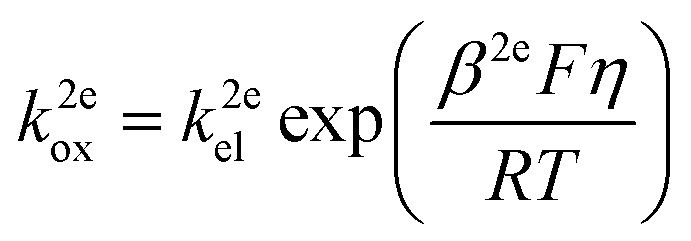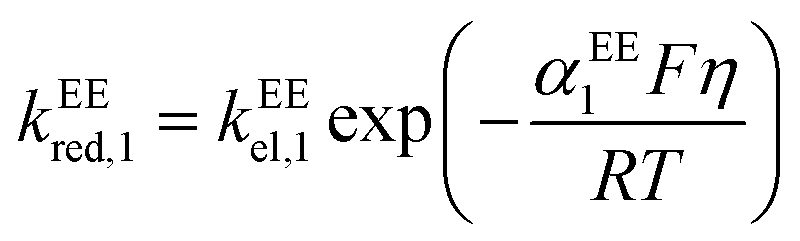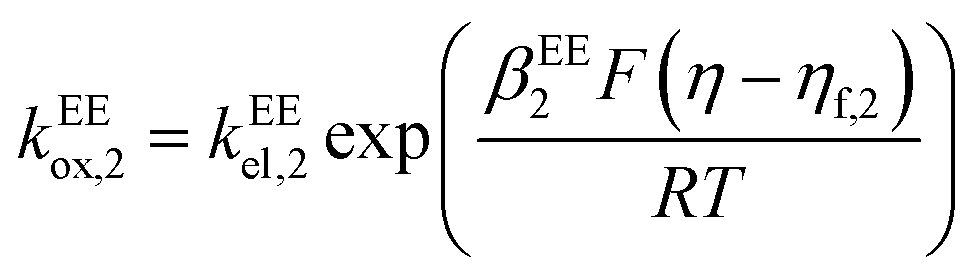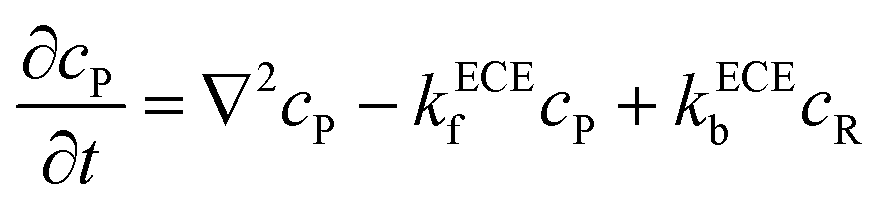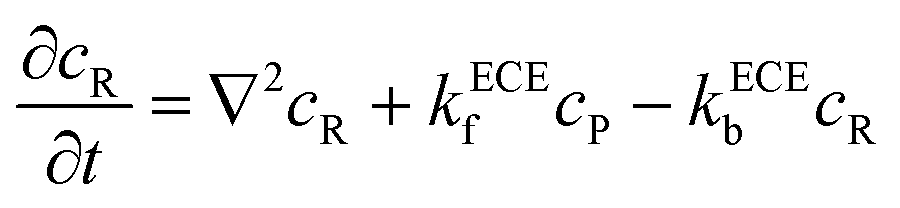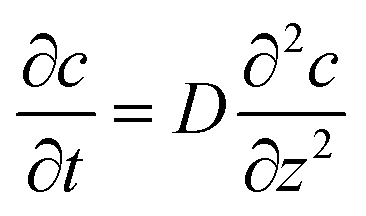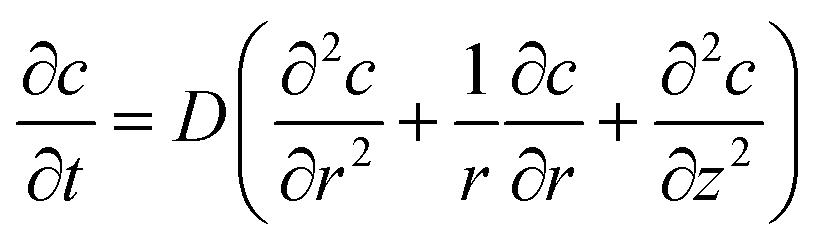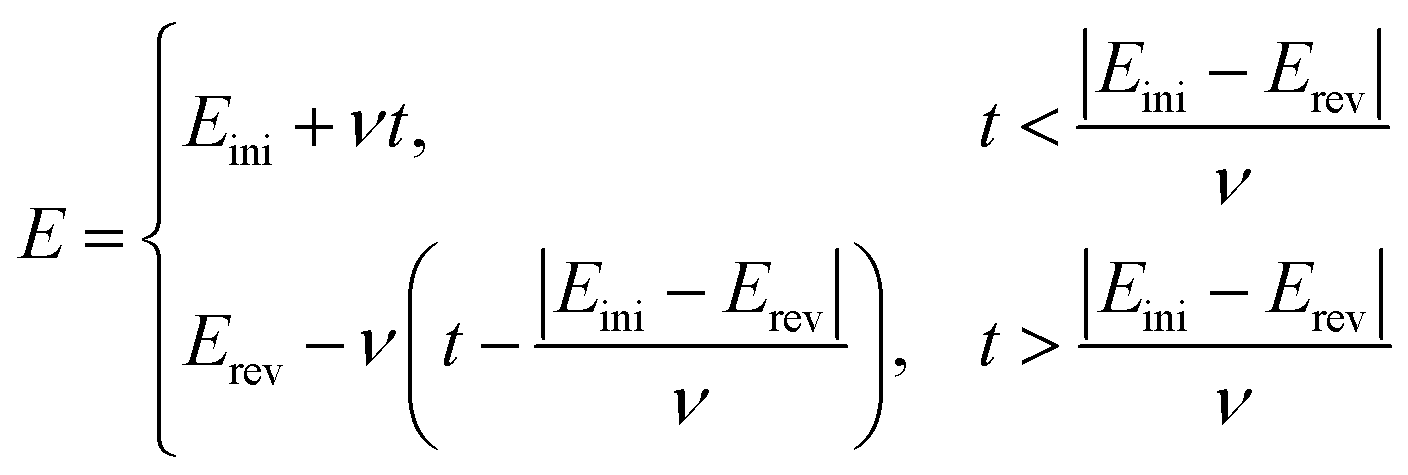Electroanalytical study of dopamine oxidation on carbon electrodes: from the macro- to the micro-scale
Received
3rd November 2017
, Accepted 22nd November 2017
First published on 4th December 2017
Abstract
The oxidation of dopamine in strongly acidic (pH = 0) solution is investigated using microdisc, microcylinder and macro-electrodes together with a range of voltage scan rates. Kinetic and mechanistic analysis over the full range of mass transport conditions show a behaviour consistent with an ECE process with a fast chemical step and in which the second electron transfer is thermodynamically more favourable than the first step. Accordingly the reaction effectively behaves as an EE process.
