
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study of non-Hammett vs. Hammett behaviour in the thermolysis and photolysis of arylchlorodiazirines†
Xing-Liang
Peng
a,
Annapaola
Migani
 b,
Quan-Song
Li
*a,
Ze-Sheng
Li
a and
Lluís
Blancafort
*c
b,
Quan-Song
Li
*a,
Ze-Sheng
Li
a and
Lluís
Blancafort
*c
aBeijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, Key Laboratory of Cluster Science of Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, 100081 Beijing, China. E-mail: liquansong@bit.edu.cn
bDepartament de Química Biològica i Modelització Molecular, IQAC-CSIC, Jordi Girona 18-26, 08034 Barcelona, Spain
cInstitut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, Facultat de Ciències, C/M. A. Capmany 69, 17003 Girona, Spain. E-mail: lluis.blancafort@udg.edu
First published on 11th December 2017
Abstract
Arylchlorodiazirines (ACDA) are thermal and photochemical precursors of carbenes that form these molecules via nitrogen elimination. We have studied this reaction with multireference quantum chemical methods (CASSCF and CASPT2) for a series of ACDA derivatives with different substitution at the aromatic ring. The calculations explain the different reactivity trends found in the ground and excited state, with good correlation between the calculated barriers and the experimental reaction rates. The ground state mechanism can be described as a reverse cycloaddition with small charge transfer from the aromatic ring to the diazirine moiety. This is consistent with the lack of correlation between the Hammett σ descriptors and the experimental rates. In contrast, the excited state reaction is the cleavage of a single C–N bond mediated by small barriers of 4–6 kcal mol−1. The reaction path goes through a conical intersection with the ground state, which facilitates radiationless decay and explains the disappearance of the transient absorption signal measured experimentally. This leads to a diazomethane intermediate that ultimately yields the carbene. Electronically, excitation to S1 is characterized initially by significant charge transfer from the phenyl ring to the diazirine. The charge transfer is reversed during the C–N cleavage reaction, and this explains the preferential stabilization of the excited-state minimum by polar solvents and electron-donating substituents. Therefore, our calculations reproduce and explain the relationship found experimentally between the Hammett σ+ parameters and the life time of S1 (Y. L. Zhang, et al. J. Am. Chem. Soc., 2009, 131, 16652–16653).
Introduction
Understanding chemical reactivity is one of the main goals of theoretical chemistry. There are many methods to confront this task, with different levels of sophistication. However, our understanding of excited-state reactivity is much less advanced than that of ground-state (thermal) chemistry. Most recent developments in excited-state theoretical and computational chemistry have focused on improving the description of energetics and dynamics, while efforts to apply reactivity descriptors to the excited state or developing specific excited-state descriptors have been scarce, in comparison.1–6 Surprisingly, the simple but very effective Hammett approach, which is mainly used to rationalize the ground-state reactivity of aromatic systems on the basis of the electron donating or accepting character of the ring substituents, has been used only in a few examples to rationalize photophysical7–12 or photochemical properties.13 In this paper we centre on one such case and show that state of the art quantum chemical methods reproduce the experimental trends and provide new insight into the molecular factors that determine the reactivity.Our reaction of interest is the nitrogen elimination of arylchlorodiazirine (ACDA) derivatives (see Scheme 1) to yield arylchlorocarbenes. This reaction proceeds thermally and photochemically. Part of the reason for our interest is that this is one of only few excited state reactions where the Hammett approach has been applied, and that it follows different trends in the ground and excited state. For the ground state there is no clear correlation between the thermal reaction rates and the Hammett σ descriptors.14 In contrast, the excited-state reaction rates show an excellent Hammett correlation with the σ+p descriptors of the aryl substituents,15 which reflect the ability of the substituents to stabilize a developing positive charge.16 To understand the difference between ground- and excited-state decomposition, we have carried out potential energy surface calculations combined with orbital and Mulliken charge analyses.
 | ||
| Scheme 1 | ||
ACDA derivatives are also of fundamental interest in the context of diazirine and carbene chemistry. Diazirines are precursors of carbenes.17,18 They yield carbenes photochemically under mild conditions.19,20 Ultrafast time-resolved laser flash photolysis of aryl diazo compounds21–23 and aryldiazirines24–26 has provided valuable information on the dynamics of the precursor excited states and the generated singlet arylcarbenes. Using ultrafast infrared spectroscopy, the singlet aryldiazomethane and arylcarbene intermediates were detected in the photolysis of phenyldiazirine, phenylchlorodiazirine and p-methoxy-3-phenyl-3-methyldiazirine in less than 1 ps upon excitation.25 The linear diazo isomers are interpreted as intermediates in the photolysis to carbenes.26 In fact, the excited states of aryl diazo compounds typically decay within 300 fs to form singlet aryl carbenes.22,23,27–29 This supports the idea that diazirines photoisomerize to diazo compounds and subsequently fragment to form carbenes in a stepwise fashion.19,30–32
The photodecomposition of diazirine has been studied also theoretically,33–40 and the more recent theoretical calculations favor the stepwise excited-state mechanism. The ground and excited states of both 3H-diazirine and diazomethane were studied with the complete active space second order perturbation (CASPT2) and complete active space self consistent field (CASSCF) methods,41 CASPT2//CASSCF, and it was proposed that the carbene is formed on the ground-state surface after decay of excited diazirine through a conical intersection, leading to diazomethane formation. The lowest singlet excited states of phenyldiazirine and phenyldiazomethane were also studied at the time-dependent (TD) density functional theory (DFT) level with the B3LYP functional, TD-B3LYP/6-31+G(d), and with restriction of identity (RI) coupled cluster (CC), RI-CC2/TZVP.32 It was found that in the ground state, direct formation of phenylcarbene from phenyldiazirine is favored along with the release of nitrogen, while on the excited state the stepwise path is preferred, i.e. isomerization into phenyldiazomethane followed by formation of phenylcarbene.
