Kinetics of the reaction of CO3˙−(H2O)n, n = 0, 1, 2, with nitric acid, a key reaction in tropospheric negative ion chemistry†
Received
17th November 2017
, Accepted 19th February 2018
First published on 20th February 2018
Abstract
A significant fraction of nitrate in the troposphere is formed in the reactions of HNO3 with the carbonate radical anion CO3˙− and the mono- and dihydrated species CO3˙−(H2O)1,2. A reaction mechanism was proposed in earlier flow reactor studies, which is investigated here in more detail by quantum chemical calculations and experimental reactivity studies of mass selected ions under ultra-high vacuum conditions. Bare CO3˙− forms NO3−(OH˙) as well as NO3−, with a total rate coefficient of 1.0 × 10−10 cm3 s−1. CO3˙−(H2O) in addition affords stabilization of the NO3−(HCO3˙) collision complex, and thermalized CO3˙−(H2O) reacts with a total rate coefficient of 6.3 × 10−10 cm3 s−1. A second solvent molecule quenches the reaction, and only black-body radiation induced dissociation is observed for CO3˙−(H2O)2, with an upper limit of 6.0 × 10−11 cm3 s−1 for any potential bimolecular reaction channel. The rate coefficients obtained under ultra-high vacuum conditions are smaller than in the earlier flow reactor studies, due to the absence of stabilizing collisions, which also has a strong effect on the product branching ratio. Quantum chemical calculations corroborate the mechanism proposed by Möhler and Arnold. The reaction proceeds through a proton-transferred NO3−(HCO3˙) collision complex, which can rearrange to NO3−(OH˙)(CO2). The weakly bound CO2 easily evaporates, followed by evaporation of the more strongly attached OH˙, if sufficient energy is available.
Introduction
The CO3˙− radical anion is an important intermediate in the tropospheric chemistry of anions.1,2 Quantitative modeling by Kawamoto et al.3 places the fractional abundance of CO3˙− core ions in the troposphere in the range of 0.9–2.3%, quite comparable to HSO4− core ions close to the ground, while the overwhelming majority of negative ions have an NO3− core. However, a significant fraction of NO3−(H2O) is formed in the reaction of CO3˙−(H2O) with HNO3.3 In recent years, ions in the troposphere have received increased attention in the form of charged aerosol particles.4 In a series of laboratory experiments, the CLOUD collaboration at CERN has recently shown that ions play an important role for aerosol nucleation and cloud formation in the troposphere.5–8
The formation of CO3˙− in atmospheric conditions was described by Fehsenfeld, Ferguson and co-workers in the 1970s.9 In the first step electrons react with O2 and an additional collision partner M to form O2−, reaction (1). Ozonide O3˙− is formed by charge transfer to an ozone molecule, reaction (2). The ozonide anion finally transfers O− to a carbon dioxide molecule, forming CO3˙− and O2, reaction (3).
| | | e− + O2 + M → O2˙− + M, M = O2, N2, Ar, H2O,… | (1) |
| | | O3˙− + CO2 → CO3˙− + O2 | (3) |
The properties of CO3˙− itself were intensively studied by spectroscopy. Matrix isolation techniques were applied by Jacox et al.,10 and a number of other groups used photodissociation techniques to characterize CO3˙−.11–14 The O2C–O− bond dissociation energy was determined by Johnson, Viggiano and co-workers via photodissociation and high level quantum chemical calculations to be 269 ± 5 kJ mol−1.15–17
Despite its key role in tropospheric anion chemistry,18 only a small number of gas phase reactions were studied.19–23 These are mostly reactions with nitrogen oxides, leading to formation of nitrate NO3−.3,24–27 Reactions of CO3−(H2O)0,1,2 with formic, acetic, propionic, butyric, glyoxylic, pyruvic and pinonic acid were investigated in a flow tube experiment by Arnold et al.28,29 They reported as initial step mostly proton transfer giving the deprotonated acids as products. For propionic acid and larger, they also observed clustering with CO3˙− and formation of radical species, e.g. CH3CH(˙)COO− from propionic acid.28,29 However, we recently showed that the reaction with formic acid proceeds mostly via oxidation of formic acid to carbon dioxide and water.30 The deprotonation product HCOO− was re-assigned to the reaction of formic acid with HCOO−(OH˙), an intermediate in the oxidation reaction that is formed in small concentrations.
A similar pattern emerged for the reaction of CO3˙− with HCl. A flow tube study by Dotan et al.24 established an upper limit for the rate of 3 × 10−11 cm3 s−1, without observing any products. Recent studies in our laboratory reveal that this reaction proceeds via formation of a short lived, very reactive Cl−(OH˙) intermediate that reacts fast with a second HCl molecule, producing HCl2− and Cl2˙− as final products.31 The rate of the first step is 4.2 × 10−12 cm3 s−1, consistent with the upper limit given by Dotan et al.24 Interestingly, hydration dramatically accelerates the rate to 2.7 × 10−10 cm3 s−1 for CO3˙−(H2O), resulting in the reactive Cl−(HCO3˙) radical species.31
Nitric acid21–23,32 and methane sulfonic acid20 show efficient reactions with CO3˙−. The gas phase ion chemistry of HNO3 was extensively investigated by Fehsenfeld and coworkers.21 They reported a reaction rate coefficient of 8 × 10-10 cm3 s−1 for reaction (4), measured by the flowing afterglow technique.
| | | CO3˙− + HNO3 → NO3− + [CO2,OH˙] | (4) |
Möhler and Arnold investigated the reaction of HNO3 with CO3˙− and CO3˙−(H2O) in a flow-reactor triple quadrupole mass spectrometer in the presence of 1.7 mbar N2 and 0.3 mbar O2 as buffer gas.22 For bare CO3˙− they observed NO3−, [NO3,OH˙]− and [CO3˙,HNO3]− as primary products, followed by clustering reactions with HNO3.22 No [NO3,OH˙]− was observed when 0.03 mbar H2O was added to the buffer gas to study the hydrated species, while [CO3˙,HNO3]− was the dominant product.22 A mechanism via an excited collision complex [(CO3˙HNO3)−]* was proposed. The complex is de-exited by collisions, or decays into NO3− or [NO3,OH˙]−.22 Catoire and co-workers used the CO3˙− + HNO3 reaction for testing their flowing afterglow setup and observed the same primary products as Möhler and Arnold.22,32 Since no mass selection was available for the reactant ions, the exact sequence of reactions generating the observed products, in particular the role of the [NO3,OH˙]− and the influence of an additional water molecule on the reactivity, remained unclear.