Introduction
Voltammetry powerfully allows the identification of the mechanism of electrochemical reactions and the determination of kinetic parameters. In particular the use of cyclic voltammetry using a macroelectrode with a semi-infinite diffusion field is widely employed. In this approach the use of a very wide range of voltage scan rates is essential to ensure a precise mechanistic assignment and to obtain reliable rate constants (both hetero- and homo-geneous), diffusion coefficients, transfer coefficients and formal potentials.1 However in practice this is easier to suggest than to implement as the onset of natural convection restricts the lower range of scan rates that can be utilized2 while the large capacitance associated with millimetre sized electrodes constrains the upper limits. Hence, in practice many studies are conducted using a restricted range of voltage scan rates, often with little more than one order of magnitude covering the measurements made.
The purpose in varying the voltage scan rate in cyclic voltammetry is to alter the thickness of the diffusion later so providing variable rates of mass transport to and from the electrode. This can also be achieved by altering the electrode geometry from the macro- to the micro-scale so that the diffusion field changes from linear to convergent (radial) with the latter creating greater mass transport fluxes.3 In the present study we use a combination of both macro and microdisc electrodes studies to investigate a complex electrochemical reaction involving multiple electron transfers and chemical (protonation/deprotonation) steps together with variable scan rates to achieve better clarity on the nature of the electrochemical reaction mechanism. Additionally, we make similar measurements with a microcylinder electrode4,5 which has intermediate character – the length of the cylinder is macro in scale whilst the radius is micron size dimensioned. We believe the study below is the first to utilise electrodes of three different geometries together with variable scan rate data in the quest for precise mechanistic clarity. The system chosen for investigation is the oxidation of dopamine in aqueous acid.
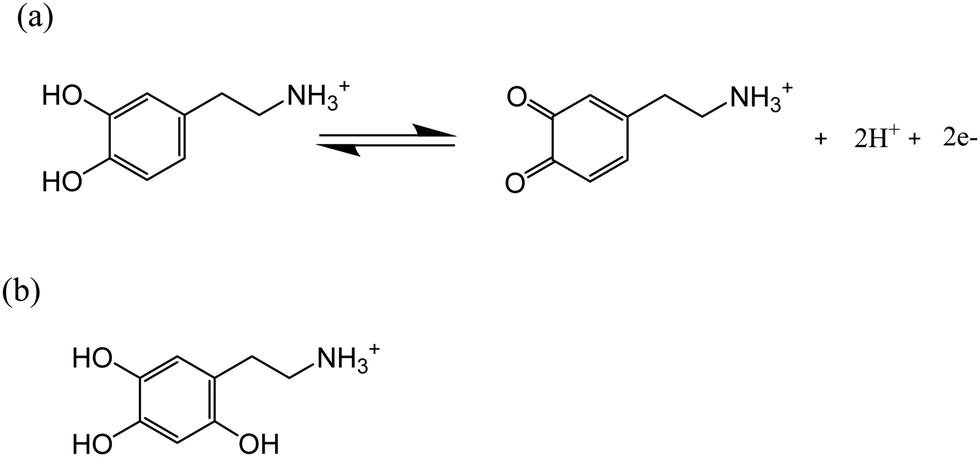
Dopamine (DA) is a neurotransmitter and has stimulated interest in chemistry due to its important physiological roles in the central nervous system.6 The oxidation pathway and mechanism of DA under physiological conditions have been extensively investigated via spectrophotometry,7 electrochemistry,8 and high-performance liquid chromatography.9 However, the DA oxidation in highly acidic conditions has hitherto received relatively little attention. It has been reported that dopamine-o-quinone (DA-Q), the oxidative product of DA, is stable in highly acidic environments (pH < 2.0)10 as the high acidity prevents the release of the proton from the amino group, which is the initial step of intramolecular cyclization.11 Therefore, the oxidation of DA in acidic conditions may result in the accumulation of DA-Q, further leading to the formation of 6-hydroxydopamine (of which the structure is shown in Scheme 1b),12 which has been reported to be toxic to the brain system13 and tissues related to melanin production.14 Thus, understanding the oxidation mechanism of DA under pH < 2.0 acidic conditions is of fundamental interest in chemistry and biology.
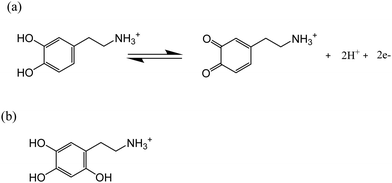 |
| | Scheme 1 (a) Oxidation of dopamine in highly acidic conditions (pH < 2.0); (b) structure of 6-hydroxydopamine. | |
The quinone/hydroquinone redox process involves changes in the protonation state of the molecules over different pH ranges.15,16 The pH dependence of the dopamine redox can be understood in terms of a two-proton two-electron (2H+, 2H−) transfer,17 as summarized using the ‘scheme-of-squares’ (Scheme 2).18 In this model a horizontal movement relates to an electron transfer and a vertical shift corresponds to a proton transfer. It is usually assumed that electron transfer is the rate-determining step whilst the protonation is at equilibrium. The mechanistic pathway depends heavily on the pH of the local environment, the pKa values of the associated intermediates, and the kinetics of each fundamental step. For the case of DA/DA-Q couples, the route has been mapped out across the full pH range in aqueous solution using carbon paste electrodes and macro-electrode cyclic voltammetry.19,20 However, the authors concluded that their model did not give a good fit at low pH.20
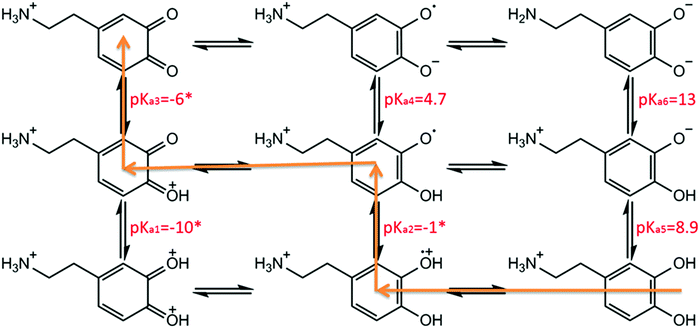 |
| | Scheme 2 Scheme-of-squares for dopamine oxidation. Note pKa5–6 values for the first and second phenolic group of dopamine are 8.9 and 13, respectively.21–23 pKa4 has been determined as 4.7 in previous literature.23 pKa2 is not known for dopamine, but it is −1.1 for duroquinone.24 pKa3 is inaccessible due to the low stability of benzoquinone in high acidic conditions,25 but it has been estimated to be −6 or more negative.16 pKa1 is not known but is probably much smaller than pKa3. Hence pKa1–3 values are assigned as −10, −1 and −6, respectively, as the same assumptions with previous literature.19,20 The orange line indicates a suggested dopamine oxidation pathway in highly acidic conditions (pH < 2.0).15 | |
Accordingly, in this present paper, we focus on the oxidation of dopamine in pH = 0 aqueous acid conditions, where a suggested pathway20 is shown in Scheme 1 (orange line), with the aim of further understanding the oxidation mechanism of DA in highly acidic environments. To this end, the present work explores for the first time the examination of the oxidation of dopamine on solid carbon electrodes with different radii, which are a macro-scale glassy carbon electrode, a carbon microcylinder electrode and a carbon microdisc electrode. This experimental data is simulated, revealing the first full theoretical study of DA oxidation across three sizes of electrodes – from 1.5 mm to 26.5 μm in radius.