Turning to our system of interest, ACDA, its ground- and excited state decomposition to yield carbenes has been studied experimentally for a series of derivatives with different aryl ring substitution (see Scheme 1, where p and m refer to substitution on the para and meta positions). The lack of a Hammett correlation for the thermolysis reaction rates was explained arguing that the mechanism involved structures with mixed diradical/zwitterionic character.14 This contrasts with the results for the photolysis of several ring-substituted analogues, which was studied with ultrafast spectroscopy in different solvents (acetonitrile, chloroform and cyclohexane) by Zhang et al.15 In this case the first excited state (S1) lifetimes of ACDA increase with solvent polarity and the electron-donating ability of X substituents, and excellent Hammett correlations between S1 state lifetimes and the substituent constants (σ+p) were established. This was interpreted in terms of a dipolar intermediate, which was confirmed by TD-DFT and RI-CC2 calculations.32 In our study, we use mutireference methods (CASSCF and CASPT2) to reproduce the reactivity trends in the ground and excited states and extract the molecular details that explain the different behaviors. In the ground state, there is only a small amount of charge transfer from the ring to the diazirine, consistent with the lack of a Hammett correlation. In contrast, the excitation induces initially a substantial intramolecular charge transfer to the ring; during the C–N cleavage reaction, the charge transfer is reversed, and this explains the stabilization of the S1 species by polar solvents and electron donating substituents on the ring.
Computational details
The calculations were carried out with the CASPT2//CASSCF approach, where the geometries of critical points, i.e. ground and first singlet excited state minima and transition structures, (S0)-Min, (S1)-Min, (S0)-TS and (S1)-TS, and S1/S0 conical intersection (S1/S0)-X, were optimized at the CASSCF level (the geometries and selected bond lengths are shown in Fig. 1), and the energies recalculated at the CASPT2 level to account for dynamic correlation. Excited-state optimizations were carried out state-averaging over S1 and S0 with equal weights and the TSs were characterized with numerical frequency calculations. The 6-31G* and ANO-S basis sets (contraction scheme 3s2p1d for all atoms except 2s1p for H) were used for CASSCF and CASPT2 calculations, respectively. The CASSCF and TD-CAM-B3LYP/6-31G* calculations were carried out with the Gaussian 0342 and Gaussian 09 packages,43 and the CASPT2 ones with Molcas 8.0 software.44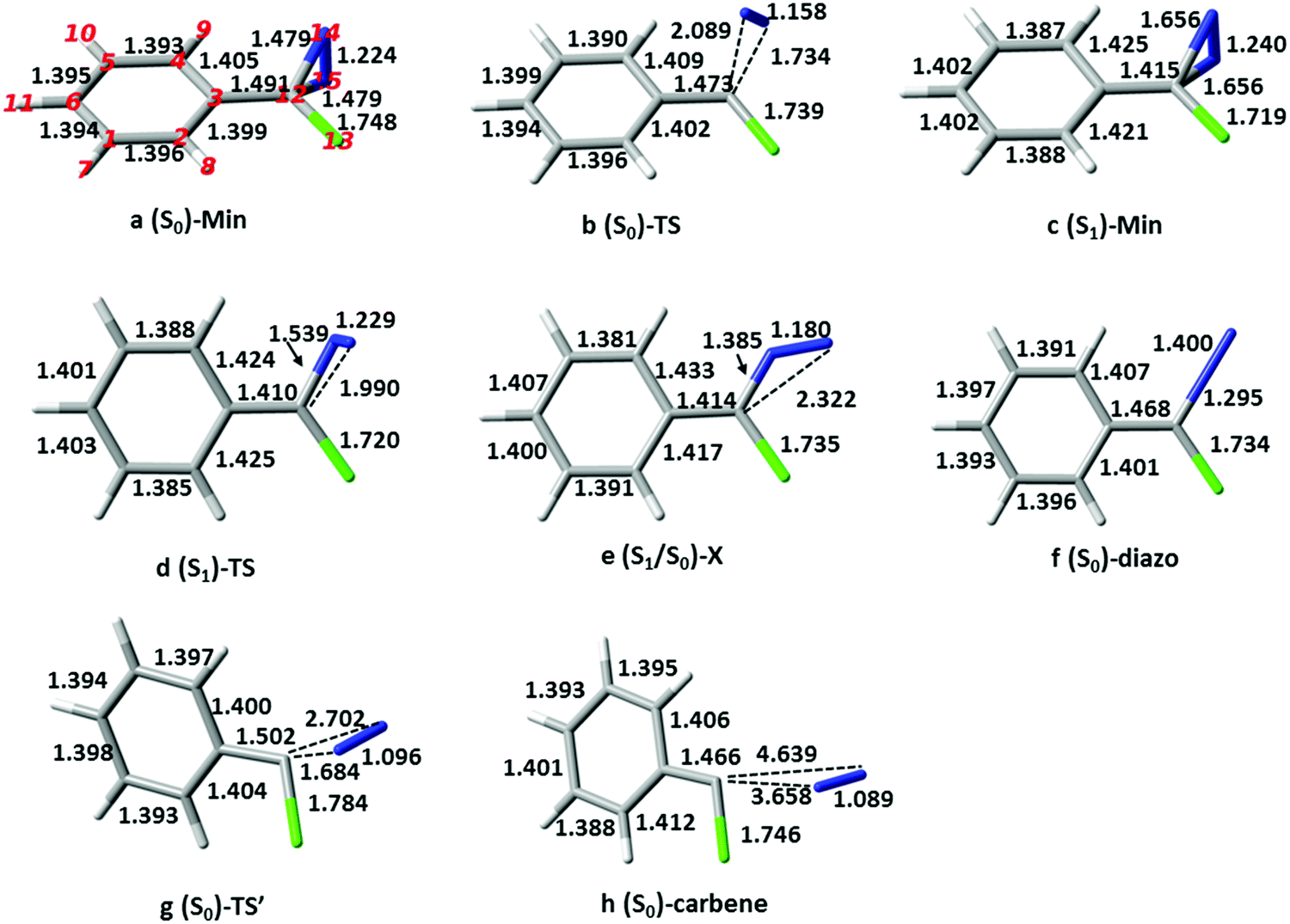 | ||
| Fig. 1 The critical structures of p-H-ACDA. | ||
The complete active space of p-H-ACDA at (S0)-Min, which has Cs symmetry (see Fig. 1 for the structure and atomic labelling), is 14 electrons in 13 orbitals. This comprises four occupied and four unoccupied π orbitals of a′′ symmetry resulting from combination of the benzene π orbitals and two orbitals localized on the C12–N14 and C12–N15 bonds; two π orbitals of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond (labelled as πNN and πNN*), of a′ and a′′ symmetry; two σ orbitals localized on the C12–N14 and C12–N15 bonds (of a′ symmetry); and the 3p orbital of Cl. This orbital was not included in the final active space because its electron occupation number in test calculations is almost 2.0. The resulting (12, 12) active space (12 electrons in 12 orbitals) for p-H-ACDA is shown in Fig. 2. An analogous active space was used for the remaining derivatives (see Tables ESI3–ESI7 in the ESI†). This active space choice provides a balanced description of the excited state and ground state because the energy degeneracy at (S1/S0)-X optimized at the CASSCF level is kept at the CASPT2 level within 0.1 eV, which validates the choice. The CASPT2 energies of optimized critical points were calculated averaging over three states with equal weights except for the vertical excitations at (S0)-Min with five states. An IPEA45 parameter of 0.0 and an imaginary level shift46 of 0.1 were used in all CASPT2 calculations.
