
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Secondary relaxation in ultrastable etoricoxib: evidence of correlation with structural relaxation†
Cristian
Rodríguez-Tinoco
 *ab,
Marzena
Rams-Baron
ab,
K. L.
Ngai
b,
Karolina
Jurkiewicz
ab,
Javier
Rodríguez-Viejo
c and
Marian
Paluch
ab
*ab,
Marzena
Rams-Baron
ab,
K. L.
Ngai
b,
Karolina
Jurkiewicz
ab,
Javier
Rodríguez-Viejo
c and
Marian
Paluch
ab
aInstitute of Physics, University of Silesia, Uniwersytecka 4, 40-007 Katowice, Poland. E-mail: cristian.rodriguez-tinoco@smcebi.edu.pl
bSilesian Center for Education and Interdisciplinary Research, 75 Pulku Piechoty 1A, 41-500 Chorzow, Poland
cGroup of Nanomaterials and Microsystems, Physics Department, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
First published on 23rd January 2018
Abstract
Secondary relaxations are fundamental for their impact in the properties of glasses and for their inseparable connection to the structural relaxation. Understanding their density dependence and aging behavior is key to fully address the nature of glasses. Ultrastable glasses establish a new benchmark to study the characteristics of secondary relaxations, since their enthalpy and density levels are unattainable by other routes. Here, we use dielectric spectroscopy at ambient and elevated pressures to study the characteristics of the secondary relaxation in ultrastable etoricoxib, reporting a 71% decrease in dielectric strength and one decade increase in relaxation time compared to the ordinary glass. Interestingly, we find an unprecedented connection between secondary and structural relaxations in ultrastable etoricoxib in exactly the same manner as in the ordinary glass, manifested through different properties, such as aging and devitrification. These results further support and extend the general validity of the connection between the secondary and structural relaxation.
Introduction
Glasses are amorphous solids wherein structural relaxation is frozen on the laboratory time-scales. At sufficiently high temperatures, a glass is transformed into the metastable supercooled liquid state through a cooperative or intermolecularly coupled structural α-relaxation process with non-exponential correlation function.1 However, motions of molecules in the glassy state are not completely frozen, and some local and shorter range molecular relaxations can still take place. Among these secondary relaxations, the fundamentally important ones are those having properties similar to and inseparable from the structural α-relaxation,1,2 called the Johari–Goldstein (JG) relaxation. For example, JG relaxations are considerably sensitive to pressure or density increase by physical aging, while more local secondary relaxations originating from intramolecular degrees of freedom are much less pressure sensitive. The nomenclature, JG relaxation, enables distinguishing the fundamentally important secondary relaxations from the trivial ones, and is also used to honor the seminal discovery of secondary relaxations in totally rigid molecules by Johari and Goldstein.3 The JG relaxation acts as the indispensable precursor of the cooperative structural relaxation.1There are several routes to produce glasses: liquid-quenching,4 cryo-milling5 or high-pressure quenching,6 among others. The discovery of ultrastable glasses (USG) by vapor deposition represents a milestone in glass research.7,8 By adjusting the deposition conditions (mainly deposition temperature and evaporation rate), it is possible to tune the properties of the glass produced, such as its kinetics, thermodynamic stability and density, over a wide range.9–11 The enthalpy of USG is equivalent to that of ordinary glasses (OG) having been aged for millions of years, such as amber.12 Recent experiments have suggested that the high mobility of molecules landing on the surface during deposition is responsible for the enhanced stability and density of USG.13,14 Some works have associated the surface mobility or the structural relaxation of ultrathin films with the primitive relaxation time, τ0 of the Coupling Model or equivalently the JG-β relaxation time τJG.15 Moreover, it was shown recently that the JG-β relaxation is a rate limiting factor of the growth of vapor deposited glasses.16 Therefore, it seems that there is a strong connection between some secondary relaxations persistent in the glassy state with the characteristics of USG, and the connection offers insight into the dynamics of USG and a new benchmark to test theories of glasses.
Recently, it was shown that the intensity of the JG-β relaxation process in vapor deposited glasses of toluene can be tuned by changing the deposition temperature during the growth process.17 A decrease of around 70% in the intensity of the secondary JG process was observed in the USG compared to the OG. Also, the relaxation time of the process was seen to be longer in the USG than in the OG. This observation agrees with the previous knowledge, according to which aging and lower cooling rates applied during the formation of OG can yield less intense and slower secondary relaxation processes.18–20 More recently, it was shown that glasses of etoricoxib prepared at high pressure exhibited a slower secondary relaxation process attributed to the measured higher density of the system.21 In this work, we study this secondary relaxation process observed in USG and OG of etoricoxib focusing on its dependence on temperature and pressure, and its connection with the structural relaxation in properties.
Experimental methods
Preparation of samples
Crystalline etoricoxib was purchased from Aopharm with purity higher than 99% and used without further purification. USG films with thickness of 30 μm were grown by thermal evaporation within a UHV setup with base pressure of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) × 10−8mbar. The growth rate was fixed at 0.25 ± 0.02nm s−1 and measured with a quartz crystal from Inficon. The deposition temperature was set at 280 K (∼0.85Tg) and controlled using a PID controlled thermal socket. A liquid nitrogen cold trap is used to improve the vacuum quality. The films were grown onto steel electrodes used for BDS and in aluminum foil for WAXS or DSC measurements (see corresponding subsection). The OG was prepared either by annealing of the USG (as indicated in the main text) or, alternatively, by melting crystalline TPD directly onto a steel electrode on a hot plate at the melting temperature (Tm = 413 K), covered by another electrode (separated by Teflon spacers with 0.1 mm of thickness) and then cooled down in a refrigerated cooper plate.