Experimental section
Chemical reagents and solutions
All chemicals were purchased from Sigma-Aldrich at reagent grade and used without further purification. Solutions were prepared using ultrapure water of resistivity not less than 18.2 MΩ cm (Millipore, Billerica, MA) at 298 K. Dopamine solutions were prepared from dopamine hydrochloride with a minimum purity of 98.5%. The buffer solution (pH = 0) was freshly prepared using 37% hydrochloric acid and measured using a Hannah pH231 pH meter (Hannah, Bedfordshire, UK). The buffer was degassed thoroughly with pure nitrogen (BOC Gases, UK) for 15 min to prevent degradation of the solution by atmospheric oxygen prior to the addition of dopamine.
Electrochemical procedures
Electrochemical experiments were conducted in a thermostatted (25.0 ± 0.5 °C) Faraday cage using a μAutolab II potentiostat (Metrohm-Autolab BV, Netherlands) and NOVA 1.10 software. All measurements were performed using a standard three-electrode setup employing three different working electrodes: carbon microdisc electrode (IJ Cambria Scientific Ltd, UK), home-fabricated carbon microcylinder electrode4 (diameter 7.0 μm, Goodfellow, Cambridge, UK) and a glassy carbon macroelectrode (3 mm diameter, CH Instruments). A saturated calomel electrode (SCE, ALS distributed by BASi, Tokyo, Japan) and a graphite rod were used as the reference electrode and the counter electrode respectively. The carbon microdisc electrode radius was calibrated as 26.5 μm electrochemically by analysing the steady state voltammetry of 1.0 mM hexaamineruthenium(III) chloride in aqueous solution containing 0.1 M KCl, using a diffusion coefficient for [Ru(NH3)6]3+ of 8.43 × 10−10 m2 s−1 at 298 K.26 Prior to each measurement, the carbon microdisc electrode and glassy carbon macroelectrode were polished using alumina of decreasing particle size (1.0, 0.3 and 0.05 μm, Buehler, IL, UK) followed by sonication in water and drying with nitrogen. Cyclic voltammetry (CV) was conducted at selected scan rates of between 25 mV s−1 to 1000 mV s−1 in pH 0.0 buffer containing 10.0 mM DA.
Theory and simulation
Reaction mechanism
Considering that the oxidation of dopamine is a two-electron-transfer reaction, the simplest model of the reaction is:The reaction rate can be expressed as:| | | jA = −k2eoxcA,surf + k2eredcB,surf | (2) |
jA (mol m−2 s−1) is the flux of the reactant A which is consumed at the electrode surface. k2eox and k2ered (m s−1) are the electron transfer rate constant of the oxidative and reductive directions for eqn (1), respectively. cA,surf and cB,surf (mol m−3) are the surface concentrations of the reactant A and the product B, respectively. Assuming that the electron transfer kinetics can be described by the Butler–Volmer equation, the electron transfer rate constants are functions of overpotential η (V), which is the difference between the applied potential E (V) and the formal potential of the electron transfer step Ef (V):27,28| | 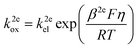 | (3) |
| | 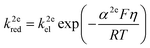 | (4) |
where kel (m s−1) is the standard heterogeneous electron transfer constant, F (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1) is the Faraday constant, R (8.3145 J mol−1) is the gas constant, T (298.15 K) is the room temperature. β2e and α2e are the “apparent” anodic and cathodic transfer coefficients of the two-electron-transfer step.
485 C mol−1) is the Faraday constant, R (8.3145 J mol−1) is the gas constant, T (298.15 K) is the room temperature. β2e and α2e are the “apparent” anodic and cathodic transfer coefficients of the two-electron-transfer step.
Assuming the solution contains sufficient supporting electrolyte,29 if the experiment time is short enough to not cause any apparent convection,2 the mass transport of the reactive species is only due to the diffusion. The concentration variation in the solution with the experiment time t can be expressed by Fick's second law:
| | 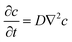 | (5) |
where
D is the diffusion coefficient (m
2 s
−1). In this work, we assume the diffusion coefficient for the reactant, the product and the intermediates are all identical. The expression of the Laplace operator is determined by the geometry of the electrode, which will be discussed below. By solving
eqn (5) with the boundary condition
eqn (2) and the boundary condition
c =
c* (
c* is the concentration in the bulk solution), the concentration profile can be calculated. The electrode current
I (A) corresponding to an applied electrode potential
E is:
| | 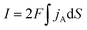 | (6) |
where
S is the area of the electrode.
