Effect of alkyl chain length on the properties of triphenylamine-based hole transport materials and their performance in perovskite solar cells†
Received
14th November 2017
, Accepted 8th December 2017
First published on 12th December 2017
Abstract
A new series of diacetylide-triphenylamine (DATPA) derivatives with five different alkyl chains in the para position, MeO, EtO, nPrO, iPrO and BuO, were synthesised, fully characterised and their function as hole-transport materials in perovskite solar cells (PSC) studied. Their thermal, optical and electrochemical properties were investigated along with their molecular packing and charge transport properties to analyse the influence of different alkyl chains in the solar cell parameters. The shorter alkyl chain facilitates more compact packing structures which enhanced the hole mobilities and reduced recombination. This work suggests that the molecule with the methoxy substituent (MeO) exhibits the best semiconductive properties with a power conversion efficiency of up to 5.63%, an open circuit voltage (Voc) of 0.83 V, a photocurrent density (Jsc) of 10.84 mA cm−2 and a fill factor of 62.3% in perovskite solar cells. Upon replacing the methoxy group with longer alkyl chain substituents without changing the energy levels, there is a decrease in the charge mobility as well as PCE (e.g. 3.29% for BuO-DATPA). The alkyl chain length of semiconductive molecules plays an important role in achieving high performance perovskite solar cells.
1 Introduction
In recent years, organic–inorganic halide perovskites have generated tremendous interest from the academic community, showing rapid improvements in their performance as absorbers in photovoltaic solar cells. Starting from 3.8% in 2009 by Miyasaka1 to a current certified record efficiency of 22.1%,2,3 perovskite-based solar cells have surpassed values of the commercialised polycrystalline silicon solar cells and are currently competitive with the established thin-film technologies. This rapid development is mainly the product of the remarkable properties of the perovskite absorber materials, e.g. MAPbX3 (MA = methylammonium; X = I, or mixed I/Br, I/Cl), such as high panchromatic absorption, large carrier diffusion length, low non-radiative recombination and easy processability, leading to great potential for low cost and large scale technologies.4–6 Different configurations for photovoltaic devices have been proposed including mesoscopic structures, planar layers and HTM-free devices. Nevertheless, the most-common standard architectures for these devices comprise six main constituents: (1) a conductive glass substrate FTO (fluorine tin oxide), (2) a compact TiO2 blocking layer, (3) mesoporous TiO2 (mp-TiO2), (4) a perovskite layer, (5) a hole transport layer and (6) gold as counter electrode. One of the main components in these devices is the hole-transporting semiconductor either in the form of a π-conjugated polymer,7,8 an inorganic salt9–11 or small organic molecule. Although many different materials have been explored, most attention has focused on small molecules due to their simplicity, ease of purification and batch-to-batch consistency. Spiro-OMeTAD12 is the most common hole transport material (HTM) in perovskite solar cells showing high conversion efficiencies. Yet, spiro-OMeTAD is expensive due to its lengthy synthetic procedure and problematic purification. To address these limitations, researchers have explored alternative HTMs structures with different motifs such as carbazole,13,14 triarylamine derivatives,15–17 spiro-based derivatives,18–20 triazatruxenes,21,22 azulenes,23 and other related examples,24,25 leading to efficiencies comparable with spiro-OMeTAD and progress on the preparation methods and cost. However, to date spiro-OMeTAD remains the preferred choice.
Triarylamines (TAA)26 are widely used as a result of the easy oxidation of the nitrogen centre and their capacity to transport positive charges via radical cations. In previous work reported by our group, a series of triphenylamine-based HTMs having two diacetylide-triphenylamine (DATPA)27 moieties and varying redox potentials was prepared by a simple synthetic route which allows easy tuning of electrochemical and other properties through varying the substituent R groups. These materials were studied as HTMs in PSCs and showed promising properties in comparison with spiro-OMeTAD. The chemical structure of the HTMs can significantly affect the hole transfer kinetics between the HTM and the perovskite and the HTM and the metal contact which affect the performance of the solar cell.28,29 The length of alkyl chain for instance, can influence the solubility, molecular ordering and charge transport of the HTMs thus affecting the performance of the solar cell.30,31
 |
| | Fig. 1 Chemical structure of HTMs used in this study. Each molecule is comprised of two bridged Ph3N moieties. | |
Although the large majority of triarylamine HTMs include methoxy groups to tune the redox potential, little research has been done to study the effect of the alkyl chain on the properties of the HTM. In previous studies Hagfeldt30 and co-workers demonstrated the importance of the substituents by tuning the position and length of the alkyl chain in four triphenylamine-based organic HTMs. In a different study, Nazeeruddin32 and co-workers studied the influence of alkyl chain length on the power conversion efficiency of a series of hole transport material. Such reports are rare however, and there are still limited comprehensive studies of the effect of alkyl chain length on material and device properties. In the case of organic photovoltaics, studies have shown both increases and decreases in power-conversion efficiency through increasing the length of alkyl chains,31,33–35 therefore greater systematic understanding of this effect for HTMs in PSCs is important Accordingly, we report a series of new diacetylide-triphenylamine substituents HTM coded as EtO-DATPA, iPrO-DATPA, nPrO-DATPA and BuO-DATPA with similar optical and electrochemical properties to the MeO-DATPA studied before.27 We have systematically investigated the effect of varying the alkyl chain in structurally-similar HTMs on material properties and solar cell performance parameters (Fig. 1).
2 Results and discussion
2.1 Synthesis
The synthetic route (Scheme 1) reported for MeO-DATPA in our previous work27 was revised and optimised to improve yields and reduce reactions times. Detailed experimental procedures are described in the ESI.† Apart from EtO-DATPA, the method of synthesis involved five stages. The first step is a simple SN2 reaction, however for EtO-DATPA 1-ethoxy-4-iodobenzene is commercially available, and this step was omitted. The triarylamine moieties were formed with a Copper Catalysed Ullmann36 coupling reaction, and subsequently the acetylene was attached with a silyl protecting group via Sonogashira cross-coupling.37 The silyl group was removed using TBAF (tetrabutylammonium fluoride), leaving the compound with a terminal alkyne. The final product was obtained by a copper-mediated oxidative homocoupling38 reaction at room temperature. The analytical and spectroscopic data for all HTMs are consistent with the formulated structures.
