
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the crystallisation temperature of very high-density amorphous ice
Josef N.
Stern
 and
Thomas
Loerting
*
and
Thomas
Loerting
*
Institute of Physical Chemistry, University of Innsbruck, A-6020 Innsbruck, Austria. E-mail: thomas.loerting@uibk.ac.at
First published on 10th April 2018
Abstract
The influence of the protocol of preparation on the crystallisation temperature TX of very high-density amorphous ice (VHDA) was studied by varying the annealing pressure (1.1, 1.6 and 1.9 GPa) and temperature (160, 167 and 175 K, respectively). TX increases by up to 4 K in the pressure range of 0.7 to 1.8 GPa for samples annealed at 1.9 GPa compared to samples annealed at 1.1 GPa. Concomitantly, secondary crystallisation channels are suppressed, indicating the absence of structural inhomogeneities. For VHDA prepared at 1.1 GPa and 1.6 GPa our results indicate such inhomogeneities, which we regard to be incompletely amorphized, distorted nanodomains of hexagonal ice that cannot be detected through X-ray diffraction experiments. VHDA prepared at high pressures and temperatures thus represents the amorphous state of water at >0.7 GPa least affected by nanocrystals that has been described so far. We expect the TX obtained for the samples prepared in this manner to be close to the ultimate limit, i.e., we do not consider it possible to raise the low-temperature border to the no-man's land notably further by changing the preparation protocol. An additional, considerable increase in this border will only be possible by working at much shorter time-scales, e.g., by employing fast heating experiments.
Introduction
The discovery of a second amorphous solid phase of water with high density in 19841 introduced a new facet to phase transitions. Besides the well-known concept of solid polymorphism2,3 – i.e., the existence of more than one crystalline form for a substance – the concept of polyamorphism had to be coined for water. In fact, both the vast variety of crystalline phases and the existence of more than one amorphous solid form for water are considered to be among its numerous anomalies.4 Before high-density amorphous ice (HDA) was discovered, lower density glassy water had been known for almost 50 years as the phase condensing from gaseous water on a very cold substrate under vacuum.5,6 The relation between low-density amorphous ice (LDA) and HDA was heavily disputed in the past, especially concerning a possible reversible first-order phase transition between the two, questioning whether or not HDA can be considered a distinct amorphous glassy phase at all.7,8 To add to the complexity Loerting et al. described a third amorphous ice phase with even higher density than HDA in 2001 named very high-density amorphous ice, VHDA.9,10 VHDA can be prepared by annealing of HDA at elevated pressures p > 1 GPa or by pressurisation of LDA at elevated temperatures.11HDA as originally prepared by pressure-induced amorphisation (PIA) of crystalline hexagonal ice Ih at low temperatures (∼77 K) by Mishima et al.1 results in a strained and unrelaxed phase and has later been referred to as “unannealed high-density amorphous ice” (uHDA).12 It is possible to relax and/or equilibrate this unrelaxed variant of HDA by isobaric annealing at low13 and higher pressures12 or by decompression of VHDA at elevated temperatures (140 K).9,14 HDA annealed at lower pressures p ≤ 0.2 GPa is of slightly lower density than uHDA and has thus been labelled and referred to as “expanded high-density amorphous ice” (eHDA).12,15 Both uHDA and eHDA are rather similar in terms of density as well as structure as demonstrated by cryoflotation and neutron diffraction experiments.9,16,17 However, they do differ substantially with respect to their state of relaxation and degree of homogeneity. Specifically, nano-scaled and distorted crystalline domains reminiscent of hexagonal ice were inferred to be the source of these inhomogeneities in the amorphous matrix by Tse et al.18–20 and Seidl et al.21 As a consequence of their nano-size they are elusive to structural characterisation such as XRD or neutron diffraction.22 One method to still identify their presence is to examine the amorphous phases’ thermal properties: at 1 bar HDA experiences a polyamorphic transition to LDA characterised by its transformation temperature Tpoly and enthalpy ΔHpoly. Compared to uHDA, Tpoly is about 20 K higher23 for eHDA and ΔHpoly is reduced by up to 300 J mol−1.24 This evidently indicates that uHDA represents an unrelaxed, high-enthalpy state in contrast to eHDA. At elevated pressures p > 0.1 GPa the polyamorphic transition is avoided and HDA crystallises instead. In a study by Seidl et al. it was demonstrated that the crystallisation temperatures TX for uHDA are considerably lower compared to those of eHDA at pressures 0.1–0.5 GPa.21 These findings have led to the conclusion that incomplete PIA of ice Ih at 77 K leaves behind distorted ice Ih within uHDA. In other words, a mechanical collapse as suggested by Tse and co-workers seems to be at the origin of the high-enthalpy nature of uHDA.18–20 These crystalline inhomogeneities then act as seeds which merely have to grow at the expense of the uHDA matrix. In relaxed eHDA seeds have to nucleate in the first place to then grow. I.e., the activation energy to grow crystals from amorphous ice is decreased due to the presence of nanocrystalline domains. Similarly, the kinetics of crystal growth are accelerated in their presence, such that they lower TX in comparison with the purely amorphous, homogeneous matrix. Furthermore, the nanocrystalline domains may allow for two competing crystallisation modes, where the first one is crystallisation of the purely amorphous material and the second one is growth of nanocrystallites. As a result, phase mixtures may crystallise from amorphous ices containing nanocrystals.
