
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The mechanism of catalase loading into porous vaterite CaCO3 crystals by co-synthesis†
A. S.
Vikulina
 *a,
N. A.
Feoktistova
bc,
N. G.
Balabushevich
b,
A. G.
Skirtach
d and
D.
Volodkin
ab
*a,
N. A.
Feoktistova
bc,
N. G.
Balabushevich
b,
A. G.
Skirtach
d and
D.
Volodkin
ab
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, NG11 8NS Nottingham, UK. E-mail: anna.vikulina@ntu.ac.uk; Tel: +44-115-848-8062
bDepartment of Chemistry, Lomonosov Moscow State University, Leninskiye Gory 1-3, 119991 Moscow, Russia
cFraunhofer Institute for Cell Therapy and Immunology, Branch Bioanalytics and Bioprocesses (Fraunhofer IZI-BB), Am Muehlenberg 13, 14476 Potsdam-Golm, Germany
dDepartment of Molecular Biotechnology, University of Ghent, Coupure Links 653, 9000 Gent, Belgium
First published on 15th March 2018
Abstract
Porous vaterite CaCO3 crystals are nowadays extensively used as high-capacity bio-friendly sacrificial templates for the fabrication of such protein-containing nano- and micro-particles as capsules and beads. The first step in the protein encapsulation is performed through loading of the protein molecules into the crystals. Co-synthesis is one of the most useful and simple methods proven to effectively load crystals with proteins; however, the loading mechanism is still unknown. To understand the mechanism, in this study, we focus on the loading of a model protein catalase into the crystals by means of adsorption into pre-formed crystals (ADS) and co-synthesis (COS). Analysis of the physico-chemical characteristics of the protein in solution and during the loading and simulation of the protein packing into the crystals are performed. COS provides more effective loading than ADS giving protein contents in the crystals of 20.3 and 3.5 w/w%, respectively. Extremely high loading for COS providing a local protein concentration of about 550 mg mL−1 is explained by intermolecular protein interactions, i.e. formation of protein aggregates induced by CaCl2 during the co-synthesis. This is supported by a lower equilibrium constant obtained for COS (5 × 105 M−1) than for ADS (23 × 105 M−1), indicating a higher affinity of single protein molecules rather than aggregates to the crystal surface. Fitting the adsorption isotherms by classical adsorption models has shown that the Langmuir and BET models describe the adsorption phenomenon better than the Freundlich model, proving the aggregation in solution followed by adsorption of the aggregates into the crystals. We believe that this study will be useful for protein encapsulation through CaCO3 crystals using the COS method.
Introduction
Novel approaches for protein encapsulation are nowadays of crucial importance because of an increased number of effective therapeutic proteins available on the market.1 Protein encapsulation into micro- and nano-particles gives an option to protect protein molecules against enzymatic cleavage (biodegradation) and other external undesirable factors such as complexation with bioactive compounds blocking the protein activity or local pH changes. At the same time, the well-defined size and shape of the protein particles allows the choice of a proper delivery route, effectively delivering the protein molecules to a target, and releasing the protein in a controlled fashion.2,3 Scientific attention has focused on the development of novel approaches to encapsulate proteins under mild conditions in order to prevent any reduction of the protein bioactivity during the encapsulation process, which is often hard to avoid using traditional encapsulation technologies that employ mechanical stress, high or low temperature, organic solvents, and surfactants.One of the most effective modern approaches is to encapsulate protein molecules into polymer-based microcapsules by means of layer-by-layer polymer adsorption onto decomposable cores followed by the core removal and formation of a multilayer capsule.4 The permeability of the multilayer capsule shell, defined by the number of layers and the nature of the polymers used, can be adjusted to provide a desired release rate of the encapsulated protein.5–12 Studies of the multilayer structure and the physico-chemical properties including permeability may help to tune the release rate.13–15 Such multilayer capsules have found a number of attractive biological applications in drug delivery, tissue engineering, etc.8,10,11,16–19 Many kinds of sacrificial cores have been used since then including melamine formaldehyde and polystyrene latex particles, silica particles, poly(lactide-co-glycolide) particles, inorganic crystals such as CaCO3, MnCO3, and CdCO3 and even erythrocytes.20,21 Nowadays, vaterite CaCO3 crystals22–24 (vaterite is a crystalline polymorph form of CaCO3) are most probably the most useful sacrificial template because of a number of advantageous characteristics over other decomposable cores that include: (i) highly developed surface areas due to porosity, (ii) bio-friendly decomposition conditions (slightly acidic pH, EDTA, or citric acid), and (iii) easy to prepare in a lab from non-expensive precursor salts such as Na2CO3 and CaCl2. It is of note that the mesoporous structure of the crystals is very attractive for loading of bio-macromolecules such as proteins that have sizes in the range of a few of nm, thus giving an option to effectively fill the internal volume of the CaCO3 crystal with pore sizes in the range of tens of nm.
The crystals are made of nanocrystallites aggregated to each other forming the crystal secondary structure. Control over the crystal growth provides one with a simple way to adjust the crystal porosity even without any additives.25,26 The surface of the nanocrystallites can be coated with different materials, for example, lipids, for protection.27 Some recent efforts have been made in the size reduction of calcium carbonate crystals that open new perspectives to employ the crystals for a wide range of administration routes in modern drug delivery.27–33
The utilization of vaterite CaCO3 crystals for the formulation of multilayer capsules has stimulated the employment of the crystals to make new delivery vehicles for the encapsulation/release of drugs and proteins. The pure crystals can be utilized themselves or they can be used as templates to assemble functional structures with encapsulated proteins34–39 as well as various hydrophilic and hydrophobic drugs.17,33,40–44 Biologically relevant particles assembled using the crystals can be made of pure proteins45–48 or biologically relevant molecules such as polyethylene glycol49,50 or temperature-sensitive polymers such as poly(N-isopropylacrylamide).51 A number of bio-applications have been reported utilizing the crystals as carriers for photodynamic therapy,52–56 transcutaneous delivery,57 delivery into the brain bypassing the blood–brain barrier,58 delivery of insulin59,60 and vaccines,61,62 and as carriers in alternative fields such as food industry.26 Crystal modification with nanoparticles allows external manipulation of the crystals63,64 that can then be utilized for targeted delivery. Besides this, the high loading capacity of CaCO3 crystals provides an opportunity for slow long-term controlled release, which is of high interest for therapeutic applications.65,66 A high content of the loaded protein would give an option to reduce the number of carriers for protein delivery (the crystals or other carriers assembled using the crystal, e.g. microcapsules). This issue is especially important for minimizing potential side effects, which may occur due to the presence of the carriers. A high loading of proteins into the crystals will also ensure a high loading capacity for the carriers assembled using the crystal. We note that other applications of the crystals including sensors67,68 or self-assembled tailor-made polymer macrostructures such as scaffolds69–71 have been reported.
Protein can effectively be loaded into the crystals using the co-synthesis approach developed a decade ago.22,72,73 It is based on trapping the protein molecules during the crystal growth simply by adding the protein to the precursor salts used to synthesize the crystals. This gives a very high efficiency of the loading, high protein content, and uniform distribution of the loaded protein molecules.74,75 A number of various proteins such as aprotinin and insulin have been successfully loaded into crystals by ADS and COS.74,75 The morphology of the crystals may affect the protein loading,76 which is of high importance for pharmacological applications.77
Although the potential for the utilization of crystals as carriers for protein loading is obvious, the loading mechanism through co-synthesis is poorly understood. Our recent works demonstrated some new findings on the interaction between protein molecules and crystals; however, the loading mechanism is still unknown. To our knowledge, there is no explanation for how the crystals can host such extremely large amounts of protein of about 10% by mass or more giving an estimated protein concentration in the crystals so high that it may not be possible to reach in solution at all. Nevertheless, recent reports indicate that the loading mechanism is tightly related to the ionic interactions between proteins and particles making the encapsulation of proteins highly dependent on the pH of reaction solutions and the isoelectric point of the protein.78,79
In this study, we focus on understanding the mechanism of protein loading into the CaCO3 crystals by co-synthesis. For this purpose, we analysed the loading into pre-formed crystals (adsorption, ADS) and the loading by co-synthesis (COS) using catalase as a model protein and employing classical adsorption theories. Analysis of the crystal internal structure and physico-chemical properties of the protein in solution and simulation of the protein loading are performed.