Because of its central role in tropospheric negative ion chemistry,1,3 we investigated the reaction of CO3˙−(H2O)n, n = 0, 1, 2, with HNO3 by FT-ICR mass spectrometry and quantum chemical calculations. Each cluster size was studied separately with mass-selected ions. The extremely low pressure in the FT-ICR instrument leads to product distributions different from the flow reactor studies, due to the absence of stabilizing collisions. A key problem in gas-phase reactivity studies with HNO3 is the inherent instability of this substance. HNO3 decomposes in concentrated aqueous solutions. In earlier studies on the reactivity of ionic water clusters,33–35 we worked with aqueous concentrated solutions of HNO3, which yields a mixture of HNO3 and H2O in the reaction region, with traces of the decomposition product HONO. The observed reaction products confirmed that the abundance of HONO was less than 3% of the HNO3 partial pressure. To identify reactions where HONO might play a role, we use quantum chemical calculations to identify thermochemically allowed reaction pathways. In addition, structural as well as thermochemical information is obtained for stationary points on the reaction potential energy surface. Together, a consistent picture of the reaction mechanism evolves.
Computational and experimental details
The ion–molecule reaction profiles were simulated with density functional theory at the M06-2X/6-311++G(d,p) level of theory employing the Gaussian09 program suite.36 The energies of all optimized geometries were corrected with zero-point energy obtained from harmonic vibration analyses. Local minima and transition structures on the potential energy surface were confirmed with absence of and presence of one imaginary frequency, respectively. The local minima associated with each transition structure were verified by the intrinsic reaction coordinate (IRC) method. Spin density distributions were evaluated at the same level of theory and shown as isosurfaces at a value of 0.01 a.u. Table S1 in ESI,† compares the used methods/basis sets with experimental values. The coordinates are given in Table S4 (ESI†).
The experiments were performed on a modified Bruker/Spectrospin CMS47X FT-ICR mass spectrometer as described in detail elsewhere.37–39 The spectrometer is equipped with a Bruker Infinity Cell, an APEX III data station, a 4.7 Tesla magnet and an external laser vaporization ion source. CO3˙− was produced in the laser vaporization ion source39–42via expansion of a helium/water/oxygen/carbon dioxide mixture using a zinc target for the production of electrons. As vaporization laser a frequency doubled Nd:YAG laser at a pulse energy of ∼5 mJ at 10 Hz was used. All produced anionic species were transferred into the ICR cell, where they can be stored for several seconds or minutes. The ion of interest, CO3˙− or CO3˙−(H2O)1,2, was then isolated via resonant excitation of unwanted ions prior to measuring the kinetics. A constant background pressure of HNO3 and H2O (concentrated aqueous solution, 70% HNO3, Sigma-Aldrich) was introduced into the ultrahigh vacuum region via a leak valve. Due to the inherent instability of HNO3, also traces of HONO were present, which are formed from the NO2˙ decomposition product on thin films of H2O and HNO3, as present in the vacuum system.43 Mass spectra were taken after different reaction delays relative to the end of the fill and isolation cycle. The intensities were extracted from the mass spectra. A rate coefficient matrix defines the allowed reaction channels and the data was fitted using a genetic algorithm that optimizes the rates from the matrix based on a pseudo-first order rate law. Relative rate coefficients were extracted from this fit. The stability of the fit was tested by systematically changing each parameter in 5 to 10 steps of ±5% from its optimized value, and re-optimizing all other parameters. The resulting error was plotted against the modified parameter. These plots are available as supporting information in Fig. S8 and S9 (ESI†). All rate coefficients reported in the results section showed stable, well-converged minima.
These pressure-dependent pseudo-first order rate coefficients are converted to pressure-independent bimolecular rate coefficients. An aqueous 70% HNO3 solution is close to the azeotropic point and the chemical composition in the gas phase is therefore close to 70% HNO3 and 30% water. As the cold cathode pressure gauge shows different sensitivity toward HNO3 and H2O, the measured pressure was corrected taking the different sensitivity into account. The HNO3 partial pressure was taken as 70% of the corrected total pressure. More details on the kinetic analysis and pressure correction are available in ESI.† The accuracy of the absolute rates is estimated to be ±40% due to uncertainties in the pressure determination. The noise level is derived from the baseline of each mass spectrum.
Results and discussion
Calculated reaction potential energy surface
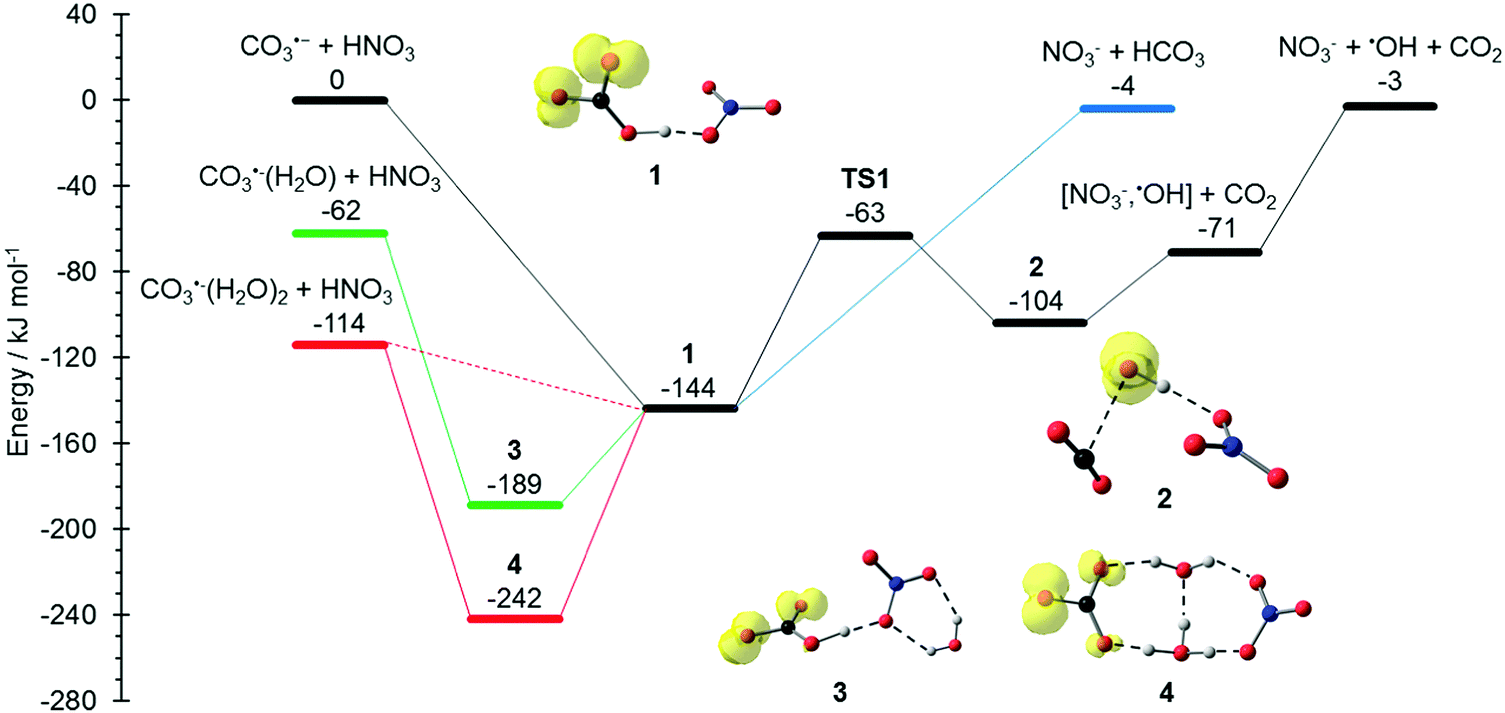
The potential energy surface (PES) corresponding to the reaction mechanisms suggested by Möhler and Arnold22 is shown in Fig. 1. The calculations show that a barrierless proton transfer takes place upon formation of the collision complex 1, which can be written as NO3−(HCO3˙). Rearrangement of the HCO3˙ unit to CO2 + OH˙ within the complex faces a significant barrier of 81 kJ mol−1viaTS1 located at −63 kJ mol−1 relative to the separated reactants. Evaporation of CO2 from complex 2, which can be written as NO3−(OH˙)(CO2), requires only 33 kJ mol−1, leading to the observed NO3−(OH˙) product. Further evaporation of the OH˙ radical requires 68 kJ mol−1, making the overall reaction with −3 kJ mol−1 below the entrance channel almost thermoneutral.
