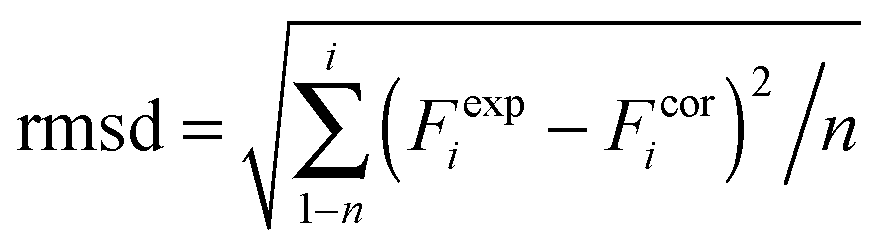Protonation/reduction dynamics at the [4Fe–4S] cluster of the hydrogen-forming cofactor in [FeFe]-hydrogenases†
Received
14th July 2017
, Accepted 29th August 2017
First published on 29th August 2017
Abstract
The [FeFe]-hydrogenases of bacteria and algae are the most efficient hydrogen conversion catalysts in nature. Their active-site cofactor (H-cluster) comprises a [4Fe–4S] cluster linked to a unique diiron site that binds three carbon monoxide (CO) and two cyanide (CN−) ligands. Understanding microbial hydrogen conversion requires elucidation of the interplay of proton and electron transfer events at the H-cluster. We performed real-time spectroscopy on [FeFe]-hydrogenase protein films under controlled variation of atmospheric gas composition, sample pH, and reductant concentration. Attenuated total reflection Fourier-transform infrared spectroscopy was used to monitor shifts of the CO/CN− vibrational bands in response to redox and protonation changes. Three different [FeFe]-hydrogenases and several protein and cofactor variants were compared, including element and isotopic exchange studies. A protonated equivalent (HoxH) of the oxidized state (Hox) was found, which preferentially accumulated at acidic pH and under reducing conditions. We show that the one-electron reduced state Hred′ represents an intrinsically protonated species. Interestingly, the formation of HoxH and Hred′ was independent of the established proton pathway to the diiron site. Quantum chemical calculations of the respective CO/CN− infrared band patterns favored a cysteine ligand of the [4Fe–4S] cluster as the protonation site in HoxH and Hred′. We propose that proton-coupled electron transfer facilitates reduction of the [4Fe–4S] cluster and prevents premature formation of a hydride at the catalytic diiron site. Our findings imply that protonation events both at the [4Fe–4S] cluster and at the diiron site of the H-cluster are important in the hydrogen conversion reaction of [FeFe]-hydrogenases.
Introduction
Hydrogenases are enzymes that catalyze hydrogen conversion in archaea, bacteria, fungi, and algae.1 Among the three types of hydrogenases only [FeFe]-hydrogenases are found in eukaryotes, for example HYDA1 of the unicellular green alga Chlamydomonas reinhardtii.1–3 HYDA1 represents the ‘minimal unit’ of biological hydrogen conversion as it exclusively binds the catalytic cofactor.3 Prokaryotic [FeFe]-hydrogenases carry additional iron–sulfur clusters and typically stem from obligate anaerobic bacteria such as Clostridium pasteurianum (CPI) or Desulvovibrio desulfuricans (DDH).4–7 [FeFe]-hydrogenases are bidirectional enzymes that catalyze H2 oxidation and H2 release from proton reduction at negligible electrochemical overpotential.8–10 Hydrogen conversion in [FeFe]-hydrogenases was proposed to be limited by the diffusion velocity of reactants11–14 and understanding the molecular principles of hydrogen conversion in [FeFe]-hydrogenases will be an important step towards the design of bio-inspired catalysts with comparable H2 release activity.15–17
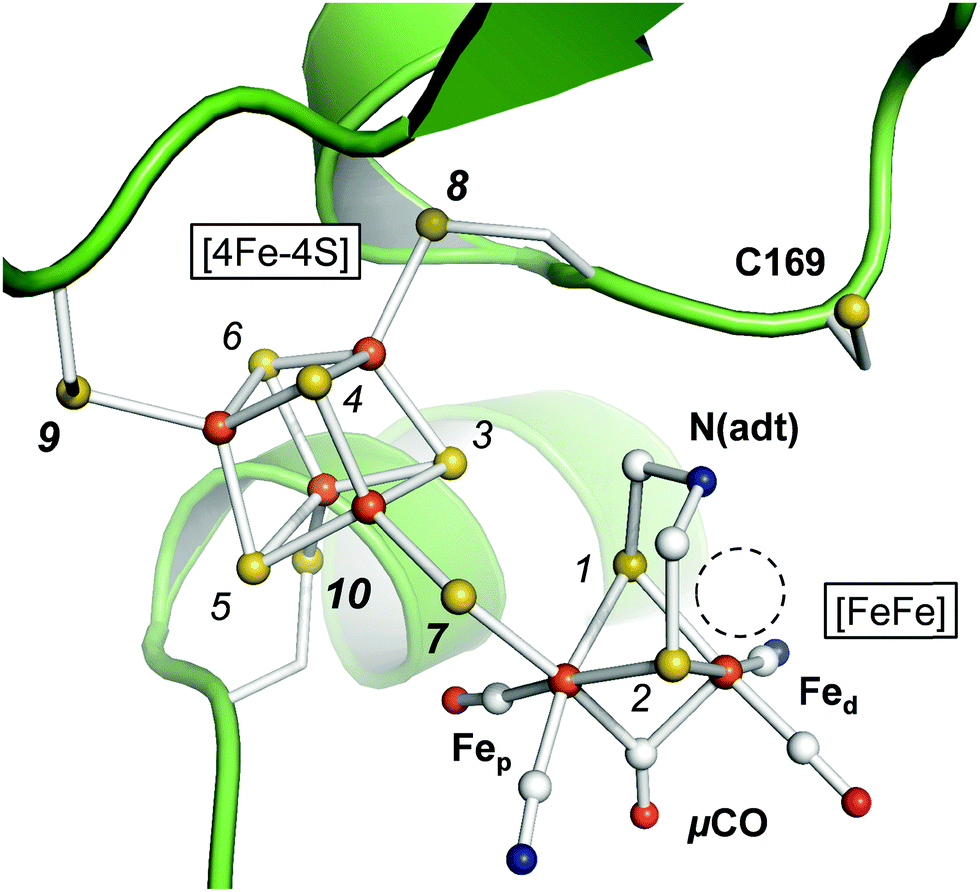
The active site cofactor of [FeFe]-hydrogenases is referred to as the H-cluster (Fig. 1). It comprises a [4Fe–4S] cluster covalently linked to a unique diiron site with a ‘proximal’ and ‘distal’ iron ion (Fep and Fed, respectively).6 An azadithiolate ligand (adt) bridges the two iron ions and introduces a potential nitrogen base in close proximity to Fed.18–20 The metal ligand sphere is complemented by a terminal carbonyl (CO) and cyanide (CN−) ligand at Fep and Fed. In the following, pCO/CN− and dCO/CN− refer to the terminal ligands in the proximal or distal position. A third carbonyl ligand is found in the Fe–Fe bridging position (μCO), for example in the active-ready, oxidized state of the cofactor (Hox).21–23 Crystal structures have shown that Fed exhibits a vacant, apical coordination site in Hox (Fig. 1). Binding of a fourth CO ligand in the CO-inhibited state (Hox-CO)24–27 has recently been proposed to result in an octahedral Fed site with an apical CN− ligand.28 Several further redox species were identified (Table 1). Two distinct one-electron reduced states have been described, Hred and Hred′, which differ in the location of the surplus electron at the H-cluster.29–31 In the two-electron ‘super-reduced’ state, Hsred, μCO adopts a terminal, non-bridging position32 and a bridging hydride (μH) was suggested.33,34 Reorientation of μCO has been discussed also for Hred.22,31,33–36 In amino acid variants of HYDA1, another two-electron reduced species has been observed. Hhyd carries a μCO ligand and was suggested to bind a terminal hydride in the apical position at Fed.37–40 It needs to be clarified whether and how the various redox species are involved in the catalytic cycle.41–45
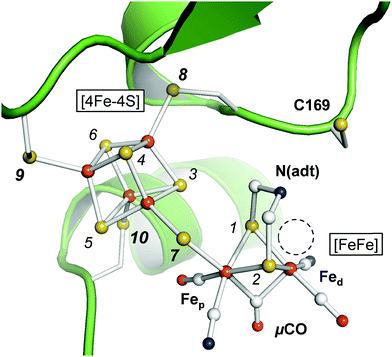 |
| | Fig. 1 Active site and catalytic cofactor in [FeFe]-hydrogenases. The crystal structure corresponds to the active-ready, oxidized state (Hox) of bacterial [FeFe]-hydrogenase CPI (PDB entry 4XDC).6 The catalytic cofactor comprises a [4Fe–4S] cluster and a diiron site, [FeFe]. Fep and Fed – proximal and distal iron ions. μCO–Fe–Fe bridging CO ligand. N(adt) – nitrogen headgroup of the adt ligand. The dashed circle highlights an apical vacancy at Fed. C169 is a conserved cysteine residue that couples the H-cluster to the proton transfer pathway. Sulfur atoms are numbered 1–10 according to the DFT model (see the ESI†). | |
Table 1 Electronic configuration of different redox species
| Redox species |
Δe− |
[4Fe–4S] cluster |
Diiron sitea |
Alternative annotation |
Ref. |
|
Formal charge is given for Fep/Fed. Only the mixed-valence state I/II is EPR-active.
|
|
Hox
|
— |
+2 |
I/II |
— |
46–48
|
|
Hox-CO
|
— |
+2 |
I/II |
— |
25–28
|
|
Hred′
|
1 |
+1 |
I/II |
H′red′, Hred |
29, 31 and 49
|
|
Hhyd
|
2 |
+1 |
II/II |
— |
37–40
|
|
Hred
|
1 |
+2 |
I/I |
HredH+ |
21, 22 and 31
|
|
Hsred
|
2 |
+1 |
I/I |
— |
32 and 33
|
Exchange of protons between the [FeFe]-hydrogenase active site and the aqueous solvent is supported by a chain of phylogenetically conserved amino acids. In HYDA1, the putative proton transfer pathway comprises residues E144, S189, E141, C169, and at least one water molecule.50–52 Side-directed mutagenesis studies identified C169 as the primary proton relay to the diiron site of the H-cluster51 and proton transfer is further facilitated by the N(adt) group (Fig. 1).18–20,35 Theoretical studies support a rapid exchange of protons via this ‘catalytic’ pathway.53–55 However, the interplay between proton transfer and electron transfer events in the hydrogen conversion reaction of [FeFe]-hydrogenases remains largely unclear.
Here, we describe in situ, real-time spectroscopy on [FeFe]-hydrogenase protein films under controlled variation of gas composition, sample pH, and reductant concentration. Shifts of the CO/CN− vibrational bands were used to study electron and proton transfer at the active site via attenuated total reflection Fourier-transform infrared spectroscopy (ATR FTIR). Vibrational band patterns were characterized for three different [FeFe]-hydrogenases, proteins with amino acid exchanges in the proton path, and five cofactor variants comprising alternative dithiolate groups. Redox and pH titrations, element and isotope exchange studies, and density functional theory (DFT) calculations identified novel H-cluster species, most likely protonated at the [4Fe–4S] cluster.
