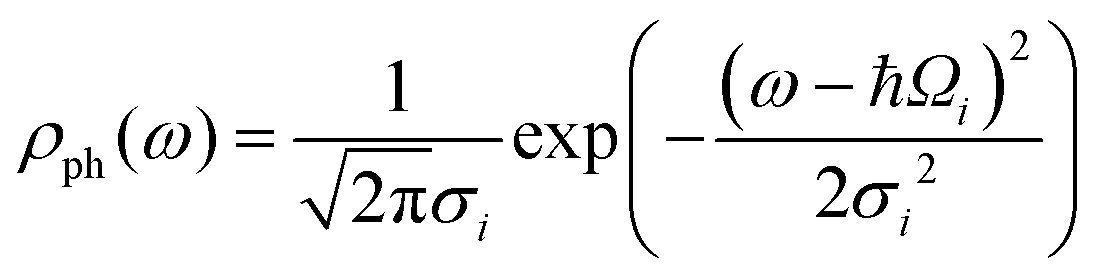 because it allows taking into account broadening effects including all possible contributions, i.e., finite temperatures, vibrational relaxation, instrumental resolution,12 and anharmonic effects due to hydrogen bonding.16 The full width of half maximum (γi) of the Gaussian vibrational DOS is given by
because it allows taking into account broadening effects including all possible contributions, i.e., finite temperatures, vibrational relaxation, instrumental resolution,12 and anharmonic effects due to hydrogen bonding.16 The full width of half maximum (γi) of the Gaussian vibrational DOS is given by 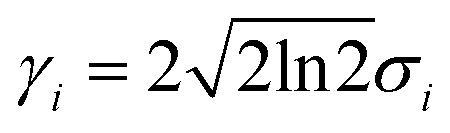 .
.
We first discuss the STM-AS for the cis ↔ cis tautomerization of d2-porphycene (Fig. 1b). The lateral tip position during the measurement is indicated in Fig. 1a, which corresponds to the side of pyrrole rings with the amine N atoms. Fig. 1c shows the current dependence of the tautomerization rate, where N changes from ∼2 to ∼1 around 260 mV. Thus, tautomerization occurs via a two-electron process and a one-electron process at the low and high voltage regimes, respectively. At higher currents, the tip–molecule distance becomes smaller and the STM tip may affect the tautomerization through the interaction with the molecule, which eventually leads to modification of the potential energy surface.11 However, the modification is expected to be rather small at the tip–molecule distances in this experiment (the tip is substantially far from the equilibrium distance of the tip–molecule potential, where the significant deformation of the potential occurs11). Hence, the influence on the reaction order should be negligible. Since the tautomerization rate is very small at low bias voltages, we used a relatively high current to measure the yields (see the lower panel of Fig. 1b) in order to observe a sufficient number of tautomerization events in a realistic time-scale of the experiment. However, according to eqn (1) and (2), Y(V) depends on the current in a multi-electron process, in contrast to a one-electron process. Therefore, the measurement current affects the Y(V) curve at low voltages. The small markers in the upper panel of Fig. 1b show the measured (raw) yields (Yraw) obtained at the used tunnelling current (Iraw) indicated in the lower panel. In order to examine the influence of the current to the Y(V) curve, we test the following normalization. The open markers in Fig. 1b are the yields (Ynorm) normalized to a specific current (Inorm: 0.1, 1, and 10 nA), which are given by 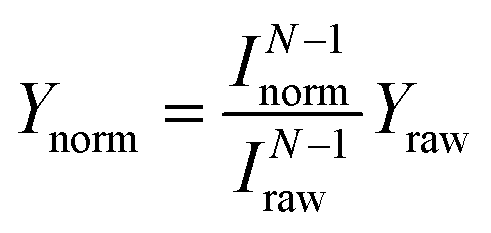 where N was obtained from a fitting analysis of the current dependence measurement (cf.Fig. 2b). These values for Inorm are in a typical range of tunnelling currents in STM. As mentioned above, we do not take into account any influence of the tip–molecule interaction that may modify the potential energy surface of the tautomerization.11 Note that this normalization was not carried out in our previous report.9 As discussed later (cf.Fig. 2c), N may be varied depending on the measured current range when one- and two-electron processes are simultaneously involved. The consistency of the normalized yield was confirmed at several bias voltages where the current dependence of the rate is available (Fig. 1c). It is clear that Y(V) at different currents exhibits a vertical shift in the two-electron regime (Fig. 1b), which leads to a critical change in Ki in eqn (5) but the other vibrational factors, namely the vibrational energy and width, are not significantly influenced in the fitting analysis.
where N was obtained from a fitting analysis of the current dependence measurement (cf.Fig. 2b). These values for Inorm are in a typical range of tunnelling currents in STM. As mentioned above, we do not take into account any influence of the tip–molecule interaction that may modify the potential energy surface of the tautomerization.11 Note that this normalization was not carried out in our previous report.9 As discussed later (cf.Fig. 2c), N may be varied depending on the measured current range when one- and two-electron processes are simultaneously involved. The consistency of the normalized yield was confirmed at several bias voltages where the current dependence of the rate is available (Fig. 1c). It is clear that Y(V) at different currents exhibits a vertical shift in the two-electron regime (Fig. 1b), which leads to a critical change in Ki in eqn (5) but the other vibrational factors, namely the vibrational energy and width, are not significantly influenced in the fitting analysis.
