
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural dynamics upon photoexcitation-induced charge transfer in a dicopper(I)–disulfide complex†
Maria
Naumova
 *abj,
Dmitry
Khakhulin‡
cd,
Mateusz
Rebarz‡
e,
Martin
Rohrmüller‡
f,
Benjamin
Dicke
bj,
Mykola
Biednov
bj,
Alexander
Britz
cd,
Shirly
Espinoza
e,
Benjamin
Grimm-Lebsanft
bj,
Miroslav
Kloz
e,
Norman
Kretzschmar
g,
Adam
Neuba
a,
Jochen
Ortmeyer§
a,
Roland
Schoch
a,
Jakob
Andreasson
ehi,
Matthias
Bauer
a,
Christian
Bressler
cd,
Wolf
Gero Schmidt
f,
Gerald
Henkel
*a and
Michael
Rübhausen
*bj
*abj,
Dmitry
Khakhulin‡
cd,
Mateusz
Rebarz‡
e,
Martin
Rohrmüller‡
f,
Benjamin
Dicke
bj,
Mykola
Biednov
bj,
Alexander
Britz
cd,
Shirly
Espinoza
e,
Benjamin
Grimm-Lebsanft
bj,
Miroslav
Kloz
e,
Norman
Kretzschmar
g,
Adam
Neuba
a,
Jochen
Ortmeyer§
a,
Roland
Schoch
a,
Jakob
Andreasson
ehi,
Matthias
Bauer
a,
Christian
Bressler
cd,
Wolf
Gero Schmidt
f,
Gerald
Henkel
*a and
Michael
Rübhausen
*bj
aDepartment of Chemistry, Paderborn University, 33098 Paderborn, Germany. E-mail: biohenkel@upb.de
bInstitut für Nanostruktur – und Festkörperphysik, Hamburg University, 20355 Hamburg, Germany. E-mail: mruebhau@physnet.uni-hamburg.de
cThe Hamburg Centre for Ultrafast Imaging, 22761 Hamburg, Germany
dEuropean XFEL GmbH, 22869 Schenefeld, Germany
eInstitute of Physics, ELI Beamlines, Academy of Sciences of the Czech Republic, CZ-18221 Prague, Czech Republic
fDepartment of Physics, Paderborn University, 33098 Paderborn, Germany
gEuropean Synchrotron Radiation Facility, 38000 Grenoble, France
hLaboratory of Molecular Biophysics, Department of Cell and Molecular Biology, Uppsala University, SE-751 24 Uppsala, Sweden
iCondensed Matter Physics, Department of Physics, Chalmers University of Technology, Gothenburg, Sweden
jCenter for Free-Electron Laser Science, Deutsches Elektronen-Synchrotron, 22607 Hamburg, Germany. E-mail: maria.naumova@desy.de
First published on 12th February 2018
Abstract
The structural dynamics of charge-transfer states of nitrogen-ligated copper complexes has been extensively investigated in recent years following the development of pump–probe X-ray techniques. In this study we extend this approach towards copper complexes with sulfur coordination and investigate the influence of charge transfer states on the structure of a dicopper(I) complex with coordination by bridging disulfide ligands and additionally tetramethylguanidine units [CuI2(NSSN)2]2+. In order to directly observe and refine the photoinduced structural changes in the solvated complex we applied picosecond pump–probe X-ray absorption spectroscopy (XAS) and wide-angle X-ray scattering (WAXS). Additionally, the ultrafast evolution of the electronic excited states was monitored by femtosecond transient absorption spectroscopy in the UV-Vis probe range. DFT calculations were used to predict molecular geometries and electronic structures of the ground and metal-to-ligand charge transfer states with singlet and triplet spin multiplicities, i.e. S0, 1MLCT and 3MLCT, respectively. Combining these techniques we elucidate the electronic and structural dynamics of the solvated complex upon photoexcitation to the MLCT states. In particular, femtosecond optical transient spectroscopy reveals three distinct timescales of 650 fs, 10 ps and >100 ps, which were assigned as internal conversion to the ground state (Sn → S0), intersystem crossing 1MLCT → 3MLCT, and subsequent relaxation of the triplet to the ground state, respectively. Experimental data collected using both X-ray techniques are in agreement with the DFT-predicted structure for the triplet state, where coordination bond lengths change and one of the S–S bridges is cleaved, causing the movement of two halves of the molecule relative to each other. Extended X-ray absorption fine structure spectroscopy resolves changes in Cu–ligand bond lengths with precision on the order of 0.01 Å, whereas WAXS is sensitive to changes in the global shape related to relative movement of parts of the molecule. The results presented herein widen the knowledge on the electronic and structural dynamics of photoexcited copper–sulfur complexes and demonstrate the potential of combining the pump–probe X-ray absorption and scattering for studies on photoinduced structural dynamics in copper-based coordination complexes.
1 Introduction
In the chemistry of life copper plays an important role being a core part of many essential metalloenzymes.1 Among copper active sites the mononuclear blue Cu site and the binuclear CuA site participate in electron transfer processes with unusually high rates.1,2 It was suggested that copper–sulfur interactions play a crucial role in their function, where the polarisable nature of sulfur serves in tuning electronic properties and reactivity.1,3 In the past few decades there have been many efforts in the field of bioinorganic chemistry for synthesising and characterising complexes which could serve as model compounds for these enzymes.4–14 In order to understand the mechanism of their function, orbital structures of many copper enzymes and their model complexes were analysed both experimentally and theoretically.15–18 Charge transfer that occurs during the catalytic cycle of a copper enzyme can be modelled by optical excitation of the metal-to-ligand or ligand-to-metal charge transfer states in a mimetic copper complex. Structural changes that accompany such photoinduced charge transfer typically occur on nanosecond timescales or faster and can thus be monitored almost exclusively by pump–probe time-resolved techniques. Hard X-ray methods, such as picosecond time-resolved K-edge X-ray absorption spectroscopy (TR-XAS) and time-resolved wide-angle X-ray scattering (TR-WAXS), are directly sensitive to the structures of excited states also in the case of dilute solutions. These techniques are still under development mainly driven by rapid progress of synchrotron sources, optical and X-ray lasers and data acquisition technologies.19–24 It is noteworthy that copper compounds were among the first systems investigated by these techniques. In particular Cu–bisphenantroline complexes have served as model compounds for X-ray as well as UV-Vis time-resolved spectroscopic studies.25–32 It was shown that upon photoexcitation with visible light the metal-to-ligand charge transfer (MLCT) transition occurs and the oxidation state of copper formally changes from +1 to +2. Such an alteration of the electronic configuration of copper – from closed-shell 3d10 to 3d9 – gives rise to significant structural changes of the complex. The excited state is subject to pseudo-Jahn–Teller distortion,33–35 and the flattening of the complex from Cu(I)-preferred pseudotetrahedral geometry in the direction of the formal Cu(II) square-planar arrangement occurs on sub-ps timescale. Another characteristic feature of copper complexes is relatively slow (in comparison with other transition metal complexes, such as Fe, Ru, Cr36) intersystem crossing (ISC) to the triplet state 3MLCT that in some cases competes with visible fluorescence due to the relaxation of 1MLCT to the ground state.37 From 3MLCT both non-radiative and emissive decays are possible, depending on the electronic structure of the particular complex due to different ligand substituents, the degree of flattening, and on the solvent. It was observed that in many Cu complexes the phosphorescence lifetime is shortened in donating solvents, and there is dispute in the literature concerning the coordination of the solvent molecule to the flattened excited state.26,27,38 Some recent TR-XAS experiments and molecular dynamic simulations on the model complex suggest the lack of complexation and rationalise the lifetime shortening by a decrease in 3MLCT energy and ISC yield in donating solvents.29 The copper complex investigated in this work bears certain similarities to Cu–bisphenantrolines, such as copper coordination by sp2-nitrogens. At the same time additional coordination by sulfur causes specific interactions influencing the excited state structure. In particular in the context of structural responses to electronic excitations within biologically relevant Cu/N/S systems, coordinated disulfide groups offer ideal conditions as electrons sink due to their empty antibonding sigma orbitals. What is more, many biorelevant and functional materials contain two or more copper atoms in a molecule. In particular, some dicopper complexes demonstrate attractive photoemissive properties making them a promising material for organic light-emitting diodes (OLEDs).39 However, to the best of our knowledge, no ultrafast studies on dicopper complexes have been reported. Both factors, Cu–S interaction and the presence of two metal atoms in the complex, make this study an interesting contribution to the emerging field of structural dynamics studies and to the knowledge of biologically-relevant ET-coupled structural changes.2 Experimental section
2.1 Materials
The investigated compound [Cu2(TMGphS-SphTMG)2]X2 (X = OTf, PF6, TMG: tetramethylguanidine) was synthesised as described in ref. 14. The details of characterisation by X-ray single crystal diffraction, elemental analysis, UV-Vis and IR spectroscopy, SQUID and cyclic voltammetry are also presented therein. Due to the presence of basic guanidine nitrogens and the labile Cu–S bonding, the choice of solvents suitable for handling the complex is limited to solvents that are polar, aprotic and non-coordinating. Following these criteria, 1,2-difluorobenzene (DFB) and dichloromethane (DCM) were chosen for the experiments. The compound is also stable at high concentrations in acetonitrile (ACN) and demonstrates better radiation damage stability in this solvent, which was also used for the experiments. Grades of the used solvents were: acetonitrile (ACN) from Carl Roth, 99.98%, Ultra LC-MS, for luminescence and TA; from Sigma-Aldrich, anhydrous, 99.8%, for TR-XAS; dichloromethane (DCM), HPLC grade, non-stabilised, from Carl Roth (stored under an inert atmosphere), for luminescence and TA; 1,2-difluorobenzene (DFB), 98%, from Thermo Fisher Scientific, Acros Organics and ChemPur, for TR-XAS and TR-WAXS. All solvents were used as purchased.2.2 Steady-state absorption and emission
UV-Vis steady state absorption spectra were measured using a Shimadzu UV-3600 spectrophotometer. The luminescence experiments were performed using an Ocean Optics 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 spectrometer as well as using a home-built time-correlated single-photon counting setup.40 The complex was excited with light pulses in the range of 360–410 nm. However, it does not show any measurable luminescence at 400–900 nm at room temperature in solution, so no more experimental details are given.
000 spectrometer as well as using a home-built time-correlated single-photon counting setup.40 The complex was excited with light pulses in the range of 360–410 nm. However, it does not show any measurable luminescence at 400–900 nm at room temperature in solution, so no more experimental details are given.