Experimental
Protein sample preparation
[FeFe]-hydrogenase HYDA1 and CPI apo-proteins were over-expressed in Escherichia coli, purified, quantitatively reconstituted in vitro with synthetic diiron complexes (Fe2(x)(CO)4(CN)2), and concentrated to 500 μM protein (∼25 g L−1).20 The typical H2 release activity was ∼800 μmol H2 mg−1 HYDA1adt min−1 and ∼2500 μmol H2 mg−1 CPIadt min−1. Analyzed cofactor variants included adt = (SCH2)2NH, odt = (SCH2)2O, sdt = (SCH2)2S, pdt = S2(CH2)3, or edt = S2(CH2)2 as bridging dithiolate groups (x) at the diiron site. Site-directed mutagenesis of HYDA1 at position 169 yielded variants C169D and C169A as described earlier.50 Selenium substitution at the [4Fe–4S] cluster was performed as recently published.56 Wildtype DDH enzyme was purified from D. desulfuricans in a functional form.57 All protein preparation and handling procedures were carried out at room temperature, under strictly anoxic conditions, and dim light.
Fourier-transform infrared spectroscopy
Fig. 2 shows a schematic drawing of the experimental ATR FTIR set-up for exposure of protein films to an aerosol with defined humidity, pH, and reactant concentration. In section (A), gasses are mixed and the flow volume is adjusted. Dry N2 carrier gas of ultra-high purity (5.0) was provided by a nitrogen generator (PN1450, Inmatec, Germany). A constant gas stream (typically 1.5 L min−1) was adjusted using digital mass flow controllers (MFC, SmartTrak, Sierra, USA). Reactant gases (12CO, 13CO, H2 from Linde, Germany) were added to the N2 stream via separate flow controllers. A 200 mbar check valve separates the dry gas section (A) from the wet gas section (B). In order to create a reactive aerosol, the gas stream was passed through a wash bottle containing 150 mL mixed buffer (50 mM Tris, MES, PIPPS, pH 3–9) and sodium dithionite (DT) and/or methyl viologen (MV). In Fig. S1 (ESI†), we demonstrate how water-soluble ingredients are transported with the aerosol stream, i.e. to reduce MV via DT enrichment. Section (C) shows the spectroscopic set-up. The aerosol was fed into a gas-tight polychlorotrifluoroethylene (PCTFE) compartment, attached on top of the ATR crystal plate and equipped with six optional gas inlets, a manometer for pressure control, and a transparent glass window for UV/vis irradiation.28 The FTIR spectrometer (Tensor27, Bruker Optik, Germany) was equipped with a triple-reflection ZnSe/Si crystal ATR cell (Smith Detection, USA) and placed in an anaerobic chamber (Coy Laboratories, USA). Infrared spectra were recorded with 80 kHz scanning velocity at a spectral resolution of 1 cm−1 from 3800 to 1200 cm−1. Under these conditions, the time-resolution of data acquisition is in the range of 1 s (one interferometer scan in the forward/backward direction). In order to determine the timescale of equilibration in the aerosol set-up, we probed a fast process, i.e. CO inhibition.28 Fig. S2 (ESI†) shows the accumulation of Hox-CO upon exchange of N2 to CO gas. Here, the Hox → Hox-CO conversion rate determined by ATR FTIR largely exceeds the value suggested by flash-photolysis IR spectroscopy (t1/2 ≈ 3 s and 15 ms, respectively).58–60 The former represents the timescale of equilibration and defines the temporal resolution of the aerosol set-up. Note that all analyzed titrations proceeded by far slowly and were therefore not limited by the experimental time resolution. ATR FTIR measurements were performed at 25 °C air-conditioning and on hydrogenase films derived by controlled dehydration and rehydration of 1 μL protein sample as reported earlier (500 μM hydrogenase in 100 mM Tris/HCl pH 8, 100 mM NaCl, and 2 mM DT if not stated otherwise). Samples of anaerobically purified and activated [FeFe]-hydrogenase typically contained the H-cluster in a mixture of the Hox, Hox-CO, and Hred states. Hox was enriched in the film under a constant stream of N2 aerosol for 30 minutes prior to the pH or redox titrations.
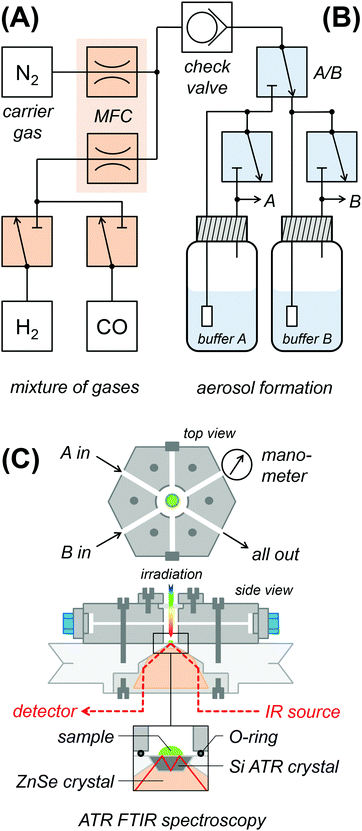 |
| | Fig. 2 Experimental set-up for ATR FTIR on protein films. (A) Various gases (e.g. H2, CO) were mixed into a constant stream of N2 carrier gas. The gas flow was controlled by digital mass flow controllers (MFC). (B) Dry gas was passed through either wash bottle A or B, comprising different buffer compositions. (C) The thereby created aerosol was fed to the headspace above the protein film. On top of the ATR reflection unit, a custom-made PCTFE chamber with gas in- and outlets and manometric pressure control capped the silicon crystal and the protein film. | |
Infrared data evaluation
A quantitative analysis of infrared spectra was performed using in-house software tools (Fig. S2, ESI†). Spectra in the 1750–2150 cm−1 frequency range were corrected for background contributions (liquid H2O) by subtraction of polynomial spline functions. The resulting spectra were normalized to unity area in the CO/CN− spectral region and the CO/CN− bands were fitted by Voigt functions (50% Gaussian and Lorentzian characters) for frequency, peak width, and relative intensity determination. A global analysis approach was used (simultaneous fitting of time-series of spectra with a common set of frequencies and relative intensities for each H-cluster species). The relative population of a specific H-cluster species at a given time point was derived as the fraction of its summed CO/CN− band intensities of the total (normalized) spectral intensity. We measured an experimental uncertainty of <25% for the determination of rate constants in pH and redox titrations (Fig. S2, ESI†).
Density functional theory calculations
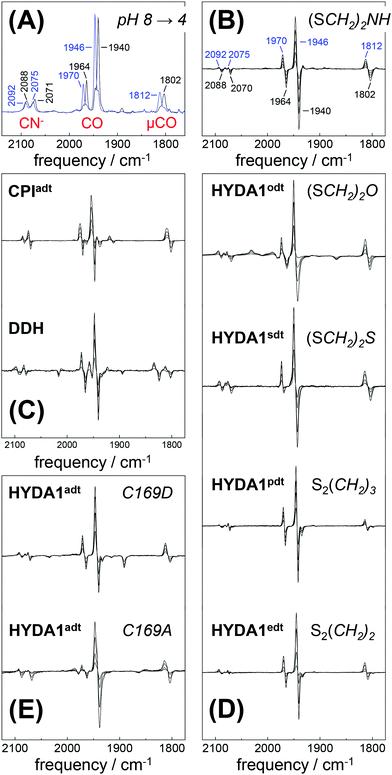
DFT calculations on model structures of the H-cluster were performed at the TPSSh functional and TZVP basis-set level of theory using Gaussian09.61–64 Starting models comprised the complete H-cluster and were constructed on the basis of the crystal structure (PDB entry 4XDC) of CPI [FeFe]-hydrogenase as described previously.28 In some cases, adjacent amino acids and water molecules were included (see below). Anti-ferromagnetic coupling was assigned to the H-cluster prior to geometry optimization by proper definition of molecular fragments (broken symmetry approach). Calculations involved the COSMO solvation model (ε = 4). H-cluster structures with varying protonation sites were calculated. During structural relaxation, the positions of the four carbon atoms of the S-CH3 groups (representing the truncated terminal cysteine ligands of the H-cluster) were restraint at their crystallographic positions.6 A maximum deviation of ∼3 cm−1 was observed for the CO and CN− band frequencies between calculations on constraint or unconstraint model structures (average deviation of ∼1.5 cm−1). Vibrational frequencies for the relaxed molecular structures were derived from normal mode analysis as implemented in Gaussian09. Summed band intensities of CO/CN− ligands from the calculations were normalized to unity for comparison with experimental data. For quantification of the correlation between experimental (exp) and calculated (cal) frequencies (F) and relative band intensities (I), the root-mean-square-deviation (rmsd) was calculated (eqn (1), n = number of CO/CN− bands). For band intensities, rmsdI values were derived using the uncorrected calculated intensities (an almost ideally linear correlation was observed for Hox: Icalox = 0.2% + 0.99Iexpox). For band frequencies, rmsdF values were calculated after correction of the calculated frequencies for a systematic deviation from ideal correlation due to the DFT theory level.61 Calculated frequencies for Hox were plotted versus the experimental Hox frequencies, a linear fit was performed yielding slope (sox = 1.15) and offset (oox = −243 cm−1) values for Hox (eqn (2)). The corrected (cor) frequencies for all H-cluster species (i) were calculated using eqn (3) (n = number of bands, i.e. 2 CN− plus 3 CO = 5 bands for, e.g., Hox) and the sox and oox values (meaning that rmsdF values for all species were directly comparable to the rmsdF for Hox).| | 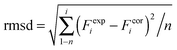 | (1) |
| | | Fcalox = oox + soxFexpox | (2) |
| | | Fcori = (Fcali − oox)/sox | (3) |
Results and discussion
IR band shifts upon acidification of [FeFe]-hydrogenase
Fully active [FeFe]-hydrogenase HYDA1 as reconstituted with the native adt cofactor19,20 was used to form a protein film on the ATR silicon crystal. The active-ready, oxidized state Hox was enriched to purity under a stream of humidified N2 at pH 8 and 2 mM sodium dithionite (DT).28 In contrast to earlier studies23,32 no build-up of reduced H-cluster species was observed, most likely due to release of H2 from the protein film. It can be assumed that the decay rate of reduced species exceeds their formation rate and the removal of H2via the N2 carrier gas precludes a re-reduction of enzyme. When the film was exposed to an acidified aerosol (pH 4, 2 mM DT), the IR band pattern of Hox was lost in favor of a novel spectrum (Fig. 3A and B). We note that the indicated pH values refer to the pH of the respective buffer used in the experiments. Based on calibration experiments, a difference of approximately ∼0.25 pH units between the buffer and the sample film was estimated (Fig. S3, ESI†). The novel pattern showed an up-shift of the CO/CN− bands by 4–6 cm−1 with similar relative intensities as in the Hox spectrum. Furthermore, the pattern comprised the characteristic low-frequency band of the μCO ligand, up-shifted by 10 cm−1. The conversion was found to be fully reversible when changing the pH in the aerosol back to alkaline values (Fig. S4 and S5, ESI†). Although the HYDA1 H2 release activity was increasingly inhibited from pH 7 to 5, no structural changes were observed in the amide region when titrating the film from pH 8 to 4 (Fig. S6, ESI†). Note that extreme conditions (pH 3, 500 mM DT) let to irreversible loss of cofactor bands (Fig. S4, ESI†). Frequency shifts by 4–10 cm−1 are not compatible with redox changes at the diiron site and the shift to higher frequencies excludes a reduction of the H-cluster, e.g. by DT. Dithionite is commonly used as a reductant to promote H2 evolution from [FeFe]-hydrogenases in solution.65 Accordingly, we attribute the observed ‘blue shift’ to a protonation event and denote the novel species HoxH (Table 2). It is important to note that the Hox → HoxH conversion was not observed in the absence of DT (Fig. S4, ESI†). This result suggests that reducing conditions are required to induce the protonation event and the shift of cofactor bands is unrelated to protonation of amino acid residues.
 |
| | Fig. 3 FTIR difference spectra for the Hox → HoxH conversion (‘blue shift’). The CO/CN− spectral region is shown. (A) Absolute spectra for the Hox → HoxH conversion in native HYDA1adt obtained at pH 8 (black trace) and pH 4 in the presence of 2 mM DT (blue trace). Assignment of bands to vibrational modes was derived from DFT calculations (CN− and CO/μCO marked in red). (B) Difference spectra obtained during Hox (negative bands) to HoxH (positive bands) conversion in HYDA1adt from the same data set as in (A). (C) Difference spectra for bacterial [FeFe]-hydrogenases CPIadt and DDHadt. (D) Difference spectra for HYDA1 cofactor variants odt, sdt, pdt, and edt. (E) Difference spectra for HYDA1adt amino acid variants C169A and C169D. Table S1 (ESI†) lists the vibrational frequencies for all analyzed variants. | |
Table 2 IR band frequencies of CO and CN− ligands in HYDA1adt
| H-Cluster species |
IR frequency/cm−1 |
Additional reference |
| CN− bands |
CO bands |
|
The bridging carbonyl (μCO) is moved to a terminal position at Fed in Hred and Hsred.