An improved model is to treat the dopamine oxidation as two one-electron-transfer steps, the so called EE mechanism:
For dopamine oxidation, the two steps correspond to two electron–proton-transfer reactions. The reaction fluxes for the species X and Z are written as:
| | | jX = −kEEox,1cX,surf + kEEred,1cY,surf | (9) |
| | | jZ = kEEox,2cY,surf − kEEred,2cZ,surf | (10) |
kEEox,1,
kEEred,1,
kEEox,2 and
kEEred,2 are the rate constants for the two electron-transfer steps, described by the Butler–Volmer kinetics:
| | 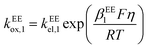 | (11) |
| | 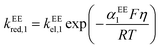 | (12) |
| | 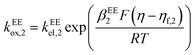 | (13) |
| | 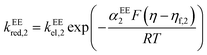 | (14) |
where the transfer coefficients of the first and the second electron transfer steps follow
αEE1 +
βEE1 = 1 and
αEE2 +
βEE2 = 1. In multi-electron-transfer models, for the convenience of calculation, the overpotential
η is defined as the difference between the applied potential
E and the formal potential of the first electron-transfer step
Ef,1. The effective overpotential for the second electron transfer step becomes
η − ηf,2, where
ηf,2 represents the formal potential difference between the first and the second electron-transfer steps. In the case of EE mechanism, the concentration variation of the reactive species in the solution is also described by the Fick's second law as
eqn (5) and the current is calculated by:
| | 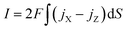 | (15) |
Based on the nine-member scheme as shown in Scheme 2, a more detailed model is the electron transfer (E)–deprotonation (C)–electron transfer (E)–deprotonation (C) mechanism. As the second deprotonation step is thermodynamically favourable, it is combined with the second electron transfer step and an ECE model is proposed as:
| | 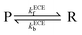 | (17) |
where
kECEf (s
−1) and
kECEb (s
−1) are the forward (deprotonation in the case of dopamine oxidation) and backward (protonation) reaction rate constants of
reaction (17). The reaction fluxes at the electrode surface of species O and R are:
| | | jO = −kECEred,1cO,surf + kECEox,1cP,surf | (19) |
| | | jR = −kECEred,2cR,surf + kECEox,2cS,surf | (20) |
The electron-transfer rate constants in the ECE model are also described as the function of
kel,
η,
α and
β as shown in
eqn (11)–(14). The mass transport of species O and S is only determined by the diffusion as shown in
eqn (5). But the deprotonation step is a homogeneous chemical reaction, which influences the concentration distribution in the solution and the mass transport equations of species P and R become:
| | 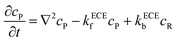 | (21) |
| | 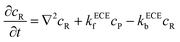 | (22) |
The current is determined by the reaction fluxes at the electrode surface:
| | 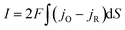 | (23) |
Electrode geometry
As mentioned in the introduction, there are three types of electrodes applied in the experiment: the macroelectrode, the microdisc electrode, and the microcylinder electrode. The electrode is modelled in the cylindrical coordinate system (r, φ, z) where the angle coordinate φ can be ignored due to the symmetry of the electrodes. Fig. 1 shows the diffusion modes at the three different electrodes.
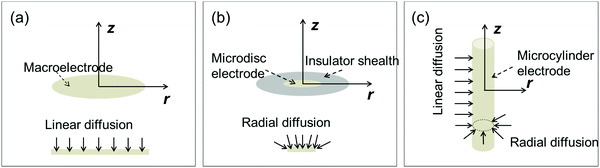 |
| | Fig. 1 Diffusion modes on three different electrodes in the two-dimensional cylindrical coordinate system. (a) Macroelectrode; (b) microdisc electrode surrounded by the insulator health; (c) microcylinder electrode. z and r are the coordinates of the cylindrical coordinate system. | |
At the macroelectrode, as the diffusion layer thickness is much smaller than the size of the electrode, the diffusion towards the plane is regarded to be linear and the flux is identical along the radial direction r parallel to the plane.30 Therefore, only the concentration variation on the direction z perpendicular to the plane needs to be taken into consideration and the diffusion equation for the macroelectrode system becomes:
| | 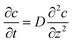 | (24) |
At the microdisc electrode, the diffusion is radial to the electrode plane and therefore the concentration is not evenly distributed along the
r direction. The diffusion equation is:
| | 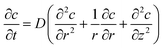 | (25) |
For the microcylinder electrode, we assume the length of the electrode is long compared to the radius. Thus only the radius of the microcylinder is at microscale and the diffusion equation is written as:
| | 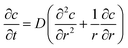 | (26) |
By solving the mass transport equation at various electrodes, the current response can be simulated as a function of applied potential. To simulate the cyclic voltammetry, the applied potential varies as a linear function of time, determined by the scan rate ν (V s−1):
| |  | (27) |
where [
Eini,
Erev] is the potential window for the cyclic voltammetry of the dopamine oxidation and
Eini <
Erev.
The mass transport equation is numerically solved by the finite difference method and the alternating direction implicit (ADI) method.31 The simulation program is written in Matlab R2017a and run on an Intel(R) Xeon(R) 3.60G CPU.
Results and discussions
The mechanism of the dopamine oxidation reaction in acidic solution is studied at carbon electrodes. Combining the experiment with the simulation models, the kinetics of the dopamine oxidation is revealed.
Analysis to the dopamine oxidation reaction
The experimental results of the dopamine oxidation reaction measured at the macro-size glassy carbon electrode are shown in Fig. 2. Fig. 2a shows cyclic voltammograms with different scan rates from 0.025 V s−1 to 1.0 V s−1. Fig. 2b are the apparent transfer coefficients referring to each voltammogram in Fig. 2a. The transfer coefficient is calculated from the slope of ln|I| vs. E in the region where the current is about 10–15% of the peak current to avoid the influence of diffusion. Fig. 2c is the relationship between the peak current, where the scan rate and the dashed line is the linear fitting of the peak current as a function of the scan rate. Fig. 2d shows the peak-to-peak separation varying with the scan rate.
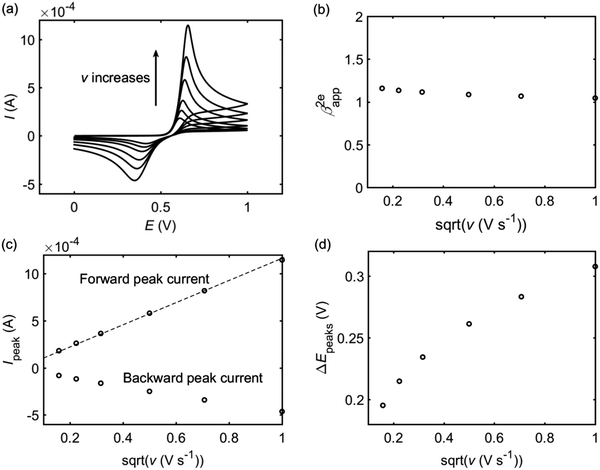 |
| | Fig. 2 Analysis of the cyclic voltammograms of the dopamine oxidation reaction with difference scan rates v (from 0.025 V s−1 to 1.0 V s−1). (a) Cyclic voltammograms; (b) apparent transfer coefficient β2eapp; (c) peak currents, where the dash line is a linear fitting of the forward peak current as a function of v1/2; (d) peak-to-peak separations. The concentration of dopamine is 10 mM and the size of the macroelectrode is 1.5 mm. | |
It is found that the transfer coefficient is close to unity in Fig. 2b, showing that if the reaction can be regarded as a simple 2e model, the electron transfer should be reversible. According to the Randles–Sevcik equation, for a reversible multi-electron-transfer reaction, the peak current can be calculated via:30
| | 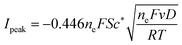 | (28) |
ne = 2 for the dopamine oxidation reaction. In
Fig. 2c, the linear fitting of the forward peak current proves that the peak current at the macroelectrode is proportional to
v1/2 and the apparent diffusion coefficient can be calculated from
eqn (28) as 5 × 10
−10 m
2 s
−1. However,
Fig. 2c shows that the backward peak is much smaller than the forward one and the peak-to-peak separation in
Fig. 2d is much larger than the theoretical value for a reversible electron transfer (57 mV), indicating that the whole reaction cannot be explained as a reversible two-electron-transfer process (A − 2e
− → B).