2.3 UV-Vis transient absorption (TA)
The TA measurements were performed in the ELI-Beamlines facility in Dolní Břežany, Czech Republic. The TA setup was based on an amplified Ti:sapphire laser, which was used to generate the pump and probe pulses. The 400 nm pump pulses at 1 kHz had a total energy of about 2.5 μJ and a diameter of about 0.4 mm at the sample position. White light continuum pulses used as a probe were generated in a CaF2 rotating plate and had a spectral range of 340–750 nm. The probe and pump beams were focused and overlapped on the quartz sample cell with 1 mm path length. Several scans were averaged and for the background removal the data recorded before excitation were subtracted from all the spectra. The instrumental response function of ∼120 fs was estimated measuring the stimulated Raman amplification signal from the solvent. All experimental data were corrected for the group velocity dispersion (GVD), and global decay analysis was used to obtain the time constants. The measurements were done on [Cu2(NSSN)2](OTf)2 and [Cu2(NSSN)2](PF6)2 in three solvents: dichloromethane, acetonitrile and 1,2-difluorobenzene. The studied compounds were dissolved at a concentration of 4–6 × 10−4 M corresponding to an OD of ∼0.4–0.6 at the pump wavelength. UV-Vis steady state spectra of the compound were registered before and after the measurements to control the integrity of the sample. More detailed technical description of the experimental setup is given in ESI.†2.4 Steady state X-ray absorption spectroscopy (XAS)
Two steady-state XAS data sets were acquired: [Cu2(NSSN)2](OTf)2 as a solid and as a solution in acetonitrile. The XAS experiments of the solid [Cu2(NSSN)2](OTf)2 at the Cu K-edge (8979 eV) were performed at the XAS beamline of the synchrotron ANKA (Ångstrømquelle Karlsruhe) in Karlsruhe (Germany) at ambient temperature. The spectra were collected in transmission mode with a maximum beam current of 180 mA by application of a Si(111) double crystal monochromator. The sample was diluted with boron nitride and a Cu foil was measured simultaneously downstream for energy calibration. The Cu K-edge XAS spectra of the 10 mM solution of [Cu2(NSSN)2](OTf)2 in acetonitrile were measured at the European synchrotron radiation facility (ESRF) beamline BM23 in Grenoble (France) with a maximum beam current of 200 mA at ambient temperature using a Si(111) double crystal monochromator. Due to the low concentration of copper, experiments were carried out in fluorescence geometry. To enable energy calibration and to exclude energy drifts of the beamline, a Cu foil spectrum was registered directly before and after the sample measurements. Energy calibration was carried out by referencing the first inflection point of the Cu foil spectrum to 8979 eV.2.5 Time-resolved (100 ps) X-ray absorption spectroscopy (TR-XAS)
The TR-XAS measurements were performed at the beamline P11 of Petra III synchrotron in Hamburg, Germany. The setup is described in detail in ref. 23. The measurements were performed on 5 mM [Cu2(NSSN)2](PF6)2 ACN and 5 mM [Cu2(NSSN)2](OTf)2 DFB solutions. Different solvents were used in order to compare the behaviour of the complex in a coordinating and a non-coordinating solvent. However, due to S/N problems the DFB XAS data could not be analysed further and therefore no further experimental details are reported. The X-ray beam for the experiment was monochromatised to energy resolution of dE/E = 1.3 × 10−4 with the Si(111) crystal and focused to 10 μm using two Kirkpatrick–Baez (KB) mirror systems. The excitation was performed using the 343 nm third harmonic of the pulsed fs laser PHAROS from Light Conversion. The laser was operated with a frequency of 65 kHz which corresponds to half of the Petra III roundtrip frequency. The sample was delivered using a cylindrical 300 μm liquid jet; laser pulse energy was 3 μJ; the spot size on the jet was ∼300 μm (adjusted to the jet diameter). The detection was done in fluorescence mode using an avalanche photo diode (APD) installed perpendicular to the X-ray beam. The normalisation was performed with the second APD which collected the X-rays scattered from a Co foil placed several cm after the jet. The absorption by copper at the energies above the absorption edge at the given concentration and jet thickness was negligible (∼0.3%) and allowed us to work in this geometry. UV-Vis steady state spectra of the compound were registered before and after the measurements to control the integrity of the sample.2.6 Time-resolved (100 ps) wide-angle X-ray scattering (TR-WAXS)
The TR-WAXS measurements were performed at beamline ID09 of the ESRF synchrotron, Grenoble, France. The description of the pump–probe setup at beamline ID09b is given elsewhere.41,42 The sample was prepared as a 4 mM solution of [Cu2(NSSN)2](OTf)2 in DFB. The sample solution was delivered via a 300 μm flat sheet jet from a sapphire nozzle providing a flow speed of approximately 3 m s−1 to ensure the complete sample exchange between consecutive pump–probe events at a repetition rate of 1 kHz of the experiment. The solution was regularly refilled with a new solvent to compensate for evaporation. Excitation was performed using the 400 nm frequency doubled output of the Ti:sapphire amplified system (Legend Elite, Coherent Inc.) with a frequency of 1 kHz and a pulse length (FWHM) of ∼1 ps. To avoid non-linear absorption and saturation effects the laser pulse energy was set to a low value of 20 μJ. Single X-ray pulses (∼100 ps FWHM, 18 keV, 3% bandwidth “pink” beam) generated from the U17 undulator were selected using a mechanical chopper system to reduce the repetition rate to 1 kHz. The size of the laser spot on the sample was measured using a pinhole and was determined to be 120 × 150 μm (h × v). The size of the X-ray beam was 90 × 60 μm (h × v). The scattered X-rays were collected using a Rayonix MX170-HS CCD detector with 3840 × 3840 pixels in 2 × 2-pixel binning. The detector was placed 38 mm behind the liquid jet. A cylindrical tungsten beamstop of 1.5 mm diameter was used to block the direct beam in front of the detector. The 2D scattering images upon 1.5 s exposure were collected for 18 different time delays between the laser and X-ray pulses in the range from −300 ps to 1 μs. After every three images a reference image was taken at a nominal delay of −3 ns corresponding to the non-excited sample. In total about 3500 images were acquired which results in approximately 150 repetitions for each pump–probe delay. The scattering contribution of the DFB solvent to the total difference scattering signal was additionally measured in a separate TR-WAXS measurement with the same setup using the solution of an azobenzene dye (4-[(4-bromophenyl)diazenyl]-N,N-diethylaniline, CAS 22700-62-5) as proposed in ref. 43.Cu Kα emission was recorded using a 1 m diameter Rowland-circle spectrometer arranged in the horizontal plane with a Si(111) analyzer crystal ((444) reflection) and a silicon drift detector with a multichannel analyser to additionally suppress the elastic background. A He-filled tank was installed in the path of emitted X-rays to avoid absorption and scattering by air. The measurements were performed by scanning the analyser Bragg angle near 79° in order to span the energy range of the Cu Kα1 peak (∼8048 eV). In total 35 scans were performed; certain spectral points were sampled 51 times.
2.7 DFT calculations
Density functional theory (DFT) calculations were carried out using the implementation of gaussian 09.44 The most frequently used hybrid functional B3LYP45–47 and a more recent meta hybrid functional TPSSh48 were applied to calculate the geometrical and electronic structures of the ground state and the lowest charge-transfer triplet state T1 (3MLCT). Excitations and geometrically relaxed singlet excited states were addressed using time dependent (TD)-DFT49 and a computationally feasible cc-pVDZ50 basis set. More precisely, the calculations were performed by solving the Casida equations within every step of a structural relaxation of the specific excited-state.51 For the evaluation of excitation spectra triple-ζ basis def2-TVZP52,53 was applied. Both basis sets included polarisation functions. Additionally, the influence of different solvents54 was examined using a continuum solvent model.55 Vibrational frequencies were checked for the ground state to ensure a true minimum. These concepts have been shown to result in reliable excited-state properties for the system within the copper-sulfur or related copper-oxygen chemistry.18,563 Results and discussion
3.1 Molecular and electronic structure
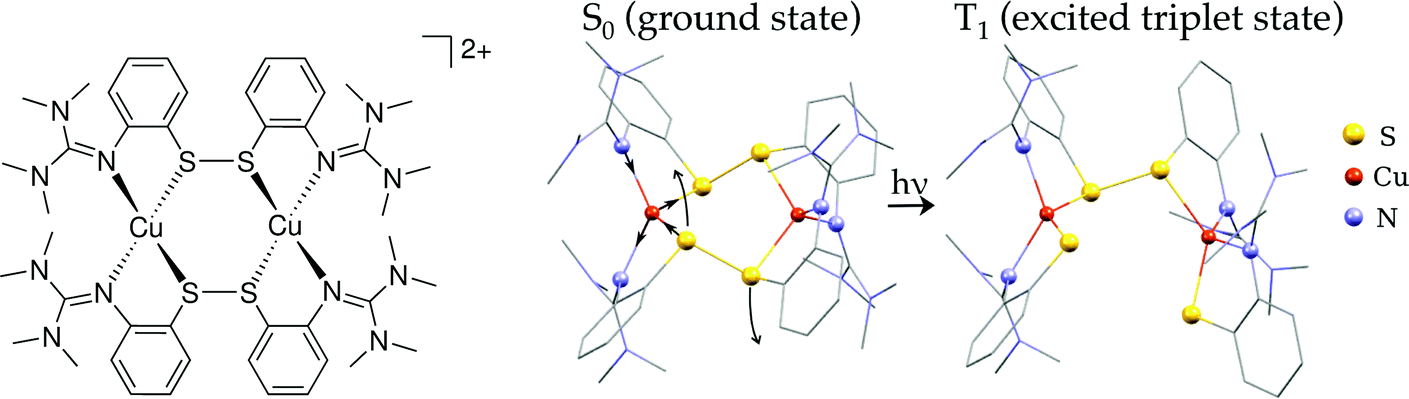 | ||
| Fig. 1 Left: Scheme of [Cu2(NSSN)2]2+; right: calculated geometries of the ground state and 3MLCT (lowest triplet state). Hydrogen atoms are omitted for visual clarity. | ||
The critical parts of the complex for its photoreactivity are Cu–N and Cu–S bonds. NTMG shows sp2 hybridisation, like Npy, and the Cu–NTMG distance determined from X-ray diffraction (av. 2.02 Å) is close to Cu–Npy coordination (2.01–2.07 Å)26,59 and shorter than typical for Cu(I)–NR3 (2.16(9) Å).60 Several examples reporting complexes existing in both Cu(I) and Cu(II) forms26,58,60 all have a shorter Cu–N bond length for Cu(II) than for Cu(I). The same trend is observed when Cu(II) is created by photoexcitation of Cu(I).26 For Cu(I)–disulfide coordination the Cu–S distance was reported to be within 2.20–2.37 Å.7,59,61 Our complex with 2.28–2.30 Å falls in this range. Cu(II) complexes tend to have 4+1 or 4+2 coordination with axial bonds elongated or shortened due to pseudo-Jahn–Teller distortion.33 For Cu(II)–disulfide bonds in axial coordination the span of covered Cu–S bond lengths is 2.68 Å to 3.28 Å in ref. 62–66. For a Cu(II)–disulfide linkage with a rare case of seemingly equal six Cu–ligand bonds67 Cu–S is within 2.46–2.57 Å, which is longer than that characteristic of Cu(I)–disulfide. However, for Cu(II) with equatorially bound radical disulfide (S2−˙) a much shorter distance of 2.27 Å was reported.68 For several complexes of Cu(I) and Cu(II) with the same aliphatic thiolate NS3 ligands Cu(I)–S bonds were reported to be 2.26 Å, whereas Cu(II)–S – was 2.37–2.38 Å.60 This broad variance in Cu(II)–S distances makes it difficult to predict the direction of the Cu–S bond length change upon photoexcitation.