In the CO-inhibited states, the coupled pCO/dCO stretching vibration gives rise to an additional IR band (Hox-CO: 2012 cm−1, HoxH-CO: 2006 cm−1). IR frequencies were determined from the spectra shown in Fig. 3, 6 and Fig. S7, S10 (ESI).
|
|
Hox
|
2088 |
2070 |
1964 |
1940 |
1802 |
21, 22 and 28
|
|
HoxH
|
2092 |
2074 |
1970 |
1946 |
1812 |
This study |
|
Hred′
|
2084 |
2066 |
1962 |
1933 |
1792 |
29, 31 and 49
|
|
Hred′H
|
2086 |
2068 |
1966 |
1938 |
1800 |
This study |
|
Hhyd
|
2088 |
2076 |
1980 |
1960 |
1860 |
40
|
|
Hred
|
2070 |
2033 |
1961a |
1915 |
1891 |
21, 22 and 31
|
|
Hsred
|
2068 |
2026 |
1953a |
1918 |
1882 |
32
|
|
Hox-CO
|
2091 |
2081 |
1968 |
1962b |
1808 |
25–28
|
|
HoxH-CO
|
2094 |
2086 |
1972 |
1966b |
1816 |
This study |
Exposure of bacterial [FeFe]-hydrogenases CPIadt and native DDH to pH 4 and 2 mM DT caused a similar ‘blue shift’ of the Hox band pattern (Fig. 3C). Formation of HoxH was not affected by the accessory iron–sulfur clusters in the prokaryotic enzymes or amino acid differences near the H-cluster. We note that HYDA1 and CPI apo-enzymes were artificially maturated19,20 whereas DDH was isolated from D. desulfuricans in the native form.57 The formation of HoxH hence is unrelated to the maturation procedure. Besides HYDA1adt, we investigated cofactor variants with a less basic (odt, sdt) or hydrophobic (pdt, edt) bridgehead group at the diiron site. For all modified enzymes, a similar Hox → HoxH conversion at pH 4 and 2 mM DT was observed (Fig. 3D). This clearly proves that the bridgehead atom does not correspond to the protonation site. However, the cofactor variant HYDA1pdt showed an approximately 20-fold slower formation of HoxH in comparison to HYDA1adt (Fig. S5, ESI†), presumably related to the much smaller H2 release activity of the pdt variant.66 Protein variants with amino acid exchanges in the proton pathway towards the diiron site were studied (C169D and C169A). Although typical IR spectra and quantitative formation of HoxH were observed in both variants (Fig. 3E), the rate of HoxH formation in the C169A variant was smaller than in wild-type HYDA1adt or in the C169D variant (Fig. S5, ESI†), ruling out that cysteine 169 functions as the protonation site in HoxH. The amino acid variant HYDA1 C169D showed H2 release activity significantly similar to native enzyme (up to 80%) while HYDA1 C169A did not evolve H2 in the presence of DT and MV. We therefore assume that H2 release activity is correlated with the formation rate of HoxH.
Proton and reductant concentration effects on HoxH formation
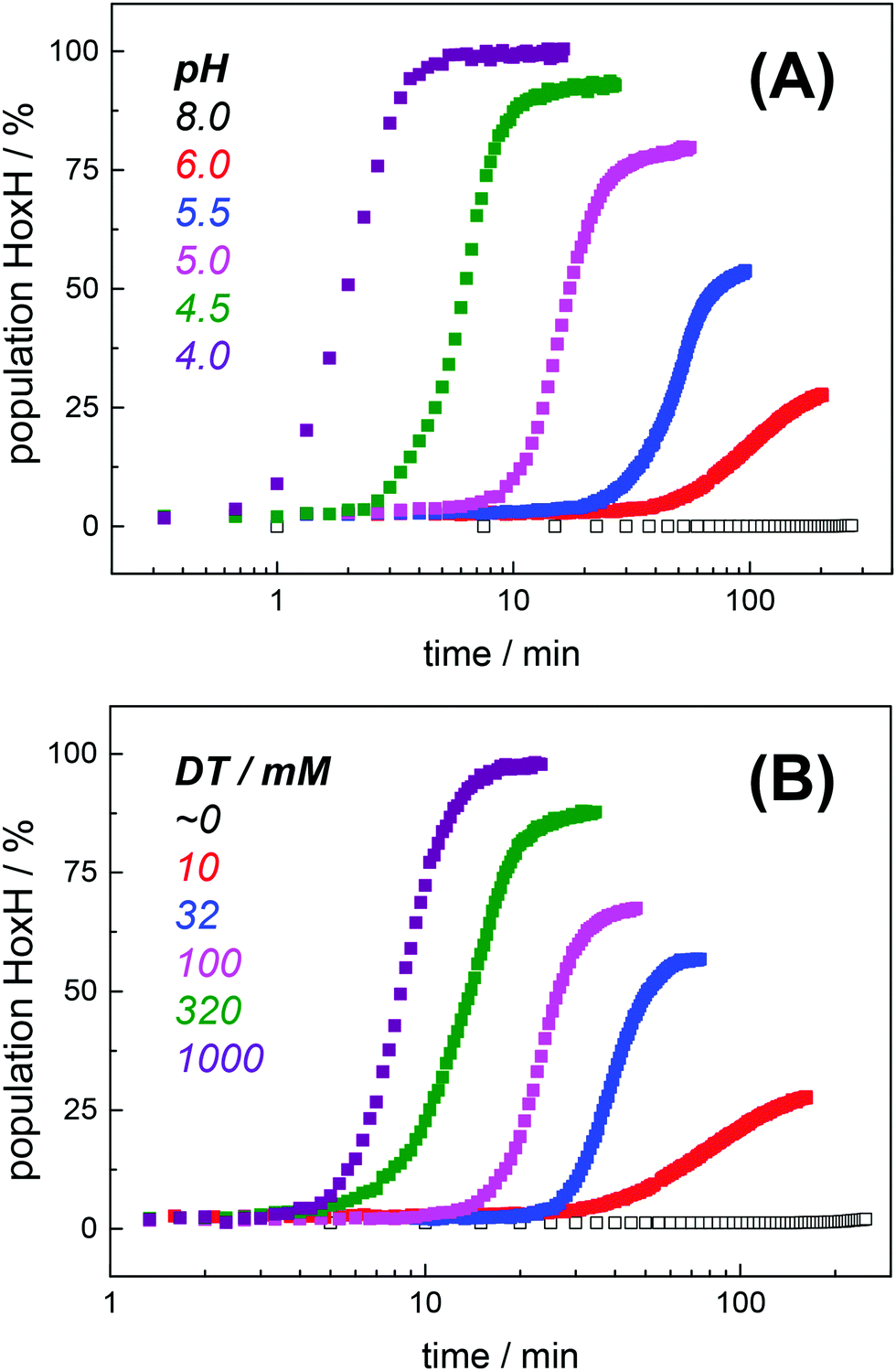
Protein concentration, hydration level, and temperature affected the Hox → HoxH conversion rate (Fig. S6, ESI†). Accordingly, a quantitative comparison demanded tight control of all experimental parameters. We determined an experimental uncertainty in the determination of conversion rates of less than 25% (Fig. S2, ESI†) and observed negligible differences in the hydration level (Fig. S6, ESI†). Accordingly, it was possible to investigate the influence of proton and DT concentration on the yield and formation rate of HoxH. Fig. 4A shows the Hox → HoxH conversion with individual protein films after a change of the aerosol from pH 8 to pH 6–4 with 10 mM DT. To elucidate the influence of DT, HoxH formation was monitored in a concentration range of 0–1000 mM DT at pH 6 (Fig. 4B). Aqueous solutions of DT were freshly prepared and the pH was checked before and after each experiment, which excluded any decomposition of DT.67,68 Clearly, the formation rate of HoxH depends on the DT concentration as well. The sigmoidal time course of the Hox → HoxH transition suggested that at least two consecutive processes are involved, i.e. proton transfer and electron transfer. In contrast, back conversion to Hox showed exponential behavior and the rate was found to be virtually independent of pH and DT (Fig. S5, ESI†). These observations might suggest that the divergent velocity of the Hox ↔ HoxH conversion is related to protein structural rearrangements in the ‘blue shift’ reaction. However, the formation of HoxH was not associated with changes in the protein structure or denaturation (Fig. S6, ESI†). Despite the high concentrations of DT used in some experiments, no enrichment of reduced H-cluster species was observed.