× 10−8mbar. The growth rate was fixed at 0.25 ± 0.02nm s−1 and measured with a quartz crystal from Inficon. The deposition temperature was set at 280 K (∼0.85Tg) and controlled using a PID controlled thermal socket. A liquid nitrogen cold trap is used to improve the vacuum quality. The films were grown onto steel electrodes used for BDS and in aluminum foil for WAXS or DSC measurements (see corresponding subsection). The OG was prepared either by annealing of the USG (as indicated in the main text) or, alternatively, by melting crystalline TPD directly onto a steel electrode on a hot plate at the melting temperature (Tm = 413 K), covered by another electrode (separated by Teflon spacers with 0.1 mm of thickness) and then cooled down in a refrigerated cooper plate.
Differential scanning calorimetry (DSC)
Calorimetric measurements were carried out using Mettler-Toledo DSC 1. Temperature and enthalpy calibrations were performed using indium and zinc standards. An aluminum crucible of 40 μL in volume was filled with the USG sample obtained after scratching from the 30 μm layer evaporated on top of aluminum foil, and closed using the crucible sealing press. The examined material was heated with rate equal to 10 K min−1 until devitrification, cooled down at the same rate to produce the OG and measured again.Wide-angle X-ray scattering (WAXS)
Wide-angle X-ray scattering measurements were performed using a Rigaku-Denki D/MAX RAPID II-R diffractometer equipped with a rotating Ag anode (λKα = 0.5608 Å), an incident beam (002) graphite monochromator and an image plate in the Debye–Scherrer geometry as a detector. USG sample was obtained after scratching from the 30 μm layer evaporated on top of aluminum foil, and the obtained powder was packed into glass capillaries with a diameter of 1.5 mm and wall thickness of 0.01 mm. The measurements were carried out at room temperature and ambient pressure. An empty capillary was also measured as a reference and subtracted from the sample signal. The acquisition time for each sample was 20 h. The X-ray beam size at the sample was 0.3 mm. The two-dimensional diffraction patterns were azimuthally integrated and converted into one-dimensional functions of intensity versus scattering vector Q (Q = 4πsinθ/λ, where: 2θ is the scattering angle and λ is the wavelength of the incident X-ray beam). The intensity functions were corrected for background, polarization, absorption, incoherent Compton scattering, and normalized to the electron units. After the data correction, the total structure factor S(Q) was computed as:where I(Q) is the coherently scattered intensity, normalized to electron units,

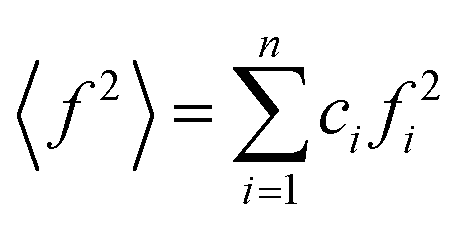 ,
, 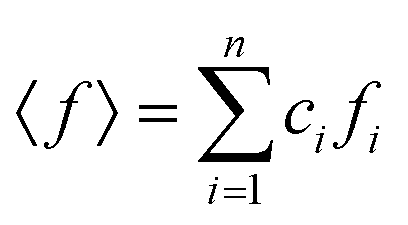 , ci and fi are the concentration and the atomic scattering factor of the i-th atomic species, respectively, and n is the number of atomic species in the sample.
, ci and fi are the concentration and the atomic scattering factor of the i-th atomic species, respectively, and n is the number of atomic species in the sample.
Broadband dielectric spectroscopy (BDS)
Isobaric dielectric measurements at atmospheric pressure were performed using a Novocontrol GMBH Alfa analyzer with frequency range from 10−2 Hz to 106 Hz. The measurement temperature was controlled by Quatro temperature controller using a nitrogen gas cryostat with accuracy better than 0.1 K. During the measurements, the samples were placed between the steel electrodes of the capacitor (15 mm diameter).Dielectric measurements at elevated pressure were performed using an automatic high-pressure system (Unipress). The sample, out of contact with compression medium by means of a Teflon casing, was fixed to the pressure chamber filled with silicon oil. The temperature during the measurement was controlled by means of a closed-circuit cooler connected to the pressure chamber.