 |
| | Scheme 1 Synthetic procedure for all HTMs. Reaction conditions: (a) K2CO3, 18-crown-6, R-Br, DMF, 100 °C, 4 h, N2 (b) CuI, 1,10-phenantroline, KOH, MePh, 4-bromoaniline, N2, 120 °C; (c) C8H16Si, CuI, piperidine, (PPh3)2PdCl2, PPh3, PhMe, N2, 90 °C; (d) 1 M TBAF, DCM, rt, N2; (e) CuCl, TMEDA, molecular sieves, DCM, air. | |
2.2 Optical and electrochemical properties
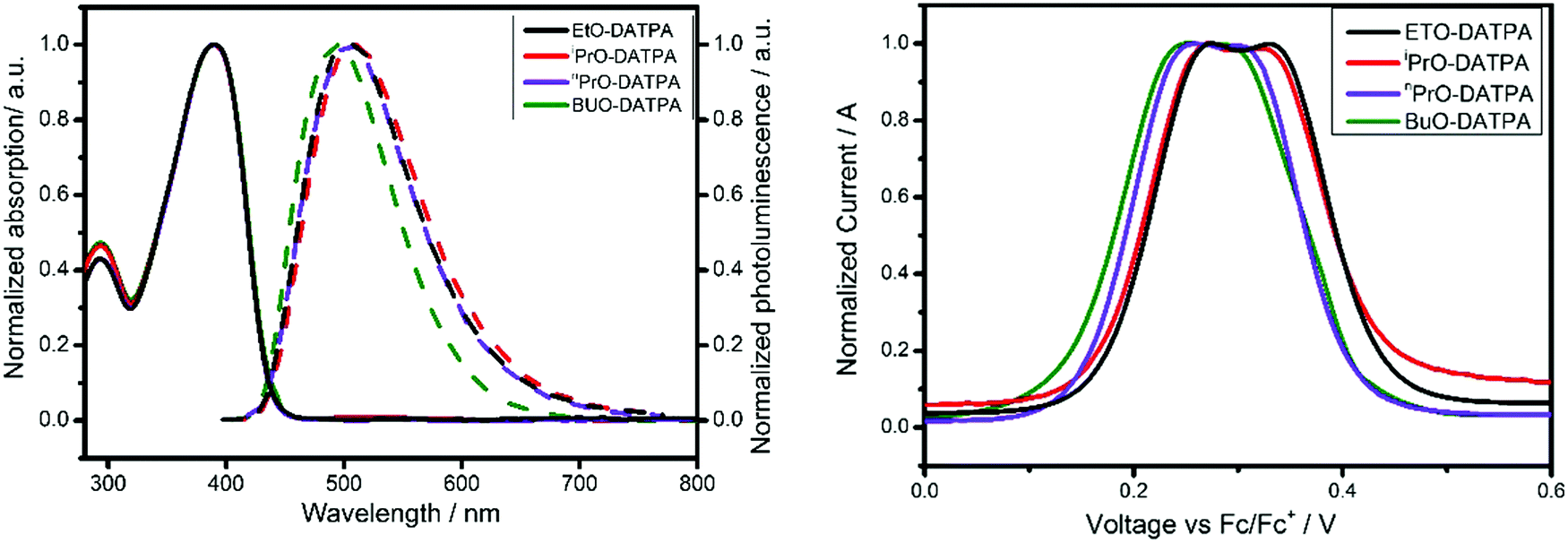
The UV-vis absorption spectra of the DATPA derivatives (EtO, nPrO, iPrO, BuO) measured in dichloromethane (DCM), are shown in Fig. 2 (left, solid line). All compounds exhibit an absorption peak at 389 nm independent of the substituent. Molar extinction coefficients were calculated for each compound using the Lambert–Beer law and results are displayed in Table 1. Additionally, photoluminescence (PL) spectra were recorded (dashed line, Fig. 2 left). The results show similar PL spectra for all DATPA derivatives with a maximum emission at 493 nm.
 |
| | Fig. 2 Left: Normalized UV-vis absorption (solid line) and emission (dashed line) of each DATPA derivative. EtO-DATPA (black line), iPrO-DATPA (red line), nPrO-DATPA (purple line) and BuO-DATPA (green line). Right: Square-wave voltammetry of EtO-DATPA (black line), iPrO-DATPA (red line), nPrO-DATPA (purple line) BuO-DATPA (green line). | |
Table 1 Summary of the optical and electrochemical properties
| HTM |
λ
max (nm) |
ε (cm−1 M−1) |
λ
em
(nm) |
E
gap
(V) |
E
ox
(V) |
E
HOMO
(eV) |
|
Excitation at λmax.
E
gap is the optical gap determined from the intersection of absorption and emission spectra.
From SWV and CV measurements and referenced to ferrocene.
E
HOMO(eV) = −5.1 − (Eox).42
|
| EtO-DATPA |
389 |
60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
504 |
2.85 |
+0.28 |
−5.38 |
|
n
PrO-DATPA |
389 |
69200 |
504 |
2.85 |
+0.26 |
−5.36 |
|
iPrO-DATPA |
389 |
64280 |
508 |
2.84 |
+0.28 |
−5.38 |
| BuO-DATPA |
389 |
65990 |
495 |
2.85 |
+0.26 |
−5.36 |
| Spiro-OMETAD |
385 |
— |
424 |
3.05 |
+0.03 |
−5.13 |
Optical gaps were determined from the intersection of the excitation and the emission spectra. Cyclic voltammograms (CV) of the DATPA series are displayed in Fig. S5 (ESI†). The redox peaks of all HTMs are chemically and electrochemically reversible, indicating excellent stability and rapid electron transfer. These materials present two oxidation peaks corresponding to one electron oxidation for each triarylamine unit. However, in cyclic voltammetry, the two oxidation process are strongly overlapped because of their similar potentials. To distinguish both processes, square-wave voltammetry (SWV) was carried out (Fig. 2, right). The similarity of the two redox potentials suggests a low interaction between the two triarylamines units. The HOMO energy level is of most importance for hole transfer from the perovskite and HTM and from molecule to molecule. The HOMO energy levels were calculated from the CV data using the following equation EHOMO = −5.1 − (Eox), where Eox is the oxidation potential of the HTM with reference to ferrocene. The extracted values are listed in Table 1. Overall these results indicate that the hole transfer from CH3NH3Pbl3 to the HTM is energetically favourable. Spiro-OMeTAD has a HOMO of −5.13 eV, which it is at higher energy than the materials reported here.