The study by Seidl et al. on HDA was extended by us to higher pressures p ≥ 0.7 GPa using VHDA.25 One of the primary aims was to examine whether the influence of (nano)crystalline remnants in uHDA on TX can be observed also at elevated pressures. We demonstrated that this is the case up to 0.8 GPa, whereas at p ≥ 1.1 GPa uHDA and VHDA become indistinguishable in terms of TX and crystallising phase mixtures. That is, at p ≥ 1.1 GPa structural inhomogeneities in uHDA seem to disappear upon its transition to VHDA. In particular, the influence of secondary crystallisation channels – i.e., transformation to more than one crystalline phase of water – distinguishes VHDA from uHDA. uHDA does not even show traces of ice XII upon crystallisation at 0.7 GPa and 0.5 K min−1, while in VHDA ∼40% of the product yield is ice XII – the crystalline phase that predominantly crystallises at 0.8–1.6 GPa.25,26 In spite of these differences, the crystallisation studies of VHDA prepared at 1.1 GPa indicate that secondary crystallisation channels are not entirely suppressed. It is, thus, the aim of the present study to investigate whether by varying its preparation conditions secondary crystallisation channels in VHDA can indeed be suppressed completely. In other words, it is our intent to probe whether or not nanocrystalline remnants can be amorphised entirely by using higher pressures and temperatures for the preparation of VHDA. This would raise the low-temperature border to the no man's land – the area in the phase diagram of water where non-crystalline phases do not exist – even further than achieved in our recent work.21,22,25 We emphasise that in our study we attempt to shift the border to the no-man's land at a given, fixed time scale, i.e., heating rate. By contrast, it is possible to shift the high-temperature border to the no-man's land to lower temperatures by working on shorter time scales27,28 or avoiding crystallisation completely29,30 by using fast or ultrafast droplet cooling techniques.
Experimental
Preparation of VHDA
The different VHDA samples were prepared from an initial state of uHDA, which was annealed at three different pressures (1.1, 1.6 and 1.9 GPa) to temperatures just prior to crystallisation (160, 167 and 175 K, respectively). To prevent friction the water was loaded into pre-cooled indium containers (with a weight of approximately 0.35 g).21 Force was applied in a uniaxial manner resulting in a semi-hydrostatic pressure distribution inside the sample. The resulting VHDA phases are labelled as VHDA1.1, VHDA1.6 and VHDA1.9 for clarity. We want to stress that the labels in this work serve the sole purpose of marking the pressure (and consequently temperature) at which VHDA was annealed.VHDA samples were subsequently quenched with liquid nitrogen to 77 K and then brought to a given pressure of 0.7–1.8 GPa. At the target pressure the amorphous ice was heated isobarically with 0.5, 5 and 30 K min−1 at 0.7 and 0.8 GPa, and with 5 K min−1 at 1.1, 1.3, 1.6 and 1.8 GPa. Crystallisation was identified from the volume curves as a step-like volume change with temperature, marking the transformation of the amorphous to a crystalline phase/phase mixture, which was then characterised ex situ by powder XRD.
Sample characterization: dilatometry, X-ray diffraction and calorimetry
Experiments were conducted by in situ volumetry using a piston cylinder setup in a material testing machine (ZWICK model BZ100/TL3S) and ex situ structural characterisation via powder X-ray diffraction (Siemens diffractometer, model D5000; Cu Kα1: 0.15418 nm, Cu Kα2 and Cu Kβ are filtered out) in θ–θ geometry as described in our previous study.25 We employ a Göbel-mirror for parallel beam optics. For calorimetric analysis a differential scanning calorimeter (Perkin Elmer, model DSC 8000) was used. Samples of approximately 10 mg were loaded into aluminium DSC crucibles. Volumetry, diffraction and calorimetry experiments were conducted in a precise and controlled thermal environment down to approximately liquid nitrogen temperature (77 K).Results
uHDA → VHDA transformation
 | ||