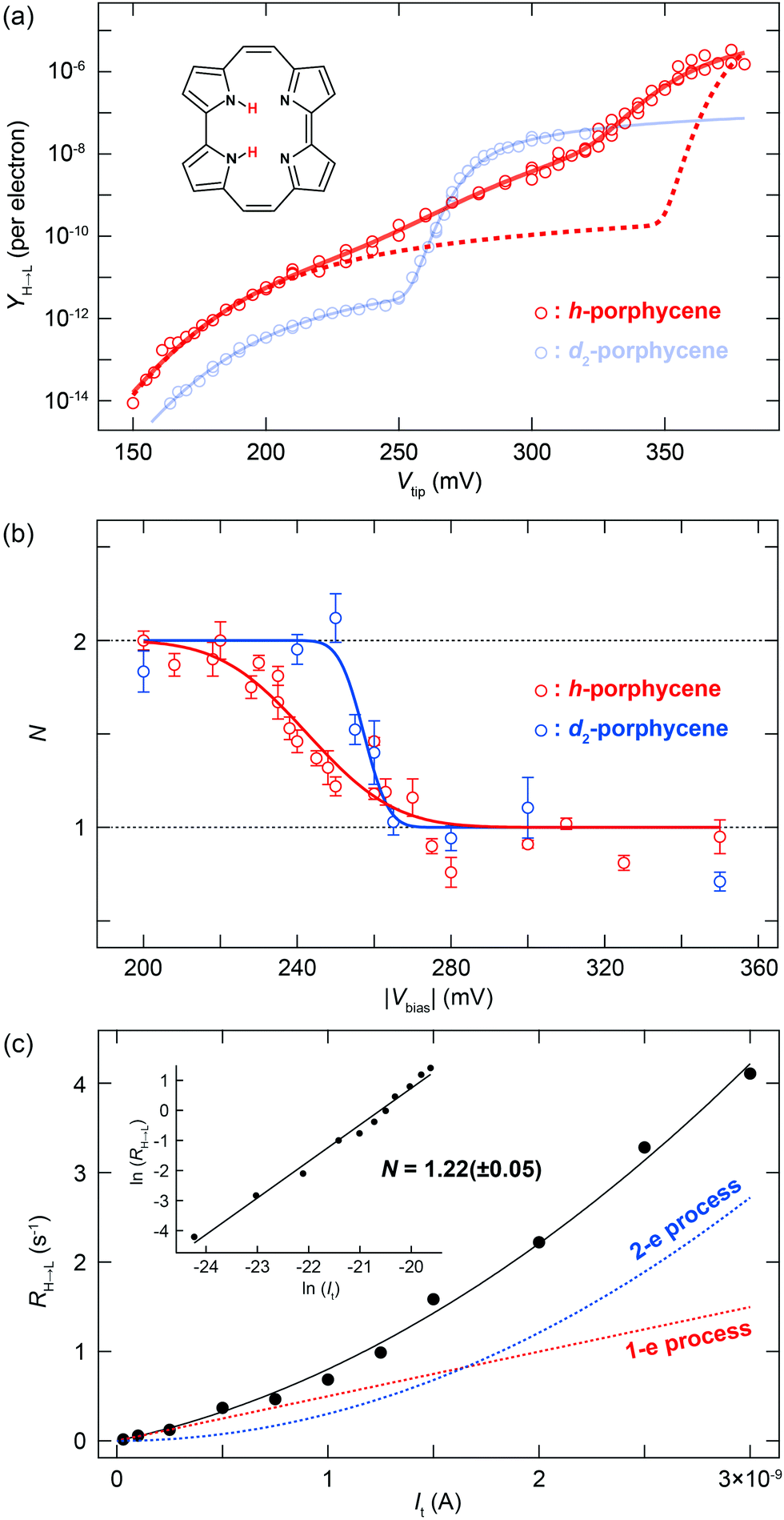 | ||
| Fig. 2 (a) Y(V) for h- and d2-porphycene on Cu(110) measured at 5 K. The yields in the N > 1 regime are normalized to a constant current (1 nA). The dashed red line represents the curve obtained from eqn (5) using the parameters: (ħΩ, σph) = (181 meV, 23.2 meV) and (375 meV, 6.8 meV). The solid red line represents the best-fitting result with the parameters listed in Table 1. The multiple data points at several bias voltages represent the yields obtained either at a different current or under different tip conditions. (b) Voltage dependence of the total reaction order (N) for h- and d2-porphycene. The experimental data (makers) are fitted to a step function convoluted with a Gaussian profile. The broadening factors (FWHM) of the Gaussian function are ∼42 and ∼12 meV for h- and d2-porphycene, respectively. (c) Current dependence of the tautomerization rate measured for h-porphycene at Vsample = −250 mV. The black solid line is the best fit using eqn (6), and the dashed lines indicate the contribution from one- and two-electron processes. The inset graph plots the logarithm of the rate and current. | ||
For d2-porphycene, Y(V) exhibits a threshold at ∼160 mV and a second steep increase at ∼260 mV. A similar result was obtained at both bias polarities. The solid line in Fig. 1b is the best-fitting result to eqn (5) with the parameters listed in Table 1. The obtained vibrational energy for the second increase of Y(V) is in excellent agreement with the calculated symmetric (anti-symmetric) N–D stretching mode, νs(as)(N–D), of 281.9 (280.9) meV. However, the vibrational DOS around the threshold voltage show a large broadening factor which ranges typically from 5 to 15 meV.13 This broad vibrational DOS may be explained by the contribution from several different vibrational excitations. Additionally, the reaction is second order (i.e., two-electron process) at this voltage range, thus multiple vibrational excitations are also involved and the reaction should comprise complex elementary processes.18 Moreover, a bunch of vibrational modes exist for porphycene below 200 meV,9 hampering a clear assignment of the involved modes. A similar threshold voltage (∼150 mV) is also observed for the tautomerization of porphycene on Cu(111).22,23 A recent theoretical study by Novko et al. proposed that the excitation of skeletal modes plays a crucial role in the reaction.24 It should also be noted that the reaction (minimum energy) path for tautomerization on Cu(110) and Cu(111) does not initially involve N–H stretching but rather a small lateral translation of porphycene along the surface.10,11,25 Therefore, the reaction coordinate should also involve low frequency modes such as the hindered translation and rotation of the molecule.