To examine the apparent contradiction between the transfer coefficient of unity and the irreversible behaviour of the backward peak, a quasi-reversible 2e mechanism is applied in the interpretation of the experimental cyclic voltammograms. Fig. 3 shows the simulation results of a quasi-reversible 2e model (blue lines) compared with the experimental data (black lines) at the macroelectrode. The diffusion coefficient and the apparent transfer coefficient calculated from the experiment are used in the 2e model. To fit the position of the current peak, the apparent formal potential is fixed as 0.5 V and the apparent electron transfer rate constant as 10−5 m s−1. In Fig. 3, the simulated forward peak current is smaller than the experimental one and the simulated backward peak current is larger. Nor can the quasi-reversible 2e simulation fit the forward and backward peak currents in the experiment, showing that the reaction of dopamine oxidation on the carbon electrode cannot be explained by a simple 2e model.
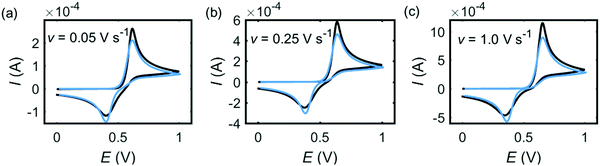 |
| | Fig. 3 Experiment (black) and 2e model simulation (blue) for dopamine oxidation on macroelectrode. In the experiment, the radius of the electrode is 1.5 mm and the concentration of dopamine is 10 mM. The scan rate is marked on each panel. In the 2e model, D2e = 5 × 10−9 m2 s−1, β2e= 1, E2ef = 0.5 V, k2eel = 10−5 m s−1. | |
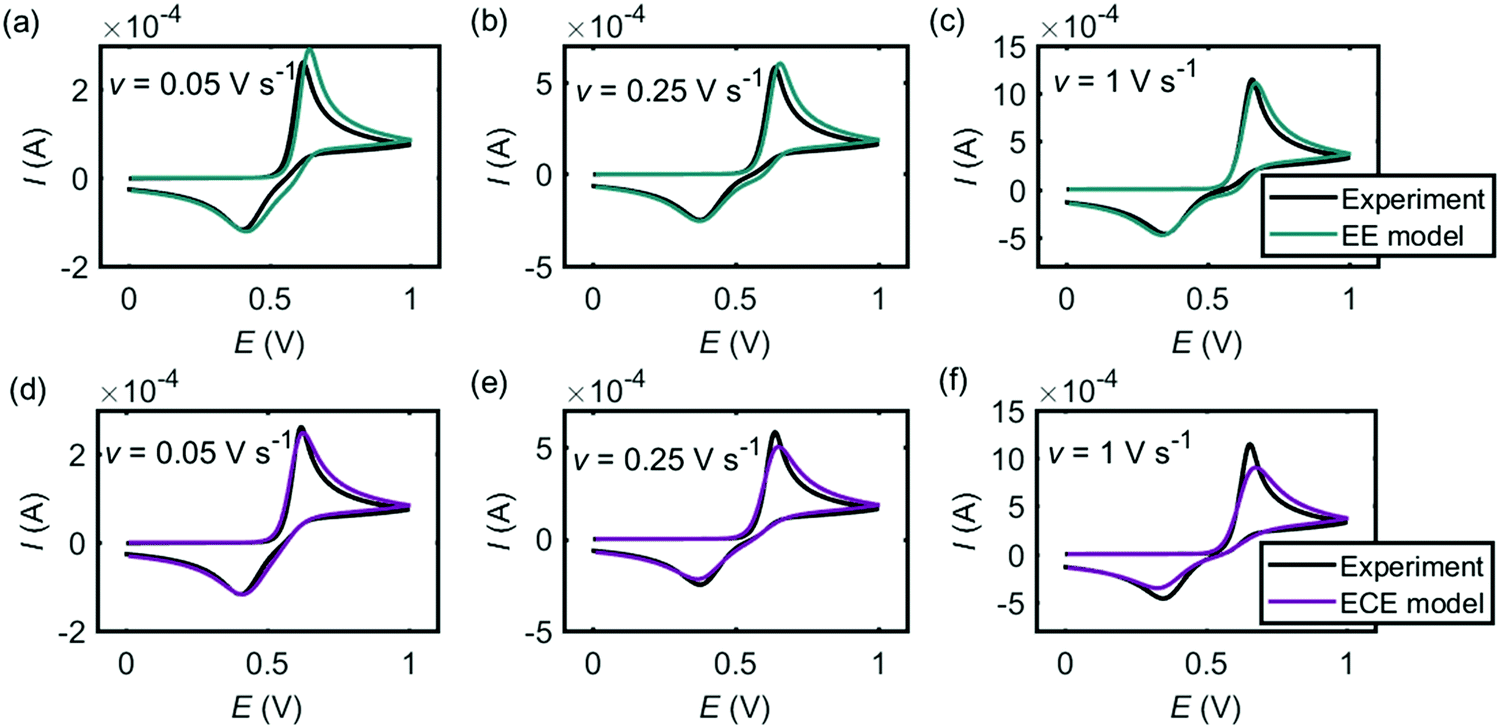
Fig. 4 shows the comparison between the experiment (black lines) and the simulation results from the EE model (Fig. 4a–c, green lines) and the ECE model (Fig. 4d–f, red lines). Both mechanisms can fit the experimental cyclic voltammograms under different scan rates. In the EE model, the key parameters are the electron-transfer rate constants kEEel,1 and kEEel,2, the formal potentials EEEf,1 and …, and the diffusion coefficient DEE. In the ECE model, apart from kECEel,1, kECEel,2, EECEf,1, EECEf,2 and DECE, the forward and backward rate constants kECEf and kECEb for the chemical step are also important. We infer that the forward wave is electrochemically reversible but as the peak-to-peak separation varies as a function of the scan rate, we assume the two separate electron-transfer steps are quasi-reversible and the transfer coefficients for each step are set to be 0.5 with fast rate constants. The other kinetic parameters are determined by fitting the experimental peak features, which are the peak current, the peak potential and the peak shape. The values of the simulation parameters used in Fig. 4 are listed in Table 1. The formal potential of the second electron-transfer step is less than the first electron-transfer step, showing that the second step is more thermodynamically favourable than the first step. The behaviour of Ef,1 < Ef,2 in the dopamine oxidation reaction is called ‘potential inversion’, which is reported to occur in several classed of compounds, including catechol, which is structurally close to dopamine.32,33 Considering the second step for both EE and ECE model is a combined electron–proton transfer, the formal potential is influenced by the deprotonation process. In the nine-member square scheme, if the reaction route from dopamine to dopamine o-quinone is electron–electron–proton–proton rather than electron–proton–electron–proton, the kinetic parameters in the second E step in the EE model and the ECE model cannot be the same as for the first step.