N bond (labelled as πNN and πNN*), of a′ and a′′ symmetry; two σ orbitals localized on the C12–N14 and C12–N15 bonds (of a′ symmetry); and the 3p orbital of Cl. This orbital was not included in the final active space because its electron occupation number in test calculations is almost 2.0. The resulting (12, 12) active space (12 electrons in 12 orbitals) for p-H-ACDA is shown in Fig. 2. An analogous active space was used for the remaining derivatives (see Tables ESI3–ESI7 in the ESI†). This active space choice provides a balanced description of the excited state and ground state because the energy degeneracy at (S1/S0)-X optimized at the CASSCF level is kept at the CASPT2 level within 0.1 eV, which validates the choice. The CASPT2 energies of optimized critical points were calculated averaging over three states with equal weights except for the vertical excitations at (S0)-Min with five states. An IPEA45 parameter of 0.0 and an imaginary level shift46 of 0.1 were used in all CASPT2 calculations.
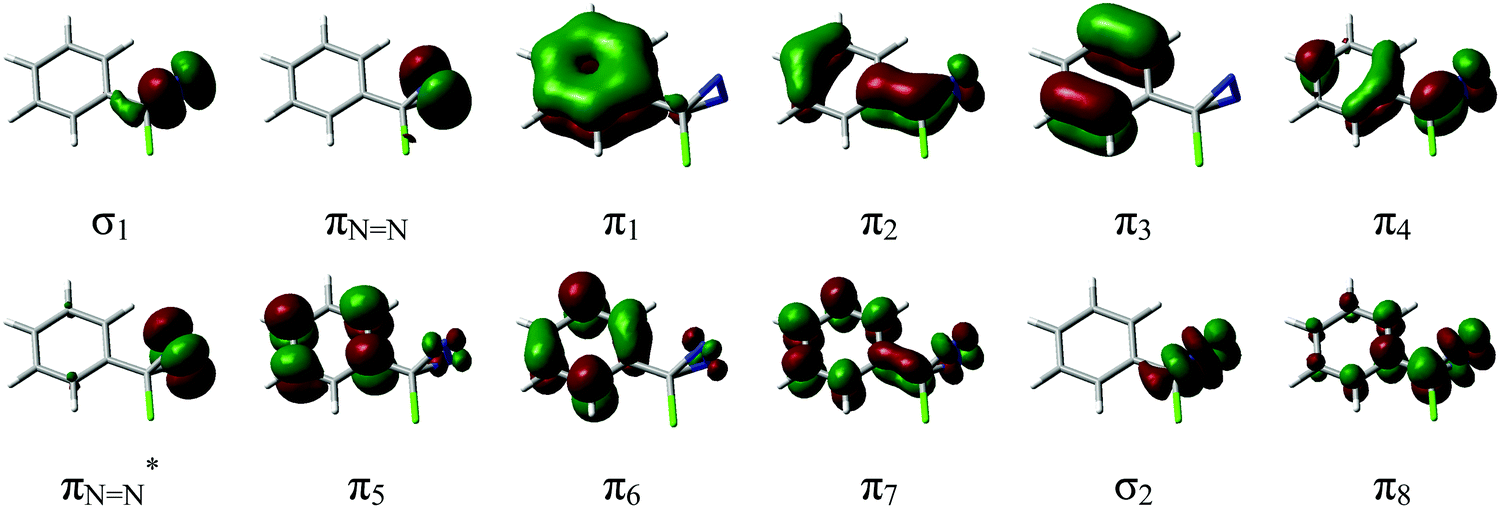 | ||
| Fig. 2 CASSCF Active space orbitals at p-H-ACDA (S0)-Min. | ||
The barriers provided in Tables 1 and 3 correspond to free energy barriers including the zero-point energy (ZPE) correction. Because of the large active spaces, the state-averaged CASSCF optimizations do not include the orbital rotation contributions to the gradients.47 This renders the CASSCF calculation of the ZPE unreliable. For this reason, the ZPE at (S0)-TS and (S1)-TS was obtained at the CAM-B3LYP and TD-CAM-B3LYP level, respectively, and the values in Tables 1 and 3 correspond to CASPT2//CASSCF energies with TD-CAM-B3LYP ZPE correction. The calculations in the solvent of interest (cyclohexane for the ground state and acetonitrile for the excited state) were carried out using the cavity-based reaction-field polarizable continuum model (PCM)48 both for the CASSCF optimizations and the CASPT2 single-points.