| Fig. 1 Volume curves for the preparation of very high-density amorphous ice at different pressures 1.1, 1.6 and 1.9 GPa. Amorphous ices are labelled by their pressure of preparation and colour coded orange (VHDA1.1), dark yellow (VHDA1.6) and cyan (VHDA1.9). Slope changes in the volume curves are marked with dotted grey lines in the case of VHDA1.1 and with vertical grey arrows in the case of VHDA1.6 and VHDA1.9. | ||
We have recently demonstrated that this onset in the change of slope shifts to lower temperatures when going to higher pressures (volume curves for uHDA in Fig. 3 in ref. 25). For 1.6 and 1.9 GPa uHDA shows densification in the whole temperature range up to 167 K and 175 K, respectively (see Fig. 1). That is, the change in slope has already shifted to <80 K, or in other words they are unannealed VHDA samples to begin with at p ≥ 1.6 GPa and 77 K. A slight flattening of the curves at 1.6 and 1.9 GPa can be noted however, starting at T > 145 K in both cases (marked in Fig. 1 with grey vertical arrows). We infer this to originate from the glass transition of the amorphous solid to the ultraviscous liquid. This is in accordance with previous dielectric spectroscopy results showing dielectric relaxation types typical of ultraviscous liquids at temperatures T ≳ 140 K and pressures p ≳ 1 GPa.32 However, since not only reversible effects but also the irreversible densification contribute to the shape of the volume curve, flattening cannot be directly assigned to the volumetric glass-to-liquid transition temperature.33
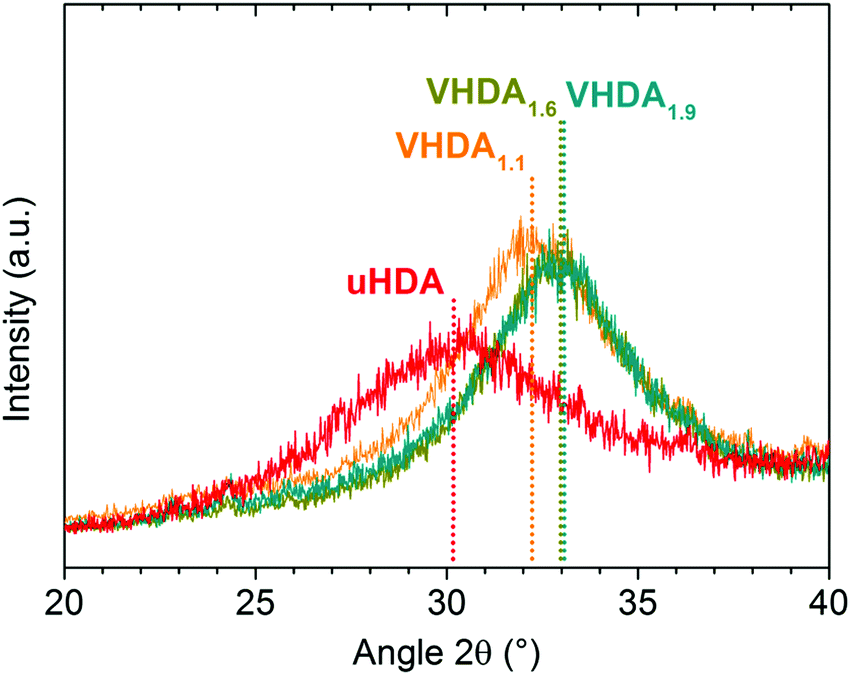 | ||
| Fig. 2 Diffractograms of unannealed high-density amorphous ice (in red) and differently prepared very high-density amorphous ices in orange (VHDA1.1), dark yellow (VHDA1.6) and cyan (VHDA1.9). | ||
In addition to the peak position also the peak width changes significantly. The uHDA halo peak is much broader than all the VHDA halo peaks (see Table 1). Assuming that the broadening reflects structural inhomogeneities34 we note a trend of decreasing inhomogeneity upon increasing the preparation pressure for VHDA (see full-width at half maximum (FWHM) listed in Table 1).
 | ||
| Fig. 3 Differential scanning calorigrams of VHDA1.1, VHDA1.6 and VHDA1.9 recorded at atmospheric pressure and employing a heating rate of 10 K min−1 (VHDA1.1 in orange, VHDA1.6 in dark yellow and VHDA1.9 in cyan). | ||
Crystallisation of VHDA at 0.7 and 0.8 GPa
To gain further insight into the nature of these differently prepared forms of VHDA their crystallisation upon isobaric heating was studied at elevated pressures using several heating rates. Fig. 4 and 5 exhibit results from individual isobaric heating experiments at 0.7 and 0.8 GPa for VHDA1.1, VHDA1.6 and VHDA1.9 at heating rates of 0.5, 5 and 30 K min−1. As has been described in the literature, transformation behaviour of the amorphous ices to crystalline phases upon isobaric heating changes both with pressure and heating rate.21,22,25,26,31 From the experiment we extract the onset crystallisation temperatures and composition of the crystallised product. | ||
| Fig. 4 Upper panel: In situ volume curves of isobaric heating runs for VHDA1.1 (orange), VHDA1.6 (dark yellow) and VHDA1.9 (cyan) at 0.7 GPa with 0.5, 5 and 30 K min−1 heating rate. Lower panel: Structural characterisation by ex situ powder X-ray diffraction of the quench-recovered samples, colour coded accordingly. Diffractograms are stacked for clarity and labelled with the composition of their crystalline phase mixtures. | ||
 | ||
| Fig. 5 Same as Fig. 4, but for experiments at a pressure of 0.8 GPa. | ||
This indicates that the crystalline phase to which VHDA converts is of higher density (mainly ice XII, some ice IV; and sometimes traces of ice V). Similar to previous work,25TX is defined by the intersection of two straight lines through linear parts in the volume curve just prior to and after the onset point of crystallisation (left upper panel, Fig. 4). The volume curves of Fig. 4 and 5 (upper panels) are aligned at the end point of crystallisation in cases of equal or very similar crystalline phase composition (see, e.g., Fig. 4 upper panel, middle and right).