Dielectric data is fitted by the Havriliak–Negami (HN) equation,
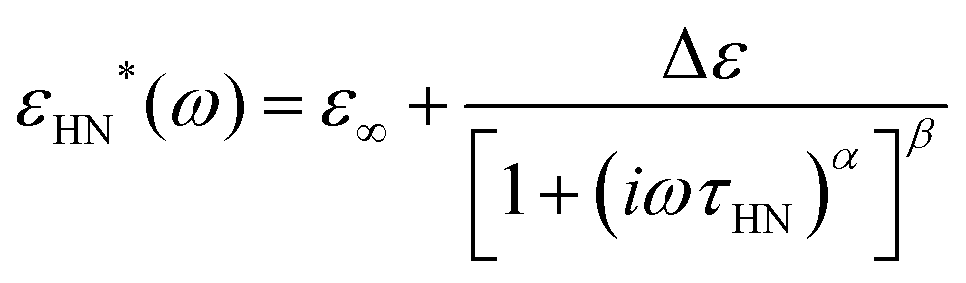
Results and discussion
The kinetic stability of the USG is elucidated in Fig. 1A, where we show the calorimetric curve of a USG vapor deposited at 280 K (∼0.85Tg) and the OG obtained after cooling the liquid at 10 K min−1. There is a 33 K increase in the onset temperature of devitrification (Ton). The area under the calorimetric peaks during the devitrification process is related to the enthalpy of the system. By integrating the heat flow data, and by matching the integrated curves in the supercooled liquid region (which is the same for OG and USG), we can determine the value of the enthalpic limiting fictive temperature (Tf′) from the intersection between the glass line and the extrapolated liquid line, as seen in the inset of Fig. 1A. For the OG, Tf′ = 327 K (in agreement with Tg = 327 K). For the USG, Tf′ = 309 K, which is 18 K below the Tg of the OG. In Fig. 1B we show the experimental structure factors of USG and OG from wide-angle X-ray scattering (WAXS) measurements. Samples are measured in powder configuration (see Methods) and hence any contribution from structural anisotropy is not observable. The total measured structure factor, S(Q), where Q is the scattering vector, can be expressed as the sum of the molecular form factors associated with the intramolecular structure (calculated from a single molecule, blue line in Fig. 1B) and the molecular packing structure factor related to intermolecular correlations. There is a slight shift towards higher Q values in the first peak of the structure factor at ∼1.45 Å−1 in the USG compared to the OG. Intramolecular correlations contribute only slightly to the first peak and, hence, the observed shift is attributed to a decrease in intermolecular distances22,23 and is calculated by the relation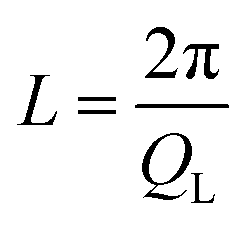 . From that shift, and assuming that the volume of the etoricoxib USG and OG is proportional to the L3, one can roughly determine an increase in density of 2–3% in the USG with respect to the OG, similar to the ∼1.5% density change observed in other USG.9,24 We note that there is also a shift in the second diffraction peak, but slightly smaller (see ESI†). The second peak is, however, strongly influenced by intramolecular structure and the relation with density is not so straightforward. Also, while no significant shift is appreciated in the region above the second peak, related with the intramolecular structure, we do observe a significant increase in the intensity of the third peak in the S(Q) measured in the USG. This may be related to a higher degree of atomic order/homogeneity within etoricoxib molecules, since this peak corresponds to intramolecular atomic correlations.
. From that shift, and assuming that the volume of the etoricoxib USG and OG is proportional to the L3, one can roughly determine an increase in density of 2–3% in the USG with respect to the OG, similar to the ∼1.5% density change observed in other USG.9,24 We note that there is also a shift in the second diffraction peak, but slightly smaller (see ESI†). The second peak is, however, strongly influenced by intramolecular structure and the relation with density is not so straightforward. Also, while no significant shift is appreciated in the region above the second peak, related with the intramolecular structure, we do observe a significant increase in the intensity of the third peak in the S(Q) measured in the USG. This may be related to a higher degree of atomic order/homogeneity within etoricoxib molecules, since this peak corresponds to intramolecular atomic correlations.
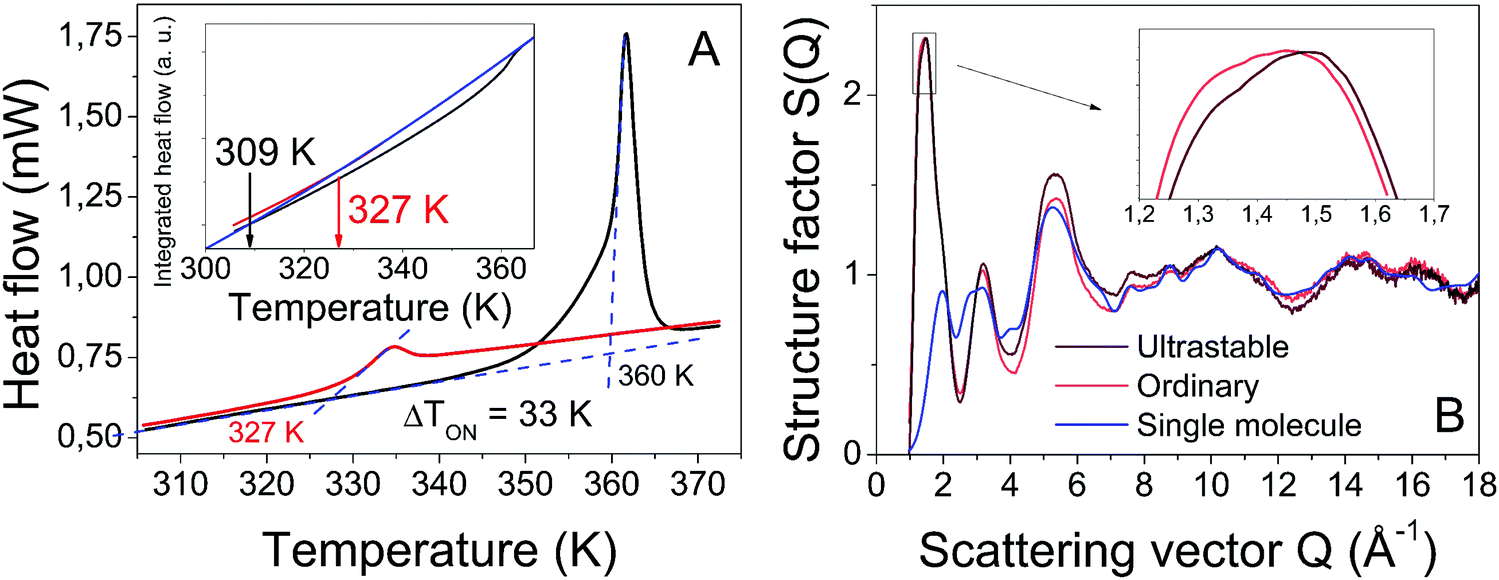 | ||
| Fig. 1 (A) Calorimetric curves measured in DSC at 10 K min−1, showing an enhancement of devitrification temperature of 33 K in the USG. In the inset, integral of heat flow data, shifted so as the liquid parts of both curves overlap. From the intersection between glassy and liquid phases we obtain TfUSG′ = 309 K and TfOG′ = 327 K. (B) Structure factors of USG and OG. The theoretical structure factor for a single etoricoxib molecule is also shown for comparison (blue line). The inset shows a zoom of the first diffraction peak. | ||