The DFT-calculated structures of the ground state show good agreement with the crystallographic data in terms of intraligand distances (see Table 1); however, the calculated Cu–ligand bond lengths are consistently longer. Xc-functional and basis set combination TPSSh/def2-TZVP results in a better match to the crystallographic distances compared to B3LYP/cc-pVDZ. Including the solvent in the calculation (DFB) results in a slight shortening of Cu–ligand distances (less than 0.01 Å) and in a better match in the UV part of the spectrum (ESI,† Fig. S1). Since TPSSh/def2-TZVP is more suitable for TM complexes and shows a better match with the crystallographic data, these calculation results were further used for data modelling.
| Bond | Crystallography, Å | DFT(TPSSh/def2-TVZP) | |
|---|---|---|---|
| S0, Å | dR(T1–S0), Å | ||
| Cu–N | 2.021 ± 0.010 | 2.070 | −0.040, 0.008 |
| Cu–S | 2.288 ± 0.011 | 2.315 | −0.079, 0.035 |
| S–S | 2.126, 2.137 | 2.180 | 0.029, 3.063 |
| Cu–Cu | 3.941 | 4.133 | 0.662 |
| S–Cph | 1.778 ± 0.012 | 1.764 | |
| N–Cph | 1.386 ± 0.013 | 1.398 | |
| N–CTMG | 1.349 ± 0.006 | 1.334 | |
| Cph–Cph | 1.411 ± 0.010 | 1.416 | |
The DFT-calculated singlet GS electronic structure is shown in Fig. 2. This complex has two close-lying LUMO orbitals with strong S-p contribution (σ* of two disulfide bridges) that are significantly lower in energy than LUMO+2 and further orbitals. Both orbitals have a large anti-bonding character equally distributed over both S–S bridges. They mainly differ by the character of Cu-3d contribution: for the LUMO it has the dxy shape, whereas for LUMO+1 − dz2. Transitions from the HOMO and the HOMO−n with strong Cu 3d contribution occur to the LUMO and the LUMO+1 and account for a broad absorption band centered at 415 nm with ε ∼ 104 M−1 cm−1. The electron is excited from an orbital located on both copper atoms, thus creating a formally Cu21.5+ state. Further in the UV region there are strong absorption bands due to intraligand transitions as well as MLCT Cu → N (ESI,† Fig. S1).
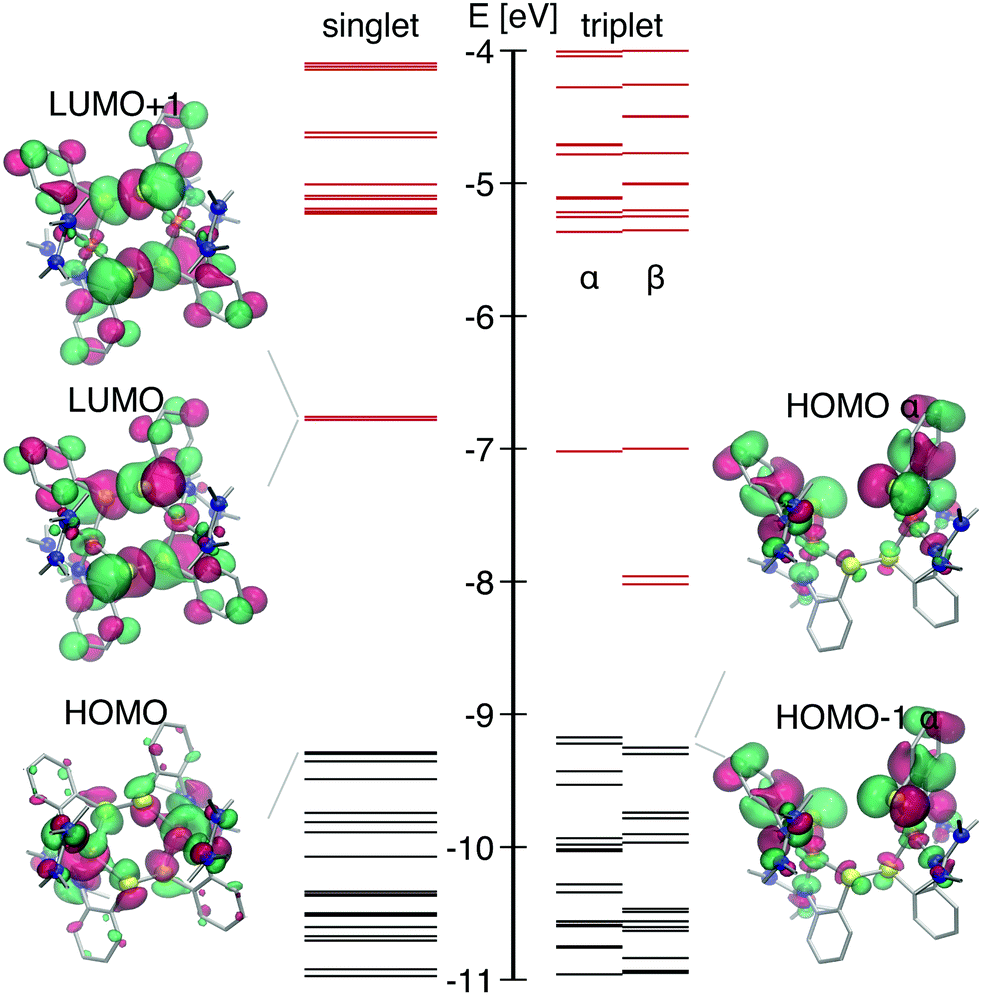 | ||
| Fig. 2 Molecule orbital spectra of S0 (left) and T1 (right) in their equilibrium structures, respectively. Occupied states are depicted in black, unoccupied – red. | ||
With the temporal resolution of the transient synchrotron-based experiments (70–100 ps) we expect that we will only observe the complex after it underwent ISC to the triplet state as it normally happens for copper complexes on a faster time-scale (<30 ps).31,34,69 The structural parameters of the calculated lowest triplet state T1 are summarised in Table 1. The orbital spectra of T1 are compared to the singlet S0 in Fig. 2. Single occupation of one antibonding disulfide σ* leads to the S–S bond breaking, causing the movement of two halves of the molecule relative to each other and increase of the Cu–S–S–Cu torsion angle from 67° to 99° (see Fig. 1). The other S–S bond is slightly elongated, and there are symmetric changes in coordination bond lengths on both copper atoms. In comparison with Cu–bisphenantroline complexes29 quite small flattening of the coordination polyhedron occurs – the angle between ligand chelate planes changes from 89° to 75° on both copper atoms.
Interestingly, the cleavage of the disulfide bridge for Cu2(NSSN)2 can be achieved not only via photoexcitation, but also chemically by addition of chloride ions.14 In the resulting Cu2(NS)2Cl2 complex both copper atoms have the oxidation state of +2, and S–S is cleaved creating two thiolate bridges.
3.2 Optical transient absorption
In order to obtain an insight into the ultrafast processes in the photoexcited complex, femtosecond transient absorption experiments were carried out upon excitation at 400 nm. Time-resolved spectra were recorded in the spectral range 350–670 nm and timescale 0–1 ns in DFB, ACN and DCM. Typical transient absorption spectra of Cu2(NSSN)2(PF6)2 in DFB at different times following excitation are depicted in Fig. 3. Quite similar transient absorption spectra were obtained for other solutions. The corresponding time constants are similar within the experimental error and are gathered in the ESI,† in Table S4.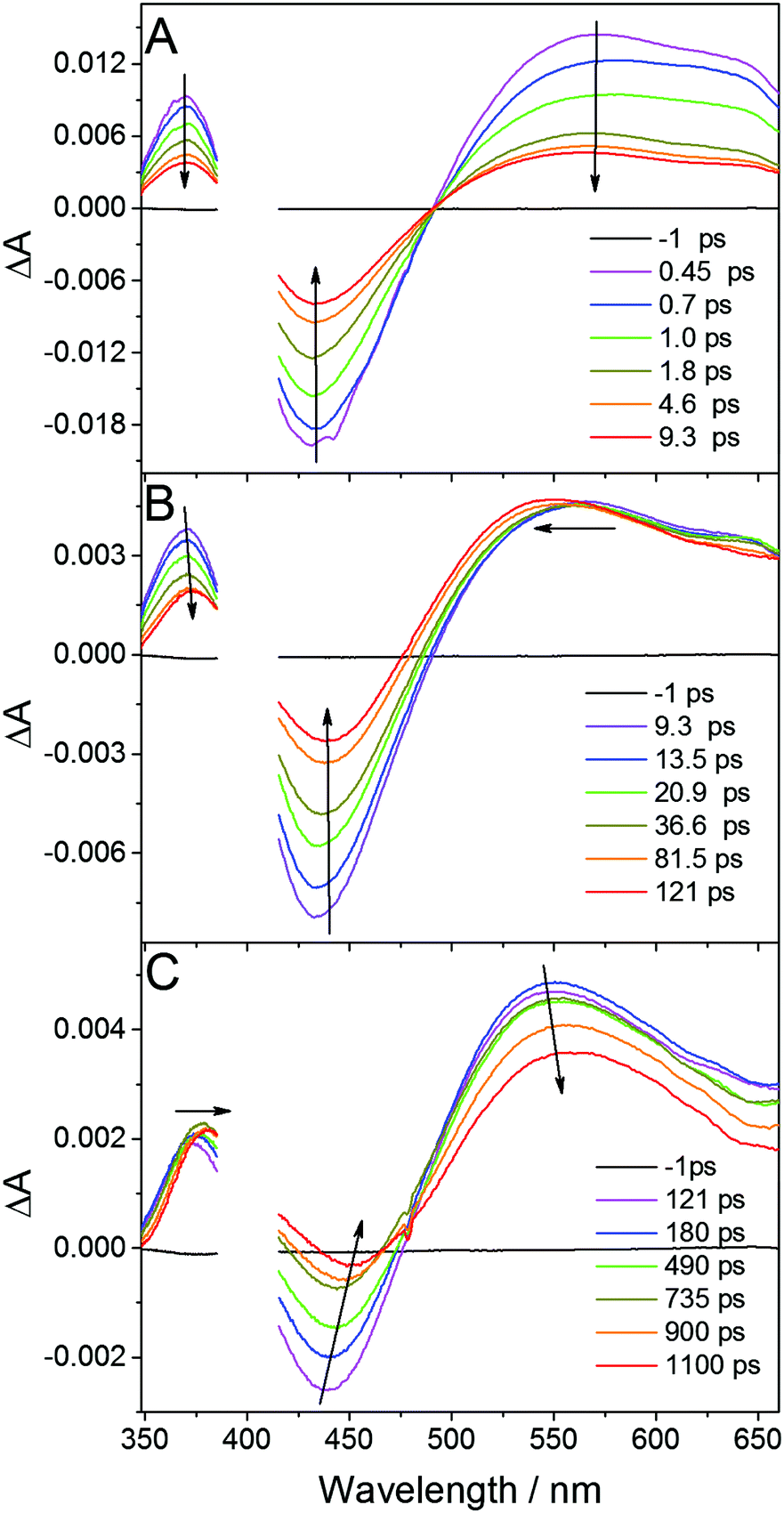 | ||
| Fig. 3 Transient absorption spectra of Cu(NSSN)2(PF6)2 in DFB recorded at different time delays after femtosecond excitation at 400 nm. | ||
Femtosecond excitation leads to the formation of a spectrum composed of three characteristic bands (spectrum at 0.45 ps, Fig. 3A). The negative band around 430 nm is located in the region of the ground state 1MLCT absorption (compare Fig. S1, ESI†) and is attributed to the ground state bleaching (GSB), whereas the positive band with the maximum at 370 nm as well as featureless broad absorption extending above 500 nm are ascribed to excited state absorption (ESA). Regarding DFT calculations and the experimental ground state absorption spectrum of the studied complex (see above), the excitation at 400 nm corresponds to the one of the higher singlet excited state Sn. Global fit analysis (see Fig. S20 in ESI†) of the time dependent spectral evolution reveals the existence of three steps (Fig. 3) characterised by three different time constants. During the first few picoseconds all the bands decrease simultaneously (Fig. 3A) by about 60% in intensity without significant shifts and shape variations revealing ultrafast deexcitation to the ground state with a time constant of about 650 fs. Since no emission was observed for the studied complex, this decay is attributed to non-radiative internal conversion (IC). It shows that the conformation in the higher excited singlet states has strong coupling with the ground state of the complex. The remaining excited species evolve with a time constant of 10.6 ps exhibiting the blueshift of the broad ESA band with a concomitant redshift and a decrease in the intensity of GSB and the second ESA band (see Fig. 3B). Such a time constant of about 10 ps was reported as intersystem crossing (ISC) to the triplet 3MLCT state in various homoleptic and heteroleptic copper complexes.34,37,70,71 The further spectral evolution exhibits a redshift and a decrease of intensity with a time constant >100 ps. This step can be ascribed to the beginning of further structural changes in the triplet state and radiationless decay to the ground state as designated in TR-XAS and TR-WAXS experiments reported below. The limited accuracy for this decay time constant is due to the limited pump–probe time delay (∼1 ns) accessible in our experiments which cannot be extended to the whole kinetics. Apart from IC and ISC, vibrational relaxation in the excited state and ground states can contribute to the observed signal. This phenomenon can take place in quite wide timescales (100 fs to 100 ps) depending on the complex structure and on the solvent. For TM complexes IC, VR and rearrangement of the solvation sphere are typically convoluted and only the average time constant for all these processes is observed.72 In order to disentangle the individual ultrafast processes the set of additional purposeful experiments would have to be performed.