The crystalline phase composition is obtained from the intensity ratio of the most intense Bragg peaks by ex situ powder XRD characterisation (at ∼80 K and p ∼ 10−1 mbar, bottom panels in Fig. 4 and 5).19,35–37 Regarding TX and taking a look into the differently prepared VHDA phases separately, trends are observed similar to the ones reported in the literature.21,22,25,26 That is, for a given amorphous phase and at a selected pressure TX increases with heating rate. For instance, TX for VHDA1.6 at 0.7 GPa changes from 162 K to 166 K and 173 K when going from 0.5 to 5 and 30 K min−1 heating rate. Also, when employing the same heating rate TX increases with pressure, e.g., VHDA1.6 from 167 to 169 K at 5 K min−1. The same can be observed for VHDA1.1 and VHDA1.9. Furthermore, TX increases going from VHDA1.1 to VHDA1.6 and VHDA1.9 under identical experimental conditions, i.e., heating with the same rate at the same pressure. VHDA1.9 crystallises at temperatures at least 2 K (and up to 6.5 K) above those of VHDA1.1 with the exception at 0.7 GPa/30 K min−1.
| VHDA1.1 | VHDA1.6 | VHDA1.9 | |
|---|---|---|---|
| 0.5 K min−1 | |||
| V/IV/XII (%) | V/IV/XII (%) | V/IV/XII (%) | |
| 0.7 GPa | 7/50/43 | —/19/81 | —/7/93 |
| 0.8 GPa | —/40/60 | —/4/96 | —/4/96 |
| VHDA1.1 | VHDA1.6 | VHDA1.9 | |
|---|---|---|---|
| 5 K min−1 | |||
| IV/XII/VI (%) | IV/XII/VI (%) | IV/XII/VI (%) | |
| 0.7 GPa | 7/93/— | 3/97/— | 1/99/— |
| 0.8 GPa | 7/93/— | 1/99/— | 1/99/— |
| 1.1 GPa | 3/97/— | —/100/— | —/97/3 |
| 1.3 GPa | 2/97/1 | —/85/15 | —/91/9 |
| 1.6 GPa | 5/94/<1 | —/85/15 | —/59/41 |
| 1.8 GPa | <1/48/51 | —/20/80 | —/2/98 |
| VHDA1.1 | VHDA1.6 | VHDA1.9 | |
|---|---|---|---|
| 30 K min−1 | |||
| IV/XII (%) | IV/XII (%) | IV/XII (%) | |
| 0.7 GPa | —/100 | 2/98 | 7/93 |
| 0.8 GPa | —/100 | <1/>99 | <1/>99 |
Crystallisation of VHDA at 1.1 to 1.8 GPa
The difference in crystallisation temperature ΔTX for differently prepared VHDA prevails also at higher pressures up to 1.8 GPa (Fig. 6 and 7). In between VHDA1.1 and VHDA1.9 ΔTX is approximately 4 K throughout the whole pressure range, employing a heating rate of 5 K min−1 (see Fig. 7). The evolution of the volume curves displayed in Fig. 6 is a reflection of the amorphous samples’ and the crystalline products’ varying densities. While VHDA1.1 apparently transforms to a denser phase upon crystallisation in all cases, VHDA1.9 in fact expands at pressures p < 1.8 GPa when crystallising. Bearing in mind that both VHDA1.1 and VHDA1.9 at pressures p < 1.6 GPa crystallise predominantly to ice XII of a fixed density, this implies that the densities of the amorphous matrices just before TX are different. That is, the amorphous matrix has not reached an equilibrium density just before TX at 1.1–1.3 GPa. At 1.8 GPa all three VHDA variants densify upon crystallisation, and in all cases the denser ice VI is now the crystalline phase primarily formed. The densification that VHDA1.1 (top panel, Fig. 6, 1.3–1.8 GPa) and VHDA1.6 (top panel, Fig. 6, 1.8 GPa) exhibit in a broad temperature range prior to the step-like transformation is due to relaxation of the amorphous matrix to a more dense one at pressures above their respective pressure of formation (1.1 GPa for VHDA1.1 and 1.6 GPa for VHDA1.6, respectively). | ||
| Fig. 6 Upper panels in situ volume curves of isobaric heating runs for VHDA1.1 (orange), VHDA1.6 (dark yellow) and VHDA1.9 (cyan) in the pressure range 1.1–1.8 GPa with a heating rate of 5 K min−1 exclusively. Lower panels results of the corresponding structural characterisation by ex situ powder X-ray diffractometry of the quench recovered samples, colour coded correspondingly. Diffractograms are stacked for clarity and labelled with the composition of their crystalline phase mixtures. | ||
 | ||
| Fig. 7 Crystallisation temperatures for differently prepared VHDAs at pressures 0.7–1.8 GPa, heated isobarically with a rate of 5 K min−1. The results of the differently prepared amorphous phases are colour coded accordingly (VHDA1.1 in orange VHDA1.6 in dark yellow and VHDA1.9 in cyan). Standard deviation is 0.2 K. Error bars are, thus, smaller than the symbol size. | ||
As demonstrated in Fig. 7 the same trend as at lower pressures (0.7 and 0.8 GPa) is observed at higher pressures 1.1–1.8 GPa regarding TX. That is, at the same pressure and heating rate TX increases with pressure (and temperature) of VHDA preparation: TX (VHDA1.1) < TX (VHDA1.6) < TX (VHDA1.9).
The reproducibility of the experimental results is rather high. Crystallisation temperatures from individual runs for the same type of experiment (i.e., isobaric heating of the same VHDA phase at the same pressure and with the same heating rate) lie within 2 K, and the standard deviation as determined from several sets of identical experiments is approximately 0.2 K.