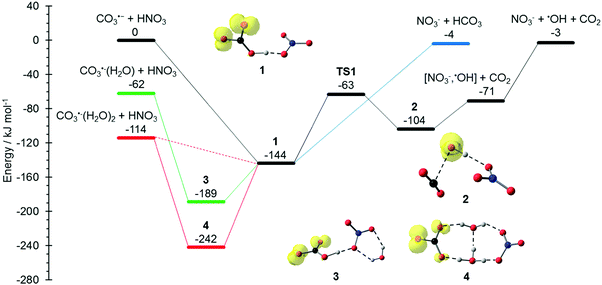 |
| | Fig. 1 Reaction potential energy surface (PES) calculated on the M06-2X/6-311++G(d,p) level of theory, with zero-point corrected energies given in kJ mol−1. More detailed results on the influence of solvation and additional reaction channels are given in the ESI,† Fig. S1–S4. | |
The water binding energies in CO3˙−(H2O)1,2 are 62 kJ mol−1 and 52 kJ mol−1 for the first and second molecule, respectively. Upon collision with HNO3, the water molecules from CO3−(H2O) and CO3−(H2O)2 evaporate due to the energy released upon formation of the collision complex, since the transition states for rearrangement prior to water evaporation lie significantly higher in energy, see Fig. S1 (ESI†). For CO3˙−(H2O), the pathway to NO3−(OH˙) + CO2 formation is still accessible, while bare NO3− formation is clearly out of reach, lying 59 kJ mol−1 above the CO3˙−(H2O) + HNO3 entrance channel. The second water molecule reduces the available energy further, only the formation of the NO3−(HCO3˙) complex 1 is thermochemically allowed, with a moderate exothermicity of −30 kJ mol−1.
Experimental results and discussion
CO3−.
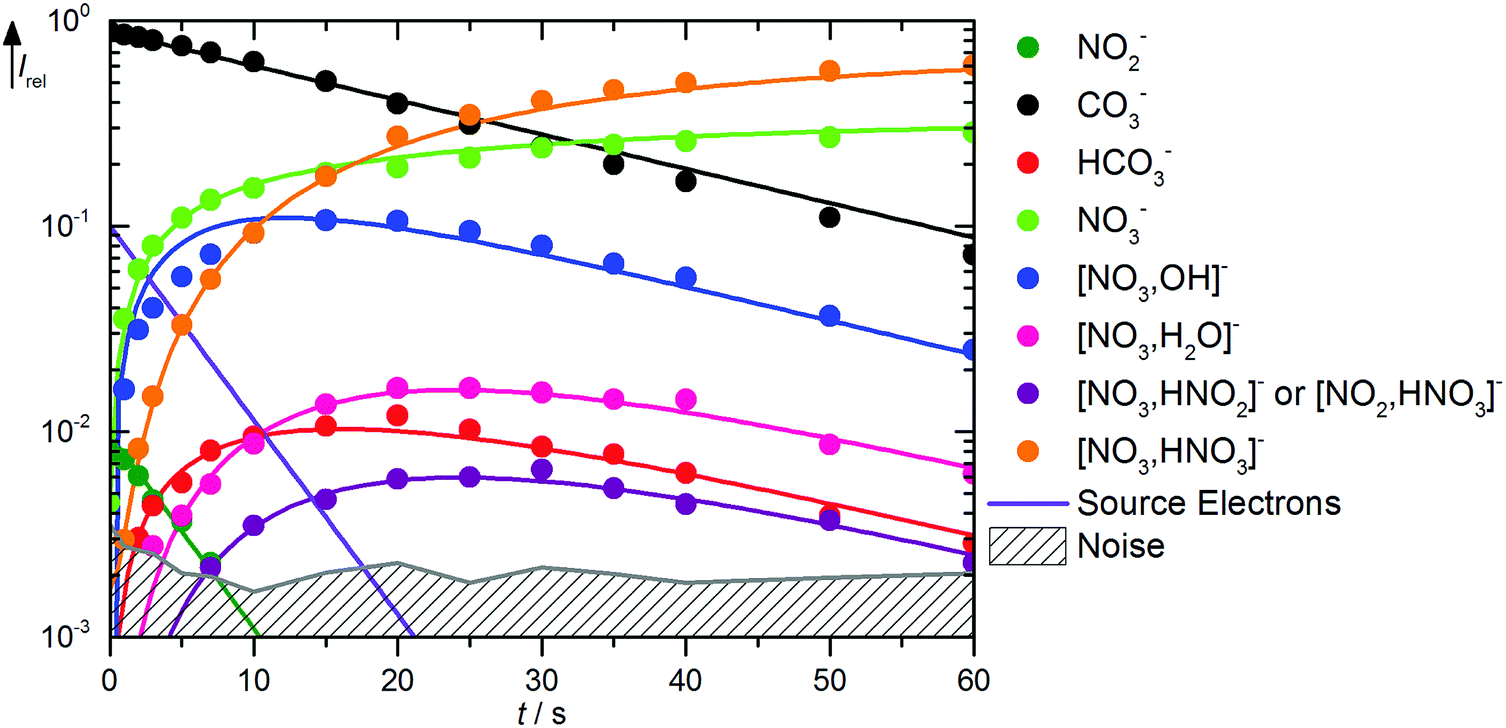
The kinetics of the reaction of CO3˙− with HNO3 is displayed in Fig. 2. A mass spectrum is available in Fig. S5 (ESI†). The decay of the CO3˙− ion intensity follows a pseudo-first order kinetics behavior. Primary reactions for pure CO3˙− with the reaction mixture lead to formation of HCO3−, NO3−, and NO3−(OH˙). Rate coefficients for all reactions are given in Table 1. Compared to the flow reactor studies, the reaction is an order of magnitude slower, with an overall rate for all reaction channels of 1.0 × 10−10 cm3 s−1. Fehsenfeld et al.21 reported a value of 8 × 10−10 cm3 s−1 in their flowing afterglow, while Möhler and Arnold22 agreed with their value of 1.3 × 10−9 cm3 s−1 within error limits with Guimbaud et al.,32 who reported 1.2 ± 0.3 × 10−9 cm3 s−1. Obviously, stabilizing collisions with the buffer gas in the flow reactor studies increase the efficiency of the reaction. The nitrate–hydroxyl radical complex arises from the reaction with gaseous HNO3 according to reaction (5). Formation of HCO3− requires a hydrogen atom transfer (HAT), which is significantly endothermic for HNO3, reaction (6). We therefore assign this small product to traces of HONO, reaction (7), which is exothermic. The calculations show a barrierless HAT from HONO to CO3−. If we assume collision efficiency for reaction (7), a partial pressure below 1 × 10−10 mbar of the HONO background is sufficient to explain the observed abundance of HCO3−.| | | CO3˙− + HNO3 → NO3−(OH˙) + CO2 ΔH0 = −71 kJ mol−1 | (5) |
| | | CO3˙− + HNO3 → HCO3− + NO3˙ ΔH0 = +48 kJ mol−1 | (6) |
| | | CO3˙− + HONO → HCO3− + NO2˙ ΔH0 = −68 kJ mol−1 | (7) |
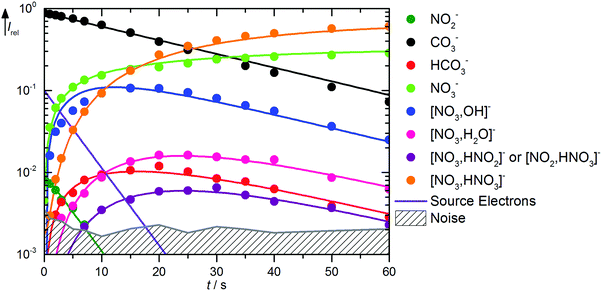 |