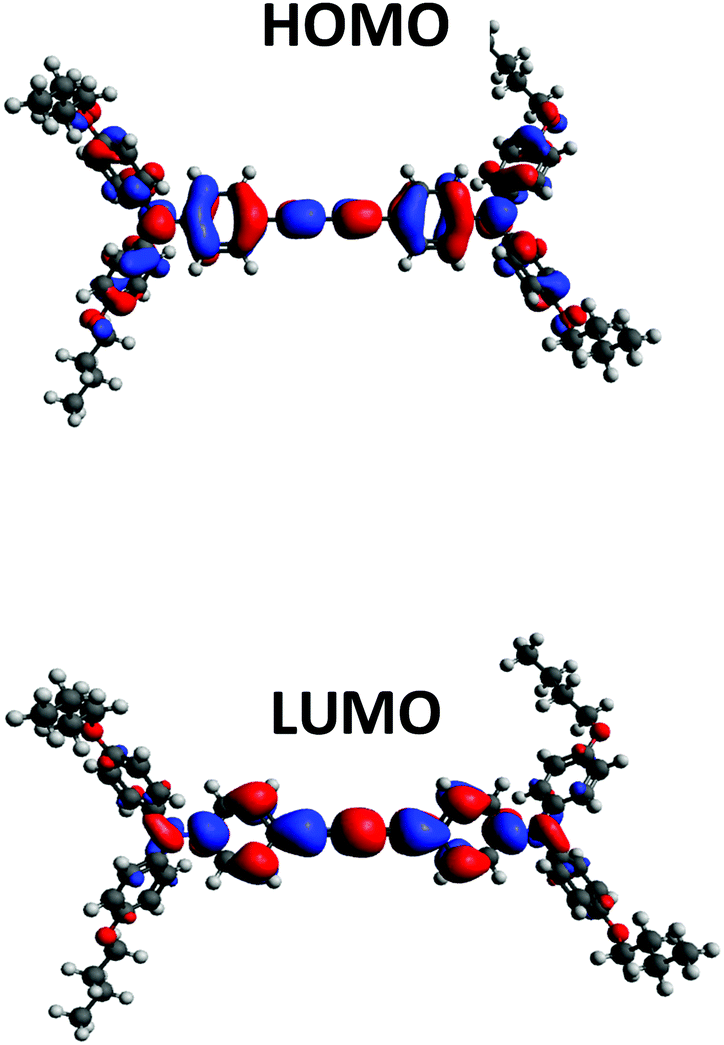
Density Functional Theory (DFT) calculations were performed to predict the electronic properties of all HTMs using Gaussian 09 with B3LYP 6-31G(d) level of theory. Fig. S1 (ESI†) shows the position of the HOMO–LUMO energy levels and their electron density on the molecules of all DATPA derivatives. The calculated trend of HOMO and LUMO matches the experimental data, and no significant difference was found among the DATPA series. The HOMO (Fig. 4) is delocalized over the π orbitals of the triphenylamine unit and the diacetylene bridge. The alkyl groups do not contribute to the HOMO energy level; therefore any change beyond the oxygen does not affect the optoelectronic properties, which coincides with the electrochemistry results. The LUMO electron density is localised on the diacetylene bridge and is also not significantly influenced by the substituents of the triphenylamine unit. A summary of the optical and electrochemical properties of all DATPA derivative is presented in Table 1. As expected, different alkyl substituents do not significantly modify the optical and electrochemical properties of the hole transport materials. This confirms that this series is appropriate to compare the structural, morphological and interface effects of differing alkyl chains without any complicating changes to the optoelectronic properties.
2.3 XRD analysis
Single crystals of the new molecules were grown by the slow solvent evaporation method to determine the crystal packing (Fig. 3) by X-ray diffraction (XRD). The compounds were found to crystallize in the monoclinic space group P21/n (EtO-DATPA, nPrO-DATPA), monoclinic space group P21/c (BuO-DATPA) with half of the formula unit present in the asymmetric unit and the other half consisting of symmetry equivalent atoms; iPrO-DATPA crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) showing two independent molecules in the asymmetric unit (Fig. S3, ESI†). In every case the molecule crystallises without the presence of any solvent within the unit cell. The EtO-DATPA molecules are packed in a layer-by-layer configuration with molecules arranged in a herringbone motif characterised by edge-to-face contacts. The intersection angle between two adjacent fragments is measured to be 72.18°. The distance between parallel fragments is too large to induce co-facial π⋯π interactions. The molecules interact mainly via C–H⋯π (3.154 Å) and C–H⋯HC formed by the CH groups of phenyls (dCH⋯HC = 2.554 Å, 2.634 Å). The EtO-DATPA molecules also form intermolecular H-bonding interactions in the crystal lattice formed by the oxygen and the hydrogen atom of the aromatic rings (dC–H⋯O–R = 2.715 Å, 2.804 Å, 2.916 Å). The crystal structure of nPrO-DATPA also exhibits a layer-by-layer herringbone pattern. The T-shaped edge-to-face and the parallel-displaced stacking arrangement predominate. Although similar, the structure of nPrO-DATPA is stabilised via π⋯π interactions. The distance between two parallel fragments and the intersection angle of adjacent molecules were measured to be 3.44 Å and 46.63° respectively. The nPrO-DATPA molecules also interact via C–H⋯π (3.086 Å) and C–H⋯O–R (shortest distance found 2.725 Å). A more complex packing is observed for iPrO-DATPA. The X-ray crystal structure reveals three crystallographically unique molecules in the asymmetric unit. Two lie on crystallographic inversion centres while the third lies entirely in general positions. Each unit is surrounded by six molecules arranged in an alternating layered pattern with an intersection angle of 77.59°. The interactions arising in the structure of iPrO-DATPA are mainly C–H⋯π (3.034 Å, 2.813 Å, and 2.875 Å) and C–H⋯O–R (shortest distance found 2.712 Å). Furthermore, C–H⋯O–R contact is also observed between the CH group of the alkyl chain and the oxygen atom (2.788 Å). The aromatic cores of the iPrO-DATPA are aligned with a very strong slipped π-stacking configuration such that no π⋯π interactions are observed. In the case of BuO-DATPA, the molecules are arranged in a zig-zag largely slipped stack packing. Similar to iPrO-DATPA, the aromatic cores have a very strong slipped π-stacking with an average distance of 4.820 Å which it is too large to induce π-orbital overlap in the crystalline structure. The bulkiness of the alkyl chain in the para position largely impacts the overlapping of the aromatic moieties resulting in the absence of face to face stacking. The crystal lattice shows C–H⋯O–R (2.665 Å), C–H⋯HC (2.270 Å, 2.304 Å) formed between one hydrogen atom of the alkyl chain and CH group of an aromatic ring. The molecules also interact via C–H⋯π (2.720 Å) formed between an aromatic centroid and the alkyl chain. The crystal packing of MeO-DATPA was reported and discussed in more detail in our previous work,27 showing a herringbone pattern, with one molecule in the asymmetric unit and a regular stacking distance between the molecules. Overall, it was observed that as the alkyl chain increases, the number of van der Waals interactions between alkyl chains also increases while the H-bonding contacts decrease. Among all of the examples, MeO-DATPA and EtO-DATPA showed the strongest and highest number of intermolecular interactions (H-bonding) while fewer H-bonds were found in the crystal lattice of BuO-DATPA but stronger van der Waals interactions within the alkyl chains. Longer alkyl chains result in the poorer stacking of the molecule and therefore weaker intermolecular interactions. Furthermore, it is known that closer molecular arrangements can facilitate charge transport in thin film devices.39 The X-ray powder diffraction patterns were experimentally obtained and compared with those calculated from their single-crystal data (Fig. S6 and S7, ESI†). The powder pattern of all DATPA derivatives showed some crystallinity, and were in agreement with the same phase as the calculated pattern from single-crystal data. A summary of the crystallographic data is depicted in the Table S4 (ESI†).