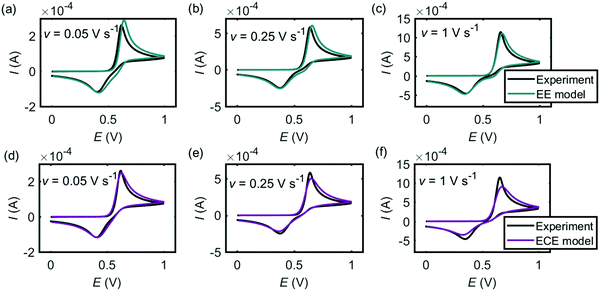 |
| | Fig. 4 Cyclic voltammograms of dopamine oxidation at the macroelectrode. (a–c) Experiment results (black) and simulations (green) from the EE model; (d–f) experiment results (black) and simulations (red) ECE model. In the experiment, the radius of the electrode is 1.5 mm and the concentration of dopamine is 10 mM. The scan rate is marked on each panel. The kinetic parameters used in the simulation are listed in Table 1. | |
Table 1 Simulation parameters of the EE model and the ECE model used in Fig. 4
| Model |
k
el,1 (m s−1) |
k
el,2 (m s−1) |
E
f,1 (V) |
E
f,2 (V) |
D (m2 s−1) |
k
f (s−1) |
k
b (s−1) |
| EE |
10−4 |
5 × 10−5 |
0.65 |
0.4 |
8 × 10−10 |
— |
— |
| ECE |
10−4 |
5 × 10−5 |
0.65 |
0.4 |
8 × 10−10 |
104 |
103 |
Fig. 5 and 6 are the cyclic voltammograms of dopamine oxidation reaction measured at the microcylinder and microdisc electrodes. Both the experiment measurement (black lines) and the simulation results from the EE model (green lines) and the ECE model (red lines) are presented. As for Fig. 4, the EE and ECE models can both fit the experiment well. But we find that not all the kinetic parameters used to fit the macroelectrode cyclic voltammograms matches the results at the microcylinder and microdisc electrodes except that the formal potentials of the two electron-transfer steps show the slight variation listed in Table 2.
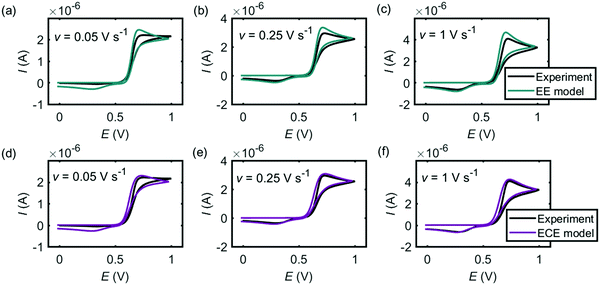 |
| | Fig. 5 Cyclic voltammograms of dopamine oxidation at the microcylinder electrode. (a)–(c) are the EE model; (d)–(f) are the ECE model. In the experiment, the radius of the microcylinder is 3.5 μm, the length is 0.75 mm and the concentration of dopamine is 10 mM. The simulation parameters used are the same as listed in Table 1, except the formal potentials. For the microcylinder electrode simulation, Ef,1 = 0.68 V and Ef,2 = 0.48 V. | |
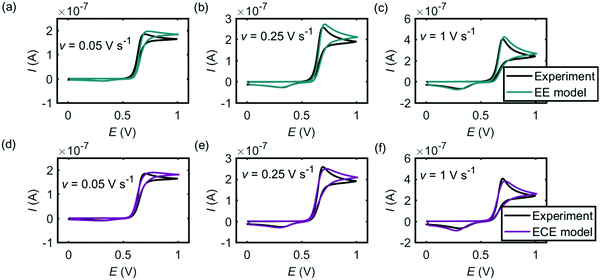 |
| | Fig. 6 Cyclic voltammograms of dopamine oxidation at the microdisc electrode. (a)–(c) are the EE model; (d)–(f) are the ECE model. In the experiment, the radius of the microdisc is 26.5 μm and the concentration of dopamine is 10 mM. The simulation parameters used are the same as listed in Table 1, except the formal potentials. For the microdisc electrode simulation, Ef,1 = 0.70 V and Ef,2 = 0.45 V. | |
Table 2 Formal potentials used in the simulation to the cyclic voltammograms
|
|
Macroelectrode |
Microcylinder electrode |
Microdisc electrode |
|
E
f,1 (V) |
0.65 |
0.68 |
0.7 |
|
E
f,2 (V) |
0.5 |
0.48 |
0.45 |
In particular, when the electrode changes from macrosize to microsize, the formal potential of the first electron-transfer step Ef,1 becomes slightly more positive and the potential difference between the first and the second electron-transfer steps |Ef,2 − Ef,1| increases. It is reported in the literature that dopamine adsorbs on the electrode and leads to the variation of the diffusion coefficient and the formal potential in the experimental measurement.34–36 The adsorption of dopamine on carbon has been proved in our previous work,37 where the electron transfer corresponding to the adsorbed dopamine rather the solution-phase dopamine was found at the potential before the oxidation of the solution-phase dopamine. In the simulation, from the macroelectrode (radius: 1.5 mm) to the microelectrode (radius: 26.5 μm), the formal potential required for the dopamine oxidation shifts slightly positively.