Discussion
Polyamorphic transitions at atmospheric pressure
As has been demonstrated for HDA in the literature the transformation to LDA at atmospheric pressure is much dependent on the amorphous ices’ state of relaxation.23,40 A transformation temperature of ∼116 K for uHDA, e.g., is 20 K below that found for eHDA when being heated with 10 K min−1 at patm.40 This reflects the unrelaxed, high-enthalpy nature of uHDA and can directly be associated with the presence of nanocrystalline domains contained in uHDA. Since the densities of uHDA and eHDA are very similar one would expect them to be roughly equal in terms of enthalpy. The difference in enthalpy, thus, requires an explanation that is not based on density – the presence of nanocrystallites provides such an explanation. By contrast, VHDA1.1 reproducibly transforms to LDA at temperatures ∼2 K higher than VHDA1.6 and VHDA1.9. This indicates that VHDA1.9 represents a high-enthalpy state at ambient pressure as well. However, because the density for VHDA1.9 is higher than the density for VHDA1.1 it is not possible to judge from these measurements on the presence or absence of nanocrystalline domains embedded in the amorphous matrix. To answer this question one needs to resort to measurements of the VHDA crystallisation kinetics.Suppression of type 1 crystallisation kinetics in VHDA1.9
Going from the lowest (0.7 GPa) to highest (1.8 GPa) pressure, the shift to ice VI as the crystalline phase predominantly formed upon crystallisation at the expense of ice XII can be observed for all differently prepared VHDA phases and is summarised in Table 2. The process of parallel crystallisation and its dependence on heating rate, but also pressure is well known for the amorphous ices. Salzmann et al. described this phenomenon in the case of HDA (uHDA) as a competition of transformation processes to different crystalline phases, being connected to different respective crystallisation kinetics.26 At a given pressure, that is depending on the heating rate, the relative yield of one crystalline phase in a mixture of crystalline phases can be influenced by choosing a low or a high heating rate. In this context Salzmann et al. labelled the processes with “type 1 kinetic behaviour” for transformation processes with slower kinetics starting at lower temperatures and “type 2 kinetic behaviour” for transformation processes related to faster kinetics starting at higher temperatures. This has also been observed in the case of VHDA (VHDA1.1).25 In fact, at lower pressures, e.g., 0.7 GPa the relative yield of ice XII in the phase mixture may change from ∼40% to ∼90% and ultimately 100% when increasing the heating rate from 0.5 to 5 and 30 K min−1 (orange diffractograms in the lower panel of Fig. 4). Here, ice XII forms favourably and in greater amounts at higher heating rates and thus apparently transforms in a process connected to faster type 2 kinetics. Ice IV (and ice V at 0.5 K min−1 heating rate) consequently transforms with slower type 1 kinetics, and its formation is entirely suppressed when heating with a high rate of 30 K min−1. Also at 0.8 GPa the suppression of ice IV formation with increasing heating rate is observable (orange diffractograms in the lower panels of Fig. 5). While at a slow heating rate of 0.5 K min−1 ice IV is still formed in considerable amounts (∼40%), at higher heating rates of 5 and 30 K min−1 its relative yield decreases to 5–10% and ultimately diminishes to zero.As has been demonstrated on the example of HDA26 the competition and interplay between transformation processes with different kinetics is not only dependent on heating rate, but also on pressure. That is, crystalline phases transforming with faster type 2 kinetics at lower pressures change their behaviour to slower type 1 kinetics when going to higher pressures with respect to other, denser crystalline phases – the formation of which is more favourable under these conditions and can now be considered of faster type 2. In the case of VHDA (VHDA1.1) it has been shown in the literature that the pronounced formation of ice IV when heating with low rates (0.5 K min−1) and at lower pressures (0.7, 0.8 GPa) decreases when going to higher pressures (Fig. 2 in ref. 25). The relation ice IV/type 1 kinetics – ice XII/type 2 kinetics is generally observed for VHDA1.1 in the whole pressure range 0.7–1.8 GPa and at all heating rates. However, at pressures p ≥ 1.3 GPa the formation of denser ice VI becomes increasingly favourable as is demonstrated in Table 2. And at 1.8 GPa the amount of ice VI formed upon crystallisation is already ∼50% when heating with a rate of 5 K min−1. In the frame of parallel transformations with different kinetic processes the formation of ice VI can now be considered of faster type 2, while crystallisation of ice IV is still of the slower type 1. Ice XII, however, must kinetically be related to a process intermediate with respect to ices IV and VI. This tendency is even more pronounced in the cases of VHDA1.6 and VHDA1.9. Already at 1.3 GPa considerable amounts (∼10% or more) in the crystalline phase mixtures are ice VI, and the formation of ice IV is entirely suppressed. And at 1.8 GPa the relative yield of ice VI after crystallisation from VHDA1.9 is almost 100%.