| | Fig. 2 Kinetics of the reaction of CO3˙− with HNO3 and H2O, with traces of HONO, at a HNO3 partial pressure of 1.4 × 10−8 mbar. The lines represent the fit with the rate coefficients from Table S1 (ESI†). Selected mass spectra are shown in Fig. S5 (ESI†). | |
Table 1 Bimolecular rate coefficients kabs for the reactions discussed in the text
| Reaction |
Rate coefficient kabs/cm3 s−1 |
|
Assuming either reaction (11) or (12).
Thermalized fraction of CO3˙−(H2O).
Cold fraction of CO3˙−(H2O); the accuracy of the absolute rates is estimated to be ±40% due to uncertainties in the pressure determination.
|
| (5) |
CO3˙− + HNO3 → NO3−(OH˙) + CO2 |
8.9 × 10−11 |
| (8/9) |
CO3˙− + HNO3 → NO3− + [CO2,OH˙] |
2.1 × 10−11 |
| (10) |
HCO3− + HNO3 → NO3− + CO2 + H2O |
2.8 × 10−10 |
| (11) |
NO3−(OH˙) + H2O → NO3−(H2O) + OH˙ |
1.2 × 10−10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
| (12) |
NO3−(OH˙) + HNO3 → NO3−(H2O) + NO3 |
5.3 × 10−11a |
| (14) |
NO3−(OH˙) + HNO3 → NO3−(HNO3) + OH˙ |
3.6 × 10−10 |
| (16) |
NO2− + HNO3 → NO3− + HONO |
7.2 × 10−9 |
| (18) |
CO3˙−(H2O) + HNO3 → NO3−(OH˙) + CO2 + H2O |
1.4 × 10−10b |
| (19) |
CO3˙−(H2O) + HNO3 → NO3−(HCO3˙) + H2O |
9.4 × 10−11c |
| 1.4 × 10−10b |
| (20) |
CO3˙−(H2O) + HNO3 → NO3− + CO2 + OH˙ + H2O |
3.8 × 10−10b |
Formation of NO3−viareactions (8) or (9) with HNO3 as neutral reactant is only slightly exothermic. Given that both reactants have low-lying vibrational modes, some extra thermal energy is available, which makes the reactions entirely plausible.
| | | CO3˙− + HNO3 → NO3− + CO2 + OH˙ ΔH0 = −3 kJ mol−1 | (8) |
| | | CO3˙− + HNO3 → NO3− + HCO3˙ ΔH0 = −4 kJ mol−1 | (9) |
According to our kinetics fit, several secondary reactions are observed. HCO3− reacts to NO3−, most likely via a straightforward proton transfer from HNO3, reaction (10).
| | | HCO3− + HNO3 → NO3− + CO2 + H2O ΔH0 = −88 kJ mol−1 | (10) |
NO3−(OH˙) either undergoes ligand exchange with H2O or HAT from HNO3 to form NO3−(H2O), reactions (11) or (12), respectively. Ligand exchange is also possible with HONO as well as HNO3, reactions (13) and (14), respectively. Since reactions (12) and (14) compete with each other, with reaction (14) being both more exothermic and mechanistically favorable, ligand exchange with water is the most plausible origin of the NO3−(H2O) product, reaction (11). Reaction (13) validates the presence of HONO in the reaction mixture.
| | | NO3−(OH˙) + H2O → NO3−(H2O) + OH˙ ΔH0 = +3 kJ mol−1 | (11) |
| | | NO3−(OH˙) + HNO3 → NO3−(H2O) + NO3 ΔH0 = −34 kJ mol−1 | (12) |
| | | NO3−(OH˙) + HONO → NO3−(HONO) + OH˙ ΔH0 = −40 kJ mol−1 | (13) |
| | | NO3−(OH˙) + HNO3 → NO3−(HNO3) + OH˙ ΔH0 = −75 kJ mol−1 | (14) |
We also observe traces of NO2−, which are assigned to dissociative electron attachment. When working with negative ions, some free electrons are usually trapped in the ICR cell,44 which react with HNO3 according to reaction (15).21 In turn, NO2− is converted with near collision rate to NO3−via proton transfer from HNO3, reaction (16).
| | | e− + HNO3 → NO2− + OH˙ | (15) |
| | | NO2− + HNO3 → NO3− + HONO | (16) |
CO3˙−(H2O).