| R | ΔGrela [kcal mol−1] | Δqb [a.u.] | k dec [10−4 s−1] | |
|---|---|---|---|---|
| Mull.c | TFVCd | |||
| a CASPT2//CASSCF including solvent and ZPE correction, see Computational details. b Difference between C12, N14 and N15 atomic charges at (S0)-Min and (S0)-TS (qTS − qMin) in vacuum. c Charges obtained from Mulliken analysis. d Charges obtained from TFVC analysis (see Computational details). | ||||
| p-CH3O | 25.1 | −0.07 | −0.05 | 6.2 |
| p-CH3 | 27.9 | −0.05 | −0.06 | 2.91 |
| p-Cl | 28.4 | −0.04 | −0.05 | 2.48 |
| p-H | 28.4 | −0.03 | −0.06 | 1.95 |
| p-NO2 | 28.6 | −0.03 | −0.02 | 2.04 |
The atomic charges presented in Tables 1 and 3 were obtained with a Hilbert-space and a real-space approach to validate that the conclusions are independent from the method used to obtain the charges. The methods of choice are Mulliken population (Tables ESI1 and ESI2, ESI†) and topological fuzzy Voronoi cells (TFVC) (Table ESI3, ESI†).49 The TFVC method is a computationally efficient real-space partition approach that gives similar results to the quantum theory of molecules in atoms.50
Results
Ground-state reactivity
The ground-state reactivity in cyclohexane has been calculated for the five derivatives for which the experimentally rates are available (see Table 1).14 The stationary structures of p-H-ACDA are shown as an example in Fig. 1 along with the selected bond parameters. (S0)-Min (Fig. 1a) has Cs symmetry with the N–N bond perpendicular to the hexatomic ring plane, and the C–N and NN bond lengths are 1.479 and 1.224 Å. These values agree with previous RI-CC2 results of 1.468 and 1.274 Å.15 The structures or the remaining derivatives are shown in the ESI† (Fig. ESI1–ESI6). The structural differences with respect to the parent compound are small.
The calculated barriers for thermal decomposition of the ACDA derivatives are provided in Table 1 together with the experimental rate constants. They range from 25 to 29 kcal mol−1, approximately, and there is a good correlation between the calculated barriers and log(k), with R2 = 0.94 (see Fig. ESI7, ESI†). The formation of the carbene is endothermic by 9–14 kcal mol−1 (see Table ESI3, ESI†). The mechanism corresponds to an asynchronous, one-step nitrogen elimination reaction with the diazirine C–N bonds stretched to approximately 1.732 and 2.091 Å (see Fig. 1b and Fig. ESI1–ESI6, ESI† for the remaining derivatives). This is in agreement with previous calculations on phenyldiazirine32 and phenylchlorodiazirine.51 Analysis of the atomic charges shows that at (S0)-TS there is only a small charge transfer from the ring to the diazo moiety compared to (S0)-Min, of less than 0.1 electron computed either with Mulliken or TFVC analysis (see the Δq values in Table 1). This explains the lack of a correlation between the Hammett substituent parameters and the thermal decomposition rate.14 It is also consistent with the description of the thermal decomposition as the reverse of a carbene attack on a nitrogen molecule,32 which would correspond to an asynchronous [1+2] cycloaddition.
Excited-state reactivity
N bond orthogonal to the plane. The small overlap between the two orbitals causes the small oscillator strength of 0.005. TD-CAMB3LYP provides similar results of 3.36 eV and oscillator strength 0.004. Importantly, the excitation features a partial intramolecular charge transfer (CT) from the phenyl ring to the diazirine nitrogen atoms (see the orbitals involved in the transition in Fig. 2). This can be also recognized from the increase in the dipole moment from 3.10 Debye at the ground state to 5.72 Debye in S1, and by an increase in the negative charge of the two diazirine nitrogens of 0.10 per atom, as measured from the Mulliken charges (see Table ESI2, ESI†). The remaining derivatives show similar S1 excitation energies of 3.17–3.42 eV (see Tables ESI4–ESI8, ESI†) and a similar charge shift to the diazirine ring (see Table ESI2, ESI†). The higher-lying states appear, for all derivatives, at about 1 eV higher than S1 at the CASPT2 level. Given that the ultrafast photolysis measurements were initiated with 375 nm excitation,15 one can assume that the photolysis involves only the S1 state. Calculations of S2 at the critical points along the photolysis path support this assumption. In line with previous studies,31 we have also not considered triplet states in our calculations because intersystem crossing seems unlikely in the short excited-state times measured experimentally. The good agreement found between the experimental data and our results support these assumptions.