The performed transient absorption experiment allows us, by judging from the relevant timescales, to conclude on the arrival of the excited complex to its triplet state (ISC). Thus, with the temporal resolution of the TR-XAS and WAXS measurements reported here we could only observe the triplet excited state. This information is important for narrowing down the set of candidate structures provided by DFT to only triplets to be considered as the starting structure for optimization.
3.3 Ground state EXAFS
Before the pump–probe experiments were performed, steady-state X-ray absorption spectra of [Cu2(NSSN)2](OTf)2 were measured in the solid state and in a 10 mM solution in acetonitrile. The EXAFS spectra of both look almost identical, which confirms that the complex is intact at this concentration in the acetonitrile solution and there are no or only negligible influences of coordinating acetonitrile molecules visible (ESI,† Fig. S2). Since the spectrum of the solid [Cu2(NSSN)2](OTf)2 has better data quality, the EXAFS analysis was carried out based on these data. The data and the fit are shown in Fig. 4. Data reduction was performed in Athena,73 EXAFS fitting was done in horae Artemis,73 using Feff85L.74 The crystallographic structure was used as the model, fitting was done in k-space for k = 3–16 Å−1. Only non-structural parameters (S02, E0, “Debye–Waller” factors σ2) were fitted, since the crystallographic structure was used to construct excited state models. The list of scattering paths and the fitting parameters are given in the ESI,† in Table S1. The fit and the extracted parameters (“Debye–Waller factors” σ2 and amplitude reduction factor S02) were further used for the fitting of TR-EXAFS data.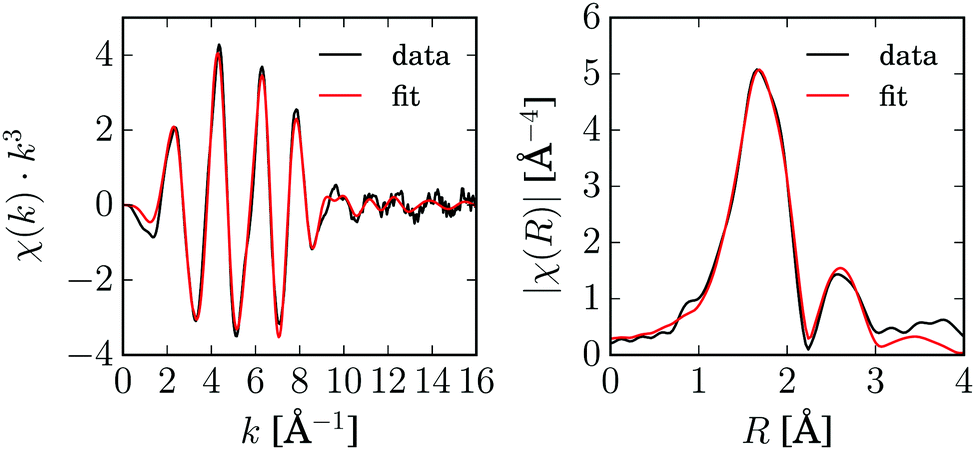 | ||
| Fig. 4 EXAFS spectra and best fit in k- and R-space of [Cu2(NSSN)2](OTf)2 (solid). | ||
In order to assure the validity of this analysis, fitting including structural parameters was performed for both solid and liquid samples as reported in the ESI.† Two different software packages (Artemis and ExCurve75) and approaches to data modeling (3D structure and shell-by-shell adding atoms) were used. The Cu–S and Cu–N distances obtained using two software packages are in agreement with each other and match the crystallographic distances within their uncertainties.
3.4 Pump–probe XAS
In order to observe the structural changes occurring upon photoexcitation, pump–probe XAS experiments were carried out. Fig. 5 shows the edge region of the Cu K-edge absorption spectrum of the complex. The edge position (maximum of the first derivative at 8981.1 eV) and shape (shoulder at 8985.5 eV, assigned as 1s → 4p transition) correspond to four-coordinated Cu(I).76,77 Upon photoexcitation with 343 nm light pulses strong changes are observed in the XANES spectrum. The transient signal, shown in Fig. 5, has a double minimum corresponding to the disappearance of two edge shoulders due to the edge shift to higher energies for Cu(II). This fact was used to estimate the excited state fraction (vide infra). At the same time a small positive preedge feature emerges at 8978 eV which in the literature is assigned to the 1s → 3d(+4p) transition which is characteristic of Cu(II) with non-centrosymmetric coordination.77–79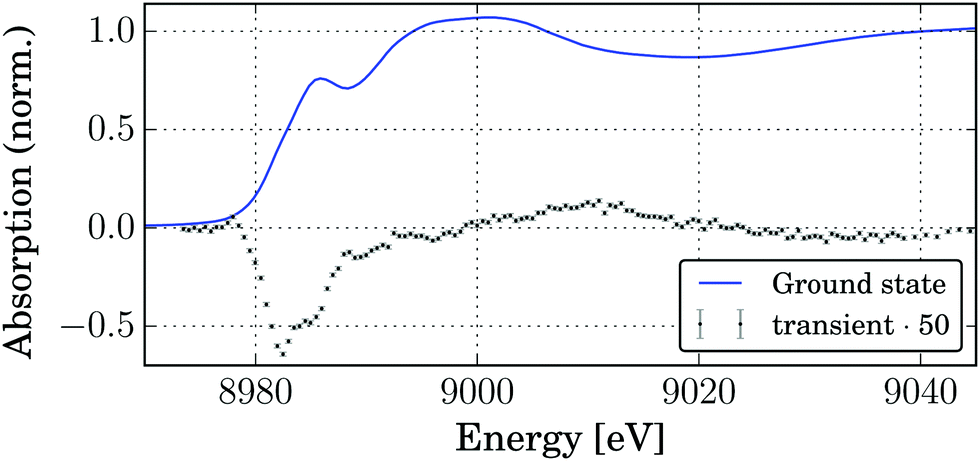 | ||
| Fig. 5 XANES spectrum of the ground state (solid Cu2(NSSN)2(OTf)2) and the transient spectrum at a delay of 80 ps (5 mM solution in acetonitrile). | ||
The laser-pumped spectrum has contributions of both ground and excited state spectra. In order to extract the ES spectrum, the knowledge of the relative contribution of the excited state spectrum (excited state fraction f) is necessary. In order to estimate it the edge shape for the GS spectrum was modelled as a sum of an error function and two pseudo-Voigt peaks centered at 8982.4 eV and 8985.4 eV (see the ESI,† Fig. S7). We expect that the transition at a lower energy (8982.4 eV) should not be present for a Cu2+ compound. However, since upon photoexcitation only one electron is transfered per two copper atoms, the electron density loss is divided between them, and a more realistic estimation is that a 50% decrease in the intensity of the edge peak at 8982.4 eV would correspond to the excited state spectrum. This condition is satisfied for f = 0.074, which is a lower-border estimation for f. For more details refer to the ESI.†
| Δχcalc.(E) = f·(χES(EES0) −χGS(EGS0)), | (1) |
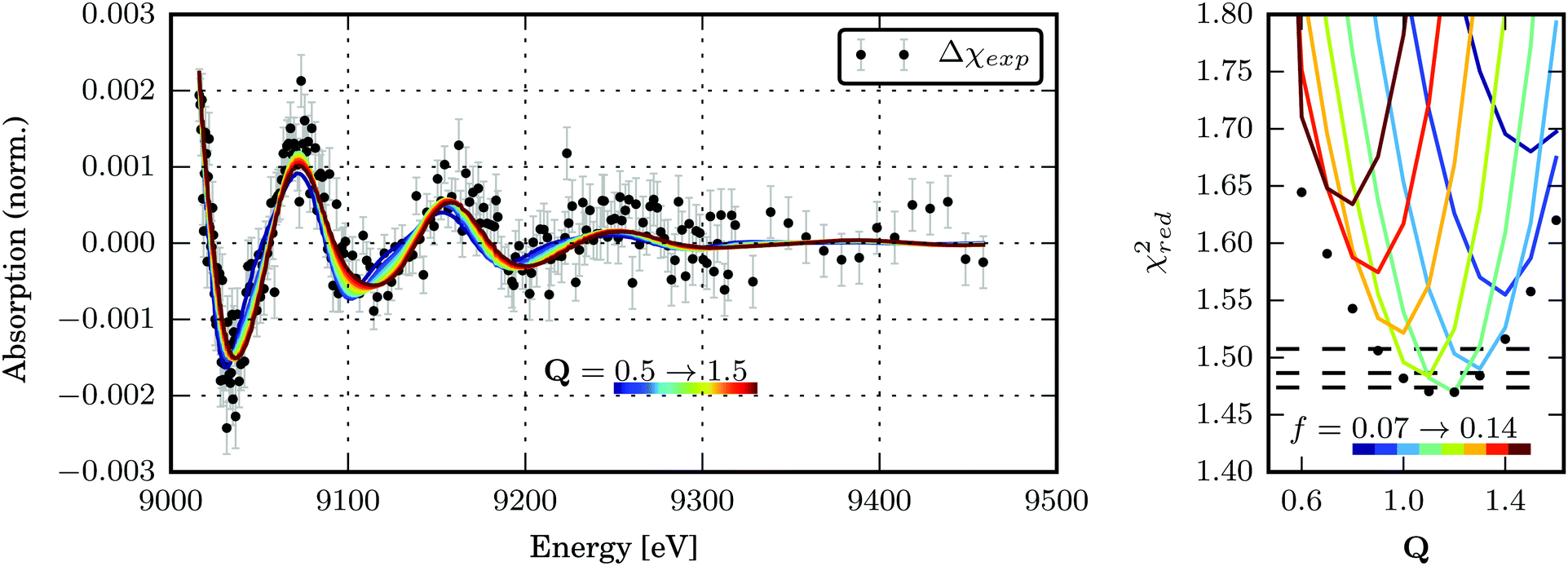 | ||
| Fig. 6 (a) Best transient EXAFS fits for structures along the Scr0 → Tcr1 transformation path Q. The best-fit parameters are given in Table S3 in the ESI.† (b) χred2 as a function of the Tcr1 structure for dE = 1.4 eV and different excited state fractions of 7–14%. The global minima of χred2 for each structure are marked with black dots. The dashed black lines denote the confidence levels of 68.3, 95.5 and 99.7% calculated with fixed optimal values of f and dE. The confidence region for (Q, dE, f) parameter space is shown in the ESI,† Fig. S10. | ||
The global minimum of χred2 is found for Q = 1.17+0.13−0.17, dE = 1.4+0.2−0.4 eV and f = 10+3.5−1%. (The errorbars cover the 68.3% confidence interval.) It corresponds to the following bond length changes: Cu–N = −0.046(6) and +0.0099(13) Å, Cu–S = +0.040(5) and −0.091(12) Å (see Fig. 8). It should be noted that a very small error bar on one of the Cu–N bond lengths (0.0013 Å) stems from our fitting model (the only structural degree of freedom is Q) and does not directly reflect sensitivity of the method. Sensitivity of EXAFS to distances to the first shell atoms can be on the order of 0.01 Å. The strongest change in the bond length happens to Cu–Scleaved: it shrinks to unusually short for a Cu–S bond length of 2.19 Å. However, a similarly short Cu–S bond (2.22 Å) is observed in the compound Cu2(NS)2Cl2 obtained from Cu2NSSN2 by chloride-induced reductive cleavage of the S–S bond.14 The values for the excited state fraction and the energy shift are in good agreement with the expectations.
According to the DFT predictions, the overall molecular structure in the triplet state is greatly perturbed compared to the GS since one of the S–S bridges is broken. In the solution phase therefore the two equal parts of the complex around the Cu atoms may significantly move with respect to each other. EXAFS is an element specific photoelectron technique with a limitation of structural sensitivity up to distances of 3–4 Å for molecules with low symmetry structures like Cu2(NSSN)2. For this complex the Cu–Cu distance is 3.9 Å in the ground state and increases in the excited state. Thus the Cu–Cu distance or large relative movements of the two parts of the Cu2(NSSN)2 molecule cannot be reliably probed via transient EXAFS only. On the other hand, the elastic scattering technique like WAXS provides a global probe of the overall structure capable of detecting large structural deformations involving long inter-atomic distances.
3.5 Time-resolved Wide-angle X-ray Scattering (TR-WAXS)