| K (mV1−n) | ħΩ (meV) | γ (meV) | n | |
|---|---|---|---|---|
| a These values are restricted around the peak and dip position observed in the dI/dV spectrum of h-porphycene in Fig. 3a. | ||||
| d2-Porphycene | ||||
| Skeletal modes | (1.53 ± 0.24) × 10−13 | 181.3 ± 3.3 | 54.5 ± 4.9 | 2 |
| ν(N–D) | (2.66 ± 0.21) × 10−7 | 274.7 ± 0.8 | 16.0 ± 2.0 | 1 |
| h-Porphycene | ||||
| Skeletal modes | (2.36 ± 0.28) × 10−12 | 168.9 ± 5.6 | 41.4 ± 5.4 | 2 |
| Overtone or combination | (3.20 ± 0.53) × 10−7 | 327a | 76.6 ± 2.8 | 1 |
| ν(N–H) | (5.04 ± 0.49) × 10−5 | 358a | 35.7 ± 1.2 | 1 |
The tautomerization is observed to be enhanced by the ν(N–D) excitation, which suggests that this reaction does not follow the reaction path since this vibrational energy does not appear in low frequency modes along the initial reaction path. The ν(N–D) energy can also be dissipated through electron–hole pair excitation in the substrate as well as intramolecular vibrational relaxation.26–28Ki in eqn (5), in principle, contains important physical parameters such as the vibrational dumping rate of the excited vibrational mode through relaxation to the reaction coordinate and/or other low frequency modes (including surface phonons) and electron–hole pair excitation.29,30 However, we were unable to extract such valuable information due to the lack of the detailed knowledge of the complex elementary process of tautomerization of porphycene on the surface.
Y(V) of h-porphycene shows a conspicuous difference from d2-porphycene (Fig. 2a). Although the threshold at ∼150 meV is similar to that of d2-porphycene, the second increase of Y(V), which should correspond to the ν(N–H) excitation, is not as obvious as ν(N–D) excitation of d2-porphycene. Instead, Y(V) exhibits a very moderate increase ranging from 200 to 380 mV. A similar moderate increase of Y(V) was also observed for the H-bonded O–H stretching mode in a water–hydroxyl complex on Cu(110), which is interpreted as vibrational broadening caused by an anharmonic potential.16 As shown in Fig. 1b, Y(V) of d2-porphycene can be reproduced with eqn (5) using two vibrational DOS, that is, excitation of skeletal modes at 181.3 meV and ν(N–D) at 274.7 meV. Using a simple harmonic approximation, the ν(N–H) energy can be estimated to be 375 meV from the ν(N–D) energy,31 which is in excellent agreement with the simulated ν(N–H) at 376 mV by DFT.9 However, if the STM-AS of h-porphycene is simulated with eqn (5) using two vibrational DOS, namely skeletal mode around the threshold voltage and ν(N–H) at 375 meV with a comparable broadening factor obtained for d2-porphycene, the resulting curve largely deviates from the experimental result (see the red dashed line in Fig. 2a).
A remarkable difference between h- and d2-porphycene is also observed in the voltage dependence of N (Fig. 2b). The transition from a two- to a one-electron process is relatively sharp for d2-porphycene, whereas it is much more gradual for h-porphycene32 and the transition spreads from 220 to 270 mV. This gradual transition suggests that the vibrational DOS of the skeletal mode and the N–H stretching are simultaneously excited in this voltage range, resulting in the non-integer N. In this situation, the total reaction rate is given by19
| R = R1 + R2 = k1It + k2It2, | (6) |
A remarkable difference between h- and d2-porphycene was also observed in the dI/dV spectrum,9 where vibrational signals appear as a characteristic peak or dip (Fig. 3a). The peak and dip at 177 and 284 mV observed for d2-porphycene nicely match the skeletal modes and the ν(N–D) excitation, respectively. These values are also consistent with the fitting parameters of Y(V) (Table 1). However, the dI/dV spectrum of h-porphycene exhibits rather complex features between 300 and 400 mV consisting of a broad peak and a dip. Furthermore, it is found that the dip includes two components in the high-resolution dI/dV spectrum (see the inset of Fig. 3a). These spectral features suggest the existence of several vibrational excitations around ν(N–H). We find that Y(V) of h-porphycene can be reproduced with eqn (5) using at least two vibrational DOS in the ν(N–H) regime where the peak and dip are observed in the dI/dV spectrum. The fitting result is indicated by the solid line in Fig. 2a and the fitting parameters are listed in Table 1.