Experimental
Materials
Calcium chloride dihydrate CaCl2·2H2O (Sigma-Aldrich, Japan); sodium carbonate Na2CO3 anhydrous (Sigma-Aldrich, Germany); catalase from bovine liver – 3809 units per mg solid (Sigma, Germany (C-1345)); 0.05 M glycine buffer (pH 9.0), 0.05 M phosphate buffer (pH 7.0), ethylenediamine-tetraacetic acid (EDTA) and hydrogen peroxide were purchased from Sigma, USA; water used in all experiments was prepared via a Millipore Milli-Q purification system and had a resistivity higher than 18.2 MΩ cm.Preparation of CaCO3 crystals
Porous crystals were prepared by rapid mixing of equal volumes of CaCl2 and Na2CO3 aqueous solutions at 22 °C. Briefly, 3 mL of 1 M CaCl2 was added to 9 mL of H2O at constant stirring, then 3 mL of 1 M Na2CO3 was rapidly added to the solution at 22 °C. After vigorous agitation with a magnetic stirrer for 40 s and incubation for 15 min, the suspension was thoroughly washed with pure water and dried at 80 °C producing crystals with the size of 4–5 μm.Protein loading into CaCO3 crystals by ADS
1.5 mL of catalase solution (0.1–2 mg mL−1) in 0.05 M glycine buffer, pH 9.0, was added to 60 mg of the dried CaCO3 crystals and incubated for 30 min followed by centrifugation for 5 min at 1000g. Then, the solution was rinsed twice and all the supernatants were collected and examined to determine the protein concentration by using UV spectroscopy at 280 nm (Lambda 35, “Perkin-Elmer”, UK) (Fig. S1, ESI†).Protein loading into CaCO3 crystals by COS
0.2 mL of 1 M CaCl2 was added to 0.6 mL of protein solution at a constant stirring rate, then 0.2 mL of 1 M Na2CO3 was rapidly added to the solution at 22 °C. After vigorous agitation with a magnetic stirrer for 40 s and incubation for 15 min, the suspension was thoroughly washed two or three times with 0.05 M glycine buffer, pH 9.0, and all the supernatants were collected and examined to determine the protein concentration using UV spectroscopy at 280 nm.Field emission scanning electron microscopy (SEM)
SEM images were recorded using a Gemini LEO 1550 electron microscope at an accelerating voltage of 3 kV. The samples were prepared by dropping the crystal suspension on a glass slide, which was dried for 1 h at 90 °C followed by conductive coating with gold palladium (5 nm).Brunauer, Emmett, and Teller (BET) analysis
To measure the surface area of the prepared crystal, N2 adsorption–desorption analysis was performed using a QUADRASORB SI-MP (Quantachrome Instruments, USA) at 77.3 K. Prior to measurement, the samples were degassed at 150 °C for 20 h. BET theory was used for the surface area analysis.Dynamic light scattering (DLS)
DLS measurements of the hydrodynamic diameter of the catalase were performed with a Zetasizer Nano ZS (Malvern Instruments Limited, Worcestershire, UK). For DLS measurements, protein solution was freshly prepared in ultrapure water or a corresponding solution. The concentration of protein in solution was 0.2 mg mL−1 and the protein was incubated in the corresponding solutions for 1–2 h followed by filtration with a 0.22 μm filter prior to measurement.Catalase specific activity
The specific activity of catalase was measured by monitoring the rate of hydrogen peroxide decomposition. A mixture of 0.040–0.100 mL of the enzyme solution containing 0.005–0.010 mg mL−1 catalase, 0.800–0.860 mL of 0.1 M phosphate buffer (pH 7.0) and 0.100 mL of 0.196 M hydrogen peroxide in water was prepared. A decrease in absorbance was registered at 240 nm. Retention of the catalase activity was calculated as the ratio between the activity of the enzyme recovered after crystal dissolution in 0.2 M EDTA and the activity of the initial catalase in phosphate buffer, pH 7.0.Results and discussion
Thermodynamics of catalase loading into the crystals
The model protein catalase was loaded into the CaCO3 crystals either by ADS75 or COS. For the ADS method, the prepared crystals were incubated in the catalase solution. The COS method is based on the addition of catalase to the precursor salts used for the crystal synthesis (sometimes called co-precipitation72). Fig. 1a shows the content of catalase in the crystals as a function of the initial catalase concentration used for the loading (c0) for the two loading methods.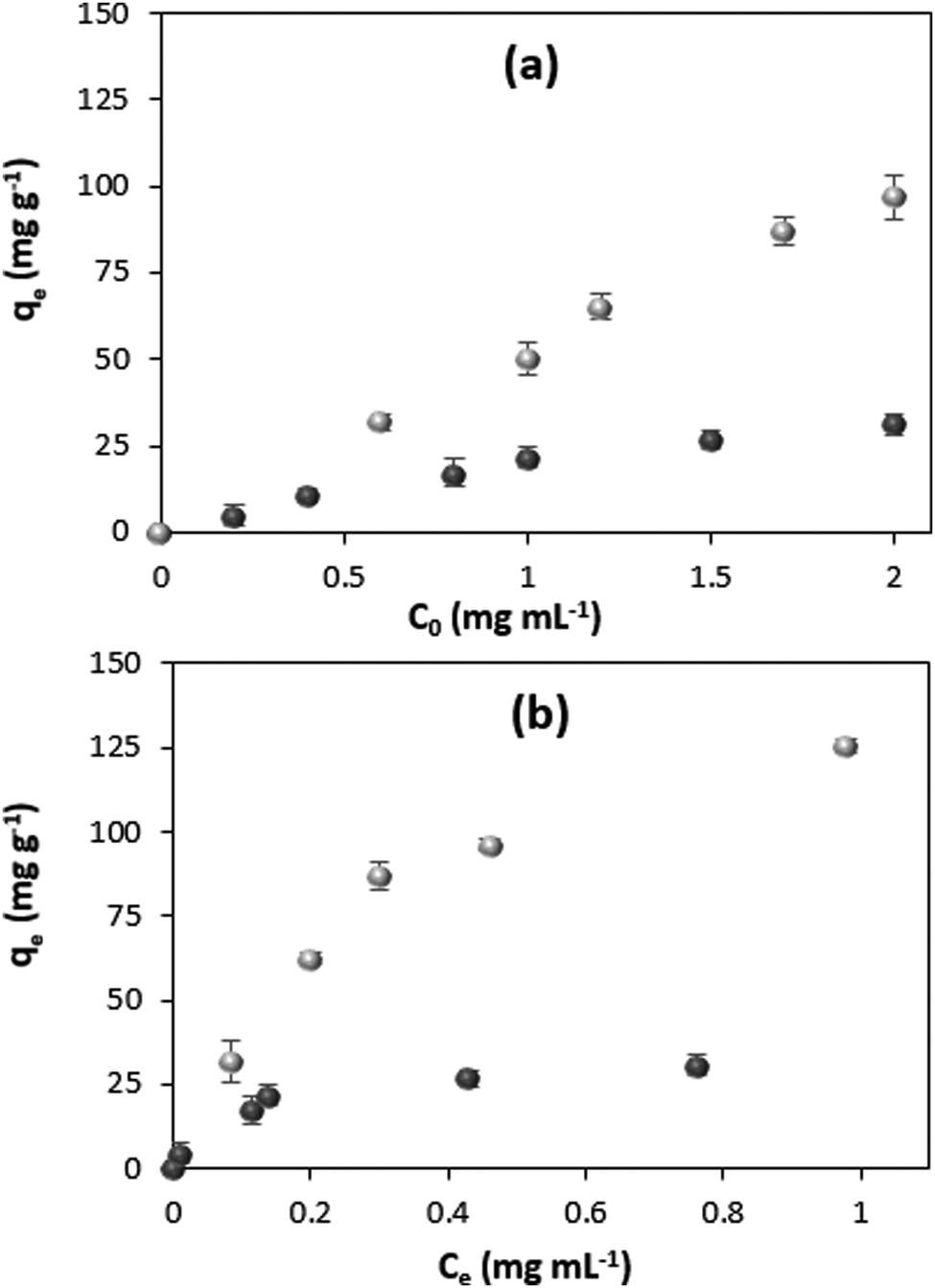 | ||
| Fig. 1 (a) Concentration dependence of adsorption capacity and (b) adsorption isotherms for protein loading by ADS (black circles) and COS (gray circles). Standard deviations (SD) are given for n = 3. Experiments are done in 0.05 M glycine buffer, pH 9.0. | ||
The amount of loaded protein under saturation conditions (equilibrium) was calculated using the following equation:
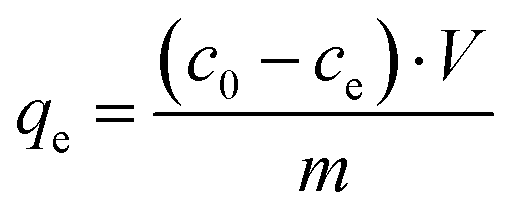 | (1) |
Both the ADS and COS methods are manifested by a high loading efficiency, which is close to 100% uptake from the catalase solutions of low concentrations and linearly decreases with the increase in the initial concentration of catalase due to the saturation of the crystals (Fig. S2, ESI†).