Influence of annealing pressure and temperature on nanocrystals embedded in VHDA
The fact that the density of the examined amorphous phases is dependent on the pressure of preparation and is related to the broad halo peak in an X-ray diffractogram is well known in the literature.9,14,41 A shift to higher angles (2θ) as demonstrated in Fig. 2 is thus a reflection of an increase in density. The higher the pressure (and temperature) of preparation and thus the density of VHDA, the higher the temperatures at which it crystallises upon isobaric heating. In comparison with VHDA annealed at 1.1 GPa (to 160 K), the resulting amorphous phases of very high density are thermally more stable against transformation by up to approximately 4–5 K (in the case of VHDA1.9) throughout the whole pressure range of 0.7–1.8 GPa. Furthermore, VHDA1.9 shows a complete suppression of secondary crystallisation channels at the lowest (0.7 GPa) as well as the highest (1.8 GPa) examined pressure. Regarding ice XII, the shift from formation with faster type 2 kinetics (ice IV being the slower type 1 kinetics co-phase at p ≤ 0.8 GPa) to slower type 1 kinetics (ice VI being the faster type 2 kinetics co-phase at p ≥ 1.1 GPa) occurs at considerably lower pressures than is the case with VHDA1.1. At 1.8 GPa the relative yield of ice VI in the product's phase mixture for VHDA1.9 is already close to 100%, whereas in the case of VHDA1.1 it is ∼50% with a mixture containing ices VI, XII and even IV. The suppression of a secondary crystallisation channel even at low heating rates as well as increased transformation temperatures (TX (VHDA1.1) < TX (VHDA1.6) < TX (VHDA1.9)) well demonstrates that the protocol of preparation for VHDA defines its homogeneity. In this context the term ‘homogeneity’ is related to the absence of crystalline remnants. As has been described in the literature these crystallites likely survive the pressure-induced amorphisation of hexagonal ice Ih when forming uHDA and are on the nano-scale, elusive to structural characterisation such as X-ray and neutron diffraction.21,22 When uHDA is annealed under pressure the nanocrystallites dissolve at least partially in the amorphous matrix prior to crystallisation. The higher the pressure of annealing the further away the system is from equilibrium conditions of the crystalline remnants. That is, the less likely the crystallites are to survive the annealing process when forming VHDA from uHDA. Or in other words, the higher the pressure and temperature HDA is annealed at, the more homogeneous the resulting VHDA.From higher to lower pressures: connecting the low-temperature boundary of no man's land
The results of our previous work on this topic25 indicated a rather smooth transition between the crystallisation lines of VHDA (VHDA1.1) and HDA (eHDA) applying intermediate heating rates (2 and 5 K min−1). The new outcomes for the more homogeneous and thermally stable VHDA1.9, however, draw a more complex picture (Fig. 8). The fact that the crystallisation temperatures of VHDA1.9 are consistently at least 4 K above those determined previously for VHDA1.1 allows for three scenarios, indicated by the numbered, dotted cyan lines in Fig. 8. In the first one the crystallisation lines connect discontinuously, marked by a pronounced kink in the boundary to the no man's land. Although such a kink would not be direct evidence, indirectly it might indicate a first-order transition between HDA and VHDA. Such a first-order relation has been described previously in isothermal compression experiments, following the polyamorphic transition path of LDA → HDA → VHDA at 125 K.11 However, it would require time scales to be slow enough for interconversion between HDA and VHDA prior to crystallisation for the considered heating rates (i.e., intermediate rates of 2 or 5 K min−1, respectively). In a second scenario the crystallisation lines may merge continuously, pointing towards a higher order transition. Also in this scenario time scales would have to allow for polyamorphic transitions prior to crystallisation. The third possible scenario would not see a conjunction of crystallisation lines at all at elevated pressures. Here, TX (HDA) and TX (VHDA) would intersect at much lower pressures. | ||
| Fig. 8 Crystallisation temperatures of eHDA (green) at p ≤ 0.5 GPa from Seidl et al.21 and differently prepared VHDA (VHDA1.1, orange and VHDA1.9, cyan) at p ≥ 0.7 GPa in the intermediate pressure regime. Dotted lines indicate possible scenarios connecting the crystallisation lines for eHDA and VHDA. Stable crystalline ice phases from the phase-diagram are marked in grey. Polymorphs (stable as well as metastable) crystallising from VHDA upon isobaric heating (black arrows) are marked by white boxes. | ||
A further interesting Gedanken experiment is the formation of eHDA via VHDA1.9 instead of the previously employed route via VHDA1.1. That is, preparing eHDA by decompressing VHDA1.9 at elevated temperatures (140 K) to lower pressures, e.g., 0.2 GPa. Comparing the crystallisation behaviour of such differently prepared eHDA variants might yield an answer to the question of whether eHDA prepared via VHDA1.1 is a homogeneous amorphous phase or if the inhomogeneities apparently prevalent in VHDA1.1 ‘survive’ the elevated-temperature decompression to eHDA. In the former case, eHDA crystallisation temperatures would be indistinguishable, no matter whether VHDA1.9 or VHDA1.1 is employed for eHDA formation. In the latter case we would expect eHDA prepared via the homogeneous VHDA1.9 to be thermally more stable against crystallisation than eHDA prepared via less homogeneous VHDA1.1. This Gedanken experiment will be realized in future studies.