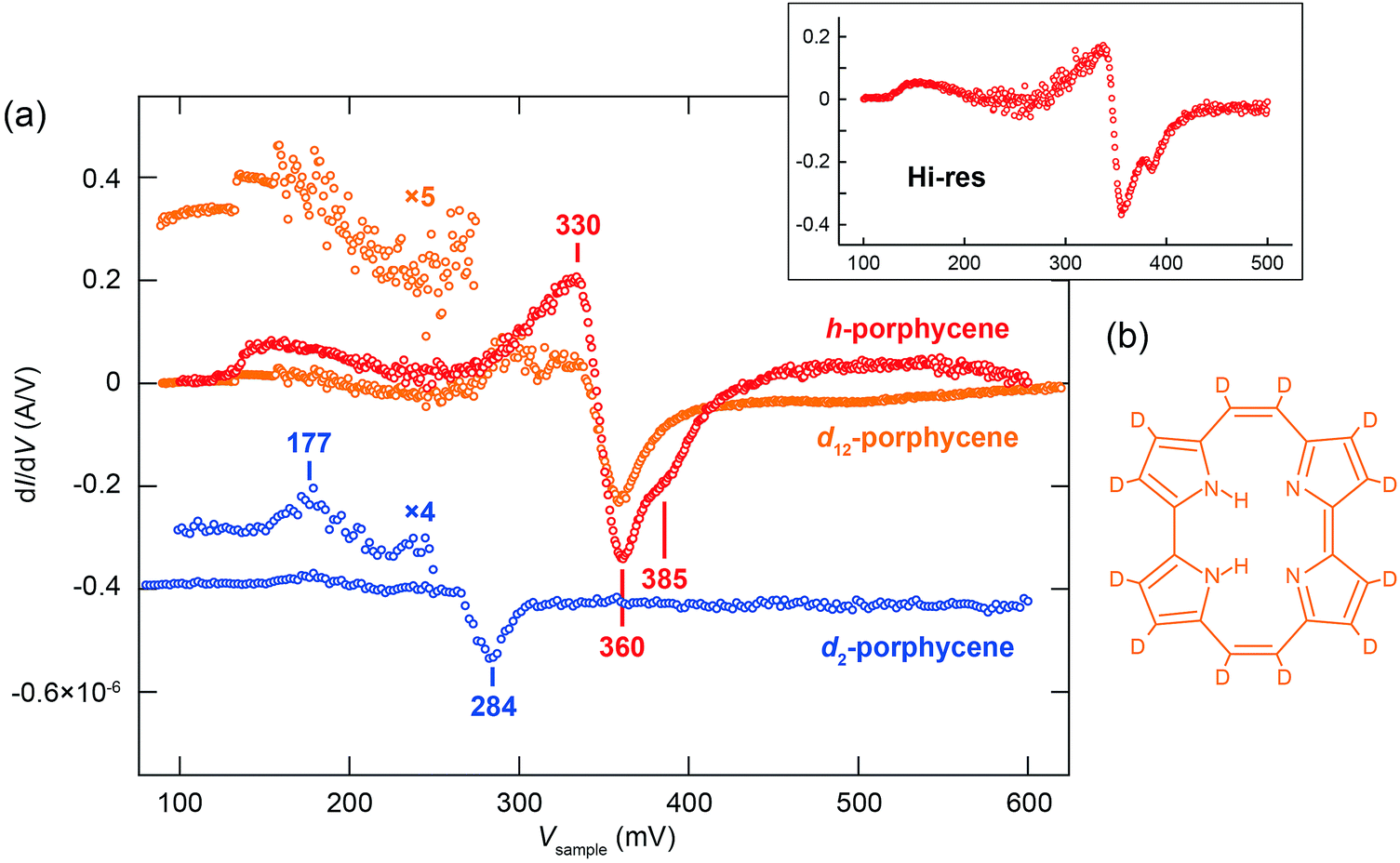 | ||
| Fig. 3 (a) dI/dV spectra for h-, d2- and d12-porphycene. The spectra over the molecule are subtracted with a background spectrum measured over the Cu(110) surface. The tip was fixed at the position indicated in Fig. 1a with the gap conditions of Vsample = 100 mV and It = 20 nA. The spectra were recorded using a lock-in amplifier with a modulation voltage of 12 mV at 710 Hz frequency. The upper inset shows the high-resolution dI/dV spectrum for h-porphycene recorded with a modulation voltage of 3 mV. (b) Chemical structures of d12-porphycene. | ||
The ν(N–H) energy found in the Y(V) and dI/dV spectra appears to be considerably red-shifted as compared to the N–H stretching mode, for example, in NH3 (417 meV35), free-base porphyrin in rare-gas matrices (412 meV36), and phthalocyanine on Ag(111) (406 meV37), indicating the presence of relatively strong H bonds in porphycene on Cu(110). A vibrational broadening and emergence of additional satellite bands for the A–H stretching mode in H bonding systems are known from IR spectroscopy.38–40 The broadening results from the anharmonic coupling of this stretching mode with other lower frequency modes,39,41–43 which leads to modulation of the A–H stretching potential and intramolecular vibrational relaxation because third- and higher-order terms of the potential are enhanced by H bonding. Furthermore, the coupling between the A–H stretching and overtone/combination modes could be enhanced by anharmonic resonance, e.g., Fermi resonance.38,42–44
Since no fundamental mode exists between 200 and 376 meV for h-porphycene on Cu(110),9 the peak at 330 mV in the dI/dV spectrum observed for h-porphycene should be assigned to overtone/combination excitation of lower vibrational modes. The absence of a peak (observed for h-porphycene) around the ν(N–D) energy region of d2-porphycene implies that the peak at 330 mV for h-porphycene appears in a similar mechanism to other H-bonding systems (as discussed above) and the coupling may be enhanced through a Fermi resonance between ν(N–H) and the overtone/combination mode(s), most likely N–H bending. Additionally, it is found that the second dip at 385 meV observed for h-porphycene is absent in d12-porphycene in which all the peripheral H atoms are replaced by D atoms but without any substitution of the inner H atoms (see the chemical structure in Fig. 3b). This indicates that the first and second dips cannot be assigned to symmetric and anti-symmetric combinations of the fundamental ν(N–H). The possibility of C–H stretching modes is also excluded because the dip is also absent in d2-porphycene. Therefore, it should also be assigned to the different overtone/combination modes from the one at 330 mV.