showing two independent molecules in the asymmetric unit (Fig. S3, ESI†). In every case the molecule crystallises without the presence of any solvent within the unit cell. The EtO-DATPA molecules are packed in a layer-by-layer configuration with molecules arranged in a herringbone motif characterised by edge-to-face contacts. The intersection angle between two adjacent fragments is measured to be 72.18°. The distance between parallel fragments is too large to induce co-facial π⋯π interactions. The molecules interact mainly via C–H⋯π (3.154 Å) and C–H⋯HC formed by the CH groups of phenyls (dCH⋯HC = 2.554 Å, 2.634 Å). The EtO-DATPA molecules also form intermolecular H-bonding interactions in the crystal lattice formed by the oxygen and the hydrogen atom of the aromatic rings (dC–H⋯O–R = 2.715 Å, 2.804 Å, 2.916 Å). The crystal structure of nPrO-DATPA also exhibits a layer-by-layer herringbone pattern. The T-shaped edge-to-face and the parallel-displaced stacking arrangement predominate. Although similar, the structure of nPrO-DATPA is stabilised via π⋯π interactions. The distance between two parallel fragments and the intersection angle of adjacent molecules were measured to be 3.44 Å and 46.63° respectively. The nPrO-DATPA molecules also interact via C–H⋯π (3.086 Å) and C–H⋯O–R (shortest distance found 2.725 Å). A more complex packing is observed for iPrO-DATPA. The X-ray crystal structure reveals three crystallographically unique molecules in the asymmetric unit. Two lie on crystallographic inversion centres while the third lies entirely in general positions. Each unit is surrounded by six molecules arranged in an alternating layered pattern with an intersection angle of 77.59°. The interactions arising in the structure of iPrO-DATPA are mainly C–H⋯π (3.034 Å, 2.813 Å, and 2.875 Å) and C–H⋯O–R (shortest distance found 2.712 Å). Furthermore, C–H⋯O–R contact is also observed between the CH group of the alkyl chain and the oxygen atom (2.788 Å). The aromatic cores of the iPrO-DATPA are aligned with a very strong slipped π-stacking configuration such that no π⋯π interactions are observed. In the case of BuO-DATPA, the molecules are arranged in a zig-zag largely slipped stack packing. Similar to iPrO-DATPA, the aromatic cores have a very strong slipped π-stacking with an average distance of 4.820 Å which it is too large to induce π-orbital overlap in the crystalline structure. The bulkiness of the alkyl chain in the para position largely impacts the overlapping of the aromatic moieties resulting in the absence of face to face stacking. The crystal lattice shows C–H⋯O–R (2.665 Å), C–H⋯HC (2.270 Å, 2.304 Å) formed between one hydrogen atom of the alkyl chain and CH group of an aromatic ring. The molecules also interact via C–H⋯π (2.720 Å) formed between an aromatic centroid and the alkyl chain. The crystal packing of MeO-DATPA was reported and discussed in more detail in our previous work,27 showing a herringbone pattern, with one molecule in the asymmetric unit and a regular stacking distance between the molecules. Overall, it was observed that as the alkyl chain increases, the number of van der Waals interactions between alkyl chains also increases while the H-bonding contacts decrease. Among all of the examples, MeO-DATPA and EtO-DATPA showed the strongest and highest number of intermolecular interactions (H-bonding) while fewer H-bonds were found in the crystal lattice of BuO-DATPA but stronger van der Waals interactions within the alkyl chains. Longer alkyl chains result in the poorer stacking of the molecule and therefore weaker intermolecular interactions. Furthermore, it is known that closer molecular arrangements can facilitate charge transport in thin film devices.39 The X-ray powder diffraction patterns were experimentally obtained and compared with those calculated from their single-crystal data (Fig. S6 and S7, ESI†). The powder pattern of all DATPA derivatives showed some crystallinity, and were in agreement with the same phase as the calculated pattern from single-crystal data. A summary of the crystallographic data is depicted in the Table S4 (ESI†).
 |
| | Fig. 3 Crystal packing of all DATPA derivatives: (a) EtO-DATPA, (b) iPrO-DATPA, (c) nPrO-DATPA and (d) BuO-DATPA. | |
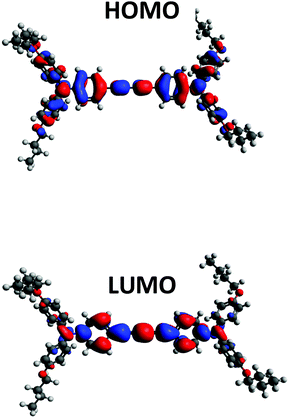 |
| | Fig. 4 Molecular orbital distribution for BuO-DATPA at B3LYP/6-31G(d) level of theory. | |
2.4 Thermal properties
Thermal properties of the HTMs were evaluated by Differential Scanning Calorimetry (DSC), and the results are presented in the ESI† (Fig. S9 and S10) The DSC results showed a glass transition temperature (Tg) of 69.5 °C and 70.7 °C for EtO-DATPA and iPrO-DATPA. nPrO-DATPA and BuO-DATPA however do not present Tg. On the other hand, lower melting points were observed for longer alkyl chains. These observations can be explained by the difference in the crystal packing, and their intermolecular interaction observed in the XRD analysis. Longer alkyl chains presented poorer stacking as the molecules are more disordered which results in weaker interactions. This explains why the BuO group showed the lowest melting point. The MeO and EtO groups have the highest melting points and the strongest intermolecular interactions. We can also observe that branched iPrO has a higher melting point than the straight-chain nPrO, although nPrO-DATPA (2080 Å3 per molecule) has a more compact structure than iPrO-DATPA (2213 Å3 per molecule). The iPrO chain typically has a smaller surface area which leads to weaker van der Waals interactions. However as we can observe in the crystal packing, iPrO shows shorter C–H⋯O–R interactions, which likely explains the higher melting point in comparison to nPrO.
2.5 Charge transport characteristics
Charge mobility is an important parameter to evaluate the properties of hole-transport materials in photovoltaic devices. To investigate the hole mobilities, OFETs (Organic Field-Effect Transistors)40 were fabricated for four of the compounds reported here (MeO-DATPA, EtO-DATPA, nPrO-DATPA and BuO-DATPA) and the mobilities were extracted from the saturation regime of the transfer characteristic curves (Fig. S8, ESI†). Details of the device fabrication and measurement are found in the Experimental section. It was not possible to fabricate a high-quality thin film for iPrO-DATPA. In general, the molecules with the shorter alkyl chains presented higher mobilities which can be attributed to stronger intermolecular interactions in the XRD analysis. A summary of the estimated hole mobilities is presented in Table 2.
Table 2 Charge transport and thermal properties
| HTM |
Hole mobility μFEa (cm2 V−1 s−1) |
T
g
(°C) |
T
m
(°C) |
|
Estimated from OFET measurements.
