
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe kinetics and mechanism of oxidation of reduced phosphovanadomolybdates by molecular oxygen: theory and experiment in concert †‡
Alexander M.
Khenkin
,
Irena
Efremenko
,
Jan M. L.
Martin
 and
Ronny
Neumann
*
and
Ronny
Neumann
*
Department of Organic Chemistry, Weizmann Institute of Science, Rehovot, 76100, Israel. E-mail: ronny.neumann@weizmann.ac.il
First published on 20th February 2018
Abstract
The reactivity of the H5PV2Mo10O40 polyoxometalate and its analogues as an electron transfer and electron transfer–oxygen transfer oxidant has been extensively studied in the past and has been shown to be useful in many transformations. One of the hallmarks of this oxidant is the possibility of its re-oxidation with molecular oxygen, thus enabling aerobic catalytic cycles. Although the re-oxidation reaction was known, the kinetics and mechanism of this reaction have not been studied in any detail. Experimentally, we show that both the one- and two-electron reduced polyoxometalate are reactive with O2, the two-electron one more so. The reactions are first-order in the polyoxometalate and O2. Solvents also have a considerable effect, protic solvents being preferred over aprotic ones. H5PV2Mo10O40 was reduced either by an electron transfer reaction (H2) or an electron transfer–oxygen transfer reaction (Ph3P). Similar rate constants and activation parameters were observed for both. DFT calculations carried out on the re-oxidation reactions strongly suggest an inner-sphere process. The process involves first the formation of a coordinatively unsaturated site (CUS) and subsequently the binding of O2 to form superoxo and then peroxo η2-O2 adducts. Most interestingly, although vanadium is the reactive redox centre as well as a necessary component for the oxidative activity of H5PV2Mo10O40, and a CUS can be formed at both Mo and V sites, O2 coordination occurs mostly at the Mo CUSs, preferably those where the vanadium centers are distal to each other.
Introduction
Molecular oxygen is a very attractive oxidant for the oxidative transformation of industrially relevant substrates. However, its direct interaction with organic compounds, particularly in the oxidation of hydrocarbons, is characterized by low selectivity because of radical chain reactions associated with the prevalence of oxo and peroxo radical intermediates. One alternative approach for use of dioxygen is its use as a terminal oxidant in oxidase type reactions, such as those catalysed by Pd compounds,1 or those catalysed by phosphovanadomolybdates, notably the H5PV2Mo10O40 of the α-Keggin structure.2–9 In the latter reactions, substrates are typically activated by electron transfer (ET) to yield either a dehydrogenated product or an oxygenated product by further oxygen transfer (OT). Over the years, reactions involving C–H, C–C and C–M bond activation as well as oxidation of sulphides have been studied.7–9 Importantly, the reduced catalyst, whose structure has been studied by EPR experiments10,11 and DFT calculations,12 can be re-oxidized by molecular oxygen. This spatiotemporal separation between the substrate oxidation and the catalyst regeneration is responsible for high selectivity of such reactions. It should be stressed, as indicated in Scheme 1, that the reaction of one equivalent of H5PV2Mo10O40 with one equivalent of substrate or reducing agent typically leads first to an observable intermediate consisting of an oxidized substrate and reduced H5PV2Mo10O40.7–12 In principle, in the presence of O2 two scenarios can be considered for a catalytic reaction that includes re-oxidation of the reduced H5PV2Mo10O40: (i) product formation as shown in Scheme 1 followed by catalyst re-oxidation; and (ii) direct aerobic oxidation of the intermediate as recently demonstrated in the oxidation of benzene.13 | ||
| Scheme 1 Oxidation of generic substrates (S) by H5PV2Mo10O40. | ||
Thus, although much is known about the mechanisms of substrate oxidation, much less is known about the reactions of reduced polyoxometalates with oxygen. It has thus been shown that a Fe(II)-substituted polyoxotungstate binds O2,14 but more generally the re-oxidation of reduced polyoxotungstates with O2 in water has been shown to proceed by an outer-sphere electron transfer (ET).15–17 The mechanism is less clear in the case of phosphovanadomolybdates. Moreover, although the redox potentials for different α-Keggin polyoxometalates show the following general trend, polyoxotungstates < polyoxomolybdates < polyoxovanadomolybdates,18 the rates of their aerobic re-oxidation are not correlated with the redox potential: polyoxotungstates (seconds) ≫ polyoxovanadomolybdates (minutes) ≫ polyoxomolybdates (days). It was suggested that aerobic re-oxidation of H6PVVVIVMo10O40 after reaction with α-terpinene occurs through inner-sphere electron transfer to oxygen,19 but other publications suggest different scenarios.20–22 Herein, we examine the mechanism of oxidation of reduced H5PV2Mo10O40 by O2 under conditions similar to practical catalytic oxidation applications. In order to understand the mechanism at the atomic and electronic level, the experimental studies were combined with density functional theory (DFT) calculations. It is known that important factors for O2 activation include, but are not limited to, redox potential, solvent, spin state, ligand architecture, and additional co-reductants.23,24 In this work we have considered the oxidation of H6PV2Mo10O40 and H7PV2Mo10O40 by molecular oxygen.