Summary and outlook
The influence of annealing pressure and temperature when preparing VHDA from uHDA on the crystallisation behaviour of VHDA upon isobaric heating at elevated pressures 0.7–1.8 GPa was studied regarding its dependence on heating rate and pressure. The amorphous phases of very high density are indexed with the pressure at which they were annealed (VHDA1.1, VHDA1.6 and VHDA1.9). It could be shown that the higher the annealing pressure and consequently temperature, the higher the thermal stability against crystallisation at all examined pressures. Crystallisation temperatures of VHDA as prepared in the past (by annealing of uHDA at 1.1 GPa to 160 K9) were consistently ∼4 K below those of VHDA annealed at 1.9 GPa to 175 K. Furthermore, the process of parallel crystallisation is almost entirely suppressed at the lowest and highest examined pressures in the case of VHDA1.9. We, thus, consider VHDA prepared at high pressures and temperatures to be the most relaxed and homogeneous amorphous matrix of very high density described so far. In the context of the discussion about nanocrystalline domains embedded in the matrix of uHDA, this implies that the amorphisation remains incomplete after annealing at 1.1 GPa, whereas it comes much closer to completion at 1.9 GPa. In other words, we regard VHDA1.9 to be close to or fully fully amorphised, nearly or entirely free from distorted, crystalline nanodomains.In the future it will be of interest to examine the transformation of the most stable amorphous phase of very high density (VHDA1.9) at pressures p < 0.7 GPa. In the authors’ previous study25 it was shown that the crystallisation temperatures of VHDA1.1 at pressures p ≥ 0.7 seem to merge perfectly with those obtained for eHDA at pressures p ≤ 0.5 GPa in the literature.21 The new results, however, indicate that the transformation of the most thermally stable form of very high density (VHDA1.9) occurs at notably higher temperatures. It would be rather interesting to see at which pressure the crystallisation temperatures of VHDA1.9 and eHDA do converge.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Benedict Neumann for all technical support. We thankfully acknowledge funding by the Austrian Science Fund FWF (bilateral international project FWF-ANR I1392). J.S. is the recipient of a DOC Fellowship of the Austrian Academy of Sciences ÖAW at the Institute of Physical Chemistry, University of Innsbruck.References
- O. Mishima, L. D. Calvert and E. Whalley, “Melting ice” I at 77 K and 10 kbar: a new method of making amorphous solids, Nature, 1984, 310, 393–395 CrossRef CAS.
- M. H. Klaproth, Bergmännische J., 1798, I, 294–299 Search PubMed.
- E. Mitscherlich, Über die Körper, welche in zwei verschiedenen Formen krystallisieren, Abhl. Akad. Berlin, 1822–1823, 43–48 Search PubMed.
- P. G. Debenedetti, Supercooled and glassy water, J. Phys.: Condens. Matter, 2003, 15, R1669–R1726 CrossRef CAS.
- E. F. Burton and W. F. Oliver, The crystal structure of ice at low temperatures, Proc. R. Soc., Ser. A, 1935, 153, 166–172 CrossRef CAS.
- E. F. Burton and W. F. Oliver, X-ray diffraction patterns of ice, Nature, 1935, 135, 505–506 CrossRef CAS.
- D. D. Klug, J. S. Tse, V. Shpakov, C. A. Tulk, I. Swainson and E. C. Svensson, et al., Transformations, dynamics, and structures of amorphous ices: is it a glass or an amorphous solid?, NATO Sci. Ser., II, 2002, 81(New Kinds of Phase Transitions: Transformations in Disordered Substances), 391–402 CAS.
- J. S. Tse, D. D. Klug, M. Guthrie, C. A. Tulk, C. J. Benmore and J. Urquidi, Investigation of the intermediate- and high-density forms of amorphous ice by molecular dynamics calculations and diffraction experiments, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 214107 CrossRef.
- T. Loerting, C. Salzmann, I. Kohl, E. Mayer and A. Hallbrucker, A second distinct structural “state” of high-density amorphous ice at 77 K and 1 bar, Phys. Chem. Chem. Phys., 2001, 3(24), 5355–5357 RSC.
- J. L. Finney, D. T. Bowron, A. K. Soper, T. Loerting, E. Mayer and A. Hallbrucker, Structure of a New Dense Amorphous Ice, Phys. Rev. Lett., 2002, 89, 205503 CrossRef CAS PubMed.
- T. Loerting, W. Schustereder, K. Winkel, C. G. Salzmann, I. Kohl and E. Mayer, Amorphous Ice: Stepwise Formation of Very-High-Density Amorphous Ice from Low-Density Amorphous Ice at 125 K, Phys. Rev. Lett., 2006, 96, 025702 CrossRef PubMed.
- R. J. Nelmes, J. S. Loveday, T. Strässle, C. L. Bull, M. Guthrie and G. Hamel, et al., Annealed high-density amorphous ice under pressure, Nat. Phys., 2006, 2(6), 414–418 CrossRef CAS.
- C. A. Tulk, C. J. Benmore, J. Urquidi, D. D. Klug, J. Neuefeind and B. Tomberli, et al., Structural Studies of Several Distinct Metastable Forms of Amorphous Ice, Science, 2002, 297, 1320–1323 CrossRef CAS PubMed.
- K. Winkel, M. S. Elsaesser, E. Mayer and T. Loerting, Water polyamorphism: reversibility and (dis)continuity, J. Chem. Phys., 2008, 128(4), 044510 CrossRef PubMed.
- O. Mishima, Polyamorphism in water, Proc. Jpn. Acad., Ser. B, 2010, 86(3), 165–175 CrossRef CAS PubMed.
- T. Loerting, M. Bauer, I. Kohl, K. Watschinger, K. Winkel and E. Mayer, Cryoflotation: Densities of Amorphous and Crystalline Ices, J. Phys. Chem. B, 2011, 115, 14167–14175 CrossRef CAS PubMed.