Difference between the EE and ECE models
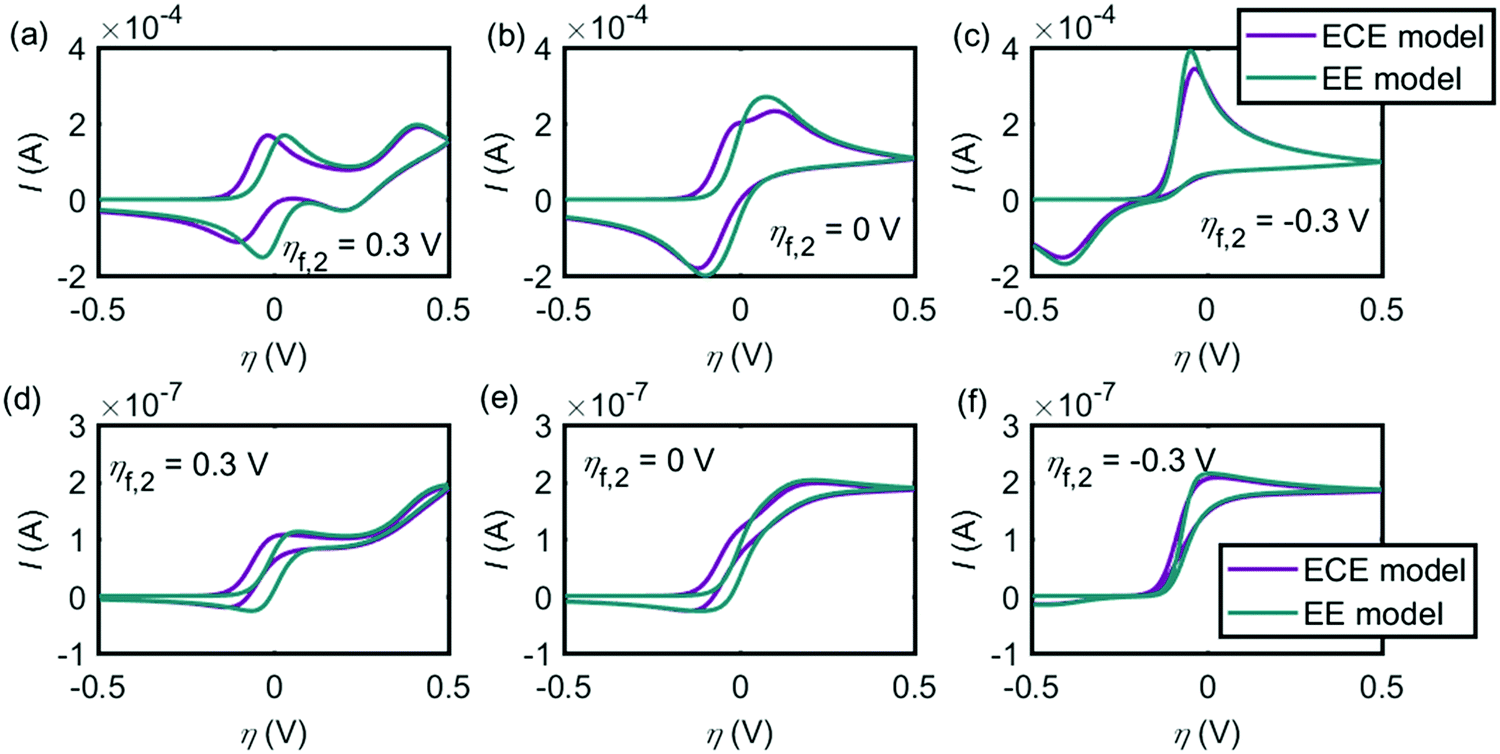
Given that both the EE and ECE mechanisms can explain the experimental data, the conditions where the two models provide similar voltammetric features are explored in this section. Fig. 7 shows the influence of the chemical reaction rate on the performance of the ECE model (red lines), compared with the EE model (green lines). The same kinetic parameters are applied for the two electron-transfer steps in both EE and ECE models. Fig. 7a simulates the cyclic voltammograms at the macroelectrode and Fig. 7b shows how the peak currents vary as a function of the rate constants in the chemical reaction. Fig. 7c and d are the results of the microdisc electrode. The sizes of the macro- and micro-electrode are the same as used in the above experiment. In Fig. 7, the pKa of the chemical step keeps −1, where kf/kb = 10. When kf varies from 1 s−1 to 105 s−1 and kb also changes from 0.1 s−1 to 104 s−1, the cyclic voltammograms of the ECE model is affected significantly by the reaction rate constants of the chemical step, at both the macroelectrode and the microelectrode. With slow reaction rates, as the chemical reaction becomes the rate-determining step, the peak current is much smaller than the value of the EE model. But when the reaction rate constants increase, the peak current becomes larger and gradually close to the value of the EE model. In Fig. 7, it is also found that when kf > 103 s−1, the influence of the chemical step on the overall ECE reaction is not observable. Therefore, the rate constants of the deprotonation step in the dopamine oxidation reaction must be large enough to avoid the deprotonation process becoming the rate-determining step.
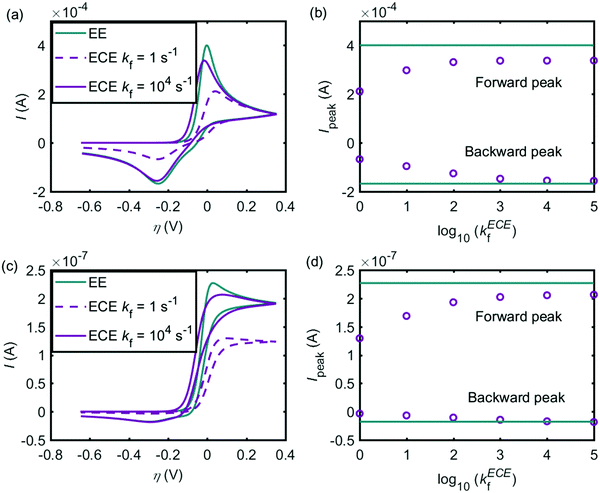 |
| | Fig. 7 Influence of kf on the performance of the ECE model (red lines), compared to the EE model (green lines). (a) Cyclic voltammograms at the macroelectrode; (b) peak currents of the ECE model under different kf, at the macroelectrode; (c) cyclic voltammograms at the microdisc electrode; (d) peak currents of the ECE model under different kf, at the microdisc electrode. The dopamine concentration is 10 mM and the scan rate is 0.1 V s−1. Except kf and kb, other simulation parameters are the same as listed in Tables 1 and 2. | |
Fig. 8 shows the influence of the potential difference between the two electron-transfer steps ηf,2 on the EE and ECE models. In Fig. 8a–c, the cyclic voltammograms with the EE model (green lines) and the ECE model (red lines) are simulated for the macroelectrode; Fig. 8d and e are the voltammograms of the microdisc electrode. It is found from Fig. 8 that at both the macroelectrode and the microelectrode, only when the potential inversion occurs (ηf,2 < 0), can the EE model and the ECE model not be distinguished. In Fig. 4–6, both the EE and ECE mechanisms are able to simulate the experiment data, indicating that the intermediate of the dopamine oxidation reaction is much more reducible than dopamine.
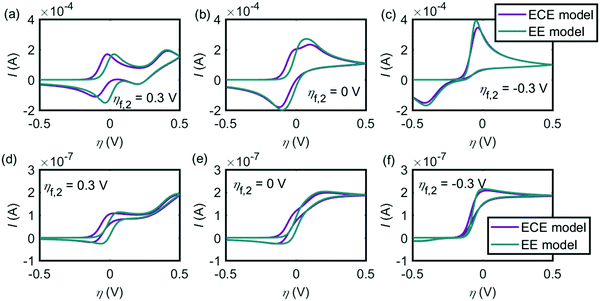 |
| | Fig. 8 Influence of the formal potential difference between the first and the second electron-transfer steps ηf,2 on the EE and ECE models. (a)–(c) are the cyclic voltammograms at the macroelectrode. (d)–(f) are at the microdisc electrode. The dopamine concentration is 10 mM and the scan rate is 0.1 V s−1. Except ηf,2, other simulation parameters are the same as listed in Tables 1 and 2. | |
Conclusions
The mechanism of dopamine oxidation reaction in acidic solution (pH = 0) is explored at three carbon electrodes of different sizes: the macroelectrode (1.5 mm radius), the microcylinder electrode (3.5 μm radius and 0.75 mm length) and the microdisc electrode (26.5 μm radius). The decrease of the electrode size changes the diffusion mode from the linear diffusion to radial diffusion. At each diffusion mode, it is found that both the EE model and the ECE model are able to explain the experimental results well. Under the nine-member square scheme, it proves that the reaction follows the electron–proton–electron transfer rather than the electron–electron–proton transfer.