Results and discussion
Kinetics of aerobic oxidation
As can be seen from Scheme 1 the reaction of H5PV2Mo10O40 (the oxidized compound, 1ox) with substrates either by an ET dehydrogenation or an ET–OT oxygenation is intrinsically a two-electron process. However, self-exchange ET between polyoxometalates is fast,2,15,17 that is, the two-electron reduced H7PV2Mo10O40 (1red2e) can readily react with 1ox to yield the one-electron reduced H6PV2Mo10O40 (1red1e). According to our calculations, this interaction is exergonic by 3.73 kcal mol−1 in acetonitrile and is favoured kinetically by the strong acidic nature of the surface protons. Therefore, the substrate/1ox ratio can be manipulated to form either 1red1e or 1red2e. Molecular hydrogen, H2, is the simplest reducing agent, since there is no product formed besides 1red1e or 1red2e. Thus, 1red1e and 1red2e can be formed by the reaction of 0.5 equivalents and 1.0 equivalent of H2 with 1ox, respectively, as in eqn (1):| H2 + 1ox → 1red2e and 1red2e + 1ox → 1red1e | (1) |
In the reaction of either the one- or two-electron reduced species (1red) with O2 in CH3CN, 1 equivalent of O2 was consumed per 2 equivalents and 4 equivalents, respectively, of 1red2e and 1red1e, as determined by volumetric measurements, Table S1 (ESI‡). Therefore, we conclude that 1red2e and 1red1e react with O2 according to eqn (2) and (3), respectively:
| 2H7PV2Mo10O40 + O2 → 2H5PV2Mo10O40 + 2H2O | (2) |
| 4H6PV2Mo10O40 + O2 → 4H5PV2Mo10O40 + 2H2O | (3) |
The kinetics of reoxidation of 1red in acetonitrile was studied spectrophotometrically, by following the disappearance of the reduced polyoxometalate blue colour at 750 nm. Typical kinetic curves are shown in Fig. S1 (ESI‡). The reaction order with respect to the concentration [1red] was determined by measuring the initial rate, −d[1red]/dt, as a function of the initial concentration of [1red], [1red]0, at a constant initial concentration of [O2]. The linear increase in the initial rate with the increase in 1red over a wide range of [1red]0 reveals the reaction to be first-order with respect to [1red] reduced by either 1 or 2 electrons, Fig. S2 (ESI‡). Similarly, the linear dependence of the initial rate on [O2]0, Fig. S3 (ESI‡), indicates that the reaction is first order with respect to O2. Therefore, the direct reaction of 1red with O2 obeys the rate law as shown in eqn (4):
| d[1red]/dt = k[1red]0[O2]0 | (4) |
From this equation, one can calculate a rate constant for 1red1e, k298 = 0.09 ± 0.02 M−1 s−1. This rate constant is two orders of magnitude lower than that for oxidation of one electron reduced polyoxotungstates in water.15 The rate constant for the reoxidation of 1red2e is k298 = 0.9 ± 0.1 M−1 s−1, which is an order of magnitude higher than that measured for 1red1e. In addition, once the blue colour associated with 1red disappeared, iodometric titration revealed that no H2O2 was present in the reaction mixture, although this does not rule out the possibility of H2O2 as a fast-reacting oxidizing intermediate. Indeed, a control experiment on the oxidation of 1red1e by H2O2 yielded a rate constant, k298 = 3.2 ± 0.8 M−1 s−1, which is significantly higher than the one with O2 as oxidant.
It is worth noting that the initial rates, V0, for the aerobic oxidation of 1red are solvent dependent – although as seen in Table 1, V0(1red2e) is always greater than V0(1red1e). Generally, the reactions are faster in protic solvents compared to aprotic ones, although addition of trifluoroacetic acid positively affects the rates in aprotic solvents, while the effect is small for protic solvents. The rate constants are not correlated with the redox potential of 1 in the various solvents.
| Solvent | V 0(1red2e) M s−1 × 107 | V 0(1red1e) M s−1 × 107 | E (V) |
|---|---|---|---|
| Conditions: 0.25 mM 1red, 1 bar air, 25 °C. The redox potentials were obtained by cyclic voltammetry (see the Experimental section) and standardized versus Fc/Fc+. Since the solubility of O2 depends somewhat on the solvent, the dependence of the rate of reoxidation of 1red is presented in terms of initial rates V0 rather than rate constants. nd – not determined. | |||
| Water | 8.4 ± 0.9 | 3.70 ± 0.4 | 0.50 |
| 0.1 M H2SO4 | 21.8 ± 2.5 | 4.48 ± 0.4 | 0.52 |
| AcOH | 15.7 ± 1.5 | 4.21 ± 0.4 | 0.38 |
| MeOH | 15.1 ± 1.6 | 2.92 ± 0.3 | nd |
| Acetonitrile | 3.6 ± 0.5 | 0.36 ± 0.06 | 0.49 |
| Sulfolane | 0.6 ± 0.1 | 0.12 ± 0.02 | 0.50 |
| DMSO | 2.8 ± 0.3 | 0.12 ± 0.02 | 0.40 |
| Diglyme | 3.1 ± 0.4 | 0.24 ± 0.03 | 0.45 |
In addition to the reduction of 1ox by H2, the reduced polyoxometalate was also formed by a typical but simple ET–OT reaction, that is, the oxidation of triphenylphosphine to the corresponding oxide, Scheme 2. At 25 °C, the ion pair formed as a result of electron transfer can be observed by EPR, Fig. S4 (ESI‡). A 31P NMR measurement at this point shows no formation of triphenylphosphine oxide, which however can easily be formed by heating the mixture to 70 °C.
 | ||
| Scheme 2 The ET–OT oxygenation of triphenylphosphine. | ||
The rate constants as well as the transition state energies found for the oxidation of 1red formed by reduction with either H2 or Ph3P are presented in Table 2. As one can see, there is some small variation in the values obtained, but they are very similar and likely within experimental uncertainty. Notable is the large negative value of ΔS#, pointing towards an organized rate-limiting state.