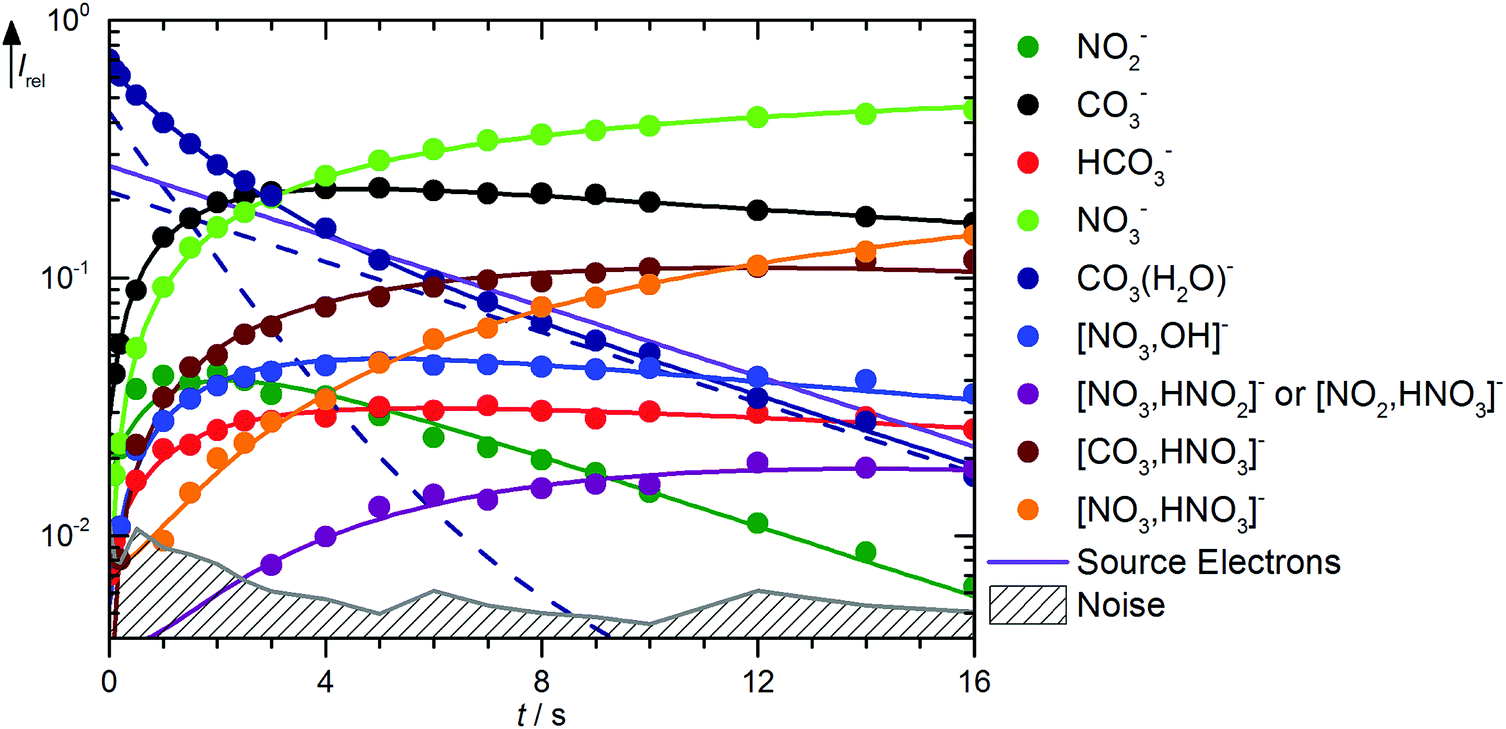
Fig. 3 shows the kinetics of the monohydrated species, mass spectra are displayed in Fig. S6 (ESI†). The scenario is quite complex, with a pronounced deviation of the CO3˙−(H2O) intensity from pseudo-first order behavior, for which a linear graph in the semi-logarithmic plot is expected. To describe the observed curvature, the population of this species is divided into two fractions, which correspond to the initially cold ions directly from the supersonic expansion, and the thermalized ions after heating by ambient black-body radiation and collision with the reaction gas. These effects are included in the kinetic model by allowing the cold fraction to be converted to the thermalized fraction, with a unimolecular rate treated as a fit parameter. Heating ultimately leads to formation of CO3˙−via black-body infrared radiative dissociation (BIRD),45–58reaction (17), with a lifetime of 3 s for the thermalized fraction. This reaction is not allowed for the cold fraction in the kinetic model, see Table S2 (ESI†). Since the major part of the CO3˙−(H2O) population is already thermalized at nominally t = 0 s, and the thermalized fraction reacts overall much faster than the cold fraction, the intensity drop of CO3˙−(H2O) is faster in the beginning, and levels off at later times because the conversion from the cold to the hot fraction becomes rate limiting, which explains the observed curvature.
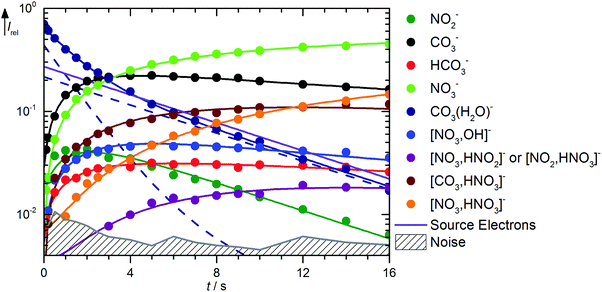 |
| | Fig. 3 Kinetics of the reaction of CO3˙−(H2O) with HNO3 and H2O, with traces of HONO, at a HNO3 partial pressure of 2.1 × 10−8 mbar. The lines represent the fit with the rate coefficients from Table S2 (ESI†). Selected mass spectra are shown in Fig. S6 (ESI†). | |
The formation of NO3−(OH˙) is observed with an appreciable rate, and can be assigned to collisions with HNO3, reaction (18). Also the transition state for CO2 formation is below the entrance channel, see Fig. 1, thus reaction (18) is fully consistent with the calculations. The same is true for the ligand exchange product NO3−(HCO3˙) formed viareaction (19). NO3− is exclusively formed from the thermalized fraction of CO3˙−(H2O) as a primary product in the kinetics. With HNO3 as the reaction partner, reaction (20) is with ΔH0 = +59 kJ mol−1 significantly endothermic. However, the thermalized fraction of CO3˙−(H2O) already contains almost enough energy for dissociation. Together with the internal energy of the HNO3 collision partner, the energy required for reaction (20) is available in the system. Reactions (18)–(20) yield a total rate coefficient of 6.3 × 10−10 cm3 s−1 for the reaction of thermalized CO3˙−(H2O) with HNO3, 37% of the flow reactor value of 1.7 × 10−9 cm3 s−1 reported by Möhler and Arnold.22 All secondary reactions as well as NO2− formation via dissociative electron attachment proceed as discussed above.
| | | CO3˙−(H2O) → CO3˙− + H2O ΔH0 = +62 kJ mol−1 | (17) |
| | | CO3˙−(H2O) + HNO3 → NO3−(OH•) + CO2 + H2O ΔH0 = −9 kJ mol−1 | (18) |
| | | CO3˙−(H2O) + HNO3 → NO3−(HCO3˙) + H2O ΔH0 = −83 kJ mol−1 | (19) |
| | | CO3˙−(H2O) + HNO3 → NO3− + CO2 + OH˙ + H2O ΔH0 = +59 kJ mol−1 | (20) |
CO3˙−(H2O)2.