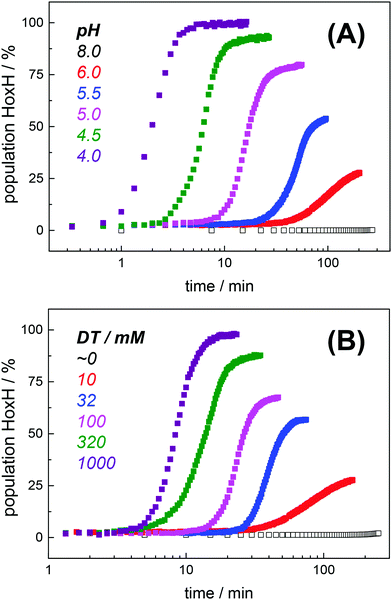 |
| | Fig. 4
Hox → HoxH conversion in HYDA1adt as a function of pH and dithionite concentration. (A) Increase of the HoxH population at a constant DT concentration of 10 mM and pH 6–4 in the aerosol. No HoxH is observed at pH 8 (open squares). (B) Increase of the HoxH population at a constant pH of 6 and varying DT concentration (10–1000 mM) in the aerosol. No HoxH is observed in the absence of DT (open squares, residual DT might still be present in the preparation). Each data point in (A) and (B) corresponds to a normalized IR spectrum for which the relative HoxH population (in %) was calculated from the respective sum of CO/CN− peak areas. | |
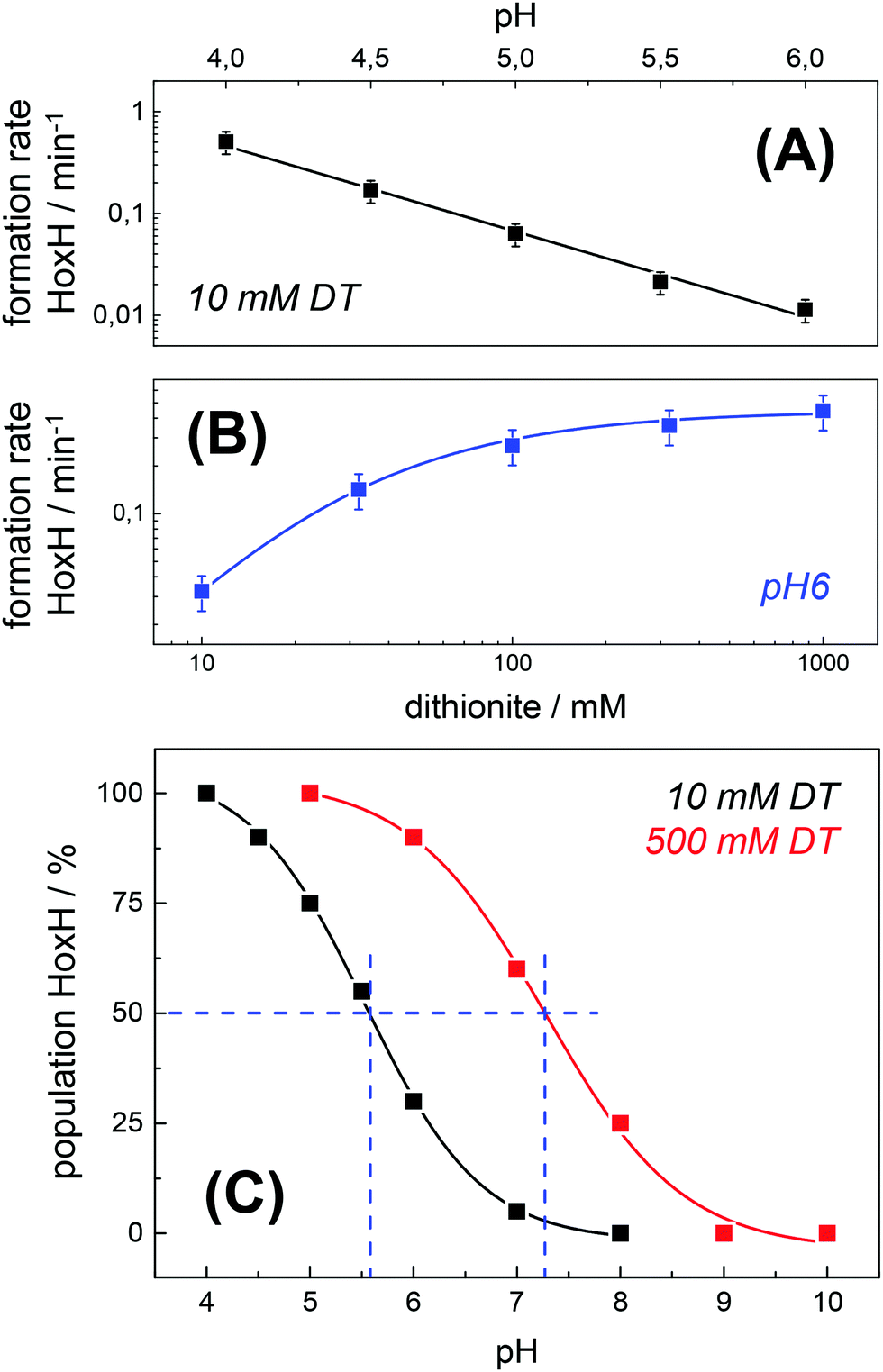
The formation rate of HoxH increased linearly with proton concentration in a pH range of 4–7 at 10 mM DT (Fig. 5A). A pH titration with a 50-fold increase of DT (500 mM) showed an overall faster Hox → HoxH conversion and linear regression from pH 3 to 7 (Fig. S4, ESI†). In contrast, saturation behavior of the HoxH formation rate was observed at excess DT concentrations (Fig. 5B). Accelerated formation of HoxH was also observed in the presence of the redox mediator MV (Fig. S5, ESI†) which is often used to optimize electron transfer from DT to hydrogenase enzymes.65Fig. 5 compares the HoxH yield for pH titrations at 10 mM or 500 mM DT, respectively. Significant formation of HoxH was observed even at alkaline pH, and the apparent pK shifted from ∼5.5 to ∼7.3 for higher DT concentrations. The titration behavior of a protonatable group should be independent of reductant concentration. The variable, apparent pK value thus excludes a specific amino acid residue as the protonation site in HoxH.
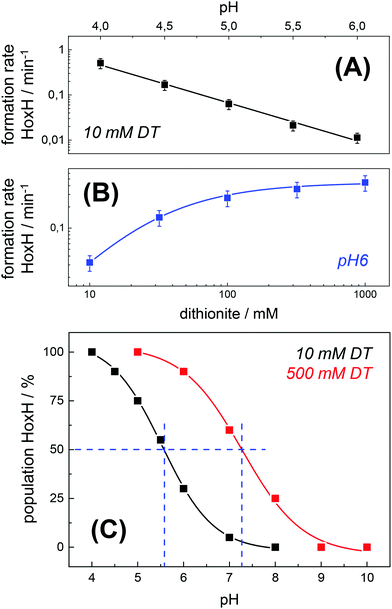 |
| | Fig. 5 Rate and yield of Hox → HoxH conversion in HYDA1adt. From the time points (t) of half-maximal HoxH formation, apparent rate constants (1/t) for the Hox → HoxH conversion were derived (see Fig. 4 for corresponding data). Error bars were estimated from multiple experiments under the same conditions (Fig. S2, ESI†). (A) HoxH formation rate as a function of pH (10 mM DT). Note the logarithmic y-axis, the lines show linear regressions. (B) HoxH formation rate as a function of DT concentration (pH 6) (the line shows a saturation curve). (C) HoxH yield as a function of pH for 10 mM (black) and 500 mM DT (red, see Fig. S4, ESI† for corresponding data). The apparent pK (blue dashes) differs by about two units. | |
Ligand exchange and isotope editing at the H-cluster
Further information on the formation of HoxH was obtained from CO-inhibition and isotopic exchange studies. HYDA1adt was quantitatively inhibited with 1% CO gas, as indicated by the typical IR bands of Hox-CO (Table 2).28 Acidification in the presence of 2 mM DT under 1% CO induced an overall similar ‘blue shift’ of the CO/CN− bands as observed for Hox (Fig. S7, ESI†). We attribute the shifted spectrum to a protonated, CO-inhibited H-cluster species, HoxH-CO. Formation of HoxH-CO implies that protonation cannot occur at the distal iron ion because its ligand sphere was completed by the additional CO ligand already.24–28 Similar to the Hox → HoxH conversion, DT was required for the formation of HoxH-CO. Exchange of the bridging sulfur (μS) against selenium (μSe) in the [4Fe–4S] cluster of the catalytic cofactor has been shown to barely affect catalytic activity.56 Selenium-substituted HYDA1adt showed unchanged Hox and HoxH IR-band patterns. Although the pK values of thiols and selenols can differ by up to three units69–71 and a protonation event at μSe of the [4Fe–4Se] cluster should influence the formation rate of HoxH, the latter was not found to be affected (Fig. S8, ESI†). Therefore, direct protonation of the bridging sulfur atoms in the [4Fe–4S] cluster seems to be less likely. Exposure of [FeFe]-hydrogenase to an aerosol containing D2O instead of H2O facilitated quantitative deuteration of the protein film (Fig. S9, ESI†). For diiron model complexes, protonation at the bridging thiolate sulfur atoms has been considered.72–74 The frequency down-shift of the S–H vibration upon deuteration75 from ∼2550 to ∼1850 cm−1 could help in identifying protonation of the dithiolate ligand at the diiron site due to vibrational coupling with the CO/CN− ligands. However, formation of HoxH in the presence of 0.5% D2SO4 and 2 mM DT did not result in altered Hox and HoxH IR-band patterns (Fig. S9, ESI†), even when HYDA1 was isolated and activated in the presence of D2O before film formation and acidification. A direct detection of SH/SD bands in the sample is unrelated to vibrational coupling with the cofactor ligands and was not attempted. These results render protonation of the thiolate sulfur atoms at the diiron site in HoxH less likely.
Reduction of HYDA1 with H2
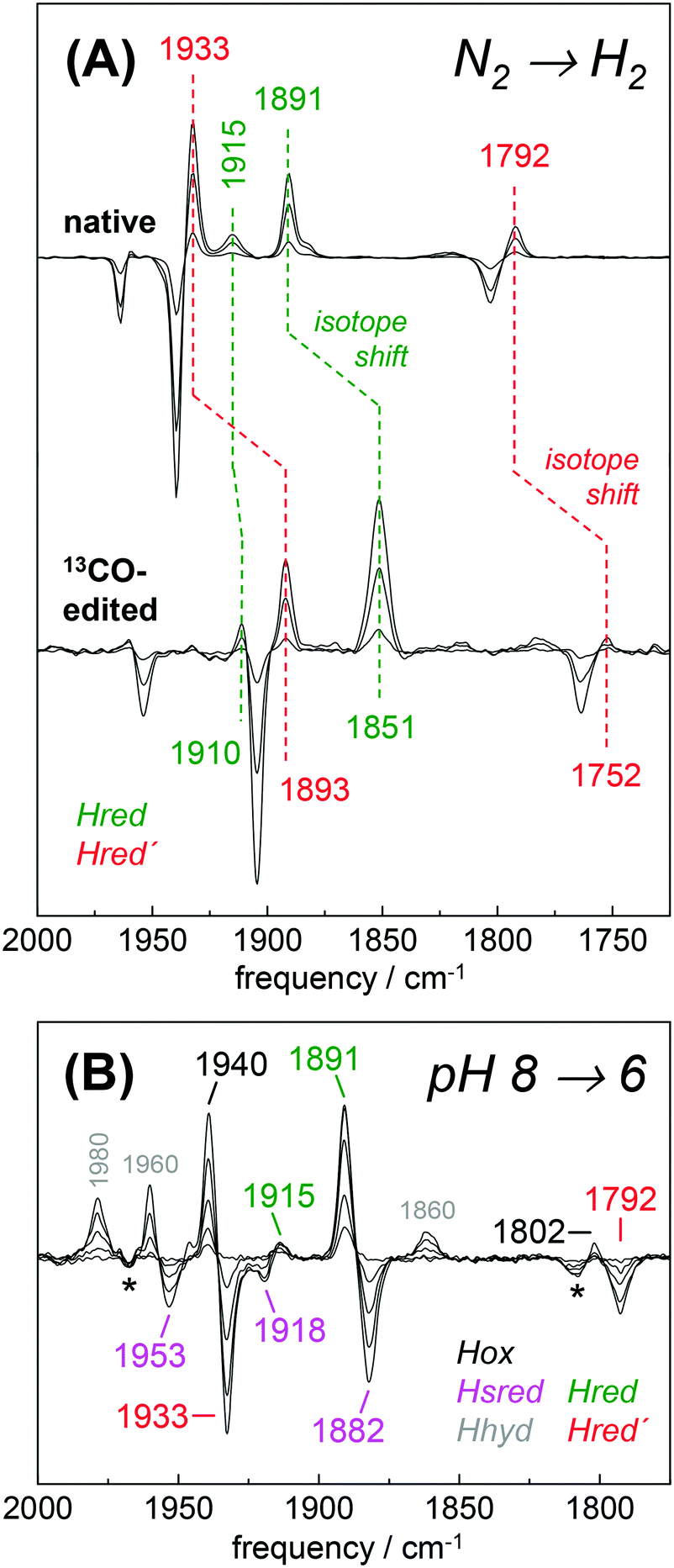
The H2 uptake activity of HYDA1adt was exploited to enrich reduced H-cluster species in the protein film. In the presence of 10% H2 the IR signature of Hox was lost in favor of band patterns mainly attributable to reduced species (Fig. 6 and Table 2). In the H2–N2 difference spectrum at pH 8, prominent CO bands at 1933 cm−1 and 1792 cm−1 resembled a pattern previously observed in reduced HYDA1pdt enzyme.29 These bands have been assigned to Hred′ in HYDA1adt recently.31,49 Interestingly, we found that Hred′ was favored over Hred when progressing from acidic to alkaline pH, and vice versa (Fig. S10, ESI†). This can be explained by the PCET reactivity in the formation of Hred and destabilization of the reduced diiron site at alkaline pH.31 For a distinct assignment of IR band patterns, we exchanged the bridging and distal 12CO ligands in HYDA1adt to 13CO, following a previously established protocol.28 Reduction of the isotope-edited enzyme with H2 facilitated direct assignment of the bands at 1891 cm−1 and 1933 cm−1 to dCO of Hred and Hred′, respectively (Fig. 6). Whether the μCO band at 1792 cm−1 belonged to Hred or Hred′ still remains unclear. To refine the assignment, HYDAadt was reduced under 10% H2 and titrated from pH 8 to pH 6 (Fig. 6 and Fig. S10, ESI†). Although the H2 concentration was kept constant, acidification caused pronounced changes in the relative population of redox species and Hsred and Hred′ were lost in favor of Hred and Hox. Due to the concomitant decrease of bands at 1933 cm−1 (dCO) and 1793 cm−1 (μCO), the experiment facilitated a clear assignment of these bands to Hred′ (Table 2). The deviating pH-dependence of bands at 1933 cm−1 and 1891 cm−1 further distinguishes Hred′ from Hred (Fig. S10, ESI†). In agreement with former observations,40 an increase of Hhyd was observed when decreasing the pH from 8 to 6 (Fig. 6). We observed no ‘blue shift’ of the marker bands of Hred (1891 cm−1) and Hsred (1882 cm−1) at acidic pH.