The spectral anomalies of ν(N–H) of porphycene on Cu(110) indicate a pronounced effect of the anharmonicity causing a strong coupling with a lower vibrational mode(s) that can modulate the H-bonding geometry within the molecular cavity. Such a coupling mechanism and vibrational line broadening of ν(N–H) were examined with quantum mechanical calculations for porphycene in the gas phase.5 Here we have investigated one anharmonic coupling term on the surface by calculating the variation of the ν(N–H) and ν(N–D) energies as a function of the displacement along a lower frequency mode (Fig. 4c). This mode causes the largest displacement of the N–H⋯N geometry and is essentially an N–H bending mode (Fig. 4b). A considerable variation occurs in the ν(N–H) energy (Fig. 4c), indicating a strong anharmonic coupling of ν(N–H) with the bending mode. However, this variation is almost negligible for ν(N–D) because the anharmonicity of the potential has lesser impact on ν(N–D) than on ν(N–H) because of the smaller zero-point motion of D than H. Finally, note that our study only indicates that there is a strong anharmonic coupling term for h-porphycene compared to d2-porphycene but is not intended to describe the anharmonic vibrational line shape and the Fermi resonance. A more complete description would require the calculation including more anharmonic terms in the coupling between the stretching mode and the bending mode as well as nuclear quantum effects.
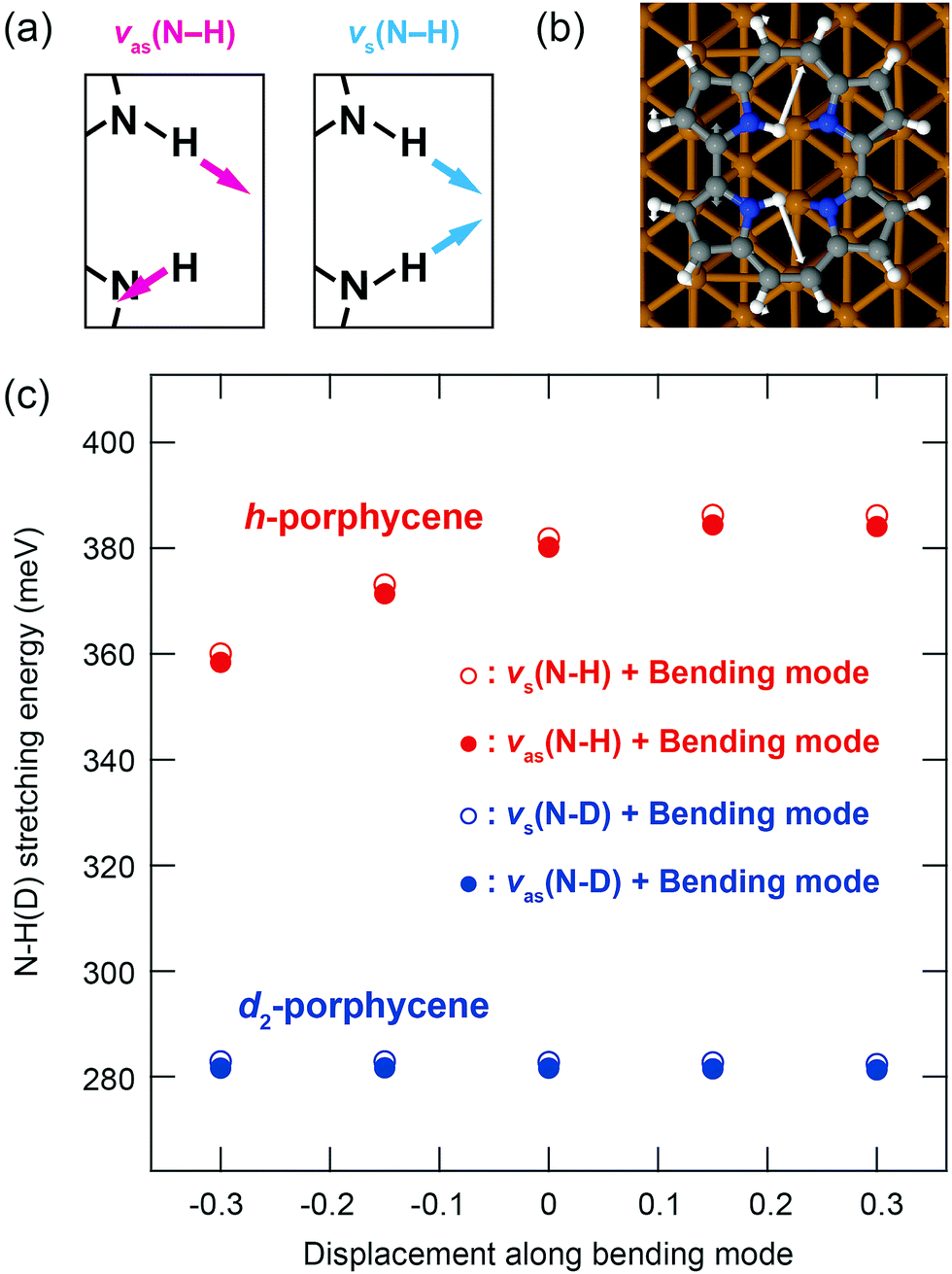 | ||
| Fig. 4 (a) Schematic of anti-symmetric and symmetric ν(N–H). (b) Calculated bending mode at 193.5 (186.6) meV for h-(d2-)porphycene on Cu(110). (c) Variation of the ν(N–H) and ν(N–D) energies as a function of the displacement along the bending mode. The horizontal axis represents the normal coordinate of the bending mode, which is normalized by its root-mean-square vibrational amplitude of the ground state. | ||
Conclusions
We presented a detailed analysis of STM-AS for the cis ↔ cis tautomerization of porphycene isotopologues on a Cu(110) surface at 5 K and discussed a pronounced anharmonicity of the N–H stretching mode. The spectral fitting analysis of STM-AS in combination with the reaction order measurements revealed the vibrational energies and broadening factors of the involved modes. The tautomerization is induced through multiple excitations of skeletal vibrational modes of porphycene and the N–H(D) stretching largely enhances the reaction. These vibrational excitations were also identified in the conductance spectroscopy, manifested as a peak or dip around the vibrational energy. We found that the N–H stretching mode shows not only a significant broadening, but also an emergence of additional vibrational features which indicate a pronounced anharmonicity and strong coupling with lower frequency modes. In contrast, these spectral anomalies were absent in the N–D stretching mode. Theoretical simulations demonstrated a strong intermode coupling of the N–H stretching with the bending mode but not for the N–D stretching mode. These results indicate a significantly different contribution of the anharmonicity between the N–H and N–D stretching modes. Our approach paves the way for studying anharmonicity in H-transfer reactions on solid surfaces at the single-molecule level.Experiments