Typically, the reversible interaction of molecules of interest with a solid substrate is quantified using adsorption isotherms by plotting the content of the adsorbed molecules in the substrate against the equilibrium concentration of the molecules (the concentration typically measured in the supernatant). The isotherm reflects the affinity of the molecules to the substrate.
The thermodynamic parameters of the adsorption process can be obtained from the isotherm. Fig. 1b shows the adsorption isotherms for catalase loading by means of ADS and COS. The ADS method results in saturation of the crystals with catalase at the protein equilibrium concentration of more than ∼0.4 mg mL−1. At the same time, the crystals loaded by COS are not saturated with the protein, even at a higher equilibrium concentration of 1 mg mL−1, much above that needed for saturation in the ADS method. The adsorption isotherms for both protein loading methods are displayed in Fig. 2 showing the clear trend towards saturation at the equilibrium concentration of about 1 and 7 mg mL−1 for ADS and COS, respectively.
 | ||
| Fig. 2 Adsorption isotherm for COS (a) and ADS (b) methods in a wide range of protein concentrations and the fitting of the isotherms with Langmuir, Freundlich and BET equations. For the experimental isotherms, the error bars are SD for n = 3. | ||
In order to quantify the affinity of catalase to the crystals, three mathematical models generally accepted for the adsorption of proteins from a liquid phase to a solid phase have been employed, namely the Langmuir, Freundlich, and Brunauer–Emmett–Teller (BET) adsorption models.80,81Eqn (2)–(4) are given as follows for these models, respectively.
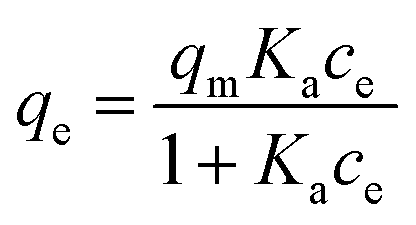 | (2) |
| qe = KF·cne | (3) |
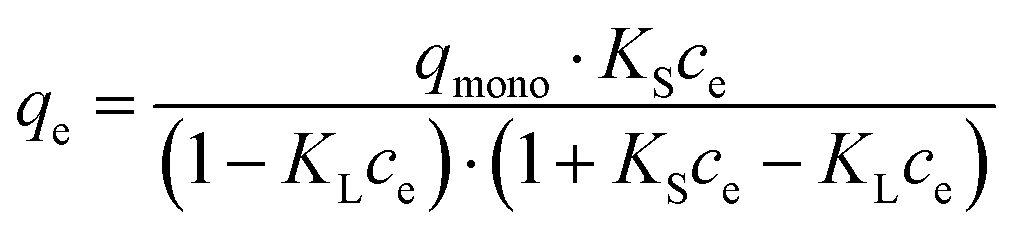 | (4) |
It is of note that the BET equation presented above is derived from the common BET equation that was originally developed for gas adsorption and was adopted for the adsorption of liquids.82,83 This equation is based on the assumption that the adsorbate molecule (protein) can be adsorbed either to the surface of an adsorbent (first layer) or to another adsorbate molecule (which is already anchored to the surface, the second layer); adsorption to the first layer is thermodynamically more favorable compared to adsorption to the second layer (KS > KL). In our case, the adsorption constant KS characterizes the binding of catalase to the CaCO3 surface and KL is devoted to catalase binding to another catalase molecule that is already adsorbed. If not specified, hereinafter, the term surface will be used to describe the overall surface of the porous vaterite CaCO3 crystals, i.e. both the outer and inner surfaces.
The three models considered in this study describe different scenarios for the adsorption of catalase. The Langmuir model can be employed for monolayer adsorption describing the formation of only one layer of the protein on the CaCO3 surface. The interactions between protein molecules are negligible, meaning that the Langmuir model does not predict any inter-protein interactions such as protein aggregation. It is also of high importance that this model is based on statistical thermodynamics and allows calculation of the adsorption equilibrium constant. In contrast, the Freundlich model of monolayer adsorption has been developed empirically. The model is typically employed for the first part of the adsorption isotherm and describes the adsorption driven by attraction (n > 1) or accompanied by repulsion (n < 1) of protein molecules. Finally, the BET model is based on an extension of the Langmuir model and it describes the polyadsorption phenomenon.
The parameters extracted by the mathematical treatment of the COS and ADS isotherms are summarized in Table 1. Obviously, the Freundlich model does not provide a good fit (R2 < 0.95) for the experimental adsorption isotherm. This indicates independent adsorption of the catalase molecules, which is not influenced by possible aggregation of catalase. On the other hand, both the Langmuir and BET models are characterized by high and close values of the coefficient of determination (R2 > 0.95). Notably, the value of the weak adsorption state equilibrium constant (KL) calculated for the BET model can be neglected compared to the high value of the adsorption constant for proteins in the strong adsorption state (KS ≫ KL). This reduces eqn (4) to eqn (2) with KS = Ka and qmono = qm and, as a result, allows the loading of catalase into the CaCO3 crystals to be described by the Langmuir model.