Determined from differential scanning calorimetry (DSC).
|
| MeO-DATPA |
1.90 × 10−4 |
76.9 |
197.3 |
| EtO-DATPA |
1.45 × 10−4 |
69.5 |
149.4 |
|
iPrO-DATPA |
— |
70.7 |
160.8 |
|
n
PrO-DATPA |
1.11 × 10−4 |
— |
148.0 |
| BuO-DATPA |
8.81 × 10−5 |
— |
118.9 |
2.6 Transient absorption spectroscopy
To further probe the performance in PSCs, the charge transfer dynamics occurring in an mp-TiO2/CH3NH3PbI3/HTM arrangement for all DATPA derivatives were obtained by ultrafast absorption spectroscopy (TAS). Samples were prepared by spin-coating of the perovskite onto a mesoporous TIO2 layer and with the HTM on top. Details of the sample preparation and measurements are described in the Experimental section. The corresponding steady-state absorption spectra are shown in Fig. S11b (ESI†), where all samples show the dominant spectral features of the perovskite layer CH3NH3PbI3, with the onset at 800 nm and the bands corresponding to the oxidised HTMs (at 1600 nm). Fig. S11a (ESI†) shows the TAS time profiles following 510 nm excitation probed at 1600 nm. All signals have a negative feature that arises from some charge separation already happening in the ground state, and the intensity of these bleaches correlates with the intensity of the band of the oxidised HTMs observed in the steady-state absorption spectra. The analysis of the normalised TAS time profiles also displays a hint of the lifetimes of the charge-separated states. From the results, MeO-DATPA presents the most long-lived signal indicating a greater charge-pair separation. Among the other DATPA derivatives, no significant differences of the lifetimes of the charge separation states could be resolved, albeit all were shorter than for MeO-DATPA.
2.7 Solar cell studies
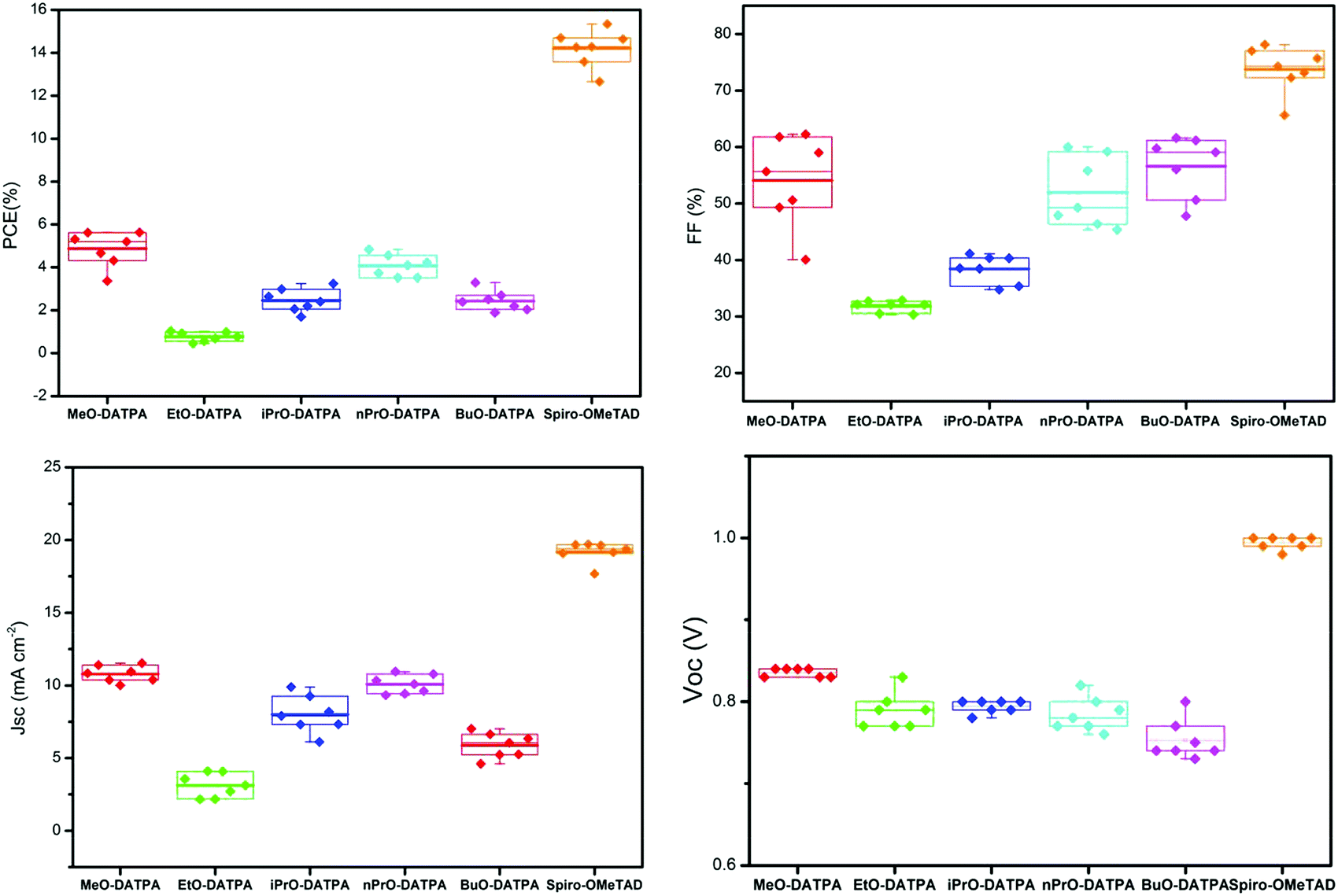
To investigate the effect of the alkyl chain of these HTMs in perovskite solar cells, we prepared a set of PSCs in the configuration FTO/bl-TiO2/mp-TiO2/CH3NH3PbI3/HTM/Au. All DATPA HTMs were doped using two times the concentration of additives typically used for spiro-OMeTAD12 which include bis(trifluoromethylsulfonyl)imide lithium salt (Li-TFSI), 4-tert-butylpyridine (tBP) and tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)-cobalt(III)tris(bis(trifluoromethylsulfonyl)imide) (FK209). Full details of the solar cell fabrication are given in the Experimental section. Current–voltage scans were recorded using an AAA-rated solar simulator calibrated against a KG5 global (AM 1.5G) solar irradiation. All devices were fabricated in a single continuous study over 7 repeat cells for each HTM to facilitate comparison. Fig. 6 shows the box plots with the mean and standard deviation of the solar cell parameters and a summary of the data can be found in Table S12 (ESI†). The J–V graphs of the champion cells are shown in Fig. 5 and the results are summarised in Table 3. Despite the identical backbone structures and HOMO energy levels, the difference in the alkyl chain length had an effect on the photovoltaic device performances. Among the DATPA series, MeO-DATPA shows the highest performance with a PCE of 5.63%. Upon increasing the length of alkyl chain the performance of the devices decreases to 4.83% for nPrO-DATPA, 3.24% for iPrO-DATPA and 2.54% for BuO-DATPA. Also noticeable is the decrease in the short circuit current values (Jsc) from 10.84 mA cm−2 for MeO-DATPA to 7.02 mA cm−2 for BuO-DATPA. These trends are in broad agreement with mobility and structural packing trends, with the exception of EtO-DATPA which gives unexpectedly poor cells. Since the HTMs in the device are doped, it is perhaps unsurprising to see some deviation from the expected trend since the morphology of the doped materials will also play a role in the final properties. The methoxy substituent presents the best semiconductive properties in comparison with the longer alkyl chains. The variance in PCE values can be attributed to the differences in the photocurrent density and fill factor which may have resulted from the higher hole mobility, greater charge-pair separation and optimal morphology. This is sustained by the results from the hole mobility data, transient absorption spectroscopy studies and XRD analyses discussed previously.