| State | CASPT2/ANO-S | TD-CAM-B3LYP/6-311G** | |||||
|---|---|---|---|---|---|---|---|
| Main configurationsa | Energyb [eV] | f | μ [D] | Main configurationsa,d | Energyb [eV] | f | |
| a Transition contribution of leading configurations. b Transition wave length [nm] in parentheses. c Ground-state dipole moment 3.10. d See ESI for the CAM-B3LYP orbitals, Fig. ESI8. | |||||||
| S1 | π4 → πNN* (0.68) |
3.35 (370) | 0.005 | 5.72 | π4 → πNN* (0.75) |
3.36 (369) | 0.004 |
| S2 |
π3 → π5 (0.35)
π4 → π6 (0.21) |
4.41 (281) | 0.002 | 3.09 |
π3 → πNN* (0.34)
π3 → π5 (0.23) π3 → π6 (0.22) |
5.24 (237) | 0.001 |
| S3 | π3 → πNN* (0.68) |
5.14 (243) | 0.008 | 11.4 | π3 → πNN* (0.53) |
5.56 (223) | 0.005 |
| S4 | π2 → πNN* (0.50) |
5.44 (228) | 0.010 | 7.50 | π2 → πNN* (0.62) |
5.87 (211) | 0.070 |
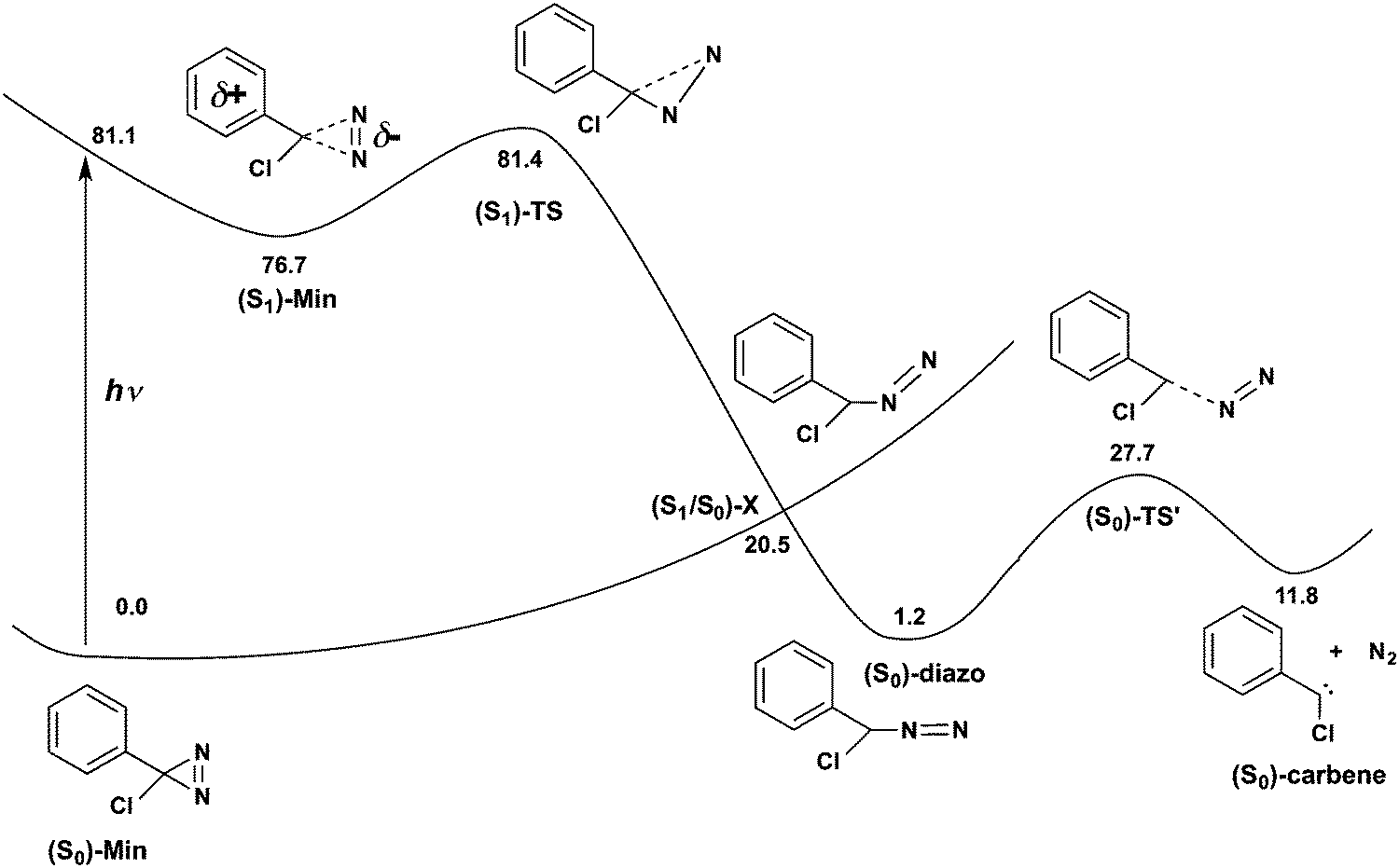 | ||
| Fig. 3 Schematic representation of the excited-state reactivity of p-H-ACDA, showing cleavage of a C–N diazirine bond to yield phenylchlorodiazomethane and further evolution to phenylcarbene. Numbers represent CASPT2//CASSCF energy in vacuum in kcal mol−1. | ||
The path continues to an open-ring, diazomethane like structure. This structure is a conical intersection with the ground state, (S1/S0)-X. The presence of the intersection explains the disappearance of the transient absorption signal in the experiments,32 since it provides a way for the molecule to deactivate to the ground state. (S1/S0)-X has a planar structure with a bent C–N–N moiety. Such a bent C–N–N group at the conical intersection is characteristic of the conical intersections found for other diazo compounds52,53 and azides.54,55 Analysis of the surface topology near the intersection shows that the crossing is connected to a single ground-state minimum (see Fig. ESI9, ESI† for details). This structure is the aryl, chloro-diazomethane minimum with a linear C–N–N group, as confirmed by an intrinsic reaction coordinate calculation. This minimum lies 1.2 kcal mol−1 higher in energy than (S0)-Min. From the diazomethane minimum, the reaction proceeds further with cleavage of the second CN bond to yield the carbene. The second C–N cleavage has a barrier of 26.5 kcal mol−1, similar to the barrier of 34.0 obtained at the RI-CC2 level.32
The calculated reaction barriers for the six derivatives measured in ref. 8 are presented in Table 3 and the Hammett correlations in Fig. 4a and b. The structures are presented in Fig. ESI1–ESI6, ESI.† In the vacuum, the barriers range from 3.8 kcal mol−1 for the most electron withdrawing CF3 substituent to 5.9 kcal mol−1 for the best electron donor, CH3O. There is a good correlation between the calculated barriers and the σ+ Hammett constants (R2 = 0.96). In acetonitrile solution, the barriers are raised by 0.1–0.5 kcal mol−1, and the correlation between the calculated barriers and the σ+ Hammett constants is maintained (R2 = 0.97). There is also a good correlation (R2 = 0.98) between the calculated barriers and the logarithm of the experimental S1 lifetimes, log(τ) (see Fig. 4b). The increase in the calculated barriers from the vacuum to the polar acetonitrile solvent is also consistent with the increase of the S1 lifetimes observed experimentally with increasing solvent polarity, going from cyclohexane to chloroform and acetonitrile.31