The time-resolved wide-angle X-ray scattering (TR-WAXS) method has been successfully applied to many systems in solution photochemistry over the past decade.22,42,82,83 The difference scattering between the laser-excited and the ground state samples contains contributions from changes in the solute structure, from rearrangements of the solvation shell and from changes in the thermodynamic state of the surrounding solvent, i.e. variations in temperature and density. The technique essentially serves as a global structural probe covering various length-scales of changing inter-atomic distances during laser-induced reactions and thus provides information complementary to the “local” element-specific X-ray absorption spectroscopy. Compared to EXAFS, the global sensitivity of WAXS offers information also on variation of large distances. A TR-WAXS experiment was carried out on Cu2(NSSN)2(OTf)2 solution in DFB as described in the experimental section. The difference scattering signal as a function of momentum transfer q at 100 ps delay is shown in Fig. 7. The momentum transfer is represented as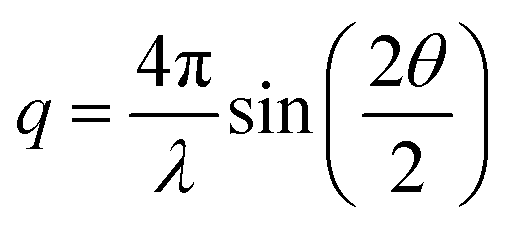 , where λ is the X-ray wavelength and 2θ is the scattering angle. Unlike XAS, the elastic X-ray scattering technique has no element selectivity and therefore is generally sensitive to all interatomic distances present in solution. Within the chosen analysis framework the laser-induced response of the solvent bulk is obtained empirically within a reference measurement as reported in the experimental part while the changes in the solute–solvent interatomic distances, or the solvent cage term, are neglected. The TR-WAXS signal component originating from the structural changes in the solute molecule was modelled using a set of candidate structures of the triplet ES and the crystallographic structure for the GS. However, the refinement of the configurational coordinate Q alone, as applied for the transient EXAFS data, is insufficient for accurate description of the solute scattering signals since changes in the overall structure of the complex have to be considered and not just in the first coordination shell of Cu as imposed by varying Q. In this respect for the Cu2(NSSN)2 complex it is essential to account for changes in relative positions of the two halves of the molecule around each copper. For each structure used in the transient EXAFS analysis, i.e. for a set of Q values along the Scr0 → Tcr1 path, one of the S–S bonds in the complex was used as a rotation axis of the two isostructural entities of the molecule relative to each other (see Fig. 1). Such modification is motivated by the structural trend of the DFT prediction for the triplet state. In this way the second structural parameter, the Cu–S–S–Cu torsion angle ϕ, is added to the refinement procedure of the TR-WAXS data. The range for ϕ variation is chosen from 52° to 144°, limited by the GS value and the arising steric hindrance of further ligand movement within the molecule, respectively. The goodness of the fit was judged by the values of the χred2 estimator. For detailed description of the signal modelling and data fitting procedures refer to the ESI.†χred2 as a function of the structural parameters demonstrates a strong dependence on the Cu–S–S–Cu torsion angle ϕ (see the ESI,† Fig. S14 and S15b). The minimum of χred2 is found at ϕ = 102° ± 2°, as compared to 99° predicted by DFT (see Fig. 8). On the other hand, the χred2 estimator does not exhibit any significant dependence on the configurational coordinate, Q (ESI,† Fig. S15a). This is explained by small relative changes in the Cu–ligand bond lengths with varying Q (see Table 1). The largest change upon the coordination bond refinement for Q = 0.5–1.5 occurs on one of the Cu–S bonds and amounts to about 0.08 Å or ∼3% of the absolute bond length. In contrast, the relative rotation of two parts of molecules, i.e. variation of ϕ, causes changes of some interatomic distances by several Å. According to the Debye equation, the elastic scattering is most sensitive to inter-atomic distances between the heaviest atoms in the molecule.82 The sensitivity roughly scales as multiplication of the two atomic numbers. Therefore, changing the Cu–Cu distance gives the strongest contribution to the difference scattering. Moreover, since this distance is relatively long (compared to the Cu–ligand distances) the limited q-range of the experiment will accommodate a larger number of structural oscillations of the scattering signal greatly increasing the fit confidence and selectivity to the Cu–Cu distance compared to the shorter Cu–ligand distances. A quantitative illustration of the changes in the interatomic distances between Cu and ligand atoms upon different structural refinement is given in the ESI,† in Fig. S18 and S19. The effect of these particular changes in the atomic position on the calculated difference scattering from the solute, ΔS(q)solute, demonstrates the contrasting sensitivity of the χred2 to variations in ϕ and Q (see the ESI,† Fig. S17). The configurational coordinate, Q, has a weak influence on ΔS(q)solute affecting mostly the signal amplitude, whereas changes in the torsion angle, ϕ, cause strong modifications in the overall signal shape.
, where λ is the X-ray wavelength and 2θ is the scattering angle. Unlike XAS, the elastic X-ray scattering technique has no element selectivity and therefore is generally sensitive to all interatomic distances present in solution. Within the chosen analysis framework the laser-induced response of the solvent bulk is obtained empirically within a reference measurement as reported in the experimental part while the changes in the solute–solvent interatomic distances, or the solvent cage term, are neglected. The TR-WAXS signal component originating from the structural changes in the solute molecule was modelled using a set of candidate structures of the triplet ES and the crystallographic structure for the GS. However, the refinement of the configurational coordinate Q alone, as applied for the transient EXAFS data, is insufficient for accurate description of the solute scattering signals since changes in the overall structure of the complex have to be considered and not just in the first coordination shell of Cu as imposed by varying Q. In this respect for the Cu2(NSSN)2 complex it is essential to account for changes in relative positions of the two halves of the molecule around each copper. For each structure used in the transient EXAFS analysis, i.e. for a set of Q values along the Scr0 → Tcr1 path, one of the S–S bonds in the complex was used as a rotation axis of the two isostructural entities of the molecule relative to each other (see Fig. 1). Such modification is motivated by the structural trend of the DFT prediction for the triplet state. In this way the second structural parameter, the Cu–S–S–Cu torsion angle ϕ, is added to the refinement procedure of the TR-WAXS data. The range for ϕ variation is chosen from 52° to 144°, limited by the GS value and the arising steric hindrance of further ligand movement within the molecule, respectively. The goodness of the fit was judged by the values of the χred2 estimator. For detailed description of the signal modelling and data fitting procedures refer to the ESI.†χred2 as a function of the structural parameters demonstrates a strong dependence on the Cu–S–S–Cu torsion angle ϕ (see the ESI,† Fig. S14 and S15b). The minimum of χred2 is found at ϕ = 102° ± 2°, as compared to 99° predicted by DFT (see Fig. 8). On the other hand, the χred2 estimator does not exhibit any significant dependence on the configurational coordinate, Q (ESI,† Fig. S15a). This is explained by small relative changes in the Cu–ligand bond lengths with varying Q (see Table 1). The largest change upon the coordination bond refinement for Q = 0.5–1.5 occurs on one of the Cu–S bonds and amounts to about 0.08 Å or ∼3% of the absolute bond length. In contrast, the relative rotation of two parts of molecules, i.e. variation of ϕ, causes changes of some interatomic distances by several Å. According to the Debye equation, the elastic scattering is most sensitive to inter-atomic distances between the heaviest atoms in the molecule.82 The sensitivity roughly scales as multiplication of the two atomic numbers. Therefore, changing the Cu–Cu distance gives the strongest contribution to the difference scattering. Moreover, since this distance is relatively long (compared to the Cu–ligand distances) the limited q-range of the experiment will accommodate a larger number of structural oscillations of the scattering signal greatly increasing the fit confidence and selectivity to the Cu–Cu distance compared to the shorter Cu–ligand distances. A quantitative illustration of the changes in the interatomic distances between Cu and ligand atoms upon different structural refinement is given in the ESI,† in Fig. S18 and S19. The effect of these particular changes in the atomic position on the calculated difference scattering from the solute, ΔS(q)solute, demonstrates the contrasting sensitivity of the χred2 to variations in ϕ and Q (see the ESI,† Fig. S17). The configurational coordinate, Q, has a weak influence on ΔS(q)solute affecting mostly the signal amplitude, whereas changes in the torsion angle, ϕ, cause strong modifications in the overall signal shape.