 |
| | Fig. 5
J–V curves (light and dark) of the champion PSCs with the DATPA series and spiro-MeOTAD HTMs. | |
 |
| | Fig. 6 Solar cells parameters over 7 repeats for each HTM. | |
Table 3 Summary of device performance for champion cells
| HTM |
PCE (%) |
J
sc (mA cm−2) |
V
oc (V) |
FF (%) |
| MeO-DATPA |
5.63 |
10.84 |
0.83 |
62.25 |
| EtO-DATPA |
1.02 |
4.10 |
0.77 |
32.19 |
|
n
PrO-DATPA |
4.83 |
10.33 |
0.79 |
59.22 |
|
iPrO-DATPA |
3.24 |
9.89 |
0.80 |
41.11 |
| BuO-DATPA |
3.29 |
7.02 |
0.77 |
61.19 |
| Spiro-OMeTAD |
15.34 |
19.67 |
1.00 |
78.13 |
3 Conclusions
In summary, we have presented the synthesis and characterisation of a new series of diacetylide-triphenylamine (DATPA) derivatives to study their function in PSCs. These molecules have the same backbone structures but different alkyl chain length in the para position, i.e., ethoxy (EtO), propoxy (nPrO), isopropoxy (iPrO) and butoxy (BuO). Although the length of the aliphatic side chain does not influence the optoelectronic properties of the molecules such as the HOMO levels, it strongly affects the charge transport properties and transfer dynamics of the molecules as well as the morphology and photovoltaic performance in PSCs. The molecules with shorter alkyl chains have a more ordered and compact structure which facilitates charge transport in the thin films. These results are in agreement with the previous work found by Hagfeldt41 and Nazeeruddin. The methoxy substituent shows the best semiconductive properties and PCE of 5.63% compared with the longer side chain. This is due to the molecular packing and faster charge transport. This work provides insight on the importance of the alkyl chain length in the design of organic semiconductors.
4 Experimental details
4.1 Chemical characterization
1H and 13C NMR spectra were recorded on a Brucker Advance 500 spectrometer (500 MHz). The deuterated solvents are indicated in the synthesis description. Chemical shifts, δ, are given in ppm, using the solvent residual as an internal standard. MS were recorded on Micro-Tof using electrospray ionisation (ESI) technique. Elemental analyses were carried out by Stephen Boyer at London Metropolitan University.
4.2 Optical characterization
Solution UV-visible absorption spectra were recorded using a Jasco V-670 UV/Vis/NIR spectrometer controlled with SpectraManager software. Photoluminescence (PL) spectra were recorded with a Fluoromax-3 fluorimeter controlled by ISAMain software. All samples were measured in a 1 cm cell at room temperature in dichloromethane as solvent. Concentrations of 2.5 × 10−5 M and 1 × 10−6 M were used for UV/Vis and PL respectively.
Crystallographic details.
Powder diffraction was performed on a Bruker Discover D8 with CuKa1/2 source and a scintillation detectior 4860.
Single yellow block-shaped crystals of EtO-DATPA were recrystallised from benzene by slow evaporation. Single orange block-shaped crystals of iPrO-DATPA were recrystallised from benzene by slow evaporation. Single yellow plate-shaped crystals of nPrO-DATPA were recrystallised from a mixture of DCM and hexane by slow evaporation. Single orange block-shaped crystals of BuO-DATPA were recrystallized from benzene by slow evaporation. A suitable crystal of EtO-DATPA (0.24 × 0.16 × 0.14), iPrO-DATPA (0.51 × 0.23 × 0.10), nPrO-DATPA (0.55 × 0.29 × 0.09) and BuO-DATPA (0.25 × 0.24 × 0.14) were selected and mounted on a MITIGEN holder in Paratone oil on a Rigaku Oxford Diffraction SuperNova diffractometer. The crystal was kept at T = 120.0 K during data collection. Using Olex2 (Dolomanov et al., 2009), the structure was solved with the ShelXS (Sheldrick, 2008) structure solution program, using the Direct Methods solution method. The model was refined with version of ShelXL (Sheldrick, 2008) using Least Squares minimisation. A summary of the data collection and structure refinement is reported in Table S4 (ESI†).
4.3 Thermal characterization
Differential scanning calorimetry (DSC) was performed on NETZSCH STA 449F1 at a scan rate of 5 K min−1 under a nitrogen atmosphere in DSC/TG aluminium pan. The measurement range was 25 °C to 250 °C.
4.4 Electrochemical characterization
All cyclic voltammetry measurements were carried out in freshly distilled CH2Cl2 using 0.3 M [TBA][BF4] electrolyte in a three-electrode system with each solution being purged with N2 prior to measurement. The working electrode was a Pt disk, the reference electrode was Ag/AgCl, and the counter electrode was a Pt rod. All measurements were made at room temperature using a μUTOLAB Type III potentiostat, driven by the electrochemical software GPES. Cyclic voltammetry (CV) measurements used scan rates of 100 mV s−1: square wave voltammetry (SWV) was carried out at a step potential of 4 mV, square wave amplitude of 25 mV, and a square wave frequency of 10 Hz, giving a scan rate of 40 mV s−1. Ferrocene was used as the internal standard in each measurement.
4.5 Computational details
All calculations were carried out using the Gaussian 09 program with Lee–Yang–Parr correlation functional (B3LYP) level of theory. All atoms were described by the 6-31G(d) basis set. All structures were input and processed through the Avogadro software package.
4.6 Organic field-effect transistors
The Ossila low-density pre-fabricated substrates (channel length 0.1 mm width 0.03 mm) with a bottom gate/bottom contact were used to fabricate OFET devices. The substrates were treated with HMDS (hexamethyldisilazane) to optimise the silicon surface property. A solution of the HTM (5 mg ml−1) in dichlorobenzene was stirred at room temperature. The Ossila substrate was covered with the solution by drop-casting. The electric characteristics of the fabricated OFETs were measured using a Keithley 2612A System SourceMeter. Mobilities were calculated using the following equation
where Z is the channel width, L the channel length, Ci the capacitance, VG the gate voltage and IDS is the drain current and  , the slope of the transfer characteristic curves in the saturation regime.40
, the slope of the transfer characteristic curves in the saturation regime.40
4.7 Transient absorption spectroscopy
Sample preparation.