| R | ΔGvacrel [kcal mol−1] | ΔGvacrel [kcal mol−1] | ΔqMin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b [a.u.] b [a.u.] |
ΔqTSc [a.u.] |
log τf |
σ
+g |
||
|---|---|---|---|---|---|---|---|---|
| Mull.d | TFVCe | Mull.d | TFVCe | |||||
| a CASPT2//CASSCF including ZPE correction, see Computational details. b Difference between C12, N14 and N15 atomic charges at (S0)-Min (ground state) and (S1)-Min (excited state) (qS1-Min − qS0-Min) in vacuum (see Table ESI2). c Difference between C12, N14 and N15 atomic charges at (S0)-Min (ground state) and (S1)-TS (excited state) (qS1-TS − qS0-Min) in vacuum. d Charges obtained from Mulliken analysis. e Charges obtained from TFVC analysis. f Life times in 10−12 s. g Ref. 56 and 57. | ||||||||
| p-CF3 | 3.8 | 4.0 | −0.16 | −0.25 | −0.08 | −0.19 | 1.03 | 0.53 |
| m-Cl | 4.4 | 4.6 | −0.16 | −0.25 | −0.08 | −0.19 | 1.34 | 0.37 |
| p-Cl | 4.6 | 4.8 | −0.17 | −0.25 | −0.09 | −0.19 | 1.70 | 0.11 |
| p-H | 4.7 | 5.1 | −0.17 | −0.26 | −0.09 | −0.19 | 1.79 | 0.00 |
| p-CH3 | 5.1 | 5.6 | −0.18 | −0.26 | −0.09 | −0.20 | 2.18 | −0.31 |
| p-CH3O | 5.9 | 6.3 | −0.18 | −0.26 | −0.09 | −0.19 | 2.88 | −0.78 |
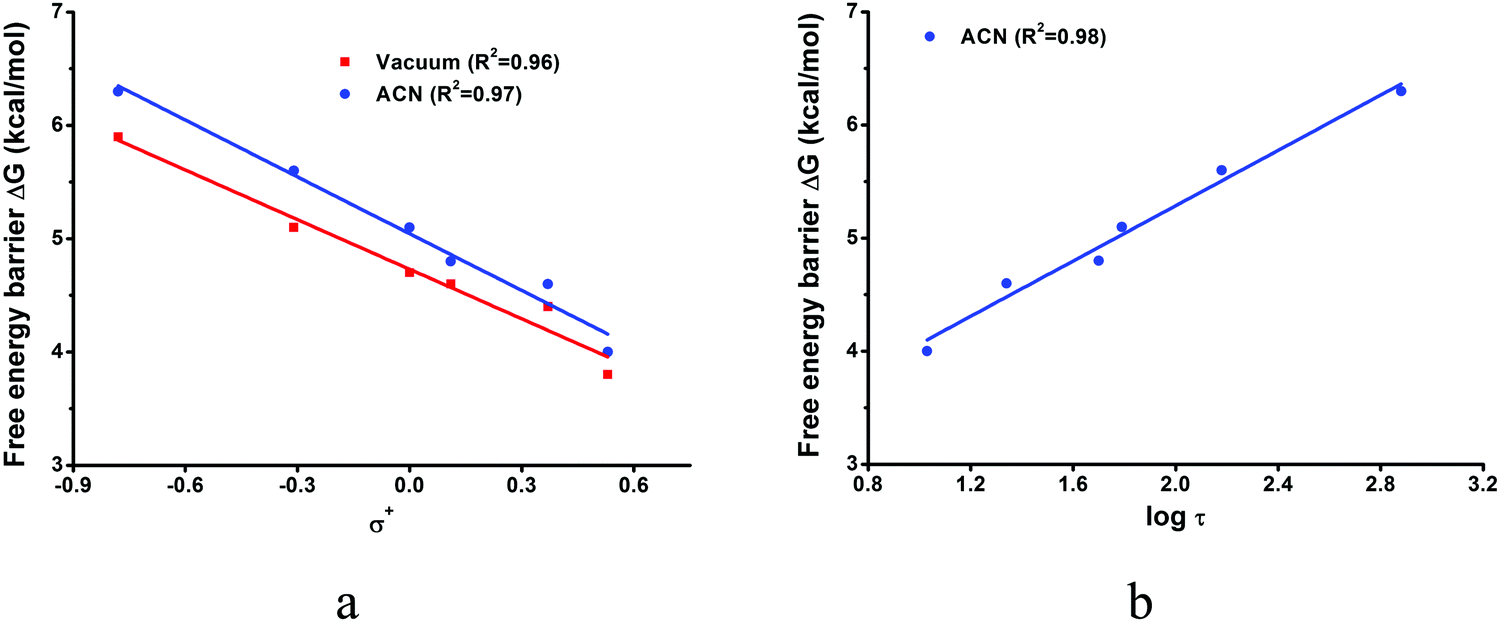 | ||
| Fig. 4 Linear correlation plot of free energy barriers for photolytic decomposition of p-R-ACDA derivatives in vacuum and acetonitrile vs. substituent constants σ+ (a) and free energy barriers in acetonitrile vs. logarithm of experimental S1 life times measured in acetonitrile8 (b). Detailed values are shown in Table 3. | ||
This behaviour can be understood examining the atomic charges at the critical points. To measure the charge transfer from the phenyl ring to the diazirine moiety induced by the excitation, we have calculated the difference between the atomic charges at (S0)-Min and (S1)-Min (ΔqMin in Table 3) and between (S0)-Min and (S1)-TS (ΔqTS). The charges have been obtained from Mulliken and real-space TFVC analyses49 (see Computational details section.) They indicate that the excitation induces a substantial transfer of negative charge from the phenyl ring to the diazirine moiety at (S1)-Min, of −0.16 to −0.18 electrons (Mulliken charges at ΔqMin in Table 3). However, during the first C–N cleavage there is a partial reverse transfer, and the relative charge transfer, ΔqTS, is reduced to −0.08 and −0.09. This trend is confirmed by the TFVC analysis, although this method gives a higher amount of charge transfer compared to the Mulliken analysis, both at (S1)-Min (−0.25 to −0.26) and (S1)-TS (−0.19). However, the reverse charge transfer at the TS is also observed with the real-space partition analysis, which validates this mechanistic feature.