| RA | 1red | k 298, M−1 s−1 | ΔH#298, kcal mol−1 | ΔS#298, cal mol−1 K−1 | ΔG#298, kcal mol−1 |
|---|---|---|---|---|---|
| Conditions: 0.50 mM 1red in CH3CN, 1 bar air. Data from Fig. S5 and S6 (ESI). | |||||
| H2 | 1red1e | 0.09 ± 0.02 | 8.4 | −32.6 | 18.3 |
| H2 | 1red2e | 0.9 ± 0.1 | 5.4 | −39.8 | 17.3 |
| Ph3P | 1red1e | 0.05 ± 0.01 | 8.3 | −36.6 | 19.2 |
| Ph3P | 1red2e | 0.7 ± 0.1 | 6.5 | −36.5 | 17.4 |
Computational results
The overall reactions of oxygen reduction by H7PV2Mo10O40 and H6PV2Mo10O40 according to eqn (2) and (3) are energetically highly favourable: ΔG298 = −57.2 and −50.3 kcal mol−1 in acetonitrile, respectively. However, the first step of the interaction, that is the abstraction of the first H-atom or coupled proton–electron transfer (CPET), is endergonic by 13.5 kcal mol−1 for 1red2e and 16.9 kcal mol−1 for 1red1e. This is in agreement with the strongly negative one-electron reduction potential of oxygen (which is −0.33 V vs. NHE in water at pH 7, 25 °C).25 The interaction between an acidic surface hydrogen of 1red1e and apolar O2 leads to a highly unstable intramolecular complex before hydrogen transfer that is 22.4 kcal mol−1 above the energy of the reactants. The subsequent activation barrier for H transfer is 29.8 kcal mol−1, prohibitively high for a room temperature reaction. See Fig. S7 (ESI‡) for optimized structures for outer sphere complexes. We were unable to locate a similar precursor complex and transition state for 1red2e using our computational approximation. In both cases the probability of the outer-sphere H-transfer is expected to be extremely low since CPET or H-atom transfers are viewed as vibrational processes that require at least weak hydrogen bonding between the reaction centres.26–28Due to the absence of spin restrictions, electron transfer (ET) is often considered the most probable initial stage of oxygen activation.29,30 Our calculations showed that in spite of the high oxidation potential of oxygen and the presence of lone pair electrons, ET and PT from the reduced polyoxometalates are unacceptably high in energy, especially in the gas phase, Table S2 (ESI‡). These results, together with the experimental observations that (a) a catalytic amount of Cu+ does not influence the rate of the oxidation reaction and (b) H2O2 was not observed as an intermediate in the reaction mixture, prompted us to consider an inner-sphere mechanism for aerobic oxidation of 1red. By way of example, we consider the (1,2) and (1,11) isomers of the [PV2Mo10O40]q− anion (Fig. 1), which have two vanadium atoms at the proximal and distal positions, respectively, to each other. These isomers were chosen because they represent two different structure types with different reactivity that have been previously observed.10–12 According to our calculations, the (1,2) isomers of 1ox and 1red1e are 1.5 and 2.2 kcal mol−1 more stable, respectively, than the (1,11) isomer. In contrast, the (1,11) isomer of 1red2e is 0.6 kcal mol−1 more stable than the corresponding (1,2) isomer.
 | ||
| Fig. 1 Surface oxygen atoms in the 1,2 (a) and 1,11 (b) isomers of the [PV2Mo10O40]q− anion. | ||
All 12 metal atoms in the Keggin structure are hexa-coordinated with oxo ligands and thus are not likely to coordinate O2. However, we have previously shown that the tendency of vanadium atoms in high oxidation states towards a reduced coordination number facilitates the formation of defect structures with coordinatively unsaturated sites (CUSs). Depending on which bonds are broken, V or Mo CUSs could be formed.12 Further calculations reported here show that reduction of the polyoxometalate tends to slightly decrease the energetic cost for such defect formation, while protonation slightly increases it, Table 3. The experimentally observed accelerating effect of protic solvents presents an additional indirect proof for CUS formation as an important stage of interaction.
| (1,2)V CUS | (1,2)Mo CUS | (1,11)Mo CUS | |
|---|---|---|---|
| The energies of 2e-reduced structures in the triplet state are shown in parentheses. The optimized structures of the V and Mo CUS are shown in Fig. S8 (ESI). | |||
| H5PV2Mo10O40 | 20.54 | 16.50 | 16.25 |
| [H5PV2Mo10O40]− | 19.58 | 17.85 | 14.10 |
| H6PV2Mo10O40 | 22.28 | 16.93 | 14.35 |
| [H6PV2Mo10O40]− | 17.64 (19.69) | 13.75 (17.94) | 15.08 (16.71) |
| H7PV2Mo10O40 | 18.64 (19.96) | 15.32 (18.86) | 15.54 (21.03) |
The bonding analysis, Fig. S9 (ESI‡), shows that one of the V–O–V bonds represents the weakest bridging bond in the 1ox (1,2) isomer. Breaking of this bond together with one of the adjacent weak Mo–O or V–O bonds leads to molybdenum- or vanadium-centred CUSs, respectively. The electron spin density in intact 1red is distributed between t1u orbitals that are nonbonding with respect to V–O and t2g orbitals that are strongly antibonding with respect to Mo–O, Fig. S10 (ESI‡). Therefore, the reduction of phosphovanadomolybdates most strongly destabilizes Mo–O bonds, making the formation of molybdenum centred CUSs more preferential. Thus, for the (1,2) isomer of 1red, the formation of a Mo CUS is preferred by 2–5 kcal mol−1 relative to its V counterpart. Moreover, Mo CUSs are usually slightly less endergonic for the (1,11) isomer, especially for 1red1e. In contrast to the intact 1red2e structures that have triplet ground states, closed-shell electronic configurations are 1.3–5.5 kcal mol−1 more stable for the defect structures. Finally, it is also notable that the presence of water as a coordinated ligand stabilizes defect structures, especially for the 1ox and 1red1e with V CUSs, Table S3 and Fig. S11 (ESI‡).
Now given that the calculations have shown that the CUSs of 1 are viable species, they can be also considered as excellent candidates for the ligation of O2 and its activation by an inner sphere reaction. Side-on η2-O2, end-on η1-O2 and η2-O2H as well as bridged M–O–O–M coordination modes of O2 at molybdenum and vanadium sites are shown in Chart 1, together with their calculated stabilities. Selected bond lengths, vibrational frequencies and atomic charges of these complexes are assembled in Table S4 (ESI‡). The most notable observation is that the side-on η2-O2 coordination to either Mo or V in defect structures was found to be the most energetically favourable for the one-electron reduced polyoxometalate. This also holds true for the two-electron reduced polyoxometalate, although here also certain M–O–O–H species were stable, albeit still less than the side-on η2-O2 species. In all considered (1,2) isomers, oxygen binding and activation is preferred at a molybdenum CUS where longer O–O bonds, lower O–O vibrational frequencies, and greater transfer of electron density were found (Table S4, ESI‡) compared to the vanadium CUS. The only exception is the end-on coordination mode, for which V superoxo complexes are slightly more stable than the corresponding Mo complexes. The (1,11) isomer possesses only Mo sites that show oxygen binding affinity qualitatively similar to that of the (1,2) isomer.