A solution of mesoporous TiO2 in terpineol (Dyesol 30 nm TiO2, weight ratio 1:2) was spin coated on a 10 mm by 10 mm glass square, with an acceleration of 12000 rpm for 30 s. The pieces of glass were cleaned with isopropanol (IPA) in a sonicator for 5 minutes before spin coating. After spin coating, the substrates were placed into a 450 °C oven for 1 hour and cooled down to room temperature before deposition of the perovskite layer. For the deposition of the perovskite layer 1 M solution of PbI2 and MeNH3I in DMSO was prepared. This solution was spin-coated onto the glass slides covered with the mesoporous oxide via a 3-step spin coating process: (i) 1000 rpm, 10 s, 2000 acc; (ii) 5000 rpm, 20 s, 2000 acc; (iii) 6000 rpm, 20 s, 2000 acc. Toluene (300 μL) was dropped on the substrates by the end of the second step. The films were then annealed at 50 °C for 20 min and at 100 °C for 25–30 min and cooled down to room temperature. A 20 mg ml−1 solution in chlorobenzene of the corresponding HTM was spin-coated onto the perovskite layer at 200 rpm for 30 s, with an acceleration of 2000.
Measurements.
UV-Vis was performed on a PerkinElmer UV/VIS Spectrometer Lambda 25. Photoluminescence spectra were recorded on a Horiba Yobin-Ybon Fluorolog-3 spectrofluorometer, using an excitation wavelength of 450 nm and slit widths of 10 nm. For pump–probe micro to millisecond transient absorption spectroscopy, films were excited by a dye laser (Photon Technology International GL-301, sub-nanosecond pulse width) pumped by a pulsed nitrogen laser (Photon Technology International GL-3300). A quartz halogen lamp (Bentham IL1) was passed through a monochromator and used to probe changes in the absorption characteristics of the film as a function of time after the laser excitation. The probe light was detected using home-built silicon (≤1000 nm) or InxGa1−xAs (>1000 nm) photodiodes and an oscilloscope. Unless otherwise stated, films were kept under flowing N2 during the measurements. All micro to millisecond transient absorption spectroscopy measurements were conducted employing 450 nm laser pulses (25 μJ cm−2).
4.8 Perovskite solar cells and characterisation
Etched FTO glass substrates (NSG Pilkington, TEC7) were cleaned sequentially in detergent, deionised water, acetone and ethanol before undergoing 10 minutes of O2 plasma treatment. A compact TiO2 layer was deposited on the glass substrates through spray pyrolysis of a 0.2 M solution of titanium diisopropoxide bis(acetylacetonate) in isopropanol at 450 °C. Upon cooling, a mesoporous layer of TiO2 nanoparticles was spin-coated from a 2:7 wt suspension of Dyesol 30NR-D paste in ethanol (4500 rpm for 30 seconds), followed by sintering at 550 °C for 30 minutes. A CH3NH3PbI3 perovskite precursor solution was prepared by dissolving 576 mg PbI2, and 199 mg CH3NH3I in a 4:1 vol solution of DMF:DMSO. 100 μl of the perovskite precursor solution was deposited onto the TiO2 films and spin-coated at 4000 rpm for 30 seconds, with 200 μl of ethyl acetate dripped onto the spinning substrate 10 seconds prior to the end of the spin-coating process. Perovskite films were annealed at 100 °C for 10 minutes. In the case of the spiro-OMeTAD HTM, a 85 mg ml−1 solution of spiro-OMeTAD in chlorobenzene was prepared with dopants including bis(trifluoromethylsulfonyl)imide lithium salt (Li-TFSI) (20 μl ml−1 of a 520 mg ml−1 solution in acetonitrile), 4-tert-butylpyridine (tBP, 30 μl ml−1) and tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)-cobalt(III) tris(bis(trifluorome-thylsulfonyl)imide) (FK209, 10 μl ml−1 of a 300 mg ml−1 solution in acetonitrile). For other HTMs, the same weight of material was dissolved in chlorobenzene with 2 times the volume of additives included. The higher dopant concentration used for the DATPA HTMs was found to be beneficial due to the deeper HOMO level of these compared with spiro-OMeTAD. The HTM solution was spin-coated onto perovskite films at 4000 rpm for 30 seconds before 80 nm thick Au contacts were thermally evaporated onto devices.
Current–voltage measurements were performed using a AAA-rated solar simulator (Oriel Sol3A) calibrated against a KG5-filtered reference diode (Oriel 91150-KG5). Solar cells were masked to 0.1 cm2 and scanned both from forward to reverse bias and vice versa at 100 mV s−1.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
RFP thanks CONACYT, Mexico for a PhD studentship. We thank EPSRC EP/H040218/1; EP/M023532/1 for financial support.
Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- Y. Zhou and K. Zhu, ACS Energy Lett., 2016, 1, 64–67 CrossRef CAS.
- NREL, efficiency-chart.png (4348 × 2415), http://https://www.nrel.gov/pv/assets/images/efficiency-chart.png, accessed 21 July 2017.
- P. Gao, M. Grätzel, M. K. Nazeeruddin, K. Zheng, A. Yartsev, T. Pascher, T. Harlang, P. Chabera, T. Pullerits, A. Stepanov, J. P. Wolf, V. Sundstrom, R. Mosca, D. G. Schlom, J. W. Ager and R. Ramesh, Energy Environ. Sci., 2014, 7, 2448–2463 CAS.
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391–402 CrossRef CAS PubMed.
- H. Oga, A. Saeki, Y. Ogomi, S. Hayase and S. Seki, J. Am. Chem. Soc., 2014, 136, 13818–13825 CrossRef CAS PubMed.
- M. Stringer, J. E. Bishop, J. A. Smith, D. K. Mohamad, A. Parnell, V. Kumar, C. Rodenburg and D. G. Lidzey, J. Mater. Chem. A, 2017, 30, 1410 Search PubMed.
- N. Yaghoobi Nia, F. Matteocci, L. Cina and A. Di Carlo, ChemSusChem, 2017, 1–10 Search PubMed.
- W. Sun, S. Ye, H. Rao, Y. Li, Z. Liu, L. Xiao, Z. Chen, Z. Bian and C. Huang, Nanoscale, 2016, 8, 15954–15960 RSC.
- C. Zuo and L. Ding, Small, 2015, 11, 5528–5532 CrossRef CAS PubMed.