All experiments were performed in an ultra-high vacuum chamber (base pressure of 10−10 mbar) equipped with a low-temperature STM (modified Omicron instrument with Nanonis Electronics). STM measurements were conducted at 5 K and the bias voltage (Vbias) was applied to the tip (Vtip) or sample (Vsample). The Cu(110) surface was cleaned by repeated cycles of argon ion sputtering followed by annealing to 700 K. STM tips were made from a tungsten or gold wire and then optimized in situ by applying a voltage pulse and controlled indentation of the tip into the surface. Porphycene molecules were deposited onto the surface at room temperature from a Knudsen cell (at a temperature between 450 and 500 K) and the sample was then transferred to the STM at 5 K.Computational methods
DFT calculations were performed using the Fritz-Haber Institute ab initio molecular simulations package (FHI-aims)45 with the Perdew–Burke–Ernzerhof (PBE) functional46 and a Monkhorst–Pack47k-grid of 3 × 3 × 1 k-points in the supercell. Long-range van der Waals forces were accounted for by the Tkatchenko–Scheffler scheme.48 An all-electron and default tight basis set was used. The Cu(110) surface was represented by a four-layer slab with a 4 × 6 surface unit cell and a >20 Å vacuum region. The two topmost layers were relaxed and the residual Cu atoms were fixed at the bulk geometry (lattice constant of 2.568 Å). The vibrations of the adsorbed molecule were calculated on a rigid substrate lattice (the Cu atoms were fixed) by diagonalising the dynamical matrix which was obtained via finite differences of the calculated forces at symmetric ionic displacements of 0.02 Å.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Mariana Rossi, Martin Wolf, Maki Kawai, and Hiromu Ueba for stimulating discussions. T. K. acknowledges the support of Morino Foundation for Molecular Science. S. V. L. is grateful for the support by the Max Planck Society, as well as by the Ministry of Education and Science of the Russian Federation, grant No.14.Y26.31.0005 (simulation of metal–organic interfaces), and grant No. K2-2017-080 in the framework of Increase Competitiveness Program of NUST “MISIS” implemented by a governmental decree dated 16 March 2013, No 211 (modelling of spectroscopy of heterogeneous systems). S. G. and J. W. acknowledge the support by the Polish National Science Center through Grants DEC-2011/02/A/ST5/00043 and DEC-2013/10/M/ST4/00069. D. B. acknowledges the Travel Grant from the UCLA Chancellor's Office. Open Access funding provided by the Max Planck Society.References
- ed. J. T. Hynes, J. P. Klinman, H.-H. Limbach and R. L. Schowen, Hydrogen-Transfer Reactions, Wiley-VHC, Weinheim, 2007 Search PubMed.