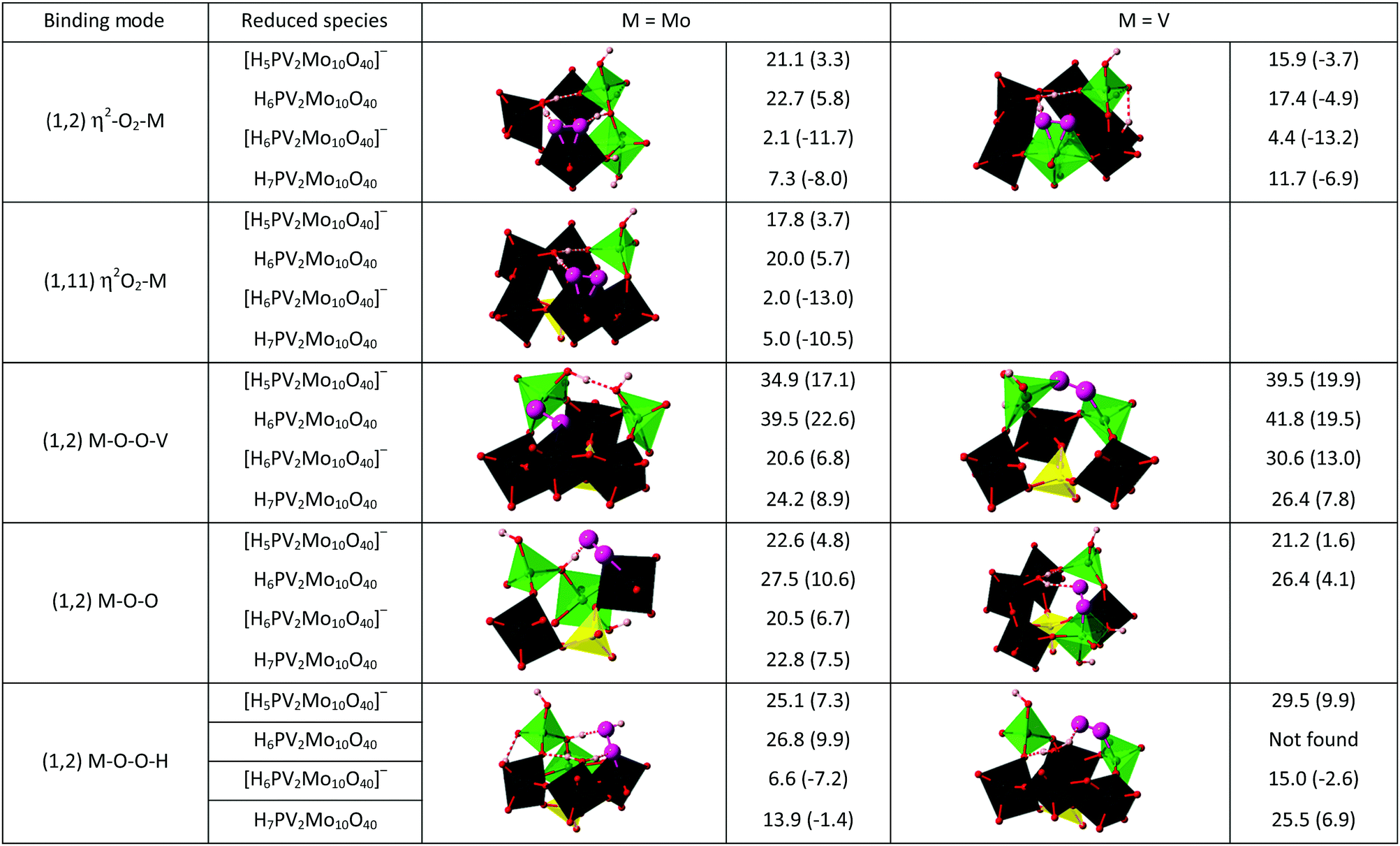 | ||
| Chart 1 Typical O2 modes of ligation to reduced species of 1 and their calculated stabilities. The free energy, ΔG298, kcal mol−1, of the species is given relative to the intact polyoxometalate and to the relevant defect structures (in parentheses). All the complexes are in the lowest possible spin state, that is singlet or doublet. These are the lowest-energy states that were found at the level of theory employed. | ||
These findings can be explained by π-repulsion caused by metal interactions with σ- and π-donating oxygen ligands, and this effect is stronger for V than for Mo because of the difference in atomic sizes.12 For superoxo complexes, the electron transfer from 1red to O2 is comparable for side-on and end-on coordination modes, resulting in quite similar stabilities of these complexes. In contrast, effective 2e-backdonation from metal to O2 in side-on peroxo complexes results in their dramatically higher stability. Indeed, for a V3+ complex with bulky amine ligands that does not suffer from π-repulsion, the side-end peroxo complex was found to be much stronger than in our case (ΔH = −75.0 ± 2.0 kcal mol−1) with a much smaller energy difference between the two isomers (4.2 kcal mol−1), favouring η2 binding.31
Having the defect structures with CUSs and their oxygen complexes in side-on and end-on binding modes as possible intermediates, the mechanism of inner-sphere oxidation 1red can be studied in more detail. The first reaction considered was the stoichiometric oxidation of four H6PV2Mo10O40, 1red1e, species by O2 on three different coordinatively unsaturated sites – Mo CUS in (1,11) and Mo and V CUS in (1,2) isomers, Fig. 2.
 | ||
| Fig. 2 Reaction profiles for the oxidation of H6PV2Mo10O40, 1red1e. All points are local minima on the corresponding potential energy surfaces. | ||
Inspection of the reaction profiles in Fig. 2 shows that defect formation and oxygen coordination are the highest energy states in acetonitrile at room temperature. For activation at vanadium, the enthalpy-controlled reaction of V CUS formation would appear to be the rate determining state of the reaction. In contrast, for the Mo CUS, entropy-controlled oxygen coordination represents the limiting state. Energetically, reactions on both Mo and V CUSs have lower barriers than that for the outer-sphere CPET (vide supra).