In Fig. 2A we show the dielectric spectra of the USG and OG measured at different temperatures in the glassy state. The OG was obtained after annealing an USG at 340 K (Tg + 13 K) for 12 h (see details in the ESI†). Alternatively, the glass is brought up to 352 K and cooled down, or obtained directly after melting of crystalline etoricoxib. In all cases, the obtained results are the same. From the relaxation map in Fig. 2B, we first note at Tg that the secondary relaxation time of the OG does not match the primitive relaxation time of the coupling model, as expected for a conventional JG β-relaxation involving the motion of the entire molecule.2 Noticeably, while the fitting of the secondary relaxation using a single HN curve is satisfactory for the OG, for the USG only the peak near the maximum is well fitted. This may be an indication of the presence of the weak and unresolved JG β-relaxation at lower frequencies in the dielectric spectra (see ESI†). In the inset of Fig. 2A, we indicate the expected position of the primitive relaxation frequency, consistent with the JG β-relaxation hidden between the α-relaxation and the observed secondary relaxation. The existence of the unresolved JG β-relaxation in etoricoxib is suggested also by its presence in celecoxib, a very similar molecule.25 Its dielectric relaxation strength in celecoxib is very weak and it was barely resolved as a bump in the loss spectra in the glassy state. Thus, it is not surprising that the JG β-relaxation cannot be resolved in etoricoxib, especially if it is next to and dominated by the strong secondary relaxation observed. The latter has relaxation time changing from Arrhenius dependence in the glassy state to a stronger T-dependence in the liquid state (see details of these data in the ESI†). This is a characteristic of JG processes. As we shall show later, this secondary relaxation has significant pressure dependence and related to the pressure dependence of the structural relaxation. From the connection of the properties of the secondary relaxation with the structural relaxation, we identify it as another JG process,2 faster than the genuine JG β-process, and refer it as the JG γ-process. The exact nature of this secondary relaxation has to wait until its motion is fully characterized by other techniques. The relatively large size of the etoricoxib molecule suggests that the observed faster JG γ-process is not a relaxation of the entire molecule, reserved for the JG β-relaxation, but involves the motion of only a part of it where the dipole moment resides. Notwithstanding, its pressure and density dependences is like that of the JG β-relaxation, indicating that it is also connected to the structural relaxation.
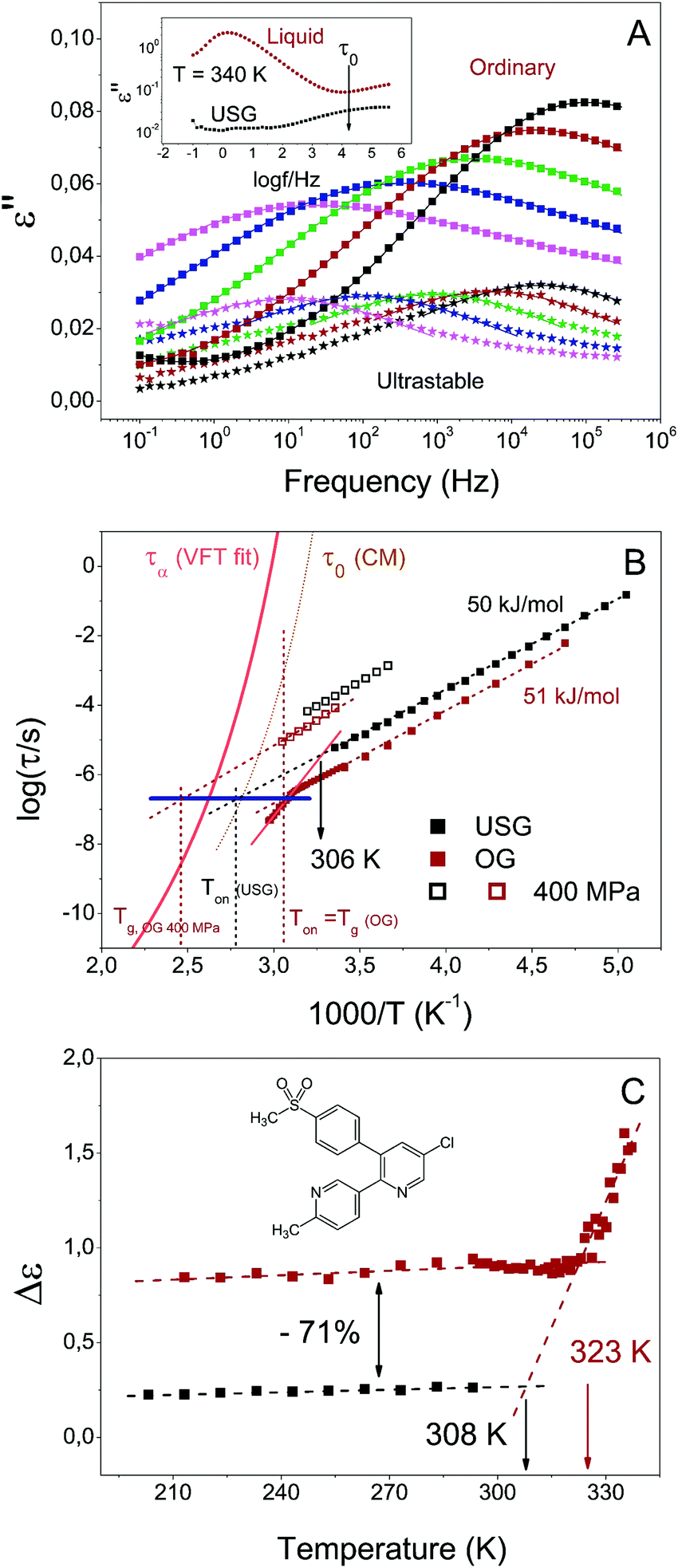 | ||
| Fig. 2 Secondary relaxation of USG and OG. (A) Dielectric loss of the secondary relaxation of USG (stars) and OG (squares), from 293 K (black) to 213 K (pink), each 20 K. In the inset, dielectric loss above Tg for the USG and transformed sample (liquid), indicating the position of the primitive relaxation, according to the coupling model. (B) Map of relaxation times for OG (red) and USG (black) etoricoxib glasses. The orange solid line corresponds to the VFT fit from τα.31 The dotted dark yellow line corresponds to the primitive relaxation time calculated from the CM and using the reported values of coupling parameters.31 The vertical dashed lines indicate the devitrification temperature at different conditions (OG, USG and OG at 400 MPa), measured at 10 K min−1. The blue horizontal line indicates the time value at which the extrapolation of the secondary relaxation time intersects the corresponding indicated devitrification temperature (C) intensity of the secondary process for the OG (and for the liquid, above Tg) and for the USG. | ||
From the data in Fig. 2, two observations can be immediately made, namely a reduction of the dielectric strength of about 71%, and an increase in secondary relaxation time of slightly less than one order of magnitude of the USG compared to the OG. The observed reduction of dielectric strength and increase of relaxation time in USG glasses was also reported for toluene, a much simpler molecule which exhibits a well resolved genuine JG β-relaxation with relaxation times in good agreement with the primitive relaxation times.17 In spite of the difference in the nature of the secondary relaxation process, the observed change in properties between OG and USG of etoricoxib is clearly analogous to that of toluene. It reinforces the interpretation of the observed secondary