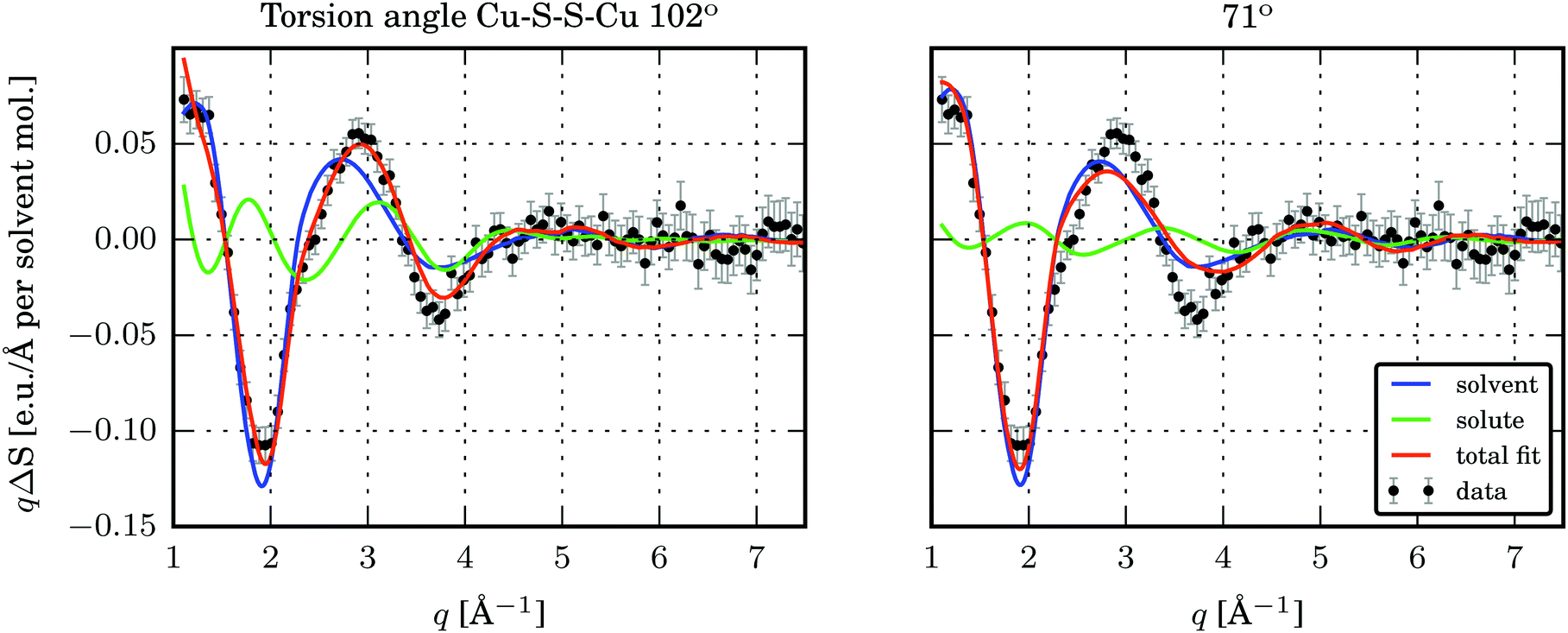 | ||
| Fig. 7 S(q) and best fits for two structures with different torsion angles Cu–S–S–Cu. Left: Torsion angle 102°, in the global minimum of χred2. Right: Torsion angle 71°. | ||
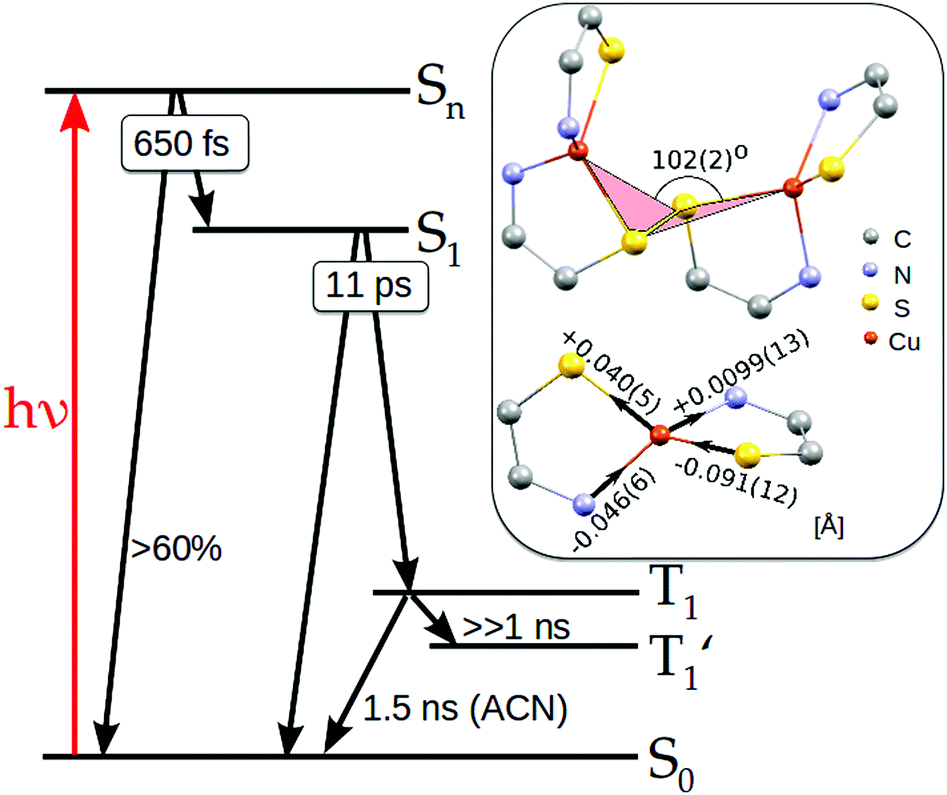 | ||
| Fig. 8 Schematic representation of the states involved in the photocycle of Cu2(NSSN)2 derived from the results of transient optical and X-ray measurements. The right panel illustrates main structural changes associated with the T1 triplet state formation as extracted from the time-resolved X-ray experiments. The bottom structure shows the changes in copper–ligand bond lengths in Å compared to the ground state structure extracted from the TR-EXAFS analysis. The top structure shows the torsion angle Cu–S–S–Cu coupled to the rotation between the two halves of Cu2(NSSN)2 around the S–S bond extracted from the TR-WAXS analysis. | ||
Comparing the two fits in Fig. 7 it appears that for the given experimental conditions the structural sensitivity is most pronounced in the q-range of 2.5–4.5 Å−1. At lower q values the solvent response dominates the signal, and starting from the q = 4.5 Å−1 no oscillations can be distinguished within the noise. Further optimisation of the experimental conditions towards using solvents with weaker heating response and maximising the excited state fraction may help in enhancing the structural sensitivity of the method providing higher confidence in refinement of the ES structure. Moreover, achieving a better signal-to-noise ratio at higher q, where contributions of solute and solvent have comparable amplitudes, would further improve the structural resolution.