The difference of the EE and ECE models is also explored. Only when the chemical step is fast and the second electron-transfer step is more favourable, the two models return similar current–potential responses. The comparison of the voltammetric features between the EE and ECE mechanisms reveals that the deprotonation of the first proton-transfer step in the dopamine oxidation is a fast reaction and the intermediate is much more reducible than dopamine.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research is sponsored by funding from the European Research Council under the European Union Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. [320403].
References
- C. Batchelor-McAuley, E. Kätelhön, E. O. Barnes, R. G. Compton, E. Laborda and A. Molina, ChemistryOpen, 2015, 4, 224–260 CrossRef CAS PubMed.
- K. Ngamchuea, S. Eloul, K. Tschulik and R. G. Compton, Anal. Chem., 2015, 87, 7226–7234 CrossRef CAS PubMed.
- J. Heinze, Angew. Chem., Int. Ed., 1993, 32, 1268–1288 CrossRef.
- J. Ellison, C. Batchelor-McAuley, K. Tschulik and R. G. Compton, Sens. Actuators, B, 2014, 200, 47–52 CrossRef CAS.
- K. Cinková, M. Clark, S. V. Sokolov, C. Batchelor-McAuley, Ľ. Švorc and R. G. Compton, Electroanalysis, 2017, 29, 1006–1013 CrossRef.
- O. Hornykiewicz, Pharmacol. Rev., 1966, 18, 925–964 CAS.
- A. N. Pham and T. D. Waite, J. Inorg. Biochem., 2014, 137, 74–84 CrossRef CAS PubMed.
- C. Díaz, C. García, P. Iturriaga-Vásquez, M. J. Aguirre, J. P. Muena, R. Contreras, R. Ormazábal-Toledo and M. Isaacs, Electrochim. Acta, 2013, 111, 846–854 CrossRef.
- J. Li and B. M. Christensen, J. Electroanal. Chem., 1994, 375, 219–231 CrossRef CAS.
- J. Segura-Aguilar and C. Lind, Chem.-Biol. Interact., 1989, 72, 309–324 CrossRef CAS PubMed.
-
G. Dryhurst, K. M. Kadish, F. Scheller and R. Renneberg, Biological electrochemistry, Academic Press, New York, 1982 Search PubMed.
- A. W. Sternson, R. McCreery, B. Feinberg and R. N. Adams, J. Electroanal. Chem. Interfacial Electrochem., 1973, 46, 313–321 CrossRef CAS.
- D. C. S. Tse, R. L. McCreery and R. N. Adams, J. Med. Chem., 1976, 19, 37–40 CrossRef CAS PubMed.
- M. M. Wick, J. Invest. Dermatol., 1978, 71, 163 CrossRef CAS PubMed.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1983, 146, 15–36 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1984, 164, 213–227 CrossRef CAS.
- P. J. Britto, K. S. V. Santhanam and P. M. Ajayan, Bioelectrochem. Bioenerg., 1996, 41, 121–125 CrossRef CAS.
- J. Jacq, J. Electroanal. Chem. Interfacial Electrochem., 1971, 29, 149–180 CrossRef CAS.
- M. R. Deakin, P. M. Kovach, K. J. Stutts and R. M. Wightman, Anal. Chem., 1986, 58, 1474–1480 CrossRef CAS PubMed.
- M. R. Deakin and R. M. Wightman, J. Electroanal. Chem. Interfacial Electrochem., 1986, 206, 167–177 CrossRef CAS.
- R. K. Boggess and R. B. Martin, J. Am. Chem. Soc., 1975, 97, 3076–3081 CrossRef CAS PubMed.
- R. B. Martin, J. Phys. Chem., 1971, 75, 2657–2661 CrossRef CAS PubMed.
- H. W. Richter and W. H. Waddell, J. Am. Chem. Soc., 1983, 105, 5434–5440 CrossRef CAS.
- E. J. Land and G. Porter, Proc. Chem. Soc., 1960, 84–85 CAS.
- H. Takashi, Bull. Chem. Soc. Jpn., 1955, 28, 483–489 CrossRef.
- Y. Wang, J. G. Limon-Petersen and R. G. Compton, J. Electroanal. Chem., 2011, 652, 13–17 CrossRef CAS.
- R. Guidelli, R. G. Compton, J. M. Feliu, E. Gileadi, J. Lipkowski, W. Schmickler and S. Trasatti, Pure Appl. Chem., 2014, 86, 245–258 CAS.
- R. Guidelli, R. G. Compton, J. M. Feliu, E. Gileadi, J. Lipkowski, W. Schmickler and S. Trasatti, Pure Appl. Chem., 2014, 86, 259–262 CAS.
- E. J. F. Dickinson, J. G. Limon-Petersen, N. V. Rees and R. G. Compton, J. Phys. Chem. C, 2009, 113, 11157–11171 CAS.
-
R. G. Compton and C. E. Banks, Understanding Voltammetry, Imperial College Press, 2nd edn, 2011 Search PubMed.
-
R. G. Compton, E. Laborda and K. R. Ward, Understanding voltammetry: simulation of electrode processes, Imperial College Press, London, UK, 2013 Search PubMed.
- Q. Lin, Q. Li, C. Batchelor-McAuley and R. G. Compton, J. Phys. Chem. C, 2015, 119, 1489–1495 CAS.
- N. A. Macías-Ruvalcaba and D. H. Evans, J. Phys. Chem. B, 2006, 110, 5155–5160 CrossRef PubMed.
- S. Corona-Avendaño, G. Alarcón-Angeles, M. T. Ramírez-Silva, G. Rosquete-Pina, M. Romero-Romo and M. Palomar-Pardavé, J. Electroanal. Chem., 2007, 609, 17–26 CrossRef.
- D. H. Kim, Y. Oh, H. Shin, C. D. Blaha, K. E. Bennet, K. H. Lee, I. Y. Kim and D. P. Jang, J. Electroanal. Chem., 2014, 717-718, 157–164 CrossRef CAS PubMed.
- S. H. DuVall and R. L. McCreery, Anal. Chem., 1999, 71, 4594–4602 CrossRef CAS.
- L. Chen, E. E. L. Tanner, C. Lin and R. G. Compton, Chem. Sci., 2018 10.1039/C7SC03672H.
Footnotes |
| † These authors contributed equally to the paper. |
| ‡ Current address: School of Engineering & Applied Sciences, Harvard University, Cambridge, MA, USA. |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*