- ed. D. Hadži, Hydrogen Bonding, Pergamon Press, London, 1959 Search PubMed.
- G. C. Pimentel and A. L. McLellan, The Hydrogen Bond, Freeman, San Francisco, 1960 Search PubMed.
- L. Antonov, Tautomerism: Methods and Theories, Wiley, 2013 Search PubMed.
- S. Gawinkowski, Ł. Walewski, A. Vdovin, A. Slenczka, S. Rols, M. R. Johnson, B. Lesyng and J. Waluk, Vibrations and hydrogen bonding in porphycene, Phys. Chem. Chem. Phys., 2012, 14, 5489–5503 RSC.
- J. Waluk, Ground- and Excited-State Tautomerism in Porphycenes, Acc. Chem. Res., 2006, 39, 945–952 CrossRef CAS PubMed.
- J. Waluk, Spectroscopy and tautomerization studies of porphycene, Chem. Rev., 2017, 117, 2447–2480 CrossRef CAS PubMed.
- P. Fita, L. Grill, A. Listkowski, H. Piwoński, S. Gawinkowski, M. Pszona, J. Sepioł, E. Mengesha, T. Kumagai and J. Waluk, Spectroscopic and microscopic investigations of tautomerization in porphycenes: condensed phases, supersonic jets, and single molecule studies, Phys. Chem. Chem. Phys., 2017, 19, 4921–4937 RSC.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Thermally and vibrationally induced tautomerization of single porphycene molecules on a Cu(110) surface, Phys. Rev. Lett., 2013, 111, 246101 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Controlling intramolecular hydrogen transfer in a porphycene molecule with single atoms or molecules located nearby, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- J. N. Ladenthin, T. Frederiksen, M. Persson, J. C. Sharp, S. Gawinkowski, J. Waluk and T. Kumagai, Force-induced tautomerization in a single molecule, Nat. Chem., 2016, 8, 935–940 CrossRef CAS PubMed.
- K. Motobayashi, Y. Kim, H. Ueba and M. Kawai, Insight into Action Spectroscopy for Single Molecule Motion and Reactions through Inelastic Electron Tunnelling, Phys. Rev. Lett., 2010, 105, 076101 CrossRef PubMed.
- Y. Kim, K. Motobayashi, T. Frederiksen, H. Ueba and M. Kawai, Action spectroscopy for single-molecule reactions – Experiments and theory, Prog. Surf. Sci., 2015, 90, 85–143 CrossRef CAS.
- B. C. Stipe, M. A. Rezaei, W. Ho, S. Gao, M. Persson and B. I. Lundqvist, Single-Molecule Dissociation by Tunnelling Electrons, Phys. Rev. Lett., 1997, 78, 4410–4413 CrossRef CAS.
- S. G. Tikhodeev and H. Ueba, Relation between inelastic electron tunnelling and vibrational excitation of single adsorbates on metal surfaces, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 125414 CrossRef.
- T. Kumagai, A. Shiotari, H. Okuyama, S. Hatta, T. Aruga, I. Hamada, T. Frederiksen and H. Ueba, Hydrogen relay reactions in real space, Nat. Mater., 2012, 11, 167–172 CrossRef CAS PubMed.
- K. Motobayashi, L. Árnadóttir, C. Matsumoto, E. M. Stuve, H. Jónsson, Y. Kim and M. Kawai, Adsorption of Water Dimer on Platinum(111): Identification of the –OH⋯Pt Hydrogen Bond, ACS Nano, 2014, 8, 11583–11590 CrossRef CAS PubMed.
- K. Motobayashi, Y. Kim, R. Arafune, M. Ohara, H. Ueba and M. Kawai, Dissociation pathways of a single dimethyl disulfide on Cu(111): Reaction induced by simultaneous excitation of two vibrational modes, J. Chem. Phys., 2014, 140, 194705 CrossRef PubMed.
- K. Motobayashi, Y. Kim, M. Ohara, H. Ueba and M. Kawai, The role of thermal excitation in the tunnelling-electron-induced reaction: Dissociation of dimethyl disulfide on Cu(111), Surf. Sci., 2016, 643, 18–22 CrossRef CAS.
- J. Oh, H. Lim, R. Arafune, J. Jung, M. Kawai and Y. Kim, Lateral Hopping of CO on Ag(110) by Multiple Overtone Excitation, Phys. Rev. Lett., 2016, 116, 056101 CrossRef PubMed.
- T. Huang, J. Zhao, M. Feng, A. A. Popov, S. Yang, L. Dunsch and H. Petek, A Molecular Switch Based on Current-Driven Rotation of an Encapsulated Cluster within a Fullerene Cage, Nano Lett., 2011, 11, 5327–5332 CrossRef CAS PubMed.
- J. N. Ladenthin, L. Grill, S. Gawinkowski, Sh. Liu, J. Waluk and T. Kumagai, Hot Carrier-Induced Tautomerization within a Single Porphycene Molecule on Cu(111), ACS Nano, 2015, 9, 7287–7295 CrossRef CAS PubMed.
- H. Böckmann, S. Liu, J. Mielke, S. Gawinkowski, J. Waluk, L. Grill, M. Wolf and T. Kumagai, Direct Observation of Photoinduced Tautomerization in Single Molecules at a Metal Surface, Nano Lett., 2016, 16, 1034–1041 CrossRef PubMed.