The reverse charge transfer increases gradually along the path to (S1/S0)-X, where there is a net transfer of positive charge compared to the ground state (see Table ESI2, ESI†). The initial transfer of negative charge and its reversal along the C–N cleavage path is consistent with the two trends that correlate the reactivity with the electron-donating capacity of the substituents and the solvent polarity. Electron donating substituents stabilize (S1)-Min, but the stabilizing effect is lost during the C–N cleavage because of the reverse charge transfer. This explains the higher barriers found with electron donating substituents. In addition, the S1 state has a higher dipole moment at the intact minimum than at the C–N broken structures, which explains the stabilization of the reactant and the increase of the barrier with increasing solvent polarity, or going from vacuum to acetonitrile. Our calculated energies are consistent with both trends.
The activated C–N cleavage path is also consistent with the experimental S1 life times of 11–760 ps measured in acetonitrile. After excitation, (S1)-Min carries an excess energy approximately equal to the difference between the excitation energy and the (S1)-Min potential energy. Initially this excess energy will be accumulated in the two C–N stretch modes activated during the excitation. However, the excess excitation energy is of similar magnitude to the barrier at the TS (see Table ESI11, ESI†). Therefore, cleavage of a single C–N bond requires intramolecular vibrational energy redistribution from the two bonds to a single one, so that (S1)-Min has enough time to equilibrate vibrationally before the cleavage. This is reflected in the S1 life times. Under these conditions, the rates follow, approximately, standard transition state theory, and the Hammett correlation can be observed.
Conclusions
Quantum chemical calculations have allowed us to rationalize the different reactivity found for ACDA derivatives in the ground and excited states. The ground-state nitrogen elimination reaction can be understood as a reverse cycloaddition with small charge transfer from the aromatic ring. In contrast, the excited-state elimination is stepwise and starts with cleavage of a single C–N bond to yield a diazomethane intermediate. This step is characterized by an initial charge transfer from the ring to the diazirine group which is reversed during the cleavage. As a result, the barrier for photolysis increases with a higher electron donating capability of the ring substituents. This is in agreement with the trend of the rates measured experimentally. While the reactivity trends can be explained from straightforward orbital and Mulliken charge analyses, the ACDA system appears as a good test ground for more sophisticated reactivity descriptors because consistent sets of data are available both for the ground and the excited state.Our calculations also show that the Hammett behaviour in the excited state follows the usual scheme from ground-state reactions, i.e. the different reaction rates are due to differential stabilization of the TS by substitution of the aromatic ring. This implies that the reaction follows traditional transition state theory, which means that the excited-state minimum is sufficiently long lived to be thermally equilibrated. The question that remains open is whether excited-state processes in the sub-ps scale, which may not necessarily obey transition state theory, can also show a Hammett behaviour. This may be the case, for instance, if the excited-state decay takes place at a conical intersection and the intersection energy is modulated by the substituent. This would be a further extension of the applicability of the Hammett approach which appears as an interesting direction for future studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Pedro Salvador (Universitat de Girona) for help with the TFVC analysis. We acknowledge financial support from the National Natural Science Foundation of China (grants 21773007 and 21303007 for QL); the Spanish Ministerio de Economía y Competitividad (Grants RYC-2011-09582 for AM and CTQ-2015-69363-P for LB); and the Departament d'Innovació, Universitats i Empresa (DIUE), Generalitat de Catalunya (Xarxa de Referència en Química Teòrica i Computacional).References
- F. Plasser, A. J. Aquino, W. L. Hase and H. Lischka, J. Phys. Chem. A, 2012, 116, 11151–11160 CrossRef CAS PubMed.
- S. A. Bäppler, F. Plasser, M. Wormit and A. Dreuw, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 90, 052521 CrossRef.
- S. Jenkins, L. Blancafort, S. R. Kirk and M. J. Bearpark, Phys. Chem. Chem. Phys., 2014, 16, 7115–7126 RSC.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- D. Casanova and A. I. Krylov, J. Chem. Phys., 2016, 144, 014102 CrossRef PubMed.
- M. Rosenberg, C. Dahlstrand, K. Kilsa and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- K. A. Kistler and S. Matsika, J. Phys. Chem. A, 2007, 111, 8708–8716 CrossRef CAS PubMed.
- X. Zhang, L.-X. Ma and X.-D. Li, Synth. Met., 2014, 198, 357–360 CrossRef CAS.
- I. K. Petrushenko and K. B. Petrushenko, Spectrochim. Acta, Part A, 2015, 138, 623–627 CrossRef CAS PubMed.
- A. Harriman and R. J. Hosie, J. Chem. Soc., Faraday Trans., 1981, 77, 1695–1702 RSC.
- P. J. Wagner, R. J. Truman and J. C. Scaiano, J. Am. Chem. Soc., 1985, 107, 7093–7097 CrossRef CAS.
- M. Sakamoto, X. C. Cai, M. Hara, M. Fujitsuka and T. Majima, J. Phys. Chem. A, 2005, 109, 4657–4661 CrossRef CAS PubMed.
- R. S. Givens and B. Matuszewski, J. Am. Chem. Soc., 1984, 106, 6860–6861 CrossRef CAS.
- M. T. H. Liu and K. Toriyama, Can. J. Chem., 1972, 50, 3009–3016 CrossRef CAS.
- Y. L. Zhang, L. Wang, R. A. Moss and M. S. Platz, J. Am. Chem. Soc., 2009, 131, 16652–16653 CrossRef CAS PubMed.
- J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, New York, 3rd edn, 1985 Search PubMed.
- R. A. Moss, Acc. Chem. Res., 2006, 39, 267–272 CrossRef CAS PubMed.
- B. Raimer and T. Lindel, Chemistry, 2013, 19, 6551–6555 CrossRef CAS PubMed.