relaxation as a JG γ-process, since it preserves the intermolecular characteristics and properties of the JG β-process. As we mentioned earlier, the secondary relaxation time above Tg follows a stronger temperature dependence compared to the glassy state (Fig. 2B). Even though the extraction of information in this frequency range (partially out from the accessible window) is very sensitive to data analysis (see ESI†), a tentative linear extrapolation of the secondary relaxation time in the liquid state intersects that of the USG at around 306 K. The same behavior is observed in Fig. 2C for the intensity of the process, i.e. the variation of intensity with temperature is stronger in the liquid state, and reaches a nearly constant value when the system is vitrified. Again, a tentative linear extrapolation of the process intensity in the liquid state intersects that of the USG at around 308 K. Interestingly, those extrapolated temperature values are compatible with the value of enthalpic Tf′ obtained from the calorimetric curve (inset of Fig. 1A). In other words, the equilibrium state between liquid and USG measured through the secondary relaxation is consistent with the equilibrium state measured with calorimetry through the structural relaxation process. Furthermore, as we show in Fig. 3, the time needed for the system at 306 K to reach the secondary relaxation time of the USG is 109.75 s or 178 years, very similar to the structural relaxation time of the liquid at that temperature (τα (306 K) = 1010.6 s). These observations are analogous to those recently reported for the JG β-relaxation process in toluene.17
 | ||
| Fig. 3 Evolution of the secondary process of an etoricoxib OG during aging at 306 K. Clearly, the relaxation time of the glass increases with time, while the intensity of the process decreases. In the inset, we plot the relaxation time as a function of aging time, showing that 109.75 s are needed to obtain a glass whose relaxation time matches the relaxation time of the USG. | ||
Noticeably, at Tg the secondary relaxation time of the OG is approximately equal to the relaxation time of the USG at Ton around 33 K higher in temperature (elucidated by the blue dashed line in Fig. 2B). Both Ton of the USG and the Tg of the OG can be defined as the temperature at which the structural α-relaxation time τα = 102 s, and critical for transformation from glass to liquid. We can then restate the previous observation by saying that the value of the secondary relaxation time τ at the temperature at which τα = 100 s is the same in USG and OG. The same observation can be made from the reported data of USG of toluene (shown in the ESI†). In that case, the increase of the isochronal JG β-relaxation temperature, ΔTβ = 6 K, is coincident with the enhancement of Ton in the USG. The choice of τα = 102 s to define Tg or Ton is arbitrary. On the other hand, the heating rate employed in the measurement of the glass determines Ton and the corresponding τα. However, according to a recent work, the enhancement of Ton between USG and OG of indomethacin and toluene is nearly independent of the heating rate26 (i.e. the distance between the black and red vertical dashed lines in Fig. 2B is independent of the heating rate employed to determine Ton). Therefore, and also taking into consideration of the very similar values of activation energy of the secondary relaxation in USG and OG, the invariance of τJG at Ton holds regardless of the arbitrary chosen τα(Ton) value. All these observations from the comparison between USG and OG are analogous to concurrent changes of the α- and the JG-β relaxation times at different pressures, where the change of Tg defined by τα(Tg) = 100 s is matched by the corresponding change in ΔTβ. This is illustrated by the data of DGEBA (diglycidyl ether of bisphenol-A)27 in ESI,† and more examples can be given.28–30 In fact, as we also show in Fig. 2B, the secondary relaxation time of the etoricoxib OG at 400 MPa and at Tg of the system at that pressure falls also onto the same blue line. All these observations draw a scenario in which the structural and secondary relaxations are strongly linked, not only from the point of view of the physical state of the system (as seen from the coincidence of the enthalpic Tf′ value and the Tf′ obtained from the secondary relaxation characteristics), but also from the point of view of its transformation from glass to liquid.