Transformation of superoxo to peroxo complexes by hydrogen transfer from another 1red1e species significantly stabilizes oxygen coordination on the Mo CUS. Protonation of coordinated oxygen and subsequent water dissociation are endergonic at this stage by 5–10 kcal mol−1. Hydrogen transfer from the third 1red1e species causes transformation from the side-on to the end-on M–OOH coordination mode, in which intramolecular proton transfer and consequent water dissociation take place. These intermediate reactions proceed with small changes in energy, and most of the complexes remain endergonic with respect to the reactants. Transfer of the fourth H-atom completes 4e O2 reduction and proceeds with strong energy gain. These energetic trends qualitatively resemble the outer-sphere oxygen reduction (Table S2, ESI‡). The last strongly exergonic stage is water dissociation and recovery of 1ox.
Although all points in Fig. 2 correspond to local minima on the corresponding potential energy surface, we believe that the activation barriers are well approximated by the calculated energies of the intermediate structures. The first reaction, formation of defect structures with CUSs, is the only individual step for which a substantial activation barrier could be anticipated. For the most unstable V CUS in the 1ox isomer, we found a late transition state that was only 3.12 kcal mol−1 higher in energy than the final defect structure. In spite of numerous efforts, we were unable to locate a saddle point on the way to other defect structures, presumably because of close geometric and energetic proximity of the local minima corresponding to the final state. Moreover, the presence of water further stabilizes the transition state. The second reaction is the formation of the η2-O2–M adducts in the absence of spin restriction for the interaction of 1red1e with oxygen in its ground state. Finally, self-exchange hydrogen transfer from other 1red1e species at the next stages of the interaction is fast and is favoured kinetically by the strong acidic nature of the surface protons, vide supra. Reorganization of the η2-O2 coordination mode to η1-OOH and the following water dissociation smoothly proceed on the energetic scale and are facilitated by hydrogen bonding (see the corresponding structures in Chart 2). Thus, we believe that the reaction profiles in Fig. 2 represent not only the thermodynamics, but also the kinetics of the 1red1e oxidation reaction.
 | ||
| Chart 2 The most stable O2 adducts to the (1,2) isomer of 1red2e with different spin states and their Gibbs free energy, O–O bond stretching vibrational frequency and Atomic Polar Tensor (APT) charges on oxygen atoms (QO). The ΔG298 values are relative to the plenary, non-defect structure. | ||
Reoxidation of 1red2e shows specific features, related mainly to the spin state of the systems. As mentioned above, the defect structures of the 2e-reduced polyoxometalates have singlet electronic configurations in the ground state, but the triplet states are only a few kcal mol−1 higher in energy, and thus, the oxygen complexes may have different multiplicities, Scheme 3. Interaction between the defect structures in the singlet state with an oxygen molecule results in the formation of a superoxo η2-O2 adduct in the triplet state. Oxygen interaction with the defect structures in the triplet state might lead to superoxo complexes in the open shell singlet (OSS) electronic configuration. In both cases, consequent intramolecular ET yields closed-shell η2-O2 peroxo complexes.
 | ||
| Scheme 3 Schematic representation of oxygen interactions with metals of various electronic states. | ||
Depending on the spin state, oxygen coordination to 1red2e features various kinds of interaction, from weak bonding (quintet) to superoxo (triplet) and peroxo (singlet), Chart 2 and Fig. S12 (ESI‡).32 For some superoxo complexes, we succeeded in locating spin symmetry-broken solutions. Calculations show that the OSS states are lower in energy and correspond to somewhat stronger O2 activation than the triplet state.
The calculated reaction profiles for the oxidation reaction of 1red2e are shown in Fig. 3. In contrast to 1red1e, water coordination does not promote defect formation in these complexes, or only barely does so. Very similar to the case for the re-oxidation of 1red1e, the two-competitive rate-limiting states in these systems are defect formation and barrier-less (spin allowed, Scheme 3) O2 coordination with formation of the triplet superoxo η2-O2 adduct. The next stage of the interaction, intramolecular ET resulting in a much more stable singlet peroxo η2-O2 complex, could potentially have a finite barrier. However, for such transition metal systems, spin crossing probabilities were shown to be close enough to unity, so that they will not materially affect reaction rates.33 The subsequent stages of the reaction are very similar to those for the re-oxidation of 1red1e and involve intermolecular H-transfers from another 1red2e species, resulting in dissociation of two water molecules and recovery of the intact Keggin structure. As in the case of 1e-reduced polyoxometalates, inspection of the reaction profiles in Fig. 3 shows that for Mo CUSs, oxygen coordination represents the highest energy state of the pathway. In contrast, for activation at vanadium, V CUS defect formation would appear to be the rate determining factor of the reaction while the following O2 coordination proceeds with some energy gain in this case.
 | ||
| Fig. 3 Reaction profiles for the oxidation of H7PV2Mo10O40, 1red2e. | ||
Although, using the broken spin-symmetry approximation, we found the OSS η2-O2 superoxo complexes to be more stable than their triplet isomers (vide infra), these reaction profiles are energetically less favourable because of the high energy of the previous stage of interaction, i.e. formation of the defect structure in the triplet state.