3.6 Lifetimes of the excited state from the time-resolved XAS and WAXS experiments
The transient spectra for both TR-XAS and TR-WAXS experiments were registered for a set of pump–probe delays. From these measurements the lifetime of the triplet excited state could be extracted. For TR-XAS the transient signal at 8982.3 eV (the biggest amplitude of the signal) was registered at delays up to 6 ns. For TR-WAXS the excited state fraction was extracted from fitting the data at pump–probe delays from 100 ps to 20 ns. Only the optimised structure of the solute ES was used in the fit, since there was no indication of the solute-related signal shape evolution within the range of delays, as shown in the ESI,† in Fig. S16. Both kinetic traces are shown in Fig. S13 in the ESI.† The data sets were fitted using a model of exponential decay to a constant offset, convolved with the Gaussian X-ray probe pulse, as described in the ESI.† TR-XAS data yield τ1 = 1.53 ± 0.04 ns lifetime for ACN solution. The lifetime in DFB, obtained from TR-WAXS, is 2.9 ± 0.7 ns. This observation is in agreement with the general trend that lifetimes of excited states of molecules in a more polar and coordinating solvent (ACN) are shorter.29 However, with the low precision of determination of the lifetime in DFB, as well as different experimental conditions for two solvents, it is difficult to make reliable conclusions about the solvent effect. In the literature about Cu–bisphenantroline complexes the question of solvent coordination in the excited state is widely discussed.29,31 The driving force for exciplex formation is flattening of coordination polyhedron and preference of Cu(II) for the 5- and 6-coordinated states. TR-EXAFS and TA measurements were performed in a coordinating solvent, acetonitrile, which potentially could form an exciplex with the excited state of Cu2(NSSN)2. However, DFT predicts only small flattening of the copper coordination polyhedron—from 89° to 75° (angle between ligand chelate planes). It does not seem to be feasible that a solvent molecule could coordinate copper in this geometry. However, we have not addressed this question in more detail.3.7 Summary and discussion of the photoexcitation cycle
Based on the results presented above, we elucidate the main steps in the photocycle and structural dynamics of solvated Cu2(NSSN)2, as shortly summarised below and in Fig. 8.The main MLCT absorption band in the UV-Vis spectrum (3d(Cu) → σ*(S–S)) shows a maximum at 410–420 nm. As evident from the UV-Vis transient absorption spectra, after exciting this MLCT transition with 400 nm photons at least 60% of the excited molecules undergo fast radiationless deexcitation to the ground state with ∼650 fs time constant. This ultrafast decay competes with transitions to the lowest singlet state S1. The remaining fraction of the excited species undergoes further evolution with a lifetime of 11 ps which we assign as ISC to the triplet state 3MLCT. In this state DFT predicts strong structural changes, which were followed by the 100 ps time-resolved synchrotron-based experiments, TR-XAS and TR-WAXS. The combination of these techniques gives access to different structural degrees of freedom of the excited state of the Cu2(NSSN)2 complex. In particular, TR-EXAFS yields the following Cu–ligand distance changes (symmetric on both copper atoms): Cu–N = −0.046(6) and +0.0099(13) Å, Cu–S = +0.040(5) and −0.091(12) Å as shown in Fig. 8. The energy shift dE = 1.4+0.2−0.4 eV and excited state fraction f = 10.0+3.5−1.0% are in agreement with estimations (0 < dE ≤ 3 eV, f ≥ 7%). The TR-WAXS fit was not sensitive to the refinement of the coordination bond lengths in the excited state. However, it showed strong sensitivity to the value of the torsion angle Cu–S–S–Cu coupled to the relative movement of the two halves of the molecule, which was beyond the sensitivity of TR-EXAFS data. This angle was found to be 102° ± 2° as shown in Fig. 8 (compared to 99° predicted by DFT). Thus, TR-XAS and TR-WAXS applied in this work complemented each other and allowed us to evaluate the most relevant structural changes occurring in the photoexcited state. For a solution at room temperature, the T1 → S0 transition follows the non-radiative pathway, therefore its lifetime could not be measured in a luminescence experiment and was extracted from both X-ray experiments in different solvents. The solution of Cu2(NSSN)2 in ACN (TR-XAS) demonstrates a lifetime of 1.53 ± 0.04 ns, while DFB solution yields a lifetime of 2.9 ± 0.7 ns (TR-WAXS), as shown in Fig. S13 in the ESI.† The fact that the resulting lifetimes are quite similar, as well as small flattening of the coordination polyhedron predicted by DFT, do not support the assumption of solvent coordination in the excited state. However, the long-lived component of the excited state transients extracted from both XAS and WAXS for the two solvents indicates the formation of another metastable state, T1′, that could be associated with partial intercalation of solvent molecules in the space between the two halves of the solute in its triplet state. In order to better address this question, a TA experiment in the ns regime (laser flash photolysis) could be performed under the same experimental conditions for different solvents. It should be noted that the performed experiments are not directly sensitive to the S–S bonding. In order to probe the cleavage and recombination of this bond directly, it could be interesting to perform a TR-XAS experiment on the sulfur K-edge as recently reported in ref. 84 and85.
4 Conclusions
In this work we studied the effect of the photoinduced metal-to-ligand charge transfer on the local environment of copper and the global shape of the molecule in a dicopper complex with disulfide and guanidine ligands Cu2(NSSN)2. Photoexcitation in the blue part of the visible spectrum populates an antibonding orbital of the disulfide bridge, causing S–S bond cleavage, which is reversible since the complex remains bound via the second disulfide bridge. The long-term air and radiation stability makes Cu2(NSSN)2 a good model to study S–S bond cleavage and recombination. At the same time, photoexcitation of a MLCT band models electron transfer from metal to ligands, which is relevant for the biological function of certain Cu–S enzymes. In particular for Cu2(NSSN)2 the charge transfer causes conversion from disulfide to thiolate coordination. Disulfide–thiolate interconversion is frequent in nature where it is found as one of the mechanisms of formation of the tertiary structure of proteins and protection of cells from oxidative stress.86 For this copper model system the disulfide–thiolate conversion can be achieved by both population of the antibonding orbital of the S–S bridge via photoexcitation and by chemical reaction with chloride ions.14This work demonstrates that combining time-resolved X-ray absorption spectroscopy and wide-angle X-ray scattering has great potential for studies of photoinduced changes in molecular systems in solution. This is one of the first studies where these two structurally-sensitive methods are applied together to investigate the photochemistry of a dinuclear copper complex providing insight into different structural aspects of the excited state.
Author contributions
M. N., M. Rüb. and G. H. designed the experiment. D. K., A. B., M. N., and N. K. performed the WAXS experiments; D. K. analysed the data. M. Reb, M. K., S. E., and M. N. performed the TA experiments; M. Reb. analysed the data. B. D., M. Bied., B. G.-L., M. N., and S. E. performed the TR-XAS experiments; M. N. analysed the data. M. Rohr. performed the DFT calculations. R. S., M. Bauer and M. N. acquired static XAS; M. N. and R. S. analysed the data. A. N. and J. O. synthesised the sample. M. N., D. K., M. Reb., M. Rohr., A. B., C. B. and M. Rüb. wrote the paper. All authors contributed to the interpretation of the results and the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Alke Meents, Dennis Goeries as well as Anja Burkhardt and other staff of beamline P11 for their support during TR-XAS measurements. The authors would like to acknowledge Sebastian Schulz, Peter Zalden, Christina Bömer and beamline scientist Michael Wulff for the support of TR-WAXS measurements at ID09b, as well as Olivier Mathon and the beamline staff of BM23 for the support of the static XAS measurements. M. N. thanks Vadim Murzin and Wolfgang Caliebe for supporting XAS measurements at beamline P64 of Petra III and Tadesse Abebaw Assefa for discussions of TR-EXAFS data analysis. M. Bauer thanks the synchrotron ANKA for provision of beamtime and Stefan Mangold for assistance during the experiment. Most of the plots were prepared using matplotlib.87 DFT calculations were performed by using grants of the Paderborn Center for Parallel Computing PC2 and the Höchstleistungs-Rechenzentrum Stuttgart. The authors acknowledge the financial support by DFG FOR1405 “BioCTDyn” and by the Federal Ministry of Education and Research of Germany (BMBF) in the framework of “Erforschung kondensierter Materie”. M. Reb., S. E., M. K. and J. A. thank ELI Extreme Light Infrastructure Phase 2 (CZ.02.1.01/0.0/0.0/15_008/0000162) and ELIBIO (CZ.02.1.01/0.0/0.0/15_003/0000447) from the European Regional Development Fund for financial support. J. A. additionally thanks the Ministry of Education, Youth and Sports as part of targeted support from the National Programme of Sustainability II with registration number LQ1606, the Röntgen Ångström Cluster and Chalmers Area of Advance, Material science.References
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- C. Belle, W. Rammal and J. L. Pierre, J. Inorg. Biochem., 2005, 99, 1929–1936 CrossRef CAS PubMed.
- M. H. M. Olsson, U. Ryde, B. O. Roos and K. Pierloot, J. Biol. Inorg. Chem., 1998, 3, 109–125 CrossRef CAS.
- R. P. Houser, J. A. Halfen, V. G. Young Jr., N. J. Blackburn and W. B. Tolman, J. Am. Chem. Soc., 1995, 117, 10745–10746 CrossRef CAS.
- R. P. Houser, V. G. Young and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 2101–2102 CrossRef CAS.
- N. D. J. Branscombe, A. J. Blake, A. Marin-Becerra, W.-S. Li, S. Parsons, L. Ruiz-Ramirez and M. Schröder, Chem. Commun., 1996, 2573–2574 RSC.
- S. Itoh, M. Nagagawa and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 4087–4088 CrossRef CAS PubMed.
- Y. Ueno, Y. Tachi and S. Itoh, J. Am. Chem. Soc., 2002, 124, 12428–12429 CrossRef CAS PubMed.
- A. I. Uraev, I. S. Vasilchenko, V. N. Ikorskii, T. A. Shestakova, A. S. Burlov, K. A. Lyssenko, V. G. Vlasenko, T. A. Kuz'menko, L. N. Divaeva, I. V. Pirog, G. S. Borodkin, I. E. Uflyand, M. Y. Antipin, V. I. Ovcharenko, A. D. Garnovskii and V. I. Minkin, Mendeleev Commun., 2005, 15, 133–135 CrossRef.
- W. Rammal, C. Belle, C. Béguin, C. Duboc, C. Philouze, J.-L. Pierre, L. L. Pape, S. Bertaina, E. Saint-Aman and S. Torelli, Inorg. Chem., 2006, 45, 10355–10362 CrossRef CAS PubMed.
- N. Roy, S. Sproules, E. Bothe, T. Weyhermüller and K. Wieghardt, Eur. J. Inorg. Chem., 2009, 2655–2663 CrossRef CAS.
- M. Gennari, J. Pécaut, S. DeBeer George, F. Neese, M.-N. Collomb and C. Duboc, Angew. Chem., Int. Ed., 2011, 50, 5662–5666 CrossRef CAS PubMed.
- A. Neuba, U. Flörke, W. Meyer-Klaucke, M. Salomone-Stagni, E. Bill, E. Bothe, P. Höfer and G. Henkel, Angew. Chem., Int. Ed. Engl., 2011, 50, 4503–4507 CrossRef CAS PubMed.
- A. Neuba, R. Haase, W. Meyer-Klaucke, U. Flörke and G. Henkel, Angew. Chem., Int. Ed. Engl., 2012, 51, 1714–1718 CrossRef CAS PubMed.
- E. I. Solomon, R. K. Szilagyi, S. Debeer George and L. Basumallick, Chem. Rev., 2004, 104, 419–458 CrossRef CAS PubMed.
- R. Sarangi, S. DeBeer George, D. J. Rudd, R. K. Szilagyi, X. Ribas, C. Rovira, M. Almeida, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 2316–2326 CrossRef CAS PubMed.
- A. Jesser, M. Rohrmüller, W. G. Schmidt and S. Herres-Pawlis, J. Comput. Chem., 2014, 35, 1–17 CrossRef CAS PubMed.
- M. Witte, U. Gerstmann, A. Neuba, G. Henkel and W. G. Schmidt, J. Comput. Chem., 2016, 37, 1005–1018 CrossRef CAS PubMed.
- L. X. Chen, Science, 2001, 292, 262–264 CrossRef CAS PubMed.
- M. Saes, C. Bressler, R. Abela, D. Grolimund, S. L. Johnson, P. A. Heimann and M. Chergui, Phys. Rev. Lett., 2003, 90, 047403 CrossRef PubMed.
- W. Gawelda, C. Bressler, M. Saes, M. Kaiser, A. Tarnovsky, D. Grolimund, S. L. Johnson, R. Abela and M. Chergui, Phys. Scr., 2005, T115, 102–106 CrossRef CAS.
- H. Ihee, M. Wulff, J. Kim and S.-i. Adachi, Int. Rev. Phys. Chem., 2010, 29, 453–520 CrossRef CAS.
- D. Goeries, B. Dicke, P. Roedig, N. Stübe, J. Meyer, A. Galler, W. Gawelda, A. Britz, P. Geßler, H. Sotoudi Namin, A. Beckmann, M. Schlie, M. Warmer, M. Naumova, C. Bressler, M. Rübhausen, E. Weckert and A. Meents, Rev. Sci. Instrum., 2016, 87, 053116 CrossRef PubMed.