| Model | Parameters | Coefficient of determination (R2) | ||
|---|---|---|---|---|
| ADS | COS | ADS | COS | |
| Langmuir | q m = 35 ± 2 mg g−1 | q m = 203 ± 5 mg g−1 | 0.987 | 0.969 |
| K a = (23 ± 1) × 105 M−1 | K a = (5 ± 1) × 105 M−1 | |||
| Freundlich | K F = 46 ± 1 | K F = 117 ± 5 | 0.932 | 0.947 |
| n = 0.5 ± 0.1 | n = 0.3 ± 0.1 | |||
| BET | q mono = 30 ± 3 mg g−1 | q mono = 206 ± 4 mg g−1 | 0.996 | 0.986 |
| K S = (29 ± 2) × 105 M−1 | K S = (4.7 ± 0.2) × 105 M−1 | |||
| K L = (36 ± 5) × 103 M−1 | K L = 405 ± 5 M−1 | |||
This finding supports the idea that the protein loading by both COS and ADS is driven by the high affinity of the protein to the CaCO3 crystals. The maximum adsorption capacity qm reaches much higher values for COS than for ADS (qm of 203 ± 5 and 35 ± 2 mg g−1, respectively). Thus, if one assumes that the pure protein solution has a density of about 1 g cm−3, the maximal protein content in the crystals is 20.3 and 3.5 w/w% for COS and ADS, respectively. Taking into account that the density of CaCO3 is much higher than for water (2.7 g cm−3),24 the estimated concentration of the catalase inside the crystals reaches values of about 550 and 95 mg mL−1 for the COS and ADS methods, respectively. The result, showing a high difference in adsorption capacity for ADS and COS, corroborates well with literature findings on the inclusion of other proteins (e.g. lysozyme, chymotrypsin) into the crystals by these methods.72 Despite the fact that the COS method can provide much higher protein loading than ADS, the affinity of catalase to the crystal surface is higher for the ADS method (Ka is 23 ± 1 × 105 M−1 for ADS against 5 ± 1 × 105 M−1 for COS). The values of the Gibbs free energy (ΔG) can be calculated from Ka giving the values of −36 ± 2 and −32 ± 7 kJ mol−1 for ADS and COS, respectively. These values indicate that the affinity of the protein to the crystals is higher for ADS compared to COS.
Modelling of catalase loading into the crystals
In order to further understand the mechanism of the protein loading into the CaCO3 crystals, it is important to estimate the theoretical adsorption capacity of the crystals and compare it with the experimentally found values. Obviously, the surface area of the crystals is the determinative factor influencing the adsorption capacity. In this study, surface area has been measured by nitrogen adsorption–desorption using the BET approach.It is known that the BET surface area and pore distribution strongly depend on the amount of loaded protein.84 Because of this reason, BET analysis was performed using the CaCO3 crystals synthesized in the absence of catalase. The very high content of the protein in the crystals prepared by COS (qm of 203 ± 5 mg g−1) will not allow for an appropriate analysis of the surface area and the porosity of the protein-loaded crystals. In addition, it has recently been shown that glycine molecules entrapped inside CaCO3 crystals during the synthesis85 may also interfere with the results of BET analysis. At the same time, we compared the SEM images of the CaCO3 crystals grown in water and in 0.05 M glycine buffer pH 9.0 and verified that their structure was identical (Fig. S3a, b and d, ESI†). That is why the BET analysis was performed for the crystals prepared in water. A summary of the BET analysis is given in Table S1 (ESI†). The crystals have a surface area of 10.4 m2 g−1 and a pore diameter in the range of 5 to 30 nm with a maximum of about 10 nm (Fig. 3). The specific surface is similar to that reported for crystals prepared under similar conditions.24
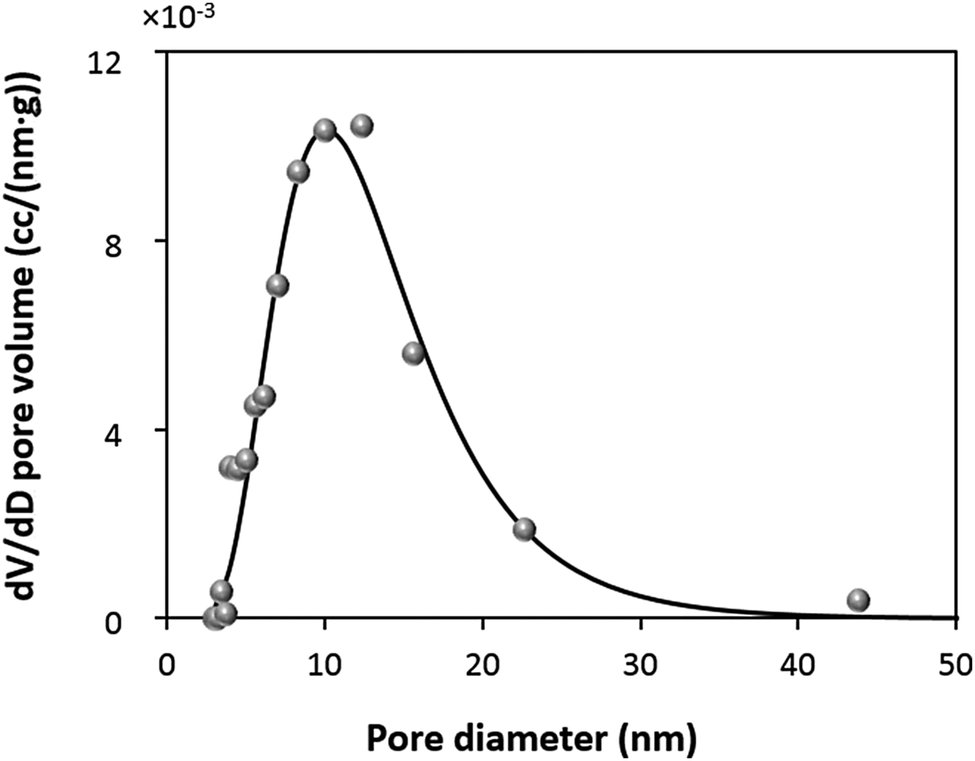 | ||
| Fig. 3 Differential pore volume distributions for 5 μm sized CaCO3 crystals determined by BET analysis using the Barret–Joyner–Halenda (BJH) model (grey circles). The fitting of the experimental data with a lognormal function (black curve) is given. Pure crystals are synthesized in water at 22 °C. | ||
The protein loading into the crystals was evaluated using three models for protein packing into the crystal pores (Fig. 4a–c). A detailed description of the models is given in Fig. S4 (ESI†). The hydrodynamic diameter and molecular weight of a catalase molecule were taken as 10.5 nm and 4.15 × 10−19 g, respectively. First, we assumed that the surface area of the CaCO3 crystals can be packed with protein molecules in a 2D plane view (model A). This assumption would mean that all the surfaces of the crystals can be occupied by a monolayer of the protein molecules (no steric limitations to diffuse into the pores, this model has been used in our previous work75).
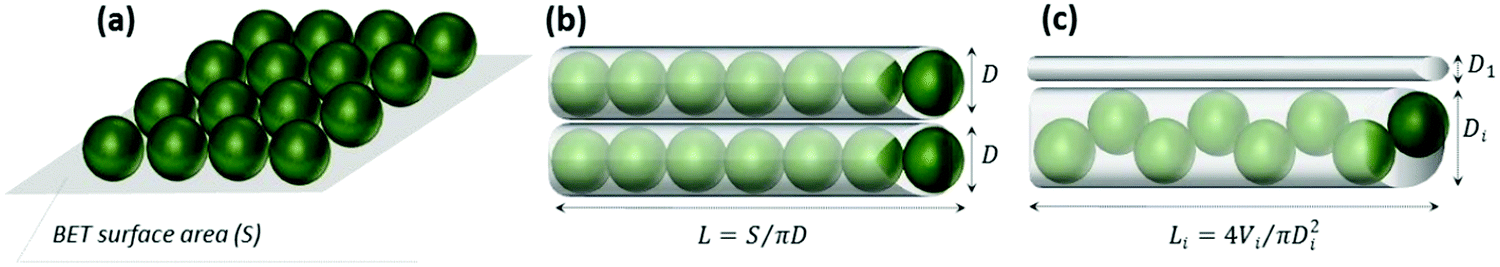 | ||
| Fig. 4 Schematic illustration of the three models (a–c) employed to estimate the theoretical maximum adsorption capacity of the CaCO3 crystals for catalase. L, D and V are the length, diameter and volume of a cylindrical pore, respectively. | ||
This results in a maximum adsorption capacity calculated as 40 mg of catalase per 1 g of the crystals. Model B assumes that the pores of the crystals have a cylindrical shape with a diameter of 10.5 nm that corresponds to the size of the protein. This allows compact protein packing in the crystal pores. Model B predicts a maximum adsorption capacity of 13 mg of catalase per 1 g of the crystals. Finally, the third model C assumes that the pores have a certain size distribution. The experimental pore distribution for this model was constructed based on the BET analysis (Fig. 3). It was fitted with a lognormal function (R2 0.955), as is shown in Fig. 3. The pore diameter of 10.5 nm was set up as a threshold for the catalase loading, meaning that the catalase molecules cannot penetrate into the pores of diameter < 10.5 nm (D1 in Fig. 4c). The pores of diameter 10.5–21.0 nm are closely packed with the protein molecules (Di in Fig. 4c). The volume fraction of pores with diameter > 21 nm was found to be less than 5% and was neglected. According to model C, up to 2 mg of catalase per 1 g of the crystals can be loaded.