In Fig. 4 we plot the secondary relaxation time of USG and OG as a function of pressure at two different temperatures. We can clearly observe that the difference in relaxation time exhibited at ambient pressure (around 1 decade) is reduced as the pressure increases. At sufficiently high pressures, both systems exhibit a similar relaxation time. This result is very similar to the one reported in a recent work performed on indomethacin USG. There, Ton was measured as a function of the applied pressure, finding that the difference in Ton approached 0 as the pressure was increased to about 300 MPa.32 By means of a phenomenological model, the authors of ref. 32 claimed that the observed behavior could be explained by a convergence of density in both systems as pressure was increased. This explanation is compatible with the finding in this work. Moreover, if the secondary relaxation times of USG and OG are approaching each other at elevated pressures (as represented in Fig. 4 as well as on the high-pressure data plotted in Fig. 2B), and considering the mentioned invariance of τJG at Ton, as observed from Fig. 2B, then necessarily Ton must also approach each other as pressure is increased, suggesting again the correspondence between structural (responsible for the devitrification at Ton) and the secondary processes. Importantly, we notice from the inset in Fig. 4 that the observed variations in relaxation time of USG with pressure are reversible.
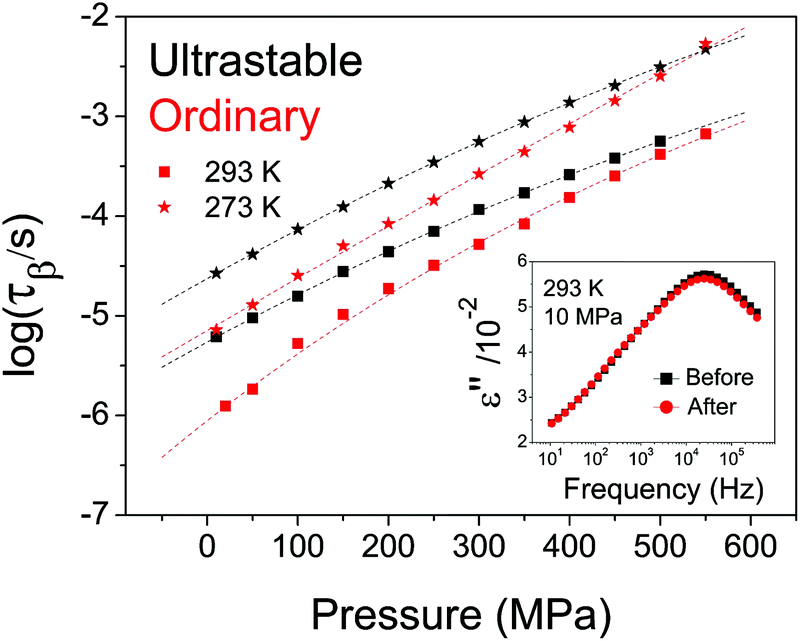 | ||
| Fig. 4 Secondary relaxation time of USG (black data) and OG (red data) as a function of pressure, at two different isotherms: 293 K (squares) and 273 K (stars). The dotted line is a guide to the eyes. In the inset, we show the dielectric spectra of an USG measured at 293 K and 10 MPa, before and after being subjected to 500 MPa, indicating that the process is completely reversible. | ||
Even though the characteristics of the structural and secondary relaxations are linked together, the way that pressure affects those characteristics is not the same in USG and OG, as seen in Fig. 4. We give three plausible explanations for this issue. (i) The especial molecular arrangement of molecules in the USG may have an influence on the effect of pressure on the characteristics of the system. (ii) The distribution of relaxation times is not the same and, therefore, application of pressure affects differently the two systems. (iii) Density may not univocally determine the relaxation time, and therefore a different density of the system may yield a different evolution with pressure. According to the reported V(p,T) data for etoricoxib,21 the density of the OG at 122 MPa is around 2.1% higher than at ambient pressure. The expected change of density in USG is ∼2–3% from WAXS measurements. From these data, we see that changes in relaxation time induced by pressurization or by ultrastability are similar, since, according to Fig. 4, the relaxation times of USG at ambient pressure and OG at 122 MPa are equal. However, we have to be cautious with this result since, according to the reported V(T) data of etoricoxib at 10 MPa,21 a change in Tf′ of 18 K corresponds to a change in density of 0.7%, meaning that other factors apart from density are responsible for the observed increase in relaxation time of USG compared to OG. We note that the exact V(T) relation in vacuum (during the formation of the USG, p = 1 × 10−12 MPa) and at 10 MPa, and therefore Δρ corresponding to ΔTf′ = 18 K, may be very different. In any case, further experiments are required to fully understand the nature of USG. It is important to remark that, notwithstanding, the origin of the different behavior of USG and OG affects both the structural and the secondary process in the same manner, emphasizing again the strong link between both processes.