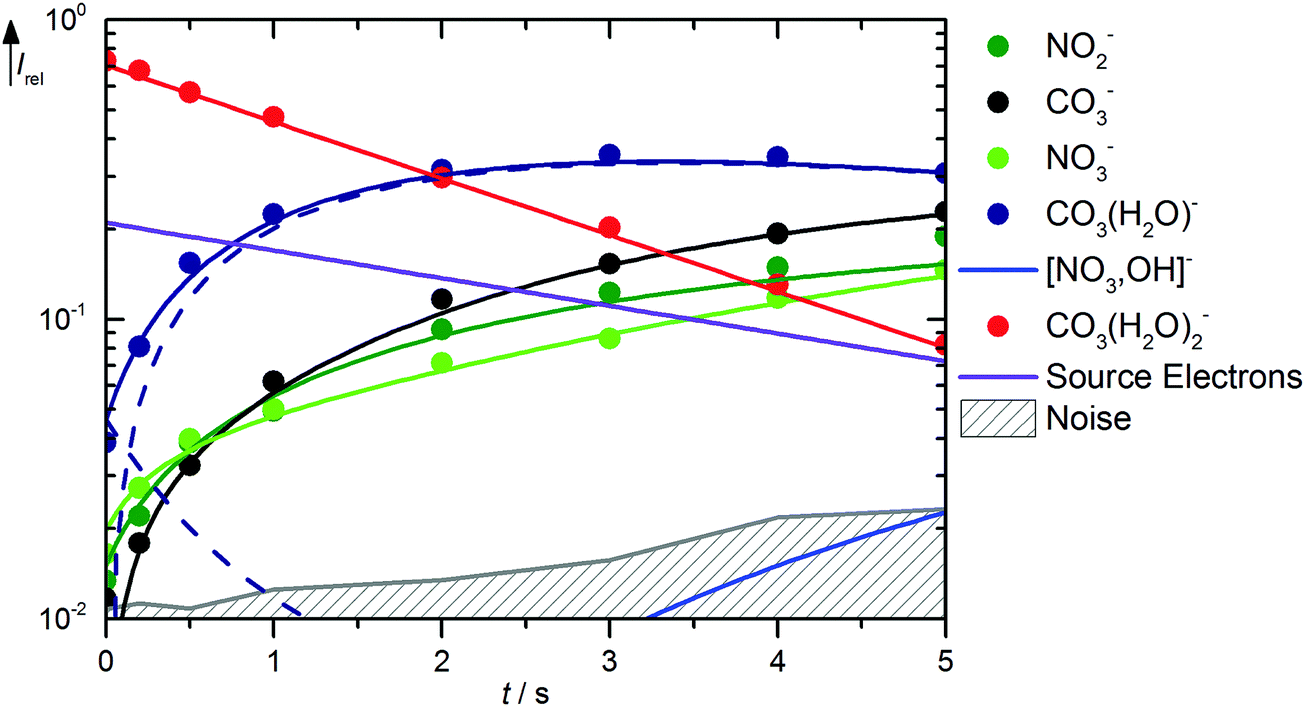
Solvation of CO3˙− with two water molecules leads to a significantly reduced reactivity with HNO3. The kinetics is shown in Fig. 4, mass spectra are available in Fig. S7 (ESI†). The major reaction channel is, as might be expected, loss of a water molecule due to BIRD, reaction (21). There is again formation of NO2−viareaction (15), but otherwise no primary products are observed from CO3˙−(H2O)2. The noise level of the kinetics, however, places a high upper limit of 6.0 × 10−11 cm3 s−1 on the rate coefficient for the direct reaction of CO3˙−(H2O)2 with HNO3. According to the thermochemical arguments discussed above, the most likely product of such a reaction would be NO3−(HCO3˙), followed by NO3−(OH˙), while direct NO3− formation this time is clearly out of reach.| | | CO3˙−(H2O)2 → CO3˙−(H2O) + H2O ΔH0 = +52 kJ mol−1 | (21) |
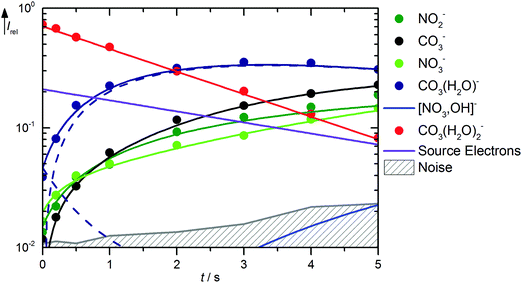 |
| | Fig. 4 Kinetics of the reaction of CO3˙−(H2O)2 with HNO3 and H2O, with traces of HONO, at a HNO3 partial pressure of 8.1 × 10−9 mbar. The lines represent the fit with the rate coefficients from Table S3 (ESI†). Selected mass spectra are shown in Fig. S7 (ESI†). | |
Conclusions
The mechanism of the reaction of HNO3 with CO3˙−(H2O)n, n = 0, 1, 2, is very similar to the previously studied HCl reaction.31 With bare CO3˙−, the reaction is relatively slow, and the proton-transfer intermediate NO3−(OH˙) is the unambiguously identified primary product, corresponding to Cl−(OH˙) in the HCl reaction. Also NO3− is directly formed, but with even lower rate. Formation of NO3−(OH˙) is accelerated for CO3˙−(H2O), but again slowed down by a second water molecule in the hydration shell. The rate is clearly dependent on the number of water molecules solvating CO3˙−, and overall significantly lower than in the flow reactor studies previously reported in the literature, indicating a strong pressure dependence.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. K. B. acknowledges startup funds from the University of Innsbruck. C. K. S. thanks Research Grants Council, Hong Kong SAR (Project no. 11300917) and City University of Hong Kong (CityU) (Project no. 7004401) for their financial support. W. K. T. acknowledges Chow Yei Ching School of Graduate Studies, CityU for his postgraduate studentships, outstanding academic performance awards, research tuition scholarships and overseas research activities award.
References
- G. Beig and G. P. Brasseur, J. Geophys. Res.: Atmos., 2000, 105, 22671–22684 CrossRef CAS.
- M. L. Huertas, J. Fontan and J. Gonzalez, Atmos. Environ., 1978, 12, 2351–2362 CrossRef CAS.
- H. Kawamoto and T. Ogawa, Planet. Space Sci., 1986, 34, 1229–1239 CrossRef CAS.
- A. Hirsikko, T. Nieminen, S. Gagné, K. Lehtipalo, H. E. Manninen, M. Ehn, U. Hõrrak, V.-M. Kerminen, L. Laakso, P. H. McMurry, A. Mirme, S. Mirme, T. Petäjä, H. Tammet, V. Vakkari, M. Vana and M. Kulmala, Atmos. Chem. Phys., 2011, 11, 767–798 CAS.