- A. Britz, T. A. Assefa, A. Galler, W. Gawelda, M. Diez, P. Zalden, D. Khakhulin, B. Fernandes, P. Gessler, H. S. Namin, A. Beckmann, M. Harder, H. Yavas and C. Bressler, J. Synchrotron Radiat., 2016, 23, 1409–1423 CrossRef PubMed.
- L. X. Chen, G. Jennings, T. Liu, D. J. Gosztola, J. P. Hessler, D. V. Scaltrito and G. J. Meyer, J. Am. Chem. Soc., 2002, 124, 10861–10867 CrossRef CAS PubMed.
- L. X. Chen, G. B. Shaw, I. Novozhilova, T. Liu, G. Jennings, K. Attenkofer, G. J. Meyer and P. Coppens, J. Am. Chem. Soc., 2003, 125, 7022–7034 CrossRef CAS PubMed.
- G. Smolentsev, A. V. Soldatov and L. X. Chen, J. Phys. Chem. A, 2008, 112, 5363–5367 CrossRef CAS PubMed.
- J. V. Lockard, S. Kabehie, J. I. Zink, G. Smolentsev, A. Soldatov and L. X. Chen, J. Phys. Chem. B, 2010, 114, 14521–14527 CrossRef CAS PubMed.
- T. J. Penfold, S. Karlsson, G. Capano, F. A. Lima, J. Rittmann, M. Reinhard, M. Rittmann-Frank, O. Braem, E. Baranoff, R. Abela, I. Tavernell, U. Rothlisberger, C. J. Milne and M. Chergui, J. Phys. Chem. A, 2013, 117, 4591–4601 CrossRef CAS PubMed.
- G. Capano, M. Chergui, U. Rothlisberger, I. Tavernelli and T. J. Penfold, J. Phys. Chem. A, 2014, 118, 9861–9869 CrossRef CAS PubMed.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282-283, 2–18 CrossRef CAS.
- G. Capano, C. J. Milne, M. Chergui, U. Rothlisberger, I. Tavernelli and T. J. Penfold, J. Phys. B: At., Mol. Opt. Phys., 2015, 48, 214001 CrossRef.
- I. B. Bersuker, Electronic structure and properties of transition metal compounds: introduction to the theory, 2010 Search PubMed.
- M. Iwamura, S. Takeuchi and T. Tahara, J. Am. Chem. Soc., 2007, 129, 5248–5256 CrossRef CAS PubMed.
- L. Hua, M. Iwamura, S. Takeuchi and T. Tahara, Phys. Chem. Chem. Phys., 2015, 17, 2067–2077 RSC.
- L. S. Forster, Coord. Chem. Rev., 2006, 250, 2023–2033 CrossRef CAS.
- Z. A. Siddique, Y. Yamamoto, T. Ohno and K. Nozaki, Inorg. Chem., 2003, 42, 6366–6378 CrossRef CAS PubMed.
- K. A. Fransted, N. E. Jackson, R. Zong, M. W. Mara, J. Huang, M. R. Harpham, M. L. Shelby, R. P. Thummel and L. X. Chen, J. Phys. Chem. A, 2014, 118, 10497–10506 CrossRef CAS PubMed.
- D. M. Zink, D. Volz, T. Baumann, M. Mydlak, H. Flügge, J. Friedrichs, M. Nieger and S. Bräse, Chem. Mater., 2013, 25, 4471–4486 CrossRef CAS.
- B. Grimm-Lebsanft, PhD thesis, Hamburg University, 2016.
- M. Wulff, A. Plech, L. Eybert, R. Randler, F. Schotte and P. Anfinrud, Faraday Discuss., 2002, 122, 13–26 RSC.
- H. Ihee, Science, 2005, 309, 1223–1227 CrossRef CAS PubMed.
- K. S. Kjær, T. B. van Driel, J. Kehres, K. Haldrup, D. Khakhulin, K. Bechgaard, M. Cammarata, M. Wulff, T. J. Sørensen and M. M. Nielsen, Phys. Chem. Chem. Phys., 2013, 15, 15003–15016 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, J. Comput. Chem., 1999, 20, 63–69 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 094107 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. Rohrmüller, A. Hoffmann, C. Thierfelder, S. Herres-Pawlis and W. G. Schmidt, J. Comput. Chem., 2015, 36, 1672–1685 CrossRef PubMed.
- A. Neuba, M. Rohrmüller, R. Hölscher, W. G. Schmidt and G. Henkel, Inorg. Chim. Acta, 2015, 430, 225–238 CrossRef CAS.
- A. Hoffmann, S. Binder, A. Jesser, R. Haase, U. Flörke, M. Gnida, M. Salomone Stagni, W. Meyer-Klaucke, B. Lebsanft, L. E. Grünig, S. Schneider, M. Hashemi, A. Goos, A. Wetzel, M. Rübhausen and S. Herres-Pawlis, Angew. Chem., Int. Ed. Engl., 2014, 53, 299–304 CrossRef CAS PubMed.
- L. G. Warner, T. O. R. Ottersen and K. Seff, Inorg. Chem., 1974, 8, 2819–2826 CrossRef.
- E. A. Ambundo, M.-V. Deydier, A. J. Grall, N. Aguera-Vega, L. T. Dressel, T. H. Cooper, M. J. Heeg, L. A. Ochrymowycz and D. B. Rorabacher, Inorg. Chem., 1999, 38, 4233–4242 CrossRef CAS.
- T. Ohta, T. Tachiyama, K. Yoshizawa, T. Yamabe, T. Uchida and T. Kitagawa, Inorg. Chem., 2000, 39, 4358–4369 CrossRef CAS PubMed.
- J. A. Thich, D. Mastropaolo, J. Potenza and H. J. Schugar, J. Am. Chem. Soc., 1974, 96, 726–731 CrossRef CAS PubMed.
- K. Miyoshi, Y. Sugiura, K. Ishizu, Y. Iitaka and H. Nakamura, J. Am. Chem. Soc., 1980, 102, 6130–6136 CrossRef CAS.
- M. L. Brader, E. W. Ainscough, E. N. Baker, A. M. Brodie and D. A. Lewandoski, J. Chem. Soc., Dalton Trans., 1990, 2089–2094 RSC.
- J. Niclos-Gutiérrez, E. Abarca-Garcia, B. Viossat, D. Nguyen-Huy, A. Busnot and J. F. Hemidy, Acta Crystallogr., 1993, C49, 19–22 Search PubMed.
- J. M. González-Pérez, D. Choquesillo-Lazarte, A. Domínguez-Martín, E. Vílchez-Rodríguez, I. Pérez-Toro, A. Castiñeiras, O. K. Arriortua, M. E. García-Rubiño, A. Matilla-Hernández and J. Niclós-Gutiérrez, Inorg. Chim. Acta, 2016, 452, 258–267 CrossRef.
- S. Fox, R. T. Stibrany, J. A. Potenza, S. Knapp and H. J. Schugar, Inorg. Chem., 2000, 39, 4950–4961 CrossRef CAS PubMed.
- J. T. York, E. C. Brown and W. B. Tolman, Angew. Chem., Int. Ed., 2005, 44, 7745–7748 CrossRef CAS PubMed.
- L. Bergmann, G. J. Hedley, T. Baumann, S. Bräse and I. D. W. Samuel, Sci. Adv., 2016, 2, e1500889 Search PubMed.
- M. Iwamura, H. Watanabe, K. Ishii, S. Takeuchi and T. Tahara, J. Am. Chem. Soc., 2011, 133, 7728–7736 CrossRef CAS PubMed.
- M. Sandroni, A. Maufroy, M. Rebarz, Y. Pellegrin, E. Blart, C. Ruckebusch, O. Poizat, M. Sliwa and F. Odobel, J. Phys. Chem. C, 2014, 118, 28388–28400 CAS.
- A. Vlček Jr., Coord. Chem. Rev., 2000, 200–202, 933–977 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- N. Binsted, EXCURV98: CCLRC Daresbury Laboratory Computer Program, CCLRC Daresbury Laboratory, Warrington, UK, 1998 Search PubMed.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- R. K. Hocking and E. I. Solomon, Struct. Bonding, 2012, 142, 155–184 CrossRef CAS.
- M. Sano, S. Komorita and H. Yamatera, Inorg. Chem., 1992, 31, 459–463 CrossRef CAS.
- Molecular Electronic Structures of Transition Metal Complexes I, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer, 2012 Search PubMed.
- W. Gawelda, V.-T. Pham, R. M. van der Veen, D. Grolimund, R. Abela, M. Chergui and C. Bressler, J. Chem. Phys., 2009, 130, 124520 CrossRef CAS PubMed.
- R. van der Veen, C. Milne, A. El Nahhas, F. Lima, V.-T. Pham, J. Best, J. Weinstein, C. Borca, R. Abela, C. Bressler and M. Chergui, Angew. Chem., Int. Ed., 2009, 48, 2711–2714 CrossRef CAS PubMed.
- M. Christensen, K. Haldrup, K. Bechgaard, R. Feidenhans'l, Q. Kong, M. Cammarata, M. L. Russo, M. Wulff, N. Harrit and M. M. Nielsen, J. Am. Chem. Soc., 2009, 131, 502–508 CrossRef CAS PubMed.
- J. H. Lee, M. Wulff, S. Bratos, J. Petersen, L. Guerin, J.-C. Leicknam, M. Cammarata, Q. Kong, J. Kim, K. B. Møller and H. Ihee, J. Am. Chem. Soc., 2013, 135, 3255–3261 CrossRef CAS PubMed.
- M. Ochmann, I. von Ahnen, A. A. Cordones, A. Hussain, J. H. Lee, K. Hong, K. Adamczyk, O. Vendrell, T. K. Kim, R. W. Schoenlein and N. Huse, J. Am. Chem. Soc., 2017, 139, 4797–4804 CrossRef CAS PubMed.
- B. E. V. Kuiken, M. R. Ross, M. L. Strader, A. A. Cordones, H. Cho, J. H. Lee, R. W. Schoenlein and M. Khalil, Struct. Dyn., 2017, 4, 044021 CrossRef PubMed.
- R. C. Fahey, in Biologically Important Thiol-Disulfide Reactions and the Role of Cyst(E)Ine in Proteins: an Evolutionary Perspective, ed. M. Friedman, Springer US, Boston, MA, 1977, pp. 1–30 Search PubMed.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Comparison of the DFT-calculated and experimental UV-Vis absorption spectra; details of the ground state EXAFS fitting; estimation of the excited state fraction in the different pump-probe experiments; TR-EXAFS data extraction and analysis; TR-WAXS: data analysis and sensitivity of the calculated transient scattering signal to the refinement of structural parameters; technical description of the setup for transient absorption in UV-Vis; comparative kinetic behavior of the sample in different solvents. See DOI: 10.1039/c7cp04880g |
| ‡ These authors contributed equally to this work. |
| § Present address: Max-Planck Institut für Kohlenforschung, 45470 Mülheim an der Ruhr, Germany. |
| This journal is © the Owner Societies 2018 |