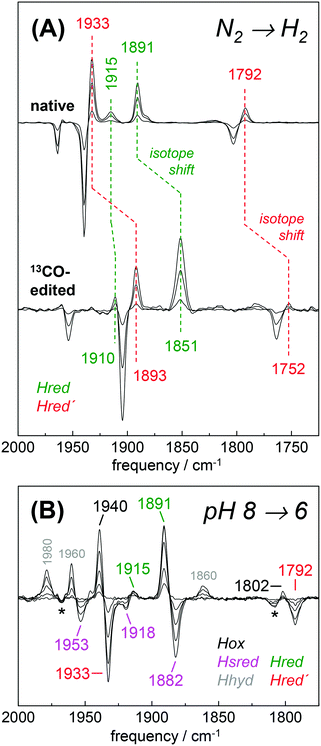 |
| | Fig. 6 Reduction of HYDA1adt in the presence of H2. (A) IR difference spectra in the CO region collected during a change from 100% N2 to 10% H2 on native (top) and 13CO-edited HYDA1adt (bottom). Spectra show an increase of Hred (green) and Hred′ (red) at the expense of Hox (black). Isotope editing of the μCO and dCO ligands leads to ∼40 cm−1 frequency down-shifts in Hox, Hred, and Hred′. (B) Difference spectra for changing the pH from 8 to 6 in the presence of 10% H2 (for absolute spectra see Fig. S10, ESI†). Here, Hred (green) and Hox (black) are populated at the expense of Hred′ (red) and Hsred (magenta). Note the additional formation of Hhyd (grey) and decrease of the minor Hox-CO population (asterisks). | |
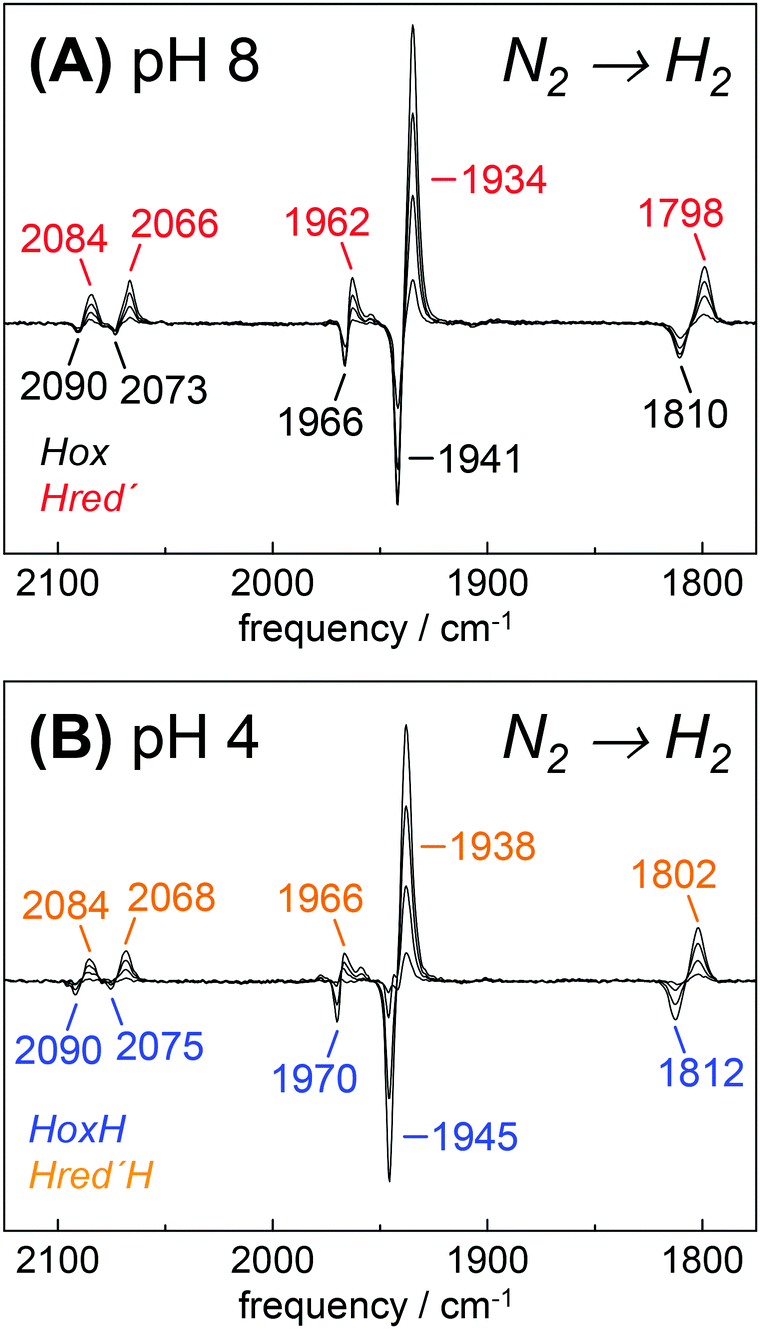
To investigate Hred′ in greater detail, we focused on HYDA1 cofactor variant pdt. This variant does not form Hred and Hsred and therefore represents an excellent model to study the relation between Hox and Hred′.29 Although HYDA1pdt has been reported to show only minimal H2 uptake activity,66 we observed quantitative Hox → Hred′ conversion under 100% H2 (Fig. 7). At pH 8, both states showed similar IR band patterns as in HYDA1adt with relative frequency shifts of less than 5 cm−1. When HYDA1pdt was purged with N2 at pH 4 and in the presence of 2 mM DT, an IR spectrum similar to the one of HoxH was observed (compare Fig. 3). Exposure to 100% H2 under acidic conditions resulted in a minor ‘blue shift’ of all CO/CN− bands with respect to Hred′ (Fig. 7). We denote the species responsible for this shifted spectrum as Hred′H (Table 2). Redox titrations indicated the presence of Hred′H in HYDA1adt as well, although this state was more difficult to detect in native enzyme due to competing population of Hred/Hsred and Hhyd (Fig. S10, ESI†).40
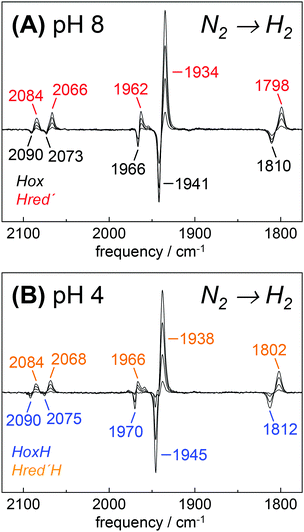 |
| | Fig. 7 Reduction of HYDA1pdt in the presence of H2. IR difference spectra collected during a change in the aerosol from N2 to H2. (A) At pH 8 and 2 mM DT, Hox (black) was found to be de-populated in favor of Hred′ (red). (B) At pH 4 and 2 mM DT, HoxH (blue) converted into Hred′H (orange). | |
When the hydrogenase concentration in the film was decreased by dilution with bovine serum albumin protein (BSA), the Hox → Hred′ conversion rate drastically dropped for relative concentrations of HYDA1pdt below 25% and no formation of reduced states was observed with 5% HYDA1 (Fig. S11, ESI†). This behavior suggests that the inter-molecular electron transfer between [FeFe]-hydrogenase molecules (disproportionation) was involved in the formation of one-electron reduced Hred′ from oxidized Hoxvia two-electron reduced species transiently formed upon reduction with H2. Inefficient inter-molecular electron transfer may explain the low rates of H2 uptake that have been reported for HYDApdt in solution.66 In contrast, the formation rate of HoxH was only slightly affected by dilution of HYDA1pdt with BSA (Fig. S11, ESI†). This observation rules out inter-molecular electron transfer as an explanation for the apparent absence of reduced species in the presence of high DT concentrations.