- J. Kirkby, J. Duplissy, K. Sengupta, C. Frege, H. Gordon, C. Williamson, M. Heinritzi, M. Simon, C. Yan, J. Almeida, J. Tröstl, T. Nieminen, I. K. Ortega, R. Wagner, A. Adamov, A. Amorim, A.-K. Bernhammer, F. Bianchi, M. Breitenlechner, S. Brilke, X. Chen, J. Craven, A. Dias, S. Ehrhart, R. C. Flagan, A. Franchin, C. Fuchs, R. Guida, J. Hakala, C. R. Hoyle, T. Jokinen, H. Junninen, J. Kangasluoma, J. Kim, M. Krapf, A. Kürten, A. Laaksonen, K. Lehtipalo, V. Makhmutov, S. Mathot, U. Molteni, A. Onnela, O. Peräkylä, F. Piel, T. Petäjä, A. P. Praplan, K. Pringle, A. Rap, N. A. D. Richards, I. Riipinen, M. P. Rissanen, L. Rondo, N. Sarnela, S. Schobesberger, C. E. Scott, J. H. Seinfeld, M. Sipilä, G. Steiner, Y. Stozhkov, F. Stratmann, A. Tomé, A. Virtanen, A. L. Vogel, A. C. Wagner, P. E. Wagner, E. Weingartner, D. Wimmer, P. M. Winkler, P. Ye, X. Zhang, A. Hansel, J. Dommen, N. M. Donahue, D. R. Worsnop, U. Baltensperger, M. Kulmala, K. S. Carslaw and J. Curtius, Nature, 2016, 533, 521–526 CrossRef CAS PubMed.
- K. Lehtipalo, L. Rondo, J. Kontkanen, S. Schobesberger, T. Jokinen, N. Sarnela, A. Kürten, S. Ehrhart, A. Franchin, T. Nieminen, F. Riccobono, M. Sipilä, T. Yli-Juuti, J. Duplissy, A. Adamov, L. Ahlm, J. Almeida, A. Amorim, F. Bianchi, M. Breitenlechner, J. Dommen, A. J. Downard, E. M. Dunne, R. C. Flagan, R. Guida, J. Hakala, A. Hansel, W. Jud, J. Kangasluoma, V.-M. Kerminen, H. Keskinen, J. Kim, J. Kirkby, A. Kupc, O. Kupiainen-Määttä, A. Laaksonen, M. J. Lawler, M. Leiminger, S. Mathot, T. Olenius, I. K. Ortega, A. Onnela, T. Petäjä, A. Praplan, M. P. Rissanen, T. Ruuskanen, F. D. Santos, S. Schallhart, R. Schnitzhofer, M. Simon, J. N. Smith, J. Tröstl, G. Tsagkogeorgas, A. Tomé, P. Vaattovaara, H. Vehkamäki, A. E. Vrtala, P. E. Wagner, C. Williamson, D. Wimmer, P. M. Winkler, A. Virtanen, N. M. Donahue, K. S. Carslaw, U. Baltensperger, I. Riipinen, J. Curtius, D. R. Worsnop and M. Kulmala, Nat. Commun., 2016, 7, 11594 CrossRef CAS PubMed.
- J. Tröstl, W. K. Chuang, H. Gordon, M. Heinritzi, C. Yan, U. Molteni, L. Ahlm, C. Frege, F. Bianchi, R. Wagner, M. Simon, K. Lehtipalo, C. Williamson, J. S. Craven, J. Duplissy, A. Adamov, J. Almeida, A.-K. Bernhammer, M. Breitenlechner, S. Brilke, A. Dias, S. Ehrhart, R. C. Flagan, A. Franchin, C. Fuchs, R. Guida, M. Gysel, A. Hansel, C. R. Hoyle, T. Jokinen, H. Junninen, J. Kangasluoma, H. Keskinen, J. Kim, M. Krapf, A. Kürten, A. Laaksonen, M. Lawler, M. Leiminger, S. Mathot, O. Möhler, T. Nieminen, A. Onnela, T. Petäjä, F. M. Piel, P. Miettinen, M. P. Rissanen, L. Rondo, N. Sarnela, S. Schobesberger, K. Sengupta, M. Sipilä, J. N. Smith, G. Steiner, A. Tomè, A. Virtanen, A. C. Wagner, E. Weingartner, D. Wimmer, P. M. Winkler, P. Ye, K. S. Carslaw, J. Curtius, J. Dommen, J. Kirkby, M. Kulmala, I. Riipinen, D. R. Worsnop, N. M. Donahue and U. Baltensperger, Nature, 2016, 533, 527–531 CrossRef PubMed.
- E. M. Dunne, H. Gordon, A. Kürten, J. Almeida, J. Duplissy, C. Williamson, I. K. Ortega, K. J. Pringle, A. Adamov, U. Baltensperger, P. Barmet, F. Benduhn, F. Bianchi, M. Breitenlechner, A. Clarke, J. Curtius, J. Dommen, N. M. Donahue, S. Ehrhart, R. C. Flagan, A. Franchin, R. Guida, J. Hakala, A. Hansel, M. Heinritzi, T. Jokinen, J. Kangasluoma, J. Kirkby, M. Kulmala, A. Kupc, M. J. Lawler, K. Lehtipalo, V. Makhmutov, G. Mann, S. Mathot, J. Merikanto, P. Miettinen, A. Nenes, A. Onnela, A. Rap, C. L. S. Reddington, F. Riccobono, N. A. D. Richards, M. P. Rissanen, L. Rondo, N. Sarnela, S. Schobesberger, K. Sengupta, M. Simon, M. Sipilä, J. N. Smith, Y. Stozkhov, A. Tomé, J. Tröstl, P. E. Wagner, D. Wimmer, P. M. Winkler, D. R. Worsnop and K. S. Carslaw, Science, 2016, 354, 1119–1124 CrossRef CAS PubMed.
- N. G. Adams, D. K. Bohme, D. B. Dunkin, F. C. Fehsenfeld and E. E. Ferguson, J. Chem. Phys., 1970, 52, 3133–3140 CrossRef CAS.
- M. E. Jacox and D. E. Milligan, J. Mol. Spectrosc., 1974, 52, 363–379 CrossRef CAS.
- J. T. Moseley, J. Chem. Phys., 1976, 65, 2512–2517 CrossRef CAS.
- R. A. Beyer and J. A. Vanderhoff, J. Chem. Phys., 1976, 65, 2313–2321 CrossRef CAS.
- D. E. Hunton, M. Hofmann, T. G. Lindeman and A. W. Castleman, J. Chem. Phys., 1985, 82, 134–150 CrossRef CAS.
- J. T. Snodgrass, C. M. Roehl, P. A. M. van Koppen, W. E. Palke and M. T. Bowers, J. Chem. Phys., 1990, 92, 5935–5943 CrossRef CAS.
- J. C. Bopp, E. G. Diken, J. M. Headrick, J. R. Roscioli, M. A. Johnson, A. J. Midey and A. A. Viggiano, J. Chem. Phys., 2006, 124, 174302 CrossRef PubMed.