The first two models (A and B) provide presumably overestimated values for the protein adsorption (40 and 13 mg of catalase per 1 g of the CaCO3 crystals) because compact packing of protein molecules for full coverage of the crystal area being planar or ideally cylindrical is hardly possible due to steric limitations. Molecules of nitrogen used for the determination of the surface area by the BET analysis are much smaller than those of catalase and steric limitations are much less pronounced for nitrogen. At the same time, model C takes into account steric limitations and this model assumes that only pores larger than the size of the protein molecule will be accessible for the protein. Thus, the maximum capacity calculated based on model C seems to be more realistic. At the same time, the experimentally found maximum adsorption capacities for both the ADS and COS methods exceed the theoretical values obtained for model C by one to two orders of magnitude (Table 1). This may be explained by the deviation from the cylindrical shape of the pores and the formation of vessel-like enlargements in the pore structure. These “vessels” inside the crystals may host much higher amounts of protein molecules. The non-uniform shape of the pore due to the “vessels” would allow many more protein molecules to be hosted in the larger pores because the pore volume scales with the pore size by three orders of magnitude. This would increase the theoretically expected loading values and explain the very high loading capacities of the crystals. For COS, the maximum loading capacity is approximately six times higher than that for ADS (Table 1).
A previous report clearly demonstrated that catalase is distributed within the entire volume of CaCO3 crystals as well as on the external surface of the crystals.75 This indicates that such a high difference between adsorption capacities found for ADS and COS loading is most likely not associated with the impregnation of the external surface of the CaCO3 crystals. In order to explain the high protein content in the crystals prepared by COS, one can assume that two possible scenarios may take place. One explanation might be related to a higher porosity of the crystals formed by COS that will make the total surface area of the crystals much larger. Another explanation can be related to the formation of protein aggregates in the pores. These two assumptions will be considered in the following sections below.
Internal structure of CaCO3 crystals
CaCO3 crystals synthesized in water at room temperature using a standard procedure24 yield rather monodispersed mesoporous crystals. The SEM images (Fig. 5a and b) show the typical morphology of the crystals and the highly developed internal structure of a broken crystal. The crystal is formed due to the secondary aggregation of spherical nanocrystallites with sizes of a few tens of nm. The pores on the outer surface of the crystals represent the space between the aggregated spherical nanocrystallites, however, this does not provide information about the internal structure of the crystal. If the crystal is broken (Fig. 5b), the internal structure can be identified, showing a channel-like structure. This is most probably due to the growth mechanism of the crystals based on the stacking of the nanocrystallites on top of each other. It is thus logical to assume that the pores of the crystals have a channel-like structure and may be described as cylinders, rather than spheres. | ||
| Fig. 5 SEM images of CaCO3 crystals. Scale bar is 2 μm for (a) and 1 μm for (b). The inset shows the internal structure of the broken crystal (enlarged 2 times). | ||
Analysis of the crystal internal structure revealed that the crystal porosity for empty (not loaded with protein) crystals is similar to that for the crystals obtained by COS (Fig. S3, ESI†).
It is more likely that catalase loading by COS is associated with protein aggregation, however, additional evidence is required. In the sections below, we further examine the protein loading processes in order to further assess our assumption.
Catalase colloidal stability during ADS and COS
The obvious difference between the loading methods is that during the COS loading, catalase is exposed to the two precursor salts (CaCl2 and Na2CO3) used for the synthesis of the crystals. For the ADS method, catalase is in contact with the already synthetized crystals. Thus, we first tested if the presence of the salts can affect the colloidal stability of the protein in solution.Fig. 6 shows the DLS volume-weighted size distribution for catalase in water and in the presence of the supernatant of the crystals, as well as 0.2 M CaCl2 and 0.2 M Na2CO3 water solutions.
 | ||
| Fig. 6 Volume weighted DLS size distribution of catalase in water solution (a), in the presence of the supernatant of the CaCo3 crystals (b), and in 0.2 M water solutions of Na2Co3 (c) and CaCl2 (d). | ||
The supernatant of the crystals does not affect the colloidal stability of the protein and only a small number of protein aggregates was detected (Fig. 6b). The presence of 0.2 M Na2CO3 does not significantly affect the colloidal stability either and only a small peak of aggregates of about 40–50 nm can be detected (Fig. 6c). Meanwhile, the presence of 0.2 M CaCl2 significantly reduces the colloidal stability and one cannot see the peak corresponding to single catalase molecules (about 10 nm); only large aggregates with average sizes of about 50 and 200 nm can be found (Fig. 6d). One should note that the concentration of the tested salts in the catalase solution was chosen to be the same as during the protein loading by COS.
The DLS results clearly demonstrate that CaCl2 induces catalase aggregation and this is most probably the reason that the protein aggregates are trapped in the crystals during the COS process, resulting in a very high loading of the protein into the crystals by COS. This means that the crystals formed by COS may have larger pores than the bare crystals in order to host protein aggregates of sizes 50 and 200 nm that exceed the average pore size of the bare crystals (about 10 nm). The number of such large pores may be, however, much smaller than those of a regular size because the capacity of the larger pores to host protein molecules will significantly exceed that of the smaller ones. This is true if the pores are completely filled by protein, as expected in the case of trapping of the protein aggregates into the pores during the COS. At the same time, the aggregation is supposed to be a reversible process with an equilibrium in formation of aggregates of various sizes. Therefore, the aggregates of smaller sizes (below 50 nm) may be enriched during the COS and trapped into the pores of regular sizes.
We suppose that the aggregation of protein molecules might play a crucial role not only for catalase loading but also for a number of other proteins for which intra- and intermolecular interactions favour protein stability. These include, for instance, insulin,86 immunoglobulin antibodies87 and other therapeutic proteins.88 Besides this, the isoelectric point of the used protein and the in-process and final pH of the formulation may have a significant effect on the protein–protein interaction. We are considering these issues in our current research and hope that our further studies will help to shed light on these questions. In this study, the isoelectric point of catalase (5.4) is below the pH value of the used buffer (pH 9.0) or pH in water solution of the crystals (pH 10.2), which ensures the negative charge of the protein.