- R. Bonneau and M. T. H. Liu, J. Am. Chem. Soc., 1996, 118, 7229–7230 CrossRef CAS.
- M. S. Platz, J. Org. Chem., 2014, 79, 2341–2353 CrossRef CAS PubMed.
- J. Peon, D. Polshakov and B. Kohler, J. Am. Chem. Soc., 2002, 124, 6428–6438 CrossRef CAS PubMed.
- J. Wang, G. Burdzinski, J. Kubicki and M. S. Platz, J. Am. Chem. Soc., 2008, 130, 11195–11209 CrossRef CAS PubMed.
- J. Wang, J. Kubicki, T. L. Gustafson and M. S. Platz, J. Am. Chem. Soc., 2008, 130, 2304–2313 CrossRef CAS PubMed.
- J. Wang, G. Burdzinski, J. Kubicki, M. S. Platz, R. A. Moss, X. L. Fu, P. Piotrowiak and M. Myahkostupov, J. Am. Chem. Soc., 2006, 128, 16446–16447 CrossRef CAS PubMed.
- Y. L. Zhang, G. Burdzinski, J. Kubicki and M. S. Platz, J. Am. Chem. Soc., 2008, 130, 16134–16135 CrossRef CAS PubMed.
- Y. L. Zhang, G. Burdzinski, J. Kubicki, S. Vyas, C. M. Hadad, M. Sliwa, O. Poizat, G. Buntinx and M. S. Platz, J. Am. Chem. Soc., 2009, 131, 13784–13790 CrossRef CAS PubMed.
- J. Wang, G. Burdzinski, T. L. Gustafson and M. S. Platz, J. Org. Chem., 2006, 71, 6221–6228 CrossRef CAS PubMed.
- J. Wang, G. Burdzinski, T. L. Gustafson and M. S. Platz, J. Am. Chem. Soc., 2007, 129, 2597–2606 CrossRef CAS PubMed.
- J. Wang, J. Kubicki, E. F. Hilinski, S. L. Mecklenburg, T. L. Gustafson and M. S. Platz, J. Am. Chem. Soc., 2007, 129, 13683–13690 CrossRef CAS PubMed.
- R. Bonneau, M. T. H. Liu, K. C. Kim and J. L. Goodman, J. Am. Chem. Soc., 1996, 118, 3829–3837 CrossRef CAS.
- M. Nigam, M. S. Platz, B. M. Showalter, J. P. Toscano, R. Johnson, S. C. Abbot and M. M. Kirchhoff, J. Am. Chem. Soc., 1998, 120, 8055–8059 CrossRef CAS.
- Y. L. Zhang, S. Vyas, C. M. Hadad and M. S. Platz, J. Phys. Chem. A, 2010, 114, 5902–5912 CrossRef CAS PubMed.
- P. L. Müller-Remmers and K. Jug, J. Am. Chem. Soc., 1985, 107, 7275–7284 CrossRef.
- F. Bernardi, M. Olivucci, M. A. Robb, T. Vreven and J. Soto, J. Org. Chem., 2000, 65, 7847–7857 CrossRef CAS PubMed.
- R. R. Pandey, Y. G. Khait and M. R. Hoffmann, J. Phys. Chem. A, 2004, 108, 3119–3124 CrossRef CAS.
- I. Fedorov, L. Koziol, A. K. Mollner, A. I. Krylov and H. Reisler, J. Phys. Chem. A, 2009, 113, 7412–7421 CrossRef CAS PubMed.
- D. S. Ahn, S. Y. Kim, G. I. Lim, S. Lee, Y. S. Choi and S. K. Kim, Angew. Chem., Int. Ed., 2010, 49, 1244–1247 CrossRef CAS PubMed.
- R. M. Mokambe, Y. G. Khait and M. R. Hoffmann, J. Phys. Chem. A, 2010, 114, 8119–8125 CrossRef CAS PubMed.
- F. Pfeiffer and G. Rauhut, J. Phys. Chem. A, 2011, 115, 11050–11056 CrossRef CAS PubMed.
- A. E. Keating, M. A. Garcia-Garibay and K. N. Houk, J. Am. Chem. Soc., 1997, 119, 10805–10809 CrossRef CAS.
- J. F. Arenas, I. López-Tocón, J. C. Otero and J. Soto, J. Am. Chem. Soc., 2002, 124, 1728–1735 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03 Rev. E1, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Rev. D1, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- F. Aquilante, L. De Vico, N. Ferré, G. Ghigo, P.-Å. Malmqvist, P. Neogrady, T. B. Pedersen, M. Pitonak, M. Reiher, B. O. Roos, L. Serrano-Andrés, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed.
- G. Ghigo, B. O. Roos and P.-Å. Malmqvist, Chem. Phys. Lett., 2004, 396, 142–149 CrossRef CAS.
- N. Forsberg and P.-Å. Malmqvist, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef CAS.
- N. Yamamoto, T. Vreven, M. A. Robb, M. J. Frisch and H. B. Schlegel, Chem. Phys. Lett., 1996, 250, 373–378 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- P. Salvador and E. Ramos-Cordoba, J. Chem. Phys., 2013, 139, 071103 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- L. A. Zapata, S. López, P. Ruiz, J. Quijano and R. Notario, Struct. Chem., 2016, 28, 597–605 CrossRef.
- Q. Li, A. Migani and L. Blancafort, J. Phys. Chem. Lett., 2012, 3, 1056–1061 CrossRef CAS PubMed.
- H. Li, A. Migani, L. Blancafort, Q. Li and Z. Li, Phys. Chem. Chem. Phys., 2016, 18, 30785–30793 RSC.
- X.-L. Peng, W.-L. Ding, Q.-S. Li and Z.-S. Li, Org. Chem. Front., 2017, 4, 1153–1161 RSC.
- J. Ghosh, S. Banerjee and A. Bhattacharya, Chem. Phys., 2017, 494, 78–89 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- L. P. Hammett, Chem. Rev., 1935, 17, 125–136 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp07281c |
| This journal is © the Owner Societies 2018 |