In summary, from the analysis of the relaxation data of the secondary JG γ-process persistent in the glassy state of USG and OG of etoricoxib we have arrived at several conclusions. Firstly, the JG process of USG, deep in the glassy state, appears to be less than one order of magnitude slower as well as less intense. This behavior is analogous to the one observed in the JG-β process of toluene glasses,17 and is attributed to the density increase of USGs resulting in slowing down of molecular rearrangements. This finding may have a strong impact in the application of USG, since secondary relaxations are responsible for many properties of glassy systems.1,33,34 Secondly, even though the observed secondary process in etoricoxib is not the unresolved JG β-relaxation, their density/pressure dependence and connection to the structural relaxation are analogous and hence it is another JG process. Likely, this JG γ-relaxation involves a substantial part of the entire molecule and is the precursor of the JG β-relaxation. Thirdly and most importantly, we have found strong evidences of a close link between the evolution in the structural and secondary relaxations as a function of thermodynamic conditions (temperature and pressure). Three different observations bring us to this conclusion. (i) At Ton of either USG or OG, the secondary relaxation time is invariant. (ii) The secondary relaxation determines an equilibrium state which is consistent with the equilibrium state measured through properties involving the structural relaxation, such as the calorimetric enthalpy or aging time. (iii) The secondary relaxation times of USG and OG seem to converge to the same value at elevated pressures, in analogy to the convergence of Ton(p) observed in USG and OG of indomethacin.32 These findings support the view that the secondary relaxation is an indispensable precursor of the structural one, and also reaffirms the importance of using the secondary relaxation of aged systems, or ideally the USG, to probe the properties of the structural relaxation, which is inaccessible by other means.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
C. R.-T., M. R.-B. and M. P. acknowledge the support from the National Science Centre through the Polonez scheme (Grant No. DEC-2015/19/P/ST3/03540/2). This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 665778. C. R.-T. also thanks Joan Ràfols-Ribé for support during the preparations of the samples.References
- K. L. Ngai, Relaxation and Diffusion in Complex Systems, Springer New York, New York, NY, 2011 Search PubMed.
- K. L. Ngai and M. Paluch, Classification of secondary relaxation in glass-formers based on dynamic properties, J. Chem. Phys., 2004, 120, 857–873 CrossRef CAS PubMed.
- G. P. Johari and M. Goldstein, Viscous Liquids and the Glass Transition. II. Secondary Relaxations in Glasses of Rigid Molecules, J. Chem. Phys., 1970, 53, 2372–2388 CrossRef CAS.
- G. P. Johari, A. Hallbrucker and E. Mayer, Thermal behavior of several hyperquenched organic glasses, J. Phys. Chem., 1989, 93, 2648–2652 CrossRef CAS.
- K. A. Graeser, C. J. Strachan, J. E. Patterson, K. C. Gordon and T. Rades, Physicochemical Properties and Stability of Two Differently Prepared Amorphous Forms of Simvastatin, Cryst. Growth Des., 2008, 8, 128–135 CAS.
- S. N. Tripathy, M. Rams-Baron, Z. Wojnarowska, J. Knapik-Kowalczuk and M. Paluch, New limits of secondary β-relaxation, Sci. Rep., 2017, 7, 43091 CrossRef CAS PubMed.
- S. F. Swallen, K. L. Kearns, M. K. Mapes, Y. S. Kim, R. J. McMahon, M. D. Ediger, T. Wu, L. Yu and S. Satija, Organic glasses with exceptional thermodynamic and kinetic stability, Science, 2007, 315, 353–356 CrossRef CAS PubMed.
- K. J. Dawson, K. L. Kearns, L. Yu, W. Steffen and M. D. Ediger, Physical vapor deposition as a route to hidden amorphous states, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15165–15170 CrossRef CAS PubMed.
- S. S. Dalal, Z. Fakhraai and M. D. Ediger, High-throughput ellipsometric characterization of vapor-deposited indomethacin glasses, J. Phys. Chem. B, 2013, 117, 15415–15425 CrossRef CAS PubMed.
- C. Rodriguez-Tinoco, M. Gonzalez-Silveira, J. Rafols-Ribe, A. F. Lopeandia and J. Rodriguez-Viejo, Transformation kinetics of vapor-deposited thin film organic glasses: the role of stability and molecular packing anisotropy, Phys. Chem. Chem. Phys., 2015, 17, 31195–31201 RSC.
- E. Leon-Gutierrez, A. Sepúlveda, G. Garcia, M. T. Clavaguera-Mora, J. Rodríguez-Viejo, T. Matsuo, L. H. Allen, T. L. Yu and S. Satija, Stability of thin film glasses of toluene and ethylbenzene formed by vapor deposition: an in situ nanocalorimetric study, Phys. Chem. Chem. Phys., 2010, 12, 14693–14698 RSC.
- T. Pérez-Castañeda, R. J. Jiménez-Riobóo and M. A. Ramos, Two-Level Systems and Boson Peak Remain Stable in 110-Million-Year-Old Amber Glass, Phys. Rev. Lett., 2014, 112, 165901 CrossRef PubMed.
- J. H. Mangalara, M. D. Marvin and D. S. Simmons, Three-Layer Model for the Emergence of Ultrastable Glasses from the Surfaces of Supercooled Liquids, J. Phys. Chem. B, 2016, 120, 4861–4865 CrossRef CAS PubMed.
- M. Tylinski, M. S. Beasley, Y. Z. Chua, C. Schick and M. D. Ediger, Limited surface mobility inhibits stable glass formation for 2-ethyl-1-hexanol, J. Chem. Phys., 2017, 146, 203317 CrossRef CAS PubMed.