The mechanism of catalase loading by the ADS and COS methods
Fig. 7 shows the proposed mechanism of catalase loading based on the findings described above. Evidently, Ca2+-induced aggregation of catalase plays a crucial role in the protein loading into the crystals by COS, which is consistent with the influence of gelatin.33 The rather high concentration of Ca2+ (on the order of 10−1 M) needed for the COS procedure results in the formation of catalase aggregates. The loading by ADS seems also to include a small number of aggregates formed in the presence of a rather low concentration of Ca2+ (about 10−5 M) in the supernatant of the crystals due to the dissociation of CaCO3 (Fig. 6b). However, the fact that adsorption isotherms constructed for both COS and ADS methods obey the Langmuir equation means that the aggregation has no effect on the isotherm shape. This suggests that the aggregates are predominantly formed in the solution and then adsorbed onto the surface of the CaCO3 crystals. This is supported by the fact that for the COS method, the protein is first dissolved in the solution of CaCl2 followed by the addition of Na2CO3. Formation of the protein aggregates on the surface of the CaCO3 crystals by consequent growth of the aggregates on the surface is not likely. It is also important to note that since Ka for COS (5 × 105 M−1) is lower than that for ADS (23 × 105 M−1), the affinity of the protein aggregates to the CaCO3 crystals is assumed to be lower than that for single protein molecules. This is logical because the aggregate is large and may not be ideally spherical, resulting in a less defined contact area for adsorption to the crystal surface.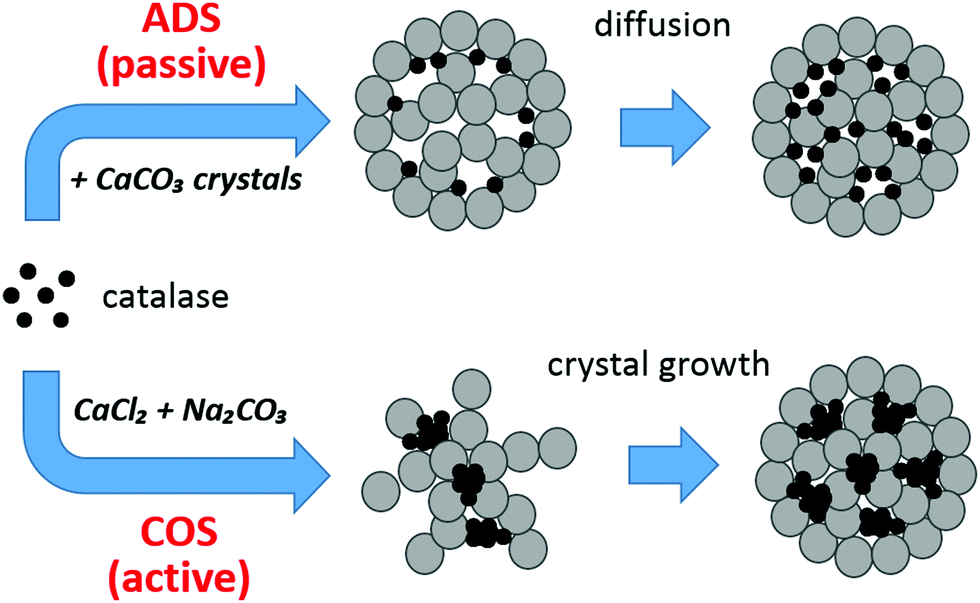 | ||
| Fig. 7 Mechanism of catalase loading into the CaCO3 crystals by ADS (passive loading) and COS (active loading). The crystals are sketched as grey porous spheres made of spherical interconnected nanocrystallites;25 catalase molecules are black circles. | ||
The aggregated state of the protein loaded by COS may dramatically affect the structure and the properties of the protein. It is known that catalase retains around 79% of its specific activity after loading into CaCO3 crystals by ADS.74 Here, we have tested the retention of catalase activity after loading into CaCO3 crystals by COS using a similar approach. Taking into account the maximum adsorption capacities found in this study, it was found that the maximum amount of active protein (after dissolution of the core with EDTA) for COS is 60.9 mg per 1 g of CaCO3 crystals while encapsulation by ADS from a solution of the same protein concentration provides 27.7 mg of active catalase per 1 g of CaCO3 crystals. These preliminary data indicate the high potential of the COS approach for loading large amounts of active protein. A more detailed study on the retention of catalase enzymatic activity is a focus for our upcoming study.
The stability of the aggregates entrapped in the CaCO3 crystals was tested by intensive washing of the crystals with 0.05 M glycine buffer pH 9.0 (Fig. S5, ESI†). It was revealed that catalase loaded by COS cannot be effectively washed out by a single washing step while catalase loaded by the ADS method is not retained in the crystals and even a single washing leads to the release of the catalase. These data support the proposed mechanism of catalase inclusion into the CaCO3 crystals by passive and active loading.
Besides this, the effectiveness of protein release as well as the influence of crystal preparation conditions (salt concentration, preparation temperature, crystallization time, etc.) on both loading and release efficiency should be tested for a better understanding of the mechanism of catalase loading. These issues will be further investigated in upcoming studies.
Conclusions
Catalase can be effectively loaded into vaterite CaCO3 crystals through ADS or COS methods. The loading capacity is very high for both methods (35 ± 2 and 203 ± 5 mg of the protein per 1 g of the crystals). This exceeds the amount required for the formation of a compact monolayer. Most likely, the monolayer is rather impossible due to the similar size of the protein and the average pore size (both about 10 nm). Such a high loading can be explained by non-homogeneous channel-like pores with “vessels” where protein molecules in large numbers can be hosted. Protein aggregates of typical sizes 50 and 200 nm are formed due to the presence of Ca2+ and are present for both methods. Rather a small concentration of Ca2+ of about 10−5 M due to the dissociation of CaCO3 during ADS gives a small number of aggregates and mostly adsorption of single protein molecules occurs. At the same time, the COS method requires a much higher Ca2+ concentration on the order of 10−1 M that results in a dominating presence of aggregates during COS. Since the isotherms of protein adsorption for both loading methods obey the Langmuir equation, the protein aggregation has no effect on the isotherm shape, proving that the aggregates are predominantly formed in the solution and then adsorbed onto the CaCO3 crystal surface. A higher affinity of catalase to the crystal surface in the case of ADS (Ka = 23 × 105 M−1) compared to that of COS (Ka = 5 × 105 M−1) also proves this assumption.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Alexander von Humboldt Foundation in the framework of the Sofja Kovalevskaja program. The authors thank Rona Pitschke and Heike Runge for SEM imaging. AV also would like to acknowledge the QR Fund from Nottingham Trent University.References
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- S. X. Yang, W. E. Yuan and T. Jin, Expert Opin. Drug Delivery, 2009, 6, 1123–1133 CrossRef CAS PubMed.
- M. L. Tan, P. F. M. Choong and C. R. Dass, Peptides, 2010, 31, 184–193 CrossRef CAS PubMed.
- E. Donath, G. B. Sukhorukov, F. Caruso, S. A. Davis and H. Möhwald, Angew. Chem., Int. Ed., 1998, 37, 2201–2205 CrossRef.
- B. G. De Geest, N. N. Sanders, G. B. Sukhorukov, J. Demeester and S. C. De Smedt, Chem. Soc. Rev., 2007, 36, 636–649 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Delcea, H. Mohwald and A. G. Skirtach, Adv. Drug Delivery Rev., 2011, 63, 730–747 CrossRef CAS PubMed.
- K. Ariga, M. McShane, Y. M. Lvov, Q. M. Ji and J. P. Hill, Expert Opin. Drug Delivery, 2011, 8, 633–644 CrossRef CAS PubMed.
- Y. Lvov, A. A. Antipov, A. Mamedov, H. Mohwald and G. B. Sukhorukov, Nano Lett., 2001, 1, 125–128 CrossRef CAS.
- Z. Tang, Y. Wang, P. Podsiadlo and N. A. Kotov, Adv. Mater., 2006, 18, 3203–3224 CrossRef CAS.
- M. N. Antipina and G. B. Sukhorukov, Adv. Drug Delivery Rev., 2011, 63, 716–729 CrossRef CAS PubMed.
- N. G. Balabushevich, V. A. Izumrudov and N. I. Larionova, Polym. Sci., Ser. A, 2012, 54, 540–551 CrossRef CAS.
- D. Volodkin and R. von Klitzing, Curr. Opin. Colloid Interface Sci., 2014, 19, 25–31 CrossRef CAS.