- D. T. Bowron, J. L. Finney, A. Hallbrucker, I. Kohl, T. Loerting and E. Mayer, et al., The local and intermediate range structures of the five amorphous ices at 80 K and ambient pressure: a Faber-Ziman and Bhatia-Thornton analysis, J. Chem. Phys., 2006, 125, 194502 CrossRef CAS PubMed.
- J. S. Tse and M. L. Klein, Pressure-induced phase transformations in ice, Phys. Rev. Lett., 1987, 58(16), 1672–1675 CrossRef CAS PubMed.
- M. Koza, H. Schober, A. Tölle, F. Fujara and T. Hansen, Formation of ice XII at different conditions, Nature, 1999, 397, 660–661 CrossRef CAS.
- G. P. Johari, On the amorphization of hexagonal ice, the nature of water's low-density amorph, and the continuity of molecular kinetics in supercooled water, Phys. Chem. Chem. Phys., 2000, 2, 1567–1577 RSC.
- M. Seidl, A. Fayter, J. N. Stern, G. Zifferer and T. Loerting, Shrinking water's no man's land by lifting its low-temperature boundary, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91(14), 144201 CrossRef.
- M. Seidl, K. Amann-Winkel, P. H. Handle, G. Zifferer and T. Loerting, From parallel to single crystallization kinetics in high-density amorphous ice, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88(17), 174105 CrossRef.
- K. Winkel, E. Mayer and T. Loerting, Equilibrated High-Density Amorphous Ice and its First-Order Transition to the Low-Density Form, J. Phys. Chem. B, 2011, 115, 14141–14148 CrossRef CAS PubMed.
- K. Winkel, Study of Amorphous-Amorphous Transitions in Water, Verlag Dr. Hut, Munich, 2011 Search PubMed.
- J. Stern and T. Loerting, Crystallisation of the amorphous ices in the intermediate pressure regime, Sci. Rep., 2017, 7(1), 3995 CrossRef CAS PubMed.
- C. G. Salzmann, E. Mayer and A. Hallbrucker, Effect of heating rate and pressure on the crystallization kinetics of high-density amorphous ice on isobaric heating between 0.2 and 1.9 GPa, Phys. Chem. Chem. Phys., 2004, 6(22), 5156–5165 RSC.
- J. A. Sellberg, C. Huang, T. A. McQueen, N. D. Loh, H. Laksmono and D. Schlesinger, et al., Ultrafast X-ray probing of water structure below the homogeneous ice nucleation temperature, Nature, 2014, 510(7505), 381–384 CrossRef CAS PubMed.
- H. Laksmono, T. A. McQueen, J. A. Sellberg, N. D. Loh, C. Huang and D. Schlesinger, et al., Anomalous Behavior of the Homogeneous Ice Nucleation Rate in “No-Man's Land”, J. Phys. Chem. Lett., 2015, 6(14), 2826–2832 CrossRef CAS PubMed.
- E. Mayer and P. Brueggeller, Vitrification of pure liquid water by high pressure jet freezing, Nature, 1982, 298(5876), 715–718 CrossRef CAS.
- E. Mayer, New method for vitrifying water and other liquids by rapid cooling of their aerosols, J. Appl. Phys., 1985, 58(2), 663–667 CrossRef CAS.
- C. G. Salzmann, T. Loerting, S. Klotz, P. W. Mirwald, A. Hallbrucker and E. Mayer, Isobaric annealing of high-density amorphous ice between 0.3 and 1.9 GPa: in situ density values and structural changes, Phys. Chem. Chem. Phys., 2006, 8, 386–397 RSC.
- O. Andersson, Relaxation Time of Water's High Density Amorphous Ice Phase, Phys. Rev. Lett., 2005, 95, 205503 CrossRef PubMed.
- M. Seidl, M. S. Elsaesser, K. Winkel, G. Zifferer, E. Mayer and T. Loerting, Volumetric study consistent with a glass-to-liquid transition in amorphous ices under pressure, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(10), 100201 CrossRef.
- S. R. Elliott, Physics of amorphous materials, Longman Group Limited, New York, USA, 1983 Search PubMed.
- H. Engelhardt and B. Kamb, Structure of ice IV, a metastable high-pressure phase, J. Chem. Phys., 1981, 75, 5887–5899 CrossRef CAS.
- B. Kamb, A. Prakash and C. Knobler, Structure of ice V, Acta Crystallogr., 1967, 22, 706–715 CrossRef CAS.
- B. Kamb, Structure of ice VI, Science, 1965, 150, 205–209 CAS.
- C. G. Salzmann, I. Kohl, T. Loerting, E. Mayer and A. Hallbrucker, Pure ices IV and XII from high-density amorphous ice, Can. J. Phys., 2003, 81, 25–32 CrossRef CAS.
- C. G. Salzmann, T. Loerting, I. Kohl, E. Mayer and A. Hallbrucker, Pure Ice IV from High-Density Amorphous Ice, J. Phys. Chem. B, 2002, 106, 5587–5590 CrossRef CAS.
- P. H. Handle, M. Seidl and T. Loerting, Relaxation Time of High-Density Amorphous Ice, Phys. Rev. Lett., 2012, 108(22), 225901 CrossRef PubMed.
- T. Loerting, K. Winkel, M. Seidl, M. Bauer, C. Mitterdorfer and P. H. Handle, et al., How many amorphous ices are there?, Phys. Chem. Chem. Phys., 2011, 13(19), 8783–8794 RSC.
| This journal is © the Owner Societies 2018 |