- D. Novko, M. Blanco-Rey and J. C. Tremblay, Intermode Coupling Drives the Irreversible Tautomerization in Porphycene on Copper(111) Induced by Scanning Tunnelling Microscopy, J. Phys. Chem. Lett., 2017, 8, 1053–1059 CrossRef CAS PubMed.
- T. Kumagai, J. N. Ladenthin, Y. Litman, M. Rossi, L. Grill, S. Gawinkowski, J. Waluk and M. Persson, Quantum tunneling in real space: Tautomerization of single porphycene molecules on the (111) surface of Cu, Ag, and Au, J. Chem. Phys., 2018, 148, 102330 CrossRef PubMed.
- B. C. Stipe, M. A. Rezaei and W. Ho, Coupling of Vibrational Excitation to the Rotational Motion of a Single Adsorbed Molecule, Phys. Rev. Lett., 1998, 81, 1263–1266 CrossRef CAS.
- T. Komeda, Y. Kim, M. Kawai, B. N. J. Persson and H. Ueba, Lateral hopping of molecules induced by excitation of internal vibration mode, Science, 2002, 295, 2055–2058 CrossRef CAS PubMed.
- M. Parschau, D. Passerone, K. H. Rieder, H. J. Hug and K. H. Ernst, Switching the chirality of single adsorbate complexes, Angew. Chem., Int. Ed., 2009, 48, 4065–4068 CrossRef CAS PubMed.
- H. Ueba, Analysis of lateral hopping of a single CO molecule on Pd(110), Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035440 CrossRef.
- T. Frederiksen, M. Paulsson and H. Ueba, Theory of action spectroscopy for single-molecule reactions induced by vibrational excitations with STM, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 035427 CrossRef.
- The square-root of the reduced mass ratio between N–H and N–D is 1.37.
- The value of N in Fig. 3b is slightly different from our previous results in ref. 9 because we repeated the measurement with the extended range of the tunnelling current to determine the reaction order.
- H. Ueba, T. Mii, N. Lorente and B. N. J. Persson, Adsorbate motions induced by inelastic-tunnelling current: Theoretical scenarios of two-electron processes, J. Chem. Phys., 2005, 123, 084707 CrossRef CAS PubMed.
- Y. E. Shchadilova, S. G. Tikhodeev, M. Paulsson and H. Ueba, Rotation of a Single Acetylene Molecule on Cu(001) by Tunnelling Electrons in STM, Phys. Rev. Lett., 2013, 111, 186102 CrossRef PubMed.
- G. Herzberg, Molecular Spectra and Molecular Structure II, Infrared and Raman Spectra of Polyatomic Molecules, Van Nostrand, Princeton, 1945.
- J. G. Radziszewski, J. Waluk and J. Michl, Site-population conserving and site-population altering photo-orientation of matrix-isolated free-base porphine by double proton transfer: IR dichroism and vibrational symmetry assignments, Chem. Phys., 1989, 136, 165–180 CrossRef CAS.
- J. Kügel, L. Klein, M. Leisegang and M. Bode, Analyzing and Tuning the Energetic Landscape of H2Pc Tautomerization, J. Phys. Chem. C, 2017, 121, 28204–28210 Search PubMed.
- S. Bratož and D. Hadži, Infrared Spectra of Molecules with Hydrogen Bonds, J. Chem. Phys., 1957, 27, 991–997 CrossRef.
- Y. Marechal and A. Witkowski, Infrared Spectra of H-Bonded Systems, J. Chem. Phys., 1968, 48, 3697–3705 CrossRef CAS.
- S. Bratos and H. Ratajczak, Profiles of hydrogen stretching IR bands of molecules with hydrogen bonds:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A stochastic theory. II. Strong hydrogen bonds, J. Chem. Phys., 1982, 76, 77–85 CrossRef CAS.
A stochastic theory. II. Strong hydrogen bonds, J. Chem. Phys., 1982, 76, 77–85 CrossRef CAS. - D. Madsen, J. Stenger, J. Dreyer, E. T. J. Nibbering, P. Hamm and T. Elsaesser, Coherent vibrational ground-state dynamics of an intramolecular hydrogen bond, Chem. Phys. Lett., 2001, 341, 56–62 CrossRef CAS.
- K. Heyne, N. Huse, J. Dreyer, E. T. J. Nibbering and T. Elsaesser, Coherent low-frequency motions of hydrogen bonded acetic acid dimers in the liquid phase, J. Chem. Phys., 2004, 121, 902–913 CrossRef CAS PubMed.
- J. Dreyer, Hydrogen-bonded acetic acid dimers: Anharmonic coupling and linear infrared spectra studied with density-functional theory, J. Chem. Phys., 2005, 122, 184306 CrossRef PubMed.
- Y. Marechal, IR spectra of carboxylic acids in the gas phase: A quantitative reinvestigation, J. Chem. Phys., 1987, 87, 6344–6353 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Ab initio molecular simulations with numeric atom-centered orbitals, Comput. Phys. Commun., 2009, 180, 2175 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Tkatchenko and M. Scheffler, Accurate Molecular van der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
Footnotes |
| † These authors contributed equally to this work. |
| ‡ Permanent address: Department of Chemistry & Biochemistry, University of California, Los Angeles, CA 90095-1569, USA. |
| This journal is © the Owner Societies 2018 |