- A. A. Antipov, G. B. Sukhorukov, S. Leporatti, I. Radtchenko, E. Donath and H. Mohwald, Colloids Surf., A, 2002, 198, 535–541 CrossRef.
- D. Kohler, N. Madaboosi, M. Delcea, S. Schmidt, B. G. De Geest, D. V. Volodkin, H. Möhwald and A. G. Skirtach, Adv. Mater., 2012, 24, 1095–1100 CrossRef CAS PubMed.
- K. Ariga, Y. M. Lvov, K. Kawakami, Q. M. Ji and J. P. Hill, Adv. Drug Delivery Rev., 2011, 63, 762–771 CrossRef CAS PubMed.
- D. Luo, M. S. Hasan, S. Shahid, B. N. Khlebtsov, M. J. Cattell and G. B. Sukhorukov, Langmuir, 2017, 33, 7982–7993 CrossRef CAS PubMed.
- D. Volodkin, A. Skirtach and H. Mohwald, in Bioactive Surfaces, ed. H. G. Borner and J. F. Lutz, Springer-Verlag Berlin, Berlin, 2011, vol. 240, pp. 135–161 Search PubMed.
- D. Volodkin, A. Skirtach and H. Mohwald, Polym. Int., 2012, 61, 673–679 CrossRef CAS.
- C. S. Peyratout and L. Dahne, Angew. Chem., Int. Ed., 2004, 43, 3762–3783 CrossRef CAS PubMed.
- B. V. Parakhonskiy, A. M. Yashchenok, M. Konrad and A. G. Skirtach, Adv. Colloid Interface Sci., 2014, 207, 253–264 CrossRef CAS PubMed.
- D. Volodkin, Adv. Colloid Interface Sci., 2014, 207, 306–324 CrossRef CAS PubMed.
- D. V. Volodkin, N. I. Larionova and G. B. Sukhorukov, Biomacromolecules, 2004, 5, 1962–1972 CrossRef CAS PubMed.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS PubMed.
- N. Feoktistova, J. Rose, V. Z. Prokopović, A. S. Vikulina, A. Skirtach and D. Volodkin, Langmuir, 2016, 32, 4229–4238 CrossRef CAS PubMed.
- M. Abebe, N. Hedin and Z. Bacsik, Cryst. Growth Des., 2015, 15, 3609–3616 CAS.
- F. Baldassarre, C. Allegretti, D. Tessaro, E. Carata, C. Citti, V. Vergaro, C. Nobile, G. Cannazza, P. D'Arrigo, A. Mele, L. Dini and G. Ciccarella, ChemistrySelect, 2016, 1, 6507–6514 CrossRef.
- B. V. Parakhonskiy, A. Haase and R. Antolini, Angew. Chem., Int. Ed., 2012, 51, 1195–1197 CrossRef CAS PubMed.
- N. G. M. Palmqvist, J. M. Nedelec, G. A. Seisenbaeva and V. G. Kessler, Acta Biomater., 2017, 57, 426–434 CrossRef CAS PubMed.
- Y. I. Svenskaya, H. Fattah, A. M. Zakharevich, D. A. Gorin, G. B. Sukhorukov and B. V. Parakhonskiy, Adv. Powder Technol., 2016, 27, 618–624 CrossRef CAS.
- D. B. Trushina, T. V. Bukreeva and M. N. Antipina, Cryst. Growth Des., 2016, 16, 1311–1319 CAS.
- E. P. Mironov, I. V. Marchenko, V. V. Artemov and T. V. Bukreeva, Colloid J., 2017, 79, 360–367 CrossRef CAS.
- A. H. Wang, Y. Yang, X. M. Zhang, X. C. Liu, W. Cui and J. B. Li, ChemPlusChem, 2016, 81, 194–201 CrossRef CAS.
- Z. E. Yilmaz, T. Cordonnier, A. Debuigne, B. Calvignac, C. Jerome and F. Boury, Int. J. Pharm., 2016, 513, 130–137 CrossRef CAS PubMed.
- M. L. De Temmerman, J. Demeester, F. De Vos and S. C. De Smedt, Biomacromolecules, 2011, 12, 1283–1289 CrossRef CAS PubMed.
- M. L. De Temmerman, J. Rejman, J. Grooten, T. De Beer, C. Vervaet, J. Demeester and S. C. De Smedt, Pharm. Res., 2011, 28, 1765–1773 CrossRef CAS PubMed.
- Z. She, M. N. Antipina, J. Li and G. B. Sukhorukov, Biomacromolecules, 2010, 11, 1241–1247 CrossRef CAS PubMed.
- C. S. Karamitros, A. M. Yashchenok, H. Mohwald, A. G. Skirtach and M. Konrad, Biomacromolecules, 2013, 14, 4398–4406 CrossRef CAS PubMed.
- P. K. Harimech, R. Hartmann, J. Rejman, P. del Pino, P. Rivera-Gil and W. J. Parak, J. Mater. Chem. B, 2015, 3, 2801–2807 RSC.
- Y. Jin, R. Yendluri, B. Chen, J. B. Wang and Y. Lvov, J. Colloid Interface Sci., 2016, 466, 254–260 CrossRef CAS PubMed.
- J. R. Lakkakula, R. Kurapati, I. Tynga, H. Abrahamse, A. M. Raichur and R. W. M. Krause, RSC Adv., 2016, 6, 104537–104548 RSC.
- N. A. N. Hanafy, M. L. De Giorgi, C. Nobile, M. Cascione, R. Rinaldi and S. Leporatti, Sci. Adv. Mater., 2016, 8, 514–523 CrossRef CAS.
- X. F. Liu, S. P. She, W. J. Tong and C. Y. Gao, RSC Adv., 2015, 5, 5775–5780 RSC.
- V. E. Bosio, M. L. Cacicedo, B. Calvignac, I. Leon, T. Beuvier, F. Boury and G. R. Castro, Colloids Surf., B, 2014, 123, 158–169 CrossRef CAS PubMed.
- D. Volodkin, Colloid Polym. Sci., 2014, 292, 1249–1259 CAS.
- S. Schmidt, M. Behra, K. Uhlig, N. Madaboosi, L. Hartmann, C. Duschl and D. Volodkin, Adv. Funct. Mater., 2013, 23, 116–123 CrossRef CAS.
- D. V. Volodkin, S. Schmidt, P. Fernandes, N. I. Larionova, G. B. Sukhorukov, C. Duschl, H. Möhwald and R. von Klitzing, Adv. Funct. Mater., 2012, 22, 1914–1922 CrossRef CAS.
- J. D. Cui, Y. M. Zhao, Z. L. Tan, Z. Cheng, P. P. Han and S. R. Jia, Int. J. Biol. Macromol., 2017, 98, 887–896 CrossRef CAS PubMed.
- M. Behra, N. Azzouz, S. Schmidt, D. V. Volodkin, S. Mosca, M. Chanana, P. H. Seeberger and L. Hartmann, Biomacromolecules, 2013, 14, 1927–1935 CrossRef CAS PubMed.
- M. Behra, S. Schmidt, J. Hartmann, D. V. Volodkin and L. Hartmann, Macromol. Rapid Commun., 2012, 33, 1049–1054 CrossRef CAS PubMed.
- N. Feoktistova, G. Stoychev, N. Puretskiy, L. Ionov and D. Volodkin, Eur. Polym. J., 2015, 68, 650–656 CrossRef.
- A. Neira-Carrillo, E. Yslas, Y. A. Marini, P. Vasquez-Quitral, M. Sanchez, A. Riveros, D. Yanez, P. Cavallo, M. J. Kogan and D. Acevedo, Colloids Surf., B, 2016, 145, 634–642 CrossRef CAS PubMed.