Density functional theory on H-cluster protonation
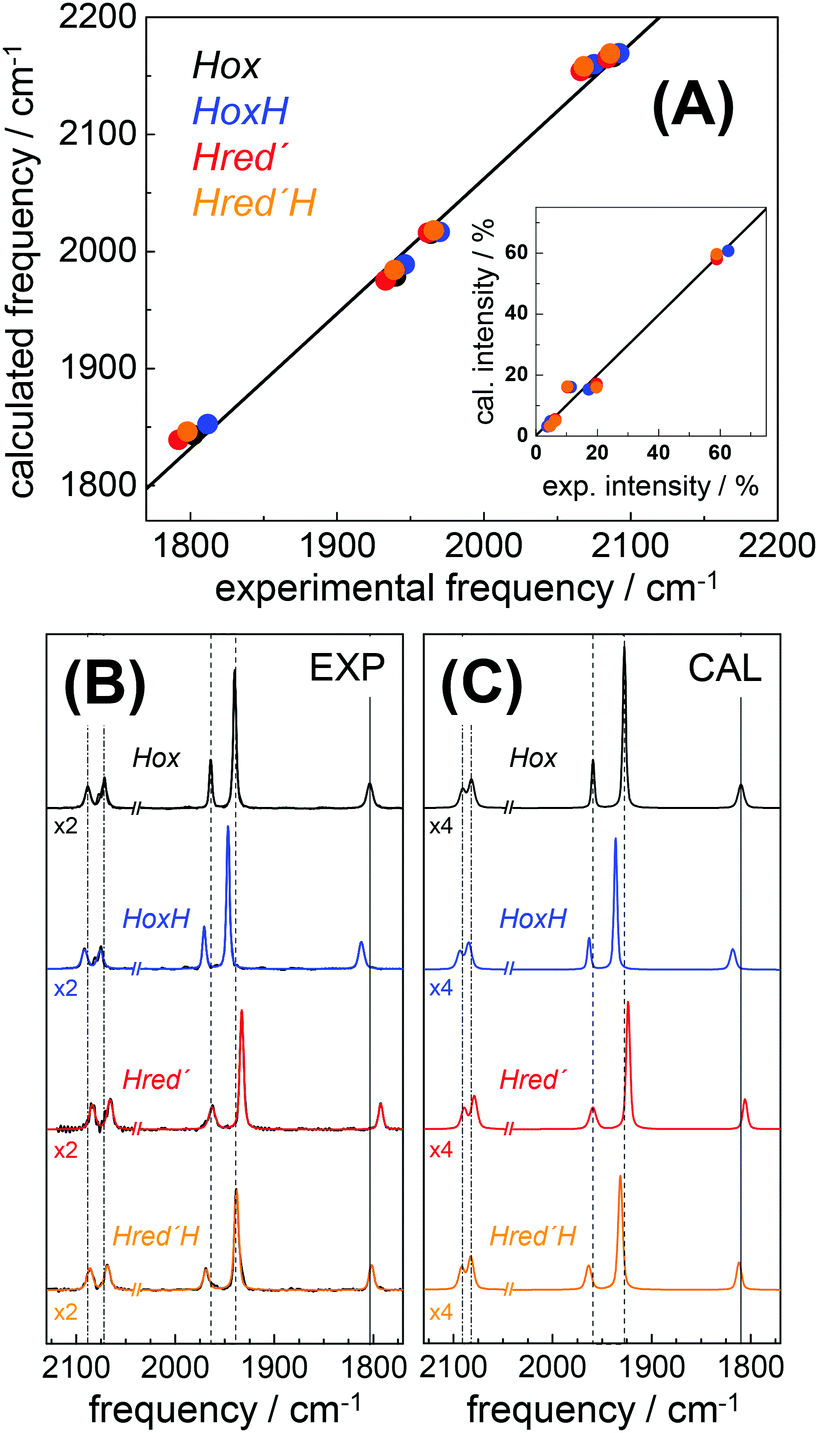
DFT calculations on H-cluster model structures (Fig. S12, ESI†) were used to explore conceivable protonation sites. Respective positions included N(adt), the μS atoms in the diiron site (S1, S2) and in the [4Fe–4S] cluster (S3–S6), as well as the sulfur atoms of the three terminal cysteine ligands (S8–S10). Good agreement between the experimental and calculated CO/CN− band patterns of Hox was obtained, as judged on the basis of the small root-mean-square-deviations (rmsd) derived from experimental and calculated band frequencies (F) and intensities (I), i.e. rmsdF ∼ 8 cm−1 and rmsdI < 3% (Fig. 8).28 We note that relative band positions and intensities of all investigated H-cluster species were largely independent of the size of the model structure and geometry-optimization restraints that were used in the calculations (Fig. S13, ESI†). Protonation at N(adt) or S1/S2 at the diiron site caused frequency up-shifts three to four fold larger than experimentally observed for HoxH (compare Table 1). The large rmsdF values (15–25 cm−1) suggested that these groups are unlikely protonation sites (Fig. S14, ESI†). Protonation at the μS atoms of the [4Fe–4S] cluster caused band shifts in better agreement with the experiment (rmsdF 8–11 cm−1). However, protonation of S3 or S5 produced a downshift of the pCO band and intensity changes less compatible with the experimental observations. Furthermore, protonation at S4 or S6 caused considerable distortion of the [4Fe–4S] cluster (e.g. >0.5 Å elongation of the S4/S6-Fe bonds), which rendered μS atom protonation at the [4Fe–4S] cluster less likely. This interpretation is in line with previous theoretical results on [4Fe–4S] clusters.76 Protonation at S10 of a terminal cysteine ligand yielded a frequency down-shift of the pCO band not observed in the experiment. In contrast, good agreement was found for protonation at S8 or S9 (rmsdF ∼ 7 cm−1). Best overall agreement with the HoxH band pattern was observed for protonation at S9 (Fig. 8). Protonation at water molecules in the vicinity of the [4Fe–4S] cluster or at the bridging cysteine S7 caused significantly larger IR band shifts than protonation at S9 (Fig. S14, ESI†). Notably, deuteration at S9 caused negligible CO/CN− band shifts (<1 cm−1), in good agreement with the experimental data of HoxH in D2O buffer and HoxH-CO (Fig. S14 and S15, ESI†).
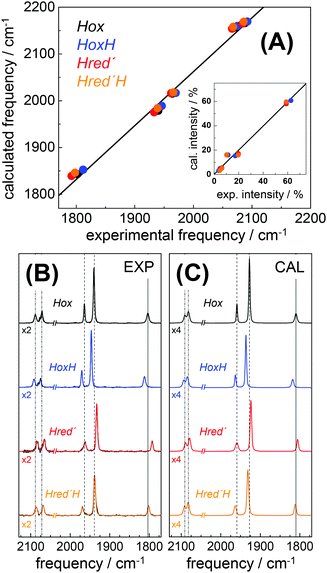 |
| | Fig. 8 Comparison of IR spectra from experiment and theory. (A) Plots of calculated (cal) vs. experimental (exp) IR frequencies and intensities (inset) for Hox, HoxH, Hred′, and Hred′H. (B) Experimental normalized IR spectra (black thin lines) and fit curves (smooth lines). (C) Calculated normalized IR spectra (from DFT at the TPSSh/TZVP level and after correction of band frequencies for systematic effects, eqn (1)–(3)). Dashed lines in (B) and (C) emphasize band shifts relative to Hox; CN− bands were scaled by factors of 2 (exp) or 4 (cal) for clarity. Experimental and calculated IR data of Hox-CO and HoxH-CO are shown in Fig. S15 (ESI†). | |
Infrared spectra were calculated for H-cluster structures of the one-electron reduced Hred′ state. A Hox-like structure with a μCO ligand and apical vacancy at the distal iron ion yielded a mean frequency down-shift of ∼35 cm−1 of the CO/CN− bands, far larger than experimentally observed (Fig. S16, ESI†). Excess frequency down-shifts (rmsdF > 30 cm−1) were also observed for protonation of the reduced H-cluster at N(adt) or S1/S2. Inclusion of an apical hydride at the distal iron ion produced excess frequency up-shifts (rmsdF ∼ 40 cm−1), in particular for the μCO ligand, resembling the μCO shift in Hhyd.37–40 Protonation at one of the sulfur atoms of the [4Fe–4S] cluster yielded small IR band down-shifts in good agreement with the experiment, but led to structural distortions, whereas protonation at a cysteine sulfur preserved the cubane geometry in Hred′. Best agreement between calculated and experimental IR frequencies of Hred′ was obtained for protonation at S9 (rmsdF ∼ 9 cm−1, see Fig. 8). These results showed that the IR spectrum of Hred′ is well compatible with a one-electron reduced H-cluster, which carries an additional proton and adopts a similar geometry to Hox. Structures for Hred′H were calculated comprising a second-site protonation. Similar trends with respect to structural changes and IR patterns in response to protonation at various sites as for HoxH and Hred′ were observed (Fig. S16, ESI†). Protonation at two cysteine S-atoms of the [4Fe–4S] cluster (i.e., S8 and S9) yielded IR frequencies in good agreement with the Hred′H spectrum (Fig. 8). Accordingly, Hred′H was attributed to a double-protonated H-cluster species with the same electron count and cofactor geometry as Hred′.
Good agreement between calculated and experimental IR band patterns for Hox/HoxH, and Hred′/Hred′H facilitated assignment of the IR bands of the CO/CN− ligands to individual vibrational modes. In addition, our simulations provided access to the spin and charge distribution of the cofactor (Fig. S17, ESI†). Protonation (i.e. at S9) and protonation/reduction affected the largely uncoupled behavior of the diatomic ligand vibrations only marginally. In the four states, the spin density distribution at the diiron site was similar, showing that the apparent mixed-valence Fe(I)Fe(II) configuration was preserved. The net spin thus resided largely on the diiron site in Hox/HoxH, in agreement with EPR data.46–48 For Hred′/Hred′H, the spin density implied that the surplus electron was localized mainly at the [4Fe–4S] cluster, as supported by the increased negative charge density on the [4Fe–4S] cluster. Anti-ferromagnetic coupling between the unpaired spins at the diiron site and at the reduced [4Fe–4S] cluster was expected to render these states EPR-silent.29 The minor charge variations at the iron ions of the diiron site correlate well with the small experimental IR band frequency differences of the CO ligands in the four states (Fig. S17, ESI†).
Protonation at the H-cluster
Real-time ATR FTIR spectroscopy on [FeFe]-hydrogenase protein films identified four states of the H-cluster that show similar CO/CN− band patterns but differ in the redox and/or protonation state (Hox/HoxH and Hred′/Hred′H). The oxidized state, Hox, has been characterized extensively. In agreement with crystallographic assignments4–7 and previous FTIR studies,21–28 DFT favored a μCO ligand and an apical vacancy at the distal iron ion in Hox.77 Our calculations suggest similar cofactor structures in all four species. Furthermore, both Hox and HoxH can be assigned to a mix-valence diiron site with an oxidized [4Fe–4S] cluster46–48 while in Hred′ and Hred′H a reduced [4Fe–4S] cluster was identified.29 The mixed-valence state clearly distinguishes Hred′ from Hred and Hsred (see Table 1). While Hox represents the unprotonated H-cluster, protonation at the diiron site has been suggested for Hred and Hsred.31–34,38 We found that a satisfactory calculation of the Hred′ band pattern also requires addition of a proton. This view is supported by the experimentally observed pH-dependence of Hred′ formation in both HYDA1pdt and HYDA1adt. However, protonation of the diiron site or at surrounding amino acid residues cannot explain the observed IR band shifts. Our computational results favor a terminal cysteine ligand of the [4Fe–4S] cluster as a common protonation site in HoxH and Hred′/Hred′H. In HYDA1, cysteine 417 (S9) is the most likely candidate. Additional protonation occurs in Hred′H, presumably at C170 (S8). Iron–sulfur clusters are typically involved in electron transfer processes, but their participation in proton transfer has been documented as well.78–82 More specifically, protonation at a terminal cysteine ligand has been proposed to be involved in the catalytic mechanism of [NiFe]-hydrogenases.83–86
Formation of HoxH and Hred′/Hred′H was found to be independent of the proton pathway to the active site that includes C169, N(adt), and Fed. The crystal structure of the HYDA1 apo-protein87 suggests that the [4Fe–4S] cluster in HYDA1 is in direct contact with bulk water while in bacterial [FeFe]-hydrogenases, the H-cluster is buried deeper in the catalytic domain.4–7 However, a chain of water molecules extends from the surface close to the cysteine that corresponds to our favored protonation site at the [4Fe–4S] cluster (Fig. S18, ESI†). Two proton trajectories thus may be present in [FeFe]-hydrogenases: the ‘catalytic’ proton pathway that shuttles protons to the diiron site50–52 and a pathway that facilitates protonation at the [4Fe–4S] cluster.