In order to bridge between the experimental and computational kinetic data, the energetic span model of Kozuch and co-workers can be applied.34–36 This model is based on Eyring's transition state theory together with the concept of rate-determining states rather than rate-determining steps. In such a way, one can retrieve the computed activation energies for oxidation of different forms of 1red on three coordinatively unsaturated sites, Table 4.
| 1red | ΔH#298, kcal mol−1 | ΔS#298, cal mol−1 K−1 | ΔG#298, kcal mol−1 | |
|---|---|---|---|---|
| 1red1e | Mo (1,2) | 14.6 | −27.2 | 22.7 |
| Mo(1,11) | 10.2 | −33.1 | 20.0 | |
| V (1,2) | 24.5 | 7.4 | 22.3 | |
| 1red2e | Mo (1,2) | 13.8 | −27.6 | 22.0 |
| Mo(1,11) | 11.5 | −28.2 | 19.9 | |
| V (1,2) | 19.3 | 2.2 | 18.6 | |
The following conclusions could be drawn from the comparison of experimental and computational data presented in this work.
(i) The experimental results showed that the oxidations with O2 of both the one- and two-electron reduced polyoxometalates are first-order in 1red and O2 with entropy-driven kinetics. The intermediate species shown in Chart 2 found by computation supported an inner sphere mechanism of these oxidation reactions with oxygen coordination as a rate determining state. Together, inspection of the reaction profiles for oxidation of 1red and kinetic parameters collected in Table 4 show that the stationary points defining the overall apparent activation barrier for Mo CUSs are always the energies of the superoxo complexes. In accordance with the experimental results, these reactions are entropy-driven. The rate-limiting state for oxygen activation on V is clearly enthalpy-driven CUS formation. This reaction pathway is higher in energy than that on the Mo CUS for 1red1e and thus is not expected to be experimentally observed for 1e-reduced polyoxometalates. For 1red2e, both reaction pathways are energetically comparable; however, Mo CUSs are more common than V CUSs in H5PV2Mo10O40 (only two out of five isomers have V atoms in adjoining positions, and even in these cases there are 7 V–O–Mo bonds per each V–O–V bond). Thus, we expect that depending on the relative number of isomers and on the reaction temperature, the kinetics of the Mo CUS or V CUS driven reaction profiles could be observed in the experiments.
(ii) It is notable that theoretically O2 activation by 1red1e is slightly preferred for the (1,11) isomer compared to the (1,2) isomer. In fact, in an EPR experiment where one can differentiate between 1red1e species with vicinal and distal vanadium atoms,10,11 we found that the species associated with distal isomers, e.g. the (1,11) isomer, were oxidized faster than the species associated with vicinal vanadium atoms.
(iii) Since the reduction of O2 to H2O is a four-electron process, additional intermolecular electron transfer and proton transfer steps are needed to split the O–O bond and form water after initial interaction of O2 with the reduced polyoxometalate. The fact that the reaction is first-order in the polyoxometalate and O2, together with the calculated energies of the intermediate hydrogen transfer reactions, clearly indicates that these latter reactions are fast relative to the O2 activation step.
(iv) For 1red2e, the experimental and computed kinetic parameters (ΔG#, ΔH# and ΔS#) are similar and also very similar to those found for 1red1e. The slight difference in the experimentally found ΔG# explains the faster rate constant found for 1red2e, but this is not so obvious from the calculated data.
Experimental
Preparation of the reduced polyoxometalate
The oxidized plenary Keggin compound, H5PV2Mo10O40, 1ox, was prepared using the published method.37 One- or two-electron reduced forms were prepared in organic solvents by reduction with H2 using a 10% Pt/C catalyst or with organic substrates. The degree of reduction was spectrophotometrically verified at 750 nm by titration with m-chloroperbenzoic acid and verified also by oxidation using a standardized solution of Br2 as oxidant. The reduced polyoxometalate was stored under argon. Precise values for the extinction coefficient ε of the one- or two-electron reduced polyoxometalates were determined by titration with m-chloroperbenzoic acid. Using this method, for 1red1e an extinction coefficient of ε750 = 1.1 + 0.2 × 103 M−1 cm−1 was found and for 1red2e, ε750 = 2.2 + 0.3 × 103 M−1 cm−1.Kinetics measurements
Reactions of reduced 1red with O2 were carried out in various organic solvents. Known amounts of 1red were treated with a solution saturated with pure O2, air or O2/Ar mixtures in spectrophotometric cuvettes. Measurements were carried out on an Agilent 8453 diode array spectrometer equipped with a temperature controller nominally accurate to 0.1 °C. Changes in absorption with time at various concentrations and temperatures were observed at 750 nm. The initial rates were derived from these absorption versus time data when plots were linear (about 10% conversion). The solubility of O2 in air-equilibrated CH3CN is 1.6 mM or 8.1 mM in O2 saturated CH3CN at 25 °C.38,39Cyclic voltammetry
Measurements were carried out at 25 °C under an argon atmosphere using an EC-Lab VCP-201 electrochemical workstation (BioLogic Science Instruments). A standard three-electrode electrochemical cell was employed with a glassy carbon disk (surface area 0.071 cm2) as the working electrode, a platinum wire as the counter electrode, and Ag wire as the reference electrode. The scan rate used was 100 mV s−1. Potentials measured versus Ag/Ag+ were converted to the Fc/Fc+ (Fc = ferrocene) scale by analysis of cyclic voltammetry of ferrocene in acetonitrile under same conditions (0.1 M Bu4NClO4 50 mM trifluoroacetic acid) in acetonitrile.Computational methods
All computations were done using a locally modified version of the Gaussian 09 package.40 Geometry optimizations and harmonic frequency evaluations were performed using the nonempirical GGA density functional41,42 PBE43 in conjunction with the PC1 basis set. The latter is a combination of Jensen's polarization consistent pc-1 basis set44–46 for the main group elements and V and the Stuttgart–Dresden–Dolg (SDD) basis set-RECP (relativistic energy-consistent pseudopotential) combination47 for molybdenum; to the latter, a single f-type polarization function was added, the exponent being the geometric average of the two f-exponents given by Martin and Sundermann.48 This combination is of double-ζ plus polarization quality. In order to speed up the calculations, we applied the density fitting (DF) approximation49–51 for calculating the two-electron (Coulomb) integrals. This approximation is known to speed up pure-DFT calculations on large systems by 1–2 orders of magnitude. The auxiliary basis sets required for this were generated by the automatic algorithm as implemented in Gaussian 09. Thus, geometries of all the complexes were optimized at the PBE-DF/PC1 level of theory without symmetry constraints. Subsequently, single-point calculations at the optimized geometries were performed using the empirical hybrid meta-GGA functional M0652 combined with the same basis set (M06/PC1), as well as using the PBE functional but combined with a larger AUG-PC1 basis set. The AUG-PC1 basis set represents a combination of the aug-pc-1 basis set on the main group elements and vanadium44–46,53,54 and the SDD basis set with added f basis functions on molybdenum. The final level of theory applied in the present work is thus the additivity approximation Ee[M06/AUG-PC1] ≈ E[PBE-DF/AUG-PC1] − E[PBE-DF/PC1] + E[M06/PC1].Bulk solvent effects of the acetonitrile medium have been calculated at the M06/PC1 level via the self-consistent reaction field (SCRF) method,55 using the continuum solvation model COSMO as implemented in Gaussian 09.56,57 Dispersion interactions within the computed structures are partially accounted for in the M06 functional. Adding the D3 empirical dispersion correction term,58,59 further improved the energies by considering long interactions. No D3(BJ) parameters for M06 are available from the Grimme group;60 as we optimized parameters for many additional functionals in the framework of a large benchmark project for noncovalent interactions,61 we found the D3(BJ) optimization for M06 to be divergent.