- K. L. Ngai, M. Paluch and C. Rodríguez-Tinoco, Why is surface diffusion the same in ultrastable, ordinary, aged, and ultrathin molecular glasses?, Phys. Chem. Chem. Phys., 2017, 19, 29905–29912 RSC.
- K. L. Ngai, L.-M. Wang and H.-B. Yu, Relating Ultrastable Glass Formation to Enhanced Surface Diffusion via the Johari–Goldstein β-Relaxation in Molecular Glasses, J. Phys. Chem. Lett., 2017, 8, 2739–2744 CrossRef CAS PubMed.
- H. B. Yu, M. Tylinski, A. Guiseppi-Elie, M. D. Ediger and R. Richert, Suppression of β Relaxation in Vapor-Deposited Ultrastable Glasses, Phys. Rev. Lett., 2015, 115, 185501 CrossRef CAS PubMed.
- R. Casalini and C. M. Roland, Aging of the secondary relaxation to probe structural relaxation in the glassy state, Phys. Rev. Lett., 2009, 102, 35701 CrossRef CAS PubMed.
- H. Yardimci and R. L. Leheny, Aging of the Johari–Goldstein relaxation in the glass-forming liquids sorbitol and xylitol, J. Chem. Phys., 2006, 124, 214503 CrossRef PubMed.
- G. Johari, Localized molecular motions of β-relaxation and its energy landscape, J. Non-Cryst. Solids, 2002, 307–310, 317–325 CrossRef CAS.
- M. Rams-Baron, Z. Wojnarowska, J. Knapik-Kowalczuk, K. Jurkiewicz, A. Burian, M. Wojtyniak, J. Pionteck, M. Jaworska, C. Rodríguez-Tinoco and M. Paluch, The dielectric signature of glass density, Appl. Phys. Lett., 2017, 111, 121902 CrossRef.
- S. R. Elliott, The origin of the first sharp diffraction peak in the structure factor of covalent glasses and liquids, J. Phys.: Condens. Matter, 1992, 4, 7661–7678 CrossRef CAS.
- S. R. Elliott, Extended-range order, interstitial voids and the first sharp diffraction peak of network glasses, J. Non-Cryst. Solids, 1995, 182, 40–48 CrossRef CAS.
- S. S. Dalal, D. M. Walters, I. Lyubimov, J. J. de Pablo and M. D. Ediger, Tunable molecular orientation and elevated thermal stability of vapor-deposited organic semiconductors, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 201421042 CrossRef PubMed.
- K. Grzybowska and M. Paluch, Molecular dynamics and physical stability of amorphous anti-inflammatory drug: celecoxib, J. Phys. Chem. B, 2010, 114, 12792–12801 CrossRef CAS PubMed.
- C. Rodríguez-Tinoco, J. Ràfols-Ribé, M. González-Silveira and J. Rodríguez-Viejo, Relaxation dynamics of glasses along a wide stability and temperature range, Sci. Rep., 2016, 6, 35607 CrossRef PubMed.
- K. L. Ngai, J. Habasaki, D. Prevosto, S. Capaccioli and M. Paluch, Thermodynamic scaling of α-relaxation time and viscosity stems from the Johari–Goldstein β-relaxation or the primitive relaxation of the coupling model, J. Chem. Phys., 2012, 137, 34511 CrossRef CAS PubMed.
- D. Prevosto, S. Capaccioli, S. Sharifi, K. Kessairi, M. Lucchesi and P. A. Rolla, Secondary dynamics in glass formers: relation with the structural dynamics and the glass transition, J. Non-Cryst. Solids, 2007, 353, 4278–4282 CrossRef CAS.
- D. Prevosto, S. Capaccioli, M. Lucchesi, P. A. Rolla and K. L. Ngai, Does the entropy and volume dependence of the structural α-relaxation originate from the Johari–Goldstein β-relaxation?, J. Non-Cryst. Solids, 2009, 355, 705–711 CrossRef CAS.
- M. Mierzwa, S. Pawlus, M. Paluch, E. Kaminska and K. L. Ngai, Correlation between primary and secondary Johari–Goldstein relaxations in supercooled liquids: invariance to changes in thermodynamic conditions, J. Chem. Phys., 2008, 128, 44512 CrossRef CAS PubMed.
- M. Rams-Baron, Z. Wojnarowska, K. Grzybowska, M. Dulski, J. Knapik, K. Jurkiewicz, W. Smolka, W. Sawicki, A. Ratuszna and M. Paluch, Toward a Better Understanding of the Physical Stability of Amorphous Anti-Inflammatory Agents: The Roles of Molecular Mobility and Molecular Interaction Patterns, Mol. Pharmaceutics, 2015, 12, 3628–3638 CrossRef CAS PubMed.
- C. Rodríguez-Tinoco, M. González-Silveira, M. Barrio, P. Lloveras, J. L. Tamarit, J.-L. Garden and J. Rodríguez-Viejo, Ultrastable glasses portray similar behaviour to ordinary glasses at high pressure, Sci. Rep., 2016, 6, 34296 CrossRef PubMed.
- H. Bin, Yu, W. H. Wang, H. Y. Bai and K. Samwer, The β-relaxation in metallic glasses, Natl. Sci. Rev., 2014, 1, 429–461 CrossRef.
- M. T. Cicerone and J. F. Douglas, β-Relaxation governs protein stability in sugar-glass matrices, Soft Matter, 2012, 8, 2983 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting data concerning data fitting procedure, isothermal transformation of USG, invariance of τβ in toluene, additional data on DGEBA at variable pressure and additional details on Fig. 1B. See DOI: 10.1039/c7cp06445d |
| This journal is © the Owner Societies 2018 |