Reaction sequence of electron and proton transfer
While the exponential HoxH → Hox conversion implies a direct deprotonation of the [4Fe–4S] cluster, the sigmoidal time course of HoxH formation suggests at least two successive reaction steps to be involved in the ‘blue shift’ reaction. HoxH was found to accumulate under H2 release conditions, that is, for increasing concentrations of the reductant (such as the one-electron donor sodium dithionite, DT). This places HoxH towards the end of a multi-step reaction cycle (Fig. S19, ESI†). Under conditions of maximal H2 release (e.g. pH 7–8, 200 mM DT as in Fig. S6, ESI†) the prevalence of Hred′ over Hred suggests Hred′ as an intermediate in the Hox → HoxH conversion. Hox dominates at alkaline pH so that we assume that deprotonation of HoxH is required to regain Hox. In contrast, the quantitative accumulation of HoxH suggests that this deprotonation was slow or impaired at low pH. Numerical simulations show that the above considerations can qualitatively reproduce the pH and DT dependence of HoxH formation (Fig. S20, ESI†). This model also consistently explains the formation of HoxH-CO due to reductive conversion of Hox-CO into Hred′, H2 release, and subsequent rebinding of CO to HoxH (Fig. S7, ESI†). It has been shown that reduction of the [4Fe–4S] cluster weakens the binding of external CO.13,29
Our data provide evidence that protonation at the same cysteine ligand of the [4Fe–4S] cluster occurred in both HoxH and Hred′. We assume that this protonation occurs from a putative donor group (BH+), such as a water molecule consistently observed in CPI and DDH structures (Fig. S18, ESI†).4–7 Deprotonation of HoxH likely involves the same molecule, now serving as a proton acceptor (B). Re-protonation of this group at acidic pH (i.e. via the adjacent chain of water molecules) would impair the deprotonation reaction so that HoxH accumulates (Fig. S19, ESI†). Hred′H is populated upon reduction and protonation of HoxH at low pH, but seems not to advance further in the reaction cycle, possibly because second-site protonation interferes with the H2 forming reactions. Further studies are required to clarify this point. Accumulation of Hred′ under H2 likely involves inter-molecular electron transfer between oxidized and (transiently formed) two-electron reduced [FeFe]-hydrogenase proteins (either in the Hsred or Hhyd states). We note that inter-molecular electron transfer seems to require a densely packed protein film and is unlikely to occur under physiological conditions.
Conclusions
In contrast to Hox and Hred′, Hred and Hsred carry an additional electron at the diiron site and lack the Fe–Fe bridging CO ligand.31–34,38 Respective ligand rotation into a terminal or “semi-bridging” position has been considered difficult to reconcile with the large hydrogen turnover rates of [FeFe]-hydrogenases.41–43 An involvement of Hred and Hsred in hydrogen turnover is certainly not excluded at present.31,38,60 However based on the differences in ligand orientation (bridging vs. terminal), these states can be considered unlikely intermediates in the reaction cycle of rapid H2 formation.34 In HYDA1adt, formation of Hred was suppressed at alkaline pH in favor of Hred′ while for acidic pH values Hred dominated over Hred′. Thus, the relative population of Hred and Hred′ in HYDA1adt critically depends on pH. This suggests that the rates of proton delivery either to the [4Fe–4S] cluster or to the diiron site may be decisive for the location of electrons at the H-cluster. In addition to the ‘catalytic’ proton pathway towards the diiron site,50–55 we propose a ‘regulatory’ proton pathway to the [4Fe–4S] cluster. Concomitant protonation and reduction at the [4Fe–4S] cluster in Hred′ stabilize the mixed-valence state and prevent ligand rotation and premature protonation at the diiron site. Whether this is a sequential or concerted process remains to be elucidated.88–90
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank J. Fontecilla-Camps (Institut de Biologie Structurale, Grenoble) for generously providing a sample of DDH protein and P. Chernev (Freie Universität Berlin) for kindly providing data analysis software. M. S. and S. T. S. thank the International Max Planck Research School (IMPRS) on Multiscale Biosystems and the Focus Area NanoScale (Freie Universität Berlin) for financial support. M. H. gratefully acknowledges funding by the Bundesministerium für Bildung und Forschung (Grant 05K14KE1). M. W. and T. H. thank the Volkswagen Foundation for funding (Grant LigH2t) and the Deutsche Forschungsgemeinschaft (DFG) for support within the Cluster of Excellence RESOLV (EXC 1069). U.-P. A. gratefully acknowledges financial support by the Fonds der Chemischen Industrie (Liebig Grant) and the DFG (Emmy Noether Grant AP242/2-1). J. D. thanks the China Scholarship Council (CSC) for support and L. K. acknowledges funding by the Fonds der Chemischen Industrie (Kekulé Mobility Fellowship).
References
- W. Lubitz, H. Ogata, O. Ru and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- P. M. Vignais and B. Billoud, Occurrence, classification, and biological function of hydrogenases: an overview, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- S. T. Stripp and T. Happe, How Algae Produce Hydrogen – News from the Photosynthetic Hydrogenase, Dalton Trans., 2009, 9960–9969 RSC.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, X-ray Crystal Structure of the Fe-Only Hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom Resolution, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- A. S. Pandey, T. V. Harris, L. J. Giles, J. W. Peters and R. K. Szilagyi, Dithiomethylether as a ligand in the hydrogenase H-cluster, J. Am. Chem. Soc., 2008, 130, 4533–4540 CrossRef CAS PubMed.
- J. Esselborn,
et al., A structural view of synthetic cofactor integration into [FeFe]-hydrogenases, Chem. Sci., 2016, 7, 959–968 RSC.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Investigating and exploiting the electrocatalytic properties of hydrogenases, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS PubMed.
- C. Greco,
et al., Combining experimental and theoretical methods to learn about the reactivity of gas-processing metalloenzymes, Energy Environ. Sci., 2014, 7, 3543–3573 CAS.
- F. A. Armstrong,
et al., Guiding Principles of Hydrogenase Catalysis Instigated and Clarified by Protein Film Electrochemistry, Acc. Chem. Res., 2016, 49, 884–892 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed.
- J. Cohen, K. Kim, P. King, M. Seibert and K. Schulten, Finding gas diffusion pathways in proteins: application to O2 and H2 transport in CpI [FeFe]-hydrogenase and the role of packing defects, Structure, 2005, 13, 1321–1329 CrossRef CAS PubMed.
- G. Goldet,
et al., Electrochemical kinetic investigations of the reactions of [FeFe]-hydrogenases with carbon monoxide and oxygen: comparing the importance of gas tunnels and active-site electronic/redox effects, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volbeda, Structure–function relationships of anaerobic gas-processing metalloenzymes, Nature, 2009, 460, 814–822 CrossRef CAS PubMed.
- C. Tard and C. J. Pickett, Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Mimicking hydrogenases: from biomimetics to artificial enzymes, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- F. Gloaguen and T. B. Rauchfuss, Small molecule mimics of hydrogenases: hydrides and redox, Chem. Soc. Rev., 2009, 38, 100–108 RSC.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz,
14N HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: evidence for a nitrogen in the dithiol bridge, Phys. Chem. Chem. Phys., 2009, 11, 6553–6554 RSC.
- G. Berggren,
et al., Biomimetic assembly and activation of [FeFe]-hydrogenases, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- J. Esselborn,
et al., Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- A. J. Pierik, M. Hulstein, W. R. Hagen and S. P. Albracht, A low-spin iron with CN and CO as intrinsic ligands forms the core of the active site in [Fe]-hydrogenases, Eur. J. Biochem., 1998, 258, 572–578 CAS.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. II. Redox properties, light sensitivity and CO-ligand exchange as observed by infrared spectroscopy, JBIC, J. Biol. Inorg. Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- D. W. Mulder,
et al., EPR and FTIR Analysis of the Mechanism of H2 Activation by [FeFe]-Hydrogenase HydA1 from Chlamydomonas reinhardtii, J. Am. Chem. Soc., 2013, 135, 6921–6929 CrossRef CAS PubMed.
- B. J. Lemon and J. W. Peters, Binding of Exogenously Added Carbon Monoxide at the Active Site of the Iron-Only Hydrogenase (CpI) from Clostridium pasteurianum, Biochemistry, 1999, 38, 12969–12973 CrossRef CAS PubMed.
- A. L. De Lacey, C. Stadler, C. Cavazza, E. C. Hatchikian and V. M. Fernandez, FTIR Characterization of the Active Site of the Fe-hydrogenase from Desulfovibrio desulfuricans, J. Am. Chem. Soc., 2000, 122, 11232–11233 CrossRef CAS.
- Y. Nicolet,
et al., Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from Desulfovibrio desulfuricans, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- Z. Chen,
et al., Infrared Studies of the CO-Inhibited Form of the Fe-Only Hydrogenase from Clostridium pasteurianum I: Examination of Its Light Sensitivity at Cryogenic Temperatures, Biochemistry, 2002, 41, 2036–2043 CrossRef CAS PubMed.
- M. Senger,
et al., Stepwise isotope editing of [FeFe]-hydrogenases exposes cofactor dynamics, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8454–8459 CrossRef CAS PubMed.
- A. Adamska-Venkatesh,
et al., New redox states observed in [FeFe] hydrogenases reveal redox coupling within the H-cluster, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS PubMed.
- C. Lambertz,
et al., Electronic and molecular structures of the active-site H-cluster in [FeFe]-hydrogenase determined by site-selective X-ray spectroscopy and quantum chemical calculations, Chem. Sci., 2014, 5, 1187–1203 RSC.
- C. Sommer,
et al., Proton Coupled Electronic Rearrangement within the H-Cluster as an Essential Step in the Catalytic Cycle of [FeFe] Hydrogenases, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- A. Adamska-Venkatesh,
et al., Identification and characterization of the ‘super-reduced’ state of the H-cluster in [FeFe] hydrogenase: a new building block for the catalytic cycle?, Angew. Chem., 2012, 51, 11458–11462 CrossRef PubMed.
- P. Chernev,
et al., Hydride Binding to the Active Site of –Hydrogenase, Inorg. Chem., 2014, 53, 12164–12177 CrossRef CAS PubMed.
- S. Mebs,
et al., Bridging Hydride at Reduced H-Cluster Species in [FeFe]-Hydrogenases Revealed by Infrared Spectroscopy, Isotope Editing, and Quantum Chemistry, J. Am. Chem. Soc., 2017, 139, 12157–12160 CrossRef CAS PubMed.
- B. E. Barton, M. T. Olsen and T. B. Rauchfuss, Aza- and oxadithiolates are probable proton relays in functional models for the [FeFe]-hydrogenases, J. Am. Chem. Soc., 2008, 130, 16834–16835 CrossRef CAS PubMed.
- W. Wang,
et al., Crystallographic characterization of a fully rotated, basic diiron dithiolate: model for the H(red) state?, Chemistry, 2013, 19, 15476–15479 CrossRef CAS PubMed.