- A. A. Viggiano, Phys. Chem. Chem. Phys., 2006, 8, 2557–2571 RSC.
- A. A. Viggiano, A. J. Midey and A. Ehlerding, Int. J. Mass Spectrom., 2006, 255, 65–70 CrossRef.
- G. Beig, S. Walters and G. Brasseur, J. Geophys. Res., 1993, 98, 12775 CrossRef.
- I. Dotan, J. A. Davidson, G. E. Streit, D. L. Albritton and F. C. Fehsenfeld, J. Chem. Phys., 1977, 67, 2874–2879 CrossRef CAS.
- N. Schoon, C. Amelynck, P. Bultinck and E. Arijs, Int. J. Mass Spectrom., 2002, 221, 209–218 CrossRef CAS.
- F. C. Fehsenfeld, C. J. Howard and A. L. Schmeltekopf, J. Chem. Phys., 1975, 63, 2835–2841 CrossRef CAS.
- O. Möhler and F. Arnold, J. Atmos. Chem., 1991, 13, 33–61 CrossRef.
- N. S. Shuman, D. E. Hunton and A. A. Viggiano, Chem. Rev., 2015, 115, 4542–4570 CrossRef CAS PubMed.
- I. Dotan, D. L. Albritton, F. C. Fehsenfeld, G. E. Streit and E. E. Ferguson, J. Chem. Phys., 1978, 68, 5414–5416 CrossRef CAS.
- M. L. Huertas and J. Fontan, Atmos. Environ., 1982, 16, 2521–2527 CrossRef CAS.
- H. Kawamoto and T. Ogawa, Planet. Space Sci., 1984, 32, 1223–1233 CrossRef CAS.
- K. Nagato, Y. Matsui, T. Miyata and T. Yamauchi, Int. J. Mass Spectrom., 2006, 248, 142–147 CrossRef CAS.
- J. Viidanoja, T. Reiner and F. Arnold, Int. J. Mass Spectrom., 1998, 181, 31–41 CrossRef CAS.
- J. Viidanoja, T. Reiner, A. Kiendler, F. Grimm and F. Arnold, Int. J. Mass Spectrom., 2000, 194, 53–68 CrossRef CAS.
- C. van der Linde, W. K. Tang, C.-K. Siu and M. K. Beyer, Chem. – Eur. J., 2016, 22, 12684–12687 CrossRef CAS PubMed.
- W. K. Tang, C. van der Linde, C.-K. Siu and M. K. Beyer, J. Phys. Chem. A, 2017, 121, 192–197 CrossRef CAS PubMed.
- C. Guimbaud, D. Labonnette, V. Catoire and R. Thomas, Int. J. Mass Spectrom., 1998, 178, 161–171 CrossRef CAS.
- J. Lengyel, J. Med, P. Slavíček and M. K. Beyer, J. Chem. Phys., 2017, 147, 101101 CrossRef PubMed.
- J. Lengyel, M. Ončák, J. Fedor, J. Kočišek, A. Pysanenko, M. K. Beyer and M. Fárník, Phys. Chem. Chem. Phys., 2017, 19, 11753–11758 RSC.
- J. Lengyel, C. van der Linde, A. Akhgarnusch and M. K. Beyer, Int. J. Mass Spectrom., 2017, 418, 101–106 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, 2016 Search PubMed.
- R. F. Höckendorf, O. P. Balaj, C. van der Linde and M. K. Beyer, Phys. Chem. Chem. Phys., 2010, 12, 3772–3779 RSC.
- P. Kofel, M. Allemann, H. Kellerhals and K. P. Wanczek, Int. J. Mass Spectrom. Ion Processes, 1986, 72, 53–61 CrossRef CAS.
- C. Berg, T. Schindler, G. Niedner-Schatteburg and V. E. Bondybey, J. Chem. Phys., 1995, 102, 4870–4884 CrossRef CAS.
- S. Maruyama, L. R. Anderson and R. E. Smalley, Rev. Sci. Instrum., 1990, 61, 3686–3693 CrossRef CAS.
- V. E. Bondybey and J. H. English, J. Chem. Phys., 1981, 74, 6978–6979 CrossRef CAS.
- T. G. Dietz, M. A. Duncan, D. E. Powers and R. E. Smalley, J. Chem. Phys., 1981, 74, 6511–6512 CrossRef CAS.
- D. de Jesus Medeiros and A. S. Pimentel, J. Phys. Chem. A, 2011, 115, 6357–6365 CrossRef PubMed.
- A. Akhgarnusch, R. F. Höckendorf and M. K. Beyer, J. Phys. Chem. A, 2015, 119, 9978–9985 CrossRef CAS PubMed.
- O. P. Balaj, C. B. Berg, S. J. Reitmeier, V. E. Bondybey and M. K. Beyer, Int.
J. Mass Spectrom., 2009, 279, 5–9 CAS.
- T. E. Cooper, J. T. O’Brien, E. R. Williams and P. B. Armentrout, J. Phys. Chem. A, 2010, 114, 12646–12655 CrossRef CAS PubMed.
- R. C. Dunbar, J. Phys. Chem., 1994, 98, 8705–8712 CrossRef CAS.
- R. C. Dunbar, Mass Spectrom. Rev., 2004, 23, 127–158 CrossRef CAS PubMed.
- R. C. Dunbar and T. B. McMahon, Science, 1998, 279, 194–197 CrossRef CAS.
- B. S. Fox, M. K. Beyer and V. E. Bondybey, J. Phys. Chem. A, 2001, 105, 6386–6392 CrossRef CAS.
- A. Kamariotis, O. V. Boyarkin, S. R. Mercier, R. D. Beck, M. F. Bush, E. R. Williams and T. R. Rizzo, J. Am. Chem. Soc., 2006, 128, 905–916 CrossRef CAS PubMed.
- E. N. Kitova, D. R. Bundle and J. S. Klassen, J. Am. Chem. Soc., 2002, 124, 5902–5913 CrossRef CAS PubMed.
- S. W. Lee, P. Freivogel, T. Schindler and J. L. Beauchamp, J. Am. Chem. Soc., 1998, 120, 11758–11765 CrossRef CAS.
- T. Schindler, C. Berg, G. Niedner-Schatteburg and V. E. Bondybey, Chem. Phys. Lett., 1996, 250, 301–308 CrossRef CAS.
- P. D. Schnier, W. D. Price, R. A. Jockusch and E. R. Williams, J. Am. Chem. Soc., 1996, 118, 7178–7189 CrossRef CAS PubMed.
- M. Sena and J. M. Riveros, Rapid Commun. Mass Spectrom., 1994, 8, 1031–1034 CrossRef CAS.
- D. Thölmann, D. S. Tonner and T. B. McMahon, J. Phys. Chem., 1994, 98, 2002–2004 CrossRef.
- R. L. Wong, K. Paech and E. R. Williams, Int. J. Mass Spectrom., 2004, 232, 59–66 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp07773d |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Wai Kit
Tang
*a,
Wai Kit
Tang