Oxygen is a triplet in its ground state while oxidized products usually have a closed-shell electronic configuration. Therefore, reactions of molecular oxygen often involve a step of intersystem crossing. Thus, we considered doublet and quartet states for 1e-reduced complexes and closed- and open-shell singlet, triplet and quintet states for 2e-reduced complexes. Spin contamination was negligible for all wave functions except for open-shell singlets, for which the broken-symmetry wave functions were contaminated by triplet state with 〈S2〉 close to 1. In this case the energy was corrected using the sum method of Cramer and Ziegler62 according to the equation
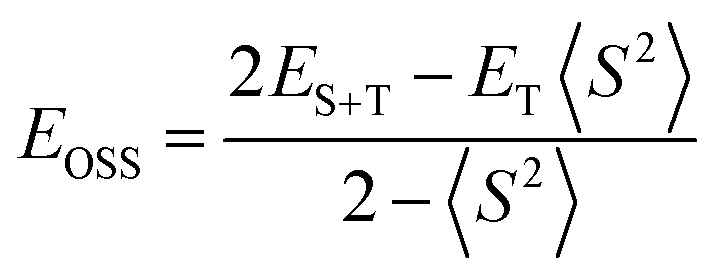
The calculations were performed for two isomers, (1,2) with adjacent vanadium atoms and (1,11) with the most distant vanadium atoms. If not stated otherwise, the energetic results present Gibbs free energy in acetonitrile at 298 K and relate to the (1,2) isomer. Presented enthalpy values involve the same corrections as ΔG, i.e. correction for AUG-PC1, Grimme's dispersion correction D3 for M06 functional and solvation correction.
Conclusions
This combined experiment and computational study provides, for the first time, important insights into the oxidation by O2 of one- and two-electron reduced H5PV2Mo10O40 species, which is one of the key steps in many aerobic catalytic cycles.7–9 The experimental kinetic studies show that the reactions are first-order in both the polyoxometalate and O2, where the rate constants are somewhat higher for the two-electron reduced species. Solvents have a considerable effect, protic solvents being preferred over aprotic solvents—presumably the protons are aiding in the formation of coordinatively unsaturated sites (CUSs). Reduction of H5PV2Mo10O40 by an ET (H2) or ET–OT (Ph3P) reaction showed similar reoxidation kinetics (rate constants and activation parameters). The entropy of activation is strongly negative, indicating a significant reorganization energy in the transition state because two species interact to form one species. DFT calculations and experiments carried out on the re-oxidation reactions suggest an inner-sphere process for O2 activation in contrast with what was observed with the isostructural tungstates.15–17 The reaction pathways proceed by the formation of a CUS, and then the binding of O2, to form a superoxo η2-O2 adduct. After formation of this superoxo η2-O2 adduct, additional inter- and intra-molecular ET and CPET steps lead to O–O bond cleavage and formation of H2O. Most interestingly, although vanadium is the reactive redox centre for oxidation of substrates (reducing agents) and a necessary component for the oxidative activity of H5PV2Mo10O40—and a CUS can be formed at both Mo and V sites—O2 coordination occurs at the Mo CUSs, preferably those where the vanadium centres are distal to each other. This may be at least a partial explanation of the unique activity of H5PV2Mo10O40, as opposed to monomeric or oligomeric vanadates, such as decavanadate where V(IV) species are inert to oxidation with O2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Israel Science Foundation Grants 763/14 and 1358/15, the Israel Ministry of Science and Technology, and the Lise Meitner Centre for Computational Quantum Chemistry. The authors would like to thank Mr Nitai Sylvetsky for assistance with editing the manuscript.Notes and references
- S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef CAS PubMed.
- I. V. Kozhevnikov, Catalysis by Polyoxometalates, Wiley, Chichester, England, 2002 Search PubMed.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef CAS.
- T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113–252 CAS.
- I. A. Weinstock, R. E. Schreiber and R. Neumann, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.7b00444.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- R. Neumann and A. M. Khenkin, Chem. Commun., 2006, 2529–2538 RSC.
- R. Neumann, Inorg. Chem., 2010, 49, 3594–3601 CrossRef CAS PubMed.
- R. Neumann, Adv. Inorg. Chem., 2017, 69, 67–90 CrossRef.
- H. Goldberg, I. Kaminker, D. Goldfarb and R. Neumann, Inorg. Chem., 2009, 48, 7947–7952 CrossRef CAS PubMed.
- I. Kaminker, H. Goldberg, R. Neumann and D. Goldfarb, Chem. – Eur. J., 2010, 16, 10014–10020 CrossRef CAS PubMed.
- I. Efremenko and R. Neumann, J. Am. Chem. Soc., 2012, 134, 20669–20680 CrossRef CAS PubMed.