- Y. I. Svenskaya, A. M. Pavlov, D. A. Gorin, D. J. Gould, B. V. Parakhonskiy and G. B. Sukhorukov, Colloids Surf., B, 2016, 146, 171–179 CrossRef CAS PubMed.
- Q. Q. Dong, J. L. Li, L. Y. Cui, H. L. Jian, A. H. Wang and S. Bai, Colloids Surf., A, 2017, 516, 190–198 CrossRef CAS.
- S. L. Yefimova, I. I. Bespalova, G. V. Grygorova, A. V. Sorokin, P. V. Mateychenko, X. Q. Cui and Y. V. Malyukin, Microporous Mesoporous Mater., 2016, 236, 120–128 CrossRef CAS.
- M. A. Vantsyan, A. A. Kochetkov, I. V. Marchenko, Y. I. Kiryukhin, B. V. Nabatov, V. V. Artemov and T. V. Bukreeva, Crystallogr. Rep., 2015, 60, 951–958 CrossRef CAS.
- E. A. Genina, Y. I. Svenskaya, I. Y. Yanina, L. E. Dolotov, N. A. Navolokin, A. N. Bashkatov, G. S. Terentyuk, A. B. Bucharskaya, G. N. Maslyakova, D. A. Gorin, V. V. Tuchin and G. B. Sukhorukov, Biomed. Opt. Express, 2016, 7, 2082–2087 CrossRef CAS PubMed.
- T. N. Borodina, D. B. Trushina, I. V. Marchenko and T. V. Bukreeva, J. Bionanosci., 2016, 6, 261–268 CrossRef.
- D. P. Liu, G. H. Jiang, W. J. Yu, L. Li, Z. Z. Tong, X. D. Kong and J. M. Yao, Mater. Lett., 2017, 188, 263–266 CrossRef CAS.
- S. Schmidt, K. Uhlig, C. Duschl and D. Volodkin, Acta Biomater., 2014, 10, 1423–1430 CrossRef CAS PubMed.
- J. L. Jia, Q. Liu, T. Y. Yang, L. Y. Wang and G. H. Ma, J. Mater. Chem. B, 2017, 5, 1611–1623 RSC.
- J. D. Snook, C. B. Chesson, A. G. Peniche, S. M. Dann, A. Paulucci, I. V. Pinchuk and J. S. Rudra, J. Mater. Chem. B, 2016, 4, 1640–1649 RSC.
- P. P. Han, Z. Y. Jiang, X. L. Wang, X. Y. Wang, S. H. Zhang, J. F. Shi and H. Wu, J. Mater. Chem. B, 2015, 3, 7194–7202 RSC.
- A. Sergeeva, R. Sergeev, E. Lengert, A. Zakharevich, B. Parakhonskiy, D. Gorin, S. Sergeey and D. Volodkin, ACS Appl. Mater. Interfaces, 2015, 7, 21315–21325 CAS.
- G. Begum, T. N. Reddy, K. P. Kumar, K. Dhevendar, S. Singh, M. Arnarnath, S. Misra, V. K. Rangari and R. K. Rana, ACS Appl. Mater. Interfaces, 2016, 8, 22056–22063 CAS.
- H. Z. Shi, L. Li, L. Y. Zhang, T. T. Wang, C. G. Wang, D. X. Zhu and Z. M. Su, CrystEngComm, 2015, 17, 4768–4773 RSC.
- N. E. Markina, A. V. Markin, A. M. Zakharevich and I. Y. Goryacheva, Microchim. Acta, 2017, 184, 3937–3944 CrossRef CAS.
- I. Stetciura, A. Yashchenok, A. Masic, E. Lyubin, O. Inozemtseva, M. Drozdova, E. Markvichova, B. N. Khlebtsov, A. A. Fedyanin, G. Sukhorukov, D. A. Gorin and D. Volodkin, Analyst, 2015, 140, 4981–4986 RSC.
- T. Paulraj, N. Feoktistova, N. Velk, K. Uhlig, C. Duschl and D. Volodkin, Macromol. Rapid Commun., 2014, 35, 1408–1413 CrossRef CAS PubMed.
- A. Sergeeva, N. Feoktistova, V. Prokopovic, D. Gorin and D. Volodkin, Adv. Mater. Interfaces, 2015, 2, 1500386 CrossRef.
- A. S. Sergeeva, D. A. Gorin and D. V. Volodkin, Langmuir, 2015, 31, 10813–10821 CrossRef CAS PubMed.
- A. I. Petrov, D. V. Volodkin and G. B. Sukhorukov, Biotechnol. Prog., 2005, 21, 918–925 CrossRef CAS PubMed.
- G. B. Sukhorukov, D. V. Volodkin, A. M. Günther, A. I. Petrov, D. B. Shenoy and H. Möhwald, J. Mater. Chem., 2004, 14, 2073–2081 RSC.
- N. G. Balabushevich, A. V. L. de Guerenu, N. A. Feoktistova, A. G. Skirtach and D. Volodkin, Macromol. Biosci., 2016, 16, 95–105 CrossRef CAS PubMed.
- N. G. Balabushevich, A. V. L. de Guerenu, N. A. Feoktistova and D. Volodkin, Phys. Chem. Chem. Phys., 2015, 17, 2523–2530 RSC.
- B. V. Parakhonskiy, A. M. Yashchenok, S. Donatan, D. V. Volodkin, F. Tessarolo, R. Antolini, H. Mohwald and A. G. Skirtach, ChemPhysChem, 2014, 15, 2817–2822 CrossRef CAS PubMed.
- P. Wuytens, B. Parakhonskiy, A. Yashchenok, M. Winterhalter and A. Skirtach, Curr. Opin. Pharmacol., 2014, 18, 129–140 CrossRef CAS PubMed.
- N. Sudareva, H. Popova, N. Saprykina and S. Bronnikov, Structural optimization of calcium carbonate cores as templates for protein encapsulation, 2014 Search PubMed.
- A. Hernandez-Hernandez, A. B. Rodriguez-Navarro, J. Gomez-Morales, C. Jimenez-Lopez, Y. Nys and J. M. Garcia-Ruiz, Cryst. Growth Des., 2008, 8, 1495–1502 CAS.
- G. Limousin, J. P. Gaudet, L. Charlet, S. Szenknect, V. Barthes and M. Krimissa, Appl. Geochem., 2007, 22, 249–275 CrossRef CAS.
- M. Králik, Chem. Pap., 2014, 68, 1625–1638 Search PubMed.
- J. Meissner, A. Prause, B. Bharti and G. H. Findenegg, Colloid Polym. Sci., 2015, 293, 3381–3391 CAS.
- A. Ebadi, J. S. S. Mohammadzadeh and A. Khudiev, Adsorption, 2009, 15, 65–73 CrossRef CAS.
- B. Nagy, A. Toth, I. Savina, S. Mikhalovsky, L. Mikhalovska, E. Geissler and K. Laszlo, Carbon, 2017, 112, 103–110 CrossRef CAS.
- A. Achour, A. Arman, M. Islam, A. A. Zavarian, A. B. Al-Zubaidi and J. Szade, Eur. Phys. J. Plus, 2017, 132 Search PubMed.
- W. Kadima, L. Ogendal, R. Bauer, N. Kaarsholm, K. Brodersen, J. F. Hansen and P. Porting, Biopolymers, 1993, 33, 1643–1657 CrossRef CAS PubMed.
- E. Sahin, A. O. Grillo, M. D. Perkins and C. J. Roberts, J. Pharm. Sci., 2010, 99, 4830–4848 CrossRef CAS PubMed.
- W. Wang, N. Li and S. Speaker, Aggregation of Therapeutic Proteins, John Wiley & Sons, Inc., 2010, pp. 119–204 DOI:10.1002/9780470769829.ch4.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp07836f |
| This journal is © the Owner Societies 2018 |