-
S. Ito, Organic-Inorganic Halide Perovskite Photovoltaics, Springer International Publishing, Cham, 2016, pp. 343–366 Search PubMed.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- H. Wang, A. D. Sheikh, Q. Feng, F. Li, Y. Chen, W. Yu, E. Alarousu, C. Ma, M. A. Haque, D. Shi, Z. S. Wang, O. F. Mohammed, O. M. Bakr and T. Wu, ACS Photonics, 2015, 2, 849–855 CrossRef CAS.
- M. Daskeviciene, S. Paek, Z. Wang, T. Malinauskas, G. Jokubauskaite, K. Rakstys, K. T. Cho, A. Magomedov, V. Jankauskas, S. Ahmad, H. J. Snaith, V. Getautis and M. K. Nazeeruddin, Nano Energy, 2017, 32, 551–557 CrossRef CAS.
- S. Lv, Y. Song, J. Xiao, L. Zhu, J. Shi, H. Wei, Y. Xu, J. Dong, X. Xu, S. Wang, Y. Xiao, Y. Luo, D. Li, X. Li and Q. Meng, Electrochim. Acta, 2015, 182, 733–741 CrossRef CAS.
- P. Agarwala, D. Kabra, F. Giordano, J.-P. Correa-Baena, M. Saliba, P. Gao, T. Matsui, J. Ko, S. M. Zakeeruddin, K. H. Dahmen, M. K. Nazeeruddin, K.-L. Liau, H.-C. Chung, C.-Y. Liu, S.-S. Sun and T. J. Chow, J. Mater. Chem. A, 2017, 5, 1348–1373 CAS.
- H. Choi, S. Park, S. Paek, P. Ekanayake, M. K. Nazeeruddin and J. Ko, J. Mater. Chem. A, 2014, 2, 19136–19140 CAS.
- P. Ganesan, K. Fu, P. Gao, I. Raabe, K. Schenk, R. Scopelliti, J. Luo, L. H. Wong, M. Grätzel and M. K. Nazeeruddin, Energy Environ. Sci., 2015, 8, 1986–1991 CAS.
- M.-H. Li, C.-W. Hsu, P.-S. Shen, H.-M. Cheng, Y. Chi, P. Chen and T.-F. Guo, Chem. Commun., 2015, 51, 15518–15521 RSC.
- M. Maciejczyk, A. Ivaturi and N. Robertson, J. Mater. Chem. A, 2016, 4, 1–22 Search PubMed.
- K. Rakstys, A. Abate, M. I. Dar, P. Gao, V. Jankauskas, G. Jacopin, E. Kamarauskas, S. Kazim, S. Ahmad, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2015, 137, 16172–16178 CrossRef CAS PubMed.
- F. J. Ramos, K. Rakstys, S. Kazim, M. Grätzel, M. K. Nazeeruddin and S. Ahmad, RSC Adv., 2015, 5, 53426–53432 RSC.
- H. Nishimura, N. Ishida, A. Shimazaki, A. Wakamiya, A. Saeki, L. T. Scott and Y. Murata, J. Am. Chem. Soc., 2015, 137, 15656–15659 CrossRef CAS PubMed.
- P. Dhingra, P. Singh, P. J. S. Rana, A. Garg and P. Kar, Energy Technol., 2016, 4, 891–938 CrossRef CAS.
- L. Calió, S. Kazim, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522–14545 CrossRef PubMed.
- M. Thelakkat, Macromol. Mater. Eng., 2002, 287, 442 CrossRef CAS.
- M. Planells, A. Abate, D. J. Hollman, S. D. Stranks, V. Bharti, J. Gaur, D. Mohanty, S. Chand, H. J. Snaith and N. Robertson, J. Mater. Chem. A, 2013, 1, 6949 CAS.
- D. Bi, L. Yang, G. Boschloo, A. Hagfeldt and E. M. J. Johansson, J. Phys. Chem. Lett., 2013, 4, 1532–1536 CrossRef CAS PubMed.
- S. Ameen, M. A. Rub, S. A. Kosa, K. A. Alamry, M. S. Akhtar, H.-S. Shin, H.-K. Seo, A. M. Asiri and M. K. Nazeeruddin, ChemSusChem, 2016, 9, 10–27 CrossRef CAS PubMed.
- J. Zhang, B. Xu, M. B. Johansson, M. Hadadian, J. P. Correa Baena, P. Liu, Y. Hua, N. Vlachopoulos, E. M. J. Johansson, G. Boschloo, L. Sun and A. Hagfeldt, Adv. Energy Mater., 2016, 6, 1502536 CrossRef.
- C. E. Song, Y. J. Kim, S. R. Suranagi, G. P. Kini, S. Park, S. K. Lee, W. S. Shin, S.-J. Moon, I.-N. Kang, C. E. Park and J.-C. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 12940–12950 CAS.
- I. Zimmermann, J. Urieta-Mora, P. Gratia, J. Aragó, G. Grancini, A. Molina-Ontoria, E. Ortí, N. Martín and M. K. Nazeeruddin, Adv. Energy Mater., 2017, 7, 1601674 CrossRef.
- J. Y. Back, T. K. An, Y. R. Cheon, H. Cha, J. Jang, Y. Kim, Y. Baek, D. S. Chung, S.-K. Kwon, C. E. Park and Y.-H. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 351–358 CAS.
- D. Deng, Y. Zhang, L. Yuan, C. He, K. Lu and Z. Wei, Adv. Energy Mater., 2014, 4, 1400538 CrossRef.
- B. P. Cherniawski, S. A. Lopez, E. K. Burnett, I. Yavuz, L. Zhang, S. R. Parkin, K. N. Houk, A. L. Briseno, M. Muccini, S. Toffanin, A. Weiß, C. Uhrich, M. Pfeiffer and P. Bäuerle, J. Mater. Chem. C, 2017, 5, 582–588 RSC.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- M. H. Vilhelmsen, J. Jensen, C. G. Tortzen and M. B. Nielsen, Eur. J. Org. Chem., 2013, 701–711 CrossRef CAS.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- C. R. Newman, C. D. Frisbie, D. A. Da Silva Filho, J. L. Brédas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436–4451 CrossRef CAS.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J.-P. Correa Baena, J.-D. Decoppet, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Sci. Adv., 2016, 2, e1501170 Search PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis of the DATPA derivatives, additional electrochemical measurments, DSC curves, X-ray structures, extended crystallographic table, powder diffraction data, calculated HOMO and LUMO energy levels of all DATPA derivatives, transfer characteristic curves for the mobility calculations, transient absorption spectroscopy curves and additional solar cell parameters. 1H and 13C for all the synthesised compounds. CCDC 1574478–1574480 and 1574719. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cp07682g |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Saif
Haque
a,
Saif
Haque




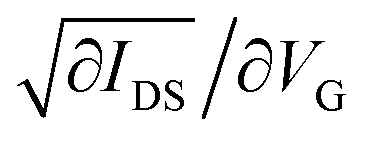 , the slope of the transfer characteristic curves in the saturation regime.40
, the slope of the transfer characteristic curves in the saturation regime.40