- B. B. Sarma, R. Carmieli, A. Collauto, I. Efremenko, J. M. L. Martin and R. Neumann, ACS Catal., 2016, 6, 6403–6407 CrossRef CAS.
- D. Barats, G. Leitus, R. Popovitz-Biro, L. J. W. Shimon and R. Neumann, Angew. Chem., Int. Ed., 2008, 47, 9908–9912 CrossRef CAS PubMed.
- Y. V. Geletii, C. L. Hill, R. H. Atalla and I. A. Weinstock, J. Am. Chem. Soc., 2006, 128, 17033–17042 CrossRef CAS PubMed.
- O. Snir, Y. Wang, M. E. Tuckerman, Y. V. Geletii and I. A. Weinstock, J. Am. Chem. Soc., 2010, 132, 11678–11691 CrossRef CAS PubMed.
- O. Snir, Y. Wang and I. A. Weinstock, Isr. J. Chem., 2011, 51, 247–259 CrossRef CAS.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219–237 CrossRef CAS PubMed.
- R. Neumann and M. Levin, J. Am. Chem. Soc., 1992, 114, 7278–7286 CrossRef CAS.
- C. D. Duncan and C. L. Hill, J. Am. Chem. Soc., 1997, 119, 243–244 CrossRef.
- J. H. Grate, J. Mol. Catal., 1996, 114, 93–101 CrossRef CAS.
- E. G. Zhizhina, V. F. Odyakov, M. V. Simonova and K. I. Matveev, Kinet. Catal., 2005, 46, 354–363 CrossRef CAS.
- S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410–11428 CrossRef CAS PubMed.
- S. Hong, Y. Lee, K. Ray and W. Nam, Coord. Chem. Rev., 2017, 334, 25–42 CrossRef CAS.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Acc. Chem. Res., 2001, 34, 273–281 CrossRef CAS PubMed.
- S. P. Neville, O. M. Kirkby, N. Kaltsoyannis, G. A. Worth and H. H. Fielding, Nat. Commun., 2016, 7, 11357 CrossRef CAS PubMed.
- G. G. Hammes, Thermodynamics and Kinetics for the Biological Sciences, John Wiley & Sons, Inc., USA and Canada, 2000 Search PubMed.
- C. Song and J. Zhang, PEM Fuel Cell Electrocatalysts and Catalyst Layers, Springer-Verlag, London, 2008, p. 88 Search PubMed.
- M. Setvin, U. Aschauer, J. Hulva, T. Simschitz, B. Daniel, M. Schmid, A. Selloni and U. Diebold, J. Am. Chem. Soc., 2016, 138, 9565–9571 CrossRef CAS PubMed.
- A. F. Cozzolino, D. Tofan, C. C. Cummins, M. Temprado, T. D. Palluccio, E. V. Rybak-Akimova, S. Majumdar, X. Cai, B. Captain and C. D. Hoff, J. Am. Chem. Soc., 2012, 134, 18249–18252 CrossRef CAS PubMed.
- P. L. Holland, Dalton Trans., 2010, 39, 5415–5425 RSC.
- R. Poli and J. M. Harvey, Chem. Soc. Rev., 2003, 32, 1–8 RSC.
- S. Kozuch and S. Shaik, J. Am. Chem. Soc., 2006, 128, 3355–3365 CrossRef CAS PubMed.
- S. Kozuch and J. M. L. Martin, ChemPhysChem, 2011, 12, 1413–1418 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- G. A. Tsigdinos and C. J. Hallada, Inorg. Chem., 1968, 7, 437–441 CrossRef CAS.
- D. T. Sawyer, A. Sobkowiak and L. L. Roberts Jr., Electrochemistry for Chemists, Wiley, New York, 2nd edn, 1995, p. 505 Search PubMed.
- Q. Li, C. Batchelor-McAuley, N. C. Lawrence, R. S. Hartshorne and R. G. Compton, J. Electroanal. Chem., 2013, 688, 328–335 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Rev. E01, Gaussian, Inc., Wallingford, CT, 2012, http://www.gaussian.com Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev. [Sect.] A, 1965, 140, 1133–1138 CrossRef.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1970, p. 230 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed . Erratum, Phys. Rev. Lett., 1997, 78, 1396.
- F. Jensen, J. Chem. Phys., 2001, 115, 9113–9125 CrossRef CAS.
- F. Jensen, J. Chem. Phys., 2002, 116, 7372–7379 CrossRef CAS.
- F. Jensen, J. Chem. Phys., 2013, 138, 014107 CrossRef PubMed.
- M. Dolg, in Modern Methods and Algorithms of Quantum Chemistry, ed. J. Grotendorst, Jülich, 2000, vol. 1, pp. 479–508 Search PubMed.
- J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408–3420 CrossRef CAS.
- B. I. Dunlap, J. Chem. Phys., 1983, 78, 3140–3142 CrossRef CAS.
- B. I. Dunlap, THEOCHEM, 2000, 529, 37–40 CrossRef CAS.
- A. B. Nadykto, H. Dua and F. Yu, Vib. Spectrosc., 2007, 44, 286–296 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- F. Jensen, J. Chem. Phys., 2002, 117, 9234–9240 CrossRef CAS.
- F. Jensen, J. Chem. Phys., 2003, 118, 2459–2463 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 112, 1995–2001 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, DFTD3 website, viahttp://www.thch.uni-bonn.de.
- B. Brauer, M. K. Kesharwani, S. Kozuch and J. M. L. Martin, Phys. Chem. Chem. Phys., 2016, 18, 20905–20925 RSC.
- W. T. G. Johnson, M. B. Sullivan and C. J. Cramer, Int. J. Quantum Chem., 2001, 85, 492–508 CrossRef CAS.
Footnotes |
| † JMLM is the Baroness Thatcher Professor of Chemistry. RN is the Rebecca and Israel Sieff Professor of Organic Chemistry. |
| ‡ Electronic supplementary information (ESI) available: Additional experimental kinetic data and computational data. See DOI: 10.1039/c7cp08610e |
| This journal is © the Owner Societies 2018 |