- D. W. Mulder,
et al., Investigations on the Role of Proton-Coupled Electron Transfer in Hydrogen Activation by [FeFe]-hydrogenase, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS PubMed.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, Identification of a Catalytic Iron-Hydride at the H-Cluster of [FeFe]-Hydrogenase, J. Am. Chem. Soc., 2016, 139, 83–86 CrossRef PubMed.
- E. J. Reijerse,
et al., Direct Observation of an Iron-bound Terminal Hydride in [FeFe]-Hydrogenase by Nuclear Resonance Vibrational Spectroscopy, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed.
- M. Winkler,
et al., Accumulating the hydride state in the catalytic cycle of [FeFe]-Hydrogenases, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- V. Fourmond,
et al., The oxidative inactivation of FeFe hydrogenase reveals the flexibility of the H-cluster, Nat. Chem., 2014, 6, 336–342 CrossRef CAS PubMed.
- C. Greco, M. Bruschi, L. De Gioia and U. Ryde, A QM/MM Investigation of the Activation and Catalytic Mechanism of Fe-only Hydrogenases, Inorg. Chem., 2007, 46, 15–16 Search PubMed.
- G. Filippi, F. Arrigoni, L. Bertini, L. De Gioia and G. Zampella, DFT Dissection of the Reduction Step in H2 Catalytic Production by [FeFe]-Hydrogenase-Inspired Models: Can the Bridging Hydride Become More Reactive Than the Terminal Isomer?, Inorg.
Chem., 2015, 54, 9529–9542 CrossRef CAS PubMed.
- M. Bruschi, P. Fantucci and L. De Gioia, Density functional theory investigation of the active site of Fe-hydrogenases. systematic study of the effects of redox state and ligands hardness on structural and electronic properties of complexes related to the [2Fe](H) subcluster, Inorg. Chem., 2004, 43, 3733–3741 CrossRef CAS PubMed.
- A. K. Justice,
et al., Chelate control of diiron(I) dithiolates relevant to the [Fe-Fe]-hydrogenase active site, Inorg. Chem., 2007, 46, 1655–1664 CrossRef CAS PubMed.
- S. P. J. Albracht, W. Roseboom and E. C. Hatchikian, The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. I. Light sensitivity and magnetic hyperfine interactions as observed by electron paramagnetic resonance, JBIC, J. Biol. Inorg. Chem., 2006, 11, 88–101 CrossRef CAS PubMed.
- C. Kamp,
et al., Isolation and first EPR characterization of the [FeFe]-hydrogenases from green algae, Biochim. Biophys. Acta, 2008, 1777, 410–416 CrossRef CAS PubMed.
- B. Bennett, B. J. Lemon and J. W. Peters, Reversible carbon monoxide binding and inhibition at the active site of the Fe-only hydrogenase, Biochemistry, 2000, 39, 7455–7460 CrossRef CAS PubMed.
- S. Katz,
et al., Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution, Chem. Sci., 2016, 7, 6746–6752 RSC.
- P. Knörzer,
et al., Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef PubMed.
- S. Morra,
et al., Site Saturation Mutagenesis Demonstrates a Central Role for Cysteine 298 as Proton Donor to the Catalytic Site in CaHydA [FeFe]-Hydrogenase, PLoS One, 2012, 7, e48400 CAS.
- M. Winkler, J. Esselborn and T. Happe, Molecular basis of [FeFe]-hydrogenase function: an insight into the complex interplay between protein and catalytic cofactor, Biochim. Biophys. Acta, 2013, 1827, 974–985 CrossRef CAS PubMed.
- A. J. Cornish, K. Gärtner, H. Yang, J. W. Peters and E. L. Hegg, Mechanism of proton transfer in [FeFe]-Hydrogenase from Clostridium pasteurianum, J. Biol. Chem., 2011, 19, 38341–38347 CrossRef PubMed.
- B. Ginovska-Pangovska,
et al., Molecular dynamics study of the proposed proton transport pathways in [FeFe]-hydrogenase, Biochim. Biophys. Acta, 2014, 1837, 131–138 CrossRef CAS PubMed.
- A. J. Cornish,
et al., Single-Amino Acid Modifications Reveal Additional Controls on the Proton Pathway of [FeFe]-Hydrogenase, Biochemistry, 2016, 55, 3165–3173 CrossRef CAS PubMed.
- J. Noth,
et al., [FeFe]-Hydrogenase with Chalcogenide Substitutions at the H-Cluster Maintains Full H2 Evolution Activity, Angew. Chem., 2016, 55, 8396–8400 CrossRef CAS PubMed.
- E. C. Hatchikian, N. Forget, V. M. Fernandez, R. Williams and R. Cammack, Further characterization of the [Fe]-hydrogenase from Desulfovibrio desulfuricans ATCC 7757, Eur. J. Biochem., 1992, 209, 357–365 CrossRef CAS PubMed.
- M. Mirmohades,
et al., Following [FeFe] Hydrogenase Active Site Intermediates by Time-Resolved Mid-IR Spectroscopy, J. Phys. Chem. Lett., 2016, 7, 3290–3293 CrossRef CAS PubMed.
- B.-J. Schultz, H. Mohrmann, V. A. Lorenz-fonfria and J. Heberle, Protein dynamics observed by tunable mid-IR quantum cascade lasers across the time range from 10 ns to 1 s, Spectrochim. Acta, Part A, 2017 DOI:10.1016/j.saa.2017.01.010.
- B. L. Greene, G. J. Schut, M. W. W. Adams and R. B. Dyer, Pre-Steady-State Kinetics of Catalytic Intermediates of an [FeFe]-Hydrogenase, ACS Catal., 2017, 7, 2145–2150 CrossRef CAS.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. Becke, Density-Functional Exchange-Energy Approximation With Correct Asymptotic-Behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
-
M. J. Frisch, et al., Gaussian 09, 2009, DOI:10.1017/CBO9781107415324.004.
- G. von Abendroth,
et al., Optimized over-expression of [FeFe] hydrogenases with high specific activity in Clostridium acetobutylicum, Int. J. Hydrogen Energy, 2008, 33, 6076–6081 CrossRef CAS.
- J. F. Siebel,
et al., Hybrid [FeFe]-Hydrogenases with Modified Active Sites Show Remarkable Residual Enzymatic Activity, Biochemistry, 2015, 54, 1474–1483 CrossRef CAS PubMed.
- W. J. Lem and M. Wayman, Decomposition of aqueous dithionite. Part I. Kinetics of decomposition of aqueous sodium dithionite, Can. J. Chem., 1970, 48, 776–781 CrossRef CAS.
- M. Wayman and W. J. Lem, Decomposition of aqueous dithionite. Part II. A reaction mechanism for the decomposition of aqueous sodium dithionite, Can. J. Chem., 1970, 48, 782–787 CrossRef CAS.
- M. Birringer, S. Pilawa and L. Flohé, Trends in selenium biochemistry, Nat. Prod. Rep., 2002, 19, 693–718 RSC.
- H. J. Reich and R. J. Hondal, Why Nature Chose Selenium, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- H. Abul-Futouh, M. El-khateeb, H. Görls, K. J. Asali and W. Weigand, Selenium makes the difference: protonation of [FeFe]-hydrogenase mimics with diselenolato ligands, Dalton Trans., 2017, 46, 2937–2947 RSC.
- U. P. Apfel,
et al., Models for the active site in [FeFe] hydrogenase with iron-bound ligands derived from bis-, tris-, and tetrakis(mercaptomethyl)silanes, Inorg. Chem., 2010, 49, 10117–10132 CrossRef CAS PubMed.
- R. Zaffaroni, T. B. Rauchfuss, D. L. Gray, L. De Gioia and G. Zampella, Terminal vs. bridging hydrides of diiron dithiolates: protonation of Fe2(dithiolate)(CO)2(PMe3)4, J. Am. Chem. Soc., 2012, 134, 19260–19269 CrossRef CAS PubMed.
- Y.-C. Liu, K.-T. Chu, R.-L. Jhang, G.-H. Lee and M.-H. Chiang, [FeFe] hydrogenase active site modeling: a key intermediate bearing a thiolate proton and Fe hydride, Chem. Commun., 2013, 49, 4743–4745 RSC.
- V. A. Lórenz-Fonfría, V. Muders, R. Schlesinger and J. Heberle, Changes in the hydrogen-bonding strength of internal water molecules and cysteine residues in the conductive state of channelrhodopsin-1, J. Chem. Phys., 2014, 141, 22D507 CrossRef PubMed.
- A. Alwaaly and R. A. Henderson, Unexpected explanation for the enigmatic acid-catalysed reactivity of [Fe4S4X4]2-clusters, Chem. Commun., 2014, 50, 4799–4802 RSC.
- L. Yu,
et al., Targeting intermediates of [FeFe]-hydrogenase by CO and CN vibrational signatures, Inorg. Chem., 2011, 50, 3888–3900 CrossRef CAS PubMed.
- H. Beinert, R. H. Holm and E. Münck, Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Structure, function, and formation of biological iron–sulfur clusters, Annu. Rev. Biochem., 2005, 74, 247–281 CrossRef CAS PubMed.
- C. T. Saouma, W. D. Morris, J. W. Darcy and J. M. Mayer, Protonation and Proton-Coupled Electron Transfer at S-Ligated [4Fe–4S] Clusters, Chemistry, 2015, 21, 9256–9260 CrossRef CAS PubMed.
- R. A. Henderson, Mechanistic Studies on Synthetic Fe–S-based Clusters and Their Relevance to the Action of Nitrogenases, Chem. Rev., 2005, 105, 2365–2437 CrossRef CAS PubMed.
- R. A. Henderson, Proton transfer to synthetic Fe–S-based clusters, Coord. Chem. Rev., 2005, 249, 1841–1856 CrossRef CAS.
- C. Fichtner, M. van Gastel and W. Lubitz, Wavelength dependence of the photo-induced conversion of the Ni-C to the Ni-L redox state in the [NiFe] hydrogenase of Desulfovibrio vulgaris Miyazaki F, Phys. Chem. Chem. Phys., 2003, 5, 5507–5513 RSC.
- M. Kampa, M. E. Pandelia, W. Lubitz, M. Van Gastel and F. Neese, A metal–metal bond in the light-induced state of [NiFe] hydrogenases with relevance to hydrogen evolution, J. Am. Chem. Soc., 2013, 135, 3915–3925 CrossRef CAS PubMed.
- E. Siebert,
et al., Resonance Raman Spectroscopy as a Tool to Monitor the Active Site of Hydrogenases, Angew. Chem., 2013, 52, 5162–5165 CrossRef CAS PubMed.
- B. L. Greene, C. Wu, G. E. Vansuch, M. W. W. Adams and R. B. Dyer, Proton Inventory and Dynamics in the Ni-S to Ni-C Transition of a [NiFe] Hydrogenase, Biochemistry, 2016, 55, 1813–1825 CrossRef CAS PubMed.
- D. W. Mulder,
et al., Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydA(ΔEFG), Nature, 2010, 465, 248–251 CrossRef CAS PubMed.
- S. Y. Reece and D. G. Nocera, Proton-coupled electron transfer in biology: results from synergistic studies in natural and model systems, Annu. Rev. Biochem., 2009, 78, 673–699 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Proton-coupled electron flow in protein redox machines, Chem. Rev., 2010, 110, 7024–7039 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Proton-Coupled Electron Transfer: Moving Together and Charging Forward, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp04757f |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Stefan
Mebs
a